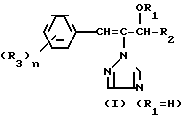
Предметом изобретения является геометрический изомер Е формы (соединение, определяемое как I А изомер в приводимом ниже описании) триазольного соединения, представленного формулой (I)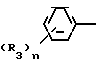 C
C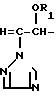 R2 (I) в которой R1 представляет атом водорода, С1-С4 алкильную, С3-С4алкенильную или 2-пропильную группу, R2 представляет С1-С6 алкильную, циклопропильную или 1-метилциклопропильную группу, элемент R3, который может иметь одинаковые или различные значения, представляет атом галогена, С1-С4 алкильную группу, галоидзамещенную С1-С3 алкильную группу, С1-С4 алкокси, фенокси, фенильную циано или нитрогруппу, и является целым числом от 0 до 3, и термин: "галоген" означает атомы хлора, брома и фтора, его соли, способ его получения и фунгицид, гербицид и/или регулятор роста растений для применения в сельском хозяйстве, и садоводстве, содержащий указанное соединение в качестве активного ингредиента.
R2 (I) в которой R1 представляет атом водорода, С1-С4 алкильную, С3-С4алкенильную или 2-пропильную группу, R2 представляет С1-С6 алкильную, циклопропильную или 1-метилциклопропильную группу, элемент R3, который может иметь одинаковые или различные значения, представляет атом галогена, С1-С4 алкильную группу, галоидзамещенную С1-С3 алкильную группу, С1-С4 алкокси, фенокси, фенильную циано или нитрогруппу, и является целым числом от 0 до 3, и термин: "галоген" означает атомы хлора, брома и фтора, его соли, способ его получения и фунгицид, гербицид и/или регулятор роста растений для применения в сельском хозяйстве, и садоводстве, содержащий указанное соединение в качестве активного ингредиента.
Каждое триазольное соединение формулы (1) имеет две геометрические изомерные формы, z-форму и Е-форму, представленные формулами
 C C
C C (III)
(III)
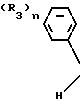
Эти два изомера отличаются друг от друга температурой плавления, спектром ядерного магнитного резонанса или газовой хроматографией, но различие между ними можно более широко и ясно охарактеризовать исходным материалом, триазольным соединением формулы (II) C
C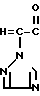 -R2 (II) в которой R2, R3 и n имеют указанные выше значения.
-R2 (II) в которой R2, R3 и n имеют указанные выше значения.
Триазольное соединение формулы (I) получают путем восстановления триазольного соединения формулы (II) с образованием триазольного соединения формулы (I)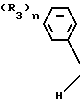 C C
C C где R1 является атомом водорода, с последующей этерификацией полученного соединения:
где R1 является атомом водорода, с последующей этерификацией полученного соединения: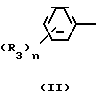 C
C -R2
-R2

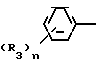 C
C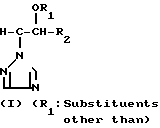
где R1, R2, R3 и n имеют указанные ранее значения.
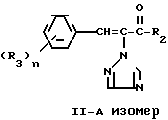
Отсюда один из двух геометрических изомеров триазольного соединения (II), одефиновый протон которого появляется в более сильном магнитном поле спектра ЯМР в дейтерохлороформе, определяется как II-A изомер, а другой, олефиновый протон которого появляется в менее сильном магнитном поле спектра ЯМР в дейтрохлороформе, определяется как изомер II-B.
Эти связи проиллюстрированы ниже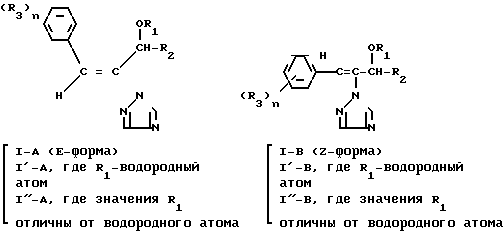
 C C
C C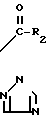

Соединения II-B опиcаны в опубликованной, еще не прошедшей экспертизу японской патентной заявке N 130661/1978 и соединения I-B описаны в опубликованной нерассмотренной (выложенной) японской патентной заявке N 41875/1979 (бельгийский патент N 870243).
Далее, соединение (I), в котором R1 является атомом водорода, полученное при восстановлении изомера II-А, определяют как I'-А изомер, соединение (I), в котором R1 является указанным заместителем, отличным от атома водорода, полученное при этерификации изомера I'-А определяют как изомер I''-А, при этом и I'-А изомер и I''-А изомер определяют обычно как I-А изомер. Соответствующие соединения, полученные из II-В изомера аналогичным образом определены как I'-В изомер, I''-В изомер и I-В изомер соответственно. Изобретение относится к I-А изомеру и II-А изомеру, который является промежуточным для I-А изомера.
До их пор было получено большое количество синтетических органических соединений, которые внесли большой вклад как сельскохозяйственные химикалии для сельскохозяйственных и садоводческих культур за счет их активности против заболеваний и вредителей, наносящих вред этим культурам. Однако, известен тот факт, что еще остается переменным значительное количество проблем, требующих своего решения. Эти проблемы иногда разрешаются, например, за счет создания новых и более желательных пестицидов, или можно считать, что их можно также решить, исследуя обычные пестициды для установления подходящих форм применения сельскохозяйственных химикалиев.
Существует немало синтетических органических соединений, которые могут также присутствовать в виде геометрических или оптических изомеров. На практике существует много случаев, в которых пестициды, содержащие эти изомеры используют на практике как сельскохозяйственные химикалии. Хорошо известно не только для сельскохозяйственных химикалиев, но также и во многих других областях, что среди многих ингредиентов, имеющих изомерные формы, наблюдаются различия в биологической активности. В настоящее время серьезное значение приобретает проблема загрязнения окружающей среды в области сельского хозяйства и садоводства, и важно ослабить эту проблему, используя один из двух изомеров, который проявляет более сильную активность. Кроме того, здесь необходимо учесть больший экономический эффект от получения этого соединения, а также при практическом применении в качестве ядохимикатов. Поэтому с этой точки зрения можно считать благоприятным факт создания изомера, обладающего более сильной активностью.
Исходя из этого изобретения провели дальнейшее исследование соединений, уже найденных ими (заявка на открытый патент Японии/Кокаи N 130661/1978, патент Бельгии N 870248 и патент Великобритании N 2004276). В результате этого было установлено, что настоящие изобретения, определяемые как I-А изомер (т. е. Е-форма), а именно один из двух геометрических изомеров триазольного соединения, представленного формулой (I), обладает более высокой фунгицидной активностью в более широком диапазоне болезней растений, а также более высоким гербицидным действием и более сильным действием, регулирующим рост растений по сравнению с соединениями, определяемыми как I-В изомер (т.е. Z -форма), другими словами, они обладают великолепными свойствами в качестве ядохимикатов.
Поэтому этим изобретением предусматривается геометрический изомер или его соль в Е-форме, имеющий формулу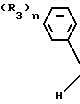 C C
C C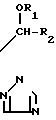 в которой R1 представляет атом водорода, С1-С4-алкильную, С3-С4алкенильную или 2-пропинильную группу, R2 представляет С1-С6 алкильную, циклопропильную или 1-метилциклопропильную группу, элемент R3, который может иметь одинаковые или различные значения, представляет атом галогена, С1-С4 алкильную группу, галоидзамещенную С1-С3 алкильную группу, С1-С4 алкокси, фенокси, фенильную, циано- или нитрогруппу, и n является целым числом от 0 до 3, или его соль.
в которой R1 представляет атом водорода, С1-С4-алкильную, С3-С4алкенильную или 2-пропинильную группу, R2 представляет С1-С6 алкильную, циклопропильную или 1-метилциклопропильную группу, элемент R3, который может иметь одинаковые или различные значения, представляет атом галогена, С1-С4 алкильную группу, галоидзамещенную С1-С3 алкильную группу, С1-С4 алкокси, фенокси, фенильную, циано- или нитрогруппу, и n является целым числом от 0 до 3, или его соль.
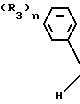
Существует также много хорошо известных триазольных соединений, описанных в патенте Великобритании N 1364619, патенте Бельгии N 345433, патентах Западной Германии N 2610022, 2654890 и 2734426, а также патенте США N 4086351. Однако отличительной особенностью изобретения является то, что получена следующая новая информация: один из двух геометрических изомеров триазольного соединения (I) отличающийся тем, что имеет и (1) двойную связь (бензилиденовая группировка) и (2) гидроксильную группу или ее простые эфиры.
 H
H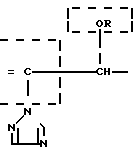 обладает гораздо более сильными характеристиками в качестве сельскохозяйственных химикалиев по сравнению с другими геометрическими изомерами. В этом плане соединения настоящего изобретения имеют структурные характеристики, отличающиеся от характеристик указанных ранее хорошо известных соединений, и, кроме того, они обладают гораздо более ценными свойствами, по сравнению с хорошо известными соединениями. Соответственно оригинальность настоящего изобретения такова, что она никак не следовала из известного ранее уровня знаний.
обладает гораздо более сильными характеристиками в качестве сельскохозяйственных химикалиев по сравнению с другими геометрическими изомерами. В этом плане соединения настоящего изобретения имеют структурные характеристики, отличающиеся от характеристик указанных ранее хорошо известных соединений, и, кроме того, они обладают гораздо более ценными свойствами, по сравнению с хорошо известными соединениями. Соответственно оригинальность настоящего изобретения такова, что она никак не следовала из известного ранее уровня знаний.
В качестве заболеваний, против которых соединения настоящего изобретения (1-А) (изомер) обладают высокой защитной активностью, можно привести пирикуляриоз риса (pyricularia oryzae), гниль оболочи риса (pellicularia sasakii), рак яблок (valsa mali), цветочная гниль яблок (Sclirotinia mali), мучнистая роса яблок (podosphaera lencotricha), парша яблони (venturia inaegualis), пятнистость яблок (Mucosphaerella ponni), пятнистость листьев у яблони (Alternaria mali), черная пятнистость груши (Alternaria kikuchiana), мучнистая роса груши (phyllactinia pyri), ржавчина груши (Gymnosporangium haralanun), парша груши (venturia Rashicola), меланрз цитрусовых (Duaporthe citri), парша цитрусовых (Elsinol fawсetti), обычная зеленая плесень цитрусовых плодов (penicillium otigitatum), синяя плесень апельсинов (penicillium italicum), коричневая гниль персиков (selerotinia cinerea), антракоз винограда (Elsinoe ampelina), гниль спелого винограда (Glomerella cingulata), серая гиль винограда (Botrytiv cinerea), мучнистая роса винограда (Uncinula necator), ржавчина винограда (phakopsora avupelopsidis), ржавчина верхушек овса (puccinia coronota), мучнистая роса ячмена (Erysiphe graminis), пятнистость листьев ячменя (Phynchosporium secalis), полосатость ячменя (Helminthosporium gramineum), рыхлая головня ячменя (ustilago huda), твердая головня ячменя (ustilago hordei), снежная гниль ячменя (Typhula incarnata), стеблевая ржавчина ячменя (puccinia graminis), листовая ржавчина пшеницы (puccinia recondita), рыхлая головня пшеницы (ustilago tritici), твердая головня пшеницы (Tilletia caries), пятнистость листьев пшеницы (Septoria tritici), пятнистость колосков пшеницы (Septoria nadorum), желтая ржавчина пшеницы (puccinia striiformis), стеблевая ржавчина пшеницы (puccinia graminis), мучнистая роса пшеницы (Erysiphe graminis), мучнистая роса огурцов (Sphaerotheca fuliginea), серная гниль огурцов (Borytis cinerea), гуммозная стеблевая гниль огурцов (Mycosphaerella melonis), гниль семянки огурцов (Sclerotinia sclerotiorum), антракоз огурцов (Colletotrichum aegenarium), лиственная гниль томатов (Cladosporium fulvum), мучнистая роса томатов (Erysiphe cichora cearum), ранняя гниль томатов (Alternaria solahi), серая гниль баклажан (Botrytis cinirea), мутовчатое увядание баклажан (verticillium albo-atrum), мучнистая роса баклажан (Erysiphe cichoracearum), мучнистая роса душистого перца (Levcillula tanrica), серая гниль земляники (Botrytis cinirea), мучнистая роса земляники (Sphaerotheca humuli), коричневая пятнистость табака (Alternaria longipes), мучнистая роса табака (Erysiphe cichoraccarum), пятнистость листьев свеклы (cercospora Beticola), пятнистость листьев зеленого горошка (cercospora personata), коричневая пятнистость листьев горошка (сercospora arachilicola) и тому подобное.
При дальнейших исследованиях противомикробной активности соединений настоящего изобретения, I-А изомера, стало очевидно, что соединения настоящего изобретения имеют также противомикробную активность против Trichophyron rubrum. Таким образом, было обнаружено, что имеется возможность использовать соединения настоящего изобретения в качестве фунгицида для медицинских целей.
Далее соединения изобретения, I-А изомер, можно также использовать в качестве регуляторов роста растений, применяемых для контроля роста полезных растений. Так, например, их можно использовать для предотвращения веретенообразного роста риса, пшеницы, дерна, деревьев для изгородей и фруктовых деревьев, а также для получения карликовых форм садовых растений, например, горшковых хризантем.
При выращивания риса и пшеницы серьезную проблему часто представляют полегание риса и пшеницы за счет внесения большего чем нужно количества удобрений или сильного ветра. Внесение же соединений настоящего изобретения в нужный момент является эффективным для контроля веса риса и пшеницы и предотвращения их полегания.
При выращивании горшечной культуры хризантем внесение соединений настоящего изобретения является полезным для повышения коммерческой ценности хризантем, так как они могут привести к укорочению длины стебля без вредного воздействия на цветы.
Соединения изобретения I-А изомеры, обладают высокой гербицидной активностью против травянистых полевых сорняков, таких, как просо петушье (Echinochloa crus-galli) высокая росичка (Digitaria sanguinalis) и щетинник зеленый (Setaria viridis), таких широколиственных полевых сорняков как зонтичные растения (Cyperus difformis Z.), марь красная (Amaranthus retroflexus), обычная марь белая (Chenopodium album), портулак огородный (portulaca oleracea) и звездачатка (Stellaria media), и таких однолетних и многолетних сорняков и болотистых поляк, как просо петушье (Echinoehloa crus-galli), лютик (Monochoria viaginalis), ротала ветвистая (Rotala oudica Koehue) (Dopatrium junceum Bulrush sp/ Seirpus jumcoides var Hotarui ohwi) и Sliuder spikerush (Ellocharis acicularis).
Если соединения изобретения вносят на поля, то они также хороши со следующих точек зрения, они обладают высокой гербицидной активностью против основных полевых сорняков, они обладают активностью как при обработке почвы до прорастания сорняков, так и при обработке листвы в начальной фазе роста, и кроме того, их можно наносить, не опасаясь нанести вред основным культурам (например рису, хлопку, сое, кукурузе, гороху, подсолнечнику, свекле), а также овощным культурам (например, латуку, редису, томатам). Если соединения изобретения наносят на болотистые поля они также демонстрируют высокую гербицидную активность против основных сорняков либо при обработке до прорастания либо при обработке листвы в начальной фазе роста, а кроме того, они совершенно безопасны для растений риса.
Далее соединения изобретения чрезвычайно полезны в качестве гербицидов не только для рисовых полей, но также для различных злаков, овощей, плодовых садов, торфяников, пастбищ, чайных плантаций, щелковичных плантаций, каучуковых плантаций, лесов и некультивируемых земельных участков.
Далее стало очевидным, что соединения изобретения характеризуются высокой безопасностью для млекопитающих и рыб, и, кроме того, их можно использовать на практике, не причиняя вреда полезным сельскохозяйственным культурам.
Триазольные соединения II-А, промежуточные для получения соединений изобретения I-А, также обладают фунгицидной активностью против различных патогенов, наносящих вред сельскому хозяйству, а также гербицидной активностью и действием в качестве регуляторов роста растений. Однако имеет место тот факт, что соединение настоящего изобретения I-А обладает гораздо более высокой активностью против широкого круга патогенов растений также, как и гораздо более высокой гербицидной активностью и активностью в качестве регуляторов роста растений, нежели соединения II-А.
Более конкретно способы получения соединений изобретения будут описаны далее.
Способ А.
Восстановление тетразольного соединения II
(R3)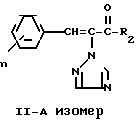


где R2, R3 и n имеют указанные ранее значения. Изомер I'-А получают, восстанавливая изомер II-А в подходящем растворителе металлогидридным комплексом (например, литийалюминийгидридом, натрийборгидридом) или алкоголятом алюминия (например, изопропилатом алюминия).
II-А изомер, подлежащий восстановлению, можно получить в чистом виде, например, осуществляя фракционую кристаллизацию или хроматографируя на колонке смесь геометрических изомеров триазольного соединения (II), полученного в соответствии со следующей схемой реакции. Изомер II-А можно также легко получить с хорошим выходом, например, облучая смесь ультрафиолетовыми лучами для проведения фотоизомеризации. Более детальные разъяснения будут приведены далее относительоно способа С и D.
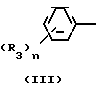 CHO +
CHO +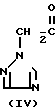 -R 2
-R 2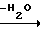
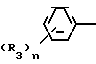 C
C -R
-R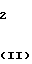 где R2, R3 и n имеют указанные ранее значения.
где R2, R3 и n имеют указанные ранее значения.
Растворители, которые можно использовать при восстановлении металлогидридным комплексом, включают, например, простые эфиры (например диэтиловый эфир, тетрагидрофуран) и спирты (например метанол, этанол, изопропанол). Если в качестве металлогидридного комплекса используют натрийборгидрид, то реакцию осуществляют добавляя в растворитель 1 моль II-А изомера и от 0,25 до 2 молей натрийборгидрида. Температуру реакции поддерживают предпочтительно, в интервале от 0оС до комнатной температуры. Используемые растворители включают например, простые эфиры (например диэтиловый эфир, тетрагидрофуран), и спирты (например этанол, метанол, изопропанол). Если в качестве металлогидридного комплекса используют литийалюминий гидрид, то реакцию осуществляют, растворяя литийалюминийгидрид в количестве от 0,25 до 0,8 моля в расчете на II-А изомер в растворителе, и, добавляя полученный раствор к раствору изомера в том же самом растворителе. Температуру реакции поддерживают, предпочтительно в интервале от -60 до 70оС. Используемые растворители включают простые эфиры (например, диэтиловый эфир, тетрагидрофуран). После завершения реакции воду или разбавленную водой кислоту добавляют к реакционному раствору, и после нейтрализации щелочью в случае необходимости, высадившиеся кристаллы собирают фильтрованием или экстрагированием органическим растворителем, умеренно растворимым в воде. Последующую обработку проводят обычными способами.
Если в качестве восстанавливающего агента используют изопропилат алюминия, то предпочтительно использовать такие растворители, как спирты (например, изопропанол) или ароматические углеводороды (например, бензол). Обычно дают возможность 1 молю II-А изомера взаимодействовать с 1-2 молями изопропилата алюминия при температуре между комнатной температурой и 100оС. Полученное соединение алюминия разлагают разбавленой серной кислотой или водным раствором гидроокиси натрия, после чего экстрагируют органическим растворителем, умеренно растворимым в воде. Последующую обработку проводят обычными способами.
Соли I'-А изомера относятся к солям, полученным из растительно- и физиологически приемлемых кислот, например галоидводородных кислот (например бромистоводородной кислоты, хлористоводородной кислоты, йодистоводородной кислоты), карбоновых кислот (например уксусной кислоты, трихлоруксусной кислоты, малеиновой кислоты, янтарной кислоты), сульфокислоты (например паратолуолсульфокислоты, метансульфокислоты), азотной кислоты, серной кислоты, и фосфорной кислоты. В случае необходимости эти соли получают обычными способами.
Способ В.
Этерификация I'-А изомера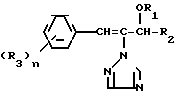

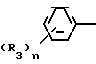 C
C R2
R2
где R1, R2, R3 и n имеют указанные ранее значения.
Соединения настоящего изобретения, I'-А изомеры, получают при взаимодействии I'-А изомеров с реакционноспособным С1-С4-алкильным, С3-С4 алкенильным или 2-пропинильным производным в подходящем растворителе в присутствии основания. Реакционноспособные производные включают, например, алкил-алкенил- или алкинил-галоиды (например метилиодид, аллилбромид, пропаргилбромид), сульфаты (например диметилсульфат, диэтилсульфат) и сульфонаты (например паратолуолсульфонат, нафталинсульфонат). Растворители включают, например, общие инертные органические растворители, например диэтиловый эфир, тетрагидрофуран, диоксан, бензол, толуол, ксилол, и диметилформамид. Эту реакцию можно проводить в присутствии воды, используя катализатор переноса фазы, известный как эксельратор реакции (например триэтилбензиламмонийхлорид, триметилбензиламмонийбромид). Основания включают, например, подходящие сильные основания (например гидриды щелочных металлов, такие как натрийгидрид, амиды щелочных металлов, например натрийамид), карбонаты (например карбонат натрия, карбонат калия) и гидрокиси щелочных металлов (например гидроокись калия, гидрокись натрия).
Эту реакцию осуществляют, смешивая I'-А изомер, реакционноспособное, С1-С4 алкил, С3-С4 алкенил или 2-пропинил-производное и основание предпочтительно в эквимолярном соотношении в подходящем растворителе. Реакцию проводят в интервале температур от 0 до 100оС и предпочтительно от 20 до 60оС. I'-А изомер с подходящим сильным основанием (например, гидридами щелочных металлов, амидами щелочных металлов) в инертном растворителе, а затем полученную соль щелочного металла подвергают взаимодействию с реакционноспособным С1-С4 -алкил, С3-С4-алкенил, или 2-пропинилпроизводным.
В некоторых случаях для выделения соединений изобретения I''-А может быть желательный следующий путь: реакционную смесь выделяют из растворителя путем выпаривания, воду и органический растворитель, умеренно растворимый в воде, добавляют к остатку, органический слой после экстрагирования выделяют и затем очищают обычными способами.
Соли I''-А изомера относятся к тем солям, которые получены с помощью физиологически приемлемых кислот, таких как, например, галоидоводородные кислоты (например бромистоводородная кислота, хлористоводородная кислота, йодистоводородная кислота), карбоновые кислоты (например уксусная кислота, трихлоруксусная кислота, малеиновая кислота, янтарная кислота), сульфоновые кислоты (например паратолуолсульфокислота, метансульфоновая кислота), азотная кислота, серная кислота и фосфорная кислота. В случае необходимости, эти кислоты получают обычными способами.
Более подробно изобретение будет проиллюстрировано со ссылкой на следующие примеры.
Если нет других указаний, то в ЯМР-спектрах в примерах указаны значения δ для дейтерохлороформа в качестве растворителя и тетраметилсилана в качестве внутреннего стандарта.
П р и м е р 1. Синтез I'-А изомера 1-(4-хлорфенил)-4,4-диметил- 2-(1,2,4-триазол-1-ил)-1-пентен-3-ола (соединение N 1) по способу А. II-А изомер (2,9 г, 0,01 моля, т.пл. 108-109оС).
1-(4-хлорфенил)-4,4-диметил-2)1,2,4- триазол-1-ил) 1-пентен-3-она (соединение I') растворили в метаноле (50 мл). К этому добавили натрийборгидрид (0,38 г, 0,01 моля), причем температуру реакции поддерживали при 20оС, или менее, охлаждая льдом. Реакционную смесь выдерживали при 20оС в течение 3 ч, а затем разложили, добавив воду (100 мл) и уксусную кислоту (1 мл). Органический слой экстрагировали этилацетатом (100 мл), полученный экстракт промыли 5% -ным водным раствором бикарбоната натрия (50 мл) и высушили над безводным сульфатом натрия. Затем растворитель удалили при пониженном давлении и полученный остаток перекристаллизовали из изопропанола до получения 2,0 г (выход 69%) I'-А изомера, точка плавления которого 153-155оС. Элементный анализ и полученный спектр ЯМР этого соединения приведены ниже.
Элементный анализ:
Рассчитано С, Н, N, Cl,
для C15H18N3OCl 61,74 6,23 14,40 12,15
Найдено 61,82 6,33 14,38 12,15
Спектр ЯМР:
8,52 (1Н, с, триазольный протон)
7.98 (1Н, с, триазольный протон)
7,30 (4Н, с, фенильный протон)
6,91 (1Н, с, олефиновый протон)
4,56 (2Н, широкий синглет, гидроксильный протон и протон метина содержащего ОН группу)
0,66 (9Н, с, бутильный протон)
Сравнительный пример 1. Синтез 1'-В-изомера 1-(4-хлорфенил)-4,4-диметил-2(1,2,4-триазол-1-ил)-1-пентен-3-ола (соединение N 1).
II-В изомер (2,9 г 0,01 моля, т.пл. 78-79оС) 1-(4-хлорфенил)-4,4-диметил-2)(1,2,4-триазол-1-ил)-1-пентен-3-она (соединение N 1') растворили в метаноле (50 мл). Этот изомер оставили реагировать с натрийборгидридом, а затем обработали таким же способом, что и в примере 1. Полученный остаток перекристаллизовали из смеси 1:10 четыреххлористого углерода и н-гексана до получения 2,2 г (выход 76%) I'-В изомера (т.пл. 116-117оС) соединения N 1. Элементный анализ и спектр ЯМР этого соединения приведены далее.
Элементный анализ:
C, H, N, Cl,
Рассчитано 61,74 6,28 14,40 12,15
для C15H18N3OCl
Найдено 61,80 6,25 14,52 12,09
Спектр ЯМР
7,92 (с, триазольный протон)
7,77 (1Н, с, триазольный протон)
7,05 (2Н, д, фенильный протон j 9 Гц)
6,58 (2Н, д, фенильный протон j 9 Гц)
6,66 (1Н, с, олефиновый протон)
4,28 (1Н, д, метиловый протон, несущий ОН группу, j 6 Гц)
3,21 (1Н, д, гидроксильный протон, j6 Гц)
0,80 (9Н, с, бутильный протон).
П р и м е р 2. Синтез I'-А изомера 3-(4-хлорфенил)-1-(1-метилциклопропил)-2-(1,2,4-триазол-1-ил) -2-пропен-1-ола (соединение N 30) по способу А.
II-А изомер (2,9 г, 0,01 моля, т.пл. 89-92оС) 3-(4-хлорфенил)-1-метилциклопропил)-2-(1,2,4-триазол-1-ил)-2- пропен-1-она (соединение 29'). Охарактеризованный по спектру ЯМР, описанному далее, растворили в метаноле (50 мл), натрийборгидрид (0,33 г 0,01 моля) добавили к этому, причем температуру реакции поддерживали 20оС или ниже, охлаждая льдом. Температуру реакции поддерживали при 20оС в течение 3 ч, а затем разлагали, добавив воду (100 мл) и уксусную кислоту (2 мл). Органический слой экстрагировали хлороформом (100 мл), экстракт промыли 5%-ным водным раствором бикарбоната натрия (50 мл), и высушили над безводным сульфатом магния. Затем растворитель удалили при пониженном давлении, а полученный осадок перекристаллизовали из смеси четыреххлористого углерода и н-гексана (1:1) 5 мл (до получения 2,4 г) выход 85% указанного в заглавии соединения.
ЯМР-спектр исходного материала II-А изомера 3-(4-хлорфенил)-1-(1-метилциклопропил)-2-(1,2,4-триазол-1-ил)-2- пропен-1-она.
8,28 (1Н, с, триазольный протон)
8,07 (1Н, с, триазольный протон)
7,32 (4Н, с, фенильный протон)
7,19 (1Н, с, олефиновый протон)
1,45-1,15 (2Н, м, метиленовый протон циклопропильной группы)
1,25 (3Н, с, метильный протон)
0,99-0,75 (2Н, м, метиленовый протон циклопропильной группы)
Сравнительный пример 2. Синтез I'-В изомера 3-(4-хлорфенил)-1-(1-метилциелопропил)-2-(1,2,4-триазол-1-ил)-2 -пропан-1-ола (соединение N 30).
II-В изомер (2 г, 0,007 моля, т.пл. 74-75оС (3-(4-хлорфенил)-1-(1-метилциклопропил)-2-1,2,4-триазол-1-ил) -2-пропен-1-она (соединение N 29), характеризующейся спектром ЯМР, приведенным далее, восстановили таким же способом, что и в примере 2, натрийборгидридом (0,27 г, 0,007 моля) в метаноле (50 мл). Таким образом получили 1,7 г (выход 85%) соединения, указанного в заглавии. Спектр ЯМР исходного материала, II-В изомера 3-(4-хлорфенил)-1-(1-метилциклопропил)-2-(1,2,4-триазол-1 -ил) -2-пропен-1-она:
8,12 (2Н, с, триазольный протон)
8,03 (1Н, с, триазольный протон)
7,55 (1Н, олефиновый протон)
7,21 (2Н, д, фенильный протон j 8 Гц)
6,81 (2Н, д, фенильный протон j 8 Гц)
1,50-1,25 (2Н, м, метиленовый протон циклопропильной группы)
1,28 (3Н, с, метильный протон)
0,90-0,65 (2Н, м, метиленовый протон циклопропильной группы).
П р и м е р 8. Синтез I''-А изомера 1-(4-хлорфенил)-4,4-диметил-3-метоси-2)-1.2.4-триазол-1-ил)- пентил (соединение N 35) по способу В.
I'-А изомер (2 г) 1-пара-хлорфенил-4,4-диметил-2(1,2,4-триазол-1- ил)-1-пентен-3-ол (соединение N 1) растворили в диметилформамиде (20 см3) и к нему добавили 65%-ный натрийгидрид в масле (0,26 г). После перемешивания в течение 1 при комнатной температуре, реакционную смесь охладили до 10оС и добавили метилиодид (1 г). После выстаивания при комнатной температуре в течение 20 ч растворитель удалили при пониженном давлении, а полученный остаток экстрагировали, добавив ледяную воду (100 г) и хлороформ (100 см3). Органический слой высушили над безводным сульфатом магния, и растворитель удалили пониженном давлении. Полученный маслянисты неочищенный продукт очистили на хроматографической колонке на силикагеле (ацетон н-гексан 1:10), и далее перекристаллизовали из смеси четыреххлористого углерода и н-гексана (1:2) до получения 1,6 г указанного в заглавии соединения (т.пл. 63-66оС).
0,99-0,75 (2Н, м, метиленовый протон циклопропильной группы)
Сравнительный пример 2. Синтез I'-В изомера 3-(4-хлорфенил)-1-(1-метилциелопропил)-2-(1,2,4-триазол-1-ил)-2 -пропан-1-ола (соединение N 30).
II-В изомер (2 г, 0,007 моля, т.пл. 74-75оС) 3-(4-хлорфенил)-1-(1-метилциклопропил)-2-1,2,4-триазол-1-ил) -2-пропен-1-она (соединение N 29'), характеризующийся спектром ЯМР, приведенным далее, восстановили таким же способом, что и в примере 2, натрийборгидридом (0,27 г, 0,007 моля) в метаноле (50 мл). Таким образом получили 1,7 г (выход 85%) соединения, указанного в заглавии. Спектр ЯМР исходного материала, II-В изомера 3-(4-хлорфенил)-1-(1-метилциклопропил)-2-(1,2,4-триазол-1-ил) -2-пропен-1-она:
8,12 (2Н, с, триазольный протон)
8,03 (1Н, с, триазольный протон)
7,55 (1Н, с, олефиновый протон)
7,21 (2Н, д, фенильный протон j 8 Гц)
6,81 (2Н, д, фенильный протон j 8 Гц)
1,50-1,25 (2Н, м, метиленовый протон циклопропильной группы)
1,28 (3Н, с, метильный протон)
0,90-0,65 (2Н, м, метиленовый протон циклопропильной группы).
П р и м е р 3. Синтез I''-А изомера 1-(4-хлорфенил)-4,4-диметил-3-метокси-2)-1, 2.4-триазол-1-ил)- пентил (соединение N 35) по способу В.
I'-А изомер (2 г) 1-пара-хлорфенил-4,4-диметил-2(1,2,4-триазол-1- ил)-1-пентен-3-ол (соединение N 1) растворили в диметилформамиде (20 см3) и к нему добавили 65%-ный натрийгидрид в масле (0,26 г). После перемешивания в течение 1 при комнатной температуре, реакционную смесь охладили до 10оС и добавили метилиодид (1 г). После выстаивания при комнатной температуре в течение 20 ч растворитель удалили при пониженном давлении, а полученный остаток экстрагировали, добавив ледяную воду (100 г) и хлороформ (100 см3). Органический слой высушили над безводным сульфатом магния, и растворитель удалили при пониженном давлении. Полученный маслянистый неочищенный продукт очистили на хроматографической колонке на силикагеле (ацетон н-гексан 1:10), и далее перекристаллизовали из смеси четыреххлористого углерода и н-гексана (1:2) до получения 1,6 г указанного в заглавии соединения (т.пл. 63-66оС).
Сравнительный пример 3. Синтез I''-В изомера 1-пара-хлорфенил-4,4-диметил-3-метокси-2-(1,2,4-триазол) -1-или-1-пентена (соединение N 35)
I'-В изомер (2 г) 1-(4-хлорфенил)-4,4-диметил-2(1,2,4- триазол-1-ил)-пентен-3-ола (соединение N 1) растворили в диметилформамиде (20 см) и к нему добавили 65% -ный натрийгидрид (0,26 г). После перемешивания при комнатной температуре в течение 1 ч реакционную смесь охладили до 10оС и добавили 1 г метилиодида. Реакционную смесь выдерживали при 10оС в течение 1 ч, а затем оставили выстаиваться при комнатной температуре в течение 16 ч. Диметилформамид удалили при пониженном давлении, и полученный остаток экстрагировали, добавив ледяную воду (100 г) и хлороформ (100 см3). Органический слой высушили над безводным сульфатом магния, и растворитель удалили при пониженном давлении. Полученный в результате неочищенный продукт очистили на хроматографической колонке на силикагеле (ацетон) н-гексана (1:10) до получения 1,0 г соединения, указанного в заглавии, в виде маслянистого продукта.
Показатель преломления nD27 1,5435.
Рассчитано С, H, N, Cl,
62,90 6,60 13,77 11,50
для C16H20N3ClO
Найдено 62,84 6,59 13,74 11,59
Соединения настоящего изобретения (I-А изомеры) полученные по способам А и В приведены в табл.1. Для сравнения рядом приведены данные для I-В изомеров. Если нет других указаний, спектры ЯМР в таблице приведены в значениях δ для CDCl3 в качестве растворителя и тетраметилсилана в качестве внутреннего стандарта. I'-А изомер и I''-А изомер обычно называют А I-А изомером, а I'-В изомер также обычно называют I-В изомером. Это общее обозначение также используется в тестовых примерах приводимых далее.
Далее приводится объяснение для получения II-А изомера триазольного соединения (II), которое является исходным материалом для I'-А изомера триазольного соединения (I).
Способ С
Изомеризация II-В изомера или смеси II-В и II-А изомеров триазольного соединения (II):
где R2, R3 и n имеют указанные ранее значения.
II-А изомер можно получить, облучая II-В изомер или смесь II-В и II-А изомеров УФ-лампами или ксеноновыми лампами, или экспериментально флюресцентными лампами или солнечными лучами, в растворителе, инертном по отношению к этим лучам. В качестве обычно используемого растворителя можно указать, например, спирты (например метанол, этанол, попанол); простые эфиры (например тетрагидрофуран, диоксан), кетоны (например ацетон, метилэтилкетон, метилизобутилкетон), алифатические углеводороды (например гексан, циклогексан, петролейный эфир) и ароматические углеводороды (например бензол, толуол, ксилол). Реакцию можно проводить при температурах, при которых обычно протекает фотоизомеризация, однако предпочтительными на практике являются температуры между 0 и 100оС. Естественно реакцию можно проводить, добавляя сенсибилизатор, используемый в обычных фотореакциях, например фенилкетоны, такие как ацетофенон и пропилфенон, однако преимущество это дает небольшое.
Далее будет проиллюстрирован способ получения триазольного соединения, представленного формулой (II)
Способ Д.
Получение смеси геометрических изомеров триазольного соединения (II) и каждого из изомеров (II-В и II-А)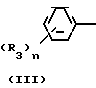 CHO
CHO  -
- -R2
-R2
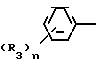 C
C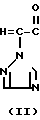 -R2
-R2
где R2, R3 и n имеют указанные ранее значения.
Триазольное соединение (II) получают, подвергая взаимодействию 1 моль кетона формулы (IV) с 1-2 молями бензальдегида формулы (III) в подходящем растворителе в присутствии основного катализатора. Основные катализаторы включают, например гидроокиси щелочных и щелочноземельных металлов (например гидроокись натрия, гидроокись калия, гидроокись кальция) алкоголята щелочных металлов (например метилат натрия, этилат натрия, метилат калия), карбонаты (например карбонат натрия, карбонат калия), ацетаты (например ацетат натрия, ацетат калия), вторичные амины (например диэтиламин, дипропиламин, пирролидин, пиперидин, морфолин) и третичные амины (например триэтиламин, трибутиламин, пиридин, пиколин, диметиланилин) и его используют в количествах от 0,01 до 10,0 молей. Растворители включают, например, спирты (например метанол, этанол), ароматические углеводороды (например бенол, толуол, ксилол), простые эфиры (например диэтиловый эфир, тетрагидрофуран, диоксан), воду и их смеси. Реакцию проводят в интервале температур до 0оС до температуры кипения растворителя.
Если в качестве основного катализатора используют ацетаты (например, ацетат натрия, ацетат калия), карбонаты (например карбонат натрия, карбонат калия) или третичные амины, то в качестве растворителя для реакции можно использовать ледяную уксусную кислоту или уксусный ангидрид.
Полученное таким образом триазольное соединение (II) является смесью двух геометрических изомеров, например, изомера II-А и изомера II-В, причем каждый из изомеров можно выделить хроматографированием на колонке или фракционной кристаллизацией. Смесь геометрических изомеров обычно содержит большее количество изомера II-В, нежели изомера II-А. Все изомеры II-А кетонового соединения представляют собой, естественно, новые соединения, а из II-В изомеров новыми соединениями являются такие, в которых R2 является I-метилциклопропильной группой.
Далее способы С и D будут проиллюстрированы более подробно со ссылкой на следующие примеры.
П р и м е р 4. Синтез 1-(4-хлорфенил)-4,4-диметил-2-(1,2,4-триазол-1-ил)-1- пентен-3-она (соединение N 1') по способу D.
α -(1,2,4-триазол-1-ил)пинаколон (50 г), безводный карбонат калия (41 г) уксусный ангидрид (200 мл) и 4-хлорбензальдегид (46,3 г) смешали, и полученную смесь нагревали при 90оС в течение 12 ч при перемешивании. После охлаждения реакционного раствора выпавший осадок отфильтровали. Полученный фильтрат по каплям добавили к теплой воде (500 мл) при 60оС для разложения уксусного ангидрида, а затем понемногу добавили карбонат калия для подщелачивания раствора. Полученный маслянистый продукт экстрагировали этилацетатом (500 мл) и органический слой высушили над безводным сульфатом натрия и затем сконцентрировали при пониженном давлении. Одну каплю полученного остатка растворили в ацетоне и ацетоновый раствор проанализировали с помощью газовой хроматографии в условиях, описанных далее. Затем было найдено, что время удеpживания для пика, соответствующего II-А изомеру, составляет 300 с, а время, соответствующее пику II-В изомера, составляет 360 с. Отношение обоих изомеров, полученное при расчете площадей пиков составило 19,8 /61,2, т.е. около 1/3.
Условия проведения газово-хроматографического анализа были следующими.
Аппаратура: газовый хроматограф Ниппон Денши 20 К, снабженный детектоpом FID.
Колонка: стеклянная колонка длиной 1 м
Жидкая фаза 5% ХЕ-60
Носитель Хромосорб W
Температура (колонка): 200оС
Температура (на воде): 240оС
Газ-носитель: газообразный азот, 1 кг/см2.
Остаток растворили в бензоле (100 мл). Полученный раствор пропустили через колонку, заполненную силикагелем с размерами частиц 100-200 мешей (1,2 кг) и провели хроматографирование смесью н-гексан/ацетон (10:1) в качестве проявляющего растворителя. Фракции, соответствующие каждому изомеру, перекристаллизовывали из четыреххлористого углерода, в результате чего получили 36 г (выход 41,6%) чистого II-В изомера), т.пл. 78-79оС (и 10 г) выход 11,5% (чистого II-А изомера) (т.пл. 108-109оС). Проявляющий растворитель н-гексан/ацетон (10/3) пропускали далее через колонку, в результате чего выделили 8 г -(1,2,4-триазол-1-ил)пинаколона. Элементный анализ и спектры ЯМР каждого изомера приведены далее. Спектры ЯМР получали в дейтерохлороформе в качестве растворителя, и химические сдвиги выражали в величинах с тетраметилсиланом в качестве внутреннего стандарта.
II-А изомер 1-(4-хлорфенил)-4,4-диметил-2-(1,2,4-триазол-1-ил)-1 -пентен-3-она (соединение N 1').
Элементный анализ
C, H, N, Cl,
Рассчитано для 62,17 5,58 14,50 12,23
C15H16N8OCl
Найдено 62,32 5,60 14,41 12,20
ЯМР-спектр:
8,11 (1Н, с, триазольный протон)
7,00 (1Н, с, триазольный протон)
7,15 (4Н, с, фенильный протон)
6,99 (1Н, с, олефиный протон)
0,99 (9Н, с, бутильный протон)
II-В изомер 1-(4-хлорфенил)-4,4-диметил-2-)1,2,4-триазоль-1-ил)-пентен-3-она (соединение N 1'):
Элементный анализ:
C, H, N, Cl,
Найдено: 62,35 5,59 14,38 12,18
Спектр ЯМР:
8,14 (1Н, с, триазольный протон)
7,98 (1Н, с, триазольный протон)
7,22 (2Н, д, фенильный протон j 8 Гц)
6,73 (2Н, д, фенильный протон, j 8 Гц)
7,49 (1Н, с, олефиновый протон)
1,22 (9Н, с, бутильный протон)
П р и м е р 5. Синтез II-А изомера 1-(4-хлорфенил)-4,4-диметил -2-(1,2,4-триазол-1-ил)1-пентен-3-она по способу С.
II-В изомер (8,0 г) 1-(4-хлорфенил)-4,4-диметил-2(1,2,4-триазол -1-ил)-1-пентен-3-она, полученный в примере 4, растворили в ацетоне (500 мл) и изолировали при 45оС под воздействием источника ультрафиолетового излучения, снабженного 500 Вт ртутной лампой высокого давления. В ходе реакции время от времени отбирали следовые количества реакционного раствора и с помощью газовой хроматографии в тех же условиях, что и в примере 4, отделяли соотношение изомеров (II-В изомер) (II-А изомер). Получили следующие результаты:
Время, мин Отношение изомеров
(II-B/II-А) 0 100/0 20 10/90 60 6/94 120 6/94
Спустя 2,5 ч реакционный раствор перенесли в 500 мл колбу в форме баклажана и удалили ацетон при пониженном давлении, в результате чего получили 7,9 г кристаллов. Полученные кристаллы перекристаллизовали из четыреххлористого углерода, в результате чего получили 6,2 г (выход 78%) кристаллов (т. пл. 108-109оС). Это соединение растворили в ацетоне и хроматографировали в описанных ранее условиях, но при этом не наблюдали пика, соответствующего II-В изомеру.
П р и м е р 6. Синтез II-А изомера из смеси геометрических изомеров 1-(4-хлорфенил)-4,4-диметил-2(1,2,4-триазол-1-ил-1 -пентен-3-она) (соединение N 1'). Реакционную смесь (10 г), содержащую II-А и II-В изомеры в отношении 1 к 3, полученную в примере 4, облучали ультрафиолетовыми лучами в тех же самых условиях, что и в примере 5. После 1,5 ч с помощью газовой хроматографии снова измеряли соотношение II-А изомера и II-В изомера, и было обнаружено, что это отношение составило около 19 к 1.
После выпаривания растворителя полученные кристаллы перекристаллизовывали из четыреххлористого углерода до получения 5,1 г II-А изомера.
П р и м е р 7. (А) Синтез 3-(4-хлорфенил)-1-(1-метил-циклопропил)-2)(1,2,4-триазол-1-ил) -2-пропен-1-она (соединение N 29') по способу С.
1-(1-метилциклопропил)-2-(1,2,4-триазол-1-ил)этан-1-он (10 г, 0,06 моля), безводный карбонат калия (8 г, 0,06 моля) и уксусный ангидрид (100 мл) смешали, и полученную смесь нагревали до 100оС в течение 6 ч при перемешивании. Образовавшийся в реакции осадок удалили фильтрованием, и полученный фильтрат сконцентрировали при пониженном давлении до получения маслянистого продукта. Этот маслянистый продукт экстрагировали хлороформом (300 мл) и полученный экстракт промыли водой, насыщенной бикарбонатом натрия (300 мл). Органический слой высушили над безводным сульфатом натрия и сконцентрировали при пониженном давлении. Одну каплю полученного остатка растворили в ацетоне, и ацетоновый раствор подвергли газовой хроматографии в условиях, описанных далее. Пик, соответствующий II-А изомеру, как было обнаружено, имеет время удерживания 250 с, а соответствующий пик II-В изомера характеризуется временем удерживании 300 с. Отношение обоих изомеров составило 19,1/63,5, т. е. около 1/3, что было рассчитано из процентных соотношений каждой из площадей.
Условия для газовой хроматографии были следующими:
Аппаратура: газовый хроматограф Ниппон Денши 20 К, снабженный детектором F1 D
Колонка: Стеклянная колонка длиной 1 м
Жидкая фаза: 5% ХЕ-60
Носитель: Хромосорб W
Температура (колонки): 181оС
Температура ввода: 240оС
Газ-носитель: газообразный азот, I кг/см2
Полученный остаток растворили в бензоле (100 мл). Полученный раствор пропустили через колонку, заполненную силикагелем (300 г) с размерами зерна 100-200 мешей, и хроматографирование провели смесью н-гексан/ацетон (10:1) в качестве проявляющего растворителя. Фракции, соответствующие каждому из изомеров, перекристаллизовали из четыреххлористого углерода до разделения двух указанных геометрических изомеров.
ЯМР-спектры каждого из изомеров приведены в табл.2.
II-А изомер: 1,7 (выход 10%)
II-В изомер: 6,7 г (выход 33%)
(В) Синтез исходного материала, 1-(1-метил-циклопропил)-2-(1,2,4-триазол-1-ил)-этан-1-она.
К смеси метил-1-метилциклопропилкетона (28 г, хорошо известное соединение, описанное в Bull. Soc. Chem. Эг. 1708(1960), хлората калия (5,8 г) и воды (70 мл) добавили 28 г брома при температуре от 40 до 50оС за 4 ч при интенсивном перемешивании, а затем реакционный раствор перемешивали в течение 2 ч при комнатной температуре. После этого, реакционный раствор экстрагировали двумя порциями эфира по 200 мл, а затем органический слой высушили над хлористым кальцием и сконцентрировали при пониженном давлении до получения 53 г неочищенного продукта. 1-(1-метилциклопропил)-1-бромэтан-1-она.
Смесь 1,2,4-триазола (18,3 г), безводного карбоната калия (37 г) и ацетонитрила (250 мл) кипятили с обратным холодильником в течение 1 ч, а затем охладили до 60оС. Неочищенный 1-(1-метилциклопропил)-2-бромэтан-1-он (53 г), полученный ранее, добавили к этой смеси за 2 ч, после чего перемешивание продолжали в течение ночи. Выпавшие из реакционного раствора остатки удалили фильтрованием, полученный фильтрат сконцентрировали при пониженном давлении. Полученный остаток экстрагировали, добавив 100 мл воды и 300 мл хлороформа, и органический слой высушили над безводным сульфатом магния и сконцентрировали при пониженном давлении. Полученный остаток экстрагировали, добавив 100 мл воды и 300 мл хлороформа, и органический слой высушили над безводным сульфатом магния и сконцентрировали при пониженном давлении. Полученный маслянистый остаток перекристаллизовали из 100 мл петролейного эфира и получили 27 г 1-(1-метилциклопропил)-2-(1,2,4-триазол-1-ил)-этан-1-она 3, выход 57% в расчете на метил-1-метилциклопропилкетон, т.пл. 57-60оС.
П р и м е р 8. Синтез II-А изомера 3-(4-хлорфенил)-1-(1-метилциклопропил)-2-(1,2,4-триазол-1-ил)-2-пропен-1-она из II-В изомера по способу С.
II-В изомер 3-(4-хлорфенил)-1-(1-метилциклопропил)-2-(1,2,4 -триазол-1-ил)-2-пропен-1-он (4 г), полученный в примере 7, растворили в 500 мл ацетона и провели изомеризацию при 45оС в течение 2 ч с помощью источника ультрафиолетового излучения, снабженного 500 Вт ртутной лампой высокого давления. Отношение II-А изомера к II-В изомеру измеряли с помощью газовой хроматографии таким же образом, как и в примере 7. Было найдено, что это отношение составило 81,2 к 18,1. Реакционный раствор сконцентрировали при пониженном давлении до получения 3,9 г кристаллов. Эти кристаллы перекристаллизовали из четыреххлористого углерода до получения 2,8 г (выход 70%) II-А изомера.
П р и м е р 9. Синтез II-А изомера 3-(4-хлорфенил)-1-(1-метилциклопропил)- 2(1,2,4-триазол-1-ил) -2-пропен-1-она из смеси геометрических изомеров.
Реакционную смесь (3 г), содержащую II-А и II-В изомеры, (II-А) II-В 1/3, полученную в примере 7, облучали ультрафиолетовыми лучами в течение 1,5 ч в тех же условиях, что и в примере 8. После этого измерили отношение II-А изомера к II-В изомеру с помощью газовой хроматографии. Было обнаружено, что это отношение изменилось с 1/3 до 7/3. После удаления растворителя выпариванием, полученные кристаллы перекристаллизовали из четыреххлористого углерода до получения 1,5 г II-А изомера.
П р и м е р 10. Синтез II-В изомера 1-(4-хлорфенил)-2-(1,2,4- триазол-1-ил)-1-гептен-3-она (соединение N 22) по способу D.
К смеси 2-гексанона (50 г) и метанола (300 мл) добавили 80 г брома при 0оС, и полученную смесь выдерживали при 10оС в течение 2 ч. К этому добавили 200 мл воды и 50 г концентрированной серной кислоты и после перемешивания в течение 16 ч добавили еще 500 мл воды. Реакционную смесь перенесли в разделительную колонку и экстрагировали 500 мл эфира. Органический слой промыли 5% -ным водным раствором карбоната калия и высушили над хлористым кальцием. Затем растворитель удалили при пониженном давлении до получения 89 г неочищенного 1-бром-2-гексанола в виде маслянистого продукта.
Смесь триазола (35 г), безводного карбоната калия (69 г) и ацетонитрила (300 мл) кипятили с обратным холодильником в течение 1 ч и оставили охлаждаться до 50оС. Неочищенный 1-бром-2-гексанон (89 г), полученный ранее, по каплям добавили к полученной смеси, которую затем перемешивали при комнатной температуре в течение 16 ч. Осадок в реакционном растворе удалили фильтрованием и растворитель удалили при пониженном давлении. К по- лученному таким образом остатку добавили 200 мл воды и 200 мл хлороформа и полученную смесь перенесли в делительную воронку, после чего эстрагировали. Органический слой высушили над безводным сульфатом магния, растворитель удалили при пониженном давлении до получения 77 г неочищенного 1-(1,2,4-триазолил)-2-гексанона в виде маслянистого продукта.
Полученный 1-(1,2,4-триазолил)-2-гексанон (20 г), безводный карбонат калия (20 г), пара-хлорбензальдегид (20 г) и уксусный ангидрид (200 мл) перемешивали и нагревали при 90оС в течение 5 ч. Затем реакционную смесь сконцентрировали при пониженном давлении, полученный остаток растворили в этилацетате (500 мл) и переместили в делительную воронку. Раствор этилацетата промыли водой, насыщенной карбонатом калия (200 мл), и выделили органический слой. Затем растворитель удалили из органического слоя при пониженном давлении, остаток ввели в воронку с силикагелем (0,5 кг силикагеля с размером 100-200 мешей) и провели хроматографирование, используя смесь н-гексан/ацетон (10: 1) в качестве проявляющего растворителя. Таким образом, получили 3,7 г II-В изомера (т.пл. 117-120оС) 1-(4-хлорфенил)-2-(1,2,4-триазол-1-ил)-1- гептен-3-она и 9 г 1-(4-хлорфенил)-2-(1,2,4-триазол-1-ил)-3-ацетокси-1,3 гептадиена (т.пл. 112-114оС).
К полученному 1-(4-хлорфенил)-2-(1,2,4-триазол-1-ил)-3-ацетокси-1,3-гептадиену (9 г) добавили концентрированную соляную кислоту (100 мл) и полученную смесь нагревали при 50оС в течение 2 ч, после чего ее вылили в ледяную воду (500 мл). Водный раствор нейтрализовали карбонатом калия и экстрагировали этилацетатом (300 мл). Органический слой высушили над безводным сульфатом магния, и растворитель удалили выпариванием. Кристаллический остаток перекристаллизовали из смеси четыреххлористый углерод (н-гексан(1: 1), в результате чего получили 6 г II-В изомера 1-(4-хлорфенил)-2-(1,2,4-триазол-1-ил)-1-гептан-3-она.
П р и м е р 11. Синтез I-А изомера 1-(2,4-дихлорфенил)-2- (1,2,4-триазол-1-ил)-4,4-диметил-1-пентен-3-ола (соединение N 2).
Первая стадия (конденсация) способ D.
Смесь α -(1,2,4-триазол-1-ил)-пинаколона (200 г) 2,4-дихлорбензальдегида (220 г) и уксусного ангидрида (700 см3) нагревали до 50оС, и к этому добавили 255 г триэтиламина. В течение 7 ч выдерживали температуру 70оС, после чего уксусный ангидрид удалили при пониженном давлении. К остатку добавили 3 л воды и образовавшиеся кристаллы отфильтровали, промыли водой и высушили. Полученный неочищенный продукт перекристаллизовали из этанола (600 см3) до получения 304 кг. II-В изомера 1-(2,4-дихлорфенил)-2-(1,2,4-триазол-1-ил)4,4-диме- тил-1-пентен -3-она (соединение N 2').
Вторая стадия (фотоизомеризация) способ С.
II-В изомер (300 г) соединения N 2', полученного на первой стадии, растворили в 2 л ацетона и подвергли изомеризации при 30оС в течение 26 ч под действием источника ультрафиолетового излучения, снабженного 500 Вт ртутной лампой высокого давления. Затем растворитель удалили при пониженном давлении до получения 300 г маслянистого продукта. С помощью газовой хроматографии было найдено, что этот продукт представляет собой смесь, состоящую на 75% из II-А изомера соединения N 2 и на 25% из II-В изомера того же соединения. Этот продукт передали на следующую стадию, не разделяя изомеры.
Третья стадия (восстановление).
300 г смеси геометрических изомеров соединения N 2', полученного из второй стадии, суспендировали в метаноле (1 кг), и к этому добавили боргидрид натрия (38 г) по частям, охлаждая реакционную смесь до 10оС. После перемешивания при комнатной температуре в течение 1 ч, реакционный раствор сконцентрировали при пониженном давлении. Полученный остаток экстрагировали, добавив 10%-ный водный раствор уксусной кислоты (2 л) и этилацетат (3 л). Выделенный органический слой промыли 5%-ным водным раствором карбоната калия (1 л) и высушили над безводным сульфатом магния (100 г). После удаления фильтрованием высушивающего агента, растворитель удалили при пониженном давлении, в результате чего получили 280 г неочищенного продукта в виде кристаллов. Этот продукт представлял собой смесь I-А и I-В изомеров соединения N 2 (отношение смеси I-А (I-В 75/25). Неочищенный продукт (280 г) перекристаллизовали из четыреххлористого углерода (600 см3), в результате чего получили 209 г указанного в заглавии соединения (I-А изомер соединения N 2). Маточный раствор от перекристаллизации сконцентрировали до половинного объема, в результате чего получили 25 г I-В изомера соединения N 2 в виде вторичных кристаллов.
II-А изомеры кетонного соединения (II), полученные способами С и D, приведены в табл. 2 наряду с II-В изомерами. ЯМР-спектры в табл.2 в таком же виде, что и в табл.1.
При практическом применении соединений изобретения полученных таким образом они могут использоваться сами по себе без других компонентов или в смеси с носителями для облегчения их применения в качестве фунгицидов, гербицидов и регуляторов роста растений. Обычно используемые препараты включают, например, дусты, смачиваемые порошки, масляные распылительные рецептуры, эмульгируемые концентраты, таблетки, гранулы, мелкие гранулы, аэрозоли и способные к истечению препараты.
Описанные выше препараты обычно содержат 01,-95,0 мас. активного ингредиента (включая другие ингредиенты смеси). Подходящее количество применяемого активного ингредиента обычно составляет 2-500 г на ар, а концентрация применяемого активного ингредиента предпочтительно лежит в интервале 0,001-1,0% Однако поскольку количество и концентрация зависят от формы препарата, времени применения, устройств для применения, заболеваний культур и их природы, эти значения могут быть уменьшены или увеличены относительно указанных интервалов.
При получении фунгицида, гербицида и регулятора роста растений изобретения примешиваются подходящие твердые носители или жидкие носители. В качестве примеров твердых носителей можно указать, например, неорганические соединения, например, глины, каолиновой группы, монтморилонитной группы и аттапульгитной группы, тальк, слюда, пирофиллит, пемза, вермикуллит, гипс, карбонат кальция, доломит, диатомовая земля, известь, апатит, цеолит, ангидрид кремневой кислоты, синтетический силикат кальция), растительные органические соединения, например порошкообразная соя, табак, порошкообразный грецкий орех, мука, порошкообразная древесина, крахмал, кристаллическая целлюлоза (синтетические или природные высокомолекулярные соединения), например петролейные смолы, аклидные смолы, поливинил хлорид, полиалкилен гликоль, кетонные смолы, эфирные смолы, копал, дамаровая смолы, а также воски (например карнаубский воск, пчелиный воск) и мочевина. отфильтровали, промыли водой и высушили. Полученный неочищенный продукт перекристаллизовали из этанола (600 см3) до получения 304 г. II-В изомера 1-(2,4-дихлорфенил)-2-(1,2,4-триазол-1-ил)4,4-диме- тил-1-пентен -3-она (соединение N 2').
Вторая стадия (фотоизомеризация) способ С.
II-В изомер (300 г) соединения N 2', полученного на первой стадии, растворили в 2 л ацетона и подвергли изомеризации при 30оС в течение 26 ч под действием источника ультрафиолетового излучения, снабженного 500 Вт ртутной лампой высокого давления. Затем растворитель удалили при пониженном давлении до получения 300 г маслянистого продукта. С помощью газовой хроматографии было найдено, что этот продукт представляет собой смесь, состоящую на 75% из II-А изомера соединения N 2 и на 25% из II-В изомера того же соединения. Этот продукт передали на следующую стадию, не разделяя изомеры.
Третья стадия (восстановление).
300 г смеси геометрических изомеров соединения N 2', полученного из второй стадии, суспендировали в метаноле (1 кг), и к этому добавили боргидрид натрия (38 г) по частям, охлаждая реакционную смесь до 10оС. После перемешивания при комнатной температуре в течение 1 ч, реакционный раствор сконцентрировали при пониженном давлении. Полученный остаток экстрагировали, добавив 10%-ный водный раствор уксусной кислоты (2 л) и этилацетат (3 л). Выделенный органический слой промыли 5%-ным водным раствором карбоната калия (1 л) и высушили над безводным сульфатом магния (100 г). После удаления фильтрованием высушивающего агента, растворитель удалили при пониженном давлении, в результате чего получили 280 г неочищенного продукта в виде кристаллов. Этот продукт представлял собой смесь I-А и I-В изомеров соединения N 2 (отношение смеси I-А (I-В 75/25). Неочищенный продукт (280 г) перекристаллизовали из четыреххлористого углерода (600 см3), в результате чего получили 209 г указанного в заглавии соединения (I-А изомер соединения N 2). Маточный раствор от перекристаллизации сконцентрировали до половинного объема, в результате чего получили 25 г I-В изомера соединения N 2 в виде вторичных кристаллов.
II-А изомеры кетонного соединения (II), полученные способами С и D, приведены в табл. 2 наряду с II-В изомерами. ЯМР-спектры в табл.2 в таком же виде, что и в табл.1.
При практическом применении соединений изобретения, полученных таким образом, они могут использоваться сами по себе без других компонентов или в смеси с носителями для облегчения их применения в качестве фунгицидов, гербицидов и регуляторов роста растений. Обычно используемые препараты включают, например, дусты, смачиваемые порошки, масляные распылительные рецептуры, эмульгируемые концентраты, таблетки, гранулы, мелкие гранулы, аэрозоли и способные к истечению препараты.
Описанные выше препараты обычно содержат 0,1-95,0 мас. активного ингредиента (включая другие ингредиенты смеси). Подходящее количество применяемого активного ингредиента обычно составляет 2-500 г на ар, а концентрация применяемого активного ингредиента предпочтительно лежит в интервале 0,001-1,0% Однако поскольку количество и концентрация зависят от формы препарата, времени применения, устройств для применения, заболеваний культур и их природы, эти значения могут быть уменьшены или увеличены относительно указанных интервалов.
При получении фунгицида, гербицида и регулятора роста растения изобретения примешиваются подходящие твердые носители или жидкие носители. В качестве примеров твердых носителей можно указать, например, неорганические соединения, например, глины каолиновой группы, монтморилонитной группы и аттапульгитной группы, тальк, слюда, пирофиллит, пемза, вермикуллит, гипс, карбонат кальция, доломит, диатомовая земля, известь, апатит, цеолит, ангидрид кремневой кислоты, синтетический силикат кальция), растительные органические соединения, например порошкообразная соя, табак, порошкообразный грецкий орех, мука, порошкообразная древесина, крахмал, кристаллическая целлюлоза (синтетические или природные высокомолекулярные соединения), например петролейные смолы, аклидные смолы, поливинил хлорид, полиалкилен гликоль, кетонные смолы, эфирные смолы, копал, дамаровая смолы, а также воски (например карнаубский воск, пчелиный воск) и мочевина.
В качестве примеров жидких носителей можно привести парафиновые или нафтеновые углеводороды (например керосин, минеральное масло, веретенное масло, белое масло), ароматические углеводороды (например бензол, толуол, ксилол, этилбензол, кумол, метилнафталин), галогенированные углеводороды (например четыреххлористый углерод, хлороформ, трихлорэтилен, монохлорбензол), о-хлортолуолэфиры (например диоксан, тетрагидрофурн), кетоны (например ацетон, метил, этил, кетон, диизобутил, кетон, циклогексан, ацетофенон, изофорон), сложные эфиры (например этил ацетат, амил ацетат, ацетат этилен гликоля, ацетат диэтилен гликоля, дибутил малеат, диэтил сукцинат), спирты (например метанол н-гексанол, этилен гликоль, диэтилен гликоль, циклогексанол, бензиловый спирт), простые эфиры спиртов (например этиловый эфир, этилен гликоля, фениловый эфир этилен гликоля, этиловый эфир этилен гликоля, этиловый эфир диэтилен гликоля, этиловый эфир диэтилен гликоля, бутиловый эфир диэтилен гликоля), полярные растворители (например диметилформамид, диметилсульфоксид и вода).
Могут использоваться поверхностно-активные агенты, применяемые для эмульгирования, диспергирования, смачивания, распределения по поверхности, связывания, регулирования дизинтеграции, стабилизации активного ингредиента, улучшения текучести и антикоррозитности, относящиеся к неионному, анионному, катионному и амфотерному типу, но обычно используют неионные и/или анионные агенты. В качестве подходящих неионных поверхностно-активных агентов могут использоваться соединения, полученные полимеризацией окиси этилена и высшего спирта (например лаурилового спирта, стеарилового спирта, олеилового спирта), окиси этилена и алкилфенола (например изооксилфенола, нонилфенола), окиси этилена и алкилнафтола (например бутилнафтола, октилнафтола) окиси этилена и высшей жирной кислоты (например пальмитиновой кислоты, стеариновой кислоты, олеиновой кислоты), окиси этилена и моно- или ди-алкил фосфата (например стеарил фосфата, дилаурил фосфата) или окиси этилена и амина (например додециламина, амида стеариновой кислоты), эфиры высших жирных кислот и полиатомного спирта (например сорбита), а также соединения, полученные полимеризацией указанных эфиров и окиси этилена, а также смешанные полимеры на основе окиси этилена и окиси пропилена.
Подходящими анионными поверхностно-активными агентами могут служить соли алкил сульфата (например натрий лаурил сульфат, аминовые соли олеил сульфата), алкилсульфонаты (например натриевая соль диоксил сульфосукцината, натрий 2-этилгексенсульфонат), а также арилсульфонаты (например натрий изопропилнафталинсульфонат, натрий изопропилнафталинсульфонат, натрий метиленбиснафталинсульфонат, натрий лигносульфонат, натрий додецилбензолсульфонат).
Кроме этого, препараты изобретения могут содержать высокомолекулярные соединения и другие вспомогательные агенты с целью усиления технологических характеристик и биологической активности. Такие высокомолекулярные соединения включают, например казеин, желатин, альбумин, клей, альгинат натрия, карбоксилметил целлюлозу, метил целлюлозу, гидроксиэтил целлюлозу и поливиниловый спирт.
Указанные выше носители и вспомогательные агенты обычно применяются сами по себе или в комбинации в соответствии с предлагаемым использованием, принимая во внимание формы препарата и технику применения.
Содержание активного ингредиента в дустах обычно составляет 1-25 мас. а остаток составляет твердый носитель.
Что касается смачиваемых порошков, то содержание в них активного ингредиента обычно составляет 25-90 мас. Остаток представляет собой твердый носитель и дисперсионно-смачивающий агент и, если необходимо, защитный коллоид, тиксотропный агент и антипенный агент добавляют в композицию.
В гранулах содержание активного ингредиента обычно составляет 1-35 мас. и большую часть остатка составляет твердый носитель. Активный ингредиент однородно смешивают с твердым носителем или его однородно фиксируют или адсорбируют на поверхности твердого носителя. Такие частицы имеют диаметр 0,2-1,5 мм.
Что касается эмульгируемых концентратов, то содержание активного ингредиента в нем составляет обычно 5-30 мас. причем эмульгатор занимает 5-20 мас. а остаток представляет собой жидкий носитель. Если необходимо, добавляют противоплесневые агенты.
Кроме этого, соединения изобретения могут использоваться в смеси с другими фунгицидами, гербицидами и регуляторами роста растений без уменьшения регулирующего эффекта каждого активного ингредиента смеси. В качестве фунгицидов могут использоваться N-(3,5-дихлофенил)-1,2-диметилциклопропан-1,2-дикарбоксимид, S-н-бутил S-п-трет.-бутилбензилдитиокарбонимидат, 0,0-диметил, 0-(2,6-дихлор-4-метилфенил) фосфортиоат, метил-1-бутилкарбамоил-1Н-бензимидазол -2-ил-карбамат, N-трихлорметилтио-4-циклогексен-1,2-дикарбоксимид, цис-N-(1,1,2,2-тетрахлорэтилтио)-4-циклогексен-1,2-дикарбоксиимид полиоксин, стрептомицин, цинк этилен-бис(дитиокарбамат), цинк диметил-тискарбамат, марганец этилен-бис(дитиокарбамат), бис (N, N-диметилтиокарбамоил (дисульфид, тетрахлоризофталонитрил, 8-гидроксихинолин, додецилгуанидин ацетат, 5,6-дигидро-2-метил-1,4-оксатиин-3-карбоксанилид. N'-дихлорфторметилтио-N', N'-диметил-N'-фенилсульфамил, 1-(4-хлорфенокси)-3,3-диметил-1-(1,2,4-триазол-1-ил)-2-бутанон, 1,2-бис(3-метоксикарбонил-2-тиоуреидо), бензол, метил N-(2,6-диметилфенил)-N-метоксиацетил-2-метилглюцинат, алюминий этилфосфит и т.д.
В качестве гербицидов могут использоваться также гербициды фенокси ряда, как 2,4-дихлорфеноксиуксусная кислота, 2-метил-4-хлорфеноксиуксусная кислота, 2-метил-4-хлорфеноксимасляная кислота и 2-метил-4-хлорфеноксиуксусная кислота (включая сложные эфиры и соли), гербициды серии дифениловых эфиров, такие как 2,4-дихлорфенил-4-нитрофениловый эфир, 2,4,6-трихлорфенил 4'-нитрофениловый эфир, 2-хлор-4-трифторметилфенил, 3'-этокси-4'-нитрофениловый эфир, 2,4-дихлорфенил, 4'-нитро-3'-метоксифениловый эфир и 2,4-дихлорфенил 3'-метоксикарбонил-4'-нитрофениловый эфир, гербициды триазиновой серии, такие как 2-хлор-4,6-бисэтиламино-1,3, 5-триазин-2-хлор-4-этиламино-6-изопропиламино-1,3,5-три- азин, 2-метилтио-4,6-бисэтиламино-1,3,5-триазин и 2-метилтио-4,6-бисизопропиламино-1,3,5-триазин, гербициды мочевинной серии, такие как 3-(3,4-дихлорфенил)-1,1-диметилмочевина, 3-(3,4-дихлорфенил)-1-метокси-1-метилмочевина-1-(а,а- диметилбензил)-3-п-толилмочевина и 1-(2-бензотиазолил)-1,3-диметилмочевина, гербициды карбаматной серии такие как изопропил N-(3-хлорфенил)-карбамат и метил N-(3,4-дихлорфенил)карбамат, гербициды тиолкарбаматной серии такие как S-(4-хлорфензил)N,N-диэтилтиокарбамат и S-этил N, N-гексаметилен тиолкарбамат, гербициды анилидной серии, такие как 3,4-дихлорпропионааилид, 2-хлор-N-метоксиметил-2', 6'-диэтилацетанилид, 2-хлор-2', 6'-диэтил-N-(бутоксиметил)ацетанилид, 2-хлор-2', 6'-диэтил-N-(н-пропоксиэтил)аце- танилид и N-хлорацетил-N-(2,6-диэтилфенил)глицин этиловый эфир, гербициды урацильной серии, такие как 5-бром 3-фтор.бутил-6-метилурацил и 3-циклогексил-5,6-триметиленуарацил гербициды ряда пиридиновых солей, такие как 1,1'-диметил-4,4'-бипиридиний хлорид, фосфорсодержащие гербициды, такие как N-(фосфонометил)глицин, N,N-бис(фосфонометил)глицин, 0-этил 0-(2-нитро-5-метилфенил) N-втор. бутил фосфороамидотиоат S-(2-метил-1-пиперидилкарбонилметил) 0,0-ди-н-пропилдимиофосфат и S-(2-метил-1-пиперидилкарбонилметил)0,0'-дифенилдитиофосфат, гербициды толуидиновой серии, такие как а,а,а-трифтор-2,6-динитро-N,N-дипропил-п-толуидин, 5-трет.бутил-3(2,4-дихлор-5-изопропоксифенил)1,3,4- оксадиазолин-2-он, 3-изопропил-1Н (-2,1,3-бензотиадиазин (3Н)-он-2,2-диоксид, а-β-нафтокси(пропионанилид, 4-(2,4-дихлор бензоил)-1,3-диметилпиразол-5-ил п-толуолсульфонат, 3-(метоксикарбониламино)фенил-3-метилфенилкарбамат, 4-амино-3-метил-6-фенил-1,2,4-триазин и т.д.
Соединения изобретения могут применяться в смеси с другими инсектицидами, что не понижает регулирующего эффекта каждого активного ингредиента смеси. В качестве таких инсектицидов могут использоваться фосфорорганические инсектициды, такие как 0, 0-диметил-0-(4-нитро-3-метилфенил)фосфортиоат, 0-(4-цианофенил)-0,0-диметилфосфортиоат, 0-(4-цианофенил)0-этилфенилфосфоно- тиоат, 0,0-диметил-S-(N-метилкарбамоилметил) фосфородитиоат, 2-метокси-4Н-1,3,2-бензодиаксафосфорин-2-сульфид и 0,0-диметил S-(1-этоксикарбонил-1-фенилметил)фосфородитиоат, и такие инсектициды перетроидной серии, как а-циано-3-феноксибензил-2-(4-хлорфенил)изовалерат, 3-феноксибензил 2,2-диметил-3-(2,2-дихлорвинил)циклопропанкарбоксилат и а-циано-3-феноксибензил 2,2-диметил 3-(2,2-дибромвинил)циклоропанкарбоксилат. Следовательно, в одно и то же время могут контролироваться одно или более заболеваний и насекомых и, кроме этого, можно ожидать синергетического эффекта из-за смешивания.
Далее полезность соединений настоящего изобретения в качестве фунгицидов, гербицидов и регуляторов роста растений для сельского хозяйства и садоводства будет более подробно проиллюстрирована на представленных ниже испытательных примерах и примерах получения.
Испытательный пример 7.
50 г смешанной почвы, содержащей морской песок, горную почву и торф помещали в цветочный горшок с диаметром в 13 см и в нем и культивировали pot-mum (разн: Snow Ridge). Через две недели pot-mum прижимали и культивировали в 3-стебельчатой форме и выростали новые почки. Через две недели после прижимания водный разбавленный раствор, содержащий заранее определенную концентрацию каждого из испытуемых соединений применяли на pot-mum, а через 42 дня после применения пестицида, исследовали эффект регулирования роста растения. Полученные результаты представлены в табл.3.
Этот эффект оценивали следующим образом.
Увеличение высоты растения рассчитывали как разность между начальной высотой растения во время применения и высотой растений на 42 день после применения и выражали индексом высоты при соответствующей разности в необработанной чаше, принятой за сто. Значения, представленные в табл.3, представляют собой среднее из трех параллельных опытов.
В качестве контрольного соединения использовали В-9 (N,N-диметиламино янтарная кислота).
Испытательный пример 9.
Действие по контролю высоты соевых и ячменных растений. В 500 мл пластмассовую чашу загружали супесчанную почву. Почву из верхней половины чаши вынимали, хорошо перемешивали с 10 мл водного разбавленного раствора эмульгируемого концентрата каждого из испытуемых соединений и возвращали в чашу. После этого сою и ячмень высеивали на обработанной почве в количестве 3 семени на чашу и 5 семян на чашу соответственно.
Сою и ячмень культивировали в стеклянной камере при 25оС и после 14 дней измеряли высоту каждого растения. По- лученные результаты представлены в табл. 4. Численные значения в табл.4 означают среднюю высоту для двух соевых растений и для трех ячменных растений, которая выражена в процентах по отношению к средней высоте и необработанной чаше, принятой за 100.
В результате было установлено, что I-А изомер изобретения демонстрирует ярко выраженный сильный эффект по контролированию высоты растения по сравнению с изомером I-В, который представляет собой контрольное соединение. Кроме этого, при использовании как изомера I-А, так и I-В никакого фитотоксического действия, такого, например, как хлороз и некроз не наблюдалось и было обнаружено, что листва остается темно-зеленой.
Испытательный пример 10.
Контролирующее действие на рост междуузлия ячменя. Ямень (разн. Coseshikoku) высевали на почве 20-го ноября и на следующий год водный разбавленный раствор эмульгируемого концентрата I-А изомера соединения I распрыскивали на листве один раз (4-го апреля) и дважды (4-го и 24-го апреля).
Ячмень в каждой чаше созревал к 28-му мая и измеряли длину междоузлия 30 стеблей ячменя на чашу. Как показано в табл.5, длина четвертого и пятого междоузлий в обработанной чаше была значительно короче по сравнению с необработанной чашей и, кроме этого, общая длина в обработанной чаше была также укорочена.
Нигде не наблюдалось фитотоксичного действия такого, как пожелтение и стерильность.
Испытательный пример 11.
Эффект контролирования роста торфа.
Почвенную смесь, из горной почвы и торфа в соотношении 3:1 помещали в чашу Вагнера площадью 1/5000 ара и 6-го декабря в чашу трансплантировали зойсию (Zoysia metrella L.).
Зойсию культивировали в теплице при 30оС при постоянном внесении удобрений и передвижении зойсии до того момента, когда ее рост становился однородным. Сразу после передвижения зойсии 9-го мая водный разбавленный раствор эмульгируемого концентрата каждого из испытуемых соединений применяли с нормой расхода 10 мл/чашу с помощью ручного распылителя.
2-го июня измеряли увеличение высоты Зойсии с целью оценки контролирования роста, вызываемого испытуемым соединением. Этот эффект выражали индексом высоты при соответствующем увеличении высоты в необработанной чаше, принятом за 100. Полученные результаты представлены в табл.6. Из табл.6 видно, что соединения изобретения обладают действием, контролирующим рост зойсии.
Пример получения 1. Дусты.
1 мас.ч. изомера I-А каждого из соединений изобретения (1)-(52), 89 мас. ч. глины и 10 мас.ч. талька хорошо смешивали до порошкообразного состояния с получением дуста, содержащего 1% активного ингредиента.
Пример получения 2. Дуст.
3 мас.ч. I-А изомера каждого из соединений изобретения (1)-(52), 67 мас. ч. глины и 30 мас.ч. талька хорошо перемешивали до порошкообразного состояния с получением дуста, содержащего 3% активного ингредиента.
Пример получения 3. Смачиваемые порошки.
30 мас. ч. I-А изомера каждого из соединений изобретения (1)-(52), 45 мас.ч. диатомитовой земли, 20 мас.ч. белого угля, 3 мас.ч. смачивающего агента (натрий лаурилсульфат) и 2 мас.ч. диспергирующего агента (кальций лигносульфонат) хорошо перемешивали до порошкообразного состояния с образованием смачиваемого порошка содержащего 30% активного ингредиента.
Пример получения 4. Смачиваемый порошок.
50 мас. ч. I-А изомера каждого из соединений изобретения (1)-(52), 45 мас.ч. диатомитовой земли, 2,5 мас. смачивающего агента (кальций алкилбензолсульфонат) и 2,5 мас. диспергирующего агента (кальций лигносульфонат) хорошо смешивали друг с другом до порошкообразного состояния с образованием смачиваемого порошка, содержащего 50% активного ингредиента.
Пример получения 5. Эмульгируемый концентрат.
10 мас. ч. I-А изомера каждого из соединений изобретения (1)-(52), 80 мас. ч. циклогексанона и 10 мас.ч. эмульгатора (полиоксиэтилен алкилариловый эфир) смешивали с образованием эмульгируемого концентрата, содержащего 10% активного ингредиента.
Пример получения 6. Гранула.
5 мас.ч. I-А изомера каждого из соединений изобретения (1)-(52), 40 мас. бентонита, 50 мас.ч. глины и 5 мас.ч. натрий лигносульфоната хорошо смешивали до порошкообразного состояния. Полученную смесь хорошо перемешивали с водой, гранулировали и сушили с образованием гранулы.
Пример получения 7. Дуст.
2 мас. ч. II-А изомера каждого из соединений изобретения (1')-(33'), 88 мас. ч. глины и 10 мас.ч. талька хорошо смешивали до образования порошкообразного состояния с получением дуста, содержащего 2% активного ингредиента.
Пример получения 8. Дуст.
3 мас. ч. II-А изомера каждого из соединений изобретения (1')-(33'), 67 мас. ч. глины и 30 мас.ч. талька хорошо перемешивали до порошкообразного состояния с получением дуста, содержащего 3% активного ингредиента.
Пример получения 9. Смачиваемый порошок.
30 мас.ч. II-А изомера каждого из соединений изобретения (1')-(33'), 45 мас.ч. диатомитовой земли, 20 мас.ч. белого угля, 3 мас.ч. смачиваемого агента (натрий лаурилсульфат) и 2 мас.ч. диспергирующего агента (кальций лигносульфонат) хорошо смешивали до порошкообразного состояния с получением смачиваемого порошка, содержащего 30% активного ингредиента.
Пример получения 10. Смачиваемый порошок.
50 мас.ч. II-А изомера каждого из соединений изобретения (1')-(33'), 45 мас. ч. диатомитовой земли, 2,5 мас.ч. смачиваемого агента (кальций алкилбензол сульфонат) и 2,5 мас.ч. диспергирующего агента (кальций лигносульфонат) хорошо перемешивали друг с другом до образования порошкообразного состояния, с получением смачиваемого порошка, содержащего 50% активного инградиента.
Пример получения 11. Эмульгируемый концентрат.
10 мас.ч. II-А изомера, каждого из соединений изобpетения (1')-(33'), 80 мас. ч. циклогексанона и 10 мас.ч. эмульгатора (полиоксиэтилен алкилариловый эфир) смешивали друг с другом с образованием эмульгируемого концентрата, содержащего 10% активного ингредиента.
Пример получения 12. Гранула.
5 мас. ч. II-А изомера каждого из соединений изобретения (1')-(33'), 40 мас. ч. бентонита, 50 мас.ч. глины и 5 мас.ч. натрий лигносульфоната хорошо перемешивали друг с другом до порошкообразного состояния. Полученную смесь смешивали с водой, гранулировали и сушили с получением гранул, содержащих 5% активного инградиента.
Пример получения 13. Смачиваемый порошок.
80 мас. ч. I-А изомера каждого из соединений изобретения (1)-(52), 15 мас. ч. диатомитовой земли, 2,5 мас.ч. смачиваемого агента (кальций алкилбензолсульфонат) и 2,5 мас.ч. диспергирующего агента (кальций лигносульфонат) хорошо перемешивали друг с другом до порошкообразного состояния с получением смачиваемого порошка, содержащего 80% активного инградиента.
Испытательный пример 19.
Эксперименты проводили по той же методике, что описана в испытательном примере 7, используя эмульгируемый концентрат с концентрацией активного ингредиента 5% (пример смешивания N 1 из препаративного примера 19) смачиваемый порошок с концентрацией активного ингредиента 15% (пример смешивания N 3 из препаративного примера 17) (табл. 8) и смачиваемый порошок с концентрацией активного ингредиента 30% (пример смешивания N 8 из препаративного примера 17) (табл. 9).
В следующих ниже таблицах представлены полученные результаты, доказывающие положительный эффект заявленных в настоящее время соединений по сравнению со сравнительными соединениями, описанными в патенте Швейцарии N 587012.
Результаты испытательного примера 19 представлены в табл.7. Испытание на замедление развития куколки pot-mum в присутствии эмульгиреумых концентратов с концентрацией активного ингредиента 5% приготовленных согласно методике примера смешивания N 1 и препаративного примера 19.
Примеры получения 14-20.
Пример получения 14. Дуст.
1 мас.ч. I-А изомера каждого из настоящих соединений (1)-(56), 2 мас.ч. белой сажи (углерода) (Si, O2 и H2O) и 97 мас.ч. глины хорошо смешивают при измельчении в порошок с получением дуста, содержащего 1% активного ингредиента.
Пример получения 15. Дуст.
5 мас.ч. I-А изомера каждого из настоящих соединений (1)-(56), 1 мас.ч. белой сажи (SiO2 и H2O) и 94 мас.ч. глины хорошо перемешивают при измельчении в порошок с получением дуста, содержащего 5% активного ингредиента.
Пример получения 16. Дуст
1 мас. ч. II-А изомера каждого из соединений изобретения (1')-(33') смешивают аналогичным образом как описано в примере получения 14 с получением дуста, содержащего 1% активного ингредиента.
Пример получения 17. Смачиваемый порошок.
1-й изомер каждого из настоящих соединений (1)-(56), смачивающий агент, диспергирующий агент и неорганический носитель хорошо смешивают с получением смачиваемого порошка, содержащего каждый активный ингредиент.
Применяемые смачивающие агенты: высшие спирты, образованные из полиоксиэтилена (например лауриловый спирт, стеариловый спирт), алкилфенолы, образованные из полиоксиэтилена (например изооктилфенон, нонилфенол), полиоксиэтилен образованные алкилнафтолы (например бутилнафтол, октилнафтол), высшие жирные кислоты, образованные из полиоксиэтилена (например пальтилиновая кислота, стеариновая кислота, олеиновая кислота), эфиры высших жирных кислот и сорбитан, эфиры высших жирных кислот, образованные из полиоксиэтилена, и сорбитан алкилсульфаты (например лаурилсульфонат натрия, олеилсульфонат натрия), алкилсульфонаты (например натриевая соль диоктилсульфосукцината, 2-этилгексенсульфонат натрия), арилсульфонаты (например изопропилнафталин сульфонат натрия, додецилбензолсульфонаты натрия) и их смеси.
Применяемые диспергирующие агенты: нафталинсульфонат натрия, продукт конденсации нафталинсульфоната натрия и формалина, литносульфоната натрия и лигносульфоната кальция.
Применяемые неорганические носители, диатомарная земля, натуральная глина (например каолинит, монтмориллониты, аттапульгиты, тальк, слюда), белая сажа (SIO2 и H2O) и их смесь.
Соотношения, при которых смешивают активный ингредиент, смачивающий агент, диспергирующий агент и неорганический носитель в соответствующих примерах смешения приведены в табл.10.
Пример получения 18. Смачиваемый порошок.
II-А изомер каждого из настоящих соединений (1')-(33') смешивают также, как описано в примере получения 17, с получением смачиваемого порошка, содержащего каждый активный ингредиент.
Пример получения 19. Эмульгируемый концентрат.
I-А изомер каждого из настоящих соединений (1)-(56), эмульгатор и растворитель смешивают с получением эмульгируемого концентрата, содержащего каждый активный ингредиент.
Применяемые эмульгаторы включают высшие спирты, образованные поликосиэтиленом (например лауриловый спирт, стеариловый спирт), алкилфенолы, образованные полиоксиэтиленом (например изооктилфенол, нонилфенол), полиоксиэтилен образованные алкилнафтолы (например бутилнафтол, октилнафтол), полиоксиэтилен образованные высшие жирные кислоты (например полмитиновая кислота, стеариновая кислота, олеиновая кислота), сложные эфиры высших жирных кислот и сорбитан, образованные полиоксиэтиленом сложные эфиры высших жирных кислот и сорбитан, алкилсульфаты (например лаурилсульфат натрия, олеилсульфат натрия), алкилсульфонаты (например натриевая соль диоктилсульфосукцината, 2-этилгексенсульфонат натрия), арилсульфонаты (например изопропилнафталин сульфонат натрия, додецилбензолсульфонат натрия) и их смеси.
Применяемые растворители включают насыщенные углеводороды (например керосин, минеральное масло, веретенное масло), ароматические углеводороды (например толуол, ксилол, этилбензол, метилнафталин), хлорированные углеводороды (например трихлорэтилен монохлорбензол, монохлортолуол, дихлорбензол), кетоны (например метилэтиловый кетон, циклогексанон, ацетофенон, изофорон), сложные эфиры (этиленгликольацетат, диэтиленгликольацетат), диметилформамид, диметилсульфоксид и их смеси.
Соотношения при смешении активного ингредиента, эмульгатора и растворителя в соответствующих примерах смешения приведены в табл.11.
Пример получения 20. Эмульгируемый концентрат.
II-А изомер каждого из настоящих соединений (1')-(33') смачивают также как описывают в примере получения 19 с получением эмульгируемого концентрата, содержащего каждый активный ингредиент.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1-ФЕНИЛ-2-(1,2,4-ТРИАЗОЛ-1-ИЛ) ПРОПЕНА E-ИЗОМЕРА И ФУНГИЦИДНАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1980 |
|
RU2039049C1 |
| РОСТРЕГУЛИРУЮЩАЯ КОМПОЗИЦИЯ | 1989 |
|
RU2017423C1 |
| ИНСЕКТИЦИДНАЯ КОМПОЗИЦИЯ ДЛЯ БОРЬБЫ С HELIOTHIS ARMIGERA И СПОСОБ УНИЧТОЖЕНИЯ HELIOTHIS ARMIGERA | 1992 |
|
RU2054870C1 |
| КОМПОЗИЦИЯ ДЛЯ ДЕЗИНФЕКЦИИ СЕМЯН | 1987 |
|
RU2011347C1 |
| ИМИНОТИАЗОЛИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ГЕРБИЦИДНАЯ КОМПОЗИЦИЯ И СПОСОБ ПОДАВЛЕНИЯ НЕЖЕЛАТЕЛЬНЫХ СОРНЯКОВ | 1992 |
|
RU2050356C1 |
| Способ получения (+) или (-) производных триазолилового спирта | 1981 |
|
SU1582987A3 |
| ЦИКЛОГЕКСАНОВЫЕ ПРОИЗВОДНЫЕ И ГЕРБИЦИДНАЯ КОМПОЗИЦИЯ | 1987 |
|
RU2054255C1 |
| Композиция для дезинфекции семян | 1987 |
|
SU1757437A3 |
| СЛОЖНОЭФИРНОЕ СОЕДИНЕНИЕ, КОМПОЗИЦИЯ ДЛЯ УНИЧТОЖЕНИЯ ВРЕДНЫХ НАСЕКОМЫХ НА ЕГО ОСНОВЕ, СПОСОБ УНИЧТОЖЕНИЯ ВРЕДНЫХ НАСЕКОМЫХ | 1998 |
|
RU2205182C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2036195C1 |
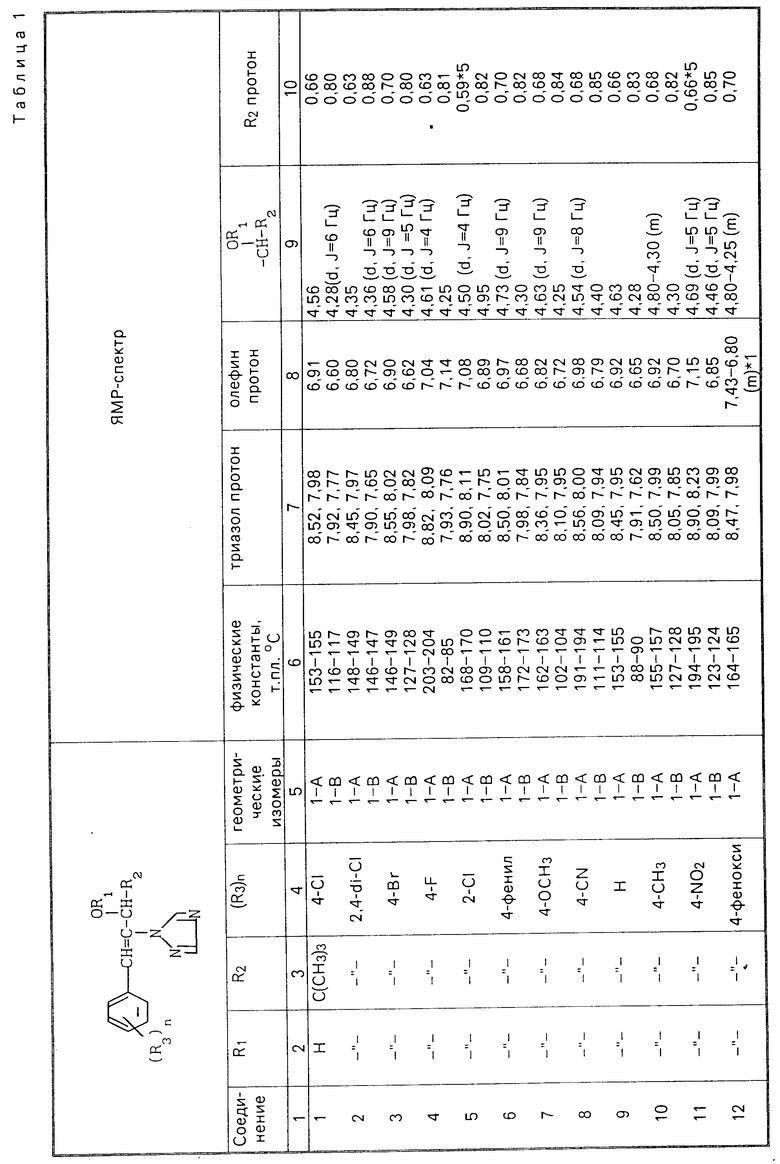
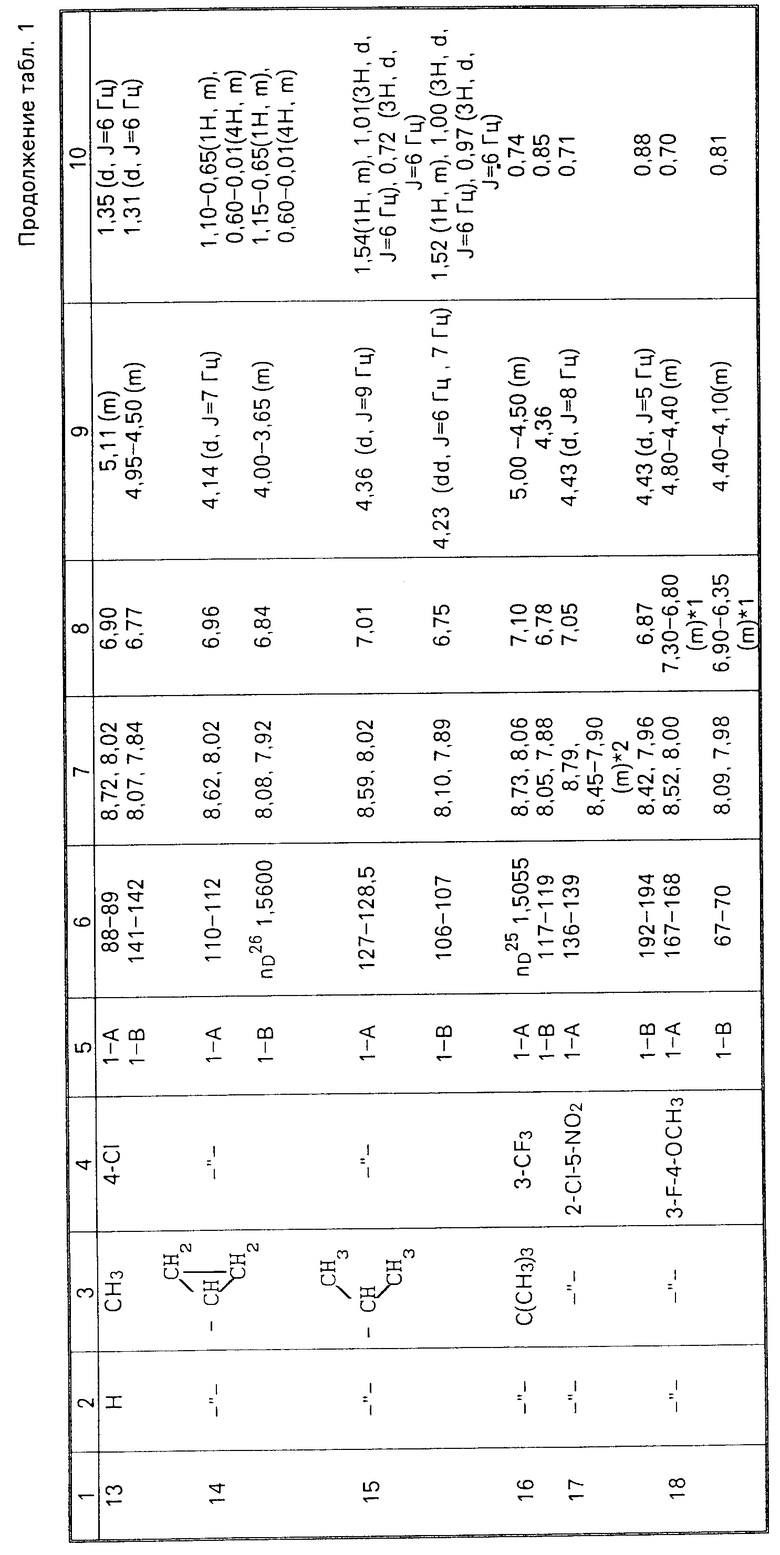
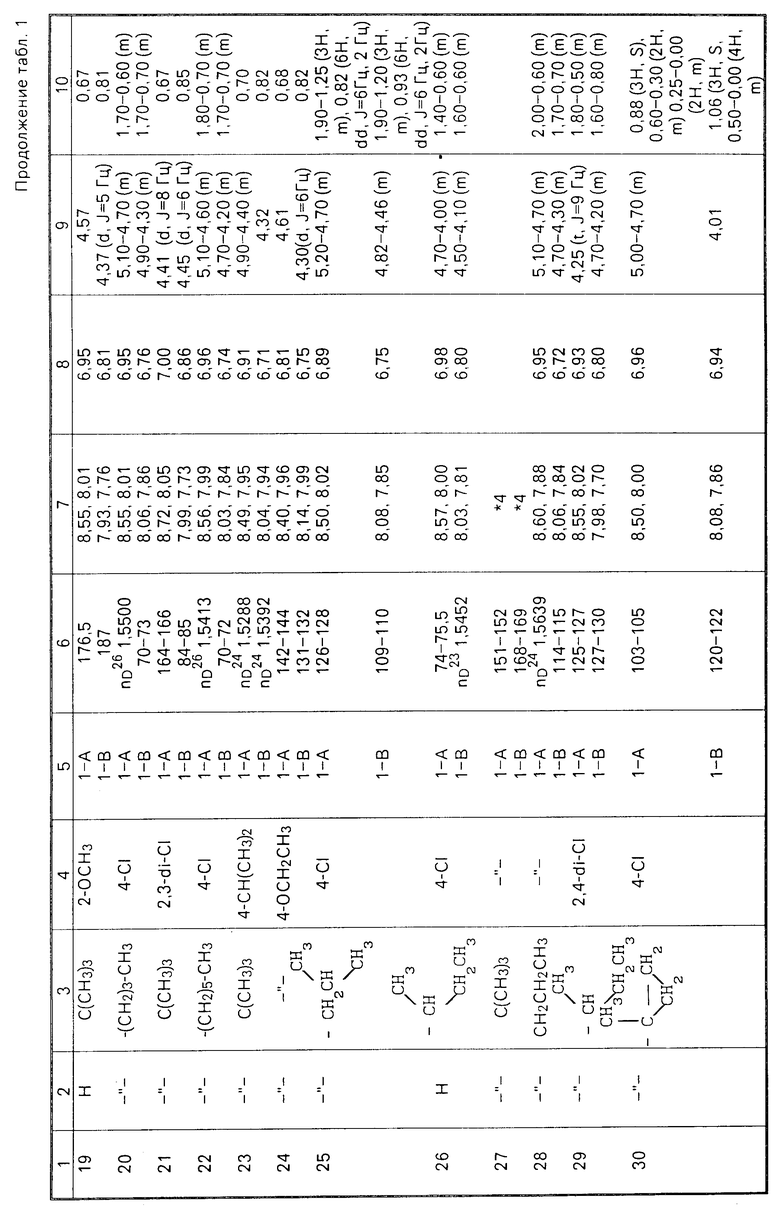
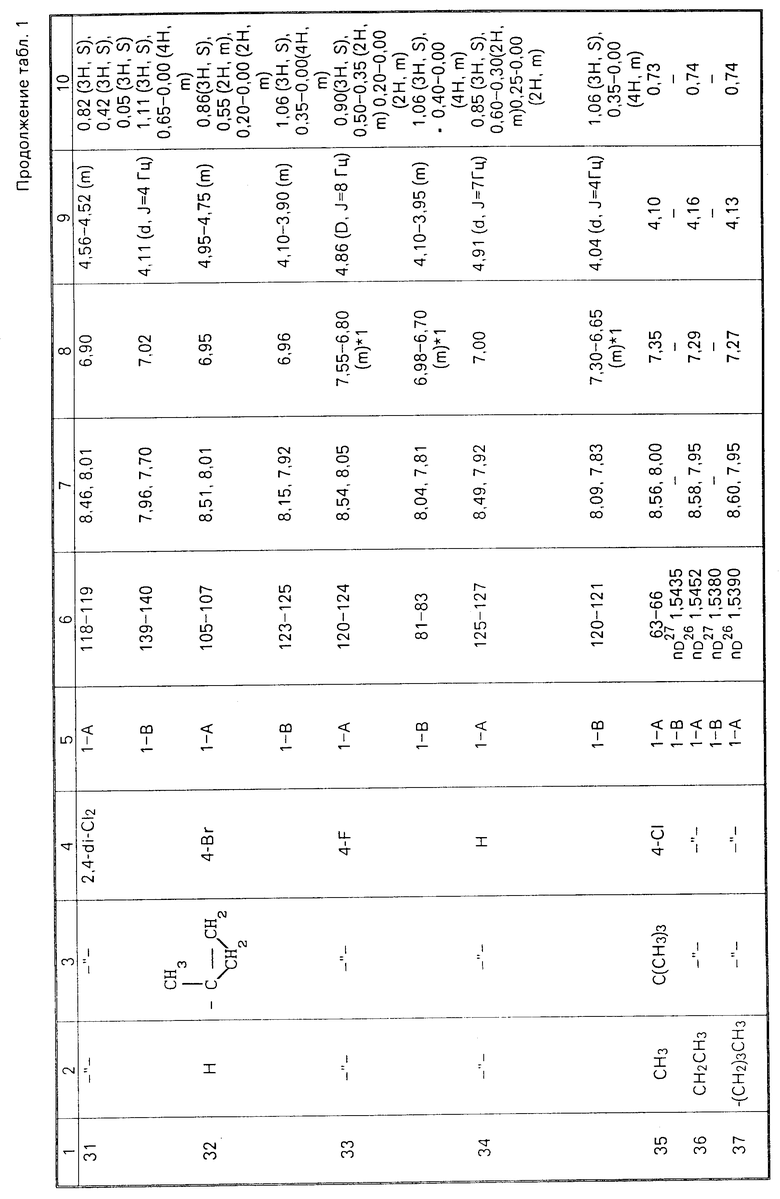
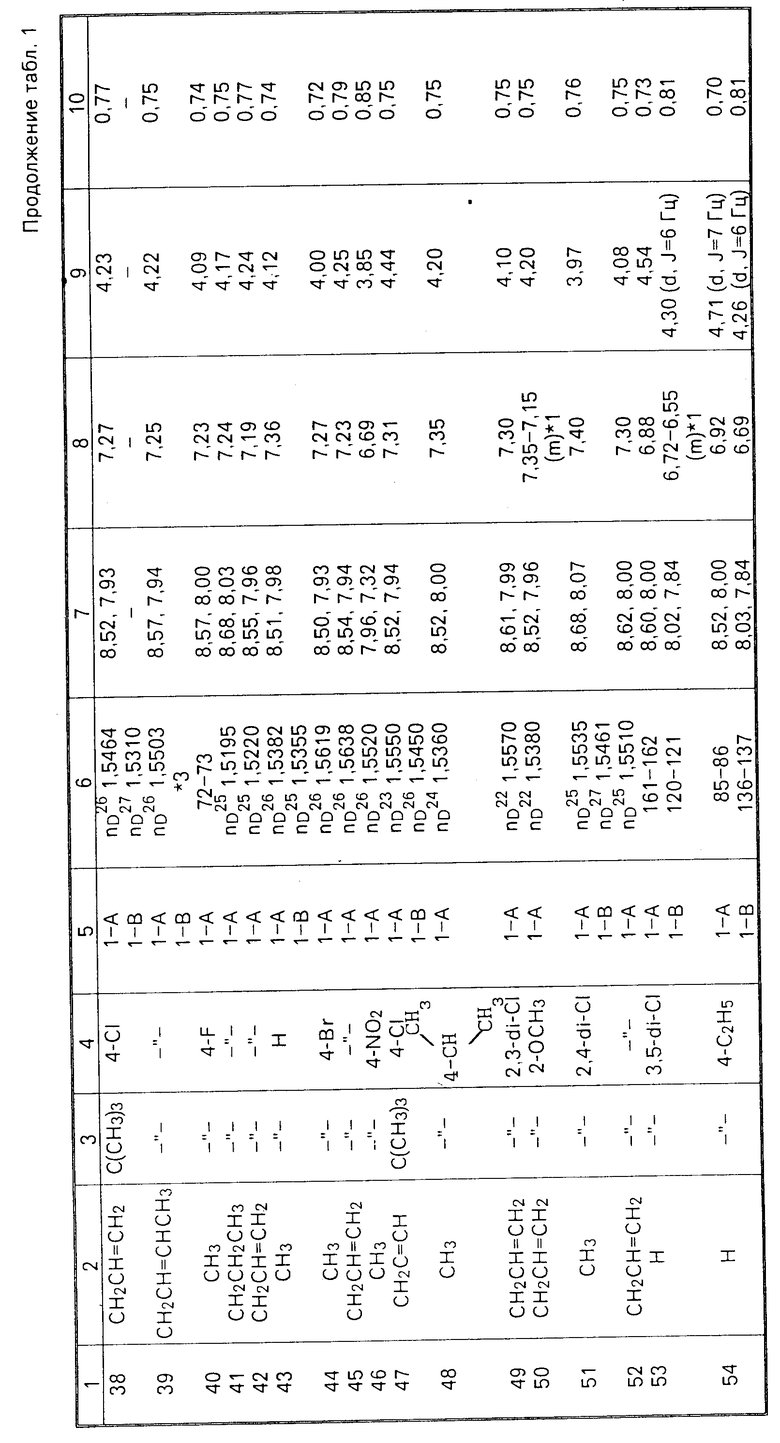
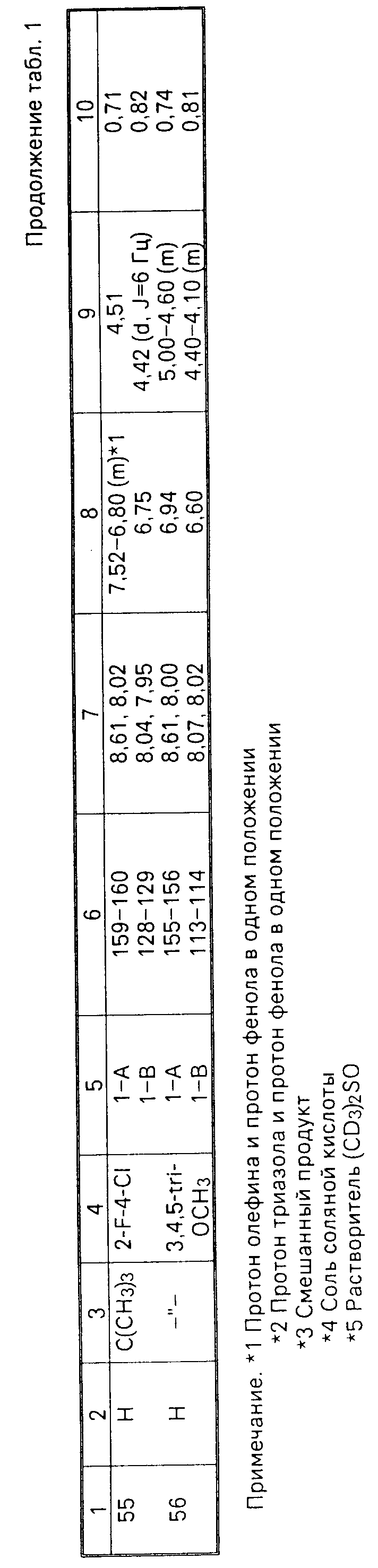
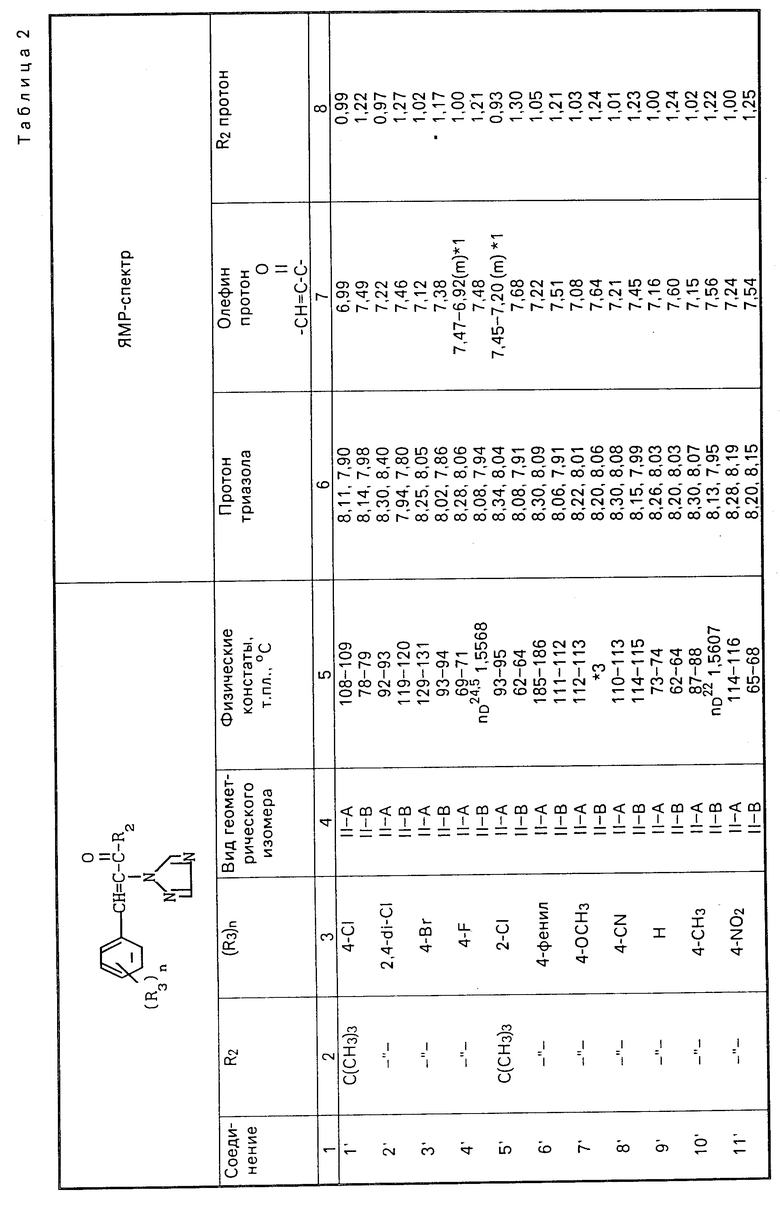
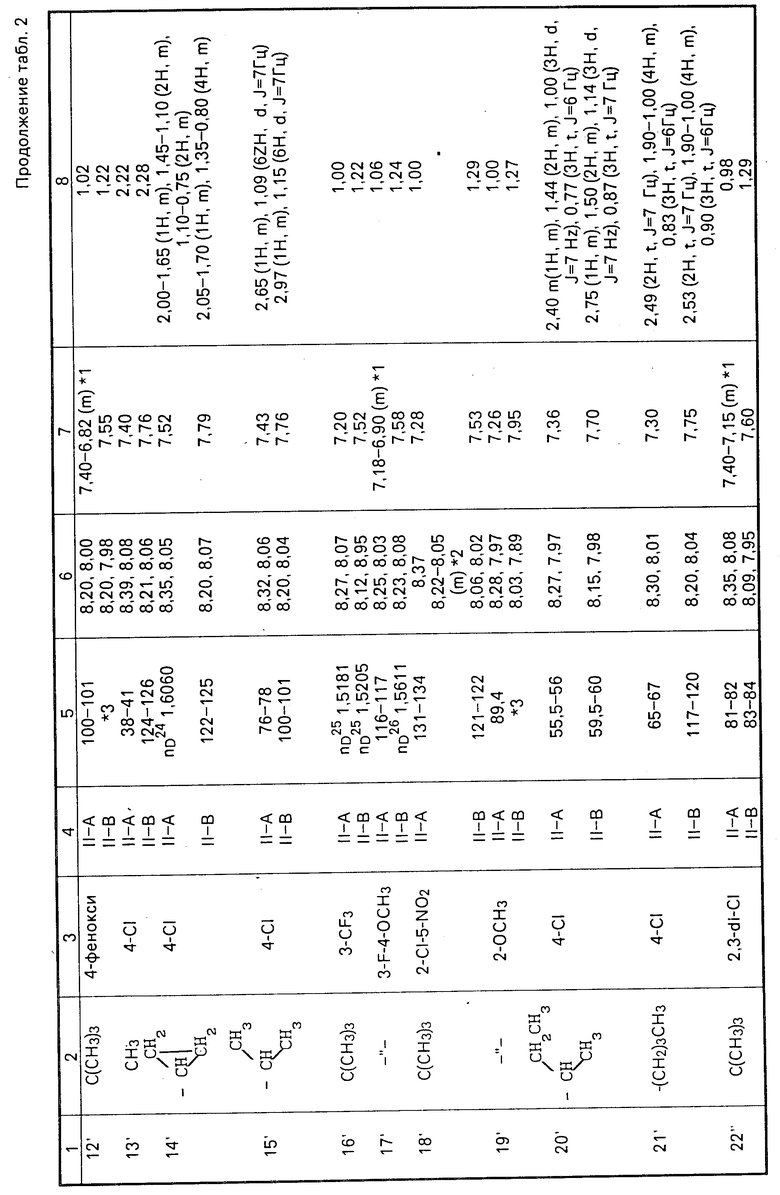
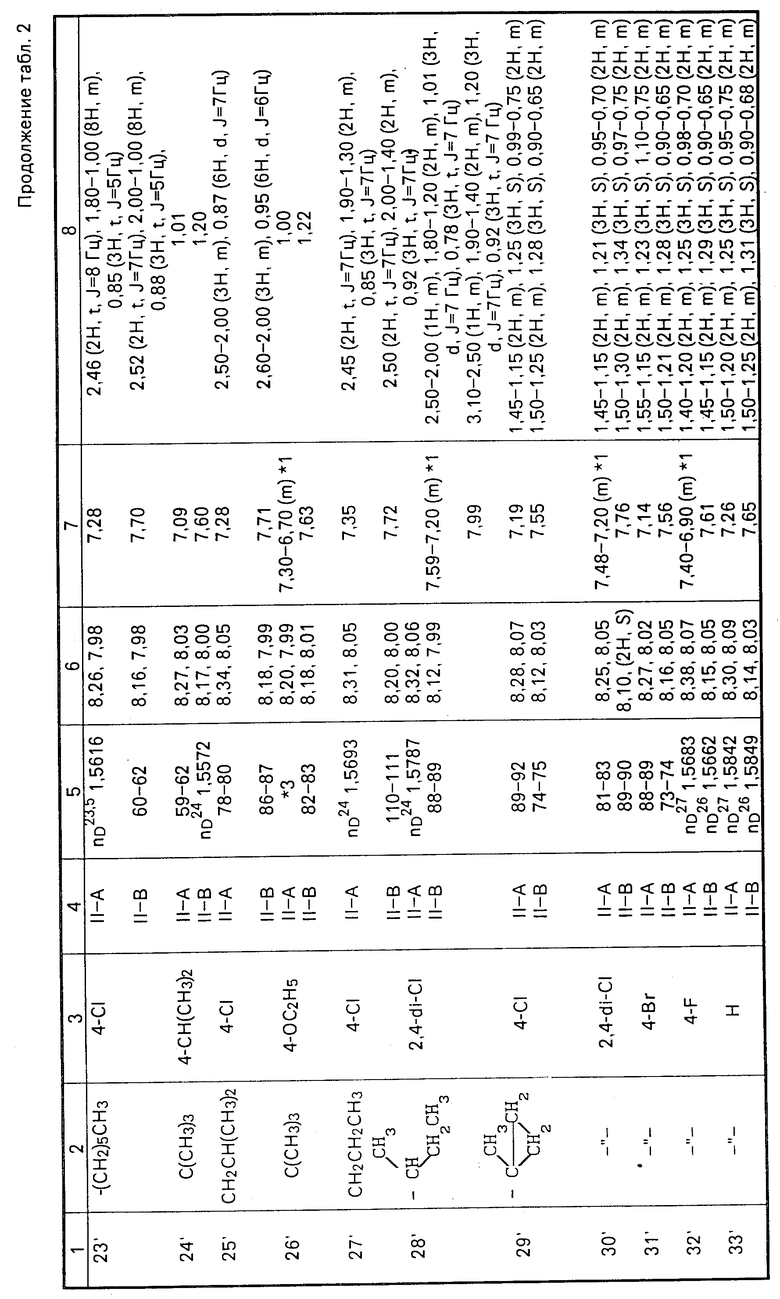
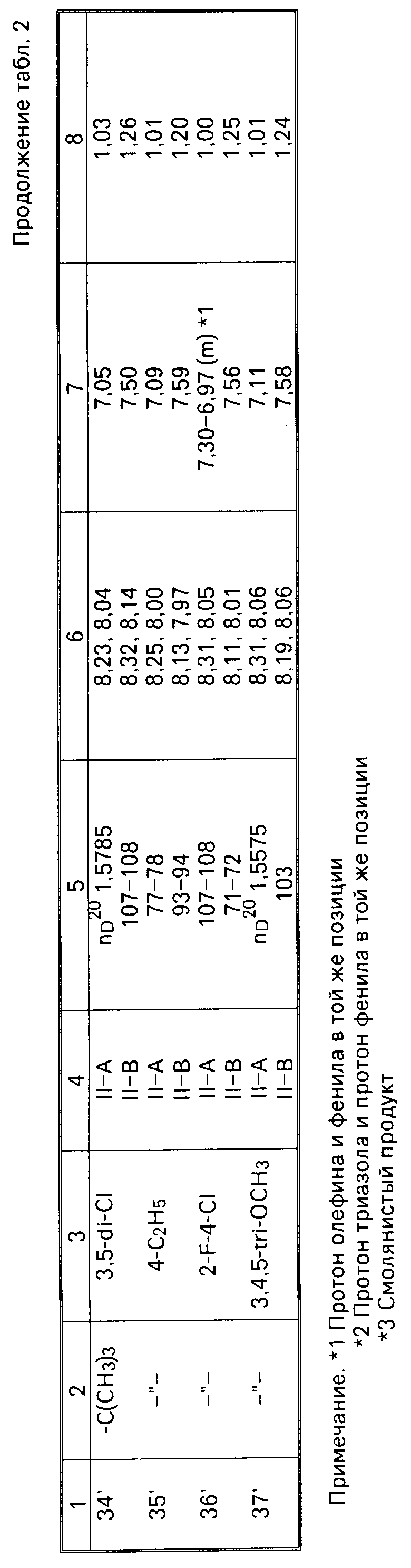
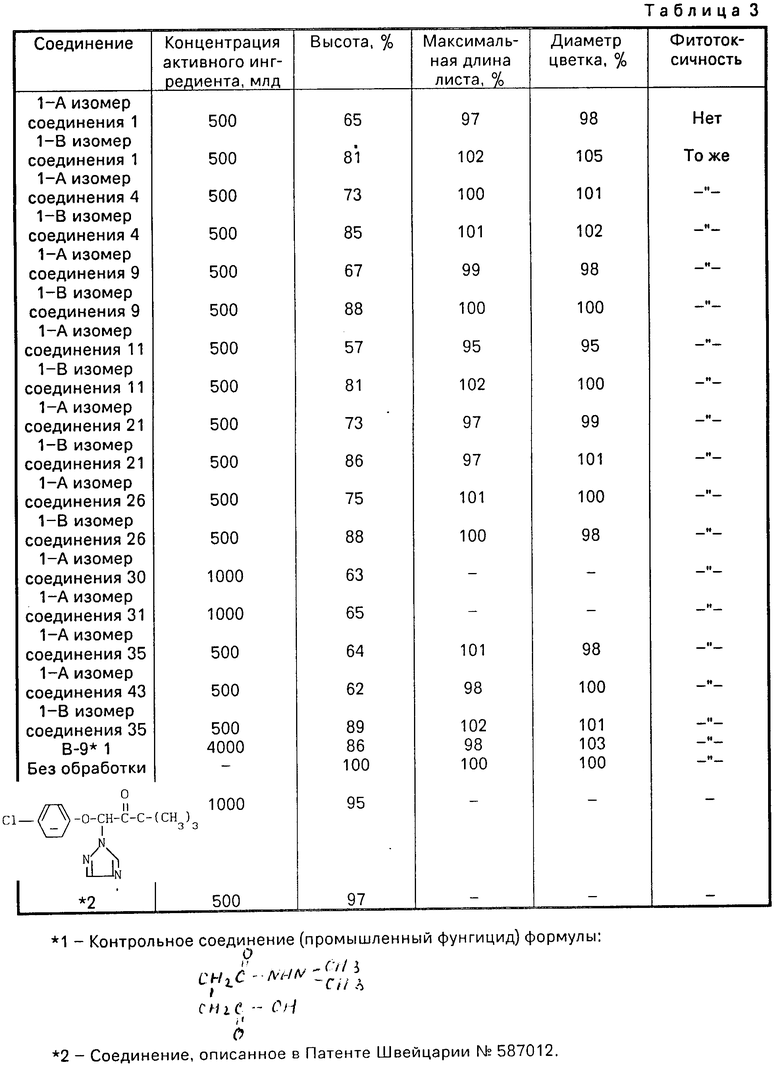
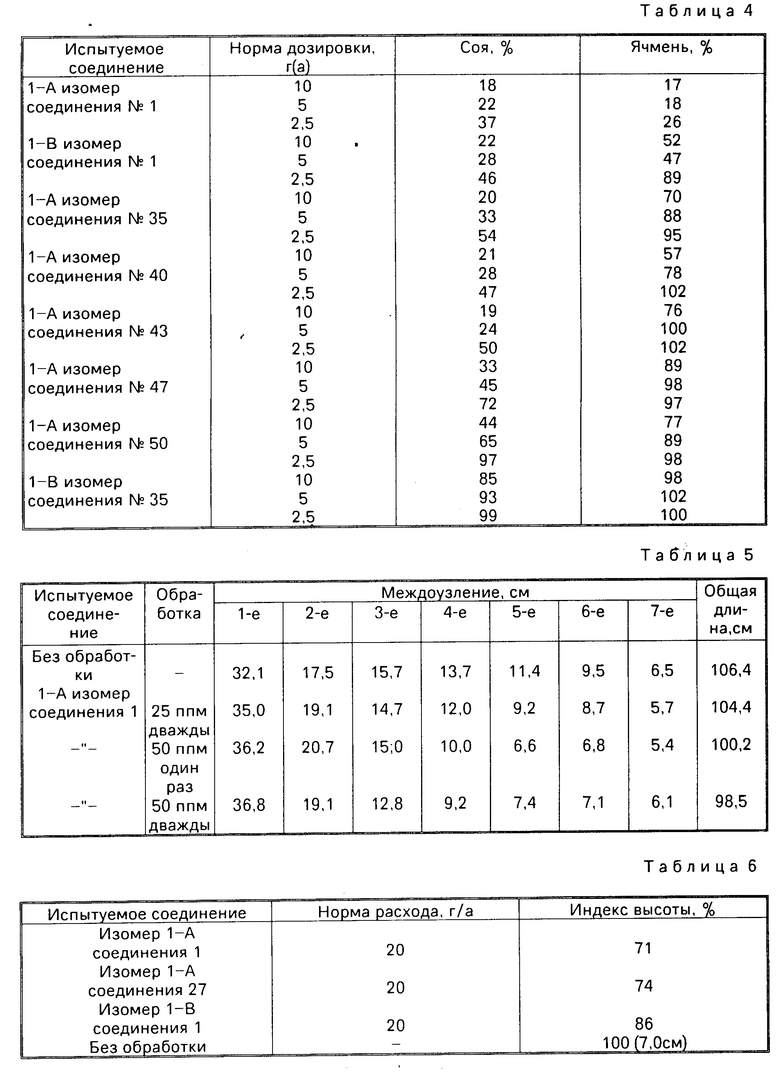
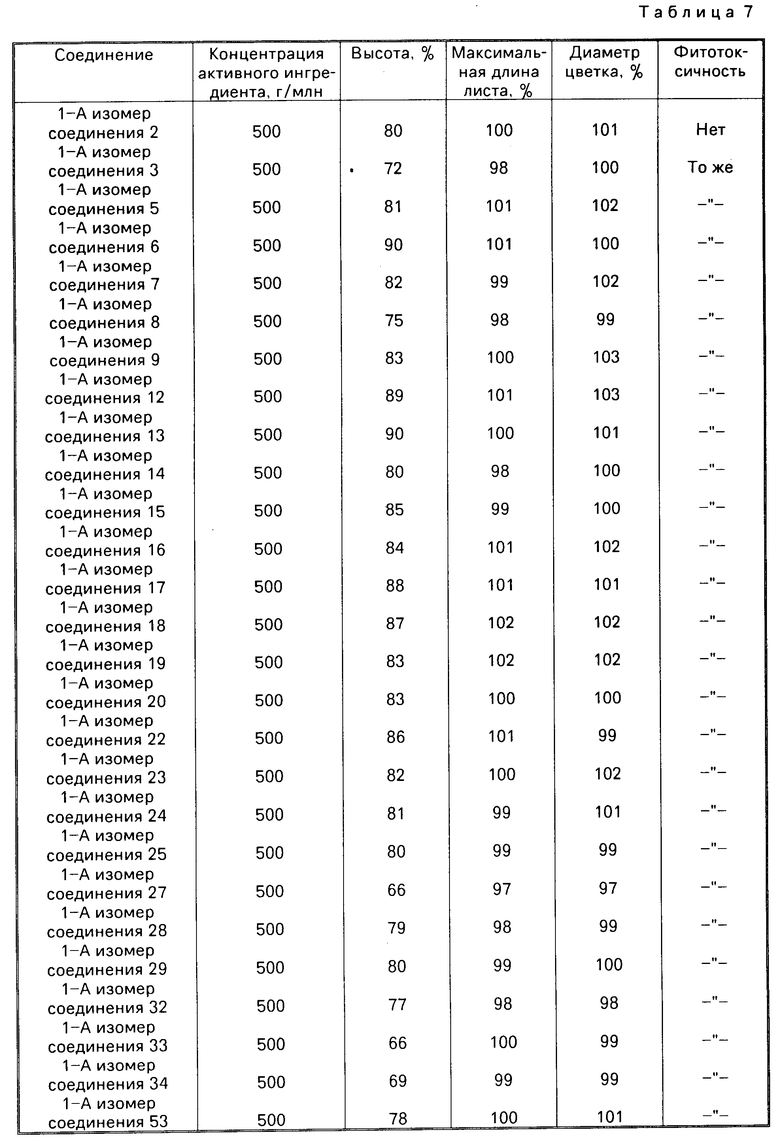
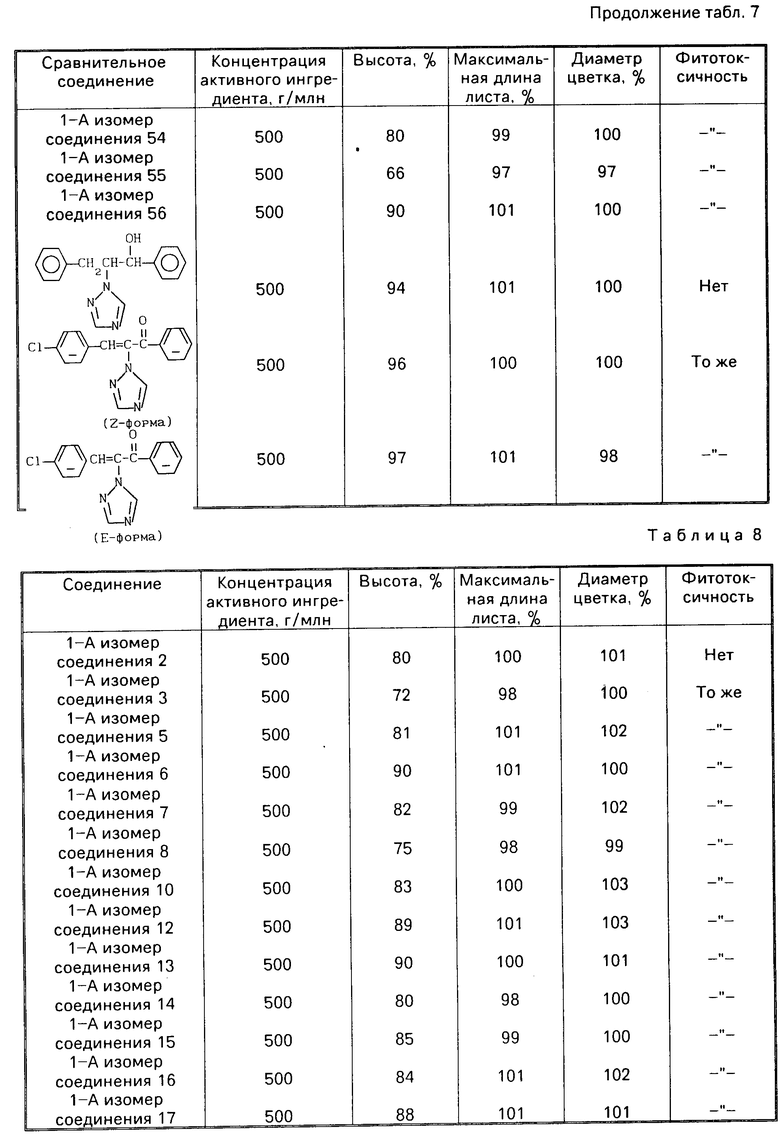
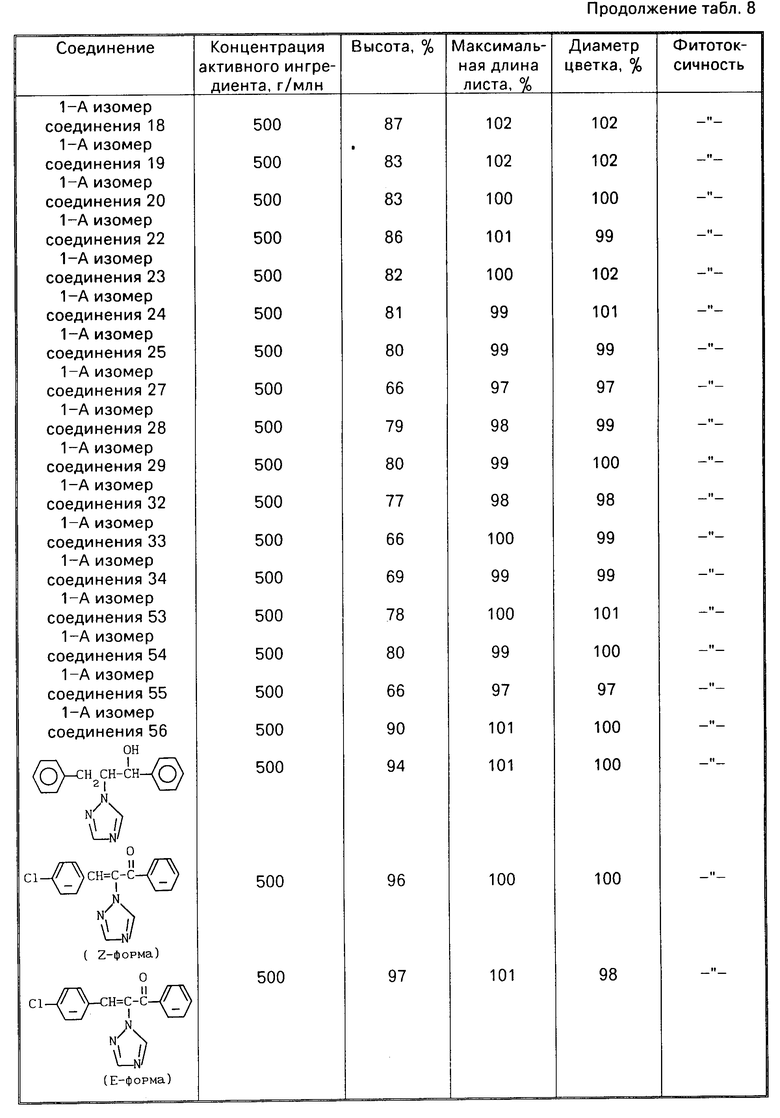
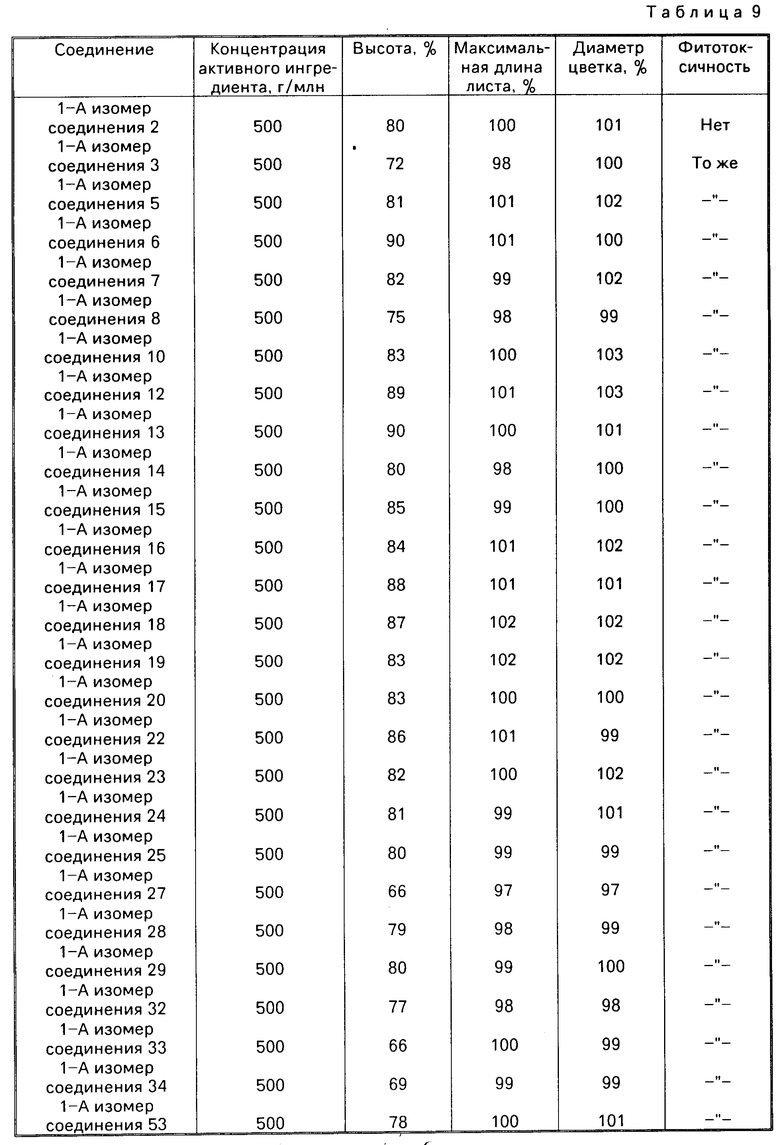
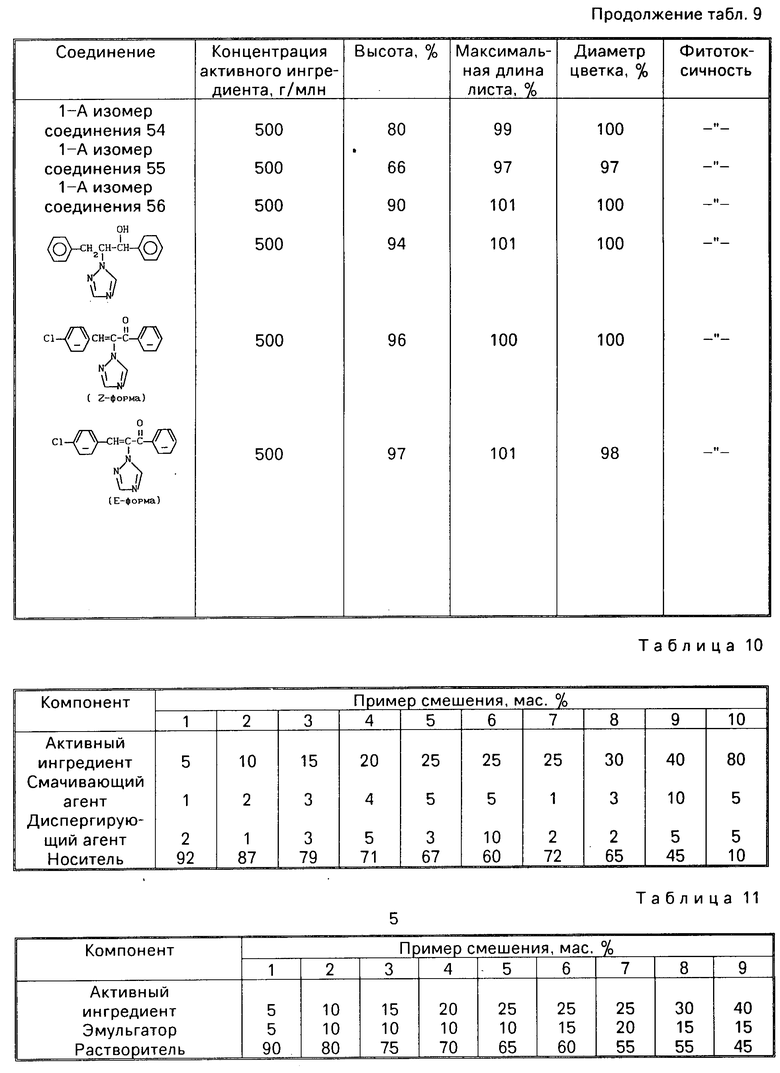
Использование: сельское хозяйство. Сущность изобретения: композиция, содержащая 1 80 мас. активного соединения-Е-изомера 1-фенил(замещенный или незамещенный)-2-(1,2,4-триазолил-1)-пропена ф-лы 1, где R2-C3-C6 алкил, циклопропил, метилциклопропил, R3 галоген, C1-C4 алкил, трифторметил, C1-C2 алкоксил, фенил, циан, фенокси- или нитрогруппа, n 0 3, или его соли, и остальное целевые добавки. Структура соединения ф-лы 1  4 з.п.ф-лы, 11 табл.
4 з.п.ф-лы, 11 табл.
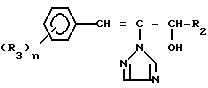
где R2 C3-C6-алкил, циклопропил или 1-метилциклопропил;
R3, одинаковые или различные, галоген, С1-С4-алкил, трифторметил, С1-С2-алкоксил, фенокси-, или циано-, или нитрогруппа, фенил; n=0-3,
или его соли при следующем соотношении компонентов, мас.
Активное вещество 1-80
Вспомогательные добавки Остальное
2. Композиция по п. 1, отличающаяся тем, что в качестве активного вещества она содержит соединение формулы I, где R2-трет-бутил.
Приоритет по признакам
06.08.79 при R2 трет-бутил, R3 галоген, n 1 или 2.
| Способ получения защитных покрытий на магнийсодержащих сплавах алюминия | 2020 |
|
RU2734426C1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
