Изобретение относится к противоопухолевым и иммунорегулирующим способностям некоторых производных арабинофуранозилпирина.
Сообщено (Blood, 61, 1983, 660; J. Clin. Invest., 74, 1984, 951 и Cancer Research, 45, 1985, 1008), что арабинофуранозилгуанин (ara G) селективно ингибирует рост Т-клеток по сравнению с B-клетками и обладает селективной цитотоксичной активностью по отношению к T-лейкозным клеткам. Ara G незначительно разрушается присутствующей в человеке нуклеозидфосфорилазы пурина (PNP), и он был предложен в качестве предлагаемого химиотерапевтического или иммунодепресного средства.
В Европейской патентной заявке N 294 114 описаны inter alia соединения формулы (I)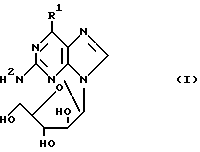
где
R1 представляет собой C1-5 алкоксигруппу (например, метокси-) или аминогруппу, которая является моно- или би- замещенной C1-5-алкилом (например, метилом) и физиологически приемлемыми их производными, для лечения или профилактики вирусных инфекций у человека, вызываемых некоторыми вирусами герпеса. Фармацевтически приемлемые сложные эфиры соединений формулы (I), как утверждают, являются предпочтительными, поскольку они обеспечивают высокие уровни соединения формулы (I) после перорального введения.
Теперь обнаружено, что соединения формулы (I), где R1 представляет собой C1-5-алкоксигруппу, и фармацевтически приемлемые их сложные эфиры, являются полезными в качестве противоопухолевых средств и, в частности являются полезными при лечении нарушений роста T-лимфатических клеток. Таким образом, они являются полезными для лечения лимфоцитарного лейкоза, злокачественной лимфомы, заболеваний аутоиммунитета (например, ревматоидный артрит, рассеянный склероз, системная красная волчанка и тип I или инсулинозависимый сахарный диабет) и в качестве иммунорегуляторов.
Соответственно изобретением является использование соединения формулы (I), где R1 представляет собой C1-5-алкоксигруппу, или фармацевтически приемлемого его сложного эфира при производстве лекарств для лечения опухолей.
В другом аспекте настоящим изобретением является способ для регулирования роста опухолей у млекопитающих, который включает введение в млекопитающего эффективного количества соединения формулы (I), в которой R1 представляет собой C1-5-алкоксигруппу, или фармацевтически приемлемого его сложного эфира.
В качестве R1 подходит метокси- или этокси-, но предпочтительной является метоксигруппа.
В другом аспекте изобретением являются новые фармацевтически приемлемые сложные эфиры соединений формулы (I), где R1 представляет собой C1-5-алкоксигруппу.
Фармацевтически приемлемые сложные эфиры вышеуказанных соединений являются практически более предпочтительными, поскольку они способны обеспечить высокие уровни ara G в плазме реципиента после перорального введения.
Обнаружено, что соединения по изобретению ферментативно превращаются в ara G в реципиенте. То что ara G и имеет ограниченную растворимость в воде, делая парентеральное введение непрактичным, а также обеспечивая низкую биологическую ценность после перорального применения в приматах, делает использование соединения по изобретению преимущественным для лечения вышеупомянутых нарушений и болезней.
Предпочтительные производные соединений по изобретению включают моно-, ди- или тризамещенные сложные эфиры арабиносахарного остатка, замещенные в позициях 2'-, 3'- и 5'- указанного остатка.
Такие предпочтительные сложные эфиры включают сложные эфиры карбоновой кислоту в которых некарбонильную часть эфирной группы выбирают из прямоцепной или разветвленноцепной группы: алкильной (например, n-пропил, t-бутил), алкоксиалкильной (например, метоксиметил), аралкильной (например, бензил), арилоксиалкильной (например, феноксиметил), арильной (например, фенил) необязательно замещенными галогеном, (C1-4-алкилом или C1-4-алкокси, нитро- или аминогруппой; сложные эфиры в виде сульфонатов, таких как алкилсульфонил; или алкиларилсульфонил (например, метансульфонил или тозилсульфонил); сложные эфиры дикарбоновых кислот (например, сукцинил) или их C1-4-алкильные сложные эфиры; сложные эфиры аминокислот (например, L-валил); и сложные эфиры в виде моно-, ди- или трифосфатов. Фармацевтически приемлемые соли этих сложных эфиров включают соли натрия, калия, NR4, где R=H или (C1-6-алкил, галиды и кислотно-аддитивные соли. В вышеуказанных сложноэфирных группах алкильные группы (включая их и в алкоксигруппах) содержат от 1 до 12 атомов углерода, а предпочтительной арильной группой является фенильная группа.
Предпочтительные сложные эфиры по изобретению включают:
1) 2-амино-6-метокси-9-(5-О-пропионил- β -D- арабинофуранозил)-  -пурин;
-пурин;
2) 2-амино-9-(5-О-бутирил- β -D- арабинофуранозил)-6-метокси- 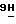 -пурин;
-пурин;
3) 2-амино-6-метокси-9-(3-О-пивалоил- β -D-арабинофуранозил)- 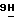 -пурин;
-пурин;
4) 2-амино-6-метокси- 9-(2-О-валерил- β -D-арабинофуранозил)-  -пурин;
-пурин;
5) 2-амино-9-(3-О- бензоил- β -D-арабинофуранозил)-6-метокси- 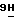 -пурин;
-пурин;
6) 2-амино-6-метокси-9-(2-О-пивалоил- β -D-арабинофуранозил)-  -пурин;
-пурин;
7) 2-амино-9-(2-О-бензоил- β -D-арабинофуранозил)-6-метокси-  -пурин;
-пурин;
8) 2-амино-6-метокси-9-(5-О-валерил- β -D-арабинофуранозил)-  - пурин;
- пурин;
9) (5-О-ацетил- β -D-арабинофуранозил)-2-амино-6-метокси- 9- 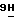 -пурин;
-пурин;
10) 2-амино-6-метокси-9-(5-О-(4-метокси-4-оксобутирил)- β -D-арабинофуранозил)- 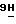 -пурин;
-пурин;
11) 9-(3,5-ди-О-ацетил- β -D-арабинофуранозил)-2-амино-6-метокси- 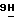 -пурин;
-пурин;
12) 9-(2,5-ди-О-ацетил- β -D-арабинофуранозил)-2-амино-6-метокси-  - пурин;
- пурин;
13) 9-(2-О-асетил- β -D-арабинофуранозил)-2-амино-6-метокси- 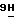 -пурин;
-пурин;
14) 9-(2,3,5-три-О-ацетил- β -D-арабинофуранозил)- 2-амино-6-метокси-9Н-пурин;
15) 2-амино-9-(5-О-изобутирил- β -D- арабинофуранозил)-6-метокси-9Н-пурин;
16) 9-(2,3-ди-О-ацетил- β -D-арабинофуранозил)-2-амино-6-метокси-9H-пурин.
3,5-ди-О-Ацетил сложный эфир (соединение 11), 5-О-(4-метокси-4-оксобутирил) сложный эфир (соединение 10) и 5-О-ацетил сложный эфир (соединение 9) являются особенно предпочтительными сложными эфирами.
Соединения по изобретению являются эффективными ara G пролекарствами, эффективного ингибитора роста Т-клеток. В изобретении поэтому сообщают способ ингибирования репликации и/или функционирования Т-клеток путем введения эффективного количества соединения по настоящему изобретению.
Влияние на рост опухоли с помощью соединения по настоящему изобретению может включать введение одного только соединения или соединения вместе с другими лекарствами в качестве подготовительного режима, предназначенного для того, чтобы удаление костного мозга предшествовало аутогенной или аллогенной пересадке костного мозга при лейкозе, лимфоме, миеломе или других злокачественностях, например, способом, аналогичным введению больших доз бусульфана и циклофосфамида перед пересадкой костного мозга при лейкозе (Santos G.W., Bone Marrow Transplant, 1989 Jan; 4 suppl. 1, 236-9).
Соединения по настоящему изобретению можно также использовать для обработки крови или костного мозга ex vivo для удаления злокачественных клеток способом, аналогичным описанному для случая 4- оксипероксициклофосфамида (Ягер А.М. с сотр.: Yeager, A.M. et al., N. Engl. J. Med., July 17 1986, 315 (3), 141-7).
Т-клетки находятся в ревматоидных суставах и могут вносить вклад в воспалительный процесс. Около 1% всего населения (женщин на 2/3 больше чем мужчин) подвержены ревматоидному артриту, и от 5 до 10% больных ревматоидным артритом теряют трудоспособность из-за болезненных глотательных суставов несмотря на полный курс лечения. Соединение по настоящему изобретению можно использовать для лечения ревматоидного артрита.
Также утверждают, что Т-клетки участвуют в мышечной дистрофии; настоящее изобретение поэтому применяют при использовании соединения по настоящему изобретению при лечении мышечной дистрофии.
Соединения формулы (I), где R1 представляет собой C1-5- алкоксигруппу, и фармацевтически приемлемые их сложные эфиры (к которым будем ссылаться как к активным ингредиентам) можно вводить любым подходящим для лечения данного состояния способом, причем подходящими способами введения являются пероральное, назальное, местное (включая трансбуккальное и подъязычное) вагинальное и парентеральное (включая подкожное, внутримышечное, внутривенное, внутрикожное, внутриоболочечное и эпидуральное). Следует понимать, что предпочтительный способ введения будет зависеть, например, от состояния реципиента.
Для каждого из вышеуказанных нарушений, болезней и показаний требуемое количество активного ингредиента (как определено выше) будет зависеть от ряда факторов, включая обостренность состояния, которое следует лечить, и личности реципиента, и будет в конечном счете в описании лечащего врача. Вообще, тем не менее, для каждого из этих использований и показаний подходящая эффективная доза будет в диапазоне от 0,1 до 250,0 мг на 1 кг веса тела реципиенту в день, предпочтительно в диапазоне от 0,1 до 100,0 мг на 1 кг веса тела в день и более предпочтительно в диапазоне от 1 до 20 мг на 1 кг веса тела в день; оптимальной дозой является доза между 5 и 15 мг на 1 кг веса тела в день (если не оговорено специально, то все веса активного ингредиента вычислены по отношению к первичному соединению формулы (I); для его солей и сложных эфиров цифры возрастут пропорционально). Нужная доза предпочтительно представляет одну, две, три, четыре или более поддозы, вводимых через подходящие интервалы времени в течение дня или недели. Эти поддозы можно вводить в форме единичной дозы, например, содержащей от 5 до 1000 мг, предпочтительно от 20 до 500 мг и более предпочтительно от 100 до 400 мг активного ингредиента на одну дозовую форму.
Следует понимать, что для процесса, который влияет на функцию Т- клеток, что требуется для лечения аутоиммунных заболеваний (например, ревматоидного артрита), величина доз будет обычно в нижней части вышеуказанного диапазона доз.
Хотя возможно введение одних активных ингредиентов, предпочтительным является представление их в виде фармацевтических форм. Эти формы по настоящему изобретению включают по крайней мере один активный ингредиент, как определено выше, вместе с одним или более подходящими их носителями и необязательно с другими терапевтическими ингредиентами. Носитель (носители) должны быть "приемлемыми" в смысле быть совместимыми с другими ингредиентами формы и не ухудшать их свойства для реципиента.
Лекарственные смеси включают формы, подходящие для перорального, назального, местного (включая трансбуккальное и подъязычное), вагинальное или парентеральное (включая подкожное, внутримышечное, внутривенное, внутрикожное, внутриоболочечное и эпидуральное) введения. Лекарственные формы удобно представлять в виде единичных доз и их можно приготавливать любым хорошо известным в области фармакологии методом. Такие методы включают этап соединения активного ингредиента и носителя, который состоит из одного или более вспомогательных ингредиентов. Вообще лекарственные формы получают однородным и основательным связыванием активного ингредиента и жидких носителей или полностью разделенных твердых носителей, или тех и других, и затем, если нужно, формовкой продукта.
Лекарственные формы по настоящему изобретению, подходящие для перорального введения, можно представлять в виде дискретных единиц, таких как капсулы, крахмальные капсулы или таблетки, которые содержат определенное количество активного ингредиента; в виде порошка или гранул, в виде растворов или суспензий в водных или неводных растворах, или в виде жидких эмульсий типа масло в воде или вода в масле. Активные ингредиенты можно также представлять в виде болюсов, электуариев или паст.
Таблетку можно изготовить путем сжатия или формовки, необязательно с одним или более вспомогательными ингредиентами. Сжатые таблетки можно приготовить путем сдавливания в подходящем устройстве активного ингредиента, находящегося в легко сжимаемой форме, такой как порошок, гранулы, необязательно в смеси со связывающим веществом (например, повидоном, желатином, оксипропилметилцеллюлозой), смазкой, инертным растворителем, консервантом, дезинтегрирующим средством (например, натрий-крахмалгликолят, поперечносшитый повидон, поперечносшитая натрий- карбоксиметилцеллюлоза), с поверхностно активным или дисперсным средством. Формованные таблетки можно получить путем формовки в подходящем устройстве смеси соединения в виде порошка, увлажненного инертным жидким разбавителем. Таблетки можно необязательно покрывать оболочкой или снабжать ядром и приготавливать так, чтобы обеспечить медленное или контролируемое высвобождение активного ингредиента при использовании, например, добавлять оксипропилметилцеллюлозу в различных пропорциях для получения желаемой скорости высвобождения лекарства.
Для лечения внешних тканей, например, рта или кожи, лекарственные формы предпочтительно применять в виде местной мази или крема, содержащих активный ингредиент в количестве, например, от 0,075 до 20 вес.%, предпочтительно от 0,2 до 15 вес.% и более предпочтительно от 0,5 до 10 вес.%. В случае приготовления в виде мази активный ингредиент можно применять либо с парафиновым, либо со смешивающимся в воде основанием. Альтернативно, активные ингредиенты можно приготавливать на основе типа масло в воде.
Если нужно, водная фаза крема может включать, например, по крайней мере 30 вес. % многоатомного спирта, т.е. спирта, имеющего две или более гидроксильные группы, такого как пропиленгликоль, бутан-1,3-диол, маннитол, сорбитол, глицерол и полиэтиленгликоль и их смесь. Лекарственные формы для местного применения могут желательно включать соединение, которое увеличивает поглощение или проникновение активного ингредиента через кожу или другие подвергаемые обработке области. Примеры подобных соединений, увеличивающих проникновение через кожу, включают диметилсульфоксид и тому подобные аналоги.
Маслянистую фазу эмульсий по изобретению можно составлять из известных ингредиентов известными способами. В то время как такая фаза может включать один только эмульгатор (или эмульгент), желательно все- таки, чтобы она включала смесь по крайней мере одного эмульгатора с жиром или маслом, или и с тем, и с другим. Предпочтительно, чтобы гидрофильный эмульгатор включался в состав совместно с липофильным эмульгатором, который действует в качестве стабилизатора. Также предпочтительно включать и масло, и жир. Вместе с этим эмульгатор (эмульгаторы) совместно или без стабилизатора (стабилизаторов) образует так называемый эмульсионный воск, и воск вместе с маслом и/или жиром образует так называемую эмульсионную основу мази, которая является маслянистой дисперсной фазой кремовых лекарственных форм.
Эмульгенты и эмульсионные стабилизаторы, подходящие для использования в лекарственных формах по настоящему изобретению, включают Tween 60, Span 80, цитостеариловый спирт, миристиловый спирт, глицерил-моностеарат и натрий-лаурил сульфат.
Выбор подходящих масел и жиров для лекарственных форм основан на достижении желательных косметических свойств, поскольку растворимость активного соединения в большинстве масел, которые вероятно применяют в фармацевтических эмульсионных препаратах, очень низка. Таким образом, крем должен предпочтительно быть обезжиренным, не оставляющим пятен и смываемым веществом, в подходящем состоянии для избежания утечки из ампул или других контейнеров. Можно использовать прямоцепные или разветвленноцепные, моно- или диосновные алкиловые сложные эфиры, такие как ди-изоадипат, изоцетилстеарат, пропиленгликолевый диэфир жирных кислот, изопропилмиристат, децилолеат, изопропилпалмитат, бутилстеарат, 2-этилгексилпалмитат или сложные эфиры со смешанными или разветвленными цепями, известные как Crodamol CAP, причем три последних эфира являются предпочтительными. Их можно использовать как в отдельности, так и совместно в зависимости от требуемых свойств. Альтернативно, можно использовать липиды с высокой температурой плавления, такие как белый мягкий парафин и/или жидкий парафин или другие минеральные масла.
Лекарственные формы, подходящие для местного применения в ротовой полости, включают таблетки, содержащие активный ингредиент с ароматизированной основой, обычно сахарозой и акацией или трагакантом; таблетки, содержащие активный ингредиент в инертном основании, таком как желатин и глицерин, или сахароза и акация; и жидкости для полоскания рта, содержащие активный ингредиент в подходящем жидком носителе.
Лекарственные формы для ректального введения могут представлять собой суппозитории с подходящим основанием, содержащим, например, масло-какао или салицилат.
Лекарственные формы, подходящие для назального введения, в которых носитель представляет собой твердую фазу, включают неизмельченный порошок, имеющий размер частиц, например, в диапазоне от 20 до 500 мкм, который вводят путем вдыхания, т.е. путем быстрой ингаляции через носовой проход порошка, находящегося в контейнере, подносимого близко к носу. Подходящие лекарственные формы, в которых носитель представляет собой жидкость, в виде, например, распрыскивателя или носовых капель, включают водные или масляные растворы активного ингредиента.
Лекарственные формы, подходящие для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или распрыскивателей, содержащих кроме активного ингредиента такие носители, которые, как известно в области фармакологии, являются подходящими.
Лекарственные формы, подходящие для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатические соединения и вещества, придающие данной лекарственной форме изотоническую совместимость с кровью реципиента; и водные и неводные стерильные суспензии, которые могут содержать суспендирующие средства и свертывающие средства. Лекарственные формы можно представлять в виде единичных доз или контейнеров, содержащих несколько доз, например, запечатанные ампулы или пробирки, и могут храниться в замороженном сухом (лиофилизированном) состоянии, требующем только непосредственно перед использованием для инъекций добавления стерильного жидкого носителя, например, воды. Неподготовленные растворы для инъекций и суспензии можно получить из стерильного порошка, гранул и таблеток, имеющих вышеописанный вид.
Предпочтительными единичными дозами лекарственных форм являются дозы, содержащие слабую дозу, дневную дозу или единичную дневную поддозу, как рассмотрено выше, или подходящую их часть, активного ингредиента.
Следует понимать, что кроме ингредиентов, частично упомянутых выше, лекарственные формы по этому изобретению могут включать другие средства, обычно применяемые в фармакологии, имеющих отношение к данному типу лекарственных форм, например, средства, подходящие для перорального введения, могут включать ароматизирующие вещества.
Изобретение также дает способ получения фармацевтически приемлемых сложных эфиров соединения формулы (I), в которой R1 представляет собой C1-5-алкоксигруппу, включающий эстерификацию соединения формулы (I). Такая эстерификация может иметь место при контактировании соединения формулы (I), в которой незащищенную гидроксигруппу эстерифицируют с помощью ацилирующего средства, обычно ацилгалидом или ангидридом, при неэкстремальной температуре от -30 до +100oC и более подходяще при температуре от -5 до +30oC, в полярном растворителе, обычно в ацетонитриле, в присутствии основания, например, триэтиламина, и затем путем удаления защищающих групп обычными способами.
Альтернативно сложные эфиры можно получать путем ферментной эстерификации, например, используя соответствующий трихлорэтиловый эфир и субтилизин в качестве инициатора реакции. Реакцию лучше проводить в растворе с основными свойствами, таком как пиридин, при неэкстремальной температуре, обычно между 10 и 50oC. Реакцию прекращают путем отфильтровывания фермента и удаления растворителя.
Соединения формулы (I) можно получать способом, описанным в Европейской патентной заявке N 294114 (где R1 представляет собой C1-5-алкокси).
Следующие примеры служат для иллюстрации получения сложных эфиров соединений формулы (I) и лекарственных форм, содержащих их.
Пример 1. Ферментативное получение сложных эфиров по настоящему изобретению.
а) 2-Амино-6-метокси-9-(5-О-пропионил- β -D- арабинофуранозил)-9Н-пурин. 2-Амино-6-метокси-9-( β -D- арабинофуранозил)-9Н-пурин (1,0 г, 3,3 мкмоль) (приготовленный, как описано в Европейской патентной заявке N 294114) суспендировали в 40 мл пиридина, содержащего 300 мкл воды и 2 мл трихлорэтилпропионата (трихлорэтилпропионат синтезировали путем добавления 19 мл пропионилхлорида (Aldrich) в течение 30 мин к 19,1 мл трихлорэтанолу (Aldrich) в 40 мл пиридина при 0oC). Продукт очищали путем тщательного промывания 2 х 100 мл частями воды, 5% NaHCO3 и H2O.
1H-ЯМР (200 МГц) CDCl3: 4,74 (s, 2H, Cl3CH2); 2,49 (q, 2H, J = 7,6 Гц, CH3CH2CO2); 1,21 (t, 3H, J = 7,6 Гц, CH3CH2CO2).
Реакцию инициировали с помощью 0,100 г субтилизина (корпорации Sigma Chemical Co., St. Louis, MO, P-5380, лот N 38F - 0356), который был активирован путем растворения 1 г фермента в 20 мл 0,1 моль/л раствора фосфата калия при pH 7,8 и лиофилизировали до сухости. После размешивания в течение 23 ч при 40oC реакцию прекращали путем отфильтровывания фермента и удаляли растворитель в условиях вакуума. Неочищенный продукт очищали с помощью хроматографии на силикагельной колонке размером 4,5 х 25 см с CH2Cl2:CH3OH (9:1) в качестве элюента. Получившуюся фракцию отстаивали и лиофилизировали от воды с получением 0,76 г нужного продукта в виде белого порошка: т.пл. 124oC; TLC (значение для тонкослойной хроматографии) Rf = 0,43 (силикагель; CH2Cl2: CH3OH (9:1): УФ λмакс ( ε , (ммоль/л)-1см-1) при pH 7,0, 278 нм (9,5).
1H-ЯМР (200 МГц, DMSO-d6): δ 7,83 (s, 1H, H8, 6,44 (s, 2H, 2-NH2); 6,14 (s, 1H, H1); 5,75 (d, 1H, J = 4,3 Гц, 2'-OH); 5,65 (d, 1H, J = 3,5 Гц, 3'-OH); 4,28 (m, 2H, H2 и H3); 4,08 (m, 2H, H5); 3,95 (s, 3H, -OCH3); 3,91 (m, 1H, H4'); 2,32 (q, 2H, J = 7,6 Гц, CH2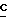 H2CO2-); 1,01 (t, 3H, J = 7,5 Гц,
H2CO2-); 1,01 (t, 3H, J = 7,5 Гц, 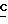 H3CH2CO2-); MC (ci) 354 (M+I), 280 (M-C2H5CO2).
H3CH2CO2-); MC (ci) 354 (M+I), 280 (M-C2H5CO2).
Аналитический расчет для C14H19N5O6•0,46H2O:
рассчитано: C 46,49; H 5,55; N 19,36;
найдено: C 46,46; H 5,52; N 19,45.
b) 9-(5-О-Ацетил- β -D-арабинофуранозил)-2-амино-6-метокси-9Н-пурин.
2-Амино-6-метокси-9- β -D-арабинофуранозил-9Н-пурин (1,0 г, 3,3 ммоль) суспендировали в 40 мл пиридина, который содержал 300 мкл воды и 1 мл трихлорэтилацетата (трихлорэтилацетат синтезировали следующим образом: 2,2,2-трихлорэтанол (19,1 мл, 197,1 ммоль) и сухой пиридин (40 мл) помещали в трехгорловую с круглым дном колбу, снабженную краном для впускания аргона, термометром, капельной воронкой, магнитной мешалкой и ванночкой со льдом (H2O). Ацетилхлорид (14,5 мл, 199,8 ммоль) помещали в капельную воронку и добавляли в течение 10 мин, поддерживая температуру ниже 25oC и размешивая в атмосфере аргона. Получившийся продукт промывали водой (2 х 100 мл), 5% NaHCO3 (2 х 100 мл) и H2O (2 х 100 мл). Органический слой высушивали над MgSO, затем отфильтровывали через ватмановскую бумагу N 1 и дистиллировали в условиях вакуума. Средний срез весом 5,18 г представлял собой требуемое вещество, загрязненное небольшим количеством уксусной кислоты.
1H-ЯМР (CDCl3): δ 4,73 (s, 2H, CH2O); 2,20 (s, 3H, CH3CO); MC (Cl, CH4); m/z 197 (M+H, C4H5O2 37Cl3); 195 (M+H, C4H5O2 37Cl2); 193 (M+H, C4H5O2O35Cl2 37Cl); 191 (M+H, C4H5O2 35Cl3); 159 (195-HCl, C4H4O2 37Cl2); 157 (193-HCl, C4H4O2 37Cl35Cl); 155 (191-HCl, C4H4O2 35Cl2); (E1): m/z 195 (M+H); 193 (M+H); 191 (M+H); 157 (193-HCl); 155 (191-HCl).
Анализ для C4H5Cl3O2 + 0,054 моль CH3COOH: C 25,35; H 2,70; Cl 54,62.
Найдено: C 25,57; H 2,72; Cl 54,66.
Реакцию инициировали 0,050 г субтилизина (компании Sigma Chemical Co., St. , Mo, P-5380, лот N 38F-0356), который был перед этим активирован путем растворения 1 г фермента в 20 мл 0,1 моль/л раствора фосфата калия с pH 7,8 и лиофилизацией до сухости. После размешивания в течение 23 ч при 40oC добавляли к реакционной смеси дополнительных 50 мг субтилизина и 2 мл трихлорэтилацетата. После размешивания при 40oC в течение дополнительных 24 ч реакцию прерывали путем отфильтровывания фермента, и растворитель удаляли в условиях вакуума. Неочищенный продукт очищали хроматографически на силикагельной колонке размером 4,5 х 25 см с CH2Cl2:CH3OH (9:1) в качестве элюента. Получившиеся фракции отстаивали и лиофилизировали из воды с получением 0,28 г нужного продукта в виде белого порошка. TLC Rf = 0,35 (силикагель; CH2Cl2: CH3OH (9: 1)); УФ λмакс ( ε , (ммоль/л)-1см-1) при pH 7,0, 279 нм (8,8).
1H-ЯМР (200 МГц, DMSO-d6): δ 7,83 (s, 1H, H8); 6,45 (s, 2H, 2-NH2); 6,14 (d, 1H, J = 3,7 Гц, H1); 5,75 (d, 1H, J = 4,5 Гц, 2'-OH); 5,65 (d, 1H, J = 3,7 Гц, 3'-OH); 4,26 (m, 2H, H2 и H3); 4,07 (m, 2H, H5); 3,94 (s, 3H, -OCH3); 3,92 (m, 1H, H4); 2,01 (s, 3H, CH3CO2-); MC (Cl) 340 (M+I); 280 (M-CH3CO2).
Аналитический расчет для C14H19N5O6•0,52H2O:
рассчитано: C 44,77; H 5,22; N 20,12;
найдено: C 44,79; H 5,21; N 20,09.
Следующие соединения были получены аналогичным способом, исходя из соответствующего трихлорэтилового эфира.
c) 2-Амино-9-(5-О-бутирил- β -D-арабинофуранозил)-6-метокси-9Н-пурин.
1H-ЯМР (200 МГц, DMSO-d6): δ 7,83 (s, 1H, H8); 6,46 (s, 2H, 2-NH2); 6,14 (d, 1H, J = 3,9 Гц, H1); 5,76 (d, 1H, J = 4,3 Гц, 2'-OH); 5,66 (d, 1H, J = 3,7 Гц, 3'-OH); 4,28 (m, 2H, H2 и H3); 4,07 (m, 2H, H5); 3,94 (s, 3H, -OCH3); 3,91 (m, 1H, H4); 2,28 (t, 2H, J = 7,2 Гц, CH3CH2CH2CO2-); 1,52 (секстет, 2H, J = 7,4 Гц, CH3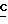 H2CH2CO2-); 0,85 (t, 3H, J = 7,3 Гц,
H2CH2CO2-); 0,85 (t, 3H, J = 7,3 Гц, 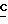 H3CH2CH2CO2-); MC (ci) 368 (M+I).
H3CH2CH2CO2-); MC (ci) 368 (M+I).
d) 2-Амино-6-метокси-9-(5-О-валерил- β -D-арабинофуранозил)-9Н-пурин.
1H-ЯМР (200 МГц, DMSO-d6): δ 7,83 (s, 1H, H8); 6,46 (s, 2H, 2-NH2); 6,15 (d, 1H, J = 3,7 Гц, H1); 5,76 (d, 1H, J = 4,2 Гц, 2'-OH); 5,65 (d, 1H, J = 3,5 Гц, 3'-OH); 4,28 (m, 2H, H2' и H3'); 4,08 (m, 2H, H5); 3,94 (s, 3H, -OCH3); 3,92 (m, 1H, H4); 2,29 (t, 2H, J = 7,1 Гц, CH3CH2CH2C 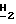 CO2-); 1,49 (m, 2H, CH3C
CO2-); 1,49 (m, 2H, CH3C 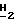 CH2CH2CO2-); 1,28 (m, 2H, CH3C
CH2CH2CO2-); 1,28 (m, 2H, CH3C 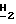 CH2CH2CO2-); 0,83 (t, 3Н, J = 7,2 Гц, C
CH2CH2CO2-); 0,83 (t, 3Н, J = 7,2 Гц, C 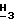 CH2CH2CH2CO2-).
CH2CH2CH2CO2-).
e) 2-Амино-6-метокси-9-(5-О-(4-метокси-4-оксобутирил)- β -D- арабинофуранозил)-9Н-пурин (в качестве исходного вещества - трихлорэтилметилсукцинат).
1H-ЯМР (200 МГц, DMSO-d6): δ 7,83 (s, 1H, H8); 6,45 (s, 2H, 2-NH2) 6,15 (d, 1H, J = 3,7 Гц, H1); 5,75 (d, 1H, J = 4,3 Гц, 2'-OH); 5,65 (d, 1H, J = 3,7 Гц, 3'-OH); 4,28 (m, 2H, H2' и H3'); 4,08 (m, 2H, H5'); 3,95 (s, 3H, -OCH3); 3,91 (m, 1H, H4'); 3,51 (s, 3H, CH3CO(O)-): 2,56 (s, 4H, -OC(O)CH2CH2C(O)O-); MC (Cl) 412 (M+I); 280 (M-C5H7O4).
Аналитический расчет для C16H21N5O8•0,40 H2O:
рассчитано: C 45,64; H 5,28; N 16,63;
найдено: C 45,62; H 5,21; N 16,67.
Пример 2. Химический синтез сложных эфиров настоящего изобретения.
a(i) 2-Амино-9-(2,5-ди-О-трет-бутилдиметилсилил- β -D-арабинофуранозил)-6- метокси-9Н-пурин. 2-Амино-9-( -β -D-арабинофуранозил)-6-метокси-9Н-пурин (10 г, 34 ммоль) добавляли в 500 мл круглодонную колбу и высушивали путем совместного выпаривания с пиридином (2 х 50 мл). Добавляли имидазол (11 г, 74 ммоль). Колбу продували аргоном и закрывали перегородкой. Добавляли сухой диметилформамид (ДМФ, 40 мл) и раствор размешивали при комнатной температуре в течение 18 ч. Тонкослойная хроматография на силикагельной колонке с ацетон : CHCl3 (1:10) выявила, что осталось около 20% исходного вещества (Rf = 0,05) и что появились три сильных особенности Rf при 0,18, 0,41 и 0,75. Добавляли дополнительно трет-бутилдиметилсилилхлорид (1,0 г, 6,6 ммоль), смесь продолжали размешивать в течение 24 ч. Последующая тонкослойная хроматография в том же растворителе показала, что все исходное вещество было израсходовано.
Затем при пониженном давлении удаляли ДМФ, остаток разделяли между этилацетатом (350 мл) и H2O (100 мл и 3 х 50 мл). Этилацетатом (100 мл) обратно экстрагировали водные слои, объединенные органические слои высушивали (MgSO4), фильтровали и концентрировали. Неочищенный продукт очищали на силикагельной флеш-колонке (5 х 25 см), элюировали пошаговым градиентом ацетона в CHCl3 (от 1:20 до 1:2). Три получаемые фракции соответствовали трем особенностям, наблюдаемым с помощью тонкослойной хроматографии. Фракция с Rf = 0,18 давала 4,0 г (23%) белого вещества в твердой фазе, идентифицированного как 2,5-дисилилированное вещество: т. пл. 180-182oC (неисправленная); УФ λмакс (95% EtOH): 248 нм и 280,8 нм; MC (E1): m/z 468 (C19H34N5O5Si2); 450 (C19H32N5O4Si2); 336 (C13H18N5O4Si); 322 (C14H24N5O2Si); 264 (C10H14N5O2Si); 222 (C8H8N5O3); 208 (C8H10N5O2); 194 (C7H8N5O2); 166 (C6H8N5O); 133 (C6H17OSi); 115 (C6H15Si); 57 (C4H9).
1H-ЯМР (CDCl3): δ 7,87 (s, 1H, H-8); 6,29 (d, 1H, H-1', J = 4,6 Гц); 4,82 (br s, 2H, NH2); 4,39-4,34 (m, 2H, H-2' и 3'); 4,07 (s, 3H, -OCH3); 3,94-3,82 (m, 3H, H-4' и 5'); 2,40 (br s, 1H, 3'-OH); 0,91 (s, 9H, (CH3)3CSi); 0,71 (s, 9H, (CH3)3CSi); 0,09 (s, 6H, (CH3)2Si); -0,02 (s, 3H, (CH3)Si); -0,24 (s, 3H, (CH3)Si).
Аналитический расчет для C23H43N5O5Si2:
рассчитано: C 52,54; H 8,24; N 13,32;
найдено: C 52,28; H 8,20; N 13,17.
a(ii) 2-Амино-9-(2,5-ди-О-трет-бутилдиметилсилил-3-О-пивалоил- β -D-арабинофуранозил)-6-метокси-9Н-пурин.
2-Амино-9-[(2,5-ди-О-трет-бутилдиметилсилил)- β -D-арабинофуранозил] - 6-метокси-9Н-пурин (2,5 г, 3,8 ммоль) отвешивали в прокаленную высушенную 250 мл круглодонную колбу. Добавляли 4-N,N-диметиламинопиридин (0,05 г, 0,4 ммоль), колбу продували аргоном и закрывали перегородкой. К реакционной смеси добавляли сухого ацетонитрила (50 мл), триэтиламина (8,0 мл) и пивалинового ангидрида (3 мл, 14,8 ммоль). После 158 ч реакционную смесь концентрировали, остаток смешивали с этилацетатом (250 мл) и экстрагировали водой (3 х 50 мл). Этилацетат высушивали (MgSO4), фильтровали и концентрировали с получением 3,8 г желтого масла, 300 г этого вещества очищали на хроматотроне (Chromatotron, Harrison Scientific), снабженном 4-мм силикагельным ротором, элюируемого с помощью смеси ацетон:CHCl3 (1 : 10). Продукт выделяли в виде прозрачной смолы (0,176 г).
MC (E1): m/z 609 (C28H51N5O6Si2); 594 (C27H48N5O5Si2); 552 (C24H42N5O6Si2); 450 (C19H32N5O4Si2); 322 (C14H24N5O2Si); 314 (C16H30O4Si); 194 (C7H8N5O2); 166 (C6H8N5O); 57 (C4H9).
1H-ЯМР (CDCl3): δ 7,93 (s, 1H, H-8); 6,25 (d, 1H, H-1', J = 3,8 Гц); 5,28 (d, 1H, H-3', J = 2,2 Гц); 5,05 (br 3, 2H, NH2); 4,30 (dd, 1H, H-2', J2',3' = 1,8 Гц); 4,10 (s, 3H, -OCH3); 4,04 (dt, 1H, H-4', J = 2,5 Гц, J = 5,9 Гц); 3,19 (d, 2H, H-5', J = 5,5 Гц); 1,27 (s, 9H, -OCOC(CH3)3); 0,75 (s, 9H, CSi(CH3)3); 0,09 (s, 6H, -Si(CH3)2); 0,02 (s, 3H, Si(CH3)); -033 (s, 3H, Si(CH3)).
Аналитический расчет для C28H51N5O6Si2• 0,75 C3H6O•0,05 CHCl3:
рассчитано: C 55,19; H 8,49; N 10,62;
найдено: C 55,32; H 8,61; N 10,53.
b) 2-Амино-6-метокси-9-(3-О-пивалоил- β -D- арабинофуранозил)-9Н-пурин.
2-Амино-6-метокси-9-[(3-О-пивалоил-2,5-ди-О-трет- бутил-диметисилил)- β -D-арабинофуранозил] -9Н-пурин (2,1 г, 3,4 ммоль) брали с ТГФ (40 мл) и охлаждали в ванночке со льдом до 5oC. Добавляли H2O (2 мл) и затем фторид тетрабутиламмония (TBAF) в виде 1 моль/л раствора в ТГФ (10 мл, 10 ммоль/л). После выдержки в течение 2 ч при 5oC добавляли дополнительно 10 мл TBAF. После 2 ч реакционную смесь еще дополнительно обрабатывали 5 мл TBAF и продолжали размешивать в течение еще 18 ч. Затем реакционную смесь разбавляли CHCl3 (40 мл) и пропускали через слой силикагеля (с зернистостью 230-400, размером 5 х 5 см) с раствором 1 : 1 ацетон : CHCl3 (500 мл). Фильтрат концентрировали и добавляли к силикагельной колонке (с зернистостью 230-400, размером 5 х 18 см). Колонку элюировали с помощью пошагового градиента ацетона с CHCl3 (от 1 : 10 до 1 : 1, ацетон : CHCl3). Из колонки получены две основные фракции, соответствующие веществам с Rf = 0,74 и 0,50 при отношении ацетон : CHCl3, равным 1 : 1. Вещество с более низким значением Rf выделяли в виде белого порошка весом 0,77 г (53%) и, как показано, оно представляло собой нужную производную 3'-О-пивалоила: т.пл. 241-243oC (неисправленная); УФ λмакс ( ε ): pH 7,00: 278,9 нм (8700) и 247,7 нм (8900); 0,1 н. раствор HCl: 287,0 (8600) и 243,7 (6800); 0,1 н. раствор NaOH: 279,2 (8900) и 247,7 (8200); MC (E1): m/z 381 (M, C16H23N5O6); 366 (C15H20N5O6); 296 (C11H14N5O5); 280 (C11H14N5O4); 250 (C10H12N5O3); 232 (C10H10N5O2); 208 (C8H10N5O2); 194 (C7H8N5O2); 165 (C6H7N5O); 136 (C5H4N4O); 85 (C5H9O).
ИК (KBr): 1733,6, 1594,7 см-1.
1H-ЯМР (Me2SO-d6): δ 7,95 (s, 1H, H-2); 6,45 (br s, 2H, NH2); 6,10 (d, 1H, H-1', J = 4,3 Гц); 6,10 (d, 1H, 2'-OH, J = 5,5 Гц); 5,16-5,10 (m, 2H, H-3' и 5'-OH); 4,23-4,20 (m, 1H, H-2'): 3,94 (s, 3H, Pur-OCH3); 3,90-3,86 (m, 1H, H-4'); 3,67-3,60 (m, 2H, 5'); 1,18 (s, 9H, C(CH3)3).
Аналитический расчет для C16H23N5O6•0,40 CHCl3:
рассчитано: C 45,90; H 5,50; N 16,32;
найдено: C 45,72; H 5,43; N 16,04.
c(i) 2-Амино-9-(3,5-ди-О-трет-бутилдиметилсилил- β -D- арабинофуранозил)-6-метокси-9Н-пурин.
2-Амино-9-( β -D- арабинофуранозил)-6-метокси-9Н-пурин (10 г, 34 ммоль) добавляли в 500 мл круглодонную колбу и высушивали путем совместного выпаривания с пиридином (2 х 50 мл). Добавляли имидазол (11 г, 160 ммоль) и затем хлорид трет-бутилдиметилсилила (11 г, 74 ммоль). Колбу продували аргоном и закрывали перегородкой. Добавляли сухого диметилформамида (ДМФ, 40 мл) и раствор размешивали при комнатной температуре в течение 18 ч. Тонкослойная хроматография на силикагеле с ацетон : CHCl3 (1 : 10) показала, что осталось около 20% исходного вещества (Rf = 0,05) и что три сильные особенности Rf проявились при 0,18; 0,14 и 0,75. Добавляли дополнительно хлорид трет-бутилдиметилсилила (1,0 г, 6,6 ммоль) и продолжали размешивать в течение 24 ч. Последующая тонкослойная хроматография в том же растворителе показала, что все исходное вещество было израсходовано.
Затем удаляли ДМФ при пониженном давлении, остаток разделяли между этилацетатом (350 мл) и водой (100 мл и 3 х 50 мл). Водные слои обратно экстрагировали с помощью этилацетата (100 мл), объединенные органические слои высушивали (MgSO4), фильтровали и концентрировали. Неочищенный продукт очищали на силикагельной флеш-колонке (5 х 25 см), элюируемой пошаговым градиентом ацетона в CHCl3 (от 1 : 20 до 1 : 2). Три полученные фракции соответствовали трем особенностям, наблюдаемым с помощью тонкослойной хроматографии. Фракция с Rf = 0,41 давала 8,0 г (45%) белого вещества в твердой фазе, идентифицированного как продукт 3,5-дисилилирования: т. пл. 88-90oC (неисправленная); УФ λмакс (95% EtOH): 247,1 нм и 280,1 нм; MC (E1): m/z 526 (M+H, C23H44N5O5Si2); 510 (C22H40N5O5Si2); 468 (C19H34N5O5Si2); 336 (C13H18N5O4Si); 301 (C13H25O4Si2); 261 (C11H11N5O3); 231 (C10H9N5O2); 208 (C8H10N5O2); 194 (C7H8N5O2); 165 (C6H7N5O); 133 (C6H17OSi); 115 (C6H15Si); 57 (C4H9).
1H-ЯМР (CDCl3) δ 8,01 (s, 1H, H-8); 6,16 (d, 1H, H-1', J = 3,1 Гц); 5,08 (br. s, 1H, 2'-OH); 4,84 (br. s, 2H, NH2); 4,31 (t, 1H, H-3', J = 1,8 Гц); 4,16-4,13 (m, 1H, H-2'); 4,05 (s, 3H, -OCH3); 4,02-3,99 (m, 1H, H-4'); 3,94 (dd, 1H, H-5', J4',5' = 3,7 Гц, J5',5'' = 11,0 Гц); 3,79 (dd, 1H, H-5'', J4',5' = 2,7 Гц, J5',5'' = 11,0 Гц); 0,94 (s, 9H, (CH3)3CSi); 0,93 (s, 9H, (CH3)3CSi); 0,17 (s, 3H, CH3Si); 0,14 (s, 3H, CH3CSi); 0,12 (s, 6H, (CH3)2Si).
Аналитический расчет для C23H43N5O5Si2:
рассчитано: C 52,54; H 8,24; N 13,32;
найдено: C 52,32; H 8,24; N 13,25.
c(ii)2-Амино-9-(3,5-ди-О-трет-бутилдиметилсилил-2-О-валерил- β -D-арабинофуранозил)-6-метокси-9Н-пурин.
2-Амино-9-[(3,5-ди-О-трет-бутилдиметилсилил)- β -D- арабинофуранозил]-6-метокси-9Н-пурин (1,3 г, 2,5 ммоль) отвешивали в обожженную высушенную 250 мл круглодонную колбу. Добавляли 4-N, N-диметиламинопиридин (0,05 г, 0,4 ммоль), колбу продували аргоном и закрывали перегородкой. Добавляли сухого ацетонитрила (30 мл) и триэтиламина (5,0 мл), раствор охлаждали в ванночке со льдом. Добавляли к реакционной смеси валериановый ангидрид (0,6 мл, 3,0 ммоль). После 18 ч при 0,5oC реакционную смесь концентрировали и остаток брали с гексан : этилацетат (1 : 1) (200 мл) и экстрагировали водой (3 х 50 мл). Органический слой высушивали (MgSO4), фильтровали и концентрировали с получением 1,7 г желтого масла. 270 мг этого вещества очищали на хроматотроне (Chromatotron, Harrison Scientific), снабженным 2-мм силикагельным ротором. Ротор элюировали смесью ацетон : CHCl3 (1 : 10). Получившийся из хроматотрона продукт представлял собой белое вещество в твердой фазе (0,21 г, 0,34 ммоль): т.пл. 105-107oC (неисправленная); MC (E1); 609 (C28H51N5O6Si2); 594 (C27H48N5O6Si2); 552 (C24H42N5O6Si2); 420 (C18H28N5O5Si); 292 (C13H18N5O3); 261 (C12H15N5O2); 231 (C10H9N5O2); 194 (C7H8N5O2); 166 (C6H8N5O); 159 (C7H25O2Si); 57 (C4H9).
1H-ЯМР (CDCl3): δ 7,92 (s, 1H, H-8); 6,39 (d, 1H, H-1', J = 5,7 Гц); 5,33 (t, 1H, H-2', J = 5,7 Гц); 4,84 (br. s, 2H, NH2); 4,60 (t, 1H, H-3', J = 5,7 Гц); 4,05 (s, 3H, OCH3); 3,93-3,80 (m, 1H, H-4' и H-5'); 2,09 (dt, 1H, C(O)CH2, J = 7,5 Гц); 1,94 (dt, 1H, C(O)CH2, J = 7,5, Гц J = 15 Гц); 1,32-1,00 (m, 4H, -CH2CH2-); 0,93 (s, (H, -SiC(CH3)3); 0,89 (s, 9H, -SiC(C3)3); 0,76 (t, 3H, -CH3, J = 7,0 Гц); 0,11 (s, 3H, Si(CH3)); 0,09 (s, 3H, -Si(CH3)); 0,09 (s, 3H, -Si(CH3)); 0,08 (s, 3H, -Si(CH3)).
Аналитический расчет для C28H51N5O6Si2:
рассчитано: C 55,14; H 8,43; N 11,48;
найдено: C 55,09; H 8,45; N 11,46.
c(iii) 2-Амино-6-метокси-9-(2-О-валерил- β -D- арабинофуранозил)-9Н-пурин.
2-Амино-9-(3,5-ди-О-трет-бутилдиметилсилил-2-О-валерил- β -D- арабинофуранозил)-6-метокси-9Н-пурин (1,4 г, 2,3 ммоль) брали с тетрагидрофураном (ТГФ, 40 мл) и охлаждали в ванночке со льдом до 5oC. Добавляли уксусную кислоту (0,06 мл, 10 ммоль) и затем фторид тетрабутиламмония (TBAF) в виде 1 моль/л раствора в ТГФ (10 мл, 10 ммоль). После выдержки при 5oC в течение 18 ч реакционную смесь разбавляли CHCl3 (40 мл) и пропускали через слой силикагеля (с зернистостью 230-400, размером 5 х 5 см) с 1 : 1 ацетон : CHCl3 (500 мл). Фильтрат концентрировали и очищали на хроматотроне, снабженном 4-мм ротором, и элюировали с помощью чистого этилацетата. Из колонки получали чистый продукт в виде белой пены в количестве 0,72 г (78%) после высушки и, как было показано, он представлял собой требуемую производную 2'-О-валерила: т. пл. 83-86oC (неисправленная); УФ λмакс ( ε ): pH 7,00: 280,0 нм (7800), 247,8 нм (8400); 0,1 н. раствор HCl: 278,6 (8000); 247,7 (7400); 0,1 н. раствор NaOH: 286,2 (7600); 244,9 (7200); MC (E1): m/z 381 (C16H23N5O6); 351 (C15H21N5O5); 292 (C13H18N5O3); 279 (C11H13N5O4); 217 (C10H11O5);
194 (C7H8N5O2); 165 (C6H7N5O); 135 (C5H5N4); 85 (C5H9O); ИК (KBr) 1745,2; 1613,3 и 1588,7 см-1.
1H-ЯМР (Me2SO-d6): δ 7,93 (s, 1H, H-2); 6,46 (br. s, 2H, NHH2); 6,26 (d, 1H, H-1', J - 5,9 Гц); 5,79 (d, 1H, 3'-OH, J = 5,1 Гц); 5,23 (t, 1H, H-2', J = 5,8 Гц); 5,02 (t, 1H, 5'-OH, J = 5,6 Гц); 4,36 (ddd, 1H, H-3', J3',3'-OH = 5,1 Гц, J2',3' = 5,7 Гц, J3',4' = 5,8 Гц); 3,93 (s, 3H, Pur-OC 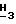 ); 3,83-3,78 (m, 1H, H-4'); 3,68-3,61 (m, 2H, H-5'); 2,09 (dt, 1H, C(O)CH2, J = 7,5 Гц, J = 15 Гц); 1,93 (dt, 1H, C(O)CH2, J = 7,5 Гц, J = 15 Гц); 1,30-0,90 (m, 4H, -CH2CH2-); 0,65 (t, 3H, -CH3, J = 7 Гц).
); 3,83-3,78 (m, 1H, H-4'); 3,68-3,61 (m, 2H, H-5'); 2,09 (dt, 1H, C(O)CH2, J = 7,5 Гц, J = 15 Гц); 1,93 (dt, 1H, C(O)CH2, J = 7,5 Гц, J = 15 Гц); 1,30-0,90 (m, 4H, -CH2CH2-); 0,65 (t, 3H, -CH3, J = 7 Гц).
Аналитический расчет для C16H23N5O6•0,15 C5H10O3:
рассчитано: C 50,41; H 6,22; N 17,29;
найдено: C 55,41; H 6,59; N 17,40.
d) (i) 2-Амино-9-(3-О-бензоил-2,5-ди-О-трет-бутилдиметилсилил- β -D-арабинофуранозил)-6-метокси-9Н-пурин.
2-Амино-9-[(2,5-ди-трет-бутилдиметилсилил)- β -D-арабинофуранозил]-6-метокси-9Н-пурин (1,5 г, 2,9 ммоль) отвешивали в обожженную высушенную 250 мл круглодонную колбу. Добавляли 4-N,N-диметиламинопиридин (0,05 г, 0,4 ммоль), колбу продували аргоном и закрывали перегородкой. К реакционной смеси добавляли сухого ацетонитрила (50 мл), триэтиламина (5,0 мл) и безангидрида (0,77 г, 3,4 ммоль). После выдержки в течение 5 ч при температуре окружающей среды реакционную смесь концентрировали, остаток брали с этилацетатом (250 мл) и экстрагировали с водой (2 х 50 мл). Этилацетат высушивали (MgSO4), фильтровали и концентрировали с получением 3,8 г желтого масла. 270 мг порцию этого вещества очищали на хроматотроне (Harrison Scientific), снабженным 4-мм силикагельным ротором. Ротор элюировали смесью ацетон : CHCl3 (1 : 10). Получившееся из хроматотрона вещество представляло собой белую твердую фазу (0,18 г, 0,29 ммоль): т. пл. 73-75oC (неисправленная); MC (E1): m/z 630 (C30H48N5O6Si2); 614 (C29H44N5O6Si2); 572 (C26H44N5O6Si2); 451 (C19H33N5O4Si2); 194 (C7H8N5O2); 179 (C6H5N5O2); 166 (C6H8N5O);
105 (C7H5).
1H-ЯМР (CDCl3); δ 8,12-8,07 (m, 2H, Ar-H); 7,92 (s, 1H, H-8); 7,63-7,45 (m, 3H, Ar-H); 6,33 (d, 1H, H-1', J = 3,7 Гц); 5,46 (t, 1H, H-3', J = 1,8 Гц); 4,79 (br. s, 2H, NH2); 4,42 (dd, 1H, H-2', J1'2' = 3,7 Гц и J2'3' = 1,7 Гц); 4,30-4,20 (m, 1H, H-4'); 4,07 (s, 3H, -OCH3); 3,99-3,95 (m, 2H, H-5'); 0,89 (s, 9H, -SiC(CH3)3); 0,76 (s, 9H, -SiC(CH3)3); 0,09 (s, 6H, -Si(CH3)2); 0,03 (s, 3H, -Si(CH3)); -0,34 (s, 3H, -Si(CH3)).
Аналитический расчет для C30H47N5O6Si2:
рассчитано: C 57,20; H 7,52; N 11,12;
найдено: C 57,08; H 7,59; N 11,05.
d) (ii) 2-Амино-9-(3-О-бензоил- β -D-арабинофуранозил)-6-метокси-9Н-пурин.
2-Амино-9-[(3-О-бензоил-2,5-ди-О-трет-бутилдиметилсилил)- β -D-арабинофуранозил] -6-метокси-9Н-пурин (1,97 г, 2,6 ммоль) соединяли с тетрагидрофураном (ТГФ, 40 мл) и охлаждали в ванночке со льдом до 5oC. Добавляли уксусной кислоты (0,6 мл, 10 ммоль) и затем фторид тетрабутиламмония (TBAF) в виде 1 моль/л раствора в ТГФ (10 мл, 10 ммоль). После выдержки в течение 18 ч при 5oC реакционную смесь разбавляли CHCl3 (40 мл) и пропускали через слой силикагеля (с зернистостью 230-400, размером 5 х 5 см) с ацетон : CHCl3 (500 мл). Фильтрат концентрировали до белой твердой фазы, которую адсорбировали в 10 г силикагеля и добавляли к силикагельной колонке (с зернистостью 230-400, размером 5 х 18 см). Колонку элюировали смесью ацетон : CHCl3 (1 : 2). Чистый продукт, получаемый из колонки, соответствовал веществу с Rf = 0,56 в ацетон : CHCl3 (1 : 1). Это вещество после высушки представляло собой 0,77 г (1,9 ммоль) белого порошка и, как было показано, являлось требуемой производной 3'-О-бензоила: т.пл. 155-157oC (неисправленная); УФ λмакс ( ε ): pH 7,00: 278,3 нм (10100), 235,2 нм (18800); 0,1 н. раствор HCl: 278,1 (91000), 245 (sh) (9600); 0,1 н. раствор NaOH: 284,8 (9600), 233,9 (18400); MC (E1): m/z 401 (M, C18H19N5O6); 296 (C11H14N5O5); 250 (C10H12N5O3); 232 (C10H10N5O2); 20 (C8H10N5O2); 194 (C7H8N5O2); 179 (C7H7N4O2); 165 (C6H7N5O); 136 (C5H4N5); 122 (C7H5O2); 105 (C7H5O); ИК (KBr): 1714,6; 1611,8 и 1591,7 см-1.
1H-ЯМР (Me2SO-d6); δ 8,06 (s, 1H, H-2); 8,02-8,00 (m, 2H, Ar-h); 7,71 (t, 1H, Ar-h, J = 7,3 Гц); 7,57 (t, 2H, Ar-H, J = 7,4 Гц); 6,45 (br. s, 2H, NH2); 6,20 (d, 1H, H-1', J = 4,3 Гц); 6,12 (d, 1H, 2'-OH, J = 5,5 Гц); 5,41 (t, 1H, H-3', J = 2,9 Гц); 5,20 (t, 1H, 5'-OH, J = 5,5 Гц); 4,43-4,37 (m, 1H, H-2'); 4,17-4,11 (m, 1H, H-4'); 3,95 (s, 3H, Pur-OC 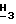 ); 3,79-3,72 (m, 2H, 5').
); 3,79-3,72 (m, 2H, 5').
Аналитический расчет для C18H19N5O6•0,60 C3H6O• 0,05 CHCl3:
рассчитано: C 53,92; H 5,16; N 15,84;
найдено: C 53,81; H 5,10; N 15,76.
e(i) 2-Амино-9-(3,5-ди-О-трет-бутилдиметилсилил-2-О-пивалоил- β -D- арабинофуранозил)-6-метокси-9Н-пурин.
2-Амино-9-(3,5-ди-О-трет-бутилдиметилсилил- β -D-арабинофуранозолил)-6-метокси-9Н-пурин (1,3 г, 2,5 ммоль) отвешивали в обожженную высушенную 250-мл круглодонную колбу. Добавляли 4-N,N-диметиламинопиридин (0,05 г, 0,4 ммоль), колбу продували аргоном и закрывали перегородкой. К реакционной смеси добавляли при размешивании при комнатной температуре сухого ацетонитрила (30 мл), триэтиламина (5,0 мл) и пивалевого ангидрида (0,6 мл, 3,0 ммоль). Через 160 ч реакционную смесь концентрировали, остаток брали с этилацетатом (250 мл) и экстрагировали с водой (3 х 50 мл). Этилацетат собирали, высушивали (MgSO4), фильтровали и концентрировали с получением 2,0 г желтого масла. 250-мг порцию этого вещества очищали на хроматотроне (Harrison Scientific), снабженном 2-мм силикагельным ротором, элюировали с помощью ацетон: CHCl3 (1:10). Получившийся из хроматотрона продукт представлял собой прозрачную смолу (0,176 г); MC (E1): m/z 609 (C28H51N5O6Si2); 594 (C27H48N5O6Si2); 552 (C24H42N5O6Si2); 420 (C18H28N5O5Si); 292 (C13H18N5O3); 261 (C12H15N5O2); 231 (C10H9N5O2); 194 (C7H8N5O2); 166 (C6H8N5O); 159 (C7H15O2Si); 57 (C49).
1H-ЯМР (CDCl3): δ 7,89 (s, 1H, H-8); 6,40 (d, 1H, H-1', J = 5,9 Гц); 5,30 (t, 1H, H-2', J = 6,0 Гц); 4,85 (br. s, 2H, NHH2Y); 4,65 (t, 1H, H-3', J = 6,0 Гц); 4,04 (s, 3H, OCH3); 3,95-3,85 (m, 1H, H-4', и H-5'); 0,92 (s, 9H, OCOC(CH3)3); 0,89 (s, 9H, SiC(CH3)3); 0,88 (s, 9H, SiC(CH3)3); 0,13 (s, 3H, SiCH3); 0,11 (s, 3H, SiCH3); 0,08 (s, 3H, SiCH3); 0,07 (s, 3H, SiCH3).
Аналитический расчет для C28H51N5O6Si2:
рассчитано: C 55,14; H 8,43; N 11,48;
найдено: C 54,97; H 8,42; N 11,10.
e(ii) 2-Амино-6-метокси-9-(2-О-пивалоил- β -D-арабинофуранозил)-9Н-пурин.
2-Амино-6-метокси-9-(3,5-О-ди-трет-бутилдиметилсилил-2-О-пивалоил- β -D-арабинофуранозил)-9Н-пурин (1,3 г, 2,0 ммоль) брали с тетрагидрофураном (ТГФ, 40 мл) и охлаждали в ванночке со льдом до 5oC. Добавляли уксусной кислоты (0,06 ммоль) и затем фторид тетрабутиламмония в виде 1 моль/л раствора в ТГФ (10 мл, 10 ммоль). После выдержки в течение 24 ч при 5oC реакционную смесь разбавляли CHCl3 (40 мл) и пропускали через слой силикагеля (с зернистостью 230-240, размером 5 х 5 см) с 1:1 ацетона: CHCl3 (500 мл). Фильтрат концентрировали и направляли в силикагельную колонку (зернистостью 230-400, размером 5 х 18 см), элюировали с ацетон:CHCl3 (1:2, 1,5 л) и затем с ацетон: CHCl3 (1:1, 1,5 л). Чистый продукт, получаемый из колонки, представлял собой белый порошок весом 0,76 г (100%) после сушки и, как было показано, представлял собой требуемую производную 2'-О-пивалоила: т. пл. 83-85oC (неисправленная); УФ λмакс ( ε ): pH 7,00: 279,7 нм (8100), 247,9 нм (8800); 0,1 н. раствор HCl: 286,6 (7300), 244,7 (6200); 0,1 н. раствор NaOH: 279,7 (8000), 248,8 (7900); MC (E1): m/z 250 (C10H12N5O3); 232 (C10H10N5O2); 217 (C10H17N5); 208 (C8H10N5O2); 194 (C7H8N5O2); 165 (C6H7N5O); 135 (C5H4N4O4); 101 (C5H9O2); 85 (C5H9O); ИК (KBr); 1734,2; 1616,3 и 2589,4 см-1;
1H-ЯМР (Me2SO-d6): δ 7,97 (s, 1H, h-2); 6,47 (br. s, 2H, NH2); 6,26 (d, 1H, H-1', J = 5,9 Гц); 5,79 (d, 1H, 3'-OH, J = 5,3 Гц); 5,23 (dd, 1H, H-2', J1',2' = 5,9 Гц, J2',3' = 5,2 Гц); 5,06 (5,1H, 5'-OH, J = 5,5 Гц); 4,37 (ddd, 1H, H-3', J3',3'-OH - 5,3 Гц, J2',3' = 5,2 Гц, J3',4' = 6,9 Гц); 3,92 (s, 3H, Pur-OC 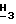 ); 3,84-3,79 (m, 1H, H-4'); 3,68-3,62 (m, 2H, H-5').
); 3,84-3,79 (m, 1H, H-4'); 3,68-3,62 (m, 2H, H-5').
Аналитический расчет для C16H23N5O6• 0,40 CHCl3:
рассчитано: C 45,90; H 5,50; N 16,32;
найдено: C 46,03; H 5,69; N 16,03.
f(i) 2-Амино-9-(2-О-бензоил-3,5-ди-О-трет-бутилдиметилсилил- β - D-арабинофуранозил)-6-метокси-9Н-пурин.
2-Амино-9-[(3,5-ди-О-трет-бутилдиметилсилил)- β -D-арабинофуранозил] - 6-метокси-9Н-пурин (1,3 г, 2,5 ммоль) отвешивали в обожженную высушенную 250-мл круглодонную колбу. Добавляли 4-N,N-диметиламинопиридин (0,05 г, 0,4 ммоль) и бензагидрид (0,67 г, 3,0 ммоль), колбу продували аргоном и закрывали перегородкой. Затем добавляли сухого ацетонитрила (30 мл) и триэтиламина (5,0 мл), смесь размешивали при комнатной температуре. После 18 ч реакционную смесь концентрировали, остаток размешивали с этилацетатом (250 мл) и экстрагировали с водой (3 х 50 мл). Этилацетат высушивали (MgSO4), фильтровали и концентрировали с получением 1,76 г желтого масла. 250 мг порцию этого вещества очищали на хроматроне (Harrison Scientific), снабженном 2-мм силикагельным ротором. Ротор элюировали смесью ацетон: CHCl3 (1:10). Получившийся из хроматрона продукт представлял собой белую твердую фазу (0,21 г): т.пл. 129-131oC (неисправленная); MC (E1): m/z 629 (C30H47N5O6Si2); 572 (C26H47N5O6Si2); 440 (C20H22N5O5Si); 312 (C15H14N5O3); 261 (C11H25O3Si2); 231 (C9H19O3Si2); 194 (C7H8N5O2); 166 (C6H8N5O); 105 (C7H5O).
1H-ЯМР (CDCl3): δ 8,07 (s, 1H, H-8); 7,67 (dd, 2H, Ar-H, J = 1,0 Гц, J = 8,2 Гц); 7,50 (tt, 1H, Ar-H, J = 2,0 Гц, J = 8,0 Гц); 7,30 (t, 2H, Ar-H, J = 7,5 Гц); 6,48 (d, 1H, H-1', J = 5,5 Гц); 5,63 (t, 1H, H-2', J = 5,5 Гц); 4,74 (t, 1H, H-3', J = 5,6 Гц); 4,68 (br. s, 2H, NH2); 3,98 (s, 3H, -OCH3); 4,00-3,80 (m, 3H, H-4' и H-5'); 0,92 (s, 9H, -SiC(CH3)3); 0,88 (s, 9H, -SiC(CH3)3); 0,12 (s, 3H, -Si(CH3)); 0,09 (s, 3H, -Si(CH3)); 0,08 (s, 3H, -Si(CH3)); 0,06 (s, 3H, -Si(CH3)).
Аналитический расчет для C30H47N5O6Si2:
рассчитано: C 57,20; H 7,52; N 11,12;
найдено: C 57,42; H 7,57; N 11,12.
f(ii) 2-Амино-9-(2-О-бензоил- β -D-арабинофуранозил)-6- метокси-9Н-пурин.
2-Амино-9-[(2-О-бензоил-3,5-ди-О-трет-бутилдиметилсилил)- β -D-арабинофуранозил] -6-метокси-9Н-пурин (1,26 г, 2,0 ммоль) размешивали в тетрагидрофуране (ТГФ, 40 мл) и охлаждали в ванночке со льдом до 5oC. Добавляли уксусной кислоты (0,06 мл, 10 ммоль) и затем фторид тетрабутиламмония (TBAF) в виде 1 моль/л раствора в ТГФ (10 мл, 10 ммоль). После выдержки в течение 24 ч при 5oC реакционную смесь разбавляли CHCl3 (40 мл) и пропускали через слой силикаагеля (с зернистостью 230-400, размером 5 х 5 см) с 1 : 1 ацетон : CHCl3 (500 мл). Фильтрат концентрировали и направляли в силикагельную колонку (с зернистостью 230-400, размером 5 х 18 см). Колонку элюировали смесью ацетон : CHCl3 (1 : 2, 1 л) и затем ацетон : CHCl3 (1 : 1, 1,5 л). Чистый продукт, получаемый из колонки, соответствовал веществу с Rf = 0,33 в ацетон : CHCl3 (1 : 1). После сушки это вещество представляло собой белый порошок весом 0,74 г (90%) и, как было показано, являлось требуемой производной 2'-О-бензоила: т.пл. 82-84oC (неисправленная); УФ λмакс ( ε ): pH 7,00: 279, 1 нм (8600); 237,5 нм (17800); 0,1 н. раствор HCl: 277,8 (10000), 245 (sh) (10800); 0,1 н. раствор NaOH: 286,1 (7600), 236,4 (17000). MC (E1): m/z (M, C18H19N5O6); 371 (C17H17N5O5); 312 (C15H14N5O3); 279 (C11H13N5O4); 237 (C12H13O5); 220 (C12H12N4); 208 (C8H10N5O2); 194 (C7H8N5O2); 165 (C6H7N5O); 135 (C5H5N5); 105 (C7H5O). ИК (KBr): 1725,3; 1613,6 и 1588,9 см-1.
1H-ЯМР (Me2SO-d6): δ 8,04 (s, 1H, H-2); 7,70-7,57 (m, 3H, Ar-H); 7,47-7,39 (m, 2H, Ar-H); 6,44 (br. s, 2H, NH2); 6,39 (d, 1H, H-1', J = 5,6 Гц); 5,90 (d, 1H, 3'-OH, J = 4,9 Гц); 5,48 (t, 1H, H-2', J = 5,3 Гц); 5,07 (t, 1H, 5'-OH, J = 5,6 Гц); 4,51 (разрешенный квартет, 1H, H-3', J3',3'-OH = 4,9 Гц, J2',3' = 5,6 Гц, J3',4' = 5,1 Гц); 3,87 (s, 3H, Pur-OC 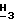 ); 3,94-3,83 (m, 1H, H-4'); 3,77-3,66 (m, 2H, H-5').
); 3,94-3,83 (m, 1H, H-4'); 3,77-3,66 (m, 2H, H-5').
Аналитический расчет для C18H19N5O6•0,20 C3H6O• 0,50 CHCl3:
рассчитано: C 48,53; H 4,41; N 14,82;
найдено: C 48,68; H 4,54; N 14,96.
g(i) 2-Амино-9-(5-О-трет-бутилдимитилсилил- β -D- арабинофуранозил)-6-метокси-9Н-пурин.
9-(β-D-арабинофуранозил)-2-амино- 6-метокси-9 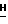 -пурин (2,0 г, 6,7 ммоль) добавляли в 500-мл круглодонную колбу и высушивали путем совместного выпаривания с пиридином (2 х 50 мл). Добавляли хлорид трет-бутилдиметилсилила (1,2 г, 8 ммоль), колбу продували аргоном и закупоривали перегородкой. Добавляли с помощью шприца и иглы сухой ацетонитрил (20 мл) и сухой пиридин (20 мл). Раствор размешивали при комнатной температуре в течение 24 ч. Тонкослойная хроматография на силикагеле с метанол : CHCl3 (1 : 10) показала, что все исходное вещество (Rf = 0,05) было израсходовано и что образовался один высокий пик Rf (Rf = 0,31). Реакционную смесь обрабатывали этанолом (2 мл) и концентрировали при пониженном давлении. Желтый остаток очищали на силикагельной колонке (с зернистостью 230-400, размером 5 х 18 см) с метанол : CHCl3 (1 : 20) в качестве элюента. Из колонки после сушки получили 1,6 г (3,9 ммоль) белого вещества в твердой фазе: т.пл. 101-103oC (неисправленная): MC (E1): m/z 412 (M+H, C17H30N5O5Si); 396 (C16H26N5O5Si); 354 (C13H20N5O5Si); 208 (C8H10N5O2); 194 (C7H8N5O2); 178 (C7H8N5O);
-пурин (2,0 г, 6,7 ммоль) добавляли в 500-мл круглодонную колбу и высушивали путем совместного выпаривания с пиридином (2 х 50 мл). Добавляли хлорид трет-бутилдиметилсилила (1,2 г, 8 ммоль), колбу продували аргоном и закупоривали перегородкой. Добавляли с помощью шприца и иглы сухой ацетонитрил (20 мл) и сухой пиридин (20 мл). Раствор размешивали при комнатной температуре в течение 24 ч. Тонкослойная хроматография на силикагеле с метанол : CHCl3 (1 : 10) показала, что все исходное вещество (Rf = 0,05) было израсходовано и что образовался один высокий пик Rf (Rf = 0,31). Реакционную смесь обрабатывали этанолом (2 мл) и концентрировали при пониженном давлении. Желтый остаток очищали на силикагельной колонке (с зернистостью 230-400, размером 5 х 18 см) с метанол : CHCl3 (1 : 20) в качестве элюента. Из колонки после сушки получили 1,6 г (3,9 ммоль) белого вещества в твердой фазе: т.пл. 101-103oC (неисправленная): MC (E1): m/z 412 (M+H, C17H30N5O5Si); 396 (C16H26N5O5Si); 354 (C13H20N5O5Si); 208 (C8H10N5O2); 194 (C7H8N5O2); 178 (C7H8N5O);
166 (C6H8N5O); 165 (C6H7N5O); 135 (C5H5N5); 57 (C4H9).
1H-ЯМР (Me2SO-d6), 300 МГц): 7,88 (s, 1H, H-8); 6,49 (br. s, 2H, NH2); 6,14 (d, 1H, H-1', J = 4,7 Гц); 5,67 (d, 1H, 2'- или 3'OH, J = 5,0 Гц); 5,57 (d, 1H, 2' - или 3'OH, J = 4,4 Гц); 4,15-4,09 (m, 2H, H-2' и 3'); 3,98 (s, 3H, -OCH3); 3,87-3,76 (m, 3H, H-4' и 5'); 0,90 (s, 9H (CH3)3CSi); 0,07 (s, 6H, (CH3)2Si);
Аналитический расчет для C17H29N5O5Si2:
рассчитано: C 49,62; H 7,10; N 17,02;
найдено: C 49,36; H 7,06; N 16,88.
g(ii) 9-(2,3-ди-О-Ацетил-5-О-трет-бутилдиметилсилил- β -D- арабинофуранозил)-2-амино-6-метокси-9Н-пурин.
2-Амино-9-(5-О-трет- бутилдимитилсилил- β -D-арабинофуранозил)-6-метокси-9Н- пурин (1,5 г, 3,5 ммоль) отвешивали в обожженную высушенную 100-мл круглодонную колбу, снабженную размешивателем. Добавляли безводный ацетонитрил (25 мл) и затем триэтиламин (5 мл). Добавляли 4-N,N-диметиламинопирин (0,05 г, 0,4 ммоль), колбу продували аргоном и закрывали перегородкой. С помощью шприца и иглы добавляли к реакционной смеси уксусный ангидрид (0,8 мл, 8,5 ммоль, очищенный дистилляцией). Реакционную смесь размешивали при температуре окружающей среды (20oC) и исследовали тонкослойной хроматографией на силикагеле с метанол:CHCl3 (1 : 20). Через 18 ч все исходное вещество (Rf = 0,34) было израсходовано и образовался сильный пик продукта (Rf = 0,77).
Реакционную смесь концентрировали, остаток смешивали с этилацетатом (250 мл) и экстрагировали с водой (3 х 50 мл). Органический слой высушивали с помощью MgSO4 (безводный), фильтровали и концентрировали с получением 1,7 г желтого масла. 210-мг порцию этого вещества очищали на хроматотроне (Harrison Scientific), снабженном 2-мм силикагельным ротором. Ротор элюировали с помощью смеси ацетон : CHCl3 (1 : 10). После просушки получали чистый продукт в виде твердой фазы (0,16 г; 0,33 ммоль): т.пл. 58-60oC (неисправленная). MC (E1): m/z 497 (C21H35N5O7Si); 438 (C17H24N5O7Si); 396 (C15H22N5O6Si); 378 (C15H20N5O5Si); 336 (C13H18N5O4Si); 318 (C13H16N5O3Si); 273 (C11H18O6Si); 250 (C10H12N5O3); 208 (C8H10N5O2); 194 (C7H8N5O2); 165 (C6H7N5O); 135 (C5H5N5).
1H-ЯМР (CDCl3, 200 МГц): 7,89 (s, 1H, H-8); 6,38 (d, H-1', J = 4,6 Гц); 5,53-4,49 (m, 2H, H-2' и 3'); 4,85 (br. s, 2H, NH2); 4,05 (s, 3H, -OCH3); 4,04-4,00 (m, 1H, H-4'); 3,94-3,91 (m, 2H, H-5'); 2,12 (s, 3H, C(O)CH3); 1,85 (s, 3H, C(O)CH3); 0,92 (s, 9H, SiC(CH3)3); 0,10 (s, 6H, Si(CH3)2).
Аналитический расчет для C21H33N5O7Si:
рассчитано: C 50,89; H 6,71; N 14,13;
найдено: C 50,70; H 6,75; N 13,91.
g(iii) 9-(2,3-ди-О-Ацетил- β -D-арабинофуранозил)-2-амино-6-метокси-9Н-пурин.
Промежуточное соединение силилирования 9-(2,3-ди-О-ацетил-5-О-трет-бутилдиметилсилил- β -D-арабинофуранозил)-2-амино-6-метокси-9Н-пурина (1,4 г, 2,9 ммоль) брали вместе с ТГФ (40 мл) и охлаждали в ванночке со льдом до 5oC. Добавляли уксусную кислоту (0,25 мл) 16 ммоль) и затем фторид тетрабутиламмония в виде 1 моль/л раствора в ТГФ (9 мл, 9 ммоль). После выдержки в течение 28 ч при 5oC тонкослойная хроматография с ацетон : CHCl3 (1 : 1) показала, что исходное вещество было израсходовано (Rf = 0,77) и образовался один низкий пик Rf (Rf = 0,32). Тонкослойная хроматография с метанол : CHCl3 (1 : 10) показала наличие одной особенности при Rf = 0,41.
Реакционную смесь пропускали через слой силикагеля (с зернистостью 230-400, размером 3 х 5 см) с ацетон : CHCl3 (1 : 1,500 мл). Фильтрат концентрировали и очищали с помощью хроматотрона, снабженного 4-мм ротором и элюируемого смесью ацетон : CHCl3 (1 : 1). Полученный продукт (0,95 г) был загрязнен гидроксидом тетрабутиламмония. Продукт очищали на хроматотроне с 4-мм ротором с тем же растворителем, что дало чистый продукт весом 0,877 г (2,3 ммоль). т.пл. 100 - 102oC (неисправленная); УФ λмакс ( ε ): pH 7,00: 279 нм (8200), 247,3 нм (9800); 0,1 н. раствор HCl: 286,7 (8500), 242,5 (6100); 0,1 н. раствор NaOH: 279,3 (8200), 248,9 (7700); MC (E1): m/z 381 (C15H19N5O7); 322 (C13H14N5O5); 292 (C12H12N5O4); 194 (C7H8N5O2); 165 (C6H7N5O);
135 (C5H5N5); 43 (C2H3O).
1H-ЯМР (Me2SO-d6, 200 МГц): δ 8,00 (s, 1H, H-8); 6,52 (br. s, 2H, NH2); 6,30 (d, 1H, H-1', J = 4,6 Гц); 5,43-5,35 (m, 2H, H-2' и 3'); 5,11 (t, 1Н, 5'-OH, J = 5,8 Гц); 4,04 (dd, 1H, H-4', J = 4,5 Гц, J = 8,6 Гц); 3,94 (s, 3H, Pur-OCH3); 3,71-3,62 (m, 2H, H-5'); 2,09 (s, 3H, C(O)CH3); 1,80 (s, 3H, C(O)CH3); ИК (KBr) 1748,4, 1613,1 и 1588,5 см-1.
Аналитический расчет для C15H19N5O7•0,40 CHCl3:
рассчитано: C 43,11; H 4,56; N 16,32;
найдено: C 43,16; H 4,69; N 16,21.
h(i) 2-Амино-9-(2,3,5-три-О-трет-бутилдиметилсилил- β -D- арабинофуранозил)-6-метокси-9Н-пурин.
2-Амино-9-( β -D-арабинофуранозил)-6-метокси-9 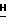 -пурин (10 г, 34 ммоль) добавляли в 500-мл круглодонную колбу и высушивали путем совместного выпаривания с пиридином (2 х 50 мл). Добавляли имидазол (11 г, 160 ммоль) и затем хлорид тетрабутилдиметилсилил (11 г, 74 ммоль). Колбу продували аргоном и закупоривали перегородкой. С помощью шприца и иглы добавляли сухой диметилформамид (ДМФ) (40 мл). Раствор размешивали при комнатной температуре в течение 18 ч. Тонкослойная хроматография на силикагеле с ацетон : CHCl3 (1 : 10) показала, что около 20% исходного вещества осталось (Rf = 0,05) и что три высоких пика Rf образовалось со значениями 0,18, 0,41 и 0,75. Поэтому добавили дополнительно хлорид тетрабутилдиметилсилила (1,0 г, 6,6 ммоль) и продолжали размешивать в течение 24 ч. Последующая тонкослойная хроматография в том же растворителе показала, что все исходное вещество было израсходовано. Затем удаляли ДМФ при пониженном давлении и остаток разделяли между этилацетатом (350 мл) и водой (100 мл и 3 х 50 мл). Водные слои обратно экстрагировали с помощью этилацетата (100 мл), объединенные органические слои высушивали (MgSO4), фильтровали и концентрировали. Неочищенный продукт очищали на силикагельной флеш-колонке (5 х 25 см) и элюировали пошаговым градиентом ацетона в CHCl3 в пропорции от 1 : 20 до 1 : 2. Трем полученным фракциям соответствовали три особенности, наблюдаемые с помощью тонкослойной хроматографии. Фракция Rf = 0,75 дала 4,0 г (6,2 ммоль, 19%) белого твердого вещества, идентифицированного как продукт трисилилирования: т.пл. 63 - 65 oC (неисправленная). УФ λмакс (95% этанол): 249,2 нм и 281,3 нм:MC (E1): m/z 640 (M, C29H57N5O5Si3); 582 (C25H48N5O5Si3); 450 (C19H32N4O4Si2); 322 (C14H24N5O2Si); 222 (C8H8N5O3); 194 (C7H8N5O2); 166 (C6H8N5O); 133 (C6H17OSi); 115 (C6H15Si); 57 (C4H9).
-пурин (10 г, 34 ммоль) добавляли в 500-мл круглодонную колбу и высушивали путем совместного выпаривания с пиридином (2 х 50 мл). Добавляли имидазол (11 г, 160 ммоль) и затем хлорид тетрабутилдиметилсилил (11 г, 74 ммоль). Колбу продували аргоном и закупоривали перегородкой. С помощью шприца и иглы добавляли сухой диметилформамид (ДМФ) (40 мл). Раствор размешивали при комнатной температуре в течение 18 ч. Тонкослойная хроматография на силикагеле с ацетон : CHCl3 (1 : 10) показала, что около 20% исходного вещества осталось (Rf = 0,05) и что три высоких пика Rf образовалось со значениями 0,18, 0,41 и 0,75. Поэтому добавили дополнительно хлорид тетрабутилдиметилсилила (1,0 г, 6,6 ммоль) и продолжали размешивать в течение 24 ч. Последующая тонкослойная хроматография в том же растворителе показала, что все исходное вещество было израсходовано. Затем удаляли ДМФ при пониженном давлении и остаток разделяли между этилацетатом (350 мл) и водой (100 мл и 3 х 50 мл). Водные слои обратно экстрагировали с помощью этилацетата (100 мл), объединенные органические слои высушивали (MgSO4), фильтровали и концентрировали. Неочищенный продукт очищали на силикагельной флеш-колонке (5 х 25 см) и элюировали пошаговым градиентом ацетона в CHCl3 в пропорции от 1 : 20 до 1 : 2. Трем полученным фракциям соответствовали три особенности, наблюдаемые с помощью тонкослойной хроматографии. Фракция Rf = 0,75 дала 4,0 г (6,2 ммоль, 19%) белого твердого вещества, идентифицированного как продукт трисилилирования: т.пл. 63 - 65 oC (неисправленная). УФ λмакс (95% этанол): 249,2 нм и 281,3 нм:MC (E1): m/z 640 (M, C29H57N5O5Si3); 582 (C25H48N5O5Si3); 450 (C19H32N4O4Si2); 322 (C14H24N5O2Si); 222 (C8H8N5O3); 194 (C7H8N5O2); 166 (C6H8N5O); 133 (C6H17OSi); 115 (C6H15Si); 57 (C4H9).
1H-ЯМР (CDCl3, 200 МГц): 7,82 (s, 1H, H-8); 6,30 (d, 1H, H-1', J = 3,5 Гц); 4,82 (br. s, 2H, NH2); 4,31 (m, 1H, H-3'); 4,10 (dd, 1H, H-2', J1',2' = 3,5 Гц и J2',3' = 1,7 Гц); 4,06 (s, 3H, -OCH3); 4,00-3,90 (m, 1H, H-4'); 3,84-3,80 (m, 2H, H-5'); 0,93 (s, 9H, (CH3)3CSi); 0,90 (s, 9H, (CH3)3CSi); 0,75 (s, 9H, (CH3)3CSi); 0,14 (s, 6H, (CH3)2Si); 0,07 (s, 3H, (CH3)Si); 0,06 (s, 3H, (CH3)Si); -0,09 (s, 3H, (CH3)Si); -0,38 (s, 3H, (CH3)Si).
Аналитический расчет для C29H57N5O5Si3:
рассчитано: C 54,42; H 8,98; N 10,94;
найдено: C 54,36; H 8,86; N 10,87.
h(ii) 2-Амино-9-(2,3-ди-О-трет-бутилдимитилсилил- β -D- арабинофуранозил)-6-метокси-9Н-пурин.
2-Амино-9-(2,3,5-ди-О-трет-бутилдимитилсилил- β -D-арабинофуранозил)-6-метокси-9 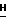 -пурин (2,0 г, 3,1 ммоль) добавляли в 250-мл круглодонную колбу и обрабатывали 80% водным раствором уксусной кислоты. Раствор нагревали при 50oC в течение 20 ч. Тонкослойная хроматография в ацетон : CHCl3 (1 : 10) показала, что все исходное вещество было израсходовано (Rf = 0,59) и остался только продукт (Rf = 0,19). Реакционную смесь концентрировали с помощью роторного испарителя, несколько раз добавляя воду (5 мл) для удаления последних следов уксусной кислоты, и затем помещали в вакуумный насос на 2 ч. Остаток очищали на силикагельной колонке (размером 5 х 18 см, с зернистостью 230 - 400) с метанол : CHCl3 (1 : 30) в качестве элюента. Из содержащего фракции продукта получили 1,2 г (2,3 ммоль) белого твердого вещества, идентифицированного как производная 2,3-дисилилирования: т.пл. 93 - 95oC (неисправленная). MC (E1): m/z 526 (C23H44N5O5Si2); 510 (C22H40N5O5Si2); 468 (C19H34N5O5Si2); 322 (C14H23N5O5Si); 306 (C13H20N5O2Si); 264 (C10H14N5O2Si); 208 (C8H10N5O2); 194 (C7H8N5O2); 166 (C6H8N5O);
-пурин (2,0 г, 3,1 ммоль) добавляли в 250-мл круглодонную колбу и обрабатывали 80% водным раствором уксусной кислоты. Раствор нагревали при 50oC в течение 20 ч. Тонкослойная хроматография в ацетон : CHCl3 (1 : 10) показала, что все исходное вещество было израсходовано (Rf = 0,59) и остался только продукт (Rf = 0,19). Реакционную смесь концентрировали с помощью роторного испарителя, несколько раз добавляя воду (5 мл) для удаления последних следов уксусной кислоты, и затем помещали в вакуумный насос на 2 ч. Остаток очищали на силикагельной колонке (размером 5 х 18 см, с зернистостью 230 - 400) с метанол : CHCl3 (1 : 30) в качестве элюента. Из содержащего фракции продукта получили 1,2 г (2,3 ммоль) белого твердого вещества, идентифицированного как производная 2,3-дисилилирования: т.пл. 93 - 95oC (неисправленная). MC (E1): m/z 526 (C23H44N5O5Si2); 510 (C22H40N5O5Si2); 468 (C19H34N5O5Si2); 322 (C14H23N5O5Si); 306 (C13H20N5O2Si); 264 (C10H14N5O2Si); 208 (C8H10N5O2); 194 (C7H8N5O2); 166 (C6H8N5O);
116 (C6H15Si); 57 (C4H9).
1H-ЯМР (CDCl3, 200 МГц); 7,82 (s, 1H, H-8); 6,45 (br. s, 2H, NH2); 6,14 (d, 1H, H-1', J = 4,1 Гц); 4,99 (t, 1H, 5'-OH, J = 4,0 Гц); 4,29 (t, 1H, H-3', J = 2,9 Гц); 4,22 (t, 1H, H-2', J = 4,5 Гц); 3,93 (s, 3H, -OCH3); 3,82 - 3,76 (m, 1H, H-4'); 3,62 - 3,58 (m, 2H, H-5'); 0,89 (s, 9H, (CH3)3SCi); 0,64 (s, 9H, (CH3)3CSi); 0,13 (s, 6H, (CH3)2Si); -0,06 (s, 3H, (CH3)Si); -0,39 (s, 3H, (CH3)Si).
Аналитический расчет для C23H43N5O5Si2:
рассчитано: C 52,54; H 8,24; N 13,32;
найдено: C 52,37; H 8,29; N 13,22.
h(iii) 2-Амино-9-(2,3-ди-О-трет-бутилдимитилсилил-5-О-изобутирил- β -D-арабинофуранозил)-6-метокси-9Н-пурин.
2-Амино-9-(2,3-ди-О-трет-бутилдимитилсилил- β -D-арабинофуранозил)-6-метокси-9 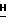 -пурин (1,3 г, 2,5 ммоль) отвешивали в обожженную высушенную 250-мл круглодонную колбу. Добавляли 4-N,N-диметиламинопиридин (0,05 г, 0,4 ммоль), колбу продували аргоном и закрывали перегородкой. Добавляли сухой ацетонитрил (25 мл) и триэтиламин (5 мл). Колбу охлаждали в ванночке со льдом и добавляли к реакционной смеси изобутириловый ангидрид (0,5 мл, 3,0 ммоль). Реакционную смесь оставляли медленно нагреваться до температуры окружающей среды (20oC). После 22 ч реакционную смесь исследовали с помощью тонкослойной хроматографии на силикагеле с метанол : CHCl3 (1 : 20). Никакого исходного вещества (Rf = 0,27) не осталось, а образовалась одна сильная особенность продукта (Rf = 0,63).
-пурин (1,3 г, 2,5 ммоль) отвешивали в обожженную высушенную 250-мл круглодонную колбу. Добавляли 4-N,N-диметиламинопиридин (0,05 г, 0,4 ммоль), колбу продували аргоном и закрывали перегородкой. Добавляли сухой ацетонитрил (25 мл) и триэтиламин (5 мл). Колбу охлаждали в ванночке со льдом и добавляли к реакционной смеси изобутириловый ангидрид (0,5 мл, 3,0 ммоль). Реакционную смесь оставляли медленно нагреваться до температуры окружающей среды (20oC). После 22 ч реакционную смесь исследовали с помощью тонкослойной хроматографии на силикагеле с метанол : CHCl3 (1 : 20). Никакого исходного вещества (Rf = 0,27) не осталось, а образовалась одна сильная особенность продукта (Rf = 0,63).
Реакционную смесь концентрировали, остаток размешивали в этилацетате (250 мл) и экстрагировали с водой (3 х 50 мл). Слой этилацетата высушивали с помощью MgSO4 (безводный), фильтровали и концентрировали с получением 1,5 г желтого масла. 200-мг порцию этого вещества очищали на хроматотроне (Harrison Scientific), снабженном 2-мм силикагельным ротором. Ротор элюировали с помощью ацетон : CHCl3 (1 : 10), получая после высушки 0,16 г (0,27 ммоль) белого вещества в твердой фазу; т.пл. 56 - 58oC (неисправленная); MC (E1): m/z 596 (C27H49N5O6Si2); 538 (C23H40N5O6Si2); 322 (C14H24N5O2Si); 318 (C13H24O5Si2); 264 (C10H16N5O2Si); 194 (C7H8N5O2); 166 (C6H8N5O);
115 (C6H15Si).
1H-ЯМР (CDCl3, 200 МГц): δ 7,84 (s, 1H, H-8); 6,34 (d, 1H, H-1', J = 3,3 Гц); 4,87 (br. s, 2H, NH2); 4,39 - 4,27 (m, 2H, H2'- и 3'); 4,20 - 4,10 (m, 3H, H-4' и 5'); 4,06 (s, 3H, -OCH3); 2,58 (септет, 1H, OC(O)C  (CH3)2, J = 7,0 Гц); 1,17 (dd, 6H, OC(O)CH(CH3)2, J = 7,0 Гц, J = 1,9 Гц); 0,94 (s, 9H, -SiC(CH3)3); 0,77 (s, 9H, -SiC(CH3)3); 0,15 (s, 3H, -Si(CH3)); 0,13 (s, 3H, -Si(CH3)); 0,09 (s, 3H, -Si(CH3)); 0,39 (s, 3H, -Si(CH3)).
(CH3)2, J = 7,0 Гц); 1,17 (dd, 6H, OC(O)CH(CH3)2, J = 7,0 Гц, J = 1,9 Гц); 0,94 (s, 9H, -SiC(CH3)3); 0,77 (s, 9H, -SiC(CH3)3); 0,15 (s, 3H, -Si(CH3)); 0,13 (s, 3H, -Si(CH3)); 0,09 (s, 3H, -Si(CH3)); 0,39 (s, 3H, -Si(CH3)).
Аналитический расчет для C27H49N5O6Si2• 0,05 C3H6O•0,10 CHCl3:
рассчитано: C 53,59; H 8,15; N 11,47;
найдено: C 53,58; H 8,23; N 11,40.
h(iv) 2-Амино-9-(5-О-изобутирил- β -D-арабинофуранозил)-6- метокси-9Н-пурин.
Промежуточное соединение силилирования 2-амино-9- (2,3-ди-О-трет-бутилдимитилсилил-5-О-изобутирил- β -D- арабинофуранозил)-6-метокси-9 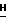 -пурина (1,2 г, 2,1 ммоль) размешивали в ТГФ (40 мл) и охлаждали в ванночке со льдом до 5oC. Добавляли уксусной кислоты (0,25 мл, 16 ммоль) и затем фторид тетрабутиламмония в виде 1 ммоль/л раствора в ТГФ (6 мл, 6 ммоль). После выдержки в течение 70 ч при 0 - 5oC, тонкослойная хроматография на смеси ацетон : CHCl3 (1 : 1) показала, что никакого исходного вещества не осталось (Rf = 0,91), а образовалась одна новая слабая особенность Rf = 0,17.
-пурина (1,2 г, 2,1 ммоль) размешивали в ТГФ (40 мл) и охлаждали в ванночке со льдом до 5oC. Добавляли уксусной кислоты (0,25 мл, 16 ммоль) и затем фторид тетрабутиламмония в виде 1 ммоль/л раствора в ТГФ (6 мл, 6 ммоль). После выдержки в течение 70 ч при 0 - 5oC, тонкослойная хроматография на смеси ацетон : CHCl3 (1 : 1) показала, что никакого исходного вещества не осталось (Rf = 0,91), а образовалась одна новая слабая особенность Rf = 0,17.
Реакционную смесь пропускали через слой силикагеля (с зернистостью 230 - 400, размером 2 х 8 см) с 1 : 1 ацетон : CH2Cl2 (1,1 л). Фильтрат концентрировали до получения светлого золотистого остатка. Этот остаток очищали на хроматотроне, снабженном 4-мм ротором с ацетон : CH2Cl2 (1 : 1) в качестве элюента. Получено белое вещество в твердой фазе (0,67 г), которое было загрязнено гидроксидом тетрабутиламмония, что определили с помощью 1H-ЯМР. Повторная очистка с той же растворяющей системой дала продукт (0,57 г), который все еще содержал небольшое количество гидроксида тетрабутиламмония. Заключительная очистка на 4-мм роторе хроматотрона с чистым этилацетатом дала после сушки 0,44 г (1,2 ммоль) чистого продукта, т.пл. 113 - 115oC (неисправленная); УФ λмакс ( ε ): pH 7,0: 278,9 нм (7800) и 247,6 нм (8300); 0,1 н. раствор HCl: 287,5 (7000) и 242,8 (5700); 0,1 н. раствор NaOH: 279,0 (7600) и 248,5 (7100); MC (E1): m/z 208 (C8H10N5O2); 194 (C7H8N5O2); 178 (C7H8N5O); 165 (C6H7N5O); 135 (C5H5N); 71 (C4H7NO).
1H-ЯМР (300 МГц, Me2SO-d6): δ 7,84 (s, 1H, H-8); 6,48 (br. s, 2H, NH2); 6,16 (d, 1H, H-1', J = 4,0 Гц); 5,77 (d, 1H, 2' или 3'-OH, J = 4,6 Гц); 5,68 (d, 1H, 2' или 3'-OH, J = 3,9 Гц); 4,35-4,23 (m, 2H, H-2' и 3'); 4,11 - 4,09 (m, 2H, H-5'); 3,96 (s, 3H, Pur-OC 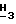 ); 3,95 - 3,92 (m, 1H, H-4'); 2,55 (септет, 1H -C(O)C
); 3,95 - 3,92 (m, 1H, H-4'); 2,55 (септет, 1H -C(O)C 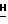 (CH3)2, J = 7,0 Гц); 0,08 (dd, 6H, -C(O)CH(C
(CH3)2, J = 7,0 Гц); 0,08 (dd, 6H, -C(O)CH(C 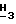 )2, J = 7,0 Гц, J = 1,8 Гц); ИК (KBr): 1734,3, 1616,0 и 1592,5 см-1.
)2, J = 7,0 Гц, J = 1,8 Гц); ИК (KBr): 1734,3, 1616,0 и 1592,5 см-1.
Аналитический расчет для C15H21N5O6• 0,20 C4H8O2:
рассчитано: C 49,29; H 5,92; N 18,19;
найдено: C 49,09; H 5,97; N 18,16.
i) 9-(2,3,5-три-О-Ацетил- β -D-арабинофуранозил)-2-амино-6- метокси-9Н-пурин.
2-Амино-9-( β -  -арабинофуранозил)-2-амино-6-метокси-9
-арабинофуранозил)-2-амино-6-метокси-9  -пурин (2,5 г, 8,4 ммоль) добавляли в 250-мл круглодонную колбу и высушивали путем совместного выпаривания с пиридином (2 х 25 мл). Остаток высушивали в вакуумном насосе в течение 2 ч. Добавляли 4-N,N-диметиламинопиридин (0,10 г, 0,8 ммоль), затем колбу продували аргоном и закрывали перегородкой. Добавляли сухой ацетонитрил (50 мл), затем триэтиламин (8,5 мл) и уксусный ангидрид (2,6 мл, 27,7 ммоль). Раствор размешивали при комнатной температуре в течение 3 ч. Тонкослойная хроматография на силикагеле с ацетон: CHCl3 (1:10) показала, что все исходное вещество (Rf = 0,05) прореагировало и что образовался один высокий пик Rf при Rf = 0,18.
-пурин (2,5 г, 8,4 ммоль) добавляли в 250-мл круглодонную колбу и высушивали путем совместного выпаривания с пиридином (2 х 25 мл). Остаток высушивали в вакуумном насосе в течение 2 ч. Добавляли 4-N,N-диметиламинопиридин (0,10 г, 0,8 ммоль), затем колбу продували аргоном и закрывали перегородкой. Добавляли сухой ацетонитрил (50 мл), затем триэтиламин (8,5 мл) и уксусный ангидрид (2,6 мл, 27,7 ммоль). Раствор размешивали при комнатной температуре в течение 3 ч. Тонкослойная хроматография на силикагеле с ацетон: CHCl3 (1:10) показала, что все исходное вещество (Rf = 0,05) прореагировало и что образовался один высокий пик Rf при Rf = 0,18.
Реакцию прерывали этанолом (2 мл), смесь концентрировали до сухости. Остаток разделяли между этилацетатом (300 мл) и 5% NaCO3 (50 мл). Органический слой промывали водой (2 х 50 мл), объединенные водные слои обратно экстрагировали с помощью этилацетата (100 мл). Окончательно объединенные органические экстракты высушивали (MgSO4), фильтровали и концентрировали. Неочищенный продукт очищали на силикагельной колонке (5 х 18 см) элюируемой смесью ацетон : CHCl3 с отношением от 1:1. После сушки получили продукт в виде белого твердого вещества весом 3,38 г (7,98 ммоль). Это вещество перекристаллизовывали из этилацетата и гептана с получением после высушки 2,97 г (7,0 ммоль); т.пл. 178-180oC (неисправленная), УФ λмакс ( ε ): pH 7,00: 279,5 нм (8500), 247,6 нм (9100); 0,1 н. раствор HCl: 287,2 (7600), 244,4 (6700); 0,1 н. раствор NaOH: 278,9 (8900), 249,0 (8300); MC (E1): m/z 424 (M+H, C17H22N5O8); 380 (C15H18N5O7); 364 (C15H18N5O5); 259 (C11H15O7); 194 (C7H8N5O2); 166 (C6H8N5O); 165 (C6H7N5O); 149 (C5H3N5O); 139 (C7H7O3); 42 (C2H2O).
1H-ЯМР (Me2SO-d6, 200 МГц): δ 7,92 (s, 1H, H-8); 6,54 (br, s, 2H, NH2); 6,35 (d, 1H, H-1', J = 4,6 Гц); 5,46-5,40 (m, 2H, H-2' и 3'); 4,41-4,20 (m, 3H, H-4' и 5'); 3,94 (s, 3H, Pur-OC  ); 2,10 (s, 3H, C(O)CH3); 2,03 (s, 3H, C(O)CH3); 1,80 (s, 3H, C(O)CH3).
); 2,10 (s, 3H, C(O)CH3); 2,03 (s, 3H, C(O)CH3); 1,80 (s, 3H, C(O)CH3).
Аналитический расчет для C17H21N5O8:
рассчитано: C 48,23; H 5,00; N 16,54;
найдено: C 48,32; H 4,93; N 16,24.
j) 9-(3,5-ди-О-Ацетил- β -D-арабинофуранозил)-2-амино-6-метокси-9Н-пурин.
2-Амино-9-(2,3,5-три-О-ацетил)- β -  -арабинофуранозил)- 6-метокси-9
-арабинофуранозил)- 6-метокси-9  -пурин (2,0 г, 4,72 ммоль) добавляли в 250-мл круглодонную колбу вместе с ацетатом натрия (1,2 г, 14,2 ммоль) и хлоридом оксиамина (0,98 г, 14,2 ммоль). Колбу продували аргоном, снабжали размешивателем и перегородкой. Добавляли сухой пиридин (25 мл), раствор размешивали при комнатной температуре в течение 7 ч. Тонкослойная хроматография на силикагеле с ацетон:CHCl3 (1 : 10) показала, что исходное вещество (Rf = 0,68) прореагировало и образовался один небольшой пик с Rf = 0,47. Реакционную смесь обрабатывали ацетоном (20 мл) и концентрировали до сухости, снова размешивали с ацетоном (100 мл) и концентрировали. Остаток смешивали с ацетоном (100 мл) и фильтровали. Осадок промывали ацетоном, фильтрата концентрировали до желтого масла. Неочищенный продукт очищали на силикагельной колонке (5 х 18 см), элюируемой смесью ацетон : CH2Cl2 (1 : 1). Продукт после сушки получался в виде белого вещества в твердой фазе весом 1,46 г (3,83 ммоль). 1Н-ЯМР-спектроскопия показала, что вещество представляло собой смесь 3,5-диацетата и 2,5-диацетата. Эти два продукта разделяли путем перекристаллизации из дихлорметана и гептана, причем сперва выкристаллизовывали 2,5-диацетат. 3,5-диацетат получали в виде белых кристаллов весом 0,796 г (2,08 ммоль) после двух перекристаллизаций: т.пл. 109 - 110oC (неисправленная); УФ λмакс ( ε ): pH 7,00: 278,9 нм (8 400), 247,9 нм (9 000); 0,1 н. раствор HCl: 286,9 (8 300), 244,5 (7 300); 0,1 н. раствор NaOH: 279,0 (8 500), 247,6 (8 000); MC (E1): m/z 381 (M+H, C15H19N5O7); 383 (C13H16N5O6); 194 (C7H8N5O2); 166 (C6H8N5O);
-пурин (2,0 г, 4,72 ммоль) добавляли в 250-мл круглодонную колбу вместе с ацетатом натрия (1,2 г, 14,2 ммоль) и хлоридом оксиамина (0,98 г, 14,2 ммоль). Колбу продували аргоном, снабжали размешивателем и перегородкой. Добавляли сухой пиридин (25 мл), раствор размешивали при комнатной температуре в течение 7 ч. Тонкослойная хроматография на силикагеле с ацетон:CHCl3 (1 : 10) показала, что исходное вещество (Rf = 0,68) прореагировало и образовался один небольшой пик с Rf = 0,47. Реакционную смесь обрабатывали ацетоном (20 мл) и концентрировали до сухости, снова размешивали с ацетоном (100 мл) и концентрировали. Остаток смешивали с ацетоном (100 мл) и фильтровали. Осадок промывали ацетоном, фильтрата концентрировали до желтого масла. Неочищенный продукт очищали на силикагельной колонке (5 х 18 см), элюируемой смесью ацетон : CH2Cl2 (1 : 1). Продукт после сушки получался в виде белого вещества в твердой фазе весом 1,46 г (3,83 ммоль). 1Н-ЯМР-спектроскопия показала, что вещество представляло собой смесь 3,5-диацетата и 2,5-диацетата. Эти два продукта разделяли путем перекристаллизации из дихлорметана и гептана, причем сперва выкристаллизовывали 2,5-диацетат. 3,5-диацетат получали в виде белых кристаллов весом 0,796 г (2,08 ммоль) после двух перекристаллизаций: т.пл. 109 - 110oC (неисправленная); УФ λмакс ( ε ): pH 7,00: 278,9 нм (8 400), 247,9 нм (9 000); 0,1 н. раствор HCl: 286,9 (8 300), 244,5 (7 300); 0,1 н. раствор NaOH: 279,0 (8 500), 247,6 (8 000); MC (E1): m/z 381 (M+H, C15H19N5O7); 383 (C13H16N5O6); 194 (C7H8N5O2); 166 (C6H8N5O);
165 (C6H7N5O); 135 (C5H5N5); 43 (C2H3O).
1Н-ЯМР (Me2SO-d6, 200 МГц): δ 7,89 (s, 1H, H-8); 6,50 (br. s, 2H, NH2); 6,18 (d, 1H, H-1', J = 4,9 Гц); 6,14 (d, 1H, 2'-OH, J = 4,1 Гц); 5,10 (t, 1H, H-3', J = 2,7 Гц); 4,42 - 4,23 (m, 3H, H-2' и 5'); 4,16 - 4,08 (m, 1H, H-4'); 3,95 (s, 3H, Pur-OC  ); 2,10 (s, 3H, C(O)CH3); 2,01 (s, 3H, C(O)CH3).
); 2,10 (s, 3H, C(O)CH3); 2,01 (s, 3H, C(O)CH3).
Аналитический расчет для C15H19N5O7•0,10 CH2Cl2:
рассчитано: C 46,52; H 4,96; N 17,96;
найдено: C 46,31; H 5,13; N 17,70.
k) 9-(2,5-ди-О-Ацетил- β -D-арабинофуранозил)-2-амино-6-метокси- 9Н-пурин.
2-Амино-9-(2,3,5-три-О-ацетил)- β - 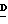 -арабинофуранозил)-6-метокси-9
-арабинофуранозил)-6-метокси-9  -пурин (2,0 г, 4,72 ммоль) добавляли в 250-мл круглодонную колбу вместе с ацетатом натрия (1,2 г, 14,2 ммоль) и хлоридом оксиамина (0,98 г, 14,2 ммоль). Колбу продували аргоном, снабжали размешивателем и перегородкой. Добавляли сухой пиридин (25 мл), раствор размешивали при комнатной температуре в течение 7 ч. Тонкослойная хроматография на силикагеле в ацетон : CHCl3 (1 : 1) показала, что исходное вещество (Rf = 0,68) прореагировало и что образовался один небольшой пик с Rf = 0,47. Реакционную смесь обрабатывали ацетоном (20 мл), концентрировали до сухости, снова смешивали с ацетоном (100 мл) и концентрировали. Остаток смешивали с ацетоном (100 мл) и фильтровали. Осадок промывали ацетоном, фильтрат концентрировали с получением желтого масла. Неочищенный продукт очищали на силикагельной колонке (5 х 18 см), элюируемой смесью ацетон : CH2Cl2 (1 : 1). Продукт получали после сушки в виде белого вещества в твердой фазе весом 1,46 г (3,83 ммоль). 1H-ЯМР-спектроскопия показала, что вещество представляло собой смесь 3,5-диацетата и 2,5-диацетата. Эти два продукта разделяли с помощью перекристаллизации из дихлорметана и гептана с выделением сперва 2,5-диацетата. 2,5-Диацетат получали в виде белых кристаллов весом 0,256 г (0,67 ммоль) после двух перекристаллизаций: т. пл. 210 - 212oC (неисправленная), УФ λмакс ( ε ) : pH 7,0: 279,2 нм (8 200), 248,3 нм (8 700); 0,1 н. раствор HCl : 287,1 (7 800), 244,3 (6 200); 0,1 н. раствор NaOH: 278,7 (8 600), 248,7 (7 900); MC (M1): m/z 381 ( M+H, C15H19N5O7); 338 (C13H16N5O6); 250 (C10H12N5O3); 217 (C9H13O5); 194 (C7H8N5O2); 166 (C6H8N5O); 165 (C6H7N5O); 157 (C7H9O4); 139 (C7H7O7); 135 (C5H5N5); 43 (C2H3O).
-пурин (2,0 г, 4,72 ммоль) добавляли в 250-мл круглодонную колбу вместе с ацетатом натрия (1,2 г, 14,2 ммоль) и хлоридом оксиамина (0,98 г, 14,2 ммоль). Колбу продували аргоном, снабжали размешивателем и перегородкой. Добавляли сухой пиридин (25 мл), раствор размешивали при комнатной температуре в течение 7 ч. Тонкослойная хроматография на силикагеле в ацетон : CHCl3 (1 : 1) показала, что исходное вещество (Rf = 0,68) прореагировало и что образовался один небольшой пик с Rf = 0,47. Реакционную смесь обрабатывали ацетоном (20 мл), концентрировали до сухости, снова смешивали с ацетоном (100 мл) и концентрировали. Остаток смешивали с ацетоном (100 мл) и фильтровали. Осадок промывали ацетоном, фильтрат концентрировали с получением желтого масла. Неочищенный продукт очищали на силикагельной колонке (5 х 18 см), элюируемой смесью ацетон : CH2Cl2 (1 : 1). Продукт получали после сушки в виде белого вещества в твердой фазе весом 1,46 г (3,83 ммоль). 1H-ЯМР-спектроскопия показала, что вещество представляло собой смесь 3,5-диацетата и 2,5-диацетата. Эти два продукта разделяли с помощью перекристаллизации из дихлорметана и гептана с выделением сперва 2,5-диацетата. 2,5-Диацетат получали в виде белых кристаллов весом 0,256 г (0,67 ммоль) после двух перекристаллизаций: т. пл. 210 - 212oC (неисправленная), УФ λмакс ( ε ) : pH 7,0: 279,2 нм (8 200), 248,3 нм (8 700); 0,1 н. раствор HCl : 287,1 (7 800), 244,3 (6 200); 0,1 н. раствор NaOH: 278,7 (8 600), 248,7 (7 900); MC (M1): m/z 381 ( M+H, C15H19N5O7); 338 (C13H16N5O6); 250 (C10H12N5O3); 217 (C9H13O5); 194 (C7H8N5O2); 166 (C6H8N5O); 165 (C6H7N5O); 157 (C7H9O4); 139 (C7H7O7); 135 (C5H5N5); 43 (C2H3O).
1H-ЯМР (Me2SO-d6, 200 МГц): δ 7,89 (s, 1H, H-8); 6,51 (br. s, 2H, NH2); 6,32 (d, 1H, H-1', J = 5,7 Гц); 5,98 (d, 1H, 3'-OH, J = 3,9 Гц); 5,18 (t, 1H, H-2', J = 2,7 Гц); 4,44 - 4,22 (m, 3H, H-3' и 5'); 4,07 - 3,97 (m, 1H, H-4'); 3,94 (s, 3H, Pur-OC  ); 2,02 (s, 3H, C(O)CH3); 1,78 (s, 3H, C(O)CH3).
); 2,02 (s, 3H, C(O)CH3); 1,78 (s, 3H, C(O)CH3).
Аналитический расчет для C15H19N5O7•0,40 CH2Cl2• 0,05 C7H16:
рассчитано: C 45,01; H 4,94; N 16,66;
найдено: C 44,92; H 4,92; N 16,51.
1(i) 9-(2-О-Ацетил-3,5-ди-О-трет-бутилдиметилсилил- β -D-арабинофуранозил)-2-амино-6-метокси-9Н-пурин.
2-Амино-9-(3,5-ди-О-трет-бутилдиметилсили- β -D-арабинофуранозил)- 6-метокси-9Н-пурин (0,5 г, 0,95 ммоль) отвешивали в обожженную высушенную 100-мл круглодонную колбу. Добавляли 4-N,N-диметиламинопиридин (0,01 г, 0,09 ммоль), колбу продували аргоном и закрывали перегородкой. Добавляли сухой ацетонитрил (25 мл) и триэтиламин (5,0 мл), раствор охлаждали в ванночке со льдом. С помощью шприца и иглы к раствору добавляли уксусный ангидрид (0,11 мл, 1,1 ммоль). Реакционную смесь исследовали с помощью тонкослойной хроматографии на силикагеле с ацетон : CHCl3 (1 : 10). После выдержки в течение 18 ч при комнатной температуре все исходное вещество (Rf = 0,20) было израсходовано, образовался один большой пик (Rf = 0,51). Реакционную смесь концентрировали, остаток размешивали в этилацетате (75 мл) и экстрагировали NaHCO (25 мл) и затем водой (2 х 25 мл). Органический слой высушивали с помощью MgSO4 (безводный), фильтровали и концентрировали с получением светлого желтого масла. Остаток очищали на силикагельной колонке (12,5 х 14 см, с зернистостью 230 - 400) элюируемой смесью ацетон : CHCl3 (1 : 4). Чистый продукт выделяли в виде белого вещества в твердой фазе (0,58 г, 1,0 ммоль); т. пл. 74 - 76oC (неисправленная); MC (E1): m/z 568 (C25H46N5O6Si2); 552 (C24H42N5O6Si2); 510 (C21H36N5O6Si2); 378 (C15H20N5O5Si): 261 (C11H25O3Si2); 250 (C10H12N5O3); 194 (C7H8N5O2); 166 (C6H8N5O).
1H-ЯМР (CDCl3, 200 МГц): δ 7,95 (s, 1H, H-8); 6,38 (d, 1H, H-1', J = 5,6 Гц); 5,33 (t, 1H, H-2', J = 5,7 Гц); 4,85 (br. s, 2H, NH2); 4,58 (t, 1H, H-3', J = 5,8 Гц); 4,05 (s, 3H, -OCH3); 3,94 - 3,80 (m, 3H, H-4' и H-5'); 1,78 (s, 1H, C(O)CH3); 0,93 (s, 9H, -SiC(CH3)3); 0,89 (s, 9H, -SiC(CH3)3); 0,11 - 0,08 (m, 12H, -Si(CH3)).
Аналитический расчет для C25H45N5O6Si2:
рассчитано: C 52,88; H 7,99; N 12,33;
найдено: C 52,67; H 7,90; N 12,23.
l(ii) 9-(2-О-Ацетил- β -D-арабинофуранозил)-2-амино-6-метокси-9Н- пурин.
Силилированное промежуточное соединение 2-амино-9-[(2-О-ацетил- 3,5-ди-О-трет-бутилдиметилсилил)- β -D-арабинофуранозил]-6- метокси-9  -пурина (0,46 г, 0,8 ммоль) отвешивали в RBF, снабженный размешивающим устройством и резиновой перегородкой. Добавляли фторид тетраэтиламмония (0,48 г, 3,24 ммоль), затем раствор ТГФ (40 мл) и уксусную кислоту (0,18 мл, 3,2 ммоль). Раствор охлаждали в ванночке со льдом до 0 - 5oC. После выдержки в течение 48 ч при 0 - 5oC тонкослойная хроматография в ацетон : CHCl3 (1 : 10) показала, что исходное вещество было израсходовано (Rf = 0,37), образовался один главный низкий пик Rf = 0,07. Реакционную смесь направляли непосредственно на силикагельную колонку (с зернистостью 230 - 400, размером 5 х 12 см) и элюировали смесью ацетон : CHCl3 (1 : 10, 300 мл) и затем ацетон : CHCl3 (5 : 4, 900 мл). Чистый продукт из колонки получали после просушки в виде белого вещества в твердой фазе (0,21 г, 0,63 ммоль) и, как было показано, оно представляло собой требуемую 2'-О-ацетил производную; т. пл. 66 - 68oC (неисправленная); УФ λмакс ( ε ): pH 7,00: 279,0 нм (8100), 247,9 нм (8400); 0,1 н. раствор HCl: 286,4 (8500), 243,4 (7000); 0,1 н. раствор NaOH: 278,8 (8700), 246,7 (8600); MC (E1): m/z 339 (M, C13N17N5O6); 296 (C11H14N5O5); 250 (C10H12N5O3); 194 (C7H8N5O2); 175 (C7H11O5); 166 (C6H7N5O). 1H-ЯМР (Me2SO-d6, 200 МГц): δ 7,94 (s, 1H, H-2) 6,49 (br. s, 2H, NH2); 6,27 (d, 1H, H-1', J = 5,5 Гц); 5,82 (d, 1H, 3'-OH, J = 5,1 Гц); 5,18 (t, 1H, H-2', J = 5,3 Гц); 5,02 (t, 1H, 5'-OH, J = 5,6 Гц); 4,33 (ddd, 1H, H-3', J3',3'-OH = 5,1 Гц, J2',3' = 5,3 Гц, J3',4' = 5,8 Гц); 3,94 (s, 3H, Pur-OC
-пурина (0,46 г, 0,8 ммоль) отвешивали в RBF, снабженный размешивающим устройством и резиновой перегородкой. Добавляли фторид тетраэтиламмония (0,48 г, 3,24 ммоль), затем раствор ТГФ (40 мл) и уксусную кислоту (0,18 мл, 3,2 ммоль). Раствор охлаждали в ванночке со льдом до 0 - 5oC. После выдержки в течение 48 ч при 0 - 5oC тонкослойная хроматография в ацетон : CHCl3 (1 : 10) показала, что исходное вещество было израсходовано (Rf = 0,37), образовался один главный низкий пик Rf = 0,07. Реакционную смесь направляли непосредственно на силикагельную колонку (с зернистостью 230 - 400, размером 5 х 12 см) и элюировали смесью ацетон : CHCl3 (1 : 10, 300 мл) и затем ацетон : CHCl3 (5 : 4, 900 мл). Чистый продукт из колонки получали после просушки в виде белого вещества в твердой фазе (0,21 г, 0,63 ммоль) и, как было показано, оно представляло собой требуемую 2'-О-ацетил производную; т. пл. 66 - 68oC (неисправленная); УФ λмакс ( ε ): pH 7,00: 279,0 нм (8100), 247,9 нм (8400); 0,1 н. раствор HCl: 286,4 (8500), 243,4 (7000); 0,1 н. раствор NaOH: 278,8 (8700), 246,7 (8600); MC (E1): m/z 339 (M, C13N17N5O6); 296 (C11H14N5O5); 250 (C10H12N5O3); 194 (C7H8N5O2); 175 (C7H11O5); 166 (C6H7N5O). 1H-ЯМР (Me2SO-d6, 200 МГц): δ 7,94 (s, 1H, H-2) 6,49 (br. s, 2H, NH2); 6,27 (d, 1H, H-1', J = 5,5 Гц); 5,82 (d, 1H, 3'-OH, J = 5,1 Гц); 5,18 (t, 1H, H-2', J = 5,3 Гц); 5,02 (t, 1H, 5'-OH, J = 5,6 Гц); 4,33 (ddd, 1H, H-3', J3',3'-OH = 5,1 Гц, J2',3' = 5,3 Гц, J3',4' = 5,8 Гц); 3,94 (s, 3H, Pur-OC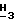 ); 3,85 - 3,79 (m, 1H, H-4'); 3,68 - 3,60 (m, 2H, H-5'); 1,76 (s, 3H, C(O)CH3).
); 3,85 - 3,79 (m, 1H, H-4'); 3,68 - 3,60 (m, 2H, H-5'); 1,76 (s, 3H, C(O)CH3).
Аналитический расчет для C13H17N5O6•0,40 CHCl3• 0,25 C3H6O:
вычислено: C 44,36; H 4,99; N 18,54;
найдено: C 44,38; H 5,27; N 18,28.
m (i) 2-Амино-9-(2,3,5-три-О-ацетил- β -D-арабинофуранозил)-6-метокси-9Н-пурин.
2-Амино-4-( β -D-арабинофуранозил)-6-метокси-9Н-пурин (1,5 г, 5,05 ммоль), триэтиламин (3,06 г, 30,3 ммоль) и уксусный ангидрид (2,06 г, 20,2 ммоль) размешивали в течение 3 ч при комнатной температуре в 15 мл сухого ДМФ в атмосфере азота. Затем раствор разбавляли этилацетатом (100 мл), органическую фазу промывали 2 х 100 мл порциями 10% NaHCO3. Растворитель удаляли путем выпаривания вращением, получая 2,05 г (96%) масла, которое выкристаллизовывалось после выстаивания в течение всей ночи: тонкослойная хроматография (силикагель) хлороформ : метанол (90 : 10) Rf = 0,63.
1H-ЯМР (DMSO): δ 1,86 (s, 3H, OAc); 2,06 (s, 3H, OAc); 2,12 (s, 3H, OAc); 3,97 (s, 3H, OCH3); 4,2 - 4,5 (m, 3H, C-4' и C-5'); 5,4 - 5,5 (m, 2H, C-2' и C-3'H); 6,38 (d, J = 4,8 Гц), 1H, C-1'H); 6,55 (широкий s, 2H, NH2) и 7,95 (s, 1H, C-8H).
m(ii) 2-Амино-9-(5-О-ацетил- β -D-арабинофуранозил)-6-метокси- 9Н-пурин.
Смесь 2-амино-9-(2,3,5-три-О-ацетил- β -D-арабинофуранозил)-6-метокси-9  -пурин (10,0 г, 23,6 ммоль), безводный ацетат натрия (19,4 г, 236 ммоль) и хлорид оксиамина (16,4 г, 236 ммоль) размешивали в течение 17 ч в 200 мл пиридина при температуре окружающей среды. Затем раствор разбавляли 50 мл ацетона, большую часть растворителя удаляли с помощью выпаривания вращением. Остающийся осадок в виде масла очищали с помощью флэш-хроматографии на колонке (размером 10х75 см, с зернистостью 230 - 400), используя CHCl3 : ацетон (1 : 1) в качестве элюента. Соответствующие фракции концентрировали с помощью вращательного выпаривания, оставляя смолянистое полутвердое вещество. Полутвердое вещество лиофилизировали из 100 мл воды, получая белый порошок; т. пл. 138 - 140oC (неисправленная); тонкослойная хроматография (силикагель) хлороформ : метанол (90 : 10) Rf = 0,27.
-пурин (10,0 г, 23,6 ммоль), безводный ацетат натрия (19,4 г, 236 ммоль) и хлорид оксиамина (16,4 г, 236 ммоль) размешивали в течение 17 ч в 200 мл пиридина при температуре окружающей среды. Затем раствор разбавляли 50 мл ацетона, большую часть растворителя удаляли с помощью выпаривания вращением. Остающийся осадок в виде масла очищали с помощью флэш-хроматографии на колонке (размером 10х75 см, с зернистостью 230 - 400), используя CHCl3 : ацетон (1 : 1) в качестве элюента. Соответствующие фракции концентрировали с помощью вращательного выпаривания, оставляя смолянистое полутвердое вещество. Полутвердое вещество лиофилизировали из 100 мл воды, получая белый порошок; т. пл. 138 - 140oC (неисправленная); тонкослойная хроматография (силикагель) хлороформ : метанол (90 : 10) Rf = 0,27.
1H-ЯМР (DMSO): δ 2,03 (s, 3H, OAc); 3,97 (s, 3H, OCH3); 4,1 - 4,4 (m, 5H, C-2', C-3', C-4' и C-5'); 5,69 (d, J = 3,4 Гц, 1H, OH); 5,78 (d, J = 4,2 Гц, 1H, OH); 6,17 (d, J = 3,6 Гц, 1H, C-1'H); 6,48 (широкий s, 2H, NH2) и 7,86 (s, 1H, C-8H).
Аналитический расчет для C13H17N5O6•H2O:
рассчитано: C 43,70; H 5,36; N 19,60;
найдено: C 43,70; H 5,34; N 19,52.
Лекарственные формы в виде таблеток. Формы А и В получали путем влажного гранулирования ингредиентов с раствором повидона с последующим добавлением стеарата магния и сжатия, мг/таблетка:
Форма А
(a) Активный ингредиент - 250
(b) Лактоза В.Р. - 210
(c) Повидон В.Р. - 15
(d) Гликоллят натрий-крахмала - 20
(e) Стеарат магния - 5 - 500
(a) Активный ингредиент - 250
(b) Лактоза В.Р. - 26
(c) Повидон В.Р. - 9
(d) Гликоллят натрий-крахмала - 12
(e) Стеарат магния - 3 - 300
Форма В
(a) Активный ингредиент - 250
(b) Лактоза В.Р. - 150
(c) Авицел РН 101 - 60
(d) Повидон В.Р. - 15
(e) Гликоллят натрий-крахмала - 20
(f) Стеарат магния - 5 - 500
(a) Активный ингредиент - 250
(b) Лактоза В.Р. - -
(c) Авицел РН 101 - 26
(d) Повидон В.Р. - 9
(e) Гликоллят натрий-крахмала - 12 - 300
Форма С
(a) Активный ингредиент - 100
(b) Лактоза В.Р. - 200
(c) Повидон В.Р. - 50
(d) Гликоллят натрий-крахмала - 5
(e) Стеарат магния - 4 - 359
Таблетки получают из вышеуказанных ингредиентов С путем влажного гранулирования с последующим сжатием. В альтернативном получении повидон В.Р. можно заменить поливинилпирролидоном.
Следующие формы D и Е получают непосредственным сжатием перемешанных ингредиентов. Лактоза в форме Е - типа прямого сжатия (Dairy Crest - "Zeparox"), мг/капсула:
Форма D
Активный ингредиент - 250
Прежелетенированный крахмал NF 25 - 250 - 400
Форма Е
Активный ингредиент - 250
Лактоза - 150
Авицел - 100 - 500
Форма F (лекарственная форма со свойством контролируемого высвобождения средства). Форму приготавливают путем влажного гранулирования градиентов (рассмотрены выше) с раствором повидона с последующим добавлением стеарата магния и сжатия, мг/таблетка:
(a) Активный ингредиент - 500
(b) Гидроксипропилметилцеллюлоза (Methocel K4M Premium) - 112
(c) Лактоза В.Р. - 53
(d) Повидон В.Р. - 28
(e) Стеарат магния - 7 - 700
Высвобождение лекарства происходит в течение времени около 6 - 8 ч и полностью завершается через 12 ч.
Лекарственные формы в виде капсул, мг/капсула
Форма А
Лекарственные формы в виде капсул приготавливают путем перемешивания ингредиентов формы D, описанной выше, и наполнением ими двух частей твердой желатиновой капсулы. Форму В (infra) получают аналогичным способом.
Форма В
(a) Активный ингредиент - 250
(b) Лактоза В.Р. - 143
(c) Гликоллят натрий-крахмала - 25
(d) Стеарат магния - 2 - 420
Форма С
(a) Активный ингредиент - 250
(b) Макрогол 4000 В.Р. - 350 - 600
Капсулы получают путем расплавления макрогола 4000 В.Р., диспергирования активного ингредиента в расплав и наполнением расплавом двух частей твердой желатиновой капсулы.
Форма D
Активный ингредиент - 250
Лецитин - 100
Арахисовое масло - 100 - 450
Капсулы получают путем диспергирования активного ингредиента в лецитин и арахисовое масло и вливания дисперсии в мягкие, эластичные желатиновые капсулы.
Форма E (капсула со свойством контролируемого высвобождения лекарственного средства). Следующие капсулы с контролируемым высвобождением средства получают путем выдавливания ингредиентов (a), (b) и (c), используя экструдер, с последующей сферонизацией выдавленных ингредиентов и сушкой. Высушенные шарики затем покрывают мембраной (d) со свойством контролируемого высвобождения средства и помещают их в две части твердой желатиновой капсулы, мг/капсула:
(a) Активный ингредиент - 250
(b) Микрокристаллическая целлюлоза - 125
(c) Лактоза В.Р. - 125
(d) Этилцеллюлоза - 13 - 513
Глазной раствор, г
Активный ингредиент - 0,5
Хлорид натрия, с аналитической чистотой - 0,9
Тиомерсал - 0,001
Очищенная вода, мл - До 100
Выбираемая величина pH - До 7,5
Лекарственная форма для инъекций, г
Активный ингредиент - 0,200
Стерильный неогнеопасный фосфатный буферный раствор (pH 7,2), мл - До 10
Активный ингредиент растворяют в большей части фосфатного буферного раствора (35 - 40oC), затем собирают в объеме и отфильтровывают через стерильный микропористый фильтр в стерильную 10-мл ампулу из янтарного стекла (тип I) и запечатывают стерильным закрытием и клеймом.
Внутримышечные инъекции, г
Активный ингредиент - 0,20
Бензиловый спирт - 0,10
Гликофурол 75 - 1,45
Вода для инъекций, q.s., мл - До 3,00
Активный ингредиент растворяют в гликофуроле. Затем добавляют бензиловый спирт, растворяют и добавляют воды до 3 мл. Смесь затем фильтруют через стерильный микропористый фильтр и запечатывают в стерильных 3-мл ампулах из янтарного стекла (тип I).
Сиропы, г
Активный ингредиент - 0,25
Раствор сорбитола - 1,50
Глицерол - 2,00
Дисперсная целлюлоза - 0,075
Бензоат натрия - 0,005
Корригент, персиковый 17.42.3169, мл - 0,0125
Очищенная вода, q.s., мл - До 5,00
Бензоат натрия растворяют в порции очищенной воды и добавляют раствор сорбитола. Добавляют активный ингредиент и диспергируют. В глицероле диспергируют уплотнитель (дисперсную целлюлозу). Две дисперсии перемешивают и доводят до требуемого объема с помощью очищенной воды. Дальнейшего уплотнения достигают при необходимости путем дополнительного диспергирования суспензии.
Суппозиторий, мг/суппозорий:
Активный ингредиент (63 мкм)* - 250
Твердый жир, BP (Witeposol H15-Dynamit Nobel) - 1700
- 1950
Одну пятую часть Witeposol H15 расплавляют в кристаллизаторе с паровой рубашкой при максимальной температуре 45oC. Активный ингредиент просеивают через 200-мкм сито и добавляют к расплавленному основанию с размешиванием, используя просеивание со срезанием пока не получится гладкая дисперсия. Поддерживая температуру смеси 45oC, добавляют к суспензии оставшийся Witeposol H25 и размешивают до получения гомогенной смеси. Всю суспензию пропускают через 250-мкм экран из нержавеющей стали, при постоянном размешивании дают охладиться до 45oC. При температуре от 38 до 40oC 2,02 г смеси помещают в подходящий пластичный расплав. Суппозитории оставляют охлаждаться до комнатной температуры.
Пессарий, мг/пессарий:
Активный ингредиент (63 мкм)* - 250
Ангидрат-декстроза - 380
Картофельный крахмал - 363
Стеарат магния - 7 - 1000
* Активный ингредиент используют в виде порошка, 90% частиц которого имеют диаметр 63 мкм или менее.
Указанные выше ингредиенты непосредственно смешивают, и пессарии приготавливают путем прямого сжатия получающейся смеси.
Селективное ингибирование роста Т-клеток. Соединения по изобретению проверяли на ингибирование роста Т-клеток (Molt 4) по сравнению с В клетками (IM9) по методу (Averett, D., Jounal of Virological Methods, 23, (1989), 263 - 276).
2-Амино-6-метокси-9-( β -D-арабинофуранозил)-9Н-пурин (соединение I) селективно ингибирует рост Т-клеток у человека (Molt 4) в противоположность B-клеткам человека (IM9). Эти данные сравнивают с селективным ингибированием ara G (табл.1).
Биологическая ценность ara G. Ara G и соединения по изобретению вводили перорально в специальных обезьян (Cynomolgus monkey), которые перед этим были изолированы на 8 ч. Каждое соединение давали двум обезьянам. Величина дозы была 94 нмоль/кг, что эквивалентно 27 мг/кг ara G. Пробы крови брали в течение следующих 24 ч, мочу собирали в течение тех же 24 ч. Концентрации ara G в экстрактах плазмы и мочи определяли с помощью жидкостной хроматографии высокого давления с обращенной фазой.
Растворимость и биологическая ценность ara G приведена в табл.2.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЭНАНТИОМЕРНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1992 |
|
RU2091386C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АКТИВНОСТЬ ПРОТИВ ВИРУСНОЙ ИНФЕКЦИИ И β D-АРАБИНОФУРАНОЗИЛ-2-АМИНО-6-МЕТОКСИ-9Н-ПУРИНЫ | 1993 |
|
RU2112765C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНЫХ СОЕДИНЕНИЙ ИЛИ ИХ ПРОИЗВОДНЫХ | 1990 |
|
RU2068849C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ ПУРИНОВЫХ АРАБИНОЗИДОВ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ ПРОИЗВОДНЫХ | 1988 |
|
RU2039752C1 |
| ЗАМЕЩЕННЫЕ БЕНЗИМИДАЗОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ОБЛАДАЮЩАЯ АНТИВИРУСНЫМ ДЕЙСТВИЕМ | 1993 |
|
RU2141952C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 2'-ДЕЗОКСИ-2'-ФТОРРИБОНУКЛЕОЗИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1990 |
|
RU2043361C1 |
| Способ получения производных гуанина или их кислотно-аддитивных фармацевтически приемлемых солей | 1988 |
|
SU1634138A3 |
| ПРОИЗВОДНЫЕ 5,6-ДИХЛОРБЕНЗИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ И СПОСОБ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИРУСНЫХ ИНФЕКЦИЙ | 1995 |
|
RU2145963C1 |
| ПРОИЗВОДНЫЕ БИЦИКЛИЧЕСКИХ АМИДОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ПАТОЛОГИЧЕСКИХ И ВОСПАЛИТЕЛЬНЫХ СОСТОЯНИЙ | 1994 |
|
RU2143422C1 |
| Способ получения производных пурина | 1984 |
|
SU1342421A3 |
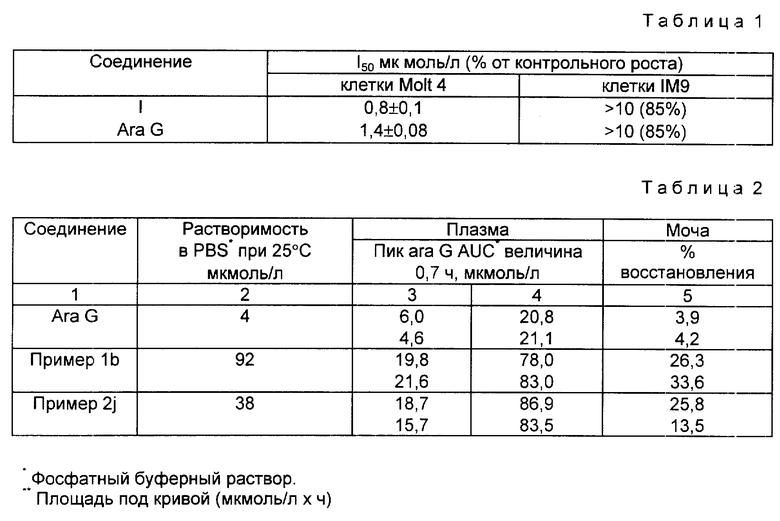
Изобретение относится к моно-, ди- или три-сложным эфирам 2-амино-6-(C1-C5-алкокси)-9-( β-D -арабинофуранозил)-9Н-пурина общей формулы (I)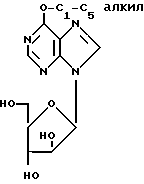
где арабинофуранозильный остаток замещен по 2'-, 3'- или 5'-положениям, а сложные эфиры образованы карбоновыми кислотами, в которых некарбонильная часть выбрана из н-пропила, трет-бутила, н-бутила, метоксиметила, бензила, феноксиметила, фенила, метансульфонила и сукцинила. Соединения формулы (I) получают этерификацией 2-амино-6-метокси-9-β-D-арабинофуранзил-9Н-пурина в присутствии фермента, при этом имеющаяся гидроксильная группа при необходимости защищена. Описаны также фармацевтические формы, содержащие соединения формулы (I) в эффективном количестве и обладающие противоопухолевым действием. 4 с. и 9 з.п.ф-лы, 2 табл.
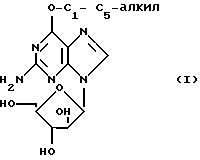
где арабинофуранозильный остаток замещен по 2'-, 3'- или 5'-положениям, а сложные эфиры образованы карбоновыми кислотами, в которых некарбонильная часть выбрана из н-пропила, трет-бутила, н-бутила, метоксиметила, бензила, феноксиметила, фенила, метансульфонила и сукцинила.
| EP, 0204251 A2, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, 0294114 A1, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Cancer Research | |||
| Железобетонный фасонный камень для кладки стен | 1920 |
|
SU45A1 |

