Изобретение относится к получению 2-пиперазинонов.
Пиперазиноны являются полезными соединениями и хорошими абсорбентами для удаления SO2 из потоков газов. Пиперазиноны и замещенные пиперазиноны известны и они могут быть приготовлены несколькими методами. N-алкил- и N,N'-диалкилпиперазиноны были приготовлены реакцией N-замещенных алкилендиаминов с 2-оксоальдегидом, что открыто в патенте США 4767860 (1), и N-замещенные гидроксиалкилпиперазиноны были приготовлены реакцией соответствующих пиперазинонов или определенных замещенных пиперазинонов с алкилен оксидами или с аналогами хлоргидрина. Эта реакция описана в патенте США 2633467 (2) и в Chemie Iherapentigue май-июнь, 1969, N 3, стр. 167-173 (3). Вышедший в последние годы патент США 4,814, 443 (4) раскрывает метод приготовления таких гидроксиалкилзамещенных соединений реакцией α, β-дикетона, а именно глиоксаля, или алкилзамещенного производного глиоксаля, с N-гидроксиалкилалкилендиамином.
В более раннем процессе, описанном в патенте США 2649450 (5) N,N'-диалкилэтилендиамин реагирует с карбонильным соединением и НСN с образованием 1,4-диалкил-2-пиперазинона. В другом процессе, описанном в том же патенте, амин взаимодействует с кетонцианогидрином. В патенте США 2700668 (6), описывается процесс создания 2-пиперазинона, в котором взаимодействуют этилендиамин и гликолонитрил.
Желательно было бы улучшить такого типа процессы сделать их менее сложными и получить хорошие выходы продукта подходящей чистоты для использования в качестве абсорбентов SO2.
Процесс создания пиперазинонов, которые были бы полезны как соединения, абсорбирующие двуокись серы, включает 2-пиперазинон и его замещенные производные. Процесс состоит в реакции 1-циано-1-гидроксиалкана с этилендиамином или его замещенными производными в водном растворе. Циановое соединение применяется в количестве, равном по меньшей мере молярному эквиваленту по отношению к диамину. К процессу предъявляется два требования: (1) использование эквимолярного или избыточного количества цианового соединения и (2) барботирование продукта инертным газом. Каждый из этих этапов является необходимым для получения продукта, который был бы эффективным в качестве абсорбента SO2.
Изобретение представляет собой процесс получения 2-пиперазинона и его замещенных производных, которые были бы применимы в качестве абсорбентов SO2. Он обладает преимуществами по сравнению с ранее известными процессами такого типа. Реакция, которая приводится в водном растворе, дает хороший выход продукта, который после барботирования является полезным без дальнейшего удорожания и затрат времени на операции очистки.
1-Циано-1-гидроксиалканы, например, 1-циано-1-гидроксиметан (гликолонитрил GH) и этилендиамин или его алкил- или гидроксиалкильные замещенные производные, например аминоэтилэтаноламин, реагируют друг с другом в эквимолекулярных количествах в водном растворе. Желательно использовать циановые соединения в небольшом избытке, так как избыток диамина в продукте нежелателен при его использовании в качестве абсорбента SO2. Кроме того, продукт 2-пиперазинон должен быть пробарботирован (продут) инертным газом, например азотом или воздухом, перед его использованием в качестве абсорбента. Инертным газом предполагается любой газ, который является инертным в условиях использования.
Таким образом, при приготовлении таких продуктов-абсорбентов SO2, должны быть соблюдены два критерия (1) цианистое соединение должно быть использовано в количестве, равном по меньшей мере молярному эквиваленту и (2) продукт реакции должен быть пробарботирован инертным газом.
Известно, что реакция циановых соединений с этилендиаминовыми соединениями протекает по следующей схеме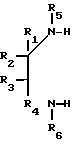 + R7-
+ R7-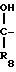 C ≡ N __→
C ≡ N __→ 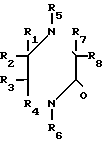 + NH3
+ NH3
где R1, R2, R3, R4, R7 и R8 являются независимо водородной или алкильной группой с 1-6 атомами углерода; R5 и R6 являются независимо водородом, алкильной группой с 1-6 углеродными атомами или гидроксиалкильной группой с 1-6 углеродными атомами.
Представленные соединения, которые являются полезными при использовании в качестве абсорбентов двуокиси серы и приготовлены в соответствии с изобретением, являются следующими соединениями 2-пиперазинон, 1,4-диметил-2-пиперазинон, 1,4-диэтил-2-пиперазинон, 1,4-ди(n-бутил)-2-пиперазинон, 4-метил-2-пиперазинон, 1,3,4-триметил-2-пиперазинон, 1-(2-гидрокси- этил)-2-пиперазинон [1-НЕР] 4-(2-гидроксиэтил)-2-пиперазинон [4-НЕР] 4-(2-гидроксиэтил)-1-метил-2-пиперазинон, 4-(2-гидрокси- этил)-3-метил-2-пиперазинон, 4-(2-гидроксиэтил)-5-метил-2-пиперазинон, 1,4-бис(2-гидроксиэтил)-2-пиперазинон, 4-(2-гидро- ксиэтил)-2-пиперазинон, 4-(2-гидроксипропил)-2-пиперазинон, 4-(2-гидроксибутил)-2-пиперазинон и 4-(2-гидроксипропил)- 6-метил-2-пиперазинон. Другие замещенные пиперазиноны могут быть приготовлены реакцией соответствующих замещенных этилендиаминов с 1-циано-1-гидроксиалканами.
Следующие опыты показывают приготовление продуктов, представляющих собой замещенные пиперазиноны, в соответствии с изобретением. Незамещенные пиперазиноны готовятся реакцией этилендиамина с 1-циано-1-гидроксиметаном, например, гликолонитрилом.
Приготовление 4-(2-гидроксиэтил)-2-пиперазинона.
Данное количество аминоэтилэтаноламина (АЕЕА) помещается в реактор, снабженный приспособлением для перемешивания, термометром, средством для нагревания и регулирования температуры и средством для добавления гликонитрильного реагента (GN). АЕЕА растворяется в деионизованной воде с образованием 50-75% -ного водного раствора. Реагент GN может быть добавлен весь одновременно или может добавляться медленно в течение определенного периода времени. Если выбирается первый метод, нагревание до нужной температуры осуществляется после его добавления. Если выбирается последний метод более медленного добавления реагента GN. АЕЕА нагревается до необходимой температуры до добавления GN. После того, как реакция (проведенная любым методом) закончена, раствор барботируют инертным газом, например азотом.
Цианистый реагент обычно используется в виде водного раствора, например гликолонитрил используется в виде раствора концентрации 40-50% Использование цианистых соединений в виде раствора не в воде, а в другом растворителе является нежелательным ни с практической, ни с экономической точки зрения. Диаминовый реагент также используется в виде водного раствора, который в случае АЕЕА может иметь значение 25-75% Целью первоначального разбавления реагентов является регулирование протекания экзотермической реакции. Температуру этой реакции следует поддерживать в интервале 70-100оС, желательно 90-100оС. Реакция проводится при атмосферном давлении. Может быть применено снижение или повышение давления, но это не дает каких-либо преимуществ.
Преимущественным методом проведения реакции является добавление цианистого соединения к диамину в определенном оптимальном интервале температур, оптимальном интервале величины рН и в течение определенного периода времени. Наиболее предпочтительными условиями проведения реакции являются добавление цианистого соединения к диамину при температуре около 100оС, при начальной величине рН около 10 и в течение периода времени не менее, чем один час или более. Когда добавление заканчивается, реакционная смесь нагревается в течение периода времени, достаточного для того, чтобы гарантировать вступление в реакцию в основном, всего количества амина. Это может быть определено посредством газохроматического анализа образца, отбираемого в ходе реакции.
Фактическое время добавления и последующего нагревания реакции определяется по крайней мере частично количеством применяемых реагентов. При более низкой температуре, например 50-80оС, требуется более длительное время добавления и нагревания реакционной смеси.
Следующие примеры представляют изобретение и процесс, который проводился либо по процедуре А, или по процедуре В:
процедура А. Соединяют вместе оба реагента при комнатной температуре и нагревают до нужной температуры реакции, поддерживая ее в течение времени, необходимого для полного протекания реакции, или
процедура В: нагревают диамин до нужной температуры, затем медленно добавляют цианистое соединение в течение времени, необходимого для его реакции с амином.
Используя процедуру А или В, добавляется достаточное количество цианистого соединения, гарантирующее остаточную концентрацию амина в конечном продукте менее, чем 0,1 мас. Процесс контролируется газо-хроматографическим методом и реакция прекращается, когда нехарактерные промежуточные пики исчезают и концентрация НЕР достигает максимума.
Примеры 1 и 2 (показанные в табл. 1) проводились в соответствии с процедурами А и В соответственно.
В двух других экспериментах, проведенных в соответствии с процедурой А, первоначальная величина рН реакционного раствора была снабжена добавлением Н2SO4 до рН 7,1 и 5,2 соответственно.
Результаты показывают, что выход в каждом эксперименте был в основном эквивалентен упомянутому, хотя отношение 4-НЕР к 1-НЕР было немного меньше при более низком значении рН.
Однако медленное добавление цианистого соединения к диамину дает лучший выход пиперазинонового продукта. Надо полагать, что более низкий выход в примере 1 обусловлен гидролизом и/или полимеризацией цианистого соединения. Из-за более низкой скорости добавления в процедуре В цианистое соединение способно быстрее прореагировать с диамином, чем подвергнуться гидролизу и/или полимеризации.
Были проведены следующие эксперименты, демонстрирующие влияние остаточного амина (пример 3) и барботирования (пример 4) на регенерационную способность пиперазинонового продукта, то есть способность удалять и улавливать SO2 из абсорбируемого растворителя. Остаточный амин присутствует в том случае, когда вместо использования эквимолярных количеств реагентов амин используется в молярном избытке или имеется избыток цианистого соединения, как это требуется в данном изобретении.
П р и м е р 3. Регенерационная способность абсорбента SO2 (остаточный аминный эффект).
Были приготовлены водные растворы 4-(2-гидроксиэтил)-2-пиперазинона, содержащие различные количества аминоэтилэтаноламина. При комнатной температуре (23оС) азот газ, содержащий 3% двуокиси серы, пропускался в течение 4 ч через крупнозернистую газовую дисперсионную трубку со скоростью 0,5 SCF Н (стандартные кубические футы в час) в раствор абсорбента известной концентрации. Небольшой образец раствора абсорбента SO2 анализировался на концентрацию ионов SO-3 и SO-4 c использованием стандартной ионной хроматографии. Из сочетания концентраций SO-3 и SO-4 рассчитывалась мощность (емкость) раствора абсорбента по отношению к SO2. Затем раствор абсорбента переносился в перегонную колбу и нагревался до кипения (около 100оС) при пропускании азота (около 0,5 S C F Н) в течение 4 ч для отгонки газа SO2 из раствора. В процессе отгонки добавлялась вода в таком количестве, чтобы возместить потерю воды при отгонке и сохранить постоянной концентрацию абсорбента в растворе. Раствор вновь анализировался на концентрацию SO3- и SO4- и разница между концентрациями в отогнанном и содержащем SO2 растворах используется для расчета регенерационной емкости раствора абсорбента. Табл. 2 показывает влияние остаточного амина на SO2, регенерируемый (улавливаемый) при регенерации.
П р и м е р 4. Регенерационая способность адсорбентов SO2 (Эффект продувки).
Образцы 4а и 4в были приготовлены в соответствии с процедурами А и В соответственно. Каждый из них был продут азотом N2 в течение 4 ч при скорости около 0,5 SCFН. Образец 4с был приготовлен в соответствии с процедурой В, но не подвергался продувке (пропусканию азота).
При комнатной температуре (23оС) газообразный азот, содержащий 3% двуокиси серы, продувался через растворы вышеописанных НЕР посредством крупнозернистой газовой дисперсионной трубки в течение четырех часов при скорости около 0,5 SCFH в раствор абсорбента известной концентрации. Образец раствора абсорбента, обогащенный SO2, анализировался, кипятился и продувался с повторением процедур, описанных в примере 3. Этот раствор вновь анализировался на концентрацию SO3- и SO4- и разница между концентрациями в отогнанном и обогащенном SO2 растворах использовалась для расчета регенерационной способности раствора абсорбента. Табл. 3 показывает разницу в регенерационной способности продукта, который был подвергнут продувке по сравнению с аналогичным продуктом, неподвергнутым продувке.
Использование: в качестве абсорбентов SO2. Сущность изобретения: продукт - производные 2-пиперазинов ф-лы 1, где R1-R4, R7 и R8 - Н и R5 и R6 - различные, атом водорода или гидрокси C1-C6 - алкил. Реагент 1: 1-циано-1-гидроксиметан. Реагент 2: этилендиамин или его производное ф-лы II, где R1-R4, R5 и R6 указано выше. Условия процесса, в водной среде при 50 - 100°С при молярном избытке 1-циано-1-гидроксиметана с последующей продувкой целевого продукта инертным газом. Молярное соотношение реагента 1 к реагенту 2, предпочтительно составляет (1,2 - 1,5): 1. В качестве инертного газа используют азот или воздух и процесс проводят при pH 5 - 10 при начальной стадии. Структура соединений ф-л 1 и 2: 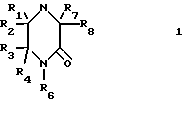
 3 табл.
3 табл.
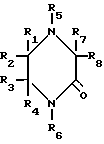
где R1 R4, R7 и R8 водород;
R5 и R6, различные, водород или гидрокси - С1-С6-алкил,
взаимодействием 1-циано-1-гидроксиметана с этилендиамином или его производным общей формулы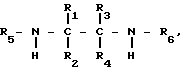
где R1 R6 имеют указанные значения,
в водной среде, отличающийся тем, что, с целью получения более чистого продукта, процесс проводят при 50 100oС при молярном избытке 1-циано-1-гидроксиметана с последующей продувкой целевого продукта инертным газом.
