Настоящее изобретение относится к новым производным дикетопиперазина, обладающим сильным и селективным антагонистическим действием на рецептор окситоцина, к способу их получения, содержащих их фармацевтическим композициям и их использованию в медицине.
Гормон окситоцин оказывает сильное сокращающее действие на матку, и его используют для стимуляции или усиления родовой деятельности. Также плотность окситоциновых рецепторов в матке значительно увеличивается в >100 раз в ходе беременности и достигает максимума при родах (преждевременных и в срок).
Преждевременные роды/родовая деятельность (в диапазоне от 24 до 37 недель) является причиной примерно 60% детской смертности/заболеваемости, и, таким образом, соединение, которое ингибирует действие окситоцина на матку, например антагонист окситоцина, было бы полезным для предотвращения или подавления преждевременной родовой деятельности.
Международная заявка на патент РСТ/ЕР02/14823 описывает класс производных дикетопиперазина, которые проявляют в особенности пригодный уровень активности в качестве селективных антагонистов окситоцинового рецептора. Предпочтительный класс описываемых здесь соединений представлен формулой А
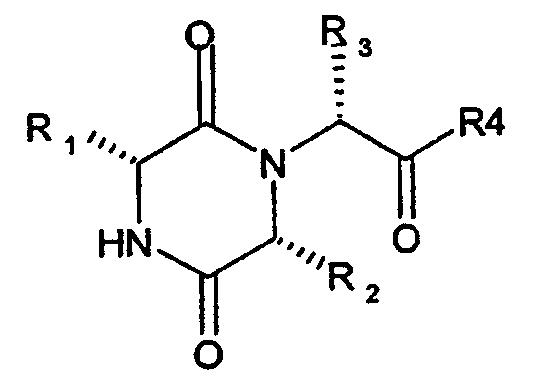 (А)
(А)
Такие соединения включают соединения, в которых среди прочих значений R1 представляет собой 2-инданил, R2 представляет собой С3-4-алкил, R3 представляет собой 5- или 6-членную гетероарильную группу, связанную с остатком молекулы через атом углерода цикла, R4 представляет собой группу NR5R6, в которой R5 и R6, каждый представляет собой алкил, например метил, или R5 и R6 вместе с атомом азота, к которому они присоединены, образуют 3-7-членный насыщенный гетероцикл, где указанный гетероцикл может содержать дополнительный гетероатом, выбранный из кислорода.
В настоящий момент заявители обнаружили новую группу селективных антагонистов рецептора окситоцина, которая обладает в особенности благоприятным фармакокинетическим профилем.
Настоящее изобретение относится, таким образом, к соединению формулы (I)
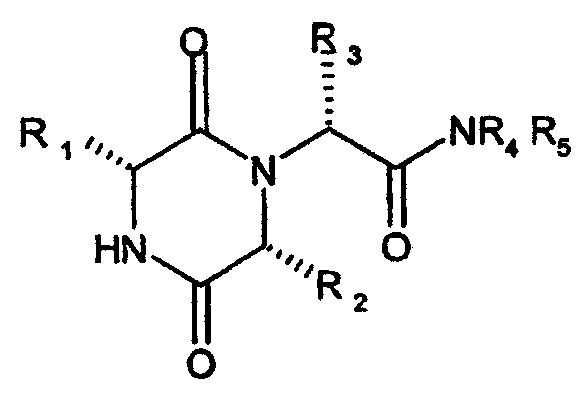 (I)
(I)
в котором R1 представляет собой 2-инданил, R2 представляет собой 1-метилпропил, R3 представляет собой 2-метил-1,3-оксазол-4-ил и R4 и R5 вместе с атомом азота, к которому они присоединены, представляют собой морфолино.
Группа R2 содержит асимметрический атом углерода, и изобретение включает в себя любой энантиомер и их смеси, включая рацемат.
Предпочтительное соединение по изобретению представляет собой соединение, получение которого конкретно описано в примере 1.
Соединения формулы (I) обладают высоким сродством к окситоциновым рецепторам в матке крыс и человека, и это может быть определено при использовании обычной методики. Например, сродство к окситоциновым рецепторам в матке крыс может быть определено при использовании методики Pettibone et al., Drug Developmental Research 30. 129-142 (1993). Соединения по изобретению также проявляют высокое сродство к человеческим рекомбинантным окситоциновым рецепторам в клетках СНО, и это может быть легко продемонстрировано при использовании методики, описанной Wyatt et al. Bioorganic & Medicinal Chemistry Letters, 2001 (11) p. 1301-1305.
Соединения по изобретению также демонстрируют благоприятный фармакокинетический профиль, включая хорошую биологическую доступность и низкий собственный клиренс при назначении внутривенно (i.v.) или перорально (p.o.), в сочетании с хорошей стабильностью по отношению к Р450 ферментам, включая 2С9, и хорошую растворимость в воде.
Соединения по изобретению, таким образом, являются пригодными для лечения или предотвращения заболеваний и/или состояний, связанных с действием окситоцина. Примеры таких заболеваний и/или состояний включают преждевременную родовую деятельность, дисменорею, эндометриоз и доброкачественную гиперплазию предстательной железы.
Соединения могут также быть пригодными для задержки родовой деятельности перед выбранным кесаревым сечением или переводом пациента в центр высокоспециализированного (третичного) лечения, лечения сексуальной дисфункции, в частности преждевременного семяизвержения, ожирения, нарушений в приеме пищи, застойной сердечной недостаточности, повышенного артериального давления, цирроза печени, нефрита или повышенного внутриглазного давления, абсессивно-компульсивных расстройств и психоневрологических расстройств. Соединения по изобретению можно также использовать для увеличения показателей плодовитости у животных, например сельскохозяйственных животных.
Изобретение таким образом описывает использование соединения формулы (I) для применения в терапии и, в особенности, использование в качестве лекарственного препарата, направленного против действия окситоцина на окситоциновый рецептор.
Изобретение также предлагает использование соединения формулы (I) для производства медицинского препарата, направленного против действия окситоцина на окситоциновый рецептор.
Согласно дополнительному аспекту изобретение также обеспечивает способ противодействия действию окситоцина на окситоциновый рецептор, включающий введение пациенту, который в этом нуждается, антагонистического количества соединения формулы (I).
Специалисту в данной области техники будет понятно, что ссылка здесь на лечение распространяется также и на профилактику, как и на лечение установленных заболеваний или симптомов.
Также будет понятно, что количество соединения по изобретению, требуемое для использования при лечении, будет изменяться в зависимости от природы подвергаемого лечению состояния, способа введения, возраста и состояния пациента и будет исключительно на усмотрении лечащего врача. Однако обычно применяемые дозы для лечения взрослого человека будут типично в диапазоне от 2 до 800 мг в день в зависимости от способа введения.
Таким образом, для парентерального введения дневная доза будет обычно составлять в диапазоне от 2 до 50 мг, предпочтительно от 5 до 25 мг в день. В случае орального введения дневная доза будет обычно находиться в диапазоне от 10 до 800 мг, например от 20 до 150 мг в день.
Требуемая доза может быть удобно представлена в виде разовой дозы или в виде разделенных доз, вводимых через подходящие интервалы, например, в виде двух, трех, четырех или более дробных доз в день.
Несмотря на то что возможно для использования вводить соединение по изобретению в виде химического вещества как такового, предпочтительно, чтобы активный ингредиент был представлен в виде фармацевтического состава.
Изобретение таким образом дополнительно обеспечивает фармацевтический состав, включающий соединение формулы (I) совместно с одним или несколькими фармацевтически приемлемыми носителями и, необязательно, другими терапевтическими и/или профилактическими ингредиентами. Носитель(и) должен быть «приемлемым» с точки зрения совместимости с другими ингредиентами состава и не являться вредным по отношению к их реципиенту.
Композиции по изобретению включают композиции в форме, специально предназначенной для орального, защечного, парентерального, ингаляционного или нагнетаемого, имплантированного или ректального введения.
Таблетки и капсулы для орального введения могут содержать обычные эксципиенты, такие как связывающие агенты, например сироп, гуммиарабик, желатин, сорбит, трагакант, крахмальный клейстер или поливинилпирролидон; наполнители, например лактозу, сахар, микрокристаллическую целлюлозу, кукурузный крахмал, фосфат кальция или сорбит; смазывающие вещества, например стеарат магния, стеариновую кислоту, тальк, полиэтиленгликоль или оксид кремния; дезинтегрирующие агенты, например картофельный крахмал или крахмал гликолят натрия, или смачивающие агенты, такие как лаурилсульфат натрия. Таблетки могут быть покрытыми согласно способам, хорошо известным в данной области техники. Жидкие составы для орального применения могут быть в форме, например, водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров или могут быть представлены в виде сухого продукта для составления с водой или другим пригодным наполнителем перед использованием. Такие жидкие составы могут содержать обычные добавки, такие как суспендирующие агенты, например сироп сорбита, метилцеллюлозу, сироп глюкозы/сахара, желатин, гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу, гель стеарата алюминия или гидрогенированные пищевые жиры; эмульгирующие агенты, например лецитин, моноолеат сорбита или гуммиарабик; неводные наполнители (которые могут включать пищевые масла), например миндальное масло, фракционированное кокосовое масло, масляные сложные эфиры, пропиленгликоль или этиловый спирт; разбавители, такие как поверхностно-активные вещества, например полисорбаты или другие агенты, такие как циклодекстрины; и консерванты, например метил или пропил п-гидроксибензоаты или аскорбиновую кислоту. Композиции могут также быть составлены в виде суппозиториев, например, содержащих традиционные основы для суппозиториев, такие как масло какао или другие глицериды.
Для защечного (буккального) введения композиции могут быть в форме таблеток или лепешек, составленных традиционным образом.
Композиции по изобретению могут быть составлены для парентерального введения при помощи инъекции или продолжительной инфузии. Составы для инъекций могут быть представлены в виде форм разовых доз в ампулах или в виде контейнеров с множеством доз с добавленным консервантом. Композиции могут быть в таких формах, как суспензии, растворы или эмульсии в масляных или водных наполнителях, и могут содержать формулирующие агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. В качестве альтернативы, активный ингредиент может быть в форме порошка для составления перед использованием с подходящим наполнителем, например стерильной апирогенной водой.
Композиции по изобретению могут содержать в диапазоне от 0,1 до 99% активного ингредиента, обычно 1-50% в случае таблеток и капсул, и 3-50% для жидких составов.
Благоприятный фармакокинетический профиль соединений по изобретению легко доказывается при использовании обычных методик измерения фармакокинетических свойств биологически активных соединений.
Соединения формулы (I) могут быть получены реакцией карбоновой кислоты (II, в которой R1, R2 и R3 имеют те же значения, как и определено в формуле I)
 (II)
(II)
или ее активированного производного с амином NHR4R5, где NR4R5 имеет значение, определенное в формуле (I), в стандартных условиях получения амидов из карбоновой кислоты или его смешанного ангидрида и амина HNR4R5.
Таким образом, амид формулы (I) может быть получен обработкой карбоновой кислоты формулы (II) активирующим агентом, таким как BOP (бензотриазол-1-илокси-трис(диметиламино)фосфоний гексафторфосфат), TBTU (2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний тетрафторборат), BOP-Cl (бис(2-оксо-3-оксазолидинил)фосфинхлорид) или оксалилхлорид в апротонном растворителе, таком как дихлорметан, необязательно в присутствии третичного амина, такого как триэтиламин, и с последующей реакцией таким образом полученного продукта с амином NHR4R5.
В качестве альтернативы, амид формулы (I) может быть получен реакцией смешанного ангидрида, полученного из карбоновой кислоты (II), с амином NHR4R5 в апротонном растворителе, таком как тетрагидрофуран. Реакцию без труда проводят при низких температурах, например примерно при -78°С. Смешанный ангидрид без труда получают реакцией карбоновой кислоты (II) с подходящим хлоридангидридом кислоты, например пиваволил хлоридом в апротонном растворителе, таком как этилацетат, в присутствии третичного органического основания, такого как триалкиламин, например триэтиламин, и при низких температурах, например примерно при -78°С.
Соединения формулы (I) можно также получать реакцией соединения формулы (III)
 (III)
(III)
(в котором R1, R2 и R3 имеют значения, определенные в формуле (I), и R6 представляет собой 2-гидроксифенил) с карбонилдиимидазолом или тиокарбонилдиимидазолом в подходящем растворителе, таком как дихлорметан, и последующей реакцией таким образом полученных продуктов с амином HNR4R5.
Соединения формулы (II) могут быть получены из соединения формулы (III), в котором R6 представляет собой 2-гидроксифенил, реакцией с карбонилдиимидазолом или тиокарбонилдиимидазолом в подходящем растворителе, таком как дихлорметан, и последующей реакцией таким образом полученного продукта с водным раствором ацетона.
Соединения формулы (III), в которых R6 представляет собой 2-гидроксифенил, могут быть получены из соответствующих соединений формулы (III), в которых R6 представляет собой 2-бензилоксифенильную группу за счет гидрогенолиза при использовании водорода и палладиевого катализатора.
Соединения формулы (III), в котором R6 представляет собой 2-бензилоксифенильную группу, без труда получают способом, описанным здесь далее. Таким образом, соединения формулы (III) могут быть получены из соединения формулы (IV)
 (IV)
(IV)
в которой R1, R2 и R3 имеют те же значения, как определено в формуле (I), R7 представляет собой 2-бензилоксифенил и R8 представляет собой N-бензилоксикарбонил, реакцией с водородом в присутствии катализатора палладий на угле и уксусной кислоты. Реакцию без труда проводят в растворителе, таком как этанол или трифторэтанол или их смесях.
Соединение формулы (IV) может быть получено реакцией аминосодержащего гидрохлорида сложного эфира (V), в котором R2 имеет то же значение, как определено в формуле (I)
 (VI)
(VI)
с альдегидом R3СНО (VI), в котором R3 имеет то же значение, как определено в формуле (I), в присутствии триэтиламина и в растворителе, таком как трифторэтанол, и затем реакцией полученного продукта с соединением (VII), в котором R1 имеет то же значение, как определено в формуле (I), и R7 представляет собой бензилоксикарбонил
 (VII)
(VII)
и изоцианид CNR6 (VIII), в котором R6 представляет собой 2-бензилоксифенильную группу, в растворителе, таком как трифторэтанол.
Заместитель R2 представляет собой 1-метилпропильную группу, и соединения формулы (I), в которых R1 представляет собой 1-метилпропильную группу, имеющую (S)- или (R)-конфигурацию, могут быть получены, исходя из сложного аминоэфира (V), в котором R2-группа имеет требуемую (S)- или (R)-конфигурацию.
Последующие примеры являются иллюстративными, но не ограничивающими вариантами осуществления настоящего изобретения.
Основные способы очистки и анализа
Аналитическую ВЭЖХ проводили на Supercosil LCABZ+PLUS колонке (3,3 см × 4,6 мм ID), при элюировании 0,1% HCO2H и 0,01 М ацетатом аммония в воде (растворитель А), и 0,05% HCO2H 5% воды в ацетонитриле (растворитель В) при использовании градиента элюирования 0-0,7 минут 0% В, 0,7-4,2 минуты 0%-100% В, 4,2-5,3 минут 100% В, 5,3-5,5 минут 0% В при скорости потока 3 мл/минуту. Масс-спектры регистрировали на Fision VG Platform спектрометре с использованием электрораспыления положительных ионов [(ES+ve с получением МН+ и М(NH4)+ молекулярных ионов] или электрораспыления отрицательных ионов [(ES-ve с получением (М-Н)- молекулярного иона] на Micromass series 2 или Waters ZQ масс спектрометре. 1Н-ЯМР спектры регистрировали при использовании Bruker DPX 400 МГц спектрометра при использовании тетраметилсилана в качестве внешнего стандарта. BiotageTM хроматография относится к очистке с использованием оборудования, поставляемого Dyax Corporation (или Flash 40i, или Flash 150i), и картриджей, заполненных KPSil. Автоматическая подготовка образцов для масс-спектроскопии относится к способам, в которых вещество очищали высокоэффективной жидкостной хроматографией на HPLCABZ +5 мкм колонке (5 см × 10 мм i.d.) с 0,1% HCO2H в воде и 95% MeCN, 5% воды (0,5% HCO2H) с использованием градиента элюирования при скорости потока 8 мл в минуту. Gilson 202-фракционный коллектор инициировали VG Platform масс-спектрометром при определении интересующей массы.
Гидрофобные фритты относятся к фильтрационным трубкам, поставляемым Whatman. SPE (твердофазная экстракция) относится к использованию картриджей, поставляемых International Sorbent Technology Ltd. ТСХ (TLC) (тонкослойная хроматография) относится к использованию пластинок для ТСХ, поставляемых Merk, покрытых силикагелем 60 F254. OasisTM относится к картриджам Waters® OasisTM для HLB экстракции, поставляемым Waters Corporation®.
Промежуточный продукт 1
2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-N-(2-гидроксифенил)-2-(2-метил-1,3-оксазол-4-ил)ацетамид
К энергично перемешиваемому раствору гидрохлорида сложного метилового эфира (D)-аллоизолейцина (5,0 г) в дихлорметане (150 мл) добавляли насыщенный раствор бикарбоната натрия (150 мл). Полученный двойной слой разделяли с использованием гидрофобной фритты и водную фазу дважды промывали дихлорметаном (50 мл). Объединенную дихлорметановую фазу разбавляли метанолом (200 мл), добавляли (2R)-[(бензилоксикарбонил)амино](2,3-дигидро-1Н-инден-2-ил)этановую кислоту (14,64 г) и смесь энергично перемешивали в течение 1 часа до получения раствора. Раствор упаривали и остаток растворяли в смеси 1:1 трифторэтанол/метанол (140 мл), затем добавляли 2-бензилоксифенилизоцианид (9,43 г) с последующим добавлением 2-метил-4-формилоксазола (5,0 г) и реакцию перемешивали в течение 4 дней при комнатной температуре. Смесь испаряли, и остаток растворяли в этаноле (500 мл), и добавляли палладий на угле (4,0 г) и уксусную кислоту (10 мл), реакционную смесь перемешивали в атмосфере водорода в течение 3 часов. Дополнительно добавляли свежий палладий на угле (4,0 г) и уксусную кислоту (20 мл) и реакционную смесь перемешивали в атмосфере водорода в течение дополнительных 16 часов. Смесь фильтровали через целит, упаривали и остаток, растворенный в этилацетате (300 мл), промывали водой (2×100 мл), насыщенным раствором бикарбоната натрия (2×100 мл) и насыщенным раствором соли (100 мл) и затем пропускали через гидрофобную фритту и выпаривали. Неочищенный продукт очищали при помощи колоночной хроматографии (силикагель), при элюировании смесью этилацетат (100% - 0%): метанол с получением 2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-N-(2-гидроксифенил)-2-(2-метил-1,3-оксазол-4-ил)ацетамида (11,8 г, 51%).
Rt ВЭЖХ = 3,2 минуты; m/z [M+H]+ = 517.
Сходным образом продукт получали из гидрохлорида сложного метилового эфира (D)-изолейцина.
Промежуточный продукт 2
2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1R)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-N-(2-гидроксифенил)-2-(2-метил-1,3-оксазол-4-ил)ацетамид
Rt ВЭЖХ = 3,17 и 3,22 минуты; m/z [M+H]+ = 517.
Промежуточный продукт 3
{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}(2-метил-1,3-оксазол-4-ил)уксусная кислота
Карбонилдиимидазол (352 мг, 1,6 экв.) добавляли к раствору 2-{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}-N-(2-гидроксифенил)-2-(2-метил-1,3-оксазол-4-ил)ацетамида (11,8 г, предварительно высушенный под вакуумом над P4O10 в течение 24 часов) в дихлорметане (20 мл) и раствор оставляли при комнатной температуре на 16 часов. Смесь упаривали и остаток растворяли в ацетоне (20 мл) и добавляли воду (20 мл) с последующим добавлением 2 н. HCl (2 мл) и смесь оставляли при комнатной температуре в течение 4,5 часов. Смесь экстрагировали этилацетатом (2×30 мл) и объединенную органическую фазу сушили над гидрофобной фриттой и выпаривали. Остаток переносили в этилацетат (30 мл), промывали 2 н. HCl (2×10 мл) и затем экстрагировали насыщенным раствором бикарбоната натрия (2×15 мл). Объединенную водную фазу подкисляли 2 н. HCl и экстрагировали этилацетатом (2×20 мл), объединенную органическую фазу промывали насыщенным раствором соли, сушили над гидрофобной фриттой и выпаривали с получением {(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}(2-метил-1,3-оксазол-4-ил)уксусной кислоты (0,355 мг, 73%) в виде белого твердого вещества.
Rt ВЭЖХ = 3,0 и 3,1 минуты; m/z [M+H]+ = 426.
Сходным образом получали из промежуточного продукта 2
{(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1R)-1-метилпропил]-2,5-диоксо-1-пиперазинил}(2-метил-1,3-оксазол-4-ил)уксусную кислоту (промежуточный продукт 4)
Rt ВЭЖХ = 3,14 минуты; m/z [M+H]+ = 426.
Пример 1
(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-1-[(1R)-1-(2-метил-1,3-оксазол-4-ил)-2-(4-морфолинил)-2-окосоэтил]-6-[(1S)-1-метилпропил]-2,5-пиперазиндион
Диизопропилэтиламин (100 мг, 3,3 экв.) pyBOP (159 мг, 1,3 экв.) и морфолин (102 мкл, 5 экв.) последовательно добавляли к раствору {(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-6-[(1S)-1-метилпропил]-2,5-диоксо-1-пиперазинил}(2-метил-1,3-оксазол-4-ил)уксусной кислоты (100 мг) в диметилформамиде (2 мл) и смесь перемешивали в течение 4 дней при комнатной температуре. Реакцию разбавляли дихлорметаном (10 мл) и добавляли 2 н. HCl (10 мл). Органическую фазу отделяли, промывали насыщенным раствором бикарбоната натрия (10 мл), сушили над фриттой и выпаривали. Остаток очищали препаративной ВЭЖХ с получением (3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-1-[(1R)-1-(2-метил-1,3-оксазол-4-ил)-2-(4-морфолинил)-2-окосоэтил]-6-[(1S)-1-метилпропил]-2,5-пиперазиндиона (9 мг) в виде бесцветного твердого вещества.
Rt ВЭЖХ = 2,8 минуты; m/z [M+H]+ = 495.
1H-ЯМР (CDCl3) δ 7,72 (с, 1Н), 7,26-7,15 (м, 4Н), 6,93 (д, 1Н), 6,30 (с, 1Н), 4,18 (д, 1Н), 4,06 (дд, 1Н), 3,70-3,30 (м, 8Н), 3,17-3,10 (м, 3Н), 2,98-2,86 (м, 1Н), 2,81-2,75 (м, 1Н), 2,49 (с, 3Н), 1,69-1,60 (м, 1Н), 1,50-1,43 (м, 1Н), 1,05-0,95 (м, 1Н), 0,80-0,75 (м, 6Н).
Сходным образом продукт получали из промежуточного продукта 4 и морфолина.
Пример 2
(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-1-[(1R)-1-(2-метил-1,3-оксазол-4-ил)-2-(4-морфолинил)-2-окосоэтил]-6-[(1R)-1-метилпропил]-2,5-пиперазиндион
Rt ВЭЖХ = 2,92 минуты; m/z [M+H]+ = 495.
Пример 3
(3R,6R)-3-(2,3-дигидро-1Н-инден-2-ил)-1-[(1R)-1-(2-метил-1,3-оксазол-4-ил)-2-(4-морфолинил)-2-окосоэтил]-6-[(1S)-1-метилпропил]-2,5-пиперазиндион
(2R)-[(бензилоксикарбонил)амино](2,3-дигидро-1Н-инден-2-ил)уксусную кислоту (35,84 г, 0,110 моль) в 500 мл круглодонной колбе обрабатывали 2,2,2-трифторэтанолом (165 мл) с последующим добавлением метанола (55 мл) и триэтиламина (11,13 г, 15,33 мл, 0,110 ммоль), суспензию перемешивали в течение 3,5 часов, пока не наблюдалось растворение. Затем раствор добавляли к гидрохлориду сложного метилового эфира (D)-аллоизолейцина (20 г, 0,110 моль) в отдельной колбе. Суспензию перемешивали, пока не наблюдалось растворение. Затем добавляли 2-метил-4-формилоксазол (12,24 г, 0,110 ммоль) с последующим добавлением 2-бензилоксифенилизоцианида (23,04 г, 0,110 ммоль). Темно-коричневую реакционную смесь затем перемешивали при 20-25°С в течение 24 часов. Раствор концентрировали отгонкой до объема около 130 мл при пониженном давлении. Раствор разбавляли дихлорметаном (200 мл) и промывали водой (2×200 мл). Органическую фазу затем растворяли N-метилпирролидиноном (460 мл) и дихлорметан удаляли при перешивании при 40°С под вакуумом в течение 2 часов. Затем добавляли уксусную кислоту (46 мл) с последующим добавлением катализатора палладий на угле (69,0 г 10 мас.% Pd, 57% воды, Johnson Matthey тип 87L) и смесь гидрировали водородом при баллонном давлении и быстром перемешивании в течение 2 часов. Реакционную смесь фильтровали, полностью промывали этилацетатом (960 мл) и промывали 3 мас./об.% водным раствором хлорида натрия (960 мл). Двухфазную смесь фильтровали и органическую фазу отделяли и промывали 3 мас./об.% водным раствором хлорида натрия (2×960 мл). Органический раствор затем разбавляли этилацетатом (200 мл) и концентрировали перегонкой при атмосферном давлении, отгоняя 385 мл растворителя. Концентрированный раствор при 20-25°С обрабатывали 1,1'-карбонилдиимидазолом (21,46 г, 0,132 моль) и перемешивали при 20-25°С в течение 1 часа, затем обрабатывали водой (290 мл) и быстро перемешивали при 20-25°С в течение 24 часов. Смеси предоставляли возможность отстояться и слой этилацетата отделяли и отбрасывали. Водную фазу промывали этилацетатом (290 мл) и смеси предоставляли возможность отстояться и водную фазу отделяли и подкисляли до рН 1-2 добавлением концентрированной хлористоводородной кислоты (18 мл). Водную фазу затем экстрагировали в этилацетат (290 мл и затем 145 мл). Объединенный раствор этилацетата затем концентрировали перегонкой при атмосферном давлении до объема примерно 93 мл. Этот раствор затем разбавляли тетрагидрофураном (62 мл), и обрабатывали триэтиламином (11,02 г, 15,20 мл, 0,109 моль), и охлаждали до -78°С. Раствор затем обрабатывали триметилацетилхлоридом (4,81 г, 4,92 мл, 39,90 ммоль) и перемешивали при -78°С в течение 7 часов. Реакционную смесь затем обрабатывали раствором морфолина (15,82 г, 15,83 мл, 0,181 моль) в тетрагидрофуране (23 мл) и перемешивали при -78°С в течение 1 часа 20 минут перед тем, как ее оставляли нагреваться до 20-25°С. Раствор затем разбавляли этилацетатом (76 мл) и промывали насыщенным водным раствором бикарбоната натрия (2×153 мл) с последующим промыванием водой (153 мл). Органический раствор затем разбавляли этилацетатом (54 мл) и упаривали до объема 69 мл при атмосферном давлении. Раствор затем охлаждали до 20-25°С, в данной точке наблюдалась кристаллизация указанного в заголовке соединения. Суспензию указанного в заголовке соединения затем дополнительно охлаждали до 0°С перед тем, как указанное в заголовке соединение отделяли фильтрованием и отсасывали досуха. Выход 8,92 г.
Фармацевтические примеры
Данные примеры иллюстрируют получение репрезентативных фармацевтических составов для введения, содержащих соединение по изобретению.
А. Состав для парентерального введения
Ингредиенты
Соединение по изобретению диспергировали в спирте и растворяли в пропиленгликоле при нагревании. Водный компонент затем добавляли при перемешивании с получением 10 мл раствора для внутривенного введения (i.v.). Данный раствор можно стерилизовать подходящими способами, такими как асептическая фильтрация или автоклавирование.
Его можно вводить в виде болюса или разводить в сосуде для инфузии, содержащем, например, нормальный физиологический раствор.
В. Капсулы для орального введения
Указанные выше ингредиенты смешивали и помещали в твердые желатиновые капсулы, каждая содержащая 100 мг.
С. Таблетки для орального введения
Указанные выше ингредиенты, за исключением стеарата магния, объединяли и смешивали. Затем добавляли стеарат магния и состав перемешивали. Состав формовали в таблетки при использовании подходящей таблетирующей машины.
Измерения активности в качестве антагониста окситоцина
Тест-буфер, использованный в ходе анализа: 50 мМ HEPES, 10 мМ MgCl2, 0,125 мг/мл BSA, рН устанавливали при 7,4 с использованием КОН. hOT-CHO мембраны подготавливали при концентрации 0,3 мг белка/мл в тест-буфере. Исследуемые соединения сначала растворяли в ДМСО (до 10 мМ) и разбавляли ДМСО (Beckman Biomek FX). 1 мкл соединения переносили на черные планшеты для анализа 384 (NUNC), используя Biomek FX. 20 мкл 1 нМ Bodipy TMR окситоцина (Perkin Elmer) в тест-буфере добавляли во все лунки (Labsystems Multidrop), затем во все лунки добавляли 20 мкл мембраны (Multidrop). Планшеты инкубировали при комнатной температуре в течение 60 минут.
Поляризацию определяли на LJL Analyst (λEx = 535 нм, λEm = 580 нМ, λдихроичная = 555 нм). Данные подгоняли по 4 параметрам логистического уравнения. Полученное значение Ki рассчитывали как IC50/5.
Указанные выше тест-соединения из примеров 1 и 2 по изобретению имели pKi величины 9,0 и 8,2 соответственно.
Соединения по изобретению, по существу, нетоксичны в терапевтически активных дозах. Так, соединение из примера 1 вводили крысам в дозах 30 мг/кг в течение 7 дней, и не было отмечено нежелательных токсикологических эффектов.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ДИКЕТОПИПЕРАЗИНЫ КАК АНТАГОНИСТЫ ОКСИТОЦИНА | 2002 |
|
RU2303032C2 |
| ПИПЕРАЗИНДИОНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРА ОКСИТОЦИНА | 2005 |
|
RU2382038C2 |
| ПРОИЗВОДНОЕ ПИПЕРИДИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2730508C2 |
| N-ДИГИДРОКСИАЛКИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 2-ОКСОИМИДАЗОЛА | 2006 |
|
RU2414456C2 |
| СП0СОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ ДИГИДРОИНДЕНАМИДА, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИИ, СОДЕРЖАЩИЕ ДАННЫЕ СОЕДИНЕНИЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗЫ | 2009 |
|
RU2528408C2 |
| ПРОИЗВОДНЫЕ ИНДАНУКСУСНОЙ КИСЛОТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2002 |
|
RU2314298C2 |
| ЗАМЕЩЕННЫЕ ТИОФЕНКАРБОКСАМИДЫ И АНАЛОГИ В КАЧЕСТВЕ АНТИБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2019 |
|
RU2799335C2 |
| ПРОИЗВОДНЫЕ БОРОНОВОЙ КИСЛОТЫ | 2018 |
|
RU2793315C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| ИНГИБИТОРЫ БЕТА-СЕКРЕТАЗЫ | 2016 |
|
RU2712272C2 |
Настоящее изобретение относится к замещенным дикетопиперазинам общей формулы I

в котором R1 представляет собой 2-инданил, R2 представляет собой 1-метилпропил, R3 представляет собой 2-метил-1,3-оксазол-4-ил и R4 и R5 вместе с атомом азота, к которому они присоединены, представляют собой морфолино. Соединения обладают антагонистическим действием на рецептор окситоцина, что позволяет их использовать в медицине. Описаны также способы получения соединений I, фармацевтическая композиция на их основе и применение. 7 н. и 4 з.п. ф-лы.

в котором R1 представляет собой 2-инданил, R2 представляет собой 1-метилпропил, R3 представляет собой 2-метил-1,3-оксазол-4-ил и R4 и R5 вместе с атомом азота, к которому они присоединены, представляют собой морфолино.
(а) взаимодействие соединения формулы (II)

в котором R1, R2 и R3 имеют значения, определенные в п.1, или его смешанного ангидрида с амином NHR4R5, в котором R4 и R5 имеют значения, определенные в формуле (I), в стандартных условиях получения амидов из карбоновой кислоты или ее смешанного ангидрида и амина.
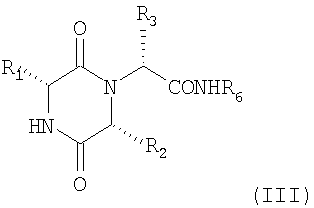
в котором R1, R2 и R3 имеют значения, определенные в п.1, и R6 представляет собой 2-гидроксифенил с карбонилдиимидазолом или тиокарбонилдиимидазолом в подходящем растворителе и последующую реакцию полученного таким образом продукта с амином NHR4R5, в котором R4 и R5 имеют значения, определенные в формуле (I).
| ВПИТЫВАЮЩЕЕ ИЗДЕЛИЕ С ЭЛАСТОМЕРНЫМ МАТЕРИАЛОМ | 2003 |
|
RU2326639C2 |
| US 5464788, 07.11.1995 | |||
| Прибор, замыкающий сигнальную цепь при повышении температуры | 1918 |
|
SU99A1 |

