Изобретение относится к новым производным индола общей формулы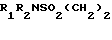
 I где R1 водород или C1-4-алкил;
I где R1 водород или C1-4-алкил;
R2 водород или С1-4-алкил;
R3 водород или С1-3-алкил,
или их фармацевтически приемлемый солям, или сольватам, обладающий 5НТ1-подобной рецепторной антагонистической активностью.
Селективная 5НТ1-подобная рецепторная антагонистическая активность и селективная сужающая сосуды активность соединений I были продемонстpированы in vitro. Кроме того эти соединения селективно сужают carotid arterial bed у анестезированных собак, при этом практически не оказывая воздействия на кровяное давление.
После непарэнтерального, включая интрадуоденальное, введения соединений I они демонстрируют повышенный показатель биоусвояемости.
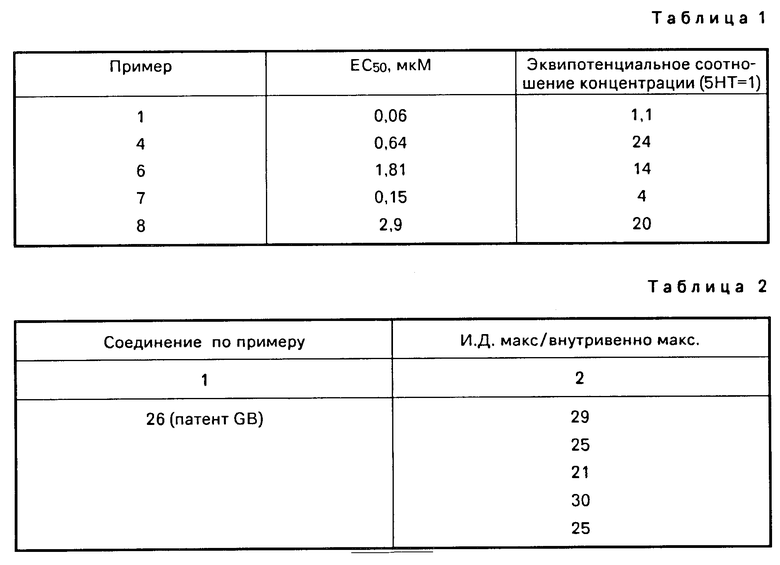
Определение величины EC50 и эквипотенциального отношения концентраций величины ЕС50 (молярная концентрация соединения необходима для достижения 50% его максимального эффекта) и эквипотенциальное концентрационное соотношение с 5-гидрокситриптамином (5НТ) были определены для соединений данного изобретения с 5НТ1-подобным рецептором, связанным с изолированной Saphenous веной собаки по известному методу.
Соединения I не оказывают значительного влияния на кровяное давление или пульс анестезированных гончих собак в дозах 0,3 мг/кг веса внутривенно.
Соединение по примеру 1 в дозе 10 мг/кг веса внутривенно не оказывает побочного эффекта у крыс.
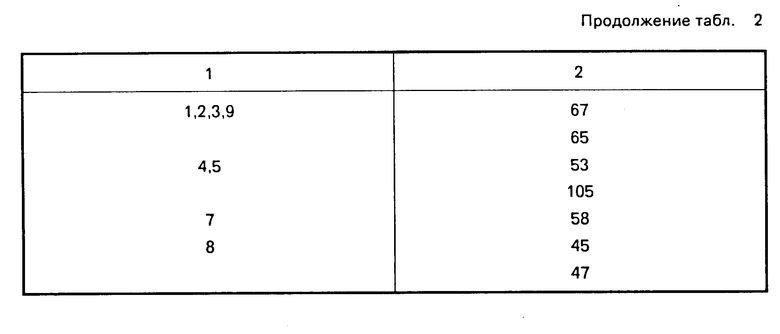
Для сравнения было взято соединение известного примера (см. патент GB N 2124210, пример 26). Биоактивность этого соединения при интрадуоденальном введении (И. Д.) анестезируемой собаке измеряли и сравнивали с соединениями данного изобретения. Результаты приведены в табл. 2.
Данные испытаний ясно показывают, что показатель биоактивности выше для соединений данного изобретения относительно известного соединения.
Промежуточное соединение 1.
N-метил-3-(1,2,3,6-тетрагидро-1-метил- 4-пиридинил)-1Н-индол- 5-этансульфонамидоксалат.
Раствор N-метил-1Н-индол-5-этансульфонамид (1,0 г) в метаноле (50 мл), содержащем гидроксид калия (5,6 г) и N-метил-4-пиперидон (1,0 мл) нагревают при кипячении с обратным холодильником в течение 24 ч, охлаждают и полученный твердый продукт отфильтровывают (1,0 г). Твердый образец (0,2 г) растворяют в горячем метанольном растворе щавелевой кислоты (0,06 г), раствор охлаждают и полученную соль осаждают добавляя этилацетат (20 мл) и сухой эфир (50 мл). Эту соль отфильтровывают и сушат в вакууме до получения указанного в заглавии соединения в виде твердого продукта (0,12 г), т.пл. 87-90оС (с усадкой).
Элементный анализ для C17H23N3O2S ˙ ˙ C2H2O4 ˙ 0,6H2O:
Найдено, C 52,2; H 5,6; N 9,5
Рассчитано, C 52,5; H 6,0; N 9,7.
Промежуточное соединение 2.
5-бром-3-(1-метил-4-пиперидинил)-1Н-индол.
Смесь 5-броминдола (39,2 г), N-метил-4-пиперидона (25,0 г) и таблеток гидроксида калия (12,0 г) в метаноле (9250 мл) перемешивают и нагревают при кипячении с обратным холодильником в течение 17 ч, а затем охлаждают до 5оС при перемешивании. Полученную смесь фильтруют. Остаток промывают последовательно метанолом, водой, снова метанолом, и эфиром и сушат в вакууме до получения промежуточного тетрагидропиридина (43,3 г) в виде порошка (т.пл. 256-261оС), который используют без дальнейшей характеристики на следующей стадии. Раствор этанольного хлористого водорода приготавливают, добавляя ацетилхлорид (20 мл) к охлажденному льдом перемешиваемому этанолу (1,31). Промежуточный тетрагидропиридин (43,2 г) растворяют в части (0,95 л) этого раствора. Гидрохлорид промежуточного соединения осаждается. Для того, чтобы снова растворить эту соль, суспензию нагревают на паровой бане и добавляют порцию 2 н. соляной кислоты (10 мл), воду (15 мл) и концентрированную (11н) соляную кислоту (10 мл). Полученный раствор добавляют к предварительно гидрированной суспензии 5% -ного оксида платины на угле (7,0 г) в этанольном растворе HCl (0,35 л вышеуказанного раствора) и полученную смесь гидрируют при комнатной температуре и атмосферном давлении до прекращения поглощения водорода. Полученную смесь фильтруют и растворитель выпаривают. Остаток повторно суспендируют в этилацетате (600 мл). Добавляют карбонат натрия (2н, 350 мл) при перемешивании и полученную смесь фильтруют. Остаток промывают водой и этилацетатом и сушат в вакууме до получения указанного в заглавии соединения (33,4 г) в виде порошка, т.пл. 160-165оС.
Промежуточное соединение 3.
5-бром-3-(1,2,3,6-тетрагидро-1-(фенил- метил)-4-пиридинил)-1Н-индол.
Свежеперегнанный 1-бензил-4-пиперидон (11,7 г) добавляют к перемешиваемому раствору 5-броминдола (11,0 г) и 2М гидроксида калия в метаноле (81 мл). Полученную смесь перемешивают при кипячении с обратным холодильником в течение 8 ч, а затем оставляют остывать до 25оС за 8 ч. Твердую часть собирают, фильтрованием промывают смесью метанола и воды (2:1, 2 x 15 мл) и сушат в вакууме при 50оС в течение 18 ч до получения указанного в заглавии соединения в виде кристаллического твердого продукта (18,6 г). Т.пл. 173-175оС (с разложением).
Промежуточное соединение 4
5-бром-3-[1-(фенилметил)-4-пипериди-нил]-1Н-индол
Раствор промежуточного соединения 3 (4,00 г) в этанольном хлористом водороде (330 мл), полученный при добавлении ацетилхлорида (1,65 мл) к этанолу (250 мл) при перемешивании, гидрируют над 5% платины на угле (3,0 г) при комнатной температуре и атмосферном давлении до завершения гидрирования. Катализатор удаляют фильтрованием. Твердую часть промывают этанолом (15 мл) и объединенные фильтраты выпаривают до получения маслянистого остатка. Остаток разделяют между 2М водным карбонатом натрия (75 мл) и этилацетатом (175 мл), фазы разделяют и водный слой снова экстрагируют этилацетатом (100 мл). Объединенные органические слои затем промывают водой (50 мл), экстрагируют насыщенным солевым раствором (50 мл), сушат над безводным сульфатом магния и растворитель выпаривают до получения указанного в заглавии соединения в виде масла (3,3 г). ТСХ на SiO2 CH2Cl2:EtOH:0,88 NH3 (100:8:1) Rg 0,44.
П р и м е р 1. N-метил-3-(1-метил-4-пиперидинил)-1Н-индол-5-этансульфорнамид.
Промежуточное соединение 1 (в виде свободного основания) (0,36 г, 0,001 моль) в абсолютированном спирте (70 мл) и безводном диметилформамиде (5 мл) гидрируют в присутствии 5% палладия на активированном угле (0,36 г) при комнатной температуре и атмосферном давлении. Спустя 20 ч поглощение водорода (25 см3, теоретически 24 см3) прекращается. Катализатор отфильтровывают, и растворитель удаляют в вакууме до получения непрозрачной смолы, которая отверждается в виде мягкого твердого вещества белого цвета (0,3 г). После очистки на хроматографе с мгновенным испарением (Сорбсил С60 силикагель, СH2Cl2(EтOH)0,88 аммиак 50:80:1) получают бесцветное масло (0,21 г), которое тщательно растирают с эфиром до получения указанного в заглавии соединения (0,17 г), т. плавления 156-158оС. ТСХ SiO2 (CH2Cl2:EtOH:0,88 аммиак 50:8:1)
Rf 0,4; детектирование по уф, 1РА.
Определение воды: найдено 0,12% мас./мас.0,02 мол.экв.
Элементный анализ для C17H25N3O2S ˙ ˙ O ˙ O2H2O
Рассчитано, C 60,8; H 7,5; N 12,5
Найдено, C 60,5; H 7,3; N 12,1
П р и м е р 2. N-метил-3-(1-метил-4-пиперидинил)-1Н-индол-5-этансульфонамид (i) (E(N-метил-2-] 3-(1-метил-4-пиперидинил)-1Н-индол-5-ил] -этенсульфонамид.
Смесь промежуточного соединения 2 (1,00 г) N-метилэтенсульфонамида (530 мг), три-орто-толилфосфина (300 мг), ацетата палладия (50 мл) и триэтиламина (730 мл) в сухом ацетонитриле (добавлен для доведения полного объема до 10 мл) перемешивают и нагревают в запаянной ампуле при 120оС в течение 1,25 ч, а затем при 80оС в течение 16 ч. Реакцию повторяют в том же объеме 10 раз. В каждом случае запаянную ампулу нагревают при 100-110оС в течение 3,5 ч. Запаянные ампулы охлаждают, содержимое объединяют и растворитель выпаривают. Остаток хроматографируют на силикагеле (450 г), используя смесь дихлорметана, этанола и аммиака (в начале 80:80:1, постепенно повышая полярность до 65: 8: 1). Фракции содержащие продукт объединяют и выпаривают до получения полутвердого вещества. Его быстро растирают в смеси циклогексана и этилацетата (1:1, 100 мл) до получения твердого продукта, который фильтруют и сушат до получения указанного в заглавии соединения (6,85 г) в виде порошка, т.пл. 190-192оС.
(ii) N-метил-3-(1-метил-4-пиперидинил)-1Н-индол-5-этансульфонамид.
Раствор продукта со стадии (i) (5,78 г) в смеси этанольного хлористого водорода, полученного при добавлении ацетилхлорида (1,71 г, 21,8 ммоль) к 1MS этанолу (400 мл) при перемешивании, и диметилформамида (300 мл), добавленного к вышеуказанному для растворения исходного материала, гидрируют при комнатной температуре и атмосферном давлении, используя 10% палладий на угле (5,00 г, 50% мас/мас с водой) в качестве катализатора до прекращения поглощения водорода. Полученную смесь фильтруют и полученный фильтрат выпаривают до получения твердого продукта. Этот твердый продукт разделяют между 2н карбонатом натрия (60 мл) и этилацетатом (200 мл), и полученную смесь нагревают до тех пор, пока не растворится твердая часть. Фазы разделяют, водную фазу экстрагируют этилацетатом (200 мл) и объединенные органические фазы промывают насыщенным рассолом (100 мл), сушат над безводным сульфатом натрия и выпаривают до получения смолы. Эту смолу кристаллизуют из этилацетата (60 мл) до получения указанного в заглавии соединения (4,30 г) в виде кристаллов с т.пл. 170-171оС.
Элементный анализ для C17H25N3O2S:
Рассчитано, C 60,9; H 7,5; N 12,5
Найдено, C 60,9; H 7,6; N 12,4
П р и м е р 3. N-метил-3-(1-метил-4-пиперидинил)-1Н-индол-5-этансульфонамид.
Раствор 4-гидразино-N-метил-бензолэтансульфонамида (0,5 г) и 1-метил-4-пиперидинацетальдегида (0,35 г) в смеси воды (10 мл) и 2н. соляной кислоты (1,0 мл, 2,00 ммоль) перемешивают в течение 2 дней при комнатной температуре. Добавляют следующую порцию альдегида (0,35 г) и перемешивание продолжают в течение еще 30 мин. Полученный раствор затем подщелачивают 8%-ным бикарбонатом натрия до рН 8 и экстрагируют хлороформом (3 х 50 мл). Объединенные органические экстракты сушат (Na2SO4) и выпаривают в вакууме до получения неочищенного гидразона в виде масла (1,0 г). Раствор гидразона (1,0 г) в хлороформе (20 мл), содержащем сложный полифосфатный эфир (10 г), нагревают при кипячении с обратным холодильником в течение 8 минут. Этот раствор выливают на лед (200 г), перемешивают в течение 2 ч, обрабатывают 2М карбонатом натрия (20 мл) и экстрагируют хлороформом (3 х 50 мл).
Объединенные органические экстракты сушат (Na2SO4), выпаривают в вакууме и остаток очищают на хроматографической колонке с мгновенным испарением (двуокись кремния 9385, 100 г), элюируя CH2Cl2:EtOH:NH3(75:8:1) до получения нечистого материала в виде желтого масла. Повторное хроматографирование с мгновенным испарением (двуокись кремния 9385, 100 г) и элюированием CH2Cl2: EtOH: NH3 (100:8:1) дает продукт в виде масла (0,05 г). Его кристаллизуют из этилацетата до получения указанного в заглавии соединения в виде твердого вещества, т. плавления 156-157оС.
ТСХ SiO2, CH2Cl2:EtOH:NH3 (50:8:1)
Rf 0,6
П р и м е р 4. N,N-диметил-3-(1-метил-4-пиперидинил)-1Н-индол-5-этансульфона- мид.
Гидрид натрия (60% мас/мас с парафином) осторожно добавляют к перемешиваемому раствору продукта примера 1 в сухом диметилформамиде (20 мл). Полученную смесь перемешивают при комнатной температуре в атмосфере азота в течение 0,25 ч, затем в поток добавляют раствор метилиодида (440 мг) в сухом диметилформамиде (1 мл). Полученную смесь перемешивают при комнатной температуре в течение 2,5 ч. Реакционную смесь гасят водой (3 мл) выпаривают в вакууме, а остаток хроматографируют на двуокиси кремния (150 г), элюируют дихлорметаном, этанолом и аммиаком (80:10:1) до получения смолы. Эту смолу быстро растирают в диэтиловом эфире и указанное в заглавии соединение кристаллизуют в виде порошка (238 мг), т. пл. 170-172оС.
ТСХ SiO2 (CH2Cl2:EtOH:NH3 50:8:1)
Rf 0,57.
П р и м е р 5.
(i) (E)-N,N-диметил-2- [3-(1-метил-4- пиперидинил)-1Н- индол-5-ил]этен- сульфонамид
Смесь 5-бром-3-(1-метил-4-пиперидинил)-1Н-индола (2,0 г), N,N-диметилэтенсульфонамид (1,184 г), три-орто-толилфосфина (0,6 г), ацетата палладия (0,1 г), триэтиламина (1,0 мл) и безводного ацетонитрила (12 мл) нагревают в двух 10-миллиметровых запаянных ампулах при перемешивании при 107оС (температура масляной бани) в течение 2,25 ч. Реакционную смесь объединяют, растворитель удаляют в роторном испарителе и остаток в виде пены очищают хроматографически, элюируя смесью дихлорметана, этанола, 0,88 аммиака (100: 80: 1). В результате роторного испарения соответствующих фракций получают продукт в виде пены (1,92 г) ТСХ SiO2 (изопропанол этанол вода 0,88 аммиак, 20:20:8:1).
Rf (основной) + 0,55 (меньший) + 0,4 (следы).
(ii) N,N-диметил-3-(1-метил-4-пиперидинил)-1Н-индол-5-этансульфонамид.
Раствор продукта со стадии (i) (5 г) в 200 мл этанола добавляют к взвеси 5% палладия на активированном угле (1,5 г) в этаноле (100 мл). Полученную смесь гидрируют при 65 пси при комнатной температуре в течение 17 ч. Смесь фильтруют и полученный фильтрат выпаривают, оставляя твердый продукт (1,0 г), который промывают изопропанолом (3 х 20 мл) до получения твердого продукта (0,8 г) т. плавления 215-225оС.
После кристаллизации из горячего этанола (60 мл) получают указанное в заглавии соединение в виде микроиголок (0,29 г), т.пл. 228-232оС.
ТСХ SiO2 (изопропанол эфир вода 0,88 аммиак, 20:20:8:1)
Rf 0,5.
П р и м е р 6. 3-(1-метил-4-пиперидинил)1Н-индол-5-этансульфонамид
(i) (E)-2-[3-(1-метил-4-пиперидинил)-1Н-индол-5-ил]-этенсульфонамид.
Смесь промежуточного соединения 2 (2,0 г), винилсульфонамида (0,88 г) ацетата палладия (100 мг), три(орто-толил)фосфина (0,60 г), триэтиламина (1,0 г) и ацетонитрила (14 мл) разделяют на две равные порции и помещают в две запаянные ампулы (10 мл) и нагревают при 100оС в течение 4 ч. В каждую ампулу добавляют дополнительное количество винилсульфонамида (0,22 г) и полученную смесь нагревают при 100оС еще 16 ч. Полученную смесь выпаривают досуха в вакууме и остаток очищают хроматографически (двуокись кремния 9385, 400 г) элюируя CH2Cl:EtOH:NH3(100:8:1) до получения указанного в заглавии соединения в виде твердого продукта (0,8 г) т.пл. 208-209оС.
(ii) 3-(1-метил-4-пиперидинил)-1Н-индол-5-этансульфонамид.
Смесь продукта со стадии (i) (0,8 г) в этанольном хлористом водороде (80 мл) гидрируют над предварительно восстановленном палладии на угле (50%-ная паста с водой, 0,8 г) до прекращения расхода водорода. Катализатор отфильтровывают, промывают горячим этанолом (50 мл) и фильтрат выпаривают в вакууме до получения неочищенного материала (0,15 г). Затем кристаллический остаток нагревают до 70оС с 2 н. соляной кислотой (200 мл), фильтруют и полученный фильтрат выпаривают досуха в вакууме (азеотропная перегонка с толуолом). Остаток объединяют с неочищенным продуктом полученным ранее, и очищают хроматографически с мгновенным испарением (двуокись кремния 9385, 100 г), элюируя СH2Cl2: EtOH: NH3 (50:8:1) до получения указанного в заглавии соединения в виде твердого продукта (0,3 г) т.пл. более 95оС (вспенивается).
ТСХ SiO2 (CH2Cl2:EtOH:NH3, 25:8:1)
Rf 0,5.
П р и м е р 7. N-метил-3-(4-пиперидинил)-1Н-индол-5-этансульфонамидгидро- хлорид.
(i) (E)-N-метил-2[3-(1-(фенилметил)-4-пиперидинил]-1H-индол-5-ил]этенсульфона- мид.
В каждой из трех запаянных ампул смесь промежуточного соединения 4 (1,10 г), N-метил-этенсульфонамида (422 мг), триэтиламина (843 мкл), три-орто-толилфосфина (242 мг) и ацетата палладия (39 мг) в сухом ацетонитриле (объем доводят до 10 мл) перемешивают и нагревают при 100оС в течение 4 ч. После охлаждения до 25оС содержание ампул объединяют, и растворитель выпаривают в вакууме при 40оС до получения маслянистого остатка.
Этот остаток очищают на хроматографической колонке на силикагеле (Merok 7229, 300 г), элюируя смесью дихлорметана, этанола, 0,88 аммиака (300:8:1 до 200:8:1 до 100:8:1). Соответствующие фракции объединяют и растворитель выпаривают в вакууме до получения указанного в заглавии соединения в виде пены (2,14 г).
ТСХ SiO2(CH2Cl2:EtOH:0,88 NH3, 200:8:1)
Rf 0,41
(ii) N-метил-3-(4-пиперидинил)-1Н-индол-5-этансульфонамид, гидрохлорид.
Раствор продукта со стадии (i) (2,14 г) в этанольном хлористом водороде (350 мл, полученный при добавлении ацетилхлорида (860 г) к этанолу (350 мл) при перемешивании гидрируют над предварительно восстановленном 10% палладии на древесном угле (6,4 г) при 25оС и давлении 1 атм в течение 18 ч. Реакционную смесь продувают азотом и добавляют раствор аммонийформата (8,2 г) в метаноле (100 мл). Полученную смесь перемешивают и доводят до кипения с обратным холодильником в атмосфере азота в течение 10 мин, охлаждают до 25оС, а затем катализатор удаляют фильтрованием. После выпаривании фильтрата в вакууме получают твердый остаток (8,5 г), который снова растворяют в воде (75 мл) и насыщают твердым хлоридом натрия. Образующийся осадок отфильтровывают, промывают ледяной водой (1,5 мл) и эфиром (10 мл) и сушат в вакууме при 45оС в течение 18 ч до получения указанного в заглавии соединения в виде кристаллического твердого продукта (640 мг), т.пл. 253-255оС.
ТСХ SiO2 (CH2Cl2:EtOH:0,88 NH3 25:8:1)
Rf 0,14.
П р и м е р 8. N-этил-3-(4-пиперидинил)-1Н-индол-5-этансульфонамид.
(i)(E)-N-этил-2-[3-(1-(фенилметил)- 4-пиперидинил]-1H-индол-5-ил]этенсульфонамид.
В каждую из двух 10 мл запаянных ампул помещают ацетат пластин (50 мл), три-орто-толилфосфин (300 мл), триэтиламин (650 мг), N-этилэтенсульфонамид (275 мг) и промежуточное соединение 4 (710 мг). Каждую смесь доводят до 110 л сухим ацетонитрилом. Ампулы нагревают при 100оС в течение 16 ч, затем оставляют при комнатной температуре на 4 дня. Содержимое запаянных ампул объединяют, и растворитель и триэтиламин удаляют в вакууме. Остаток обрабатывают хроматографически на двуокиси кремния (205 мг, merck 9385), элюируя дихлорметаном, этанолом и аммиаком (100:8:1) до получения пенообразного вещества (759 мг). Эту пену кристаллизуют из горячей смеси этилацетата и циклогексана до получения указанного в заглавии соединения (582 мг) в виде микрокристаллов, т.пл. 178-180оС.
(ii) N-этил-3-(4-пиперидинил)-1Н-индол-5-этансульфонамид.
Раствор продукта со стадии (i) (370 мг) в этанольном хлористом водороде, полученный при добавлении ацетилхлорида (105 г, 1,34 ммоль) к 1MS этанолу (50 мл) при перемешивании, гидрируют над предварительно восcтановленным 10% -ным оксидом палладия на угле (50% мас/мас с H2O; 1,13 г), при комнатной температуре и атмосферном давлении до тех пор, пока не прекращается расход водорода. Полученную смесь фильтруют и полученный фильтрат выпаривают до получения пены (280 мг), которую растворяют в метаноле (4 мл). Добавляют карбонат натрия (2н. 1 мл) и растворитель выпаривают. Остаток разделяют между водой (10 мл) и этилацетатом (50 мл). Водную фазу экстрагируют этилацетатом (50 мл) и объединенные органические фракции сушат (Na2SO4) и выпаривают до получения смолы (235 мг), которая кристаллизуется из смеси этилацетата и эфира (10 мл, главным образом, этилацетата) до получения указанного в заглавии соединения (104 мг) в виде порошка, т.пл. 95-100оС.
ТСХ SiO2 (CH2Cl2:EtOH:NH3, 25:5:1)
Rf 0,3.
П р и м е р 9. N-метил-3-(1-метил-4-пиперидинил)-1Н-индол-5-этансульфонамид, гидрохлорид.
Раствор продукта примера 1 (50 мг) в горячем этаноле (0,5 мл) добавляют к этанольному хлористом у водороду, полученному добавлением ацетилхлорида (33 мг, 0,420 ммоль) к этанолу (1 мл) при комнатной температуре в потоке при перемешивании, при комнатной температуре. Твердый продукт выкристаллизовывается из пеpвоначально прозрачного раствора. Полученную суспензию перемешивают и охлаждают до 5оС за 15 мин, затем фильтруют с подсосом. Остаток промывают небольшим количеством этанола, затем сушат при 60оС в вакууме в течение 1 ч до получения указанного заглавии соединения (44 мг) в виде микрокристаллов, т.пл. 237-239оС.
ТСХ SiO2 (CH2Cl2:EtOH:NH3, 50:8:1)
Rf 0,45.
Использование: в качестве веществ, обладающих 5HT1 подобной рецепторной антагонистической активностью. Сущность изобретения: продукт: производные индола общей формулы I; где R1 и R2 водород или C1-4 алкил; R3 водород или C1-3 алкил, или их фармацевтически приемлемые соли, или сольваты. 10 з. п. ф-лы. 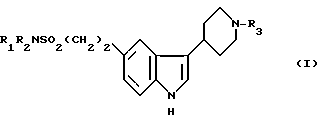
Производные индола общей формулы
где R1 и R2 водород или С1 С4-алкил;
R3 водород или С1 С3-алкил,
или их фармацевтически приемлемые соли или сольваты.
Приоритет по признакам:
14.06.88 все значения радикалов R1 R3.
| СПОСОБ ИЗМЕРЕНИЯ ЛИНЕЙНОЙ СКОРОСТИ | 1995 |
|
RU2124210C1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
