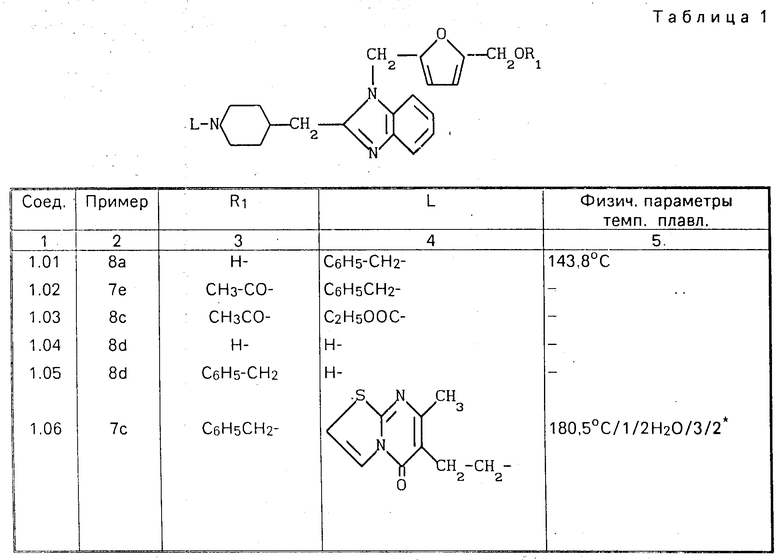
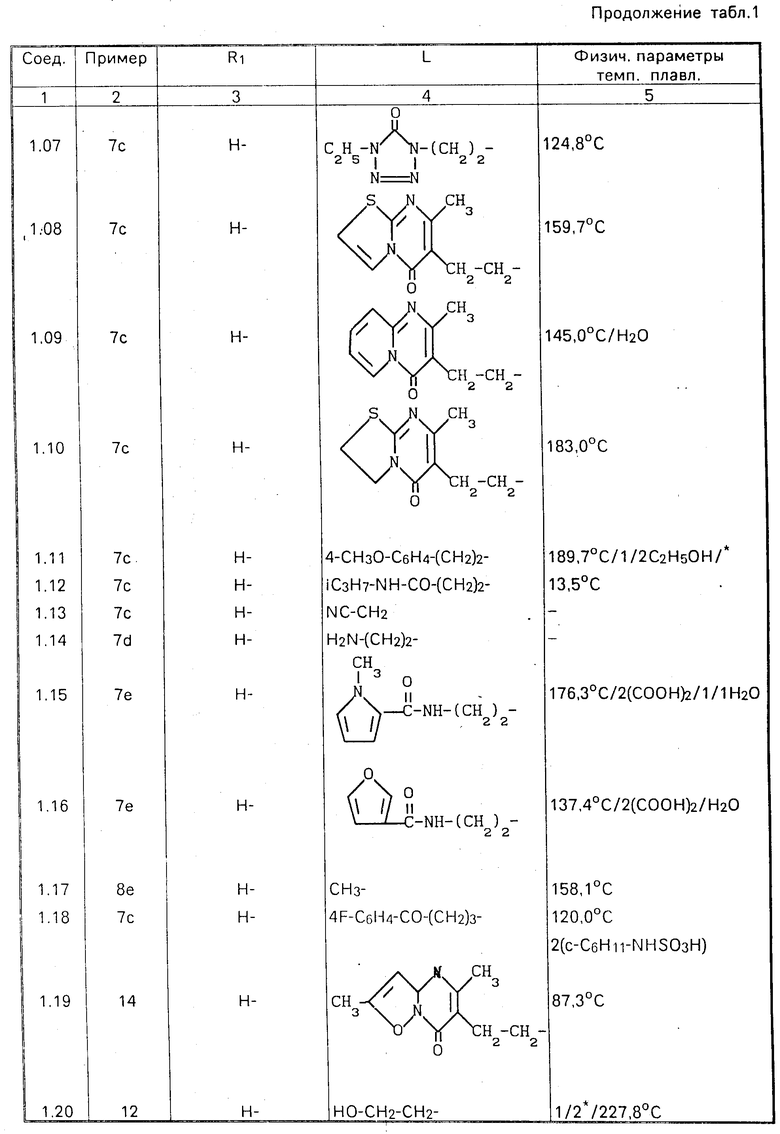
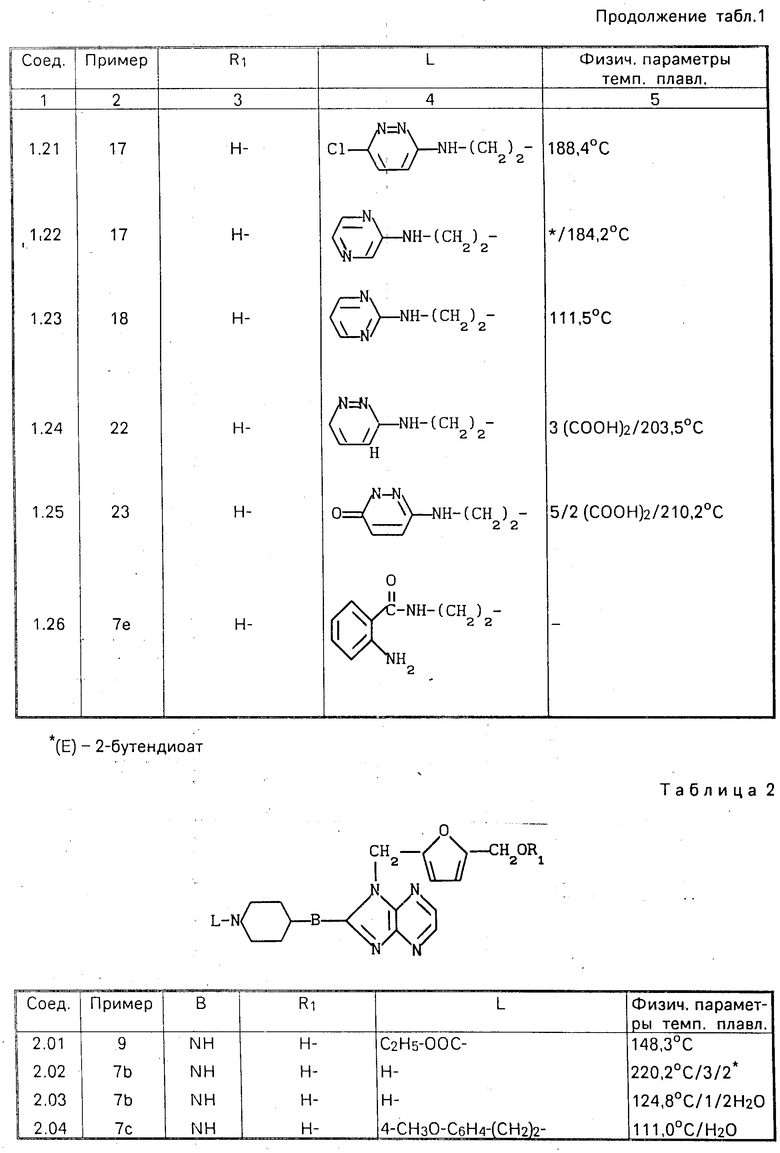
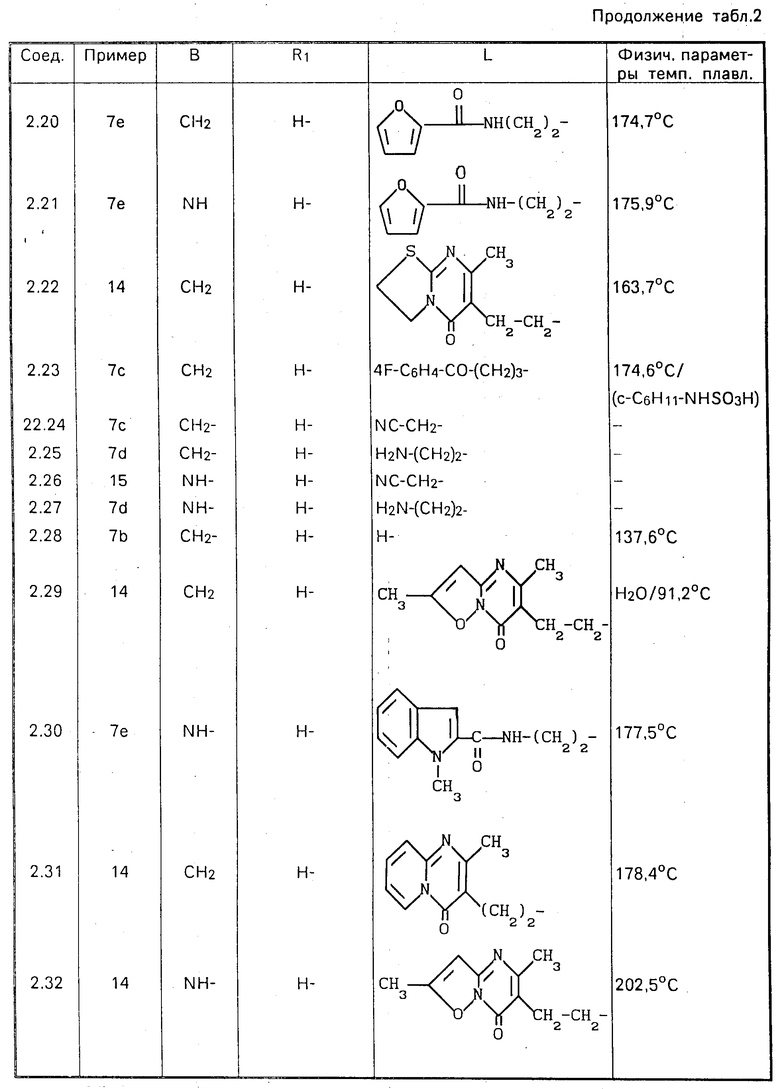
Изобретение относится к соединениям формулы I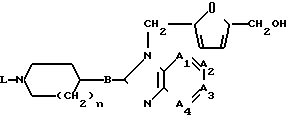 (I) или фармацевтически приемлемую соль присоединения кислот его или стереоизомерная форма соединения, где
(I) или фармацевтически приемлемую соль присоединения кислот его или стереоизомерная форма соединения, где
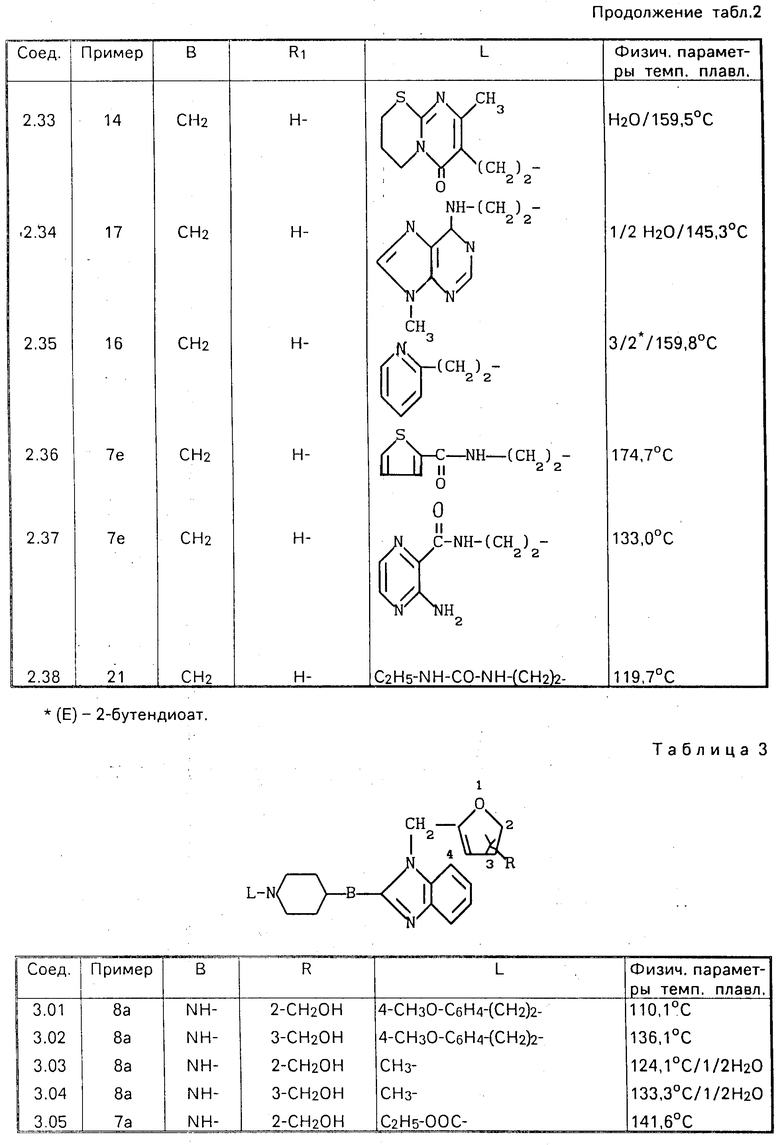
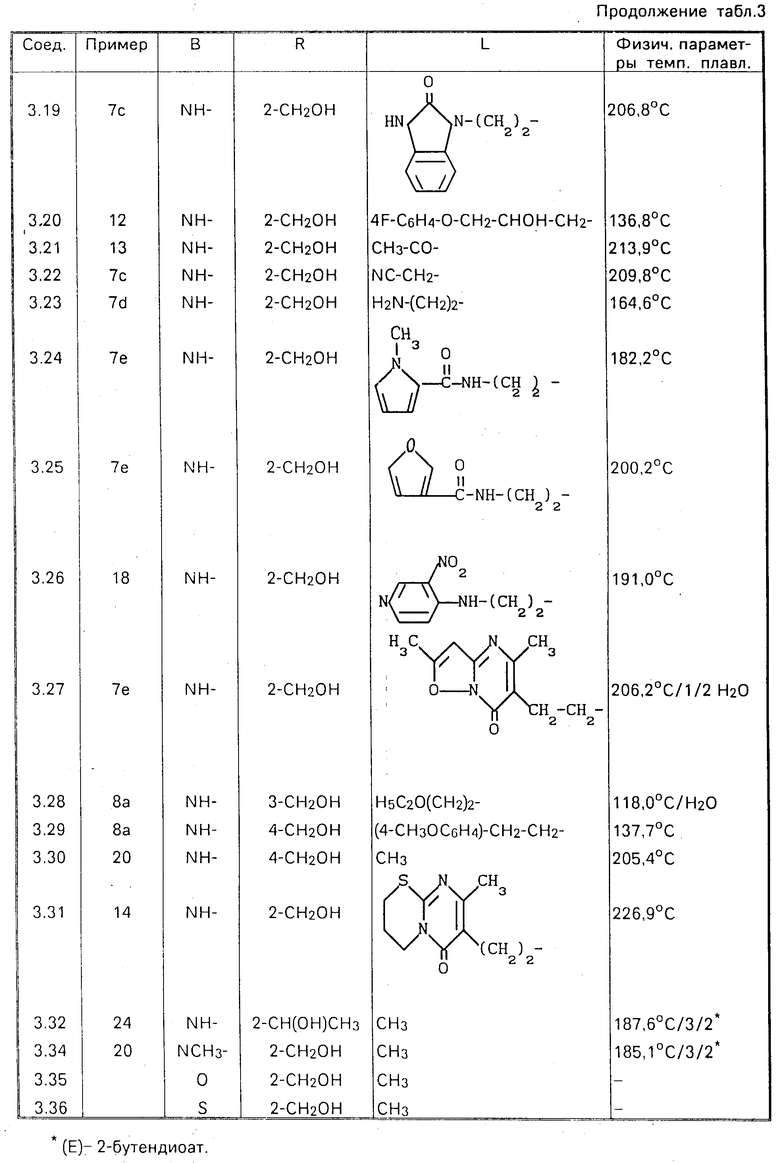
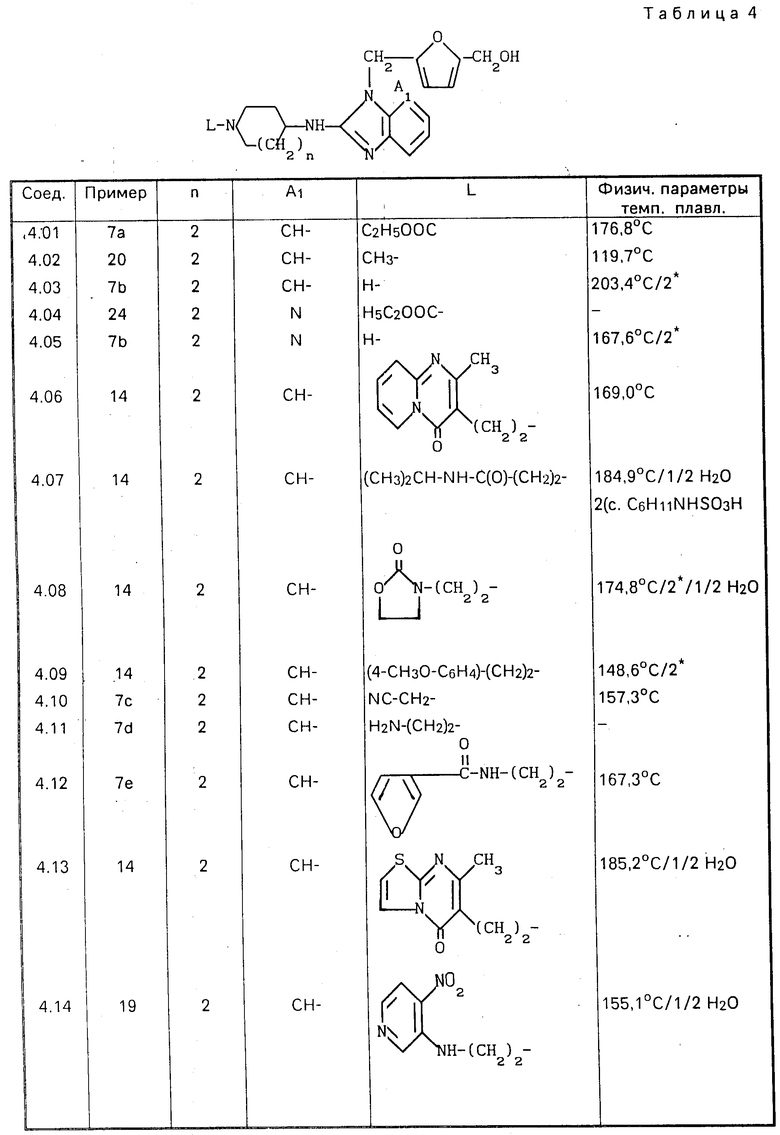
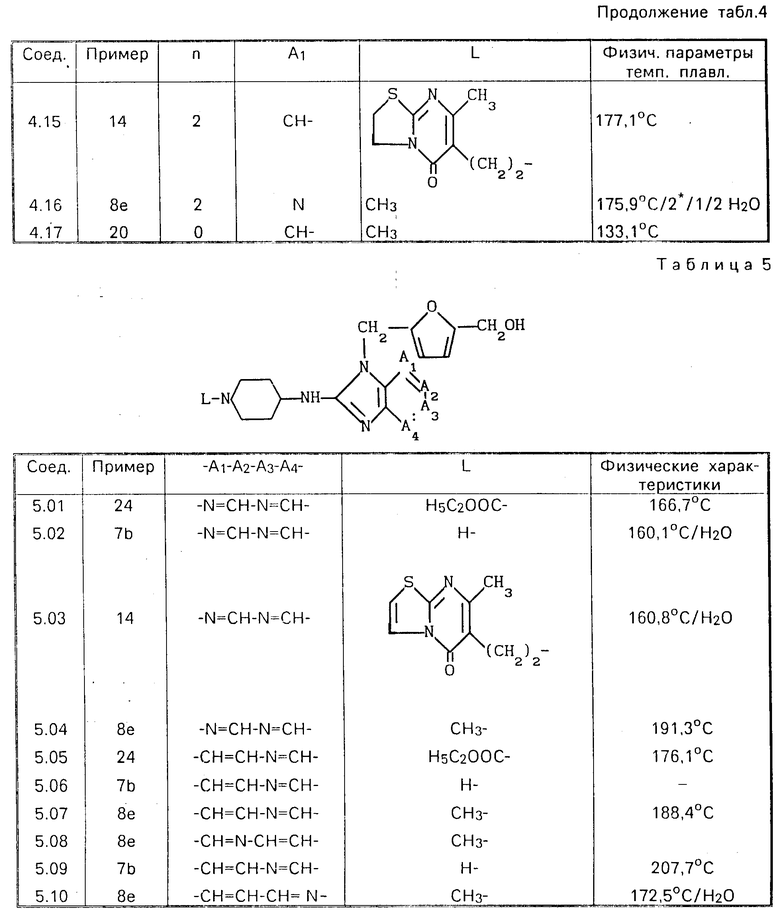
-А1 = А2 - А3 = А4 - двухвалентный радикал, имеющий формулу
-СН=СН-СН=СН- (а-1)
-N=СН-СН=СН- (а-2)
-СН=СН-СН=N (а-5) или
-N=СН-N=СН- (а-6),
n=1 или 2
В - NR4 или СН2
R4 - водород или С1-С6 алкил
L - водород, С1-С6 алкил, С1-С6 алкилоксикарбонил, или радикал формулы
-Alk - R5 (b-1),
-Alk - Y - R6 (b - 2),
-Alk - Z1 - C(=X) - Z2 - R7 (b-3), или
-СН2 - СНОН - СН2 - О - R8 (b-4), где R5-циано, фенил необязательно замещенный С1-С6 алкилокси; пиридинил; 4,5-дигидро-5-оксо-1-Н-тетразолил; 2-оксо-3-оксазолидинил; 2,3-дигидро-2-оксо-1-Н-бензимидазолил; или бицикличный радикал формулы (с-4-а)
G где G2 - CH=CH-CH=CH-, -S-(CH2)3,- -S-(CH2)/2-, -S-CH=CH- или -CH=C(CH3)-O-;
где G2 - CH=CH-CH=CH-, -S-(CH2)3,- -S-(CH2)/2-, -S-CH=CH- или -CH=C(CH3)-O-;
R6 - C1-C6-алкил, пиридинил необязательно замещенный нитро; пиримидинил; пиразинил; пиридазинил; необязательно замещенный галогеном, 2,3-дигидро-3-оксопиридазинил; или 9-метил-6-пуринил;
R7 - С1-С6-алкил; галофенил; 1-метил-1Н-пирролил; фуранил, тиенил, или аминопиразинил;
R8 - галофенил;
Y - O или NH;
Z1 или Z2 каждый независимо NH или прямая связь Х-O
каждый Аlk независимо - С1-С6 алкандиил.
Вышеупомянутые фармацевтически приемлемые кислые аддитивные соли включают терапевтически активные нетоксичные аддитивные соли, которые могут образовывать соединения формулы (I). Указанные соли могут быть получены путем обработки соединений формулы (I), находящимися в основной форме, подходящими кислотами, например неорганическими, такими как галогенводородные, в частности хлористоводородная, бромистоводородная и т.д., серная, азотная, фосфорная и т.д., или органическими кислотами, такими как уксусная, пропановая, оксиуксусная, 2-оксипропановая, 2-оксопропановая, диэтановая, дипропановая, дибутановая (Z)-2-дибутановая, (Е)-2-дибутановая, 2-оксидибутановая, 2,3-диоксидибутановая, 2-окси-1,2,3-пропантрикарбоновая, метансульфоновая, бензолсульфоновая, 4-метилбензолсульфоновая, циклогексансульфаминовая, 2-оксибензойная, 4-амино-2-оксибензойная и т.п. кислоты. И наоборот, соли путем их обработки щелочью могут быть переведены в свободные основания.
Под кислотно-аддитивными солями имеются в виду также гидраты и аддитивные соединения с растворителями, которые могут образовывать соединения формулы (I). Примерами таких соединений являются, в частности, гидраты, алкоголяты и т.п.
Соединения в соответствии с настоящим изобретением могут содержать несколько асимметрических атомов углерода. Каждый из этих хиральных центров может быть обозначен стереохимическими символами R и S.
Индивидуальные стереоизомеры соединений формулы (I) могут быть получены известными способами. Диастереоизомеры могут быть разделены с помощью физических методов, например селективной кристаллизацией, или хроматографических способов, в частности противоточным разделением, жидкостной хроматографией и т.д. Энантиомеры могут быть разделены с помощью известных способов разделения, например путем селективной кристаллизации их диастереомерных солей с хиральными кислотами. Чистые стереоизомеры могут быть также получены из соответствующих стереохимически чистых исходных материалов при условии стереоспецифического протекания реакции. Предпочтительно для получения чистых стереоизомеров синтезировать их стереоселективными методами. Предпочтительно при осуществлении этих методов использовать энантиомерно чистые исходные материалы. Стереохимические формы соединений формулы (I) безусловно составляют объем настоящего изобретения.
В целях упрощения написания структурных формул некоторых соединений и полупродуктов в нижеследующих примерах часть молекулы, содержащая имидазольную группу, сконденсированную с бензольным, пиридиновым или пиримидиновым кольцом, в дальнейшем будет обозначаться символом Q.

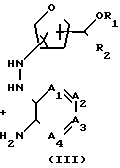
Соединения формулы (I) в общем случае могут быть получены путем взаимодействия полупродукта формулы (II) с соответствующим образом замещенным диамином формулы (III).
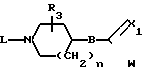
 _____→(I)
_____→(I)
В этой и последующих схемах реакций W означает соответствующую отщепляющуюся группу, например атом галогена, в частности атом хлора, брома или иода, С1-С6-алкилокси-группу, С1-С6-алкилтио, арилокси- или арилтио-группу, а Х1 - O, S или NH.
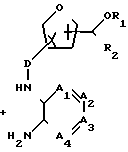
Производные формулы (II), у которых В означает СН2, а W - атом галогена, могут быть получены in situ, например путем галогенирования соответствующей карбоновой кислоты тионилхлоридом, треххлористым фосфором, фосфорилхлоридом, полифосфорной кислотой и т.п. Реакцию между соединениями формул (II) и (III) можно проводить в среде подходящего инертного растворителя, например углеводорода, в частности бензола, гексана и т.п., простого эфира, в частности 1,1' -оксибисэтана, тетрагидрофурана и т.п., кетона, в частности 2-пропанона, 2-бутанона, 2-бутанона и т.п., спирта, в частности метанола, этанола, 2-пропанола, 1-бутанола и т.п., галогенированного углеводорода, в частности трихлорметана, дихлорметана и т.п., органической кислоты, в частности уксусной, пропановой кислот и т.п., диполярного апротонного растворителя, в частности N,N-диметилформамида, N,N-диметилацетамида и т. п. , или их смесей. В зависимости от природы растворителя и W может оказаться целесообразным добавлять к реакционной смеси основание, к примеру из таких, которые обычно используются для проведения реакций N-алкилирования, и/или иодистую соль, например иодид щелочного металла. Повышение температуры и перемешивание могут увеличить скорость реакции. В некоторых случаях в результате реакции между (II) и (III) вначале может образовываться промежуточное соединение формулы (II-a), которое затем может быть циклизировано в целевое соединение формулы (I) in situ или, при желании, после выделения и очистки.
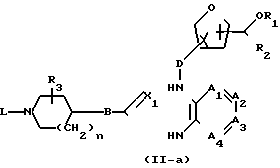 _____→(I)
_____→(I)
Соединения формулы (I) могут быть также получены путем взаимодействия полупродукта формулы (V) с последующей известной реакцией замещения. В формуле (IV) и далее М означает атом водорода (в том случае, если В имеет иное, чем СН2, значение) или щелочной или щелочноземельный металл, например литий или магний (если В означает СН2).
L N
N B
B M
M  (I)
(I)
Аналогичным образом соединения формулы (I) могут быть получены путем взаимодействия промежуточного соединения формулы (VI) с промежуточным соединением формулы (VII), у которого М имеет вышеприведенное определение. В формуле (VI) и далее W1 означает соответствующую отщепляющуюся группу, например атом галогена, в частности атом хлора, брома и т.д., или сульфонилокси-группу, в частности метансульфонилокси-, 4-метилбензолсульфонилокси-группы и т.п.
L N
N W1
W1 (I)
(I)
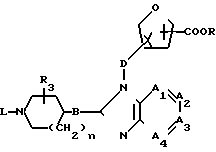
Соединения формулы (I), у которых В означает -СН2- (эти соединения описываются формулой (I-a), могут быть также получены путем взаимодействия между промежуточными соединениями формул (VIII) и (IX) или между промежуточными соединениями формул (Х) и (XI).
L N
N
 L
L N
N Q
Q
Реакции между соединениями формул (IV), (VI), (VIII) и (Х) и соответственно соединениями формул (V), (VII), (IX) и (XI) целесообразно проводить в среде инертного в условиях реакции растворителя, например ароматического углеводорода, в частности бензола, метилбензола и т.п., простого эфира, в частности 1,4-диоксана, 1,1' -оксибисэтана, тетрагидрофурана и т. п. , галогенированного углеводорода, в частности трихлорметана и т.п., N,N-диметилформамида, N, N-диметилацетамида, нитробензола, диметилсульфоксида, 1-метил-2-пирролидона и т.п. В том случае, если М означает атом водорода, таким растворителем может быть также С1-С6-алканол, например метанол, этанол, 1-бутанол и т.п., кетон, например 2-пропанон, 4-метил-2-пентанон и т. п. В некоторых случаях, в частности, когда В означает гетероатом, целесообразным может оказаться добавление соответствующего основания, например карбоната или гидрокарбоната щелочного металла, в частности карбоната или гидрокарбоната натрия и т.п., гидрида натрия или органического основания, например N,N-диэтилэтанамина или N-(1-метилэтил)-2-пропанамина, и/или добавление иодистой соли, предпочтительно иодида щелочного металла. Некоторое повышение температуры и перемешивание могут увеличить скорость реакции.
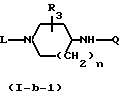
Соединения формулы (I), у которых В означает -NR4- (такие соединения описываются формулой (I-b)), могут быть также получены путем взаимодействия промежуточных соединений формулы (XII) и формулы (VII), в которой В-М означает радикал-NR4-H- (эти промежуточные соединения описываются формулой (VII-a)) и последующего известного N-алкилирования.
L N
N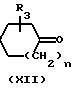
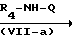 L
L N
N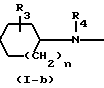 Q
Q
Реакцию между соединениями формул (XII) и (VII-a) целесообразно проводить путем смешения реагентов в среде подходящего инертного в условиях реакции растворителя с соответствующим восстановителем. Предпочтительно вначале проводят взаимодействие кетона формулы (XII) с промежуточным соединением формулы (VII-a) с образованием энамина, который можно выделить и подвергнуть дополнительной очистке и затем подвергнуть восстановлению. Подходящими растворителями для проведения этой реакции являются, например, вода, С1-С6-алканолы, в частности метанол, этанол, 2-пропанол и т.п., простые эфиры, в частности 1,4-диоксан и т.п., галогенированные углеводороды, в частности трихлорметан и т.п., диполярные апротонные растворители, в частности N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и т.п., или их смеси. Подходящими восстановителями являются, например, гидриды металлов или комплексов металлов, в частности боргидрид натрия, цианоборгидрид натрия, литийалюминийгидрид и т. п. Можно в качестве восстановителя использовать и водород в присутствии подходящего катализатора, например палладия или платины на активированном угле и т.п. Для того чтобы предупредить нежелательное гидрирование некоторых функциональных групп в реагентах и продуктах реакции, может оказаться целесообразным добавлять к реакционной смеси соответствующий канализаторный яд, например тиофен и т.п.
Соединения формулы (I-b), у которых В означает -NH- (эти соединения могут быть описаны формулой (I-b-I), могут быть также получены путем реакции циклодесульфуризации соответствующей мочевины формулы (II-a). Такая мочевина описывается формулой (II-a-I) и может быть получена путем конденсации изотиоцианата формулы (XIII) с диамином формулы (III).
Такую циклодесульфуризацию можно осуществить путем взаимодействия соединения формулы (II-a-I) с соответствующим галогеналкилом, предпочтительно иодметаном, в среде подходящего инертного в условиях реакции органического растворителя, например С1-С6-алканола, в частности метанола, этанола, 2-пролпанола и т.п. По другому варианту циклодесульфуризацию можно проводить путем взаимодействия соединения формулы (II-a-I) с подходящим для этой цели оксидом металла или солью, например оксидом или солью Hg(II) или Pb(II), в частности HgO, HgCl2, Hg(OAc)2, PbO или РВ(ОАс)2, в присутствии подходящего растворителя и последующих известных процедур. В некоторых случаях может оказаться целесообразным добавлять к реакционной смеси небольшие количества серы. В качестве циклодесульфуризирующего агента можно использовать, в частности, и метандиимиды.



Соединения формулы (I) могут быть также получены путем N-алкилирования промежуточного соединения формулы (XV) соответствующим алкилирующим агентом формулы (XIV).
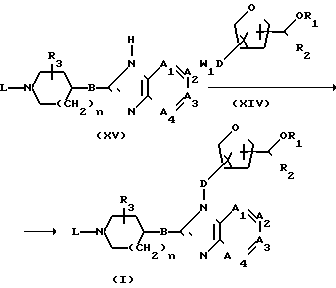
Указанную реакцию N-алкилирования целесообразно проводить в среде инертного в условиях реакции растворителя, например воды, ароматического углеводорода, в частности бензола, метилбензола, диметилбензола и т.п., алканола, в частности метанола, этанола, 1-бутанола и т.п., кетона, в частности 2-пропанона, 4-метил-2-пентанона и т. п. , простого эфира, в частности тетрагидрофурана, 1,4-диоксана, 1,1'- оксибисэтана и т.п., диполярного апротонного растворителя, например, N,N-диметилформамида, N,N-диметилацетамида, диметилсульфоксида, нитробензола, 1-метил-2-пирролидона и т. п. , или их смесей. Для связывания кислоты, образующейся при протекании реакции, целесообразно добавлять к реакционной смеси подходящее основание, например карбонат, гидрокарбонат, алкоксид, гидрид, амид, гидроксид или оксид щелочного или щелочноземельного металла, в частности карбонат или гидрокарбонат натрия, карбонат калия, метоксид или этоксид натрия, трет. бутоксид калия, гидрид натрия, амид натрия, гидроксид натрия, карбонат кальция, оксид кальция и т.п., или органическое основание, например третичный амин, в частности N,N-диэтилэтанамин, N-(1-метилэтил)-2-пропанамин, 4-этилморфолин, пиридин и т.п. В некоторых случаях предпочтительно добавлять иодистую соль, в частности иодид щелочного металла. Некоторое повышение температуры и перемешивание позволяют увеличить скорость реакции. Кроме того, может оказаться целесообразным проводить указанное N-алкилирование инертного газа, например не содержащего кислорода аргона или азота.
По другому варианту N-алкилирование можно осуществлять в известных условиях с использованием катализаторов, переносчиков между фазами. Такие условия включают перемешивание реагентов с подходящим основанием, при желании в атмосфере инертного газа в соответствии с вышеуказанным определением, в присутствии подходящего межфазного катализатора, например галоида, гидроксида, гидросульфата триалкилфенилметиламмония, тетраалкиламмония, тетраалкилфосфония, тетраарилфосфония и других подобных катализаторов.
Соединения формулы (I), у которых R1 означает атом водорода (такие соединения описываются формулой (I-c)), могут быть получены также путем конденсирования фуранового производного формулы (XVI) с альдегидом R2-СНО формулы (XVII) в присутствии подходящего кислого или основного катализатора.
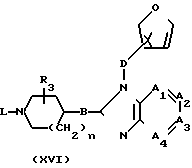

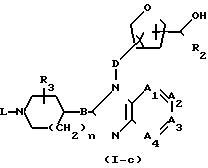
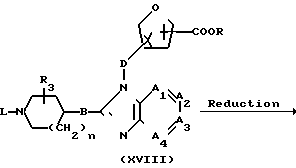
Указанные соединения формулы (I-c) могут быть также получены путем восстановления производного карбоновой кислоты формулы (XVIII), в которой R означает атом водорода, алкил или арил, восстановителем, например литийалюминийгидридом, боргидридом лития, боргидридом натрия и т.п., в среде инертного в условиях проведения реакции растворителя, например простого эфира, в частности тетрагидрофурана, 1,1'-оксибисэтана, 1,4-диоксана и т.п. , или путем взаимодействия указанной карбоновой кислоты формулы (XVIII) или ее соли с металлоорганическим соединением, в частности С1-С6-алкиллитием, и восстановления образующегося в результате кетона восстановителем, например литийалюминийгидридом, боргидридом лития, боргидридом натрия и т.п., в среде инертного в условиях проведения реакции растворителя, например простого эфира, в частности тетрагидрофурана, 1,1'-оксибисэтана, 1,4-диоксана и т.п.
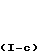
Соединения формулы (I), у которых L имеет иное значение, чем атом водорода (L в этом случае обозначается как L1, а указанные соединения описываются формулой (I-d)), могут быть также получены путем N-алкилирования соединения формулы (I), у которого L означает атом водорода (такие соединения описываются формулой (I-e)), алкилирующим агентом формулы (XIX).
H N
N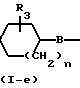 Q
Q  L
L N
N Q
Q
Указанную реакцию N-алкилирования целесообразно проводить обычным для таких случаев образом, как это описано выше при получении соединений формулы (I) из исходных соединений формул (XIV) и (XV).
Соединения формулы (I-d), у которых L означает С3-С6-циклоалкил, С1-С12-алкил, радикал формулы (b-1), (b-2) или (b-3) (эти радикалы могут быть представлены как радикал L2Н, а указанные соединения описываются формулой (I-d-I), могут быть также получены путем редуктивного N-алкилирования соединения формулы (I-e) кетоном или альдегидом формулы L2=O (ХХ), причем указанное соединение формулы L2=O представляет собой промежуточное соединение формулы L2Н2, у которого два атома водорода, находящиеся у одного атома углерода, замещены = O, а L2 представляет собой двухвалентный радикал с двумя связями у одного атома углерода, включающий С3-С6-циклоалкилдиен, С1-С12-алкилидан, R5-С1-С6-алкилиден, R6-Y-C1-C6-алкилиден и R7 - Z2-C(= X) - Z1-C1-C6-алкилиден.
(I-e L2= 0
L2= 0  L2H
L2H N
N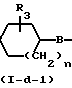 Q
Q
Указанную реакцию восстановительного N-алкилирования целесообразно проводить таким же образом, как это описано при получении соединений формулы (I-b) из исходных соединений формулы (VII-a) и (XII), в частности путем каталитического гидрирования.
Соединения формулы (I), у которых L означает радикал формулы (b-2), а R6-арил или Het (R6 в этом случае может быть обозначен как R6-а, а указанные соединения описаны формулой (I-d-2), могут быть также получены путем алкилирования соединений формулы (I), у которых L означает радикал формулы (b-2), а R6 означает атом водорода (эти соединения могут быть описаны формулой (I-d-3) реагентом формулы (XXI).
H-Y-Alk N
N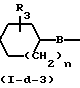 Q
Q 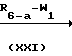 R6-a-Y-Alk
R6-a-Y-Alk N
N Q
Q
Аналогичным образом соединения формулы (I-d-2) могут быть получены путем обработки соединения формулы (I-d-4) реагентом формулы (XXII).
W′-Alk N
N Q
Q  R6-a-Y-Alk
R6-a-Y-Alk N
N Q
Q
Реакции алкилирования соединения формулы (I-d-3) соединением формулы (XXI) и соединения формулы (I-d-4) соединением формулы (XXII) целесообразно проводить в среде инертного органического растворителя, например ароматического углеводорода, в частности бензола, метилбензола, диметилбензола, кетона, в частности 2-пропанона, 4-метил-2-пентанона, простого эфира, в частности 1,4-диоксана, 1,1' -оксибисэтана, тетрагидрофурана, или диполярного апротонного растворителя, в частности N,N-диметилформамида, N,N-диметилацетамида, диметилсульфоксида, нитробензола, 1-метил-2-пирролидинона и т. п. Для связывания кислоты, образующейся в результате реакции, к реакционной смеси можно добавлять соответствующее основание, например карбонат или гидрокарбонат щелочного металла, гидрид натрия или органическое основание, например N,N-диэтилэтанамин или N-(1-метилэтил)-2-пропанамин. Некоторое повышение температуры может увеличить скорость реакции.
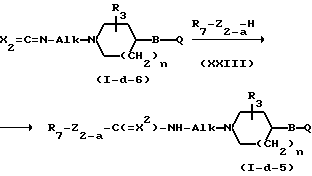
Соединения формулы (I), у которых L означает радикал формулы (-b-3), Z1 - означает NH, Z2 имеет иное значение, чем простая связь, а Х имеет иное значение, чем NR11 (Z2 и Х в этом случае могут быть обозначены соответственно Z2-a и Х2, а указанные соединения описаны формулой (I-d-5), могут быть получены путем взаимодействия изоцианата (Х2=О) или изотиоцианата (Х2=S) формулы (I-d-6) с соединением формулы (XXIII).
Соединения формулы (1), у которых L означает радикал формулы (b-3), Z2 означает NH, Z1 имеет иное значение, чем простая связь, а Х имеет иное значение, чем NR11(Z1 и Х в этом случае могут быть обозначены как Z1-a и Х2), а соединения описаны формулой (I-d-7), могут быть получены путем взаимодействия изоцианата (Х2=О) или изотиоцианата (Х2=S) формулы (XXIV) с соединением формулы (I-d-8).

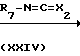
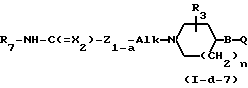
Реакции между соединениями формул (XXIII) и (I-d-6) или (XXIV) и (I-d-8), как правило, можно проводить в среде подходящего, инертного в условиях реакции растворителя, например простого эфира, в частности тетрагидрофурана и т.п., галогенированного углеводорода, в частности трихлорметана и т.п. Скорость реакции можно увеличить путем повышения температуры.
Соединения формулы (1), у которых L означает радикал формулы (b-3), Z2 - простую связь, Z1 имеет иное значение, чем простая связь, а Х имеет иное значение, чем NR11 (Z1 и Х в этом случае обозначены соответственно Z1-a и Х2, а указанные соединения описываются формулой (I-d-9), могут быть получены путем взаимодействия соединения формулы (XXV) или его реакционноспособного функционального производного с соединением формулы (I-d-8).
HZ1-a-Alk N
N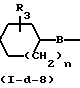 Q
Q 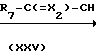 R7-C(=X2)-Z
R7-C(=X2)-Z Alk
Alk N
N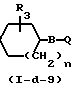
Реакцию между соединениями формул (XXV) и (I-d-8) можно проводить известным образом, как обычно проводят реакции этерификации или амидирования. Так, например, карбоновая кислота может быть переведена в ее реакционноспособное производное, например ангидрид или галоидангидрид, которое затем подвергают взаимодействию с соединением формулы (I-d-8). По другому варианту реакцию между (XXV) и (I-d-8) проводят в присутствии подходящего реагента, способного образовывать амиды или сложные эфиры. Таким реагентом может быть, например, N,N-метантетраилбис(циклогексамин), 2-хлор-1-метилпиридинийиодид и т. п. Эти реакции целесообразно проводить в среде подходящего растворителя, например простого эфира, в частности тетрагидрофурана, галогенированного углеводорода, в частности дихлорметана или трихлорметана, диполярного апротонного растворителя и т.д. Целесообразным может оказаться добавление к реакционной смеси основания, например N,N-диэтилэтанамина и т. п.
Соединения формулы (I), у которых L означает радикал формулы L3-С2-С6-алкандиил, где L3 означает арил, Het, арилсульфонил или радикал формулы R7 - Z2-C(=X)- (указанные соединения могут быть описаны формулой (I-d-10), могут быть также получены путем реакции присоединения соединения формулы (I-e) к соответствующему алкену формулы (XXVI).
H N
N Q
Q  L3-C2-6аикандиид
L3-C2-6аикандиид  N
N
Соединения формулы (I), у которых L означает 2-окси-С2-С6-алкил или радикал формулы (b-4) (указанные соединения описываются формулой (I-d-11), могут быть получены путем взаимодействия соединения формулы (I-n) с эпоксидом формулы (XXVII), где R14 означает атом водорода, С1-С4-алкил или радикал R8-O-СН-2-.
H N
N Q
Q R14-CHOH-CH
R14-CHOH-CH N
N
Реакцию между соединением формулы (I-e) и соединением формулы (XXVI) или (XXVII) можно проводить при перемешивании и, при желании, нагреве, в среде инертного в условиях реакции растворителя, например кетона, в частности 2-пропанона, 4-метил-2-пентанона, простого эфира, в частности тетрагидрофурана, 1,1' -оксибисэтана, спирта, в частности метанола, этанола, 1-бутанола, диполярного апротонного растворителя, в частности N,N-диметилформамида, N,N-диметилацетамида, и т.д.
Соединения формулы (I), у которых R5, R6 или R7 означают Het, могут быть получены известными способами, использующимися для получения гетероциклических кольцевых систем, или другими аналогичными способами. Некоторые из таких способов циклизации описаны, например, в патенте США N 4695575 и в приведенных в нем ссылках, в частности в патентах США N 4335127, 4342870 и 4443451.
Соединения формулы (I) могут быть также переведены в другие соединения этой же формулы известными способами трансформации функциональных групп. Соединения формулы (I), содержащие в качестве заместителя циано-группу, могут быть переведены в соответствующие амины путем перемешивания и, при желании, нагревания исходного цианопроизводного в водородсодержащей среде в присутствии подходящего катализатора, например платины на активированном угле, никеля Ренея и т.п. Подходящими растворителями являются, например, метанол, этанол и т.п. Аминогруппы могут быть переведены в соответствующие изоцианатные группы путем обработки исходного соединения CS2, при желании в присутствии N, N-метантетраилбис(циклогексамина). Амино-группы могут быть алкилированы или ацилированы известными способами, например путем N-алкилирования, N-ацилирования, редуктивного N-алкилирования и т.д. Соединения формулы (I), содержащие амино-группу, замещенную радикалом арил-СН2, могут быть подвергнуты гидрогенолизу путем обработки исходного соединения водородом в присутствии подходящего катализатора, например палладия или платины на активированном угле и т.п., в спиртовой среде.
В случае всех предыдущих и нижеследующих операций продукты реакции могут быть выделены из реакционной смеси и при желании подвергнуты дополнительной очистке известными способами.
Некоторые полупродукты и исходные материалы, использующиеся для осуществления вышеописанных способов, являются известными соединениями, которые могут быть получены известными способами, использующимися для получения этих или аналогичных соединений. Другие же описанные соединения являются новыми. Некоторые способы получения этих соединений более подробно будут описаны в нижеследующих примерах.
Исходные материалы, например промежуточные соединения формул (II), (IV), (VI), (VIII), (X), (XII), (XIII), (XV) и (XVI) могут быть получены с помощью способов, аналогичных тем, которые описаны, например, в патентах США N 4219559, 4556660, 4634704, 4695569, 4695575, 4588722, 4835161 и 4897401, а также в европейских патентах А-0206415, 0282133, 0297661 и 0307014.
Полупродукты формулы (III) могут быть получены из исходного ароматического соединения с заместителями атомом галогена и нитрогруппой у соседних атомов углерода формулы (XXVIII), путем взаимодействия его с соответствующим амином формулы (XXIX), осуществляемого известным способом восстановления нитро- в амино-группу.
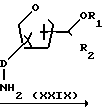
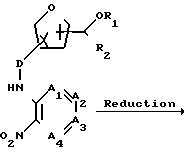
Полупродукты формул (V), (VII), (IX) и (XI) могут быть получены из полупродуктов формулы (III) известным способом переведения ароматических соединений, замещенных амино-группами у соседних атомов углерода, в бензимидазолы, имидазопиридины и/или пурины.
Полупродукты формулы (XVIII) могут быть получены путем N-алкилирования полупродукта формулы (XV) соответствующим образом замещенного производного фуранкарбоновой кислоты формулы (XXX), у которого Р означает атом водорода, алкил или арил, как это описано выше при получении соединений формулы (I) из полупродуктов формул (XV) и (XIV).


Соединения формулы (I), их фармакологически приемлемые кислые аддитивные соли и стереоизомеры обладают ценными фармакологическими свойствами. Так, в частности, они являются активными антигистаминными агентами, что может быть убедительно продемонстрировано, например, результатами тестов "Защита крыс от вызывающего их гибель соединения 48/80", "Антагонизм гистамина у морских свинок" и "Ascaric Allergy-тесты на собаках". Помимо антигистаминной активности некоторые из заявляемых соединений обладают также антагонистической активностью по отношению к серотонину, что можно продемонстрировать с помощью теста "желудочные заболевания крыс, индуцируемые соединением 48/80".
Благодаря своей антигистаминной и серотонинной активности соединения формулы (I) и их кислые аддитивные соли могут с успехом использоваться для лечения аллергических заболеваний, таких, как, например, аллергические насморки, аллергические конъюнктивиты, аллергическая крапивница, аллергическая астма и т.п.
Поскольку предлагаемые соединения обладают антиаллергической активностью, на их основе можно готовить различные фармацевтические препараты. Для получения антиаллергических композиций в соответствии с настоящим изобретением эффективное количество заявляемого соединения в виде основания или кислой аддитивной соли, играющих роль активного компонента, тщательно смешивают с фармацевтически приемлемым носителем, который может находиться в самой различной форме в зависимости от вида получаемой композиции. Предпочтительно выпускать такие фармацевтические композиции в виде единичных доз, которые можно было бы вводить орально, ректально, через кожу или парентерально. Например, при получении композиций для орального введения можно использовать любые из обычно применяемых фармацевтических ингредиентов, в частности воду, гликоли, масла, спирты и т.д. (при получении жидких композиций, таких как суспензии, сиропы, эликсиры и растворы) или твердые носители, такие как крахмалы, сахара, каолин, скользящие добавки, связующие, диспергаторы и т.д. (при получении порошков, пилюль, капсул и таблеток). Благодаря простоте введения таблетки и капсулы являются наиболее предпочтительными оральными единичными дозами, в которых, естественно, применяются твердые фармацевтические носители. В парентеральных композициях в качестве носителя обычно применяют (по меньшей мере частично) стерилизованную воду, хотя, например, для облегчения растворимости, носитель может содержать и другие компоненты. Для получения растворов для инъекций в качестве носителя можно использовать, например, физиологический раствор, раствор глюкозы или смесь этих растворов. На основании предлагаемых соединений могут быть также получены суспензии для инъекций, содержащие подходящие жидкие носители, диспергаторы и т.п. В композициях для введения через кожу носитель может содержать агент, способствующий проникновению активного вещества, и/или подходящий смачиватель, при желании в комбинации с небольшими количествами подходящих добавок любой природы, не оказывающих вредного действия на кожу. Эти добавки могут облегчать введение композиций через кожу и/или оказаться полезными при их получении. Эти композиции можно вводить различными способами, например чреcкожно, путем нанесения в виде пятна или в виде мази. Кислые аддитивные соли соединений формулы (I) благодаря их более высокой растворимости в воде по сравнению с соответствующими основными формами могут, очевидно, использоваться для получения водных композиций.
Предпочтительно, в частности, выпускать перечисленные фармацевтические композиции в виде единичных доз. Это облегчает их прием и дозировку. Под используемым в описании и формуле изобретения определением "единичные дозы" имеются в виду приготовленные в виде физически дискретных порций единичные дозы, каждая из которых содержит определенное количество активного компонента, способного вызывать нужный терапевтический эффект, в комбинации с соответствующим фармацевтическим носителем. Примерами таких единичных доз являются таблетки (включая таблетки с надрезом или таблетки с оболочкой), капсулы, пилюли, порошки в пакетиках, облатки, растворы или суспензии для инъекций, дозировки жидких композиций объемом в чайную или столовую ложку, а также сегрегированные упаковки, содержащие множество таких доз.
Настоящее изобретение относится также к способу лечения теплокровных животных, страдающих вышеуказанными аллергическими заболеваниями, путем введения антиаллергически эффективного количества соединения формулы (I) или его фармацевтически приемлемой кислой аддитивной соли.
Специалисты по лечению аллергических заболеваний теплокровных животных легко могут определить эффективное количество из приведенных ниже результатов испытаний. Предполагается, что антиаллергически эффективное количество активного вещества должно составлять от примерно 0,001 до примерно 100, предпочтительно от примерно 0,01 до примерно 1 мг/кг веса тела.
Нижеследующие примеры иллюстрируют настоящее изобретение, но никоим образом не ограничивают его объем. Если это не оговорено, все приведенные части являются весовыми.
А. Получение полупродуктов.
П р и м е р 1. Смесь 28,8 ч. этил-4-(1Н-бензимидазол-2-иламино)-1-пиперидинкар- боксилата (полученного, как это описано в примере XIV патента США N 4219559), 33,9 ч. этил-5-хлорметил-2-фуранкарбоксилата, 15,9 ч. карбоната натрия и 282 ч. N,N-диметилформамида перемешивали в течение двух ночей при 70оС. Реакционную смесь затем выливали в воду и образующийся продукт экстрагировали метилбензолом. Экстракт промывали водой, сушили, фильтровали и упаривали. Остаток подвергали очистке с помощью колоночной хроматографии на силикагеле (подвижная фаза: смесь CHCl3 и CH3OH в соотношении 97: 3). Элюат нужной фракции упаривали и остаток перемешивали в 1,1' -оксибисэтане. Выпадающий осадок отфильтровывали и высушивали. В результате получали 31,2 ч. (70,8%) этил-4-[[1-[[5-(этоксикарбонил)-2-фуранил]метил] -1Н-бензимидазол-2-ил] - амино] -2-пиперидинкарбоксилата. Т. пл. 136,0оС (полупродукт 1).
Аналогичным образом 4-(1Н-бензимидазол-2-иламино)гексагидро-1Н-азепин-1-карбоксилат (полученный таким же образом, как это описано в примере 9 европейского патента N 0297661, опубликованного 4.01.1989) переводили в этил-4-[[1-[[-5- (этоксикарбонил)-2-фуранил]метил]-1Н-бензими- дазол-2-ил] амино]гексагидро-1Н-азепин-1-карбоксилат (полупродукт 2) и этил-3-(1Н-бензимидазол-2-иламино)-1-пирроли- динкарбоксилат моногидрохлорид (по-лученный таким же образом, как это описано в примере 8 европейского патента N 0297661, опубликованного 4.01.1989) в этил-3-[[1-[[5-(этоксикарбонил)-2-фуранил] - метил] -1Н-бензимидазол-2-ил] амино] -1-пирролидинкарбоксилат (полупродукт 3).
П р и м е р 2. К 470 ч. N,N-диметилформамида добавляли порциями 17,3 ч. дисперсии гидрида натрия в минеральном масле (50% ) и 91,6 ч. 2-[[1-(фенилметил)-4-пиперидинил] метил] -1Н-бензимидазола (полученного таким же образом, как это описано в примере 16 патента N 4695575), при перемешивании, в атмосфере азота. После перемешивания в течение часа к смеси по каплям, при охлаждении добавляли 67,9 ч. этил-5-хлорметил-2-фуранкарбоксилата. Перемешивание продолжали в течение еще часа, после чего к реакционной смеси добавляли воду. Полученный продукт экстрагировали метилбензолом и экстракт промывали водой, высушивали, фильтровали и упаривали. Остаток высушивали путем азеотропной перегонки и метилбензолом (дважды). В результате получали 119 ч. (86,6%) этил-5-[[2-[[1-(фенилметил)-4-пиперидинил]]метил-2-фуранкарбоксилата (полупродукт 4). Таким же образом, используя соответствующие исходные материалы, получали
метил-5-[[2-[[1-[2-(4-метоксифенил)этил]- 4-пиперидинил]амино]-1Н-бензимидазол-1- -ил]метил]-2- фуранкарбоксилат, т.пл. 124,3оС (полупродукт 5);
этил-5-[[2-[[1-[2-(4-метоксифенил)этил] -4-пиперидинил-] амино] - 1Н-бензимидазол-1-ил] метил] -3-фуранкарбоксилат, т. пл. 121,9оС (полупродукт 6);
метил-5-[[-2-[(1-метил-4-пиперидинил)-амино]-1Н-бензимидазол-1-ил] метил]-2-фуранкарбоксилат, т. пл. 169,5оС (полупродукт 7) ;
этил-5-[[2-[(1-метил-4- пиперидинил)амино]-1Н-бензимидазол-1-ил]метил] -3-фуранкарбоксилат (Е)-2-бутендиоат (1:2). Т.пл. 200,9о (полупродукт 8);
этил-2-[[2-[[1-[2-(4-метоксифенил)этил] -4-пиперидинил] амино]-1Н- бензимидазол-1-ил] метил] -3-фуранкарбоксилат (полупродукт 9); этил -4-[[1-[[3-(этоксикарбонил)-2-фуранил] метил] -1Н-бензимидазол- 2-ил]амино]- 1-пиперидинкарбоксилат/ т. пл. 162,3оС (полупродукт 10);
этил-4-[[1-[[2-(метоксикарбонил)-3-фура- нил]метил]-1Н-бензимидазол-2- ил] амино] -1-пиперидинкарбоксилат (полупродукт 11) и метил-3-[[2-[[1-[2-(4-метоксифенил)этил] -4-пиперидинил] амино]-1Н- бензимидазол-1-ил]метил]-2-фуранкарбоксилат (полупродукт 12).
П р и м е р 3. а) Смесь 55 ч. N-(2-фуранилметил)-3-нитро-2- пиридинамина, 2 ч. 4%-ного раствора тиофена в метаноле и 400 ч. метанола, насыщенного аммиаком, подвергали гидрированию при нормальном давлении и комнатной температуре в присутствии 4 ч. 5%-ного платинового катализатора на активированном угле. После поглощения расчетного количества водорода катализатор отфильтровывали и фильтрат упаривали, в результате чего получали 48 ч. N2-(2-фуранилметил)-2,3-пиридиндиамина (полупродукт 13).
в) Смесь 54 ч. этил-4-изоцианато-1-пиперидинкарбоксилата, 48 ч. полупродукта (13) и 450 частей тетрагидрофурана перемешивали в течение ночи при кипячении с обратным холодильником, после чего реакционную смесь упаривали и остаток перекристаллизовывали из смеси 2-пропанона и 2,2'-оксибиспропана. В результате получали 76 ч. (75%) этил-4-[[2-[([2-(фуранилметил)амино]-3-пиридинил]аминотиоксоме- тил]амино] -1-пиперидинкарбоксилата, т.пл. 132,7оС (полупродукт 14).
Аналогичным образом этилгексагидро-4-изотиоцианато-1Н-азепин-1- карбоксилат (полученный по способу в соответствии с примером 9 европейского патента N 0297661, опубликованного 4 января 1989 года) переводили в этилгексагидро-4-[[[[2-[[[5-(оксиметил)-2-фуранил]метил]амино]-3- пиридинил]амино] тиоксометил]амино]-1Н- азепин-1-карбоксилат (полупродукт 15).
с) Смесь 74 ч. полупродукта (14), 96 ч. оксида ртути (11), 0,1 ч. серы и 800 ч. этанола перемешивали в течение 3 ч при кипячении с обратным холодильником. Реакционную смесь затем фильтровали через диатомную землю и фильтрат упаривали. Остаток перекристаллизовывали из ацетонитрила, получая в результате 52,5 ч. (79%) этил-4-[[3-(2-фуранилметил)-3Н-имидазо[4,5-b]пиридин-2-ил]амино]-1- пиперидинкарбоксилата. Т.пл. 149,2оС (полупродукт 16).
П р и м е р 4. а) Смесь 16,3 ч. 4,6-дигидро-5-пиперидинамина, 14 ч. 5-(аминометил)-2-фуранметанола, 12 ч. N,N-диэтилэтанамина и 200 ч. воды перемешивали в течение 10 ч при кипячении с обратным холодильником. Реакционную смесь затем упаривали и остаток подвергали очистке с помощью колоночной хроматографии на силикагеле с использованием в качестве подвижной фазы смеси CH2Cl2 и CH3OH(NH3) при соотношении 95:5. Элюат фракции, содержащей целевой продукт, упаривали и остаток перекристаллизовывали из ацетонитрила. Продукт отфильтровывали и высушивали, получая в результате 19,5 ч. (76,6%) 5-[[(5-амино-6-хлор-4-пиримидинил)-амино] метил] -2-фу- ранметанола. Т.пл. 136,7оС (полупродукт 17).
в) Смесь 18,5 ч. полупродукта (17), 1 ч. 4%-ного раствора тиофена в метаноле, 119 ч. метанола и 10 ч. оксида кальция гидрировали при нормальном давлении и комнатной температуре в присутствии 4 ч. 10%-ного палладиевого катализатора на активированном угле. После поглощения расчетного количества водорода катализатор отфильтровывали и фильтрат упаривали, получая в результате 15,9 ч. (100%) 5-[[(5-аино-4-пиримидинил)амино]метил]-2-фуран- метанола (полупродукт 18).
П р и м е р 5. а) Смесь 15,9 ч. 4-хлор-3-нитропиридина, 12,7 ч. 5-(аминометил)-2-фуранметанола, 13,3 ч. N,N-диэтилэтанамина и 745 ч. трихлорметана перемешивали в течение 3 ч. при температуре кипения с обратным холодильником. После охлаждения реакционную смесь промывали водным раствором К2СО3, высушивали, фильтровали и упаривали. Остаток перекристаллизовывали из ацетонитрила. Целевой продукт отфильтровывали и высушивали. В результате получали 17,76 ч. (71,3%) 5-[[(3-нитро-4-пиридинил)амино]-метил]-2-фуранметанола. Т.пл. 134,9оС (полупродукт 19).
в) Смесь 17,3 ч. полупродукта (19), 1 ч. 4%-ного раствора тиофена в метаноле и 158 ч. метанола гидрировали при нормальном давлении и комнатной температуре в присутствии 1 ч. 5%-ного платинового катализатора на активированном угле. После поглощения расчетного количества водорода катализатор отфильтровывали и фильтрат упаривали. Остаток перекристаллизовывали из ацетонитрила. Продукт отфильтровывали и высушивали, получали в результате 12,7 ч. (83,9% ) 5-[[(3-амино-4-пиридинил)амино]-метил]-2-фуран- метанола (полупродукт 20).
с) Смесь 14,3 ч. этил-4-изотиоцианато-1-пиперидинкарбоксилата, 12,7 ч. полупродукта (20) и 188 ч. N,N-диметилформамида перемешивали в течение ночи при 60оС, после чего реакционную смесь упаривали, получая в результате 25,1 ч. (100% ) этил-4-[[[[4-[[[5-(оксиметил)-2-фуранил]метил]амино]- 3-пиридинил] амино]тиоксометил]амино]-1-пиперидинкарбоксилата (полупродукт 21).
Проводя процесс таким же образом, полупродукт (18) переводили в 4-[[[[4-[[[5-(оксиметил)-2-фуранил]метил]амино]-5-пирими-динил]амино] тиоксометил]амино]-1-пиперидинкарбоксилата (полупродукт 22).
П р и м е р 6. а) К раствору 25 ч. 1-(фенилметил)-4-пиперидинацетонитрила в 178 ч. тетрагидрофурана добавляли по каплям при комнатной температуре 37,97 весов. части хлорформиата. Реакционную смесь перемешивали в течение 15 ч при комнатной температуре и затем упаривали. Остаток растворяли в 90 ч. этилацетата и раствор промывали последовательно 3 н. HCl, NaHCO3 и насыщенным раствором NaCl. Растворитель отгоняли и остаток перегоняли при температуре 100-110оС и давлении 6,7 Па. В результате получали 18,7 ч. (81,4%) этил-4-(цианометил)-1-пиперидинкарбоксилата (полупродукт 24).
в) Смесь 18,32 ч. полупродукта (24), 4,30 ч. этанола и 74,5 ч. трихлорметана охлаждали на ледяной бане и при охлаждении пропускали через нее в течение 30 мин хлористый водород. Реакционную смесь оставляли затем в холодильнике на 48 ч, после чего упаривали. Остаток растирали со 142 ч. 1,1' - оксибисэтана. Продукт высушивали в вакууме, в результате получали 14,2 ч. (54,1% ) этил-4-(2-этокси-2-иминоэтил)-1-пиперидин- карбоксилат моногидрохлорида (полупродукт 25).
Проводя процесс таким же образом, 1-(фенилметил)-4-пиперидинацетонитрил переводили в Q этил-1-(фенилметил)-4-пиперидинэтанимидат дигидрохлорид (полупродукт 26).
В. Получение целевых соединений.
П р и м е р 7. а) К перемешиваемой смеси 4,4 ч. полупродукта (1) и 133,5 ч. тетрагидрофурана добавляли по каплям в атмосфере азота 5 мл раствора тетрагидробората лития в тетрагидрофуране (2М). Перемешивание продолжали в течение ночи, кипятя смесь с обратным холодильником, после чего добавляли к ней последовательно 2-пропанон и уксусную кислоту. Полученную реакционную массу упаривали, остаток растворяли в воде и подщелачивали К2СО3. Образующийся продукт экстрагировали дихлорметаном, экстракт высушивали, фильтровали и упаривали. Остаток перекристаллизовывали из 4-метил-2-пентанона, получая в результате 2,08 ч. (52,2%) этил-4-[[1-[[5-(оксиметил)-2-фуранил] метил] -1Н-бензимидазол-2- ил]амино]-1-пиперидинкарбоксилата. Т. пл. 141,6оС (соединение 3.05).
в) Смесь 75,7 ч. соединения (3,05), 106,5 ч. гидроксида калия и 390 ч. 2-пропанола перемешивали в течение ночи при температуре кипения с обратным холодильником. После охлаждения реакционную смесь упаривали и остаток растворяли в воде. Образующийся продукт экстрагировали дихлорметаном и экстракт фильтровали через диатомную землю, а затем упаривали. Остаток перекристаллизовывали из 2-пропанона, получая в результате 46,5 ч. 5-[[2-(4-пиперидиниламино)-1Н-бензимидазол-1- ил] метил]-2-фуран-метанола. Т.пл. 156,3оС (соединение 3.11).
с) Смесь 4,53 ч. хлорацетонитрила, 16,26 ч. соединения (3.11), 8 ч. карбоната натрия и 141 ч. N,N-диметилформамида перемешивали в течение 2 ч при комнатной температуре. Реакционную смесь выливали затем в воду и образующийся продукт экстрагировали трихлорметаном. Экстракт высушивали, фильтровали и упаривали. Остаток перемешивали в 2,2 '-оксибиспропане и выпадающий в осадок продукт отфильтровывали. В результате получали 17,71 ч. (96,9%) 4-[[1-[[5-(оксиметил)-2-фуранил]метил]-1Н-бензимидазол-2-ил]-1- пиперидинацетонитрила. Т.пл. 209,8оС (соединение 3.22).
d) Смесь 16,6 ч. соединения (3.22) и 790 ч. метанола, насыщенного аммиаком, гидрировали при нормальном давлении и комнатной температуре в присутствии 6 ч. никеля Ренея. После поглощения расчетного количества водорода катализатор отфильтровывали и фильтрат упаривали. Остаток перекристаллизовывали последовательно из ацетонитрила и 2-пропанола, получая в результате 7,4 ч. 5-[[2-[[1-(2-аминоэтил)-4-пиперидинил]амино]-1Н-бензимидазол-1-ил]- метил]-2-фуранметанола. Т.пл. 164,6оС (соединение 3.23).
е) К перемешиваемому раствору 1,38 ч 1-метил-1Н-2-пирролкарбоновой кислоты, 2,81 ч. 2-хлор-1-метилпиридинийиодида, 2,2 ч. N,N-диэтилэтанамина и 199,5 ч. дихлорметана добавляли раствор 3,7 ч. соединения (3.23) в смеси дихлорметана и N, N-диметилацетамида. После перемешивания в течение 3 ч реакционную смесь выливали в воду. Образующийся продукт экстрагировали дихлорметаном, экстракт высушивали, фильтровали и упаривали. Остаток подвергали очистке с помощью колоночной хроматографии на силикагеле, используя в качестве подвижной фазы смесь CHCl3 и CH3OH, насыщенный аммиаком, в соотношении 95:5. Фракцию элюата, содержащую целевой продукт, упаривали и остаток перекристаллизовывали из ацетонитрила. В результате получали 2,25 ч. (47,2% ) N-[2-[4-[[1-[[5-(оксиметил)-2-фуранил] метил]-1Н-бен-зимидазол-2- ил] амино] -1-пипери-динил] этил]-1-метил-1Н-пиррол-2-карбокса- мида. Т.пл. 182,2оС (соединение 3.24).
П р и м е р 8. К перемешиваемой смеси 12 ч. литийалюминийгидрида и 445 ч. тетрагидрофурана добавляли при температуре кипения, в атмосфере азота по каплям раствор 137 ч. полупродукта (4) в тетрагидрофуране. Кипячение смеси с обратным холодильником продолжали в течение еще часа, после чего ее охлаждали и добавляли к ней последовательно этилацетат, 42 ч. 15%-ного раствора NaOH (по каплям) и 36 ч. воды. Полученную смесь фильтровали, фильтрат упаривали и остаток растворяли в воде. Образующийся продукт экстрагировали дихлорметаном, экстракт высушивали, фильтровали и упаривали. Остаток перекристаллизовывали из ацетонитрила, в результате чего получали 64,1 ч. (51,4% ) 5-[[2-[[1-(фенилметил)-4-пиперидинил]метил]--1Н-бензимидазол-1-ил] метил]- 2-фуранметанола. Т.пл. 143,8оС (соединение 1.01).
в) К перемешиваемой смеси 14,1 ч. 2-хлор-1-метилпиридинийиодида, 11,5 ч. N, N-диэтилэтанамина и 282 ч. N,N-диметилформамида добавляли по каплям при комнатной температуре 3,3 части уксусной кислоты. После перемешивания в течение часа к реакционной смеси добавляли 41,5 ч. соединения (1.01), перемешивание продолжали в течение ночи и затем выливали смесь в воду. Образующийся продукт экстрагировали дихлорметаном, экстракт высушивали, фильтровали и упаривали, получая в результате 40 ч. (87,4%) 5-[[2-[[1-(фенилметил)-4-пиперидинил] метил] -1Н-бензимидазол-1-ил] метил]-2-фуранметанолацетата (сложный эфир) (соединение 1.02).
с) К перемешиваемой смеси 45,8 ч. соединения (1.02) и 261 ч. метилбензола добавляли по каплям при температуре кипения 12 ч. этилхлорформиата. Кипячение смеси с обратным холодильником продолжали в течение еще часа. После охлаждения к реакционной смеси добавляли воду. Образующийся продукт экстрагировали метилбензолом. Экстракт высушивали, фильтровали и упаривали, получая в результате 44,0 ч. (100%) этил-4-[[1-[[5-(ацетилокси)метил]-2-фуранил]метил]- 1Н-бензимидазол-2-ил]метил]-1- пиперидинкарбоксилата (соединение 1.03). Полученное соединение содержало, кроме того, некоторое количество побочного продукта, а именно этил-4-[[1-[[5-[(фенил)метокси] метил] -2-фуранил] метил] -1Н- бензимидазол-2-ил]метил]-1-пиперидинкарбоксилата.
d) Смесь 44,0 ч. соединения (1.03), 56 ч. гидроксида калия и 234 ч. 2-пропанолда перемешивали в течение ночи при кипячении с обратным холодильником. После этого реакционную смесь упаривали и остаток растворяли в воде. Образующийся продукт экстрагировали дихлорметаном и экстракт высушивали, фильтровали и упаривали. Остаток подвергали очистке с помощью колоночной хроматографии на силикагеле, используя в качестве подвижной фазы смесь CHCl3 и CH3OH, насыщенный аммиаком, в соотношении 95:5 80:20). Элюат второй фракции упаривали, получая в результате 27,5 ч. (84,5%) 5-[[2-(4-пиперидинилметил)-1Н-бензимидазол-1-ил]метил]-2- фуранметанола (соединение 1.04).
После упаривания первой фракции получали 9 ч. 2-[[5-(фенилметокси)метил]-2-фуранил]метил]-2-(4-пиперидинилметил)-1Н- бензимидазола (соединение 1.05).
е) Смесь 3,25 ч. соединения (2.04), 2 ч. полиоксиметилена, 2 ч. 4%-ного раствора тиофена в метаноле и 119 ч. метанола гидрировали при нормальном давлении и комнатной температуре в присутствии 2 ч. палладиевого катализатора на активированном угле. После поглощения расчетного количества водорода катализатор отфильтровывали и фильтрат упаривали. Остаток растворяли в дихлорметане и раствор промывали водным раствором NH4OH. Органический слой высушивали, фильтровали и упаривали. Остаток последовательно перекристаллизовывали из 4-метил-2-пентанона и ацетонитрила, получая в результате 1,72 ч. (50,7%) 5-[[2-[(1-метил-4-пиперидинил)метил]-1Н-бензимидазол-1-ил]метил] -2- фуранметанола. Т.пл. 158,1оС (соединение 1.17).
П р и м е р 9. Через перемешиваемую смесь 8,4 ч. полупродукта (16), 5,05 ч. 40%-ного раствора формальдегида и 4,25 ч. пиперидина продували хлористый водород до тех пор, пока все твердое вещество не переходило в раствор. Полученный раствор перемешивали в течение уикэнда, после чего обрабатывали аммиаком. Образующийся продукт экстрагировали трихлорметаном. Экстракт высушивали, фильтровали и упаривали. Остаток подвергали очистке с помощью колоночной хроматографии на силикагеле, используя в качестве подвижной фазы смесь CHCl3 и CH3OH в соотношении 97:3 98:2. Фракции элюата, содержащие целевой продукт, упаривали и остаток перекристаллизовывали из смеси 4-метил-2-пентанона и 2,2'-оксибиспропана, получая в результате 1,0 часть (12,5%) этил-4- [[3-[[5-(оксиметил)-2-фуранил]метил]-3Н-имидазо-[4,5-b]пиридин-2-ил] амино]-1-пиперидинкарбоксилата. Т.пл. 148,3оС (соединение 2.01).
П р и м е р 10. Смесь 105,9 ч. полупродукта (25), 197,5 ч. 5-[[(3-амино-2-пиридинил)амино] метил] -2-фуранметанола и 470 ч. N,N-диметилформамида перемешивали в течение 3 дней при комнатной температуре. Реакционную смесь выливали затем в воду, подщелачивали К2СО3 и подвергали экстракции дихлорметаном. Экстракт высушивали, фильтровали и упаривали, а остаток растворяли в 435 ч. метилбензола. К раствору добавляли небольшое количество 4-метилбензолсульфокислоты и перемешивания смесь в течение 2 ч при температуре кипения. После охлаждения и подщелачивания водным раствором К2СО3 продукт экстрагировали дихлорметаном и экстракт высушивали, фильтровали и упаривали. В результате получали 156 ч. (100%) этил-4-[[3-[[5-(оксиметил)-2-фуранил] метил] -3Н-имида-зо[4,5-b] пиридин-2-ил] метил] -1-пиперидинкарбоксилата (соединение 2.13).
П р и м е р 11. Смесь 20 ч. соединения (3.05) и 237 ч. метанола подкисляли серной кислотой до рН 1 при перемешивании. Перемешивание продолжали в течение ночи при температуре кипения. После охлаждения реакционную смесь подщелачивали метанолом, насыщенным аммиаком, и упаривали. Остаток растворяли в воде и продукт экстрагировали дихлорметаном. Экстракт высушивали, фильтровали и упаривали. Остаток подвергали очистке с помощью колоночной хроматографии на силикагеле, используя в качестве подвижной фазы смесь CHCl3 и CH3OH в соотношении 97:3. Фракцию элюата, содержащую целевой продукт, упаривали, получая в результате 29 ч. (100% ) этил-4-[[1-[[5-(метоксиметил)-2-фуранил] метил] -1Н-бензимидазол- -2- ил]амино]-1-пиперидинкарбоксилата (соединение 3.06).
П р и м е р 12. Смесь 2 ч. [(4-фторфенокси)метил]оксирана, 3,26 ч. соединения (3.11) и 39 ч. 2-пропанола перемешивали в течение 48 ч при температуре кипения. Реакционную смесь затем упаривали и остаток подвергали очистке с помощью колоночной хроматографии на силикагеле, используя в качестве подвижной фазы смесь CHCl3 и CH3OH, насыщенного аммиаком, в соотношении 97: 3. Фракцию элюата, содержащую целевой продукт, упаривали и остаток перекристаллизовывали из смеси ацетонитрила и 2,2 '- оксибиспропана, получая в результате 2,80 ч. (56,6%) α- [(4-фторфенокси)метил]-4[[1-[[5-(оксиметил)-2-фуранил] метил] -1Н- бензимидазол-2-ил]амино]-1-пиперидинэ-танола. Т.пл. 136,8оС (соединение 3.20).
П р и м е р 13. К перемешиваемой и охлаждаемой (до 0оС) смеси 3,3 ч. соединения (3.11), 1,01 ч. 1,1' -оксибисэтана и 94 ч. N,N-диметилформамида добавляли по каплям раствор 0,8 ч. ацетилхлорида в N,N'-диметилформамиде. Реакционной смеси давали нагреться до комнатной температуры и упаривали. Осадок кипятили 4 раза в трихлорметане, декантируя после каждого кипячения. Объединенные жидкие фазы высушивали, фильтровали и упаривали. Остаток подвергали очистке с помощью колоночной хроматографии на силикагеле, используя в качестве подвижной фазы смесь CH2Cl2 и CH3OH в соотношении 95:5. Фракцию элюата, содержащую целевой продукт, упаривали и остаток перекристаллизовывали из метанола, получая в результате 0,8 ч. (21,7%) 1-ацетил-N-[1-[[5-(оксиметил)-2-фуранил] метил]-1Н-бензимидазол-2- ил]-4-пиперидинамина. Т.пл. 213,9оС (соединение 3.21).
П р и м е р 14. Смесь 3,7 ч. 7-(2-бромэтил)-3,4-дигидро-8-метил- 2Н, 6Н-пиримидо-[2,1-b] тиазин-6-он моногидробромида, 3,2 ч. соединения (3.11), 2,1 ч. карбоната натрия и 160 ч. 4-метил-2-пентанона перемешивали в течение ночи при температуре кипения. Реакционную смесь затем упаривали и остаток растворяли в воде. Продукт экстрагировали дихлорметаном и экстракт высушивали, фильтровали и упаривали. Остаток подвергали очистке с помощью колоночной хроматографии на силикагеле, используя в качестве подвижной фазы смесь CH2Cl2 и CH3OH в соотношении 95:5. Фракцию элюата, содержащую целевой продукт, упаривали и остаток перекристаллизовывали из ацетонитрила. Продукт отфильтровывали и высушивали, получая в результате 1,5 ч. (28,0%) 3,4-дигидро-7-[2-[4-[[1-[[5-(оксиметил)-2-фуранил] метил] -1Н-бензимидазол- 2-ил] амино]-1-пиперидинил]этил]-8-метил-2Н, 6Н-пиримидо[2,1- b][1,3]-тиазин-6-она. Т.пл. 226,9оС (соединение 3.31).
П р и м е р 15. Смесь 3,3 ч. 2-хлорацетонитрила, 13 ч. соединения (2.03), 5 ч. N,N-диэтилэтанамина и 94 ч. N,N-диметилформамида перемешивали в течение 4 ч при комнатной температуре. После добавления карбоната калия реакционную смесь разбавляли водой, перемешивали в течение непродолжительного времени и образующийся продукт экстрагировали дихлорметаном. Экстракт высушивали, фильтровали и упаривали. Остаток перемешивали в 1,1' -оксибисэтане, фильтровали и высушивали, получая в результате 10,85 ч. (74%) 4-[[3-[[5-(оксиметил)-2-фуранил] метил] -3Н-имидазо-[4,5-b]пиридин-2-ил]амино] -1-пцетонитрила (соединение 2.26).
П р и м е р 16. Смесь 3 ч. 2-этенилпиридина, 3,7 ч. соединения (2.28) и 122 ч. 1-бутанола перемешивали в течение ночи при кипячении с обратным холодильником. Реакционную смесь затем упаривали и остаток подвергали очистке с помощью колоночной хроматографии на силикагеле, используя в качестве подвижной фазы смесь CH2Cl2, CH3O H и СH3ОH, насыщенного аммиаком, при соотношении 90:10:0 90:8:2. Фракцию элюата, содержащую целевой продукт, упаривали и остаток переводили в (Е)-2-бутендиоат (2:3) в этаноле. Эту соль дважды перекристаллизовывали из этанола, полу-чая в результате 2,14 ч. (35,3%) 5-[[2-[[1-[2-(2-пиридинил)этил] -4-пиперидинил]метил]3Н- -имида-зол[4,5- b] -пиридин-3-ил] метил]-2-фуранметанол (Е)-2-бутендиоата (2:3). Т.пл. 159,8оС (соединение 2.35).
П р и м е р 17. Смесь 4,5 ч. 3,6-дихлорпиридазина, 11,1 ч. соединения (1.14) и 3.2 ч. карбоната натрия перемешивали в течение получаса при 150оС. После охлаждения реакционную смесь разбавляли водой. Образующийся продукт экстрагировали трихлорметаном и экстракт высушивали, фильтровали и упаривали. Остаток кипятили в ацетонитриле. После охлаждения осадок отфильтровывали и высушивали, получая в результате 9,72 ч. (67,4%) 5-[[2-[[1-[2-[(6-хлор-3-пиридазинил)амино] этил]-4-пипери- динил]метил]- 1Н-бензимидазол-1-ил]метил]-2-фуранметанола. Т.пл. 188,4оС (соединение 1.21).
П р и м е р 18. Смесь 1,1 ч. 4-хлор-3-нитропиридина, 2,5 ч. соединения (3.23), 1 ч. карбоната натрия и 39,5 ч. этанола перемешивали в течение ночи при комнатной температуре, после чего добавляли к реакционной смеси воду и образующийся продукт экстрагировали дихлорметаном. Экстракт высушивали, фильтровали и упаривали. Остаток перекристаллизовывали из смеси ацетонитрила и этанола, получая в результате 1,9 ч. (56,8%) 5-[[2-[[1-[2-[(3-нитро-4-пиридинил)амино] этил] -4-пиперидинил]амино]- 1Н-бензимидазол-1-ил]метил] -2-фуранметанола. Т.пл. 191,0оС (соединение 3.26).
П р и м е р 19. Смесь 1,74 ч. 4-хлор-3-нитропиридина, 4,22 ч. соединения (4.11), 1,11 части N,N-диэтилэтанамина и 149 ч. трихлорметана перемешивали в течение ночи при комнатной температуре. Реакционную смесь затем промывали водным раствором К2СО3, водный слой отделяли и проводили из него экстракцию дихлорметаном. Объединенные органические слои высушивали, фильтровали и упаривали. Остаток подвергали очистке с помощью колоночной хроматографии на силикагеле, используя в качестве элюента смесь CH2Cl2, CH3OH и CH3OH, насыщенного аммиаком, в соотношении 90:10:1. Фракцию элюата, содержащую целевой продукт, упаривали и остаток перекристаллизовывали из смеси 4-метил-2-пентанона и этанола. Продукт отфильтровывали и высушивали, получая в результате 1,2 ч. (21,2%)-5-[[2-[[гексагидро-1-[2-[(3-нитро-4-пиридинил)амино] этил] -1Н- азепин-4-ил]амино]-1Н-бензимидазол-1-ил]-метил] -2-фуранметанол полугидрата. Т.пл. 155,1оС (соединение 4.14).
П р и м е р 20. К раствору 1,3 ч. литийалюминийгидрида в 89 частях тетрагидрофурана добавляли раствор 4,6 ч. полупродукта (3) в тетрагидрофуране. После кипячения в течение 2 ч с обратным холодильником реакционную смесь обрабатывали этилацетатом, добавляли к ней по каплям при перемешивании 7,9 ч. 15%-ного раствора NaOH и 4,8 ч. воды, фильтровали через диатомную землю и фильтрат упаривали. Остаток подвергали очистке с помощью колоночной хроматографии на силикагеле, используя в качестве подвижной фазы смесь CH2Cl2, СH3ОH и CH3OH, насыщенного NH3, в соотношении 90:5:5. Фракцию элюата, содержащую целевой продукт, упаривали и остаток перекристаллизовывали из смеси ацетонитрила и 2,2'-оксибиспропана (дважды), получая в результате 0,65 части (18,4%) -5-[[2-[(гексагидро-1-метил-1Н-азепин-4-ил) амино] -1Н-бен-зимидазол-1-ил]метил]-2- фуранметанола (соединение 4.02).
П р и м е р 21. Смесь 2,5 ч. 3-бром-N-(1-метилэтил)пропанамида, 3,26 части соединения (3.11), 1,26 ч. гидрокарбоната натрия и 39.5 ч. этанола перемешивали в течение ночи при температуре кипения с обратным холодильником. Реакционную смесь затем упаривали, остаток растворяли в воде, продукт экстрагировали смесью трихлорметана и этанола, экстракт высушивали, фильтровали и упаривали. Остаток перекристаллизовывали из ацетонитрила, получая в результате 3,20 ч. (72,8%) 4-[[1-[[5-(оксиметил)-2-фуранил]метил]-1Н-бензимидазол-2-ил] амино] -N-(1-метилэтил)-1-пиперидинпропанамида. Т. пл. 139,7оС (соединение 3.12).
П р и м е р 22. Смесь 4,36 ч. соединения (1.21), 1 ч. 4%-ного раствора тиофена в метаноле, 198 ч. метанола и 2 ч. оксида кальция гидрировали при нормальном давлении и комнатной температуре в присутствии 2 ч. палладиевого катализатора на активированном угле. После поглощения расчетного количества водорода катализатор отфильтровывали и фильтрат упаривали. Остаток растворяли в воде и продукт экстрагировали дихлорметаном. Экстракт высушивали, фильтровали и упаривали. Остаток подвергали очистке с помощью колоночной хроматографии на силикагеле, используя в качестве элюента смесь CH2Cl2 и CH3OH, насыщенного NH3, в соотношении 95:5. Фракцию элюата, содержащую целевой продукт, упаривали и остаток переводили в соль этандиоат (1:3) в смеси метанола и этанола. Продукт отфильтровывали и высушивали, получая в результате 4,51 ч. (69,9%) 5-[[2-[[1-[2-(3-пиридазиниламино)этил]-4-пи- перидинил]метил]-1Н-бензимидазол-1- ил]метил]-2-фуранметанол этандиоата (1: 3). Т.пл. 203,5оС (соединение 1.24).
П р и м е р 23. Смесь 3,4 ч. соединения (1.21), 0,82 ч. ацетата натрия и 73,5 ч. уксус- ной кислоты перемешивали в течение 4 ч при температуре кипения с обратным холодильником. Реакционную смесь затем упаривали и остаток растворяли в воде. После подщелачивания К2СО3 раствор подвергали экстракции дихлорметаном и экстракт высушивали, отфильтровывали и упаривали. К остатку добавляли 3,5 ч. гидроксида калия и 39 ч. 2-пропанола и смесь перемешивали в течение 3 ч при температуре кипения с обратным холодильником, после чего растворитель отгоняли и добавляли к остатку воду. Образующийся продукт экстрагировали 1-бутанолом и экстракт высушивали, фильтровали и упаривали. Остаток подвергали очистке с помощью колоночной хроматографии на силикагеле, используя в качестве элюента смесь CH2Cl2 и CH3OH, насыщенного NH3, в соотношении 90:10. Фракцию элюата, содержащую целевой продукт, упаривали и остаток переводили в соль этандиоат (2:5) в этаноле. Продукт отфильтровывали и высушивали, получая в результате 2,20 ч. (45,6%) 6-[[2-[4-[[1-[[5-(оксиметил)-2-фуранил] метил] -1Н-бензимида- зол-2-ил]метил]-1-пиперидинил] этил] амино- 3(2Н)-пиридазинон этандиоата (2:5). Т.пл. 210,2оС (соединение 1,25).
П р и м е р 24. Смесь 22,4 ч. полупродукта (15), 13 ч. оксида ртути, (II) столовой ложки серы и 178 ч. тетрагидрофурана перемешивали в течение 3 ч. при температуре кипения с обратным холодильником. Реакционную смесь затем фильтровали и фильтрат упаривали. Остаток распределяли между H2SO4 и дихлорметаном. Органический слой высушивали, фильтровали и упаривали, а остаток подвергали очистке с помощью колоночной хроматографии на силикагеле, используя в качестве элюента смесь CH2Cl2 и C2HCOH в соотношении 95: 5. Фракцию элюата, содержащую целевое соединение, упаривали, получая в результате 9,9 части (47,9%) этилгексагидро-4-[[3-[[5-(оксиметил)-2-фуранил] метил] -3Н-имидазо[4,5-b] - пиридин-2-ил]амино]-1Н-азепин-1-карбок-силата (соединение 4.04).
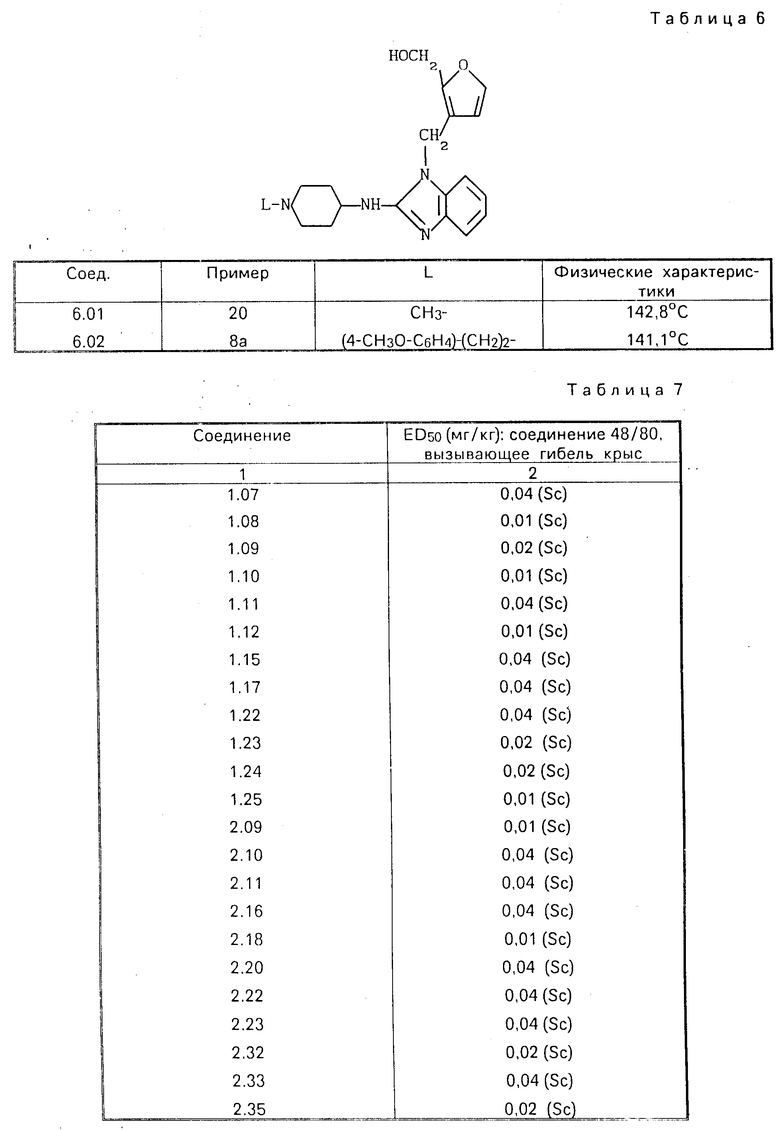
Все новые соединения, перечисленные в табл.1-6, были получены описанным способом.
C. Фармакологичеcкие примеры.
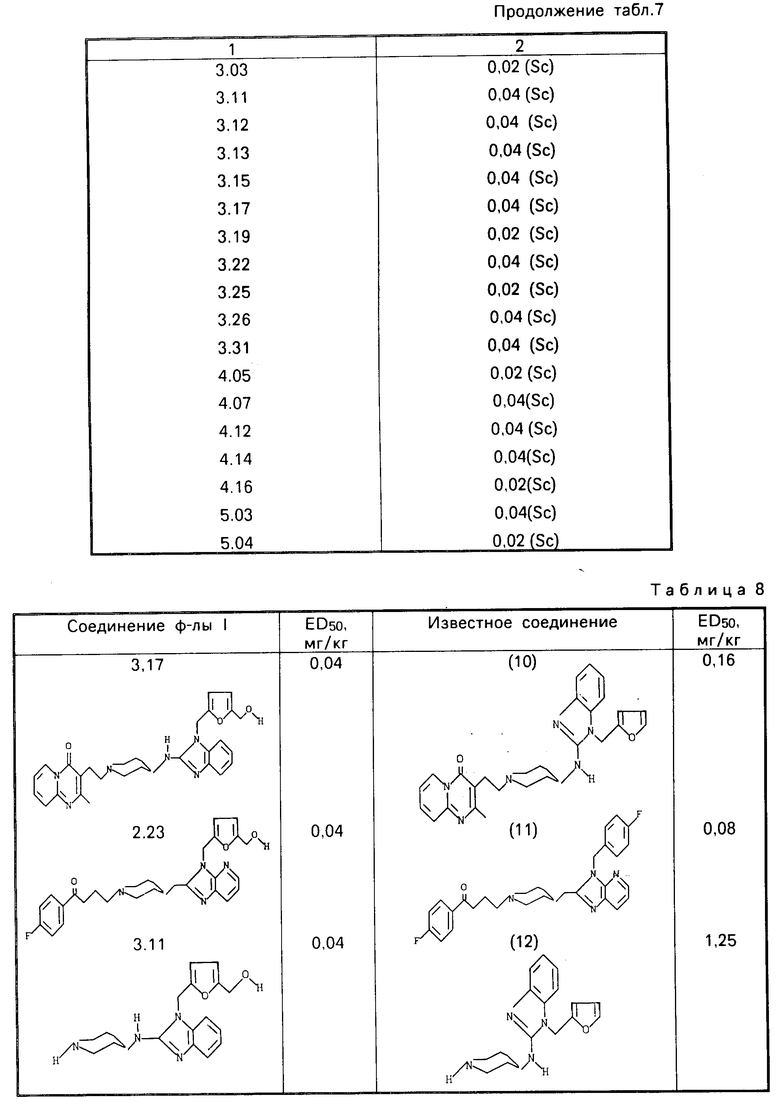
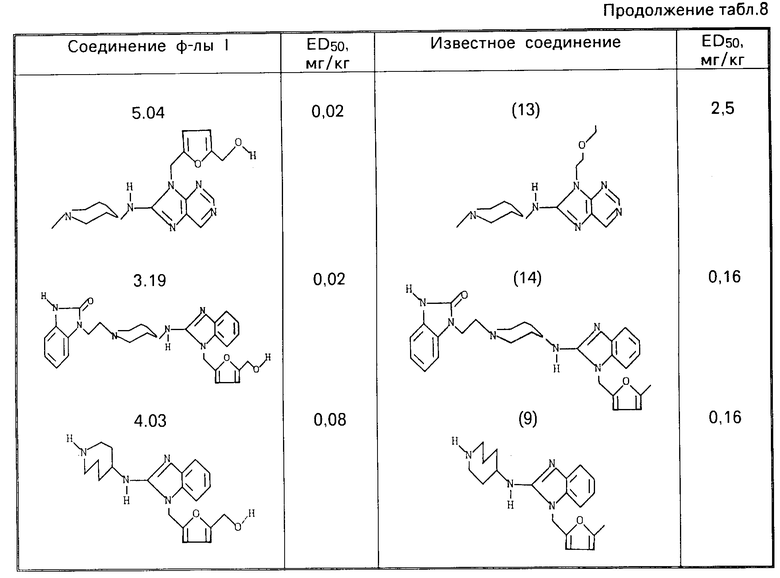
П р и м е р 25. Антигистаминная активность соединений формулы (I) может быть продемонстрирована в тесте "Защита крыс от вызывающего их гибель соединения 48/80", описанном в патенте США N 4556660, который включен в ссылки к настоящей заявке. Полученные результаты приведены в табл.7. Термин (sc) означает подкожное введение. Значения ЕД50приведены в табл.8.
D. Примеры на композиции.
П р и м е р 26. Капли для орального применения.
500 ч. активного вещества растворяли в 0,5 л 2-оксипропановой кислоты и 1,5 л полиэтиленгликоля при 60-80оС. После охлаждения до 30-40оС к полученному раствору добавляли 35 л полиэтиленгликоля и смесь хорошенько перемешивали. После этого к ней добавляли раствор 1750 ч. сахарина натрия в 2,5 л очищенной воды и при перемешивании 2,5 л Cocoa flavor и полиэтиленгликоль до общего объема 50 л. В результате получали раствор капель с содержанием активного вещества 10 мг/мл. Приготовленный раствор разливали в подходящие емкости.
П р и м е р 27. Раствор для орального применения.
Растворяли 9 ч. метил-4-оксибензоата и 1 ч. пропил-4-оксибензоата в 4 л кипящей очищенной воды. В 3 л этого раствора растворяли вначале 10 ч. 2,3-диоксидибутановой кислоты и затем 20 ч. активного вещества. Приготовленный раствор объединяли с остальной частью первого раствора и добавляли к смеси 12 л 1,2,3-пропантриола и 3 л 70%-ного раствора сорбитола. 40 ч. сахарина натрия растворяли в 0,5 л воды и добавляли к приготовленному раствору 2 мл малинового и 2 мл крыжовенного экстрактов. Этот раствор объединяли с приготовленным ранее раствором и добавляли воду до общего объема 20 л. В результате получали раствор для орального применения с содержанием активного вещества 5 мг на чайную ложку (5 мл). Полученный раствор разливали в подходящие емкости.
П р и м е р 28. Капсулы.
20 ч. активного вещества, 6 ч. лаурилсульфата натрия, 56 ч. крахмала, 56 ч. лактозы, 0,8 ч. коллоидного диоксида кремния и 1,2 ч. стеарата магния энергично перемешивали и полученной смесью заполняли 100 соответствующим образом отвержденных желатиновых капсул. Содержание активного вещества в одной капсуле составляло 20 мг.
П р и м е р 29. Таблетки с оболочкой.
Получение основы таблеток
Смесь 100 частей активного вещества, 570 ч. лактозы и 200 ч. крахмала тщательно перемешивали, после чего увлажняли раствором 5 ч. додецилсульфата натрия и 10 ч. поливинилпирролидона (Kollidon-К 90 ) примерно в 200 мл воды. Влажную порошкообразную смесь просеивали, высушивали и снова просеивали. После этого к ней добавляли 100 частей микрокристаллической целлюлозы (A vicel
) примерно в 200 мл воды. Влажную порошкообразную смесь просеивали, высушивали и снова просеивали. После этого к ней добавляли 100 частей микрокристаллической целлюлозы (A vicel ) и 15 ч. гидрированного масла (Steretex
) и 15 ч. гидрированного масла (Steretex ). Массу перемешивали и прессовали из нее таблетки. В результате получали 10000 таблеток с содержанием в каждой таблетке 10 мг активного вещества.
). Массу перемешивали и прессовали из нее таблетки. В результате получали 10000 таблеток с содержанием в каждой таблетке 10 мг активного вещества.
Оболочка.
К раствору 10 ч. метилцеллюлозы (Methocel 60 HG ) в 75 мл денатурированного этанола добавляли раствор 5 ч. этилцеллюлозы (Ethocel 22 cps
) в 75 мл денатурированного этанола добавляли раствор 5 ч. этилцеллюлозы (Ethocel 22 cps ) в 150 мл дихлорметана, а затем 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола. Расправляли 10 ч. полиэтиленгликоля и растворяли их в 75 мл дихлорэтана. Этот раствор добавляли к первоначально приготовленному раствору и затем добавляли к смеси 2,5 части октадеканоата магния, 5 ч. поливинилпирролидона и 30 мл концентрированной цветной суспензии (Opaspray K-1-2109
) в 150 мл дихлорметана, а затем 75 мл дихлорметана и 2,5 мл 1,2,3-пропантриола. Расправляли 10 ч. полиэтиленгликоля и растворяли их в 75 мл дихлорэтана. Этот раствор добавляли к первоначально приготовленному раствору и затем добавляли к смеси 2,5 части октадеканоата магния, 5 ч. поливинилпирролидона и 30 мл концентрированной цветной суспензии (Opaspray K-1-2109 ) и перемешивали всю массу. Таблетки покрывали приготовленной таким образом смесью в предназначенном для этого устройстве.
) и перемешивали всю массу. Таблетки покрывали приготовленной таким образом смесью в предназначенном для этого устройстве.
П р и м е р 30. Раствор для инъекций.
1,8 ч. метил-4-оксибензоата и 0,2 ч. пропил-4-оксибензоата растворяли в примерно 0,5 л предназначенной для инъекций кипящей воды. После охлаждения до примерно 50оС к раствору при перемешивании добавляли 4 ч. молочной кислоты, 0,05 ч. пропиленгликоля и 4 ч. активного вещества. Раствор охлаждали до комнатной температуры и добавляли к нему воду, предназначенную для инъекций, до общего объема 1 л. В результате получали раствор с содержанием активного вещества 4 мг/мл. Раствор стерилизовали путем фильтрации (u.s.P. XVII, с. 811) и заполняли им стерильные емкости.
П р и м е р 31. Свечи.
3 ч. активного вещества растворяли в растворе 3 ч. 2,3-диоксидибутановой кислоты в 25 мл полиэтиленгликоля 400. Расплавляли 12 ч. поверхностно-активного вещества (SPAN ) и триглицерина (Witepsol 555
) и триглицерина (Witepsol 555 ) до общего количества 300 ч. и перемешивали расплав с ранее приготовленным раствором. Приготовленную таким образом смесь разливали в формы при 37-38оС. В результате получали 100 свеч с содержанием активного вещества в каждой свече 30 мг/мл.
) до общего количества 300 ч. и перемешивали расплав с ранее приготовленным раствором. Приготовленную таким образом смесь разливали в формы при 37-38оС. В результате получали 100 свеч с содержанием активного вещества в каждой свече 30 мг/мл.
П р и м е р 32. Раствор для инъекций
Перемешивали 60 ч. активного вещества и 12 ч. бензилового спирта. К смеси добавляли кунжутное масло до общего объема 1 л. В результате получали раствор с содержанием активного вещества 60 мг/мл, который стерилизовали, и заполняли им стерильные емкости.


Использование: в медицине в качестве антиаллергического агента. Сущность изобретения: продукт-оксиалкилфурановые производные ф-лы I, приведенный в тексте описания,- фармацевтически приемлемая соль присоединения кислот его или стереоизомерного соединения, где -A1-A2-A3-A4- двухвалентный радикал, имеющий ф-лу: -CH=CH-CH=CH-(a-1), -N=CH-CH=CH-(a-2), -CH=CH-CH=N-(a-5) или -N= CH-N=CH-(a-6), n-1 или 2B-NR4 или CH2 , R4 -H или C1-C6 алкил, L-H, C1-C6 алкил, C1-C6 алкилоксикарбонил, или радикал ф-лы -Alk-R5 (в-1;) -Alk-Y-R6 (в-2), -Alk-Z1-C(-X)-Z2-R7 (в-3), или -CH2-CHOH-CH2-O-R8 , где R5 циано группа, фенил, необязательно замещенный C1-C6 алкилокси; пиридинил; 4,5-дигидро-5- оксо-1-н-тетразолил; 2-оксо-3-оксазолидинил; 2,3-дигидро-2-оксо -1-н-бензимидазолил; или бицикличный радикал ф-лы (с-4-а), где G2 -CH= CH-CH= CH-, -S-(CH2)3- , -S(CH2)2- -S-CH= CH- или -CH=C(CH3)-O- группа R6-C1-C6 алкил; пиридинил необязательно замещенный нитро группой; пиримидинил; пиразинил; пиридазинилб необязательно замещенный галогеномМ 2,3-дигидро -3-оксопиридазинил; или 9-метил -6-пуринил; R7-C1-C6-алкил ; галоидфенил; 1-метил-1Н-пирролил; фуранил, тиенил, или аминопиразинил; R8-галоидфенил, Y-O или NH, Z1 или Z2 каждый независимо NH или прямая связь, X-O, каждый Alk независимо C1-6 алкандиил. 1 з.п. ф-лы, 8 табл.
ОКСИАЛКИЛФУРАНОВЫЕ ПРОИЗВОДНЫЕ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТ, ИЛИ ИХ СТЕРЕОХИМИЧЕСКИЕ ИЗОМЕРНЫЕ ФОРМЫ, ОБЛАДАЮЩИЕ АНТИАЛЛЕРГИЧЕСКОЙ АКТИВНОСТЬЮ И АНТИАЛЛЕРГИЧЕСКАЯ КОМПОЗИЦИЯ.

| Паровоз для отопления неспекающейся каменноугольной мелочью | 1916 |
|
SU14A1 |
| Г. Г. Гарифзянов,И. М. Колесников и И. X. Бикбулатов | 0 |
|
SU295742A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Механическая топочная решетка с наклонными частью подвижными, частью неподвижными колосниковыми элементами | 1917 |
|
SU1988A1 |
