Изобретение относится к способам получения замещенных производных амина общей формулы I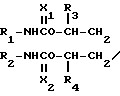 N-A-Y где R1 и R2 каждый представляет собой низший алкил;
N-A-Y где R1 и R2 каждый представляет собой низший алкил;
R3 и R4 каждый означает атом водорода или низшая алкильная группа;
A - углеродная цепь, имеющая два или более углеродных атомов, которая может образовывать циклическое и/или ароматическое кольцо;
X1 и X2 каждый означает атом кислорода или атом серы;
Y - фталимидогруппа, заключающемуся в том, что осуществляют взаимодействие соединения формулы: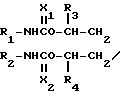 N-A-OH где R1, R2, R3, R4 и A каждый определен выше, с фталимидом.
N-A-OH где R1, R2, R3, R4 и A каждый определен выше, с фталимидом.
Аритмия представляет собой одно из заболеваний, часто наблюдаемых особенно у людей пожилого возраста, и в серьезных состояниях она представляет опасность для жизни. В последние годы быстро возрастает число коронарных сердечных заболеваний, и следовательно, делом серьезной озабоченности стали контрмеры против фатальных случаев аритмии, являющейся следствием этих заболеваний.
В качестве терапевтических агентов для лечения аритмии разработано и клинически используется большое разнообразие фармацевтических препаратов (например, дизопирамид). Поскольку причины сердечных аритмий являются настолько сложными, что постоянно ведется поиск противоаритмических агентов, которые являются эффективными против относительно многих типов аритмий и дают меньше нежелательных побочных эффектов, потому что обычные противоаритмические агенты различны по эффективности в зависимости от симптомов.
Изобретение направлено на способ получения соединений формулы Iи их солей, полезных в качестве против аритмических агентов.
В качестве низшего алкила, представленного формулой I, упоминается, например, алкильная группа с прямой или разветвленной цепью, имеющая примерно 1-6 атомов углерода, таких как метил, этил, н-пропил, н-бутил, н-пентил, н-гексил, изо-пропил, изо-бутил, изо-пентил, изо-гексил, втор-бутил, трет-бутил, трет-пентил и неопентил.
Соединение формулы I может иметь в зависимости от обстоятельств асимметричный атом углерода в молекуле, и в данном случае в сферу настоящего изобретения включаются каждый изомер и их смесь.
Примеры солей соединения 1 включают фармацевтически приемлемые соли, например соль с неорганической кислотой, такой как соляная кислота, бромистоводородная кислота, иодистоводородная кислота, серная, фосфорная или азотная кислота; и соли с органическими кислотами, такими как уксусная, молочная, винная, бензойная кислота, лимонная кислота, меносульфоновая, этансульфоновая бензолсульфоновая или толуолсульфоновая кислота. Среди них предпочтительными являются соли с неорганической кислотой, такой как соляная, бромистоводородная или иодистоводородная кислота.
Соли соединения I могут в некоторых случаях получаться с помощью метода получения соединения I, но они могут при необходимости получаться с помощью добавления к соединению I кислоты.
Соединения I и их соли обладают отличной противоаритмической активностью, и они являются полезными в качестве профилактических и терапевтических агентов для лечения аритмии. Соединения I и их соли могут безопасно назначаться млекопитающим для приема орально или неорально, в порошкообразной или жидкой форме как они есть, или в виде подходящей фармацевтической композиции. Наряду с другими факторами, дозировка зависит от субъектов, симптомов заболевания или способов назначения, и в случае внутривенной инъекции для профилактики и лечения аритмии удобно назначать соединение I или его соль при одной дозе, в количестве обычно примерно 0,01-20 мг/кг массы тела, предпочтительно в количестве примерно 0,1-10 мг/кг массы тела при приеме от одного до примерно пяти раз в день, предпочтительно примерно от одного до трех раз в день. В случае орального назначения для профилактики и лечения аритмии удобно назначать соединение I или его соль в единичной дозе в количестве обычно примерно 0,5-100 мг/кг массы тела примерно 1-3 раза в день.
Назначаемые фармацевтические композиции содержат эффективное количество соединения I или его соли и фармацевтически приемлемый носитель, эксципиент или разбавитель, и они формулируются в виде дозировочных форм, подходящих для орального или неорального назначения. В качестве носителя, эксципиента и разбавителя применяются обычные вещества, используемые в области фармацевтических препаратов. В качестве дозировочных форм упоминаются агент для инъекций (внутривенных инъекций, включая капельное введение, подкожные инъекции, внутримышечную инъекцию и др.), таблетки, капсулы, порошок, пилюли, гранулы, жидкости, медицинские свечи и другие.
Эти фармацевтические композиции могут содержать любые другие активные ингредиенты, если только они не вызывают побочные взаимодействия с соединением I или его солью.
П р и м е р ы. Примеры получения описывают изобретение более подробно, но следует понимать, что настоящее изобретение не должно ограничиваться ими.
Пример получения 1.
Дигидрохлорид 1-амино-3-бис(н-бутилтиокарбамоилоксиэтил)-аминопропана (16)
1) Синтез 1-фталимидо-3-бис(н-бутилтиокарбамоилоксиэтил)аминопропана (15).
Соединение (8) [1,462 г (5,0 ммолей)], синтезированное в примере получения 4-1), и н-бутилизотиоцианат [3,0 мл (27,3 ммоля)] нагревались в запаянной трубке при 130оС в течение 2 сут. Реакционная смесь охлаждалась и концентрировалась при пониженном давлении. Сырой продукт, полученный таким образом, очищался с помощью хроматографии на колонке (силикагель: 80 г; элюент: н-гексан/этилацетат = 2: 1), давая желаемый продукт (15) 1,034 г (39,6%) (бесцветное маслянистое вещество).
ТСХ (силикагель; н-гексан/этилацетат (1:1): Rf = 0,34
ЯМР (90 МГц, CDCl3) δ: 0,93 (6Н, м), 1,11-1,96 (20Н, М.), 2,51-3,09 (6Н, м.), 3,16-4,70 (10Н, м.), 6,06 (2Н, шир.), 7,65-7,93 (4Н, м.).
ИК (пленка) см-1: 3300, 2945, 2910, 2850, 1760, 1700, 1520, 1460, 1390, 1360, 1330, 1180, 755, 720.
2) Синтез дигидрохлорида 1-амино-3-бис(н-бутилтиокарбамоилоксиэтил)аминопро- пана (16).
Соединение [1,0,34 г (1,98 ммоля)], синтезированное в 1), растворялось в метаноле (35 мл). К раствору добавлялся гидразингидрат [0,383 мл (7,91 ммоля)] , и смесь нагревалась с обратным холодильником в атмосфере азота. Реакционная смесь охлаждалась и концентрировалась при пониженном давлении. К остатку добавлялся хлороформ, и нерастворимые вещества удалялись, а затем маточная жидкость концентрировалась при пониженном давлении. Полученный таким образом сырой продукт очищался с помощью хроматографии на колонке (силикагель: 40 г; элюент: метанол/конц. аммиачная вода = 40:1); давая свободное основание [180 мг (23,2%)]. Данное свободное основание обрабатывалось при охлаждении льдом эфиром, насыщенным хлористым водородом, давая целевой продукт (16) [213 мг (бесцветный порошок)].
(Свободное основание).
ТСХ (силикагель; метанол/конц. аммиачная вода 19:1): Rf = 0,26.
ЯМР (90 МГц, CDCl3) δ: 0,94 (6Н, м), 1,11-1,81 (12Н, м.), 2,51-3,01 (8Н, м.), 3,14-3,67 (4Н, м.), 4,44 (4Н, м.).
ИК (KBr) см-1: 3225, 2925, 2850, 1510, 1460, 1410, 1190.
Пример получения 2.
Дигидрохлорид N,N-бис(н-бутилкарбамоилоксиэтил)-2-(4-фторфенил)этиленди- амина (72).
1) Синтез 2-[N, N-бис(н-бутилкарбамоилоксиэтил)амино] -1-(4-фторфенил)этанола (70).
Смесь 4-фторэпоксистирола [1,17 г (8,47 ммоля)] и N,N-бис(н-бутил-карбамоилоксиэтил)-амина [2,53 г (8,47 ммоля)] перемешивалась при 110оС на протяжении ночи. После охлаждения неочищенный продукт подвергался хроматографии на колонке с использованием силикагеля и элюировался смесью гексанэтилацетат (1: 1), давая спиртовое соединение (70) [3,37 г (90,1%)] в виде коричневого маслянистого вещества.
ИК (четкий) см-1: 320 (шир), 1700 (шир.), 1600.
ЯМР (90 МГц, CDCl3) δ: 0,7-1,1 (6Н, м.), 1,1-1,9 (8Н, м.), 2,44 (1Н, дд. , I = 10, 15 Гц), 2,6-3,0 (5Н, м.), 3,15 (4Н, кв., I = 6 Гц), 3,7-4,3 (4Н, м. ), 4,58 (1Н, дд., I = 3, 10 Гц), 4,95 (2Н, м), 7,00 (2Н, т., I = 9 Гц), 7,34 (2Н, дд., I = 6,9 Гц).
2) Синтез N-[[2-[N',N'-бис(н-бутилкарбамоилоксиэтил)амино-1-(4-фторфенил)]этил]фталими да (71).
Диэтилазодикарбоксилат [1,41 мл (9,16 ммоля)] добавлялся по каплям при перемешивании при комнатной температуре, к раствору соединения (70) [3,37 г (7,63 ммоля)], синтезированного на стадии 1), фталимида [1,35 г (9,16 ммоля)] и трифенилфосфина [2,40 г (9,16 ммоля)] в безводном тетрагидрофуране (90 мл) и смесь перемешивалась в течение 30 мин при комнатной температуре. Растворитель отгонялся, и остаток подвергался хроматографии на колонке с использованием силикагеля и элюировался смесью гексана и этилацетата (2:1), давая фталимидо соединение (71) [2,57 г (59,0%)] в виде желтого маслянистого вещества.
ИК (четкий) см-1: 3325 (шир.), 1770, 1710 (шир.), 1600.
ЯМР (90 МГц, CDCl3) δ: 0,7-1,1 (6Н, м.), 1,1-1,7 (8Н, м.), 2,82 (4Н, т. , I = 6 Гц), 2,8-3,3 (5Н, м.), 3,86 (1Н, дд., I = 11,14 Гц), 4,00 (4Н, т., I = Гц), 4,86 (2Н, м.), 5,45 (1Н, дд., I = 5,11 Гц), 7,00 (2Н, т., I = 9 Гц), 7,40 (2Н, дд., I = 6,9 Гц), 7,4-8,0 (4Н, м).
3) Синтез дигидрохлорида N,N-бис(н-бутилкарбамоилоксиэтил)-2-(4-фторфенил)эти- лендиамина (72).
Раствор соединения (71) [2,06 (3,61 ммоля)], синтезированного на стадии 2), и гидразингидрата [0,21 мл (4,33 ммоля)] в метаноле (15 мл) нагревался в течение 2 ч при кипячении с обратным холодильником. После охлаждения растворитель отганялся, и к остатку добавлялся хлороформ. Осадки отфильтровывались, и фильтрат концентрировался при пониженном давлении. Остаток подвергался хроматографии на колонке с использованием силикагеля и элюировался смесью метанола и этилацетата (1:10), давая свободное аминовое соединение 1,30 г (81,7%) в виде желтого маслянистого вещества.
ИК (четкий) см-1: 3320 (шир.), 1700 (шир.), 1600.
ЯМР (90 МГц, CDCl3) δ, 0,7-1,1 (6Н, м.), 1,1-1,7 (8Н, м.), 2,47 (1Н, дд., I = 10, 13 Гц), 2,72 (1Н, дд., I = 5, 13 Гц), 2,82 (4Н, т., I = 6 Гц), 3,15 (44, кв., I = 6 Гц), 3,98 (1Н, дд., I = 5,10 Гц), 4,14 (4Н, т., I = 6 Гц), 5,03 (2Н, м.), 7,00 (2Н, т., I = 9 Гц), 7,35 (2Н, дд., I = 6,9 Гц).
Указанное свободное аминовое соединение [1,30 г (2,95 ммоля)] растворялось в 3,5 М растворе хлористого водорода в метаноле, и растворитель отгонялся с получением желаемого продукта (72) [1,38 г (74,4% в расчете на соединение 71)] в виде желтого маслянистого вещества.
Пример приготовления 41.
Дигидрохлорид 1-амино-2-бис(н-бутилкарбамоилоксиэтил)амино-2-фенилэтан (107).
1) Синтез 2-фталимидо-1-фенилэтанола (104).
2-Амино-1-фенилэтанол [5,0 г (36,45 ммоля)] и N-карбоэтоксифталимид [7,99 г (36,45 ммоля)] растворялись в метиленхлориде (40 мл). К раствору добавлялся триэтиламин [5,08 мл (36,45 ммоля)] и смесь перемешивалась в течение 3 ч при комнатной температуре. Реакционная смесь концентрировалась при пониженном давлении и неочищенный продукт, полученный таким образом, перекристаллизовывался из смеси н-гексан - метиленхлорид, в результате получался желаемый продукт (104).
[8,01 г (83,5%, бесцветные кристаллы)].
ТСХ [силикагель; CHCl3 метанол (40/1)]: Rf = 0,50.
ЯМР (90 МГц, CDCl3) + CD3OD) δ: 3,90 (2Н, м.), 5,04 (1Н, д.д.), 7,14-7,57 (5Н, м.), 7,62-8,00 (4Н, м.).
2) Синтез 1-бром-2-фталимидо-1-фенилэтана (105).
Соединение [5,266 г (20 ммоля)] синтезированное в 1) и тетрабромид углерода [7,959 г (24 ммоля)] растворялись в хлороформе (80 мл). К раствору при охлаждении добавлялся трифенилфосфин [6,295 г (24 ммоля)] и затем смесь нагревалась в течение 3 ч при температуре дефлегмации. После охлаждения реакционная смесь концентрировалась при пониженном давлении и остаток очищался хроматографией на колонке (силикагель: 150 г; элюент: хлороформ), в результате получался желаемый продукт (105) [6,62 г (100%, желтые кристаллы)].
ТСХ [силикагель; н-гексан/этилацетат (1/1)]: Rf = 0,70.
ЯМР (90 МГц, CDCl3) δ: 4,32 (2Н, м), 5,48 (1Н, т.), 7,14-8,07 (9Н, м.).
3) Синтез 1-бис(-н-бутилкарбамоилоксиэтил)амино-2-фталимидо-1-фенилэтан (106).
Соединение [1,321 г (4 ммоля)], синтезированное в 2), триэтиламин [0,42 мл (3 ммоля)] и соединение [910 мг (3 ммоля)], синтезированное в примере получения 8-3) добавлялись к толуолу (10 мл). Смесь нагревалась при 100-130оС в течение 3 дней. После охлаждения к смеси добавлялась вода и последняя подвергалась экстракции хлороформом. Органический слой сушился над безводным карбонатом калия и затем растворитель отгонялся при пониженном давлении. Неочищенный продукт очищался хроматографией на колонке (силикагель: 60 г; элюент: н-гексан/этилацетат = 1/1), в результате получался желаемый продукт (106) [779 мг (47,0%, бесцветное маслянистое вещество)].
ТСХ [силикагель; н-гексан/этилацетат (1/1)]: Rf = 0,40.
ЯМР (90 МГц, CDCl3) δ: 0,90 (6Н, н.), 1,40 (3Н, м.), 2,42-2,93 (4Н, м. ), 3,08 (4Н, кв. ), 3,70-4,58 (7Н, м.), 5,02 (2Н, шир.), 7,31 (5Н, с.), 7,57-7,93 (4Н, м.).
ИК (пленка) см-1: 3320, 2950, 2915, 2855, 1765, 1705, 1520, 1464, 1400, 1250, 1110, 1020, 760, 725, 715, 705.
4) Синтез дигидрохлорида 1-амино-2-бис(н-бутилкарбамоилоксиэтил)-амино-2-фенилэтана (107).
Соединение [770 мг (1,393 ммоля)], синтезированное на стадии 3) растворялось в метаноле (10 мл). К раствору добавлялся гидразингидрат (0,25 мл) и смесь нагревалась в течение 1 ч при температуре дефлегмации в струе азота. После охлаждения реакционная смесь концентрировалась при пониженном давлении. К остатку добавлялся хлороформ и нерастворимые вещества удалялись. Маточная жидкость концентрировалась при пониженном давлении. Полученный таким образом неочищенный продукт очищался хроматографией на колонке (силикагель: 25 г; элюент: метанол/концентрированная аммиачная вода = 240 (1) для получения свободного амина [461 мг (78,4%, бесцветный маслянистый продукт)]. Свободный амин обрабатывался при охлаждении этиловым эфиром, ненасыщенным хлористым водородом для получения желаемого продукта (107) [541 мг (бесцветный порошок)].
ТСХ [силикагель; метанол/концентрированная аммиачная вода (240/1)]: Rf = 0,30.
ЯМР (90 МГц, CDCl3) δ: 0,93 (6Н, м.), 1,43 (8Н, м.), 2,44 (3,33 (10Н, м.), 3,66 (1Н, м.), 4,11 (4Н, м.), 5,24 (2Н, шир.), 7,33 (5Н, м.).
ИК (пленка) см-1: 3315, 2950, 2925, 2855, 1700, 1560, 1250.
Использование: замещенные производные амина ф-лы I R1-NHCO(X1)CH(R3CH2)-N(A-Y)-CH2 _→ _→ CH(R4)NHCO(X2)-R2 обладают противоаритмической активностью, где R1 и R2 каждый обозначает низший алкил; R3 и R4 каждый обозначает водород или низший алкил; A - углеродная цепь, имеющую два или более атомов углерода, которая может образовывать циклическое или ароматическое кольцо, X1 и X2 каждый - атом кислорода или сера; Y - фталимидогруппа. Сущность изобретения: соединения ф-лы 1 получают взаимодействием соединения ф-лы II R1-NHCO(X1)CH(R3CH2)-N(A-OH)-CH2 _→ _→ CH(R4)NHCO(X2)-R2 с фталимидом.
СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ ПРОИЗВОДНЫХ АМИНА общей формулы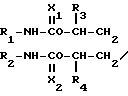 N-A-Y
N-A-Y
где R1, R2 - каждый низший алкил;
R3 и R4 - каждый водород или низший алкил;
А - углеродная цепь, имеющая два или более атомов углерода, которая может образовывать циклическое или ароматическое кольцо;
Х1 и Х2 - каждый кислород или сера;
Y - фталимидогруппа,
отличающийся тем, что соединение общей формулы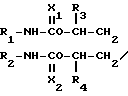 N-A-OH
N-A-OH
где R1 - R4, А имеют указанные значения,
подвергают взаимодействию с фталимидом.
| j | |||
| Med | |||
| Chem | |||
| Приспособление для склейки фанер в стыках | 1924 |
|
SU1973A1 |
