Изобретение относится к некоторым замещенным пуриновым арабинозидам и их приемлемым с физиологической точки зрения производным, в частности сложным эфирам и их использованию с целью лечения некоторых ДНK-вирусных заболеваний.
Вирус Варицелла-герпес зостер (ВВЗ), который вызывает сифилис у цыпленка и опоясывающий лишай, является ДНК-вирусом семейства герпес. Ветряная оспа (сифилис у цыпленка) является основным заболеванием, вызываемым ВВЗ у хозяина без иммунной реакции; в общем случае оно является неострым заболеванием маленьких детей, которое проявляется в виде лихорадки и часоточной сыпи. Герпес зостер (опоясывающий лишай) является рецидивной формой заболевания, имеющего места у взрослых, которые ранее подвергались инфицированию вирусом Варицелла-зостер. Клинические симптомы этой инфекции характеризуются невралгией и везикулярной кожной сыпью, которая имеет одностороннее и кожное распределение. Распространение воспаления может привести к параличу или судорогам и кроме, если в последующем развивается менингит.
У пациентов с иммунодефицитом этот вирус может распространяться, вызывая серьезные, часто фатальные заболевания. Причиной иммунодефицита могут быть лекарственные препараты, используемые при лечении пациентов с трансплантантом или при лечении злокачественных неоплазм, или заболевания такие, как СПИД, которые разрушают иммунную систему, в результате чего пациент становится уязвимым для инфекций, которые в других случаях не являлись бы столь опасными.
Цитомегаловирус (ЦМБ) является еще одним вирусом семейства герпес. Инфекция может быть приобретена в детстве или в молодости и в плоде, причем внутриматочное заражение является видимо, самой распространенной формой заражения, но до 90% врожденных инфекций являются асимптоматичными при родах. Предварительное заражение матери в течение беременности рассматривается в общем случае в качестве наибольшего риска для неродившегося ребенка, в то время, как повторная активация инфекций плода является в общем случае клинически редкой. Клинические эффекты простираются от смерти и тяжелого заболевания (микроцефалия, увеличение печени или селезенки, желтуха, задержка умственного развития) через прекращение заболевания к его быстрому развитию, восприимчивости к легочным и ушным инфекциям, до отсутствия очевидных эффектов заболевания. У молодых людей инфекция может протекать незамеченной или проявляться в виде лихорадки, вызванной воспалением желез, подобно заболеванию, полученному в результате близкого физического контакта.
Серьезные инфекции могут также иметь место в результате реактивации "дремлющего" вируса у пациентов с неэффективной иммунной системой, как это описано для ВВЗ-инфекций. Такие инфекции приводят к более высокой заболеваемости и смертности от воспаления оболочки глаза, воспаления легких и кишечно-желудочных расстройств.
Было установлено, что некоторые пурин-арабино нуклеозиды, описанные подробно ниже, отличающиеся присутствием групп, замещенных в 2- и 6-позициях пуринового кольца, обладают высокой активностью против вирусных инфекций человека, в частности, тех, которые вызваны вирусом Варицелла зостер (ВВЗ) или цитомегаловирусом (ЦМВ).
Некоторые замещенные пурин-арабино нуклеозиды, в частности 9-β -D-арабинофуранозил-6-метокси-9Н-пури, 9-β -D-арабинофуранозил-6-пирролидино-9Н-пурин, 9-β -D-арабинофуранозил-6-метиламино-9Н-пурин и 9-β -D-арабинофуранозил-6-диметиламино-9Н-пурин, которые описаны ниже, при их применении для лечения ВВЗ и ЦМВ-инфекций описаны ранее в J. Org. Chem. том 27, 3274-9 (1962), Caner Jreatment Rep. 60(10), 1567-84 (1976), Tetrahedron 40(4), 709-13, (1984), Canada J. Biochem 43(1), 1-15 (1965), J. Med. Chem. 12, 498-504, (1969), J. Biol. Chem. 251(13), 4055-61 (1976), Ann. N.J. Acad.Sci, 284, 81-90, (1977), в Европейском патенте 002192, патентах США 3 666 856, 4 371 613, 3758684.
Таким образом, в соответствии с изобретением предлагается способ получения соединения формулы I: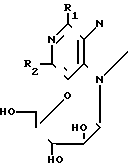 (I) где R1 представляет галоген, метокси, этокси, пропокси; амино-группу, монозамещенную метилом или С3-С6-циклоалкилом, или двузамещеную С1-5-алкилом; или R1 означает азотсодержащее гетероциклическое кольцо, содержащее 4-7 атомов углерода и связанное с пуриновым радикалом через атом азота; R2 представляет водород, галоген или амино при условии, что когда R2 означает водород, R1 не означает метокси, метиламино, диметиламино, пиперидино или пирролидино; а когда R2 означает аминогруппу, R1 не означает метиламино и его приемлемые с физиологической точки зрения производные для лечения или профилактики вирусных инфекций у человека, вызванных ВВЗ или ЦМВ.
(I) где R1 представляет галоген, метокси, этокси, пропокси; амино-группу, монозамещенную метилом или С3-С6-циклоалкилом, или двузамещеную С1-5-алкилом; или R1 означает азотсодержащее гетероциклическое кольцо, содержащее 4-7 атомов углерода и связанное с пуриновым радикалом через атом азота; R2 представляет водород, галоген или амино при условии, что когда R2 означает водород, R1 не означает метокси, метиламино, диметиламино, пиперидино или пирролидино; а когда R2 означает аминогруппу, R1 не означает метиламино и его приемлемые с физиологической точки зрения производные для лечения или профилактики вирусных инфекций у человека, вызванных ВВЗ или ЦМВ.
Предпочтительными соединениями формулы (I) являются соединения, в которых:
(a) R2 является водородом, и
(b) R1 является С1-5-алкокси, особенно метокси для фармацевтически приемлемых производных или
(с) R1 является С1-5-алкиламино, особенно диметиламино для фармацевтически приемлемых производных или
(d) R1 является галогеном, особенно йодом.
Следующие соединения являются предпочтительными соединениями в соответствии с изобретением ввиду их сильной противовирусной активности против ВВЗ или ЦМВ:
1) 9-β -D-арабинофуранозил-6-этокси-9Н-пурин,
2) 9-β -D-арабинофуранозил-6-йод-9Н-пурин,
3) 9-β -D-арабинофуранозил-2-амино-6-йодпурин,
4) 9-β -D-арабинофуранозил-2-хлор-6-метиламино-9Н-пурин,
5) 9-β -D-арабинофуранозил-6-циклопропиламино-9Н-пурин,
6) 9-β -D-арабинофуранозил-6-этилметиламино-9Н-пурин,
7) 9-β -D-арабинофуранозил-2-амино-6-метокси-9Н-пурин,
8) 9-β -D-арабинофуранозил-6-н-пропокси-9Н-пурин.
Приемлемые с физиологической точки зрения производные соединения общей формулы (I), отличные от 2' 3', 5' -триацетатных и трибензиловых производных соединений формулы (I), в которой R1 является хлором или фтором R2 является хлором, фтором, водородом или амино, используются в медицинской терапии, в частности для лечения вирусных инфекций человека, вызванных ВВЗ или ЦМВ.
Приемлемыми с фармацевтической точки зрения производными соединений формулы (I) являются любые приемлемые с фармацевтической точки зрения простые эфиры, соли, сложные эфиры или соли таких сложных эфиров, или любое другое соединение, котороое после применения к человеку способно дать (непосредственно или косвенно) соединение формулы (I) или его противовирусный метаболит или остаток.
Приемлемые с фармацевтической точки зрения сложные эфиры упомянутых соединений формулы (I) являются особенно предпочтительными, так как они способны обеспечить высокие концентрации основного соединения в плазме пациента после стоматического применения.
Пуриновые нуклеозиды формулы (I) и их производные в дальнейшем именуются как соединения, являющиеся предметом изобретения, или активные ингредиенты.
В соответствии с еще одним, предпочтительным, аспектом изобретения предлагается использование соединения, являющегося предметом изобретения, с целью получения медицинского препарата для лечения или профилактики вирусных инфекций человека, вызванных ВВЗ или ЦМВ.
Кроме того, в соответствии с изобретением предлагается способ лечения или профилактики ВВЗ и ЦМВ инфекций у человека, который содержит применение к упомянутому человеку эффективного количества соединения, являющегося предметом изобретения.
Способ включает ингибирование репликации ВВЗ или ЦМВ-вирусов в клетке-хозяине млекопитающих, который содержит применение эффективного количества для ингибирования репликации вируса соединения формулы (I) или его приемлемого с фармацевтической точки зрения производного к инфицированным клеткам.
Примеры клинических заболеваний, вызванных ВВЗ и ЦМВ инфекциями, которые могут быть обработаны в соответствии с изобретением, включают перечисленные заболевания.
Соединения формулы (I) и их приемлемые с фармацевтической точки зрения производные (в дальнейшем именуемые вместе как активные ингредиенты) могут быть применены любым способом, соответствующим заболеваниям, к таким способам относятся стоматический, прямокишечный способы, через нос, местный способ (включая буккальный и под язык), влагалищный и парентеральный (включая подкожный, внутримышечный, внутривенный, внутрикожный и эпидуральный) способы. Предпочтительный способ может варьироваться в зависимости, например, от состояния больного.
Для каждого из указанных использований и указаний количество требующегося активного ингредиента (как он определен выше) будет зависеть от различных факторов, включающих серьезность заболевания, подлежащего лечению, и совместимость с реципиентом, и будет в большей степени зависеть от решения лечащего врача. В общем случае для каждого из таких применений и указаний соответствующая эффективная доза будет изменяться в области от 0,1 до 250 мг на 1 кг массы тела реципиента в день, в предпочтительном варианте в области от 0,1 до 100 мг на 1 кг массы тела в день, а в наиболее предпочтительном варианте в области от 1 до 20 мг на 1 кг массы тела в день; оптимальная доза составляет примерно 15 мг на 1 кг массы тела в день (если не оговорено противное, все веса активного ингредиента рассчитаны для основного соединения формулы (I): для его солей и сложных эфиров эти числа должны быть увеличены пропорционально). Искомую дозу в предпочтительном варианте разбивают на две, три, четыре, или более поддоз, которые применяют через соответствующие промежутки времени на протяжении дня. Такие поддозы могут быть применены в формах единичных доз, например, содержащих от 5 до 1000 мг, в предпочтительном варианте от 20 до 500 мг, а в наиболее предпочтительном варианте от 100 до 400 мг активного ингредиента на форму единичной дозы.
Несмотря на то, что можно применять только активные ингредиенты, в предпочтительном варианте их применяют в фармацевтических формах, формы (композиции) в соответствии с изобретением содержат, по крайней мере, один активный ингредиент, как он определен выше, вместе с одним или несколькими приемлемыми носителями его, а также, возможно, другие терапевтические ингредиенты. Носитель (носители) должен быть "приемлемым" в том смысле, что он должен быть совместимым с другими ингредиентами композиции и не должен оказывать неблагоприятное воздействие на реципиента.
К упомянутым формам относятся формы, пригодные для стоматического, прямокишечного применения, применения через нос, местного (включая буккальное и под щеку) применения, влагалищного или парентерального (включая подкожное, внутримышечное, внутрикожое и эпидуральное) применения. Такие формы могут быть изготовлены в форме единичной дозы и могут быть получены любым из хорошо известных приемов в области фармацевтики. К таким приемам относится стадия приведения в контакт активного ингредиента с носителем, который включает один или несколько вспомогательных ингредиентов. В общем случае такие формы получают при помощи равномерного и тщательного перемешивания активного ингредиента с жидкими носителями или тонко измельченными твердыми носителями, или с тем и другим, а затем придают смеси необходимую форму.
Формы, в соответствии с изобретением, для стоматического применения могут иметь вид дискретных единиц, таких как капсулы, пилюли или таблетки, каждая из которых содержит заранее определенное количество активного ингредиента; как порошок или гранулы; как раствор или суспензия в водной жидкости или неводной жидкости; или как жидкая эмульсия типа масло в воде или жидкая эмульсия типа вода в масле. Активному ингредиенту может быть также придана форма шарика, лекарственной кашки или пасты.
Таблетка может быть получена при помощи прессования или формовки, возможно с использованием одного или нескольких вспомогательных ингредиентов. Прессованные таблетки могут быть получены при помощи прессования, с использованием соответствующего устройства, активного ингредиента в свободно текущей форме, такой как порошок или гранулы, может быть смешанного со связывающим агентом (например, повидоном, желатином, оксипропилметилцеллюлозой), смазывающим материалом, инертным разбавителем, консервирующим агентом, разрыхляющим агентом (например, гликоллат крахмал натрием, сшитым повидоном, сшитой натрийкарбоксиметилцеллюлозой), поверхностно- активным или диспергирующим агентом. Формованные таблетки могут быть изготовлены при помощи формования в соответствующем устройстве смеси порошкообразного соединения, смоченного инертным жидким разбавителем. Таблетки могут быть покрыты оболочкой или снабжены бороздкой, и им может быть придана такая форма, которая обеспечивала бы медленное или контролируемое высвобождение активного ингредиента, используя, например, оксипропилметилцеллюлозу в варьируемых пропорциях с тем, чтобы обеспечить необходимый профиль высвобождения.
При инфицировании глаза или других внешних тканей, например рта и кожи, композиции в предпочтительном варианте применяют в виде местных мазей или кремов, содержащих активный ингредиент в количестве, например, от 0,075 до 20 в/в в предпочтительном варианте от 0,2 до 15 в/в а в наиболее предпочтительном варианте от 0,5 до 10 в/в Если упомянутой формой является мазь, активные ингредиенты могут быть использованы вместе либо с парафиновой, либо со смешивающейся с водой основой мази. В качестве альтернативы активные ингредиенты могут быть использованы для изготовления крема на основе масла в воде.
Если это необходимо, водная фаза крема может включать, по крайней мере, 30 в/в многоатомного спирта, а именно спирта, содержащего две и более гидроксильные группы, такого как пропилен гликоль, бутан-1,3-диол, маннит, сорбит, глицерин и полиэтилен гликоль, и их смеси. Местные формы могут при желании содержать соединение, которое увеличивает поглощение или проникновение активного ингредиента через кожу или другие пораженные области. Примеры таких агентов, ускоряющих проникновение через кожу, включают диметилсульфоокись и связанные с ней аналоги.
Масляная фаза эмульсий в соответствии с изобретением может быть сформована из известных ингредиентов при помощи известного приема. Несмотря на то, что эта фаза может содержать только эмульгатор (известный также под другим названием эмульгент), в предпочтительном варианте она содержит смесь, по крайней мере, одного эмульгатора с жиром или маслом или с тем и другим. В предпочтительном варианте гидрофильный эмульгатор включается вместе с линофильным эмульгатором, который действует как стабилизатор. В предпочтительном варианте она также включает вместе масло и жир. Вместе эмульгатор (эмульгаторы) со стабилизатором (стабилизаторами) или без него образуют так называемый эмульгирующий воск, и этот воск вместе с маслом и/или жиром образует так называемую эмульгирующую основу мази, которая образует диспергированную в масле фазу крема.
Эмульгаторы и стабилизаторы эмульсии, пригодные для использования в форме в соответствии с изобретением, включают Твин 60, Спэн 80, кетостеариловый спирт, миристиловый спирт, моно-стеарат глицерила и лаурил-сульфат натрия.
Выбор соответствующих масел или жиров для композиции основан на достижении необходимых косметических свойств, так как растворимость активного соединения в большинстве масел, подобных тем, что используются в формах фармацевтических эмульсий, очень низка. Так крем в предпочтительном варианте должен быть нежирным, не оставляющим пятен и смываемым продуктом, имеющим соответствующую консистенцию, которая позволяла бы избежать вытекания из труб или других контейнеров. Можно использовать одно- или двухосновные алкиловые сложные эфиры с линейной или разветвленной цепью такие, как диизоадипат, изоцетил стеарат, пропилен гликолевый сложный диэфир жирных кислот кокосового ореха, изопропил миристат, децил олеат, изопропил палминат, бутил стеарат, 2-этилгексил палминат или смесь сложных эфиров с разветвленной цепью, известная как Кродамол КАП, причем последние три являются предпочтительными сложными эфирами. Они могут быть использованы отдельно или в комбинаци, в зависимости от необходимых свойств. В качестве альтернативы можно использовать липиды с высокой температурой точки плавления, такие как белый мягкий парафин и/или жидкий парафин, или другие минеральные масла.
Композиции, пригодные для местного применения к глазам, включают также глазные капли, в которых активный ингредиент растворен или суспендирован в соответствующем носителе, в частности водном растворителе для активного ингредиента. Активный ингредиент в предпочтительном варианте содержится в таких формах в концентрации от 0,5 до 20% в предпочтительном варианте от 0,5 до 10% например, в концентрации примерно 1,5 в/в
Композиции, пригодные для местного применения в ротовой полости, включают лепешки, содержащие активный ингредиент в приятной на вкус основе, в общем случае в сахаре и акации или трагаканте; пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин, или сахар и акация; и мекстуру для ротовой полости, содержащую активный ингредиент в соответствующем жидком носителе.
Композиции для прямокишечного применения могут иметь форму суппозиториев на подходящей основе, содержащей, например, кокосовое масло или салицилат.
Композиции, предназначенные для применения через нос, в которых носитель является твердым веществом, включают крупный порошок, имеющий размер частиц, например, в области от 20 до 500 мкм, который применяют при помощи приема, содержащего контейнер, а именно при помощи быстрого вдыхания через носовую полость из контейнера с порошком, который придерживают вблизи носа. К подходящим композициям, в которых носителем является жидкость, для применения, например, в форме аэрозоля для носа или капель для носа, относятся водные или масляные растворы активного ингредиента.
Формы, предназначенные для влагалищного применения, могут иметь вид пессариев, тампонов, кремов, гелей, паст, пен или аэрозолей, содержащих наряду с активным ингредиентом носители, применение которых в этой области техники хорошо известно.
Формы, предназначенные для парентерального применения, включают водные и неводные стерильные растворы для инъекций, которые могут включать антиоксиданты, буферные материалы, бактериостаты и растворенные вещества, которые придают форме изотонные свойства относительно крови предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загущающие агенты. Эти формы могут иметь вид единичной дозы или нескольких доз, например, герметически закрытые ампулы и пузырьки, и могут храниться в высушенных замораживанием (лиофилизованных) условиях, требующих добавления только стерильного жидкого носителя, например воды для инъекций непосредственно перед использованием. Неподготовленные растворы и суспензии для инъекций могут быть получены из стерильных порошков, гранул и таблеток указанного типа.
Предпочтительными формами единичной дозы являются формы, содержащие ежедненую дозу или единичную ежедневную поддозу, как она была определена выше, или соответствующие ее доли активного ингредиента.
Наряду с перечисленными ингредиентами формы, являющиеся предметом изобретения, могут включать другие агенты, применение которых в зависимости от типа формы хорошо известно, например, агенты предназначенные для стоматического применения, могут включать придающие приятный вкус агенты.
В соответствии с изобретением предлагается способ получения соединения формулы (I) или его приемлемого с фармацевтической точки зрения производного, в частности его сложного эфира, заключающийся во взаимодействии соединения формулы (II)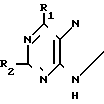 (II) в которой R1 и R2 уже были определены, с соединением формулы (III):
(II) в которой R1 и R2 уже были определены, с соединением формулы (III):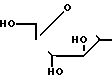 X (III) в которой Х представляет пиримидиновое или пуриновое основание (отличное от соединения формулы II), и, возможно, последующее или одновременное с этим в том случае, когда полученным в результате соединением является соединение формулы (I), превращение его в его приемлемое с фармацевтической точки зрения производное, или в том случае, когда полученный в результате соединением является приемлемое с фармацевтической точки зрения производное, превращение его в другое, приемлемое с фармацевтической точки зрения, производное соединение формулы (I).
X (III) в которой Х представляет пиримидиновое или пуриновое основание (отличное от соединения формулы II), и, возможно, последующее или одновременное с этим в том случае, когда полученным в результате соединением является соединение формулы (I), превращение его в его приемлемое с фармацевтической точки зрения производное, или в том случае, когда полученный в результате соединением является приемлемое с фармацевтической точки зрения производное, превращение его в другое, приемлемое с фармацевтической точки зрения, производное соединение формулы (I).
Что касается данного способа, то Х в предпочтительном варианте является урациловым основанием. Эта реакция может быть осуществлена, например, при помощи обработки соединений формул (II) и (III) ферментом, таким как фермент фосфорилаза, например уридин фоосфорилаза или пурин нуклеозид фосфорилаза, или их смесью в предпочтительном варианте в присутствии фосфатной соли при рН в области 5,0-9,0 и температуре в области от 15 до 90оС, в предпочтительном варианте в области от 40 до 60оС.
Приемлемые с физиологической точки зрения сложные эфиры и соли соединений формулы (I) могут быть получены при помощи известных приемов, например, сложные эфиры могут быть получены при помощи этерификации основного соединения с использованием подходящего ацилгалида или ангидрида. В качестве альтернативы сложные эфиры могут быть получены при помощи замещения соответствующей замещаемой группы, например галида подходящей карбоновой кислоты или при помощи разрыва соответствующего ангидронуклеозида основного соединения при помощи подходящей карбоновой кислоты или ее соли.
Приводимые ниже примеры служат иллюстрацией изобретения, поэтому их не следует рассматривать в качестве ограничения изобретения.
П р и м е р 1. 9-β -D-Арабинофуранозил-6-метиламино-9Н-пурин.
6-Тиол-9-β -D-арабинофуранозил-9Н-пурин (Рейст Е. Дж. и др. J. Org. Chem. 27, (1962), стр. 3274-3279) (0,35ммоль, 100 мг) и 5 мл абсолютного метанола соединяли и охлаждали до температуры -10оС, обеспечивая при этом защиту от влаги. В течение 2 мин через суспензию осторожно барботировали газообразный хлор. Полученный в результате раствор перемешивали в течение 5 мин при температуре 10оС, затем через холодный раствор в течение 15 мин барботировали сухой азот до тех пор, пока не будет удален избыточный хлор. В реакционную смесь добавляли 2 мл 40%-ного водного раствора метиламина, которую затем нагревали в автоклаве из нержавеющей стали при температуре 115оС, поддерживаемую в течение 4,5 ч. Реактор охлаждали до 0оС и содержимое выпаривали до сухого состояния, обеспечивая 88% выход соединения из заголовка примера. После рекристаллизации в воде проба имела температуру точки плавления 201,5-202,5оС.
Результаты анализа: рассчитано дляС11Н13N5O4
Рассчитано,С 47,0; Н 5,38; N 24,9.
Найдено, С 47,2; Н 5,72; N 25,2
П р и м е р 2. 9-β -D-Арабинофуранозил-6-диметиламинопурин
6-Диметиламинопурин (Сигма Кэмикэл Ко. Сент-Луис, МО) (6,4 ммоль, 1,04 г) и урацил арабинозид (Торренс, П.Ф. и др. J. Med. Chem. 22 (3), 1979, с. 316-319) (8 ммоль, 1,96 г) соединяли в 0,412 л 5 мМ раствора фосфата калия, рН 8,0, с 0,02% азида калия. Добавляли очищенную пурин нуклеозид фосфорилазу (3260 единиц) и уридин фосфорилазу (810 единиц), раствор перемешивали при температуре 35оС. Через 59 дней реакцию подвергали лиофилизации. Остаток суспендировали в 250 мл воды и перемешивали при комнатной температуре в течение 1 ч. Твердые материалы отделяли фильтрацией и фильтрат хранили при температуре 3оС.
Через 72 ч осадок собирали и соединяли с предыдущей лепешкой. Все это добавляли в 100 мл 95%-ного этанола в воде, нагревали до кипения и фильтровали. Растворитель удаляли под вакуумом, а остаток подвергали хроматографии на смоле типа БИОРад П-2 в 30% н-пропанол (вода o/o), один раз используя колонну с размерами 7,5 х 90 см и еще два раза колонну 5 х 90 см. В результате этой процедуры получали 0,12 г 6-диметиламинопурин-9-β -D-арабинофуранозида в форме 0,5 гидрата.
Результаты анализа: рассчитано для С12Н17N5O4 ˙0,5 Н20.
Рассчитано, С 47,36; Н 5,96; N 23,01
Найдено, С 47,23; Н 5,59; N 22,75
ЯМР- и масс-спектры согласуются с такой структурой.
П р и м е р 3. 9-β -D-Арабинофуранозил-6-метоксипурин.
6-Метоксипурин (фирма Сигма Кэмикэл Ко. Сент-Луис, МО), (6,6 ммоль, 1 г) и урацил арабинозид (Торренс, П. Ф. и др. J. Med. Chem. 22 (3), 1979, (10,1 ммоль, 2,45 г) суспендировали в 575 мл 10мМ фосфата калия, 0,04% растворе азида калия, рН 7,8, содержащем 10% н-пропанола (o/o). Добавляли очищенную уридин фосфорилазу (560 м. е.) и пурин нуклеозид фосфорилазу (10000 м.е.) Кренитцкий Т.Э. и др. Biochemistry, 20, стр. 3615, 1981 и патент США N 4 381 444 и раствор перемешивали при температуре 35оС. Тридцать дней спустя реакцию фильтровали. Фильтрат подщелачивали до рН 10,5 при помощи гидрата окиси аммония и подвергали хроматографии на колонне 2,5 х 7 см, содержащей смолу Доуекс-1-формиат. Смолу элюировали 30% н-пропанол/вода (o/o).
Фракции, содержащие продукт, соединяли, а растворитель удаляли под вакуумом. Смолу растворяли в 30% н-пропанол/вода (o/o) и подвергали хроматографии на колонне типа Био-Рад П-2 (7,5 х 90 см). Содержащие продукт фракции соединяли и после лиофилизации получали 0,922 г 6-метоксипурин-9-β -D-арабинофуранозида в виде дигидрата.
Результаты анализа: рассчитано для С11Н14N4O5 ˙2Н2О
Рассчитано, С 41,51; Н 5,70; N 17,70
Найдено, С 41,46; Н 5,74; N 18,13
ЯМР- и масс-спектры согласуются с такой структурой.
П р и м е р 4. 9-β -D-Арабинофуранозил-6-этоксипурин.
6-Этоксипурин (фирма Сигма Кэмикэл Ко. Сент-Луис, МО) (3,05 ммоль, 0,5 г) и урацил арабинозид (6,09 ммоль, 1,48 г) суспендировали в 100 мл 10 мМ фосфата калия, 0,04% растворе азида калия при рН 7,4. Добавляли очищенную уридин фосфорилазу (6000 м. е.) и пурин нуклеозид фосфорилазу (8400 м.е.) Кренитцкий, Т. Э. и др. Biochemistry, 20, стр. 3615, 1981 и патент США N 4 381 444 и суспензию перемешивали при температуре 35оС.
Через 168 ч добавляли еще 18000 единиц уридин фосфорилазы и 75 600 единиц пурин нуклеозид фосфорилазы. Через семь дней реакцию фильтровали, а фильтрат подвергали хроматографии на колонне, содержащей смолу Доуекс-1-гидроксид (2,5 х 8 см). Колонну элюировали смесью 90% метанол/вода (o/o), фракции, содержащие продукт, соединяли, а растворитель удаляли под вакуумом. Остаток растворяли в 30%-ном н-пропаноле и воде (o/o) и подвергали хроматографии на колонне, содержащей Био Рад П-2 (5 х 90 см). Продукт элюировали смесью 30% н-пропанол/вода (о/о). Фракции, содержащие продукт, соединяли, а растворитель удаляли под вакуумом, в результате чего получали 0,363 г 6-этоксипурин-9-β -D-арабинофуранозида, который идентифицировали как 0,3 гидрат.
Результаты анализа: рассчитано для С12Н16N4O5 ˙0,3 H2O
Рассчитано, С 47,78; Н 5,55; N 18,57
Найдено, С 47,99; Н 5,54; N 18,40.
ЯМР- и масс-спектры согласуются с этой структурой.
П р и м е р 5. 9-β -D-Арабинофуранозил-6-йодпурин.
6-Йодпурин (фирма Сигма Кэмикэл Ко. Сент-Луис, МО) (4 ммоль, 1 г) растворяли в 15 мл 1,2-диметоксиэтана при нагревании. Добавляли 50 мл раствора урацил арабинозида (10,1 ммоль) в 10 мМ фосфата калия, 0,04% раствор азида калия, рН 7,4. Добавляли очищенную уридин фосфорилазу (6800 м.е.) и пурин нуклеозид фосфорилазу (12 000 м.е.) и реакцию перемешивали при температуре 35оС. Через 21 день добавляли еще 4800 единиц уридин фосфорилазы и 20 000 единиц пурин нуклеозид фосфорилазы. Через девяносто дней реакцию фильтровали, а растворитель удаляли под вакуумом. Остаток растворяли в 100 мл воды, нагревали паром, а затем фильтровали. Фильтрат подвергали хроматографии на колонне, содержащей смолу КСАД-2 (5 х 35 см). Эту колонну элюировали 2 л воды, а азтем 2 л этанола. Фракции, содержащие продукт, соединяли. а растворитель удаляли под вакуумом. Остаток растворяли в смеси 30% н-пропанол/вода (о/о) и подвергали хроматографии на колонне, содержащей Био-Рад П-2 (5 х 90 см).
Продукт элюировали смесью 30% н-пропанол/вода (о/о). Фракции, содержащие продукт, соединяли, а растворитель удаляли под вакуумом. Остаток растворяли в 30% н-пропанол/вода (о/о) и подвергали хроматографии на колонне типа Сефадекс Г-10 (5 х 90 см). Эту колонну элюировали смесью 30% н-пропанол/вода (о/о). Фракции, содержащие продукт, соединяли и после удаления растворителя под вакуумом получали 0,253 г 6-йодпурин-9-β -D-арабинофуранозида, который идентифицировали как 1,5 гидрат.
Результаты анализа: рассчитано для С10Н11JN4О4 ˙1,5 H2O
Рассчитано, С 29,65; Н 3,48; N 13,83; J 31,32
Найдено, С 29,43; Н 3,53; N 13,66; J 31,20
ЯМР- и масс-спектр согласуются с такой структурой.
П р и м е р 6. 8-β -D-Арабинофуранозил-2-амино-6-йодпурин.
2-Амино-6-йодпурин (фирма Сигма Кэмикэл, Сент-Луис, МО) (25,5 ммоль, 6,75 г) и урацил арабинозид (61,9 ммоль, 15,1 г) соединяли в 0,31 л 10мМ фосфата калия, рН 6,9, с 0,02% азида калия. Добавляли очищенную пурин нуклеозид фосфорилазу (17 000 единиц) и уридин фосфорилазу (2000 единиц), и раствор перемешивали при температуре 37оС. Через 18 дней добавляли еще 5700 единиц уридин фосфорилазы. Через пятьдесят семь дней реакцию фильтровали, а фильтрат подвергали хроматографии на колонне, содержащей смолу КСАД-2 (8 х 11 см). Продукт элюировали последовательным градиентом этанол/вода (о/о) следующим образом: 0,35 л 10% 1 л 20% 1 л 50% 0,2 л 95% Фракции, содержащие продукт, соединяли, а этанол удаляли под вакуумом. Остаток растворяли в 30% н-пропанол/вода (о/о) и подвергали хроматографии на колонне (7,5 х 90 см), содержащей смолу Био-Рад П-2. В результате получали 1,1 г 2-амино-6-йодпурин-9-β -D-арабинофуранозида в виде 0,5 гидрата.
Результаты анализа: рассчитано для С10Н12N5О4 ˙0,5 Н2О
Рассчитано, C 29,87; Н 3,26; N 17.41
Найдено,С 29,86; Н 3,29; N 17,39.
ЯМР- и масс-спектр подтверждали эту структуру.
П р и м е р 7. 9-β -D-Арабинофуранозил-6-пирролидинопурин.
6-Пирролидинопурин (фирма Сигма Кэмикэл Ко. Сент-Луис, МО) (2,6 ммоль), 0,5 г и урацил арабинозид (5,29 ммоль, 1,29 г) суспендировали в 100 мл 10 мМ фосфата калия, 0,04% растворе азида калия при рН 7,4. Добавляли очищенную уридин фосфорилазу (6000 м. е.) и пурин нуклеозид фосфорилазу (8400 м.е.) Кренитцкий,Т.А. и др. Biochemistry, 20, стр. 3615, 1981 и патент США N 4 381 444), и суспензию перемешивали при температуре 35оС. Через двадцать дней реакцию фильтровали и фильтрат подвергали хроматографии на колонне, содержащей смолу Доуекс-1-гидроксид (2,5 х 8 см). Продукт элюировали из колонны при помощи смеси 90% метанол/вода (о/о). Фракции, содержащие продукт, соединяли, а растворитель удаляли под вакуумом. Остаток растворяли в 50 мл 30% н-пропанола и воды (о/о) и подвергали хроматографии на колонне типа Био-Рад П-2 (5 х 90 см). Продукт элюировали с использованием 30% н-пропанол/вода (о/о). Фракции, содержащие продукт, соединяли, а растворитель удаляли под вакуумом, в результате чего получали 0,573 г 6-пирролидинопурин-9-β -D-арабинофуранозида.
Результаты анализа: рассчитано для С14Н19N5O4.
Рассчитано, С 52,33; Н 5,96; N 21,79.
Найдено, 52,60; Н 6,09; N 21,51
ЯМР- и масс-спектры подтверждают структуру.
П р и м е р 8. 9-β -D-Арабинофуранозил-2-хлор-6-метиламинопурин.
Раствор 2,6-дихлор-2',3',5'-три-0-бензил-9-(β-D-арабинофуранозил)пурина (Келлер Ф. и др. J. Org. Chem. 32, стр. 1644, 1967, Монтгомери, Дж. Э. и Хьюсон, К. Дж. J. Med. Chem. 12, стр. 498, 1967 5,92 г, 10 ммоль) в бензоле (35 мл раствора 0,6 г метиламина на 10 мл бензола) выдерживали при комнатной температуре в герметически закрытом автоклаве в течение 4 дней. Реактор охлаждали льдом, открывали и содержимое фильтровали, чтобы удалить хлоргидрат метиламина. Растворитель удаляли под вакуумом, в результате чего получали масло, которое соединяли с маслом реакции с теми же количествами, но проведенной при температуре 125оС. Общий вес составлял 11,4 г. В результате тонкослойной хроматографии (ТСХ) устанавливали, что материал представляет собой смесь исходного материала, моно- и диметиламиносоединений. Масло подвергали хроматографии на 285 г силикагеля, используя 30% ацетона и 70% циклогексана (объемы). Компоненты, вытекающие ниже исходного материала (ТСХ, силикагель, 3 7, ацетон циклогексан), собирали, а растворитель удаляли под вакуумом. Выход: 4,8 г 2-хлор-6-метиламино- 2',3',5'-три-0-безил-9-(β -D-арабинофуранозил)пурина в виде масла. Порцию в 1,1 г этого материала в 40 мл 2-метоксиэтанола добавляли в хлорид палладия (0,87 г), который предварительно восстанавливали в аппарате Парра. Смесь подвергали гидрогенизации при давлении 50 фунтов/кв. дюйм (3,515 кг/см2) в течение 30 мин в атмосфере водорода, которую заменяли после первых 15 мин. Катализатор удаляли фильтрацией через слой Целита и промывали метанолом. Фильтрат нейтролизовали добавлением Доуекс-1 (НСО3). Смолу удаляли фильтрацией и промывали метанолом. Фильтрат выпаривали под вакуумом, а остаток растирали с хлороформом. Сырой продукт промывали горячей водой, растворяли в горячем метаноле, фильтровали, охлаждали и твердое вещество собирали. В результате кристаллизации из кипящей воды получали продукт в виде гидрата.
Выход: 44,5 мг; температура точки плавления: 224-225оС.
Результаты анализа: рассчитано для С11Н14N5О4Сl˙Н2О
Рассчитано, С 39,58; N 20,98; Н 4,84;
Найдено, С 39,27; N 20,83; Н 5,16.
П р и м е р 9. 9-β -D-Арабинофуранозил-6-циклопропиламинопурин.
6-Циклопропиламинопурин, полученный при помощи нуклеофильного замещения группы хлора на 6-хлорпурине (фирма Сигма Кэмикэл Ко. Сент-Луис, МО) с использованием циклопропиламина в ацетонитриле (2,85 ммоль, 0,5 г) и урацил арабинозид Торренс, П. Ф. и др. J. Med, Chem. 22 (3), 1979, стр. 316-319 (5,71 ммоль, 1,39 г суспендировали в 100 мл 10 мМ фосфата калия, 0,04% растворе азида калия с рН 7,4. Добаляли очищенную уридин фосфорилазу (6000 м.е.) и пурин нуклеозид фосфорилазу (8 400 м.е.) Кренитцкий и др. Biochemistry, 20, стр. 3615, 1981 и патент США N 4 381 444 и суспензию перемешивали при температуре 35оС. Через 120 ч реакцию фильтровали и фильтрат подвергали хроматографии на колонне, содержащей смолу Доуекс-1-гидроксид (2,5 х 10 см). Колонну элюировали смесью 90% метанол/вода (о/о). Фракции, содержащие продукт, соединяли, а растворитель удаляли под вакуумом. Остаток растворяли в 30% н-пропаноле и воде (о/о), и подвергали хроматографии на Био-Рад П-2 (5 х 90 см). Колонну элюировали 30% н-пропанол/вода (о/о). Осадок из реакции повергали рекристаллизации из горячего метанола, в результате чего получали 0,0352 г соединения, которое идентифицировали как моногидрат 6-циклопропиламинопурин-9-β -D-арабинофуранозида. Фильтрат после рекристаллизации подвергали хроматографии на смоле Био-Рад П-2 (5 х 90 см), как это было описано. Фракции, содержащие продукт, из обеих колонн соединяли, а растворитель удаляли под вакуумом, в результате чего получали 0,342 г 6-циклопропиламинопурин-9- β -D-арабинофуранозида, который имел форму 0,8 гидрата с 0,3 С3Н8О.
Результаты анализа: рассчитано для С13Н17N5О4 ˙1 Н2О
Рассчитано, С 48,00; Н 5,89; N 21,53.
Найдено, С 48,05; Н 5,89; N 21,55.
ЯМР- и масс-спектры подтверждают эту структуру.
П р и м е р 10. 9-β -D-Арабинофуранозил-6-этил(метил)аминопурин.
6-Этил(метил)аминопурин получали при помощи нуклеофильного замещения группы хлора на 6-хлорпурине (фирма Сигма Кэмикэл, Сент-Луис, МО) при помощи 6-этил(метил)амина в ацетонитриле. 6-Этил (метил)аминопурин (2,8 ммоль, 0,5 г) и урацил арабинозид (5,6ммоль, 1,38 г) суспендировали в 575 мл 10 мМ фосфата калия, 0,04% растворе азида калия, рН 7,4, содержащем 10% н-пропанола (о/о). Добавляли очищенную уридин фосфорилазу (6 000 м.е.) и пурин нуклеозид фосфорилазу (8 400 м.е.) (Кренитцкий и др. Biochemistry, 20, стр. 3615, 1981 и патент США N 4 381 444), и раствор перемешивали при температуре 37оС. Через девятнадцать дней реакцию фильтровали и фильтрат подвергали хроматографии на колонне 2.5 х 13см, содержащей смолу Доуекс-1-гидроксид. Смолу элюировали смесью 90% метанол/вода (о/о). Фракции, содержащие продукт, соединяли и растворитель удаляли под вакуумом. Остаток растворяли в 30% н-пропанол/вода (о/о) и подвергали хроматографии на колонне типа Био-Рад П-2 (7,5 х 90 см). Содержащие продукт фракции соединяли и после лиофилизации получали 0,680 г 6-этил(метил)аминопурин-9-β- D-арабинофуранозида.
Результаты анализa: рассчитано дляС13Н19N5О4
Рассчитано, С 50,48; Н 6,19; N 22,64.
Найдено, С 50,36; Н 6,25; N 22,52
ЯМР и масс-спектры согласуются с такой структурой.
П р и м е р 11. 9-β-D-Арабинофуранозил-2-амино-6-метоксипурин.
2-Амино-6-метоксипурин, полученный при помощи нуклеофильного замещения группы хлора на 2-амино-6-хлорпурине (фирма Сигма Кэмикэл, Сент-Луис, МО) при помощи метанола с гидридом натрия в тетрагидрофуране (6,4 ммоль, 1,05 г) соединяли с 35 мл раствора урацил арабинозида (7,04 ммоль 1,75 г) в 10 мМ фосфате калия и 7% н-пропаноле (o/o). рН обеспечивали на уровне 6,75. Добавляли очищенную пурин нуклеозид фосфорилазу (18 000 единиц) и уридин фосфорилазу (1020 единиц), и раствор инкубировали при температуре 37оС. Через 26 дней реакцию фильтровали и фильтрат подвергали хроматографии на колонне из смолы Доуекс-1-формиат (2 х 7 см) после доведения рН до 10,5 с использованием концентрированного гидрата окиси аммония. Колонну элюировали с использованием 7% смеси н-пропанол/вода (о/о) и фракции, содержащие продукт, соединяли, а растворитель удаляли под вакуумом. Остаток экстрагировали 25 мл воды и фильтрат отделяли от твердого материала центрифугированием. Верхний слой после выдерживания при окружающей температуре давал кристаллы 2-амино-6-метокси-9-β-D-арабинофуранозида, которые давали после сушки под вакуумом 0,327 г продукта.
Результаты анализа: рассчитано для С11Н15N5О5
Рассчитано, С 44,44; Н 5,09; N 23,56
Найдено, С 44,49; Н 5,13; N 23,52.
ЯМР- и масс-спектры подтверждали эту структуру.
П р и м е р 12. 9-β-D-Арабинофуранозил-6-н-пропоксипурин.
6-н-Пропоксипурин (5,6 ммоль, 1 г), (фирма Сигма Кэмикэл, Сент-Луис, МО), соединяли с 545 мл раствора урацил арабинозида (10,1 ммоль) в 10 мМ фосфата калия и 7% н-пропаноле (о/о). Добавляли очищенную уридин фосфорилазу (680 м. е. ) и пурин нуклеозид фосфорилазу (12 000 м.е.), и реакцию перемешивали при температуре 35оС. Реакцию фильтровали через 58 дней и фильтрат хранили при температуре 3оС в течение 20 ч. Полученный в результате осадок собирали центрифугированием, растворяли в 30% н-пропанол/вода (о/о) и подвергали хроматографии на колонне из Доуекс-1-формиата (2,5 х 5 см), после того как рН обеспечивали на уровне 10,5 с использованием концентрированного раствора гидрата окиси аммония.
Колонну элюировали смесью 30% н-пропанол/вода (о/о) и фракции, содержащие продукт, соединяли и растворитель удаляли под вакуумом. Остаток растворяли в н-пропаноле и подвергали хроматографии на колонне, содержащей Био-Рад П-2 (5 х 90 см). Колонну элюировали при помощи 30% н-пропанол/вода (о/о). Содержание продукт фракции соединяли и после удаления растворителя под вакуумом остаток растворяли в воде и подвергали хроматографии на колонне, содержащей Био-Рад П-2 (5 х 90 см). Колонну элюировали водой. Содержащие продукт фракции соединяли и после лиофилизации получали 0,758 г 6-н-пропоксипурин-9-β -D-арабинофуранозида в форме моногидрата.
Данные анализа: рассчитано для С13Н18N4О5 ˙1,0 Н2О
Рассчитано, С 47,56; Н 6,14; N 17,06
Найдено, С 47,63; Н 6,13; N 17,11.
ЯМР- и масс-спектры согласуются с этой структурой.
П р и м е р 13. 9-(5-0-Бензоил-β-D-арабинофуранозил)-6-метокси-9Н-пурин.
9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин примера 2 (0,283 г, 1,0 ммоль) растворяли в безводном диметилацетамиде (5,0мл), быстро охлаждали до 4оС и добавляли бензоил хлорид (0,155г, 1,1 ммоль). Смесь перемешивали в течение 24 ч в атмосфере аргона, затем давали возможность медленно нагреться до комнатной температуры. Затем при комнатной температуре добавляли еще 1,01 эквивалента бензоил хлорида и перемешивание продолжали еще в течение 24 ч. Реакцию быстро прерывали, сливая на 50 мл смеси лед-вода, экстрагировали с использованием СНСl3 (3 х 30 мл) и органические экстракты сушили (сульфат магния), Остаток после выпаривания подвергали очистке при помощи оперативной хроматографии на силикагеле (25,0 г, 2,5 х 15 см) последовательным градиентом от СНСl3 до СНСl3: ацетон 1:1. Фракции, содержащие продукт с Rf 0,21 (силикагель, СНСl3: ацетон 1:1) соединяли, в результате чего получали 92 мг целевого соединения: температура точки плавления: 202-204оС.
Результаты анализа: рассчитано для С18Н18N4О6
Рассчитано, С 55,96; Н 4,70; N 14,50
Найдено, С 56,04; Н 4,74; N 14,40.
ЯМР- и масс-спектр подтверждают приведенную структуру.
П р и м е р 14. 6-Метокси-9-[5-0-(-4-метилфенилсульфонил)-β-D-арабинофурано- зил]-9Н-пурин.
4-Толуолсульфонил хлорид (0,312 г, 1,63 ммоль) сразу же после рекристаллизации и 9-( β-D-арабинофуранозил)-6-метокси-9Н-пурин примера 3 (0,308 г, 1,09 ммоль) суспендировали в безводном ацетонитриле (25 мл), охлажденном до 3оС в ледяной ванне, и добавляли пиридин (5,0 мл), чтобы растворить нуклеозид. Раствор перемешивали в атмосфере аргона при температуре 3оС в течение 1 ч, затем выдерживали при температуре -15оС в течение 42 ч. Реакцию быстро прекращали добавлением 5% NaHCO3 (3 мл), выпаривали до приблизительно 10 мл, затем снова выпаривали с 95% этанолом. Остаток абсорбировали на минимальном количестве силикагеля и добавляли в колонну на силикагеле для оперативной хроматографии (25,0 г, 2,5 х 15 см) в СН2Cl2:MeOH (15:1). В результате элюирования таким растворителем (450 мл), затем CH2Cl2:MeOH (10:1, 550 мл) получали три УФ-абсорбирующих материала. Фракции, содержащие материал с Rf 0,42 (силикагель, СН2Cl2: MeOH 10: 1), собирали, а затем подвергали очистке на трех последовательных пластинках из силикагеля для препаративной хроматографии, первую с использованием СН2Сl2:MeOH (10:1) и ацетон: CH2Cl2 (1: 1) на следующих двух пластинках, чтобы получить 88 мг целевого материала в виде прозрачного стекла: температура точки плавления: 177-181оС.
Результаты анализа: рассчитано для С18Н20N4O7S˙ 0,15Н2О
Рассчитано, С 49,23; Н 4,66; N 12,76.
Найдено, C 49,28; H 4,71; N 12,71.
ЯМР- и масс-спектры согласуются с такой структурой.
П р и м е р 15. 6-Метокси-9-(5-0-метилсульфонил-β-D-арабинофуранозил)-9Н-пу- рин.
9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин примера 3 (0,600 г, 2,13 ммоль) суспендировали в сухом ацетонитриле (40 мл) и добавляли безводный пиридин (8 мл). Колбу охлаждали до -3оС в ванне лед-соль и добавляли метан сульфонил хлорид (0,16 мл, 2,13 ммоль). Через 25 мин раствор быстро охлаждали водой (3 мл) и концентрировали до объема 10 мл, затем несколько раз выпаривали с несколькими добавлениями этанола, выдерживая температуру в области ниже 38оС в течение всего времени. Остаточный пиридин удаляли вакуумным насосом. Остаток наносили на колонну из силикагеля для оперативной хроматографии (25,0 г, 2,5 х 15см), уравновешенную в СН2Cl2:MeOH (15:1). Эту колонну сначала элюировали 150 мл этого растворителя, а затем при помощи СН2Cl2:MeOH (10: 1, 450 мл). Фракции, содержащие материал с Rf 0,34 (силикагель, СН2Сl2: MeOH 10:1), давали 0,448 г продукта (57%) в виде белой пены: температура точки плавления: 177-181оС (разлож.).
Результаты анализа: рассчитано для С12Н16N4О7Si˙0,5 Н2О
Рассчитано, С 39,02; Н 4,64; N 15,17
Найдено, С 39,02; Н 4,66; N 15,08.
ЯМР- и масс-спектры подтверждают эту структуру.
П р и м е р 16. 9-[5-0-(4-Метилбензоил)- β -D-арабинофуранозил]-6-метокси-9Н-пурин.
9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин примера 3. (0,283 г, 1,0 ммоль) суспендировали в безводном ацетонитриле (10,0 мл), добавляли пиридин (2,3 мл), чтобы получить полное растворение, затем добавляли 4-метилбензоил хлорид (0,170 г, 1,1 ммоль). Смесь перемешивали при комнатной температуре в течение 24 ч в атмосфере аргона, реакцию быстро прекращали добавлением изопропанола (5 мл) и выпаривали до сухого состояния, а затем снова выпаривали с этанолом (2 х 10 мл).
Далее остаток подвергали очистке с использованием оперативной хроматографии на силикагеле (25,0 г, 2,5 х 15см) при помощи последовательного градиента от СНС3 до CHCl3:ацетон 1:1. Фракции, содержащие продукт с Rf 0,41 (силикагель, СНСl3:ацетон 1:1), соединяли, в результате чего получали 132 мг целевого соединения. Температура точки плавления: 127-128оС.
Результаты анализа: рассчитано для С19Н20N4O6 ˙0,5 Н2О
Рассчитано, С 55,74; Н 5,17; N 13,69
Найдено, С 55,48; Н 5,36; N 13,64.
ЯМР- и масс-спектры подтверждали вышеупомянутую структуру.
П р и м е р 17. 9-[5-0-(4-Хлорбензоил)- β-D-арабинофуранозил]-6-метокси-9Н- пурин.
9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин примера 3 (0,283 г, 1,0 ммоль) суспендировали в безводном ацетонитриле (10,0 мл), затем добавляли пиридин (2,3 мл), чтобы получить полное растворение, наконец, 4-хлорбензоил хлорид (0,193 г, 1,1 моль). Смесь перемешивают при комнатной температуре в течение 24 ч в атмосфере аргона, быстро прекращали реакцию добавлением изопропанола (5 мл) и выпаривали до сухого состояния, затем снова выпаривали с этанолом (2 х 10 мл). Остаток далее подвергали очистке при помощи оперативной хроматографии на силикагеле (25,0 г, 2,5 х 15 см) с использованием последовательного градиента от СНСl3 до СНСl3:ацетон (1:1). Фракции, содержащие продукт с Rf 0,37 (силикагель, СНСl3:ацетон 1:1), соединяли, в результате чего получали 105 мг целевого соединения: температура точки плавления: 122-124оС.
Результаты анализа: рассчитано для С18Н17CIN4O6 ˙0,5 Н2О
Рассчитано, С 50,30; Н 4,22; N 13,04.
Найдено, С 50,20; Н 4,28; N 12,94.
ЯМР- и масс-спектры подтверждали приведенную структуру.
П р и м е р 18. 9-[5-0-(4-Метоксибензоил)-β-D-арабинофуранозил]-6-метокси-9Н- пурин.
9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин примера 3 (0,283 г, 1,0 ммоль) суспендировали в безводном ацетонитриле (10,0мл), добавляли пиридин (2,3 мл), чтобы осуществить полное растворение, затем добавляли 4-метоксибензоилхлорид (1,1 ммоль). Смесь перемешивали при комнатной температуре в течение 24 ч в атмосфере аргона и реакцию быстро прекращали добавлением изопропанола (5 мл) и выпаривали до сухого состояния, затем снова выпаривали с этанолом (2 х 10 мл). Остаток затем подвергали очистке с использованием оперативной хроматографии на силикагеле (25,0 г, 2,5 х 15 см) с использованием последовательного градиента от СНСl3 до СHСl3:ацетон 1:1. Фракции, содержащие продукт с Rf 0,30 силикагель, СНСl3:ацетон 1:1 cоединяли, чтобы получить 110 мг целевого соединения: температура точки плавления: 195-197оС.
Результаты анализа: рассчитано дляС19Н20N4O7 ˙0,25 Н2О
Рассчитано, С 52,22; Н 4,91; N 13,31.
Найдено, С 54,22; Н 4,94; N 13,30.
ЯМР- и масс-спектры были согласованы с такой структурой.
П р и м е р 19. 5-Метокси-9-(5-0-фенилацетал-β -D-арабинофуранозил)-9Н-пурин.
9-(β-D-арабинофуранозил)-6-метокси- 9Н-пурин примера 3 (0,300 г, 1,06 ммоль) суспензировали в ацетонитриле (25 мл), затем добавляли безводный пиридин (5 мл). Раствор охлаждали до 3оС в ледяной ванне и добавляли финилацетил хлорид (0,20 мл, 1,5 ммоль). После перемешивания при температуре 3оС в течение 1 ч, раствор охлаждали до -15оС и выдерживали при этой температуре 40 ч. Реакцию быстро прекращали добавлением 5% NaHCO3 (3 мл), концентрировали до 10 мл, а затем выпаривали при добавлении несколько раз этанола. Остаток переносили в смесь СН2Сl2:MeOH (15:1) и наносили на колонну из силикагеля для оперативной хроматографии (25,0 г, 2,5x х 15 см), расположенную в том же растворителе, элюировали 500 мл этого же растворителя, затем смесью (15: 1) СН2Сl2:метанол (15: 1, 500 мл). Фракции с Rf 0,38 (силика, 10:1, СН2Сl2: метанол) давали 0,128 г сырого продукта с примесями, имеющими более высокие Rf. В результате последующей очистки при помощи препаративной тонкослойной хроматографии с использованием СН2Сl2:MeOH 10:1, а затем при помощи второй пластинки с использованием смеси ацетон:CH2Cl2 1:1 получали 0,102 г целевого продукта в виде белой пены: температура точки плавления: 74-75оС.
Результаты анализа: рассчитано для С19Н20N4О6 ˙0,1 Н2О
Рассчитано, С 56,74; Н 5,06; N 13,93.
Найдено, 56,74; Н 5,11; N 13,90.
ЯМР- и масс-спектры согласуются с упомянутой структурой.
П р и м е р 20. 6-Метокси-9-(5-0-фенилоксиацетил-β -D-арабинофуранозил)-9Н-пурин.
9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин примера 3 (0,322 г, 1,14 ммоль) суспендировали в сухом ацетонитриле (25 мл), затем добавляли безводный пиридин (5 мл). Раствор охлаждали до 3оС в ледяной ванне и добавляли феноксиацетил хлорид (0,24 мл, 1,7 ммоль). После перемешивания в течение 2 ч при температуре 3оС реакцию быстро прекращали 5%-ным раствором NaHCO3 (2 мл), концентрировали до объема 10 мл и выпаривали после нескольких добавлений этанола. Остаток подвергали обработке на оперативном хроматографе на силикагеле (25,0 г, 2,5 х 15 см), в смеси СН2Сl2:метанол 15:1). В результате элюирования 400 мл этого растворителя, затем смесью 10:1 CH2Cl2:метанол (500 мл) и 9:1 CH2Cl2:метанол (200 мл) получали 0,213 г сырого продукта. Этот материал подвергали рекристаллизации из метанола, после чего получали 0,101 г (20% ) белого кристаллического материала: температура точки плавления 193-195оС.
Результаты анализа: рассчитано для С19Н20N4O7 ˙0,05 Н2О
Рассчитано, С 54,69; Н 4,85; N 13,43.
Найдено, С 54,69; Н 4,89; N 13,40.
ЯМР- и масс-спектры подтверждают указанную структуру.
П р и м е р 21. 6-Метокси-9-(5-0-метоксиацетил-β -D-арабинофуранозил)-9Н-пирин.
9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин примера 3 (0,300 г, 1,06 ммоль) суспендировали в ацетонитриле (25мл), а затем добавляли безводный пиридин (5 мл). Раствор охлаждали до -3оС в ванне лед-соль и добавляли метоксиацетил хлорид (0,10мл, 1,1 ммоль). После перемешивания в ванне лед-соль в течение 2 ч реакцию прекращали 5% NaHCO3 (2 мл), объем концентрировали до 10 мл, а затем выпаривали после нескольких добавлений этанола. Остаток наносился на силикагель колонны для оперативной хроматографии (25,0 г, 2,5 х 15 см), содержащейся в 10:1 смеси СН2Сl2:МеОН. В результате элюирования 500 мл этого растворителя, затем 9:1 смесью СН2Сl2:MeOH (300 мл) получали 0,086 г сырого продукта. Этот материал подвергали рекристаллизации из МеОН и воды, после чего получали 0,035 г (9,3%) белых кристаллов: температура точки плавления: 137-139оС.
Результаты анализа: рассчитано для С14Н18N4O7 ˙0,5 Н2О
Рассчитано, С 46,28; Н 5,27; N 15,42.
Найдено, С 46,33; Н 5,27; N 15,37
ЯМР- и масс-спектры подтверждают указанную структуру.
П р и м е р 22. 9-[5-0-(4-Нитробензоил)- β-D-арабинофуранозил]-6-метокси-9Н- пурин.
9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин примера 3 (0,283 г, 1,0 ммоль) суспендировали в безводном ацетонитриле (10,0 мл), добавляли пиридин (2,3 мл), чтобы осуществить полное растворение, затем 4-нитробензоил хлорид (0,205 г, 1,1 ммоль). Смесь перемешивали при комнатной температуре в течение 24 ч в атмосфере аргона, реакцию быстро прекращали добавлением изопропанола (5 мл) и выпаривали до сухого состояния, затем еще два раза выпаривали с этанолом (2 х 10 мл). Остаток затем подвергали очистке с использованием оперативной хроматографии на силикагеле (25,0 г, 2,5 х 15 см) и последовательного градиента от СНСl3до CHCl3:ацетон 1:1). Фракции, содержащие продукт с Rf 0,37 (силикагель, СНСl3:ацетон 1:1) соединяли, в результате чего получали 105 мг целевого соединения: температура точки плавления 202-203оС.
Результаты анализа: рассчитано для С18Н17N4О8.
Рассчитано, С 50,12; Н 3,97; N 16,24.
Найдено, С 50,21; Н 4,02; N 16,16
ЯМР- и масс-спектры подтверждают указанную структуру.
П р и м е р 23. 6-Метокси-9-(5-0-пентаноил- β-D-арабинофуранозил)-9Н-пурин.
9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин примера 3 (0,852 г, 3,01 ммоль) суспендировали в сухом ацетонитриле (75 мл) и добавляли сухой пиридин (15 мл). Раствор охлаждали в ванне лед-вода и добавляли пентаноил хлорид (0,4 мл, 3,31 ммоль). Реакцию быстро прекращали спустя 2 ч добавлением 3 мл метанола и выпаривания до прозрачного, вязкого масла. Остаток подвергали очистке с использованием оперативной хроматографии на силикагеле и последовательного градиента от СНСl3 до СНСl3:MeOH 9:1. Продукт с выходом 330 мг содержал примеси соответствующего сложного 2'-эфира (см. пример 32) и неизвестный материал с более низким Rf. Затем остаток подвергали очистке при помощи обращенно-фазовой препаративной жидкостной хроматографии (высоко эффективной) (ВЭЖХ) (Аллтех С18, 10 х 25 мм, размер частиц 10 мкм 70% Н2О: 30% CH3CN, 4,0 мл/мин), приготавливая раствор 10 мг/мл в том же растворителе с объемом впрыскивания 1,0 мл. Остаток снова выпаривали с ацетоном, в результате чего получали 260 мг белой ломкой пены (24%): температура точки плавления: 85-95оС.
Результаты анализа для С16Н22N4O6x x0,05(СН3)2CО˙0,55 Н2О.
Рассчитано, С 51,16; Н 6,22; N 14,78.
Найдено, С 50,98; Н 6,03; N 14,63.
ЯМР- и масс-спектр подтверждают указанную структуру.
П р и м е р 24. 9-[5-0-(4-Аминобензоил- β-D-арабинофуранозил)]-6-метокси-9Н-пу- рин.
9-[5-0-(4-Нитробензоил)- β-D-арабинофуранозил]-6-метокси-9Н-пурин примера 22 (0,350 г, 0,81 ммоль) суспендировали в этаноле (100,0 мл) и добавляли 10% палладий на углероде (0,100 г). После попеременной откачки и заполнения системы водородом реакцию встряхивали на установке Парра под давлением 50 фунтов на кв.дюйм(3,515 кг/см2) в течение 3 ч. Затем смесь фильтровали через Целит и фильтрат выпаривали до сухого состояния. Остаток после выпаривания суспендировали в метаноле и фильтровали, чтобы получить целевое соединение в виде белого твердого вещества (0,285 г, 88%): температура точки плавления: 198%-200оС.
Результаты анализа для С18Н19N5O6x x0,3 H2O
Рассчитано, С 53,15; Н 4,86; N 17,22.
Найдено, С 52,96; Н 4,64; N 17,07
Масс- и ЯМР-спектры подтверждают указанную структуру.
П р и м е р 25. 6-Метокси-9-(5-0-пропионил-β -D-арабинофуранозил)-9Н-пурин.
9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин примера 3 (0,847 г, 3,0 ммоль) суспендировали в безводном ацетонитриле (30,0 мл) и добавляли пиридин (9 мл), чтобы осуществить полное растворение. После охлаждения до 5оС добавляли пропионил хлорид (0,305 г, 3,3 ммоль) и смесь перемешивали в течение ночи в атмосфере аргона при одновременном нагревании до 13оС. После прекращения реакции изопропанолом (5 мл) и выпаривания до сухого состояния, дополнительного выпаривания с этанолом (2 х 10 мл) остаток подвергали очистке с использованием оперативной хроматографии на силикагеле (25,0 г, 2,5 х 15 см) и последовательного градиента от СНCl3 до СНСl3:ацетон 1:1. Фракции, содержащие продукт с Rf 0,38 (силикагель, СНСl3:ацетон 1:1 соединяли и этот материал снова подвергали очистке с использованием хроматографии на силикагеле под давлением среды (тандем колонн, 1,5 х 25,0 см и 1,5 х x100,0 см; СНСl3: ацетон 3: 1), в результате чего получали 0,114 г целевого соединения в виде белого твердого вещества: температура точки плавления: 62-64оС.
Результаты анализа: рассчитано для С14Н18N4O6 ˙0,5 СН3ОН
Рассчитано, С 50,68; H 5,76; N 15,25.
Найдено, С 50,72; Н 5,80; N 15,28.
ЯМР- и масс-спектры подтверждают указанную структуру.
П р и м е р 26. 9-(5-0-Бутаноил-β-D-арабинофуранозил)-6-метокси-9Н-пурин.
9-(β-D-арабинофуранозил)-6-метокси- 9Н-пурин примера 3 (0,847 г, 3,0 ммоль) суспендировали в безводном ацетонитриле (30,0 мл) и добавляли пиридин (9,0 мл), чтобы осуществить полное растворение. После охлаждения до 5оС добавляли бутирил хлорид (0,352 г, 3,3 ммоль) и смесь перемешивали в течение ночи в атмосфере аргона при одновременном нагревании до 13оС. После прекращения реакции изопропанолом (5 мл) и выпаривания до сухого состояния, последующих выпариваний с этанолом (2 х 10 мл) остаток подвергали очистке с использованием оперативной хроматографии на силикагеле (25,0 г, 2,5 х 15 см) и последовательного градиента от СНСl3 до CHCl3:ацетон 1:1. Фракции, содержащие продукт с Rf 0,38 (силикагель, CHCl3:ацетон 1:1), соединяли. Этот материал подвергали очистке еще раз с использованием хроматографии при давлении среды на силикагеле (тандем колонн, 1,5 х 25,0 см и 1,5 х 100,0см; CHCl3: ацетон 2: 1), в результате чего получали 267мг целевого соединения в виде белого твердого вещества: температура точки плавления 108-110оС.
Результаты анализа для С15Н20N4O6
Рассчитано, С 51,13; Н 5,72; N 15,90
Найдено, С 51,21; Н 5,73; N 15,81.
ЯМР- и масс-спектр подтверждали указанную структуру.
П р и м е р 27. 9-[5-0-(2,2-Диметилпропионил)-β-D-арабинофуранозил]-6-меток- си-9Н-пурин.
9-[β-D-Арабинофуранозил] -6-метокси- 9Н-пурин примера 3 (0,850 г, 3,01 ммоль) суспендировали в сухом ацетонитриле (75 мл) и добавляли пиридин (15 мл). Раствор охлаждали до 3оС в ледяной ванне и добавляли 2,2-диметилпропионил хлорид (0,41 мл, 3,3 ммоль). После перемешивания при температуре 3оС в течение 6 ч реакцию быстро прекращали с использованием МеОН (2 мл), концентрировали до объема 10 мл, а затем несколько раз выпаривали после нескольких добавлений этанола. Остаток наносили на колонну для оперативной хроматографии на силикагеле (20,0 г, 2,5 х 12,0 см), помещенную в СН2Сl2:МеОН 10: 1. В результате элюирования с использованием 300 мл этого растворителя получали 0,464 г исходного материала и 0,553 г сырого продукта.
В результате еще одной очистки при помощи оперативной хроматографии (двуокись кремния, 2,5 х 10,0 см), элюируя градиентом от СН2Cl2 до МеОН в СН2Cl2 1: 9, получали 0,419 г (38%) прозрачного стекла: температура точки плавления: 73-76оС.
Результаты анализа для С16Н22N4O6x x0,5 Н2О
Рассчитано, С 51,19; Н 6,18; N 14,93.
Найдено, 51,43; Н 6,06; N 14,91
ЯМР- и масс-спектр согласуются с указанной структурой.
П р и м е р 28. 9-(5-0-Ацетил-β-D-арабинофуранозил)-6-метокси-9Н-пурин.
9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин примера 3 (0,850 г, 3,01 ммоль) суспендировали в сухом ацетонитриле (75 мл), затем добавляли безводный пиридин (15 мл). Раствор охлаждали до 3оС в ледяной ванне и добавляли ацетилхлорид(0,24 мл, 3,4 ммоль). После перемешивания в ванне лед соль в течение получаса реакцию прерывали метанолом (2 мл), концентрировали до объема 10 мл, затем выпаривали несколько раз после нескольких добавлений этанола. Остаток подвергали очистке с использованием оперативной хроматографии на силикагеле (25,0 г, 2,5 х 15 см), элюируя смесью СН2Сl2:MeOH (10:1, 300 мл), чтобы получить в результате 0,710 г сырого продукта. Вторая колонна (25,0 г, 2,5 х 15 см) после элюирования смесью СН2Cl2:MeOH (15:1) давала 0,610 г прозрачного стекла, которое все-таки содержало небольшое количество примесей с более высокими Rf. Последующая очистка продукта при помощи препаративной тонкослойной хроматографии с использованием СН2Сl2:MeOH 9:1, затем при помощи загрузки плоских слоев на колонну для оперативной хроматографии (20,0 г 2,5 х 12 см) в СН2Сl2 и элюируя градиентом МеОН в СН2Сl2получали целевой продукт в виде белой пены (0,308 г, 31%): температура точки плавления: 64-67оС.
Результаты анализа для C13H16N4O6 x x0,5 Н2О
Рассчитано, С 46,85; Н 5,14; N 16.81
Найдено, С 47,04; Н 5,12; N 16,72.
ЯМР- и масс-спектры согласуются со структурой.
П р и м е р 29. 6-Метокси-9-[5-0-(2-метилпропионил)-β-D-арабинофуранозил] 9Н-пурин.
9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин примера 3 (0,500 г, 1,77 ммоль) суспендировали в сухом ацетонитриле (25 мл), добавляли безводный пиридин (5 мл). Раствор охлаждали до 3оС в ледяной ванне и добавляли изобутирил хлорид (0,21 мл, 2,0 ммоль). После перемешивания при температуре 3оС в течение 3 ч реакцию быстро прекращали при помощи МеОН (2 мл), концентрировали до объема 10 мл, а затем выпаривали несколько раз после нескольких добавлений этанола. Остаток наносили на колонну для оперативной хроматографии на силикагеле (25,0 г, 2,5 х 15 см), помещенную в СН2Cl2:MeOH 10:1. После элюирования с использованием 300 мл этого растворителя получали 0,167 г исходного материала и 0,221 г сырого продукта. В результате последующей очистки продукта при помощи препаративной тонкослойной хроматографии с СН2Сl2:MeOH 9: 1, последующей загрузки плоских слоев на колонну для оперативной хроматографии на силикагеле (16,0 г, 2,5 х 10 см), помещенную в СН2Сl2. Колонну элюировали градиентом МеОН в СН2Сl2, в результате чего получали 0,127 г прозрачного стекла (20%): температура точки плавления 68-71оС.
Результаты анализа: рассчитано для С15Н20N4O6 ˙0,25 H2O 0,05 МеОН
Рассчитано, С 50,43; Н 5,82; N 15,63.
Найдено, С 50,49; Н 5,83; N 15,62.
ЯМР- и масс-спектры подтвердили указанную структуру.
П р и м е р 30. 6-Метокси-9-[2-0-(2,2- диметилпропионил)-β-D-арабинофуранозил] 9Н-пурин
9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин примера 3 (298,5 мг, 1,06 ммоль), 4-диметиламинопиридин (1 мг, 10 ммоль), ацетонитрил (20 мл) и пиридин (5 мл) добавляли в трехгорлую колбу с круглым дном, снабженную термометром, клапаном для подачи аргона, магнитной мешалкой, холодильником и рубашкой. Добавляли триметилуксусный ангидрид (0,22 мл, 1,06 ммоль) и раствор нагревали до температуры 40оС до 26 ч. Реакцию быстро прекращали в этот момент добавлением 2 мл воды и выпаривали до прозрачного масла. Остаток подвергали очистке при помощи оперативной хроматографии на силикагеле с использованием последовательности градиентов от СНСl3 до СНСl3 MeOH 9:1. Этот материал далее подвергали очистке с использованием препаративной тонкослойной хроматографии и СНСl3 MeOH 9:1, а затем выпаривали с треххлористым углеродом и ацетоном, в результате чего получали 90 мг белой хрупкой пены.
Результаты анализа: рассчитано для С16Н22N4O6 ˙0,05 (CH3)2CO ˙0,45 CCl4. Рассчитано, С 45,47; Н 5,13; N 12,78.
Найдено, С 45,67; Н 5,27; N 12,67
ЯМР- и масс-спектры подтверждают упомянутую структуру.
П р и м е р 31. 6-Метокси-9-[(2,3,5-три-0-ацетил)-β -D-арабинофуранозил] -9Н-пурин.
9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин примера 3 (1,009 г, 3,57 ммоль), 4-диметиламинопиридин (0,0098 г, 80 моль), триэтиламин (0,59 мл, 4,2 ммоль) и безводный ацетонитрил (52 мл) добавляли в трехгорлую колбу с круглым дном, снабженную термометром, входом для аргона, магнитной мешалкой и сухой ванной лед ацетон. После того как белый раствор охлаждали до температуры -20оС, добавляли уксусный ангидрид (2,4 мл, 25,4 ммоль). Раствор мгновенно становился прозрачным. Через 5 мин реакцию прекращали добавлением МеОН (5 мл) и выпаривали до прозрачного вязкого масла. В результате обработки с использованием оперативной хроматографии на силикагеле с градиентом СНСl3 и МеОН получали продукт в виде прозрачного вязкого масла. При помощи растворения этого материала в минимальном количестве ацетона, разбавления водой и лиофилизации выделяли 1,03 г (71%) продукта в виде белого порошка.
Результаты анализа для С17Н20N4O8
Рассчитано, С 50,00; Н 4,94; N 13,72
Найдено, С 49,80; Н 5,09; N 13,50
ЯМР- и масс-спектры подтверждали вышеуказанную структуру.
П р и м е р 32. 6-Метокси-9-(2-0-пентаноил- β-D-арабинофуранозил)-9Н-пурин.
Метод 1. 9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин примера 3 (283 мг, 1,0 ммоль), ацетонитрил (20 мл) и пиридин (5 мл) добавляли в трехгорлую колбу с круглым дном, снабженную термометром, клапаном для подачи аргона и магнитной мешалкой. Добавляли валерьяновый ангидрид (0,2 мл, 1,0 ммоль) и раствор перемешивали при температуре 20оС в течение 3 ч. Реакцию быстро прекращали водой (2 мл) и выпаривали до прозрачного масла, затем очищали с использованием оперативной хроматографии на двуокиси кремния и последовательности градиентов от СНСl3 до СНСl3:MeOH 9:1. В результате получали 110 мг сложного 2'-эфира с примесями сложного 5'-эфира (см. пример 23), материала с меньшим Rf. Этот материал далее подвергали очистке при помощи препаративной тонкослойной хроматографии на двуокиси кремния с СНСl3:MeOH 9:1 и совместного выпаривания с ацетоном, в результате чего получали 70 мг сложного 2'-эфира в виде прозрачного стекла.
Результаты анализа для С16Н22N4O6x x0,5 (CH3)2CO.
Рассчитано, C 53,16; Н 6,37; N 14,17.
Найдено, С 52,89; Н 6,37; N 13,96.
ЯМР- и масс-спектры согласуются с указанной структурой.
Метод 2. 5-Метокси-9-[3,5-0-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)-β-D-ара- бинофуранозил]-9Н-пурин примера 40 (2,5 г, 4,8 ммоль) добавляли в колбу с круглым дном емкостью 250 мл вместе с 4-диметиламинопиридином (0,06 г, 0,47 ммоль). Добавляли сухой ацетонитрил (30 мл) и триэтиламин (2,65 мл), и через воронку для непрерывной подачи загружали ацетонитрил (20 мл) и пентановый ангидрид (1,13 мл, 5,7 ммоль). Реакционную смесь охлаждали до 3оС в ледяной ванне в потоке аргона. Медленное добавление раствора пентанового ангидрида осуществляли в течение 2 ч. Затем реакционную смесь обрабатывали метанолом (10 мл) и концентрировали при пониженном давлении. Остаток переносили в СНСl3 (200 мл) и экстрагировали водой (2 х x50 мл). Соединенные водные слои снова экстрагировали СНСl3 (25 мл) и соединенные органические слои сушили (сульфат магния), фильтровали и концентрировали, в результате чего получали 3,44 г сырого продукта.
Сырой 6-метокси-9-[2-0-пентаноил-3,5-0-(1,1,3,3-тетраизопропилдисилоксан-1,3- диил)-β-D-арабинофуранозил]-9Н-пурин растворяли в тетрагидрофуране (120 мл) и воде (3 мл). Раствор охлаждали до 3оС в ледяной ванне и добавляли 1,0 М раствор фторида тетрабутиламмония в тетрагидрофуране (6,0 мл). Через 1,5 ч добавляли насыщенный раствор хлорида аммония (2 мл) и реакционную смесь непосредственно пропускали на очистительную колонну из силикагеля (5,0 х 8,0 см), находящуюся в хлорформе. Колонну элюировали хлороформом (200 мл), затем смесью 1: 1 ацетон:хлороформ (400 мл) и все фракции, содержащие продукт, соединяли и концентрировали. Финальную очистку осуществляли на колонне для оперативной хроматографии на силикагеле (5,0 х 15 см), элюировали последовательным градиентом ацетона в СНСl3, в результате чего получали 1,17 г (67%) прозрачного липкого стекла с примесями пентановой кислоты.
Результаты анализа для С16Н22N4O6 x x0,2 C5H10O2.
Рассчитано, С 52,79; Н 6,25; N 14,48
Найдено, С 52,97; Н 6,38; N 14,60.
П р и м е р 33. 9-(2-0-Бутаноил-β-D-арабинофуранозил)-6-метокси-9Н-пурин.
9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин примера 3 (283 мг, 1,0 ммоль), ацетонитрил (20 мл) и сухой пиридин (5 мл) добавляли в трехгорлую колбу с круглым дном, снабженную термометром, входом для аргона, магнитной мешалкой, и охлаждали до температуры 4оС. Добавляли масляный ангидрид (170 л, 1,0 ммоль) и раствор перемешивали при 5оС в течение 6 ч в атмосфере аргона. В этот момент реакцию прекращали добавлением 2 мл воды и выпаривали до прозрачного масла. Реакционную смесь подвергали очистке при помощи оперативной хроматографии с использованием последовательного градиента от СНСl3 до 1:1 СНСl3 ацетон. Этот материал далее подвергали очистке при помощи препаративной тонкослойной хроматографии на силикагеле с использованием смеси 9:1 CHCl3:МеОН, в результате чего получали 90 мг сложного 2'-эфира в виде прозрачного масла.
Результаты анализа для С15Н20N4О6x x0,3 Н2O.
Рассчитано, С 50,36; Н 5,80; N 15,66.
Найдено, 50,36; Н 5,81; N 15,66.
ЯМР- и масс-спектры соответствовали указанной структуре.
П р и м е р 34. 6-Метокси-9-[2-0-(2-метилпропионил)-β-D-арабинофуранозил]-9Н- пурин.
9-(β-D-арабинофуранозил)-6-метокси- 9Н-пурин примера 3 (290,0 мг, 1,03 ммоль), ацетонитрил (20 мл) и пиридин (5 мл) добавляли в трехгорлую колбу с круглым дном, снабженную термометром, входом для аргона, магнитной мешалкой, холодильником и рубашкой. Добавляли изомасляный ангидрид (170 л, 1,0 ммоль) и раствор нагревали рубашкой до температуры 30оС на 4 ч. Реакцию быстро прекращали в этот момент добавлением 2 мл воды и выпаривали до прозрачного масла. Остаток подвергали очистке при помощи оперативной хроматографии на силикагеле с использованием последовательности градиентов от СНСl3 до СНСl3 MeOH 9 1. Фракции, содержащие продукт с Rf 0,54 (силикагель, СНCl3 MeOH 20: 1), далее подвергали очистке при помощи препаративной тонкослойной хроматографии на силикагеле с СНСl3 MeOH 9:1. В результате получали 90,0 мг сложного 2'-эфира в виде прозрачного стекла.
Результаты анализа для С15Н20N4O6 x x0,1 С3Н6О
Рассчитано, С 51,31; Н 5,80; N 15,64.
Найдено, С 51,41; Н 5,81; N 15,72.
ЯМР- и масс-спектры подтверждали указанную структуру.
П р и м е р 35. 9-[3-0-Бензоил-β-D-арабинофуранозил(6-метокси-9Н-пурин-9)2,3-ангидро-β-D-лик софуранозил]-6-метокси- 9Н-пурин (получали по примеру 36) (0,264 г, 1,0 ммоль) растворяли в безводном этаноле (20,0 мл), нагревали до дефлегмации и добавляли бензоат аммония (0,209 г, 1,5 ммоль) в атмосфере аргона. Дополнительно 1,5 ммоль бензоата аммония добавляли через 24 ч и 35 ч. Реакцию выпаривали до сухого состояния через 48 ч при дефлегмации. Остаток после выпаривания подвергали очистке с использованием оперативной хроматографии на силикагеле (25,0 г, 2,5 х 15 см) и смеси СНСl3 ацетон 3:1. Фракции, содержащие продукт с Rf 0,52 (силикагель, СНСl3:ацетон 1:1) соединяли, в результате чего получали 138 мг целевого соединения. Температура точки плавления 180-182оС.
Результаты анализа, рассчитанные для С18Н18N4O6.
Рассчитано, С 55,96; Н 4,70; N 14,50
Найдено, С 55,90; Н 4,71; N 14,44.
ЯМР- и масс-спектры подтверждали упомянутую структуру.
П р и м е р 36. 9-(2,3-Ангидро-β-D-ликсофуранозил)-6-метоксипурин.
Трифенилфосфин (5,902 г, 22,5 ммоль) растворяли в 1,4-диоксане (138 мл) и нагревали до температуры 70оС. В раствор добавляли 9-(β-D-арабинофуранозил)- 6-метокси-9Н-пурин примера 3 (4,234 г, 15,0 ммоль), смесь перемешивали в течение 10 мин и по каплям в течение 10 мин добавляли диэтилазодикарбоксилат (3,919 г, 22,5 ммоль) в 1,4-диоксане (50 мл). Реакцию перемешивали при температуре 70оС в течение 1 ч, охлаждали до комнатной температуры и выпаривали до высоковязкого коричневого масла. Этот материал подвергали очистке на оперативной хроматографии на силикагеле (188,0 г, 5,0 х x21,0 см), элюируя смесью СНСl3 ацетон (5 1, 3,6 л) и СНСl3 ацетон (3:1, 2,0 л). Фракции, содержащие материалы с Rf 0,24 (двуокись кремния, СНСl3 ацетон, 1:1), соединяли и выпаривали до сухого состояния. Остаток очищали с использованием оперативной хроматографии на двуокиси кремния (250,0 г, 5,0 х 28,0 см), элюируя при помощи Е ОАс, в результате чего получали 2,452 г (62%) целевого соединения в виде белой пены: температура точки плавления 144-145,5оС.
Результаты анализа, рассчитанные для С11Н12N4О4 ˙0,25 C3H6 O˙0,05 CНСl3. Рассчитано, С 49,78; Н 4,80; N 19,68.
Найдено, С 49,80; Н 4,95; N 19,87.
ЯМР- и масс-спектры подтверждали указанную структуру.
П р и м е р 37. 6-Метокси-9-[2-0-(4-метоксибензоил)-β-D-арабинофуранозил] 9Н-пурин. 6-Метокси-9-[2-(4-метоксибензоил)-3,5-0-]1,1,3,3-тетраизопропилдисилоксан-1, 3- диил]-β-D-арабинофуранозил]-9Н-пурин (полученные по примеру 43) (0,86 г, 1,3 ммоль) растворяли в ТГФ (40 мл) и добавляли воду (1 мл). Раствор охлаждали до 3оС и добавляли 1,0 М раствор фторида тетрабутиламмония в ТГФ (6,3 мл). Через 30 мин добавляли дополнительно 5 мл воды, реакцию уменьшали на половину и пропускали непосредственно через колонну из силикагеля для оперативной хроматографии (25,0 г, 2,5 х 15 см), расположенную в СН2Сl2. Колонну элюировали СН2Сl2 (200 мл), затем смесью СН2Сl2:MeOH (20: 1, 400 мл) и все фракции, содержащие материал с Rf 0,20 (силика, СН2Сl2 MeOH 20:1), соединяли, в результате чего получали 0,570 г белой пены. Финальную очистку осуществляли на колонне для оперативной хроматографии на силикагеле (5,0 х 15 см), расположенной в СНСl3.
После элюирования последовательным градиентом ацетона в хлороформе получали 0,325 г (60%) продукта в виде белой пены: температура точки плавления 71-75оС.
Результаты анализа, рассчитанные для С19Н20N4O7 ˙0,3 CHCl3 ˙0,25 С3Н6О Рассчитано, С 51,60; Н 4,71; N 12,00.
Найдено, С 51,56; Н 4,70; N 11,95
ЯМР- и масс-спектры согласованы с указанной структурой.
П р и м е р 38. 6-Метокси-9-[(2-0-(4-метилбензоил)]-β -D-арабинофуранозил]-9Н-пурин.
6-Метокси-9-[2-(4-метилбензоил)-3,5-0- [ 1,1,3,3-тетраизопропил дисилоксан-1,3-диил)-β-D-арабинофуранозил] -9Н-пурин (получение по примеру 42) (0,85 г, 1,3 ммоль) растворяли в ТГФ (40 мл) и воде (1 мл). Раствор охлаждали до 3оС и добавляли 1,0 М раствор фторида тетрабутиламмония в ТГФ (5,0 мл). Через 30 мин добавляли дополнительно 5 мл воды, реакционный объем уменьшали на половину и пропускали непосредственно через колонну из силикагеля для оперативной хроматографии (20,0 г, 2,5x х 12 см), расположенную в СН2Сl2. Колонну элюировали с использованием СН2Cl2 (200 мл), затем смесью СН2Сl2 MeOH (15: 1, 400 мл) и все фракции, содержащие материал с Rf 0,20 (двуокись кремния, СН2Сl2:MeOH 20:1), соединяли.
Сырой продукт наносили на вторую колонну из силикагеля для оперативной хроматографии (25,0 г, 2,5 х 18 см), расположенную в смеси ацетон CH2Cl2 (1: 1). В результате элюирования тем же растворителем получали 0,633 г продукта, который затем подвергали очистке при помощи препаративной тонкослойной хроматографии (двуокись кремния, ацетон CH2Cl2 1:1) и оперативной хроматографии (25,0 г, 2,5 х 15 см). Колонну элюировали последовательностью градиентов ацетона в СНСl3, в результате чего получали 0,243 г (47%) прозрачного стеклa: температура точки плавления 69-73оС.
Результаты анализа, рассчитанные для С19Н20N4O6 ˙0,4 C3H6O ˙0,25 CHCl3 Рассчитано, C 54,17; H 5,03; N 12,36
Найдено, С 54,00; Н 5,08; N 12,30
ЯМР- и масс-спектр подтверждали указанную структуру.
П р и м е р 39. 9-[2-0-(4-Хлорбензоил)- β-D-арабинофуранозил]-6-метокси-9Н-пурин.
9-[2-4-Хлорбензоил-[3,5-0-(1,1,3,3-тетра- изопропилдисилоксан-1,3-диил)-β-D-ара- бинофуранозил] -6-метокси-9Н-пурин (получение по примеру 44) (1,07 г, 1,7 ммоль) растворяли в ТГФ (40 мл) и воде (1 мл). Раствор охлаждали до температуры 3оС и добавляли 1,0 М раствор фторида тетрабутиламмония в ТГФ (4,5 мл). Через 30 мин добавляли еще 5 мл воды и реакционную смесь пропускали непосредственно через колонну из силикагеля для оперативной хроматографии (20,0 г, 2,5 х 12 см), расположенную в СН2Cl2. Эту колонну элюировали СН2Cl2 (200 мл), затем смесью СН2Cl2:MeOH (20:1, 400 мл) и все фракции, содержащие материал с Rf 0,20) двуокись кремния, СН2Сl2:MeOH 20:1), соединяли. Сырой продукт наносили на колонну из силикагеля для оперативной хроматографии (30,0 г, 2,5 х 18 см), расположенную в смеси ацетон:СНCl3(1: 1). После элюирования тем же растворителем получали 0,269 г продукта, который дополнительно подвергали очистке с использованием препаративной тонкослойной хроматографии (двуокись кремния, ацетон:CHCl3 (2:3) и оперативной хроматографии (25,0 г, 2,5 х 15 см). Колонну элюировали последовательностью градиентов ацетона в СНСl3. Аналитическую пробу получали при помощи переноса продукта в безводный простой эфир и осаждения петролейным эфиром (40-60оС), и лиофилизации осадка, в результате чего получали 0,082 г (12%) белого порошка:температура точки плавления 76-81оС.
Результаты анализа, рассчитанного для С18Н17N4O6Cl ˙1,20 H2O ˙0,35 C3H6О Рассчитано, C 49,45; Н 4,68; N 12,11
Найдено, С 49,78; Н 4,38; N 11,88,
ЯМР- и масс-спектры подтверждают указанную структуру.
П р и м е р 40. 6-Метокси-9-[3,5-0-(1,2,3,3-тетраизопропил-1,3-дисилоксан-1,3- диил)- β-D-арабинофуранозил]-9Н-пурин.
9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин по примеру 3 (1,0 г, 3,54 ммоль) и имидазол (0,965 г, 14,2 ммоль) растворяли в сухом диметилформамиде (10 мл), и раствор охлаждали до температуры 3оС. Затем добавляли 1,3-дихлор-1,1,3,3-тетраизопропилдисилоксан (1,35 мл, 3,90 ммоль) и смесь перемешивали в атмосфере аргона в течение 3 ч. Затем реакцию прекращали добалением воды (1 мл) и концентрировали при пониженном давлении. Остаток разделяли между этилацетатом (150 мл) и водой (50 мл), и слой этилацетата сушили (сульфат магния), фильтровали и концентрировали. Сырой продукт переносили в этилацетат и подвергали очистке с использованием оперативной хроматографии на силикагеле (25,0 г, 2,5 х 15 см), в этилацетате. Из фракции c Rf 0,70 (двуокись кремния, этилацетат) получали 1,48 г (80%) целевого материала в виде белого твердого вещества.
УФмакс. EtOH 248,4 нм.
ЯМР- и масс-спектры подтверждали упомянутую структуру.
П р и м е р 41. 6-Метокси-9-[2-0-(2-ами- нобензоил)-β-D-арабинофуранозил]-9Н-пурин.
9-(β-D-Арабинофуранозил)-6-метокси- 9Н-пурин по примеру 3 (0,283 г, 1,0 ммоль), ацетонитрил (15 мл), п-карбоксиантраниловый ангидрид (0,189 г, 1,1 ммоль) и бикарбонат натрия (84 мг, 1,0 ммоль) добавляли в трехгорлую колбу с круглым дном, снабженную термометром, входом для аргона, магнитной мешалкой, холодильником и рубашкой для подогрева. Суспензию дефлегмировали в течение 3,5 ч, а затем добавляли второй эквивалент п-карбоксиантранилового ангидрида. После дефлегмации еще в течение 1 ч реакцию охлаждали до комнатной температуры, фильтровали и фильтрат выпаривали до пенистого стекла. Остаток подвергали очистке с использованием оперативной хроматографии на силикагеле и последовательного градиента от СНСl3 до 9:1 смеси СНСl3:MeOН. В результате получали 53,1 мг сложного 2'-эфира в виде хрупкого стекла: температура точки плавления 91-93оС.
Результаты анализа, рассчитанные для С18Н19N5O5 ˙0,3 H2O ˙0,2 СН4О
Рассчитано, С 52,90; Н 4,98; N 16,95.
Найдено, С 53,23; Н 4,98; N 17,21.
ЯМР- и масс-спектры согласуются с указанной структурой.
П р и м е р 42. 6-Метокси-9-[2-0-[4-метилбензоил(-3,5-0-)1,1,3,3-тетраизопропил- дисилоксан-1,3-диил]-β-D-арабинофурано- зил]-9Н-пурин.
6-Метокси-9-[3,5-0-(1,1,3,3-тетраизопропил- дисилоксан-1,3-диил)-β-D-арабино фуранозил]-9Н-пурин по примеру 40 (1,0 г, 1,9 ммоль) добавляли в 100 мл трехгорлую колбу с круглым дном с 4-диметиламинопиридином (0,02 г, 0,16 ммоль), ацетонитрилом (25 мл) и триэтиламином (0,4 мл) и раствор охлаждали в ледяной ванне до температуры 3оС на 5 мин. Добавляли толуолхлорид (0,30 мл, 2,3 ммоль) и смесь перемешивали 3 ч при температуре 3оС. Затем добавляли дополнительное количество триэтиламина (0,5 мл) и толуол хлорида (0,5 мл), и смесь нагревали до комнатной температуры. Через 7 ч смесь обрабатывали метанолом (2 мл), концентрировали при пониженном давлении и остаток наносили на колонну из силикагеля для оперативной хроматографии (2,5 х 15 см), в СН2Сl2. Колонну элюировали смесью СН2Сl2 MeOH (10:1, 400 мл), в результате чего получали 0,90 г (74%) целевого продукта.
Анализ, рассчитанный на С31Н46N4O7Si2
Рассчитано,C 57,92; Н 7,21; N 8,71.
Найдено, C 57,69; N 7,43; N 8,40
ЯМР- и масс-спектр подтверждают указанную структуру.
П р и м е р 43. 6-Метокси-9-[2-0-[4-метилбензоил(-3,5-0-)1,1,3,3-тетраизопропил- дисилоксан-1,3-диил]-β-D-арабинофурано- зил]-9Н-пурин.
6-Метокси-9-[3,5-0-(1,1,3,3-тетраизопро- пилдисилоксан-1,3-диил)-β-D-арабинофу- ранозил] -9Н-пурин по примеру 40, (1,0 г, 1,9 ммоль) добавляли в 100 мл колбу с круглым дном с 4-диметиламинопиридином (0,02 г, 0,16 ммоль), ацетонитрилом (25 мл) и триэтиламином (0,4 мл), затем добавляли анозоил хлорид (0,35 мл, 2,5 ммоль). Через 4 ч при комнатной температуре реакционную смесь обрабатывали метанолом (2 мл) и концентрировали при пониженном давлении. Остаток наносили на колонну из силикагеля для оперативной хроматографии (25,0 г, 2,5 х x15 см) в СН2Cl2:MeOH (20:1). В результате элюирования тем же растворителем получали 0,930 г (74%) целевого продукта.
Анализ, рассчитанный для С31Н46N4O8Si2 ˙0,35 Н2О
Рассчитано, С 55,97; Н 7,08; N 8,42.
Найдено, С 56,02; Н 7,01; N 8,32.
П р и м е р 44. 9-[2-0-(4-Хлорбензоил )-3,5-0-(1,1,3,3-тетраизопропилдисилоксан-1,3-диил)- β-D-арабинофуранозил]-6-метокси- 9Н-пурин.
6-Метокси-9-[3,5-0-(1,1,3,3-тетраизопро- пилдисилоксан-1,3-диил)-β-D-арабинофу- ранозил] -9Н-пурин по примеру 40 (1,0 г, 1,9 ммоль) добавляли в 250 мл колбу с круглым дном с 4-диметиламинопиридином (0,02 г, 0,16 ммоль), ацетонитрилом (25 мл) и триэтиламином (0,4 мл) и раствор охлаждали в ледяной ванне при температуре 3оС в течение 5 мин. Добавляли 4-хлорбензоил хлорид (0,31 мл, 2,5 ммоль) и реакционную смесь удаляли из ледяной ванны. После выдерживания в течение 8 ч при комнатной температуре раствор обрабатывали МеОН (3 мл) и концентрировали при пониженном давлении. Остаток переносили в СН2Сl2 и наносили на колонну из силикагеля для оперативной хроматографии (25,0 г, 2,5 х 15 см) в таком же растворителе. Колонну элюировали при помощи СН2Cl2 (200 мл), затем смесью СН2Cl2:MeOH (20:1, 400 мл), в результате чего получали 1,05 г (82%) продукта, однородность которого подтверждали с использованием тонкослойной хроматографии (двуокись кремния, СН2Cl2:MeOH 20:1, Rf 0,44).
Анализ, рассчитанный для С30Н43N4O7СlSi2
Рассчитано, С 54,32; Н 6,53; N 8,45
Найдено, С 54,48; Н 6,72; N 8,28.
П р и м е р 45. 5'-Монофосфатный сложный эфир 9-β-D-арабинофуранозил-6-диметиламин-9Н-пурина.
5'-Монофосфатный сложный эфир 9-β-D-арабинофуранозил-6-диметиламин-9Н-пурина синтезировали при помощи переноса концевой фосфатной группы 5'-трифосфата аденозина (АТФ) в 5'-ОН-группу 9-β D-арабинофуранозил-6-диметиламин-9Н-пурина, катализируемого тимидинкиназой (ТК), выделенной из вируса Варицелла зостер (ВВЗ) (Файф, Molecular Pharmac. 21, стр. 432, 1982).
В 0,5 мл 0,02 М буфера трис-оксиметил/аминометан-НСl, рН 7,5, добавляли 50 нмоль 9- β-D-арабинофуранозил-6-диметиламин-9Н-пурина, 55 нмоль АТФ (фирма Сигма Кэмикэлз, Сент-Луис, МО), 55 нмоль MgCl2 и 0,005 м.е. ТК ВВЗ. Раствор инкубировали при температуре 21оС в течение 2,5 ч, а затем фильтровали через фильтр "Центрикон 10" (УМ-мембрана с отсечкой мол.м. 10К, полученная от фиры Амикон Инк. Дэнвер, МА), чтобы удалить протеин. Продукт анализировали на диапазон поглощения в ультрафиолетовом свете с использованием анионообменной тонкослойной хроматографии, который варьировался с Rf общим для монофосфата.
П р и м е р 46. Арабинозид 5' -монофосфат-6-метоксипурина.
Нуклеозид, 6-метоксипуринарабинозид, 1,5 г (4,9 ммоль) растворяли в 15 мл триэтилфосфата и 4,8 мл триэтиламина. Этот раствор охлаждали до -10оС. При перемешивании добавляли 0,91 мл оксихлорида фосфора. Через 10 мин добавляли дополнительное количество (0,3 мл), оксихлорида фосфора. Ееще через 10 мин реакцию прерывали добавлением 100 мл охлажденного льдом 0,5 М раствора триэтиламина в воде. Все указанные реагенты получали от фирмы Алдрич.
Компоненты окончательной реакционной смеси разделяли при помощи ионообменной хроматографии на QАЕ-Сефадекс (фирма Фармация). Компоненты элюировали градиентом 50 мМ-1М бикарбоната аммония, 5'-монофосфат-6-метокси- пуринарабинозида составлял только примерно 10% от материала, элюированного из колонны. Чтобы удалить неорганические ортофосфаты из соединения, использовали ионообменную хроматографию на смоле БиоРАД-АГ-1Х8 с последовательным градиентом 100 мМ-750 мМ бикарбоната аммония. Фракции, содержащие это соединение, выпаривали один раз под вакуумом, чтобы удалить избыточное количество бикарбоната аммония, затем подвергали лиофилизации.
6-Метоксиарабинозид-5'-монофосфат получали с 3% выходом (0,14 ммоль, 50 мг). УФ-спектр (макс. 250 нм и мин 221 нм при рН 7) и отношение основание/фосфат (1,00/1,08) находились в согласии со структурой соединения. Оно может быть расщеплено в нуклеозид с использованием щелочной фосфатазы и 5'-нуклеозидазы.
П р и м е р 47. 6-Метоксипурин арабинозид 5'-трифосфат.
5'-Монофосфат-6-метоксипурин арабинозида по примеру 46 (36 мг, 93 ммоль) растворяли в 2 мл гексаметилфосфорамида. Добавляли карбонилдиимидазол (82 мг, 506 ммоль) и смесь перемешивали в течение 2 ч при комнатной температуре. Добавляли метанол (0,032 мл, 800 ммоль) и смесь перемешивали в течение 20 мин. Добавляли пирофосфат трибутиламммония (220 мг, 470 ммоль от фирмы Сигма), который предварительно растворяли 1,5 мл гексаметилфосфорамида, и смесь перемешивали в течение 18 ч при комнатной температуре. Реакцию прекращали добавлением 50 мл воды. Все упомянутые реагенты получали от фирмы Алдрич, если не указаны другие. 5'-Трифосфат подвергали очистке с использованием ионообменной хроматографии на QAE Сефадекс (фирма Фармация), элюируя последовательным градиентом 50-800 мM бикарбоната аммония. Фракции, содержащие соединение, выпаривали один раз под вакуумом, чтобы удалить избыток бикарбоната аммония, затем снова растворяли в воде и выдерживали замороженным при температуре -20оС.
5'-Трифосфат-6-метоксипуринарабино- зида получали с 60% выходом (57 моль, 30 мг), УФ-спектр (макс. 250 нм и мин 223 нм при рН 7) и отношение основание/фосфат (1,00/3,10) находились в согласии со структурой соединения. Оно может быть расщеплено в нуклеозид при помощи щелочной фосфатазы и в монофосфат при помощи фосфодиэстеразы 1.
П р и м е р 48. 6-Диметиламино-9[(2-0-валерил)-β -D-арабинозил]-9Н-пурин.
6-Диметиламинопуринарабинозид (0,501 г, 1,67 ммоль) растворяли в 20 мл пиридина и 20 мл ДМФ. Температуру раствора доводили до 4оС и медленно добавляли валерьяновый ангидрид (0,43 мл, 2,17 ммоль, фирма Алдрич Кэмикэл Ко. Милуоки, ВИ), растворенный в 5 мл пиридина и 5 мл ДМФ. Температуру раствора обеспечивали на уровне 40оС и через 2 ч растворитель удаляли под вакуумом. Остаток обрабатывали на колонне из силикагеля для оперативной хроматографии, 4,8 х 20 см при помощи 150 мл каждой из следующих смесей дихлорметана:метанола (о/о): 100: 0, 98:2, 96:4, 94:6, 92:8, 90:10. Фракции, содержащие продукт, собирали и после лиофилизации получали 0,195 г 6-диметиламино-9- (2-0-валерил- β-D-арабинозил)-9Н-пурина: TCX Rf 0,55/силикагель, 9:1 (дихлорметан:метанол).
Анализ для С17Н25N5O5 ˙0,5 H2O
Рассчитано, C 52,57; Н 6,75; N 18,03
Найдено, C 52,61; Н 6,77; N 17,99.
ЯМР- и масс-спектры согласуются с указанной структурой.
П р и м е р 49. 6-Диметиламино-9-(2,3,5-триацетил-β -D-арабинозил)-9Н-пурин.
6-Диметиламинопуринарабинозид (3,00 г, 10,0 ммоль) растворяли в 20 мл ацетонитрила и уравновешивали при температуре 4оС 45 мл пиридина. В реакционную смесь по каплям добавляли ацетил хлорид (2,85 мл, 40,08 ммоль, фирма Алдрич Кемикэл Ко. Милуоки, ВИ) в 7 мл ацетонитрила. Через 5 ч растворитель удаляли под вакуумом при температуре ниже 45оС, а остаточный материал обрабатывали высоковакуумным насосом в течение 24 ч. Высушенный остаток далее подвергали обработке с использованием оперативной хроматографии на колонне из силикагеля, 4,8 х 25 см с использованием смеси 9:1 (дихлорметан метанол). Фракции, содержащие продукт, собирали и после лиофилизации получали 0,445 г 6-диметиламино-9-(2,3,5-триацетил-β -D-арабинозил)-9-пурина.
ТСХ Rf 0,61 (силикагель, 9:1 дихлорметан метанол).
Анализ, рассчитанный для С18Н23N5О7
Рассчитано, C 51,30; H 5,50; N 16,62.
Найдено, С 51,40; Н 5,55; N 16,54.
ЯМР- и масс-спектры подтверждают указанную структуру.
П р и м е р 50. Композиции в форме таблеток.
Получали следующие композиции А и В при помощи влажного гранулирования ингредиентов с раствором позидона, затем добавляли стеарат магния и смесь прессовали (см. табл. 1).
Таблетки получали из упомянутых ингредиентов (С) при помощи влажного гранулирования и последующей прессовки. При альтернативном получении повидон Б.Ф. можно заменить поливилпирролидоном.
Приводимые композиции D и Е получали при помощи прямой прессовки смешанных ингредиентов. Лактоза в композиции Е имеет тип для прямой прессовки (фирма Дейри Крест "Зепарокс").
Композиция D мг/капсулу: Активный ингредиент 250 Предварительно жела- тинизированный крахмал НФ15 150
____
400
Композиция Е, мг/капсулу: Активный ингредиент 250 Лактоза 150 Авицел 100
____
500
Композиция F (для регулируемого высвобождения композиции).
Композицию получали при помощи влажного гранулирования ингредиентов с раствором повидона, а затем добавляли стеарат магния и прессовали, мг/таблетку: а) Активный ингредиент 500 b) Оксипропилметил- целлюлоза 112 (Метоцел К4М Премиум) с) Лактоза Б.Ф. 53 d) Повидон Б.Ф. 28 е) Стеарат магния 7
____
700
Высвобождение лекарственного препарата имеет место в течение примерно 6-8 ч и заканчивается после 12 ч.
П р и м е р 51. Композиция в форме капсулы
Композиция А.
Композицию в форме капсулы получали при помощи смешения ингредиентов из композиции 1 в примере 50 и заполнения смесью жесткой желатиновой капсулы, состоящей из двух частей. Композицию получали аналогичным образом.
Композиция, В, мг/капсулу: а) Активный ингредиент 250 b) Лактоза Б.Ф. 143 с) Гликоллат натрий крахмал 25 d) Стеарат магния 2
_____
420
Композиция С, мг/капсулу: а) Активный ингредиент 250 b) Макрогол 4000 Б. Ф. 350
___
600
Капсулы получали расплавлением макрогола 4000 Б.Ф. диспергированием активного ингредиента в расплаве и заполнением расплавом жестких желатиновых капсул, состоящих из двух частей.
Композиция J, мг/капсулу: Активный ингредиент 250 Лецитин 100 Арахисовое масло 100
___
450
Капсулы получали диспергированием активного ингредиента в лецитине и арахимовом масле и заполнением дисперсией мягких, эластичных желатиновых капсул.
Композиция Е (регулируемое высвобождение).
Следующую композицию для капсулы с регулируемым высвобождением получали при помощи экструзии ингредиентов a, b и с с использованием экструдера, затем из полученного материала получали шарики и сушили. Высушенные шарики затем покрывали оболочкой (d), регулирующей высвобождение, которую помещали в жесткую желатиновую капсулу, состоящую из двух частей.
Композиция Е, мг/капсулу: а) Активный ингредиент 250 b) Микрокристаллическая целлюлоза 125 с) Лактоза Б.Ф. 125 d) Этилцеллюлоза 13
___
513
П р и м е р 52. Офтальмологический раствор. Активный ингредиент, г 0,5 Хлорид натрия аналити- ческого сорта, г 0,9 Тиомерсал, г 0,001 Очищенная вода, мл До 100 рН обеспечивается на уровне 7,5
П р и м е р 53. Композиция для инъекций. Активный ингредиент, г 0,200 Стерильный, не содержащий пирогенов, фосфатный буфер (рН 9,0), мл До 10
Активный ингредиент растворяли в большей части фосфатного буфера (35-40оС), затем доводили до необходимого объема и фильтровали через стерильный микропористый фильтр в 10 мл стеклянный пузырек янтарного цвета (тип I), который закрывали стерильной пробкой и герметической крышкой.
П р и м е р 54. Внутримышечная инъекция. Активный ингредиент, г 0,20 Бензиловый спирт, г 0,10 Гликофурол 75, г 1,45 Вода для инъекций, мл До 3,00
Активный ингредиент растворяли в гликофуроле. Затем добавляли бензиловый спирт и осуществляли растворение, затем добавляли воду до 3 мл. Затем смесь фильтровали через стерильный микропористый фильтр и герметически закрывали в стерильные 3 мл стеклянные пузырьки янтарного цвета (типа I).
П р и м е р 55. Сиропная суспензия Активный ингредиент, г 0,25 Раствор сорбита, г 1,50 Глицерин, г 2,00 Диспергируемая целлюлоза, г 0,075 Бензоат натрия, г 0,005 Вкусовое вещество, Пич 17.42.3169, мл 0,0125 Очищенная вода до объема, мл 5,00
Бензоат натрия растворяли в части очищенной воды и добавляли раствор сорбита. Добавляли и диспергировали активный ингредиент. В глицерине диспергировали загущающее вещество (диспергируемую целлюлозу). Обе эти дисперсии смешивали и доводили до необходимого объема при помощи очищенной воды. Большее загущение обеспечивали, если это необходимо, увеличением среза суспензии.
П р и м е р 56. Суппозиторий, мг/сусппозиторий: Активный ингредиент (63 м) 250 Тугоплавкий жир. Б.Ф. (Уитепсол Х15-Динамит Нобель) 1700
____
1950
Активный ингредиент использовали в виде порошка, в котором, по крайней мере 90% частиц имеют диаметр 63 м и меньше.
Одну пятую часть Уитепсола Х15 расплавляли в чашке, снабженной рубашкой, через которую пропускали пар, при температуре 45оС максимум. Активный ингредиент просеивали через сито с диаметром отверстий 200 м и добавляли в расплавленную основу при перемешивании, используя сильверсон, снабженный срезающей головкой до тех пор, пока не будет получена однородная дисперсия.
Поддерживая температуру смеси на уровне 45оС, добавляли оставшийся Уитепсол Х15 в суспензию и при помощи перемешивания добивались получения однородной смеси. Суспензию полностью пропускали через сито из нержавеющей стали с диаметром отверстий 200 м и при непрерывном перемешивании, давая возможность смеси остыть до 40оС. При температуре в области от 38 до 40оС 2,02 г смеси заполняли подходящие пластмассовые формы. Далее суппозиториям давали возможность охладиться до комнатной температуры.
П р и м е р 57. Пессарии, мг/пессарий: Активный ингредиент 63 м 250 Безводная декстроза 380 Картофельный крахмал 363 Стеарат магния 7
____
1000
Указанные ингредиенты непосредственно перемешивали и пессарии получали при помощи прямой прессовки полученной в результате смеси.
П р и м е р 58. Противовирусная токсичность и испытание на биопригодность.
Определение противовирусной активности против вируса Барицелла-Зостер.
Ингибиторный эффект соединений на репликацию ВВЗ (штамм Ока) исследовали при помощи процедуры ЭЛИЗА (Берковиц, Ф.Е. и Левин, И,Дж. 1985), Antimicrob. Agents and Chemother.), которую модифицировали следующим образом. Инфицирование начинали в присутствии лекарственного препарата, а не перед его добавлением. В конце третьего дня инкубирования лекарственного препарата и вируса с неинфицированными клетками (человеческие диплоидные фибробласты, штамм MRG-5) пластины с 96 углублениями подвергали центрифугированию в течение 5 мин со скоростью 200g, чтобы осадить отделившиеся клетки перед фиксацией глютаральдегида. В модифицированной ЭЛИЗЕ в качестве второго антитела использовали конъюгированный щелочной фосфатазой античеловеческий IgG. Скорость расщепления пара-нитрофенил фосфата рассекающей щелочной фосфатазой определяли при помощи процедуры, описанной в (Тадепалли, С.М. Квини, Р.П. и Аверетт, Д.Р. 1986, Antimicrob, Agents and Chemother, 29, стр. 93-98), Неинфицированные клетки использовали для того, чтобы получить "чистые" скорости реакции, которые вычитали из скоростей, полученных в присутствии вируса. Этот анализ пригоден для обнаружения вирусного потомства в культурах, которые первоначально были инфицированы от 15 до 3600 инфицирующими частицами на углубление.
Исследование ингибирования роста неинфицированных клеток млекопитающих.
Способность предлагаемых соединений ингибировать рост клеток Д98 (человека) и L-клеток (мыши) измеряли при помощи определения количества клеток с различными концентрациями соединения (Ридеут, Дж.Л. и др. 1982, J. Med. Chem, 25, стр. 1040-1044). Затем это количество клеток сравнивали с количеством, полученным в эксперименте без соединения. Счет клеток осуществляли либо при помощи прямого подсчета частиц после трипсинизации монослоя, либо при помощи спектрофотометрического определения количества живого штамма, составляемого клетками.
Сравнимые результаты получали при помощи обеих процедур. Анализ данных. Концентрацию соединения, приводящую к 50% от контрольных значений (IС50) рассчитывали либо при помощи прямой интерполяции из графиков log концентрации соединений относительно процента контрольного значения, либо с использованием компьютерной программы, которая осуществляет анализы в соответствии с одним и тем же алгоритмом. В этих расчетах использовали данные в области от 20 до 80% от контрольного (см. табл. 2).
Анализ и данные относительно биопригодности.
Две крысы вида Лонг-Эванс получали дозу соединения при помощи трубки, вставленной внутрь желудка, по примеру 3 10 мг/кг или молярные эквиваленты соединений примеров 16, 24 и 32.
Животных помещали в метаболические клетки и собирали мочу в периоды от 0 до 24 ч и от 24 до 48 ч после введения дозы. Мочу анализировали при помощи обращенно-фазовой высокоэффективной жидкостной хроматографии. Результаты дозы, выделенной из мочи в течение 48 ч. Пример Доза, 3 4,9/2,5 16 11 24 7,9 32 15,9
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АКТИВНОСТЬ ПРОТИВ ВИРУСНОЙ ИНФЕКЦИИ И β D-АРАБИНОФУРАНОЗИЛ-2-АМИНО-6-МЕТОКСИ-9Н-ПУРИНЫ | 1993 |
|
RU2112765C1 |
| МОНО-, ДИ- ИЛИ ТРИСЛОЖНЫЕ ЭФИРЫ 2-АМИНО-6-(С - С-АЛКОКСИ-9-(βD-АРАБИНОФУРАНОЗИЛ)-9Н-ПУРИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2114860C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНЫХ СОЕДИНЕНИЙ ИЛИ ИХ ПРОИЗВОДНЫХ | 1990 |
|
RU2068849C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 2'-ДЕЗОКСИ-2'-ФТОРРИБОНУКЛЕОЗИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1990 |
|
RU2043361C1 |
| ЭНАНТИОМЕРНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1992 |
|
RU2091386C1 |
| Способ получения нуклеозидов или их фармацевтически приемлемых сложных эфиров | 1988 |
|
SU1779256A3 |
| СПОСОБ ПОЛУЧЕНИЯ ИМИДАЗОПИРИДАЗИНОВ | 1989 |
|
RU2017741C1 |
| Способ получения 2,4-диамино-5-(замещенных) пиримидинов | 1983 |
|
SU1306473A3 |
| Способ получения 2,4-диамино-5-(замещенных)пиримидинов | 1983 |
|
SU1318148A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПИРИДИНОВЫХ НУКЛЕОЗИДОВ | 1989 |
|
RU2036199C1 |
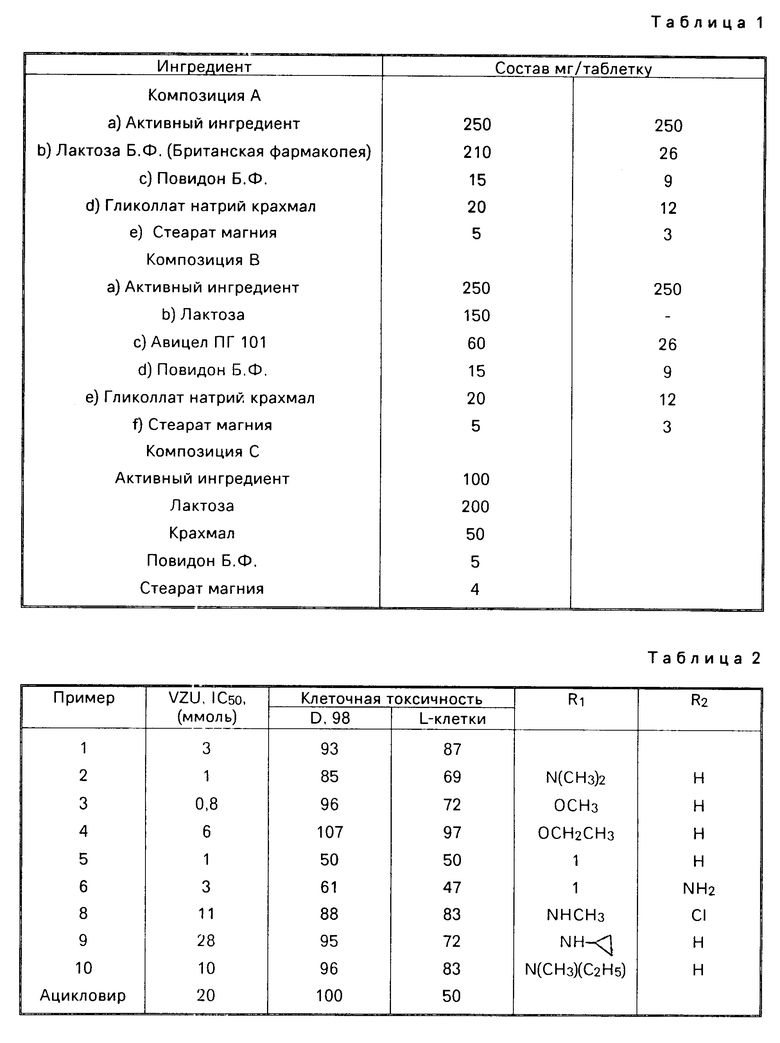
Использование: в медицине, в частности в способе получения замещенных пуриновых арабинозидов, обладающих противовирусной активностью: вирус Варицелла зостер или цитомегаловирус. Сущность изобретения: продукт замещенные пуриновых арабинозидов ф-лы I: где R1 галоген, метокси, этокси, пропокси; аминогруппа, монозамещенная метилом или C C-циклоалкилом, либо дизамещенная C C-алкилом; или R1 -азотосодержащий гетероцикл; R2-водород, галоген или аминогруппа; при условии, что, когда R2-водород, то R1 не является метокси, метиламино, диметиламино, пиперидино или пирролидиногруппой; и когда R2 - аминогруппа, то R1 не является метиламиногруппой, и/или его фармацевтически приемлемые производные, за исключением 2,3,5-триацетата и 2,3,5-трибензил-производных соединений ф-лы I, где R1 хлор или фтор и R2 хлор, фтор, водород или аминогруппа. Реагент 1: соединение ф-лы II. Реагент 2: соединение ф-лы III. 2 табл. Структура соединений ф-л I(см. ниже), II, III: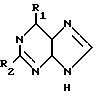
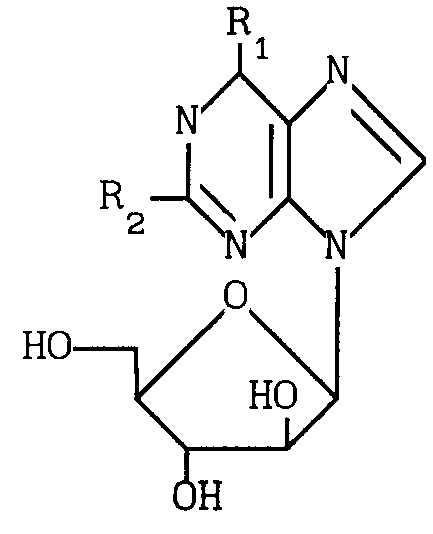
Способ получения замещенных пуриновых арабинозидов общей формулы I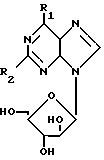
где R1 галоид, метокси, этокси, пропокси, аминогруппа, однозамещенная метилом или C3 C6-циклоалкилом, либо двузамещенная C1 C5-алкилом, или азотсодержащее гетероциклическое кольцо, содержащее 4 7 атомов углерода, связанное с пуриновым радикалом через атом азота;
R2 водород, галоид или аминогруппа,
или его фармацевтически приемлемых производных, отличных от 2′, 3′, 5′ -триацетатных и 2′, 3′, 5′ -трибензиловых производных, где R1 хлор или фтор, R2 хлор, фтор, водород или амино, при условии, что когда целевой продукт не является фармацевтически приемлемым производным, как отмечено выше, а R2 водород, R1 не может означать метокси, метиламино, диметиламино, пиперидино или пирролидино, и когда R2 аминогруппа, R1 не может означать метиламино,
отличающийся тем, что соединение общей формулы II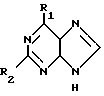
где R1 и R2 имеют указанные значения,
подвергают взаимодействию с соединением общей формулы III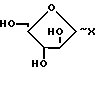
где X пиримидиновое или пуриновое основание,
за исключением оснований указанной общей формулы II и, в случае необходимости, если полученное соединение представляет собой соединение общей формулы I, превращают его в фармацевтически приемлемое производное или, если полученное соединение представляет фармацевтически приемлемое производное, превращают его в другое фармацевтически приемлемое производное.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Патент США N 4371613, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
