Изобретение относится к химии и технологии получения гетероциклических соединений пиримидинового ряда, а именно 4,6-диметил-N-( β-оксиэтил)-2-оксо-1,2-дигидропиримидина, который под названием ксимедон рекомендован к применению в медицине в качестве противоожогового препарата иммунного действия [1]
Известен способ получения ксимедона [2] состоящий в превращении гидрохлорида 2-окси-4,6-диметилпиримидина в натриевую соль 2-окси-4,6-диметилпиримидина, при нагревании которой с этиленхлоргидрином получали ксимедон. Основным недостатком данного способа являлось низкое качество ксимедона. Даже после четырехкратной перекристаллизации из смеси метанола с хлороформом температура плавления продукта составила 134,5-135,5оС. Согласно требованиям временной фармакопейной статьи (ВФС) температура плавления ксимедона должна быть в пределах 138-142оС. С целью повышения качества ксимедона был разработан усовершенствованный вариант указанного способа, положенный в основу регламента [3] принятый за прототип. Процесс по прототипу осуществляется в три стадии (схема 1).
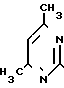
Схема 1
CO(NH2) 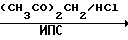
_____→ 
 +
+
+
 +
+ 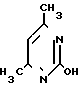
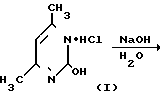
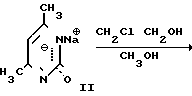
На первой стадии мочевину обрабатывают ацетилацетоном в среде изопропанола (ИПС) в присутствии соляной кислоты при температуре кипения. После охлаждения смеси отфильтровывают выпавший гидрохлорид 2-окси-4,5-диметилпиримидина (I), промывают ИПС и сушат. Выход I-78% На второй стадии I обрабатывают водным раствором гидроксида натрия при 105оС, сливают в кристаллизаторы, охлаждают, переносят вручную затвердевшую массу натриевой соли 2-окси-4,6-диметилпиримидина (II) на вакуумную воронку, отжимают от маточника и сушат под вакуумом при 80-90оС до влажности не более 0,5% Выход II 85% на I.
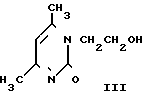
На третьей стадии к нагретому до кипения раствору этиленхлоргидрина в абсолютном метаноле дозируют II при 50-55оС в течение 6 ч, после чего дают выдержку при кипении в течение 20 ч. Массу охлаждают, фильтруют от хлорида натрия, отгоняют метанол и этиленхлоргидрин, добавляют ацетон. Образовавшиеся кристаллы технического ксимедона красно-коричневого цвета отфильтровывают, промывают ацетоном, сушат, кристаллизуют из ИПС. Получают ксимедон красного цвета, содержащий 4-8% примесей (в основном 2-окси-4,6-диметилпиримидин). Выход ксимедона на II 51% Фармакопейный ксимедон белого цвета получают, пропуская хлороформный раствор кристаллизованного из ИПС ксимедона через колонку, заполненную 2,5 м.д. окиси алюминия и 0,1 м.д. активированного угля. Из очищенного раствора отгоняют 80% взятого хлороформа, добавляют к остатку ацетон, ксимедон отфильтровывают, промывают, сушат. Выход фармакопейного ксимедона в пересчете на мочевину или на ацетилацетон 20,0%
Недостатками процесса по прототипу являются: низкий выход целевого продукта; использование высокотоксичных веществ (этиленхлоргидрина и метанола) и дефицитной окиси алюминия; многостадийность процесса; сложность техпроцесса и наличие большого количества ручных операций; большой расход сырья.
Целью изобретения является повышение выхода ксимедона, упрощение технологии его получения, снижение расхода сырья.
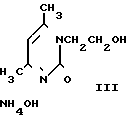
Цель достигается тем, что, нагревая мочевину с избытком этаноламина при 110-125оС в течение 6,5-1,5 ч соответственно с барботажем азота, получают этанолмочевину, не выделяя или выделяя ее из реакционной массы, нагревают с ацетилацетоном и соляной кислотой в среде изопропанола, отделяют образовавшийся кристаллический гидрохлорид 4,6-диметил-N (β-оксиэтил)-2-оксо-1,2-дигидропиримидин (гидрохлорид ксимедона) от маточника и обрабатывают его щелочным реагентом в водной среде или водном хлороформе с последующим выделением ксимедона.
Обработку гидрохлорида ксимедона в водной среде проводят гидроксидом натрия, карбонатом натрия, бикарбонатом натрия до рН среды не более 9,0 (предпочтительно рН 8,0) с последующей отгонкой воды, экстракцией сухого остатка хлороформом и выделением ксимедона из хлороформа известным приемом.
Обработку гидрохлорида ксимедона в водном хлороформе проводят карбонатом натрия, бикарбонатом натрия, водным аммиаком при соотношении 1 м.д. гидрохлорида ксимедона: 0,4 об.д. воды: 12 об.д. хлороформа с последующим отделением хлороформного экстракта и выделением из него ксимедона известным приемом.
Химизм процесса можно представить схемой 2.
Схема 2
CO(NH2)2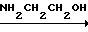 NH2CONHCH2CH2OH
NH2CONHCH2CH2OH 

П р и м е р 1. Получение гидрохлорида ксимедона.
Смесь 65,7 г 97,5%-ного этаноламина (64,0 г или 1,05 моля в пересчете на 100% -ное вещество) и 60,0 г (1 моль) мочевины нагревают до 115оС и при этой температуре и барботаже азота выдерживают в течение 4,5 ч. Смесь охлаждают и при температуре не выше 80оС к реакционной массе сливают 350 мл ИПС и 105,8 г 94,5% -ного технического ацетилацетона (100 г или 1,0 моль в пересчете на 100% -ное вещество). К полученному раствору при 78-80оС дозируют 120 мл 36% -ной соляной кислоты (1,4 моля) в течение часа. По окончании дозировки выдерживают реакционную массу при кипении (82-84оС) в течение 2,5 ч, затем охлаждают до 10-15оС, выпавшие кристаллы гидрохлорида ксимедона отфильтровывают и промывают дважды по 70 мл ИПС, сушат при 90оС. Выход гидрохлорида ксимедона составляют 159,4 г (77,9% в пересчете на мочевину или ацетилацетон). Массовая доля основного вещества 99,6% (данные потенциометрического титрования).
По температуре плавления 232оС (с разл.), ИК-спектру УФ-спектру и Rf продукт идентичен гидрохлориду ксимедона, полученному встречным синтезом из ксимедона по прототипу и соляной кислоты в среде изопропанола.
Получение ксимедона. Полученный гидрохлорид ксимедона (159,4 г) растворяют при перемешивании и температуре 40-50оС в 200 мл (1,25 м.д.) воды и при этой температуре дозируют 40% -ный раствор гидроксида натрия до рН 8,0 (расход гидроксида натрия около 31,5 г). Из подщелоченного раствора отгоняют воду под вакуумом. К сухому остатку добавляют 1275 мл (8 об.д) хлороформа, смесь перемешивают при 45-50оС в течение часа, охлаждают до 20-25оС, отфильтровывают хлорид натрия. Из хлороформенного раствора ксимедона отгоняют избыточный хлороформ до объема кубового остатка 400-450 мл, добавляют 640 мл ацетона при перемешивании, дают выдержку при 15-20оС и перемешивают в течение часа. Кристаллы ксимедона отфильтровывают, промывают 160 мл ацетона, сушат при 80-90оС в течение 2-3 ч. Получают 131,2 г (78,1%) ксимедона с tпл= 141,0-141,8оС, м.д. основного вещества 99,3% остальные показатели соответствуют требованиям ВФС.
П р и м е р ы 2-9. Показывают влияние условий синтеза этанолмочевины (мольного соотношения этаноламина и мочевины, температуры и времени реакции, барботажа азота) на выход и качество гидрохлорида ксимедона и ксимедона. Порядок ведения процесса по описанию в примере 1.
П р и м е р 10. Проведение процесса с выделением из реакционной массы этанолмочевины. Смесь 65,7 г (1,05 м на 100%) 97,5% этаноламина и 60 г (1 м) мочевины при барботаже реакционной массы азотом нагревают в течение 4,5 ч при 115оС, охлаждают до 80оС, добавляют 95 мл изопропанола, нагревают до кипения, охлаждают до 15-20оС и выдерживают при перемешивании 1,5 ч. Выпавшие кристаллы этанолмочевины отфильтровывают, промывают 2 раза по 50 мл свежего изопропанола, сушат.
Получают 68,8 г (66,1%) этанолмочевины с tпл=84-88оС.
Получение гидрохлорида ксимедона. 68,8 г (0,66 м) этанолмочевины растворяют в 230 мл изопропилового спирта и добавляют 70 г (66,15 г в пересчете на 100% -ное вещество; 0,66 м) 94,5%-ного технического ацетилацетона, нагревают и при 78-80оС дозируют 80 мл 36%-ной соляной кислоты в течение часа. Дают выдержку в течение 2,5 ч при кипении реакционной массы (82-84оС), затем охлаждают в течение 1,5 ч до 10-15оС, при этом выпадают кристаллы гидрохлорида ксимедона. Суспензию перемешивают 0,5 ч, гидрохлорид ксимедона отфильтровывают и промывают дважды по 45 мл ИПС, подсушивают на фильтре 0,5 ч и сушат при 90оС.
Выход 107,15 г (79,2%) на ацетилацетон), tпл=232оС (с разл.). М.д. основного вещества по данным потенциометрического титрования 99,7% ИК-спектр идентичен ИК-спектру гидрохлорида ксимедона, полученного из ксимедона, полученного из ксимедона по способу-прототипу. Далее гидрохлорид ксимедона используют в синтезе ксимедона без очистки.
Получение ксимедона. 107,15 г гидрохлорида ксимедона растворяют при перемешивании и температуре 40-50оС в 135 мл воды, дозируют 40%-ный раствор гидроксида натрия до рН 8,0 (примерно 21,0 г 100% NaOH), отгоняют воду под вакуумом. К сухому остатку добавляют 860 мл хлороформа, перемешивают 1 ч при 45-50оС, охлаждают до 20-25оС, отфильтровывают хлорид натрия. Из фильтрата отгоняют хлороформ до объема кубового остатка 270-320 мл, добавляют 430 мл ацетона, выдерживают 1 ч при 15-20оС. Ксимедон отфильтровывают, промывают 100 мл ацетона, сушат при 80-90оС в течение 2-3 ч. Получают 69,1 г (78,5%) ксимедона с tпл=140,0-140,8оС, м.д. основного вещества 99,1% остальные показатели соответствуют требованиям ВФС.
П р и м е р ы 11-21. Получение гидрохлорида ксимедона проводится как в примере 1.
Выделение ксимедона из гидрохлорида ксимедона щелочными реагентами гидроксидом натрия, карбонатом натрия и бикарбонатом натрия при различных значениях рН подщелоченного водного раствора проводится как в примере 1.
Гидроксид натрия при подщелачивании гидрохлорида ксимедона используют как в примере 1 в виде 40%-ного водного раствора, а карбонат и бикарбонат натрия в твердом виде.
П р и м е р 22. Выделение ксимедона из гидрохлорида ксимедона карбонатом натрия в среде водного хлороформа.
К суспензии 20,45 г (0,1 м) гидрохлорида ксимедона (пример 1) в 245 мл (12 об. д. ) хлороформа добавляют 8,2 мл (0,4 м.д.) воды и присыпают 5,8 г (0,55 м) карбоната натрия. Дают выдержку при 45оС в течение 1,5 ч при интенсивном перемешивании. Отделяют хлороформ декантацией от неорганических солей, затем в делительной воронке отделяют водный слой. Из образовавшегося раствора ксимедона в хлороформе отгоняют 190 мл хлороформа, в кубовый остаток добавляют 82 мл (4 об.д.) ацетона. Перемешивают 1,5 ч при 15оС, отфильтровывают ксимедон, промывают 15 мл ацетона, сушат.
Выходного ксимедона 12,6 г (75,0%), tпл=139,4-140,2оС, м.д. основного вещества 99,4% Остальные показатели соответствуют ВФС.
П р и м е р ы 23-29. Процесс проводится как в примере 22 с изменением количеств карбоната натрия, воды и хлороформа.
П р и м е р 30. Выделение ксимедона из гидрохлорида ксимедона гидрокарбонатом натрия в среде водного хлороформа.
К суспензии 20,45 г (0,1 м) гидрохлорида ксимедона (пример 1) в 245 мл (12 об. д. ) хлороформа добавляют 8,2 мл (0,4 об.д.) воды и 9,25 г (0,11 м) гидрокарбоната натрия, перемешивают 1,5 ч при 45оС, отделяют хлороформ от смеси солей и воды, отгоняют из него 190мл хлороформа, в кубовый остаток добавляют 82 мл (4 об. д.) ацетона. Перемешивают 1,5 ч при 15оС, отфильтровывают ксимедон, промывают 15 мл ацетона, сушат. Выход ксимедона 12,9 г (76,8% ), tпл= 139,5- -140,4оС, м.д. основного вещества 99,4% остальные показатели соответствуют ВФС.
П р и м е р 31. Выделение ксимедона из гидрохлорида ксимедона гидроксидом натрия в среде водного хлороформа.
К суспензии 20,45 г (0,1 м) гидрохлорида ксимедона (пример 1) в 245 мл (12 об.д.) хлороформа добавляют при перемешивании раствор 4,0 г (0,1 м) гидроксида натрия в 3 мл воды. Перемешивают 1 ч при 45оС, отделяют хлороформенный раствор ксимедона от водной фазы и твердого хлорида натрия. Из хлороформенного раствора ксимедона отгоняют 210 мл хлороформа, добавляют 82 мл (4 об.д.) ацетона, охлаждают до 15оС, дают выдержку 1,5 ч, отфильтровывают ксимедон, промывают 15 мл ацетона, сушат при 80-90оС 2 ч. Выход 3,4 г (50%), tпл=132,2-138,4, м.д. основного вещества 98,6%
П р и м е р ы 32-33. Процесс проводят аналогично описанному в примере 31, но с использованием различных количеств гидроксида натрия.
П р и м е р 34. Выделение ксимедона из гидрохлорида ксимедона водным аммиаком в среде хлороформа.
К суспензии 20,45 г (0,1 м) гидрохлорида ксимедона (пример 1) в 245 мл (12 об. д. ) хлороформа добавляют 8,9 мл (0,4 об.д) 25,4%-ного аммиака (1,2 м), перемешивают 1 ч при 45оС, отделяют хлороформенный раствор ксимедона от воды и хлорида аммония. Добавляют 10,4 г безводного сульфата натрия, перемешивают 3 ч, осушитель отфильтровывают. Из хлороформенного раствора ксимедона отгоняют 210 мл хлороформа, добавляют 82 мл (4 об.д.) ацетона, охлаждают до 15-20оС, дают выдержку 1,5 ч, ксимедон отфильтровывают, сушат. Выход 13,1 г (77,9%).
1,0 г (6,1%) ксимедона содержится в хлороформенно-ацетоновом маточнике, остальные потери связаны с высокой растворимостью ксимедона в водной фазе.
П р и м е р 35 проведена аналогично примеру 34 без осушки хлоформенного раствора ксимедона перед выделением из него целевого продукта.
Из представленных примеров и схем 1, 2 можно сделать следующие выводы.
Общие признаки предполагаемого изобретения и прототипа.
Использование в процессе одинаковых реагентов (мочевины, ацетилацетона, соляной кислоты).
Использование реакции образования пиримидинового цикла из уреидного компонента, ацетилацетона и соляной кислоты при температуре кипения.
Выделение ксимедона из хлороформного раствора высаживанием ацетоном.
Отличительные признаки предлагаемого изобретения.
В прототипе используют мочевину в качестве уреидного компонента, нагревая ее с ацетилацетоном и соляной кислотой в изопропаноле. При этом образуется гидрохлорид 2-окси-4,6-диметилпиримидин.
В предлагаемом изобретение мочевину нагревают с избытком этаноламина и образующуюся при этом этанолмочевину (с выделением или непосредственно в виде реакционной массы) используют в качестве уреидного компонента в реакции с ацетилацетоном и соляной кислотой в среде изопропанола при температуре кипения. При этом образуется гидрохлорид ксимедона. В дальнейшем процесс выделения ксимедона из его гидрохлорида, кроме конечной стадии выделения ксимедона из хлороформного раствора, отличен от прототипа. Принципиальное различие способов видно и при сопоставлении схем 1 и 2.
Ведение процесса нагрева мочевины с избытком этаноламина при 110-125оС в течение 6,5-1,5 ч с барботажем в реакционную массу азота обеспечивает высокое качество образующейся при этом этанолмочевины и отсутствие примеси в ней мочевины, что позволяет осуществить дальнейший процесс получения гидрохлорида ксимедона и ксимедона без выделения этанолмочевины (примеры 1-9).
При отсутствии избытка этаноламина (примеры 3, 4) в реакционной массе содержится непрореагировавшая мочевина, которая при реакции с ацетилацетоном образует примесь гидрохлорида 2-окси-4,6-диметилпиримидина (схема 1), и эта примесь при подщелачивании образует свободное основание 2-окси-4,6-диметилпиримидин, для удаления которого требуется использование колонки с окисью алюминия. Небольшой же избыток этаноламина не образует нерастворимых в реакционной среде примесей при получении гидрохлорида ксимедона и удаляется при фильтрации и промывке изопропанолом гидрохлорида ксимедона.
Температура и время процесса определены кинетикой по выделению аммиака.
Использование в качестве уреидного компонента непосредственно этанолмочевины, выделенной из реакционной массы при синтезе эталономочевины (пример 10) приводит к усложнению общей схемы процесса и перерасходу сырья (мочевины, этаноламина, изопропанола).
Выделение ксимедона подщелачиванием водного раствора гидрохлорида ксимедона до рН не выше 9,0 (предпочтительно рН 8,0) с последующей отгонкой воды и экстракцией сухого остатка хлороформом и дальнейшей обработкой хлороформного раствора известным приемом обеспечивает максимальный выход и качество ксимедона (примеры 1 и 11-21). При рН ниже 8,0 не достигается полнота нейтрализации (рН водного раствора ксимедона 8,2), а при рН 9,5 и выше выделенный ксимедон не удовлетворяет требованиям ВФС по массовой доле основного вещества и примесей, температуре плавления и цвету. Появление примесей и связанной с их присутствием желтой окраски при рН 9,5 обусловлено свойством самого ксимедона, что доказано при нагревании ксимедона в воде при рН≥9,5; при этом ксимедон вступает в реакцию самоконденсации.
Использование водно-хлороформной среды при выделении ксимедона из гидрохлорида ксимедона (примеры 22-35) позволяет существенно упростить технологический процесс ксимедона, так как при этом исключается отгонка воды досуха. Осуществление процесса отгонки воды досуха сложно в технологическом оформлении, требует нагрева и вакуума.
При использовании водно-хлорофоpмной среды снижаются требования к рН среды, так как отсутствует нагрев водной фазы. В то же время применение таких жестких щелочных реагентов как гидрохлорид натрия нецелесообразно (примеры 31-33) вследствие прохождения побочной реакции с хлороформом.
Использование хлороформа обусловлено тем, что этот растворитель единственный, достаточно хорошо растворяющий ксимедон (требуется 5 об.д. хлороформа на 1 м.д. ксимедона при 20оС) и не смешивающийся с водой.
При этом количество хлороформа должно быть не менее 12 об. д. для исключения значительных потерь ксимедона с водой, в которой он растворяется очень хорошо (требуется 0,8 об.д. воды на растворение 1 м.д. ксимедона).
По причине высокой растворимости в воде оптимальное количество ее в процессе должно быть 0,4 м.д. на 1 м.д. гидрохлорида ксимедона.
При м. д. воды менее 0,4 снижается выход ксимедона вследствие того, что при этом невозможно достигнуть растворения гидрохлорида ксимедона, а частично гетерогенный процесс осуществить до конца не удается и выход ксимедона снижается (пример 24). Увеличение количества воды более 4 об.д. также приводит к снижению выхода ксимедона, но уже за счет потерь ксимедона с водой после отделения ее от хлороформного слоя (примеры 25, 26).
Использование водного аммиака требует дополнительной осушки хлороформного раствора ксимедона, но при этом достигается такой же выход, как и при использовании приема нейтрализации гидрохлорида ксимедона в воде без хлороформа с последующей отгонкой воды (примеры 34, 35).
Несмотря на использование в процессе значительного количества хлороформа, он достаточно экономичен, так как перед окончательным выделением ксимедона из хлороформа высаживанием ацетоном значительное количество хлороформа (80% от взятого) отгоняется и может быть повторно использовано в последующих операциях техпроцесса.
Из приведенных выше примеров, схемы процесса и пояснений можно сделать следующие заключения.
Технология получения ксимедона по предлагаемому способу значительно проще, чем по прототипу.
В оптимальном варианте по предлагаемому способу выход ксимедона в расчете на мочевину или ацетилацетон составляет 54,8% а по прототипу в расчете на те же компоненты 20%
В процессе по предлагаемому способу существенно уменьшается расход сырья.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ N-(БЕТА-ГИДРОКСИЭТИЛ)-4,6-ДИМЕТИЛДИГИДРОПИРИМИДОНА-2 | 2009 |
|
RU2407530C1 |
| Способ получения производных пенама | 1978 |
|
SU974936A3 |
| Способ получения производных 4-амино-2-пиперидинохиназолина или их солей с фармацевтически приемлимыми кислотами | 1980 |
|
SU895291A3 |
| Способ получения 2,3-дизамещенных 6-азаиндола | 1977 |
|
SU687075A1 |
| СПОСОБ ПОЛУЧЕНИЯ 2,4,7-ТРИАМИНО-6-ФЕНИЛПТЕРИДИНА | 1993 |
|
RU2047616C1 |
| 5-( ω - АМИНОАЦИЛ)-5,10-ДИГИДРО-11H-ДИБЕНЗО[B, E] [1,4]-ДИАЗЕПИН-11-ОНЫ ИЛИ ИХ СОЛИ, ОБЛАДАЮЩИЕ ПРОТИВОАРИТМИЧЕСКОЙ АКТИВНОСТЬЮ | 1989 |
|
RU2026862C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЛАППАКОНИТИН ГИДРОБРОМИДА И ЛАППАКОНИТИНА | 2016 |
|
RU2641967C1 |
| Хинуклидил-3-диарал(гетерил) карбинолы, проявляющие антигистаминную, антисеротониновую и антиаллергическую активность и способ их получения | 1974 |
|
SU495310A1 |
| СПОСОБ ПОЛУЧЕНИЯ D-(+)-БИOTИHA | 1989 |
|
RU1626647C |
| Способ получения 3,3',3'',3'''-(3,8,13,17-тетраметилпорфирин-2,7,12,18-тетраил) тетрапропионовой кислоты (копропорфирина) | 2017 |
|
RU2644674C1 |
Использование: в медицинской промышленности. Сущность изобретения: способ получения N-( b -гидроксиэтил)-4,6-диметил-дигидропиримидона-2. Реагент 1: мочевина. Реагент2: этаноламин в избытке, нагревают при 110-125°С в течение 6,5 1,5 ч с барботажем в реакционную массу азота, образующуюся при этом этанолмочевину выделяют и обрабатывают ацетилацетоном и соляной кислотой в среде изопропанола, полученный кристаллический продукт выделяют в виде хлоргидрата или основания путем обработки хлороформенного раствора хлоргидрата щелочным реагентом. 3 з.п.ф-лы.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Лабораторный регламент на производство лекарственного средства "Ксимедон", Утв.зам.директора ИОФХ КФАН СССР 15.10.88, Казань, 1988, с.94. | |||
