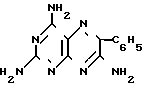
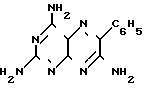
Изобретение относится к химии и технологии получения гетероциклического соединения 2,4,7-триамино-6-фенилптеридина, который используется в качестве калийсберегающего диуретика "Триамтерен" или в качестве компонента диуретического медпрепарата "Триампур".
Триамтерен впервые был получен конденсацией 2,4,6-триамино-5-нитрозопиримидина (ТАНП) с цианистым бензилом (ЦБ) в среде этилцеллозольва в присутствии основания метилата натрия (1. Spickett R.G, W. Timis G.M. J. Chem. Soс. 1954, N 8, р. 2887) с выходом 55%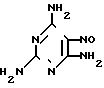 + C6H5CH2CN
+ C6H5CH2CN 
Недостатками процесса по способу (1) являются невысокий выход триамтерена и использование дорогостоящих и дефицитных компонентов метилата натрия и этилцеллозольва.
Известен также способ получения триамтерена, в котором вместо ЦБ используется соль фенилбензилпиридиний кетона в присутствии цианида щелочного металла (2. Пат.ФРГ 1 274585):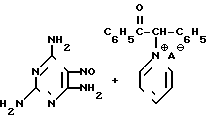

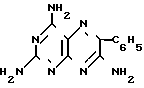
Возможно получение триамтерена из тетрааминопиримидина и бензоилцианида в кипящем ДМСО в присутствии щелочного агента (3. Пат. Японии 25914/67):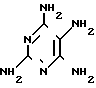 + C6H
+ C6H CN ___→
CN ___→ 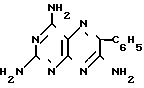
Выходы в способах (2,3) около 60% общими их недостатками являются малая доступность используемых вместо ЦБ реагентов.
За прототип предлагаемого технического решения взят наиболее приемлемый с нашей точки зрения способ, реализованный в опытном производстве ВНИХФИ (4. Опытно-промышленный регламент на производство триамтерена. М. ВНИХФИ, 1989). Он заключается в нагревании ЦБ, ТАНП и карбоната калия в мольном соотношении 1: 0,77: 0,83 в ДМФА (16,8 м.д. на 1 м.д. ЦБ) при 145-150оС в течение 7 ч с последующей очисткой образовавшегося триамтерена через ацетат кристаллизацией из 35%-ной уксусной кислоты (26 об.д. на 1 м.д. триамтерена) и регенерацией триамтерена обработкой водным аммиаком (0,78 об.д. на 1 м.д. триамтерена).
Схема реакции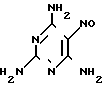 + C6H5CH2CN
+ C6H5CH2CN 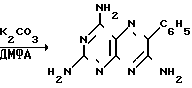
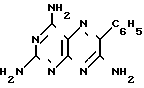
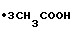
Выход технического триамтерена составляет 78,3 фармакопейного триамтерена 68,8% на ТАНП, или 53,0% на ЦБ.
Недостатками процесса по способу (4) являются:
жесткие требования к сырью по содержанию влаги (не более 0,1% в ДМФА, 0,3% в карбонате калия и ТАНП);
сложность его технологического оформления, связанная с высокой температурой процесса (для ее обеспечения необходим либо пар высокого давления, либо контур с высокотемпературным теплоносителем);
сложность регенерации ДМФА;
большой расход сырья.
При содержании влаги в сырье, более указанного и температуре ниже 145оС, увеличивается время процесса и снижается выход триамтерена (4).
Целью заявляемого способа является упрощение техпроцесса получения триамтерена, снижение расхода сырья и расширение сырьевой базы.
Поставленная цель достигается использованием в процессе отличных от прототипа компонентов и условий его проведения.
Сущность предлагаемого способа состоит в нагревании ЦБ, ТАНП и гидроксида натрия в мольном соотношении 1:0,95:(0,25-0,5) в 8-10 м.д. бутанола на 1 м.д. ЦБ при 110-115оС в течение 8-12 ч с последующим выделением и очисткой триамтерена.
Вместо ТАНП, гидроксида натрия и бутанола предлагается также использовать в реакции с ЦБ реакционную массу, полученную нагреванием гуанидиниевой соли изонитрозомалононитрила (ГС) и гидроксида натрия в молярном соотношении (1,0-1,05): (0,25-0,5) на 1 моль ЦБ в 8-10 м.д. бутанола на 1 м.д. ЦБ при 110-155оС в течение 0,5-1,0 ч с последующим добавлением в горячую реакционную массу ЦБ и дальнейшим продолжением процесса при 110-115оС в течение 8-12 ч. выделением и очисткой триамтерена.
Схема процесса по предлагаемому способу
(NC)2C=NOH·(H2N)2C=NH 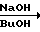
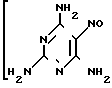
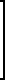
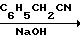
_____→ 
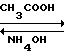
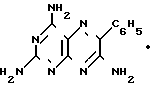 3CH3COOH
3CH3COOH
П р и м е р 1. Получение триамтерена из 2,4,6-триамино-5-нитрозопиримидина.
117 г (1 моль) ЦБ добавляют при перемешивании к смеси 146,3 г (0,95 молей) ТАНП, 16 г (0,4 моля) гидроксида натрия в 1170 (10 м.д. на 1 м.д. ЦБ) бутанола. Смесь нагревают при температуре 110-115оС в течение 10 ч.
(Окончание выдержки контролируется методом ТСХ до исчезновения ТАНП Rf 0,4, элюент ЭА: MeOH: CH3COOH8:1:1). Реакционную массу охлаждают до 20оС, осадок отфильтровывают, промывают водой до рН 7, сушат при 60-70оС.
Выход технического триамтерена 203,9 г (80,6% на ЦБ).
Очистка триамтерена проводится по способу по прототипу (4).
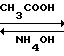
К 5300 мл (26 об.д. на 1 м.д. технического триамтерена) 35%-ной уксусной кислоты присыпают 203,9 г технического триамтерена. Реакционную массу нагревают до 100-105 оС и дают выдержку при этой температуре 10-15 мин до полного растворения триамтерена. Раствор охлаждают до 80-90оС, присыпают 1,3 г (0,07 м.д.) активированного угля. Реакционную массу нагревают до 100-105оС, дают 10-минутную выдержку, уголь отфильтровывают через нагретую воронку. Фильтрат охлаждают до 20оС в течение 2-3 ч. Осадок ацетата триамтерена отфильтровывают.
К 2915 мл (14,3 об.д.) воды приливают 163 мл (0,8 об.д.) 25%-ного аммиака, присыпают ацетат триамтерена. Суспензия перемешивается в течение 1 часа при температуре 20-25оС (рН не менее 10).
Осадок фармакопейного триамтерена отфильтровывают, промывают водой, сушат при 60-70оС.
Получают 173,3 г (68,5% на ЦБ, 72,1% на ТАНП).
Массовая доля основного вещества 100,0 Продукт удовлетворяет все требованиям Временной фармакопейной статьи.
П р и м е р 2. Получение триамтерена из ГС.
154 г (1 моль) ГС и 16 г (0,4 моля) гидроксида натpия в виде порошка или мелких гранул в 1170 г (10 м.д. на 1 м.д. ЦБ) бутанола нагревают при перемешивании при 110-115оС в течение 1 ч. В процессе нагрева выпадает осадок ТАНП. В горячую реакционную массу сливают 117 г (1 моль) ЦБ и дают выдержку при 110-115оС в течение 10 ч. Реакционную массу охлаждают до 20оС, осадок отфильтровывают, промывают водой до рН 7, сушат при 60-70оС,
Выход технического триамтерена 203 г (80,2% на ЦБ).
Очистка проводится аналогично описанному в примере 1.
Выход фармакопейного триамтерена 172,5 г (68,2%) на ЦБ. Массовая доля основного вещества 99,8%
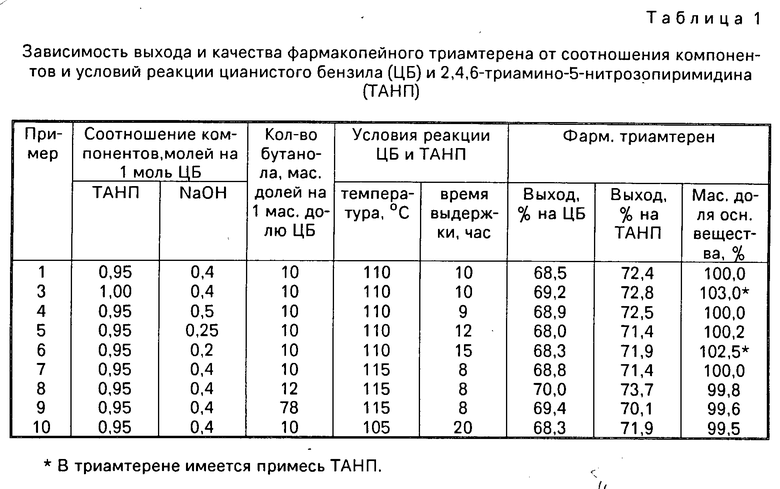
П р и м е р ы 3-10 выполнены по описанию, приведенному в примере 1, с другим соотношением компонентов, температурой и временем процесса. Результаты экспериментов приведены в табл.1.
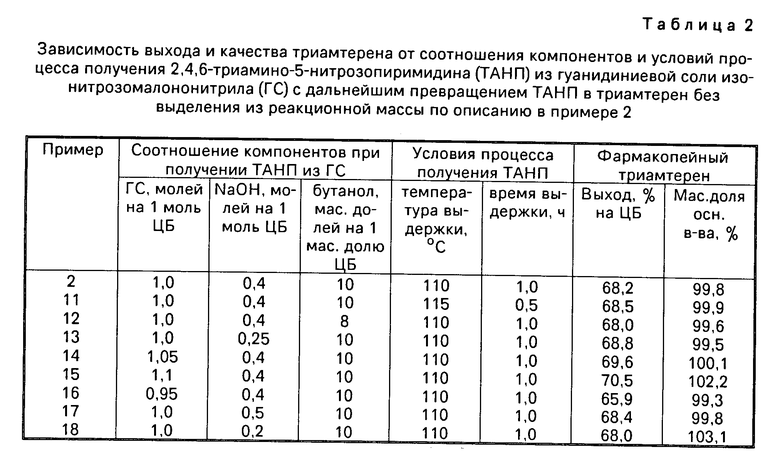
П р и м е р ы 11-18 выполнены по описанию, приведенному в примере 2, с вариацией соотношения компонентов и условий процесса получения триамтерена. Результаты приведены в табл.2.
Анализ предлагаемого способа (примеры 1-18) и способа по прототипу позволяет сделать следующие выводы.
Общие признаки способов состоят в использовании реакции циклизации ЦБ и ТАНП для получения триамтерена в присутствии основания в среде органического растворителя.
Отличительные признаки способов состоят в том, что:
в реакции циклизации ЦБ и ТАНП использован в качестве основания гидроксид натрия, а в качестве растворителя бутанол в отличных от прототипа соотношениях, температуре и времени процесса;
в реакции циклизации ЦБ и ТАНП для получения триамтерена вместо ТАНП, гидроксида натрия и бутанола использована реакционная масса, полученная нагреванием ГС и гидроксида натрия в среде бутанола в заявляемых соотношениях, температуре и времени процесса.
В обосновании выбранных условий процесса по предлагаемому способу отметим следующее.
Молярное соотношение ЦБ и ТАНП выбрано 1:0,95 (табл.1), так как при эквимолярном соотношении не обеспечивается полнота превращения ТАНП (пример 3). Это можно объяснить частичным разложением ЦБ в щелочной среде. В примере 3 массовая доля основного вещества больше 100% что обусловлено примесью ТАНП.
Молярное соотношение ЦБ и гидроксида натрия выбрано в пределах 1: (0,25-0,5), так как при использовании менее 0,25 молей гидроксида натрия (пример 6) несмотря на значительное увеличение времени процесса не удается обеспечить полноту превращения ТАНП до триамтерена. Полученный в таких условиях триамтерен очистить от примеси ТАНП не удается. Использование в процессе более 0,5 молей гидроксида натрия нецелесообразно, так как при этом лишь незначительно уменьшается только время процесса без изменения выхода триамтерена.
Температура процесса циклизации ЦБ и ТАНП выбрана в пределах 110-115оС, так как обеспечить большую температуру при атмосферном давлении невозможно, а при меньшей температуре увеличивается время процесса (пример 10).
Время процесса циклизации при 110-155оС взято в пределах 8-12 ч, так как за это время обеспечивается полнота реакции, которая легко контролируется по ТСХ и визуально по переходу малиновой окраски ТАНП в желтую триамтерена.
Количество бутанола принято в пределах 8-10 м.д. на 1 м.д. ЦБ исходя лишь из обеспечения приемлемой текучести реакционной массы, так как при количестве бутанола менее 8 м.д. перемешать реакционную массу и слить ее из реактора после завершения процесса практически невозможно. В указанных пределах выход и качество триамтерена изменяются мало.
Использование в реакции циклизации с ЦБ вместо исходных компонентов ТАНП, гидроксида натрия и бутанола реакционной массы, полученной нагреванием ГС и гидроксида натрия в бутаноле и содержащей те же компоненты реакции, позволяет значительно расширить сырьевую базу процесса получения триамтерена, так как ТАНП по- лучают из ГС (5. Франц.патент. 1.364.734, 1964). При выделении ТАНП из реакционной массы процесс получения триамтерена существенно усложняется и снижается выход триамтерена за счет потерь ТАНП с отработанным растворителем.
Соотношение компонентов при предварительном получении ТАНП из ГС (табл. 2) выбраны с учетом найденных в примерах таблицы 1 условий для дальнейшего превращения ТАНП в триамтерен после добавления в реакционную массу ЦБ. На 1 моль ЦБ берется 1,00-1,05 молей ГС, так как при этом обеспечивается максимальный выход триамтерена и высокое его качество. При большем количестве ГС (пример 15) триамтерен не удовлетворяет требованиям ВФС по массовой доле основного вещества, так как в этом случае образуется больше ТАНП, чем необходимо для реакции с ЦБ. Поэтому молярное соотношение ГС и гидроксида натрия выбрано в пределах (1,00-1,05):(0,25-0,50) на 1 моль ЦБ и количество бутанола 8-10 м. д. на 1 м.д. ЦБ. Температурный интервал превращения ГС в ТАНП взят 110-115оС, так как этот интервал используется в дальнейшем процессе и в этом интервале реакция превращения ГС завершается за 0,5-1,0 ч.
Новизна предлагаемого изобретения состоит в том, что
в реакции циклизации ЦБ и ТАНП использованы другие по сравнению с прототипом основание и растворитель и условия процесса, в совокупности обеспечивающие уменьшение температуры процесса;
показана возможность использования для получения триамтерена ТАНП, образующегося в реакционной массе после нагревания ГС в бутаноле в присутствии гидроксида натрия вместо очищенного ТАНП.
Из сопоставления предлагаемого и по прототипу способов можно сделать следующие выводы:
процесс по предлагаемому способу осуществляется проще, чем по прототипу, так как температура процесса 110-115оС может быть достигнута обогревом паром, в прототипе специальным теплоносителем.
Технологически проще осуществляется и регенерация бутанола. Так, 80% от взятого бутанола регенерируется отгонкой из маточника.
Расширяется сырьевая база для синтеза триамтерена, так как используются ГС, бутанол и гидроксид натрия вместо ТАНП, ДМФА и карбоната калия.
Если учесть, что промышленное производство ТАНП отсутствует, то преимущества предлагаемого способа становятся очевидными.
Уменьшается расход сырья на получение 1 кг триамтерена (см.дополнительные материалы).
Таким образом, поставленную цель можно считать достигнутой.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 2,4,6-ТРИАМИНО-5-НИТРОЗОПИРИМИДИНА | 1993 |
|
RU2054423C1 |
| СПОСОБ ПОЛУЧЕНИЯ N-( β -ГИДРОКСИЭТИЛ)-4,6-ДИМЕТИЛДИГИДРОПИРИМИДОНА-2 | 1992 |
|
RU2044730C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2,4,6-ТРИАМИНО-5-НИТРОЗОПИРИМИДИНА | 1989 |
|
RU1624955C |
| 4-ТРИМЕТИЛСИЛОКСИМЕТИЛФОСФИНИЛ -2- ТРИМЕТИЛСИЛОКСИ -2-БУТЕННИТРИЛ В КАЧЕСТВЕ ПОЛУПРОДУКТА СИНТЕЗА 4-МЕТИЛГИДРОКСИФОСФИНИЛ-2- КЕТОБУТАНОВОЙ КИСЛОТЫ, ОБЛАДАЮЩЕЙ ГЕРБИЦИДНОЙ АКТИВНОСТЬЮ, И СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 1992 |
|
RU2024536C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИНАТРИЕВОЙ СОЛИ БИС (N-НИТРОЗОГИДРОКСИЛАМИНО)МЕТАНА | 1990 |
|
RU2027702C1 |
| СПОСОБ ПОЛУЧЕНИЯ 4-АМИНО-1,2,4-ТРИАЗОЛА | 1992 |
|
RU2036912C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОХЛОРИДА-4-АМИНО-3-ФЕНИЛБУТАНОВОЙ КИСЛОТЫ | 1993 |
|
RU2072984C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1,2-БИС(2-АМИНОЭТОКСИ)БЕНЗОЛА | 1995 |
|
RU2120937C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРЕТ-БУТИЛАЦЕТИЛЕНА | 2002 |
|
RU2238260C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ АВИБАКТАМА | 2018 |
|
RU2722625C1 |
Использование: в медицине в качестве калийсберегающего диуретика "Триамтерен" или в качестве компонента диуретического медпрепарата "Триампур". Сущность изобретения: получение 2, 4, 6-триамино-6-фенилптеридина (1)-медпрепарата триамтерена ведут нагреванием цианистого бензила (2), 2, 4, 6-триамино-5-нитрозопиримидина (3) и гидроксида натрия (4) в молярном соотношении 1 0,95 (0,25-0,5) в 8 10 м.д. бутанола (5) на 1 м.д. 2 при 110 115°С в течение 8 12 ч с последующим выделением и очисткой 1. Вместо компонента 3 5 в реакции с 2 используется также реакционная масса, полученная нагреванием гуанидиниевой соли изнитрозомалононитрила (6) и 4 в молярном соотношении (1,0 1,05) (0,25 0,5) на 1 моль 2 в 8 10 м.д. 5 при 105 115°С в течение 0,5 1,0 ч, причем 2 добавляется в горячую реакционную массу с дальнейшим проведением процесса при 110 115°С в течение 8 12 ч. Выход 1 на 2 68 70% м. д.основного вещества 99,5 - 100% При использовании 6 выход 1 на 2 68 69% м.д. основного вещества 99,3 100,1% 1 з. п. ф-лы, 2 табл.
| Опытно-промышленный регламент на производство триамтерена, М.: ВНИХФИ, 1989. |
