Настоящее изобретение касается способа получения N-метил-3-(1-метил-4-пиперидинил)-1H-индол-5-этансульфонамида и его физиологически приемлемых солей, а также используемого для их получения нового промежуточного продукта, N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-3-пиридинил)-1H-индол- 5-ил] этенсульфонамида, и способов его получения.
N-метил-3-(1-метил-4-пиперидинил)-1H-индол-5-этансусльфонамид, который может быть представлен формулой I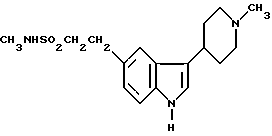
и его физиологически приемлемые соли и сольваты известны (GB2208646). Это соединение проявляет избирательную сосудосуживающую активность и показано для использования при лечении мигрени.
Известны (GB2208646) способ получения предложенных соединений, который включает восстановление соответствующего производного 3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-индол-5-этансульфонамида, и способ, который включает восстановление соответствующего производного 3-(1-метил-4-пиперидинил)-индол-5-этенсульфонамида. Способ, который включает восстановление производного 3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-индол-5-этенсульфонамида, не описан.
Задача, на решение которой направлена заявленная группа изобретений, заключается в создании способа получения соединения формулы I из нового диенового промежуточного соединения, обеспечивающего хороший выход и высокую степень чистоты получаемого продукта, а также в разработке способов получения указанного промежуточного соединения.
Данная задача решается тем, что предложен способ получения N-метил-3-(1-метил-4-пиперидинил)-1H-индол-5-этансульфонамида формулы I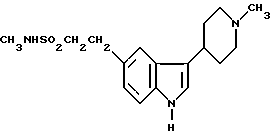
или его физиологически приемлемой соли, отличающийся тем, что восстановлению подвергают соединение формулы II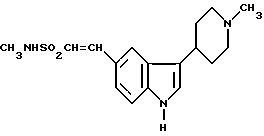
в присутствии водорода, катализатора из благородного металла и растворителя.
Предпочтительно, чтобы катализатор из благородного металла представлял собой палладий, оксид палладия, никель Рэнея, платину, оксид платины или родий, желательно нанесенные на уголь, более предпочтительно, чтобы катализатор из благородного металла представлял собой оксид палладия на угле. Наиболее предпочтительно, чтобы катализатор из благородного металла представлял собой 10%-ный оксид палладия на угле.
Целесообразно оксид палладия добавлять в реакционный сосуд в виде влажной пасты.
Восстановление целесообразно осуществлять в растворителе, включающем воду или спирт, простой эфир, сложный эфир или амид или их смесь. Предпочтительные растворители представляют собой воду, метанол, этанол, диоксан, этилацетат, диметилформамид или их смесь.
Восстановление целесообразно осуществлять при температуре от 10 до 50oC.
Вышеуказанная задача решается также тем, что предложен N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H-индол-5-ил] этенсульфонамид для использования в качестве промежуточного соединения.
Для решения поставленной задачи предложен способ получения N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H-индол-5-ил] этенсульфонамида, при котором соединение формулы III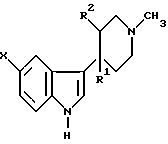
или его соль, где R1 представляет собой гидрокси-группу, и R2 представляет собой водород, или R1 и R2 вместе образуют двойную связь, X представляет собой уходящий атом, такой как атом галогена, например атом брома, или уходящую группу, например трифлатную (CF3SO3) группу, конденсируют с N-метил-винилсульфонамидом формулы IV
CH2=CHSO2NZCH3
где Z представляет собой водород или аминозащитную группу, и возможно, если необходимо и/или желательно, снимают защиту с полученного таким образом защищенного производного.
Предложен также способ получения N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H-индол-5-ил] этенсульфонамида, при котором метилируют соединение формулы VII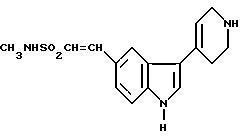
Предложен способ получения N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил-)-1H-индол-5-ил] этенсульфонамида, при котором соединение формулы X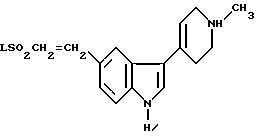
где L представляет собой приемлемую уходящую группу, конденсируют с метиламином.
Кроме того, предложен способ получения N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H-индол-5-ил] этенсульфонамида, при котором соединение формулы XII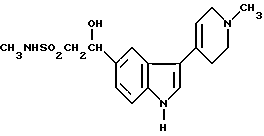
дегидратируют в присутствии кислоты или основания.
Также предложен способ получения N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H-индол-5-ил] этенсульфонамида, отличающийся тем, что соединение формулы VIII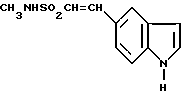
подвергают взаимодействию с соединением формулы VI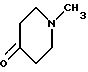
И наконец, предложен способ получения N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H-индол-5-ил] этенсульфонамида, отличающийся тем, что дегидратируют соединение формулы XVI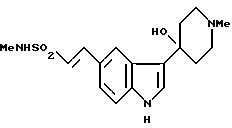
Типичные аминозащитные группы, используемые при реализации первого из предложенных способов получения соединения формулы (II), хорошо известны специалистам и могут быть использованы обычным способом (см. например, "Protective Groups in Organic Chemistry". Ed. J.F.W. McOmie (Plenum Press 1973), "Protective Groups in Organic Synthesis", T.W. Green (John Wiley 7& Sons 1981). Так, например, аминозащитные группы включают в себя третичный бутил, силил, например триметилсилил, аралкильные группы и ацильные группы. Удаление таких групп может быть достигнуто обычными методами.
При реализации этого способа реакцию обычно осуществляют в присутствии палладиевого катализатора, такого как, например, палладий или оксид палладия на угле, или соль или комплекс палладия. Соли палладия, которые могут быть использованы как катализаторы, включают соли органических кислот, такие как ацетаты, или соли неорганических кислот, такие как хлориды или бромиды. Комплексы палладия включают, например, тетрахлорпалладат лития или нуль-валентные комплексы, такие как бис(дибензилиденацетон) палладий и тетракис(трифенилфосфин)палладий. Предпочтительным катализатором является ацетат палладия.
Желательно, чтобы реакция могла быть осуществлена в присутствии основания, такого как триэтиламин или три-н-бутиламин, карбоната щелочного металла, такого как карбонат натрия, гидрокарбоната щелочного металла, такого как гидрокарбонат натрия, или ацетата щелочного металла, такого как ацетат калия, желательно вместе с катализатором фазового переноса, таким как хлорид тетрабутиламмония.
Данная реакция, возможно, может быть осуществлена в присутствии фосфина, например триарилфосфина, такого как трифенилфосфин или три-о-толилфосфин или фосфинированный полистирол или бидентатный лиганд, такой как дифенилфосфин-(CH2)x-дифенифосфин, где x представляет собой целое число 2, 3 или 4. Фосфин должен присутствовать, когда этот процесс осуществляют с соединением формулы III, в которой X представляет собой атом брома.
Упомянутую реакцию можно проводить в присутствии или в отсутствии растворителя. Можно использовать безводную или водную среду, включающую один или более растворителей. Соответствующие растворители включают нитрилы, например ацетонитрил, спирты, например метанол, амиды, например диметилформамид или диметилацетамид, 1-метил-2-пирролидон или гексаметилфосфорамид, или воду. Эту реакцию можно удобно осуществлять при температуре от 25 до 200oC, предпочтительно от 75 до 150oC, например от 80 до 110oC.
Некоторые соединения формулы III известны, и их получение описано (GB 2208646).
Так, например, соединения формулы III могут быть получены конденсацией соединения формулы V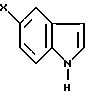
с пиперидоном формулы VI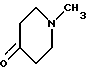
в подходящей реакционной среде в присутствии кислоты или основания, удобно при температуре от 0 до 120oC. Соединения формулы III, в которой R1 и R2 вместе образуют двойную связь, предпочтительно получают в присутствии основания, такого как гидроксил калия, при повышенной температуре, например при температуре кипения реакционной смеси с обратным холодильником. Напротив, соединения формулы III, в которой R1 является гидрокси-группой, и R2 - водородом, предпочтительно получают в присутствии основания, такого как гидроксид калия, при комнатной температуре. Эту реакцию можно удобно осуществлять в подходящем растворителе, таком как спирт, например метанол или этанол.
Метилирование соединения формулы VII, лежащее в основе следующего способа получения соединения II, может быть осуществлено при использовании обычных методов.
Так, например, соединение формулы II может быть получено метилированием соединения формулы VII восстановительным аминированием с использованием водного формальдегида и борогидрида натрия в подходящем растворителе, таком как метанол, или с использованием водного формальдегида и муравьиной кислоты при 100oC (условия Эшвейлер-Кларка).
Альтернативно эта реакция может быть осуществлена при использовании подходящего метилирующего агента, такого как метилгалогенид, метилтозилат или диметилсульфат. Метилирование может быть удобно проведено в инертном органическом растворителе, таком как амид, например диметилформамид, простой эфир, например тетрагидрофуран, спирт, например метанол или технический денатурат, или нитрил, например ацетонитрил, предпочтительно в присутствии основания. Подходящие основания включают, например, карбонаты щелочных металлов, такие как карбонат натрия, или гидрокарбонаты щелочных металлов, такие как гидрокарбонат натрия или калия. Реакцию метилирования удобно осуществлять при температуре от 25 до 100oC.
Соединение формулы VII может быть получено взаимодействием соединения формулы VIII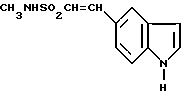
с соединением формулы IX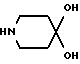
с использованием соответствующих условий, как описано ранее для получения соединений формулы III из соединений формулы V и VI.
Процесс конденсации соединения формулы X, приемлемые уходящие группы которого (L) включают, например, атомы галогенов, таких как хлор, и арилокси-группы, такие как фенокси-группа, с метиламином может быть осуществлен в соответствующей реакционной среде, такой как амид, например, диметилформамид, или простой эфир, например тетрагидрофуран, нитрил, например ацетонитрил, галогеналкан, например дихлорметан, или их смеси, желательно в присутствии органического основания, такого как пиридин или триэтиламин, или неорганического основания, такого как карбонат кальция или бикарбонат натрия. Обычно реакцию проводят при температуре от -70 до 150oC.
Соединения формулы X могут быть получены взаимодействием соединения формулы III, в которой R1 и R2 вместе образуют двойную связь, с соединением формулы XI
CH2 = CHSO2Y
где Y представляет собой уходящую группу L, как это было определено ранее, или группу, чувствительную к замещению уходящей группой L, например гидрокси-группу, используя соответствующие условия, как описано выше для получения соединения формулы II из соединений формулы III и IV.
Так, например, соединение формулы X может быть получено взаимодействием соединения формулы III, где R1 и R2 вместе образуют двойную связь, с соединением формулы XI, где Y представляет собой гидрокси-группу, за которым следует взаимодействие с галогенирующим агентом, таким как PCl5 или SOCl2, с использованием обычных методик.
Соединение формулы XII, дегидратирование которого дает соединение формулы II, может быть получено взаимодействием соединения формулы XIII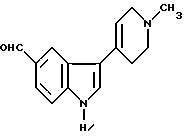
с соединением формулы XIV
CH3SO2NHCH3
в присутствии сильного основания, такого как н-бутиллитий.
Соединение формулы XIII может быть получено взаимодействием соединения формулы III, в которой R1 и R2 вместе образуют двойную связь, с соединением формулы XV
HCON(CH3)2
в присутствии литийалкильного реагента.
Взаимодействие соединения формулы VIII с соединением формулы VI, приводящее к образованию соединения формулы II, может быть осуществлено при использовании соответствующих условий, как описано выше для получения соединений формулы III из соединений формулы V и VI.
Дегидратирование соединения формулы XVI, дающее соединение формулы II, осуществляют в присутствии кислоты или основания, такого как гидроксид калия.
Соединение формулы XVI может быть получено взаимодействием соединения формулы VIII с соединением формулы VI, при использовании соответствующих условий, как описано ранее для получения соединений формулы III, в которых R1 является гидрокси-группой, и R2 является водородом, из соединений формулы V и VI.
В случае, когда хотят выделить соединение I в виде физиологически приемлемой соли, это может быть осуществлено обычными способами, например обработкой соответствующей кислотой в подходящем растворителе. Сольваты соединения I обычно можно получить кристаллизацией или перекристаллизацией из соответствующего растворителя.
Далее настоящее изобретение иллюстрируют следующими неограничивающими примерами. Все температуры указаны в oC. ТД означает технический денатурат. ДМФ означает N,N-диметилформамид.
Промежуточное соединение 1
5-бром-3-(1,2,3,6-тетрагидро-1-метил-пиридинил)-1H-индол
Способ A
Смесь 5-броминдола (1 кг), 1-метил-4-пиперидона (692 мл) и гидроксида калия (30,6 г) в ТД (6,0 л) кипятили с обратным холодильником в атмосфере азота в течение 18 ч. Эту суспензию охладили до 5-10oC, выдержали при этой температуре в течение 15 мин и отфильтровали. Осадок на фильтре промыли метанолом (300 мл), затем водой (800 мл) и высушили в вакууме при 50o. Получили продукт в виде белого твердого вещества (1,40 кг, 94% от теории).
ЯМР: - 2,31 δ (3H) s; 2,54 δ (2H + ДМСО-d5 m; 2,61 δ (2H) m; 3,08 δ (2H) m; 6,12 δ (1H) m; 7,27 δ (1H) d of d, J = 8,5 Гц, 1,9 Гц, 7,40 δ (1H) d, J = 8,5 Гц; 7,50 δ (1H) s; 7,98 δ (1H) d, J = 1,9 Гц; 11,4 δ (1H) широкий s.
Способ B
Смесь 5-броминдола (5,0 г), гидрохлорида 4-дигидрокси-1-метилпиперидина (5,72 г) и гидроксида калия (2,28 г) в 1-пропаноле (45 мл) кипятили с обратным холодильником в атмосфере азота в течение 5 ч. Суспензию охладили до комнатной температуры и профильтровали. Осадок на фильтре промыли 1-пропанолом (2 х 5 мл), затем водой (2 х 10 мл) и сушили в вакууме при 50oC в течение ночи. Получили продукт в виде твердого белого вещества (5,7 г, 76% от теории).
ЯМР: - 2,32 δ (3H), s; 2,54 δ (2H + ДМСО-d5 m; 2,61 δ (2H) m; 3,08 δ (2H) m; 6,11 δ (1H) m; 7,27 δ (1H) d of d, J = 8,5 Гц, 1,9 Гц, 7,40 δ (1H) d, J = 8,5 Гц; 7,49 δ (1H) s; 7,96 δ (1H) d, J = 1,9 Гц; 11,4 δ (1H) широкий s.
Промежуточное соединение 2
5-бром-3-(4-гидрокси-1-метил-4-пиперидинил)-1H-индол
Смесь 5-броминдола (490 г), 1-метил-4-пиперидинона (339 мл) и гидроксида калия (15 г) в ТД (3 л) перемешивали в атмосфере азота при комнатной температуре в течение 24 ч. Эту смесь охладили до 7o и профильтровали. Осадок на фильтре промыли этанолом (300 мл), затем водой (800 мл) с получением беловатого порошка, который сушили в вакууме при 50o в течение 24 ч (563,8 г, 73% от теории).
ЯМР: 1,89 δ (2H) m; 2,03 δ m;2,24 δ (3H) s; 2,55 δ (закрыт ДМСО-d5 m; 4,70 δ (1H) s; 7,19 δ (1H) d of d, J = 8,7 Гц, 2,0 Гц; 7,24 δ (1H) d, J = 2,3 Гц; 7,35 δ (1H) d, J = 8,7 Гц, 7,98 δ (1H) d, J = 2,0 Гц; 11,08 δ (1H) широкий s.
Промежуточное соединение 3
(E)-N-метил-2-(1H-индол-5-ил)этенсульфонамид
Смесь N-метилэтенсульфонамида (45 г), 5-броминдола (60 г), ацетата палладия (0,9 г), три-о-толилфосфина (18,6 г) и триэтиламина (90 мл) в изопропаноле (300 мл) нагревали при 85o в атмосфере азота в течение 18 ч. Реакционную смесь охладили до комнатной температуры, профильтровали и осадок на фильтре промыли пропанолом (30 мл). Объединенные промывные жидкости и фильтрат концентрировали в вакууме с получением желто-коричневого вещества (160 г). Этот продукт очищали хроматографически на колонке из силикагеля. Элюировали сначала смесь этилацетат/циклогексан (1:1), затем этилацетатом, и получили продукт (26,4 г, 36% от теории) в виде желтого порошка.
ЯМР: - 2,55 δ (3H) d, J = 4,9 Гц; 6,51 δ (1H) m; 6,97 δ (1H) d, J = 15,5 Гц; 7,00 δ (1H) m; 7,43 δ (1H) d, J = 15,5 Гц, 7,42 δ (1H) d, J = 2,8 Гц; 7,45 δ (1H) d, J = 8,6 Гц; 7,49 δ (1H) d of d, J = 8,6 Гц, 1,5 Гц; 7,90 δ (1H) s; 11,35 δ (1H) широкий s.
Промежуточное соединение 4
(E)-N-метил-2-[3-(4-гидрокси-1-метил-4-пиперидинил)- 1H-индол-5-ил] этенсульфонамид
Смесь (E)-N-метил-2-(1H-индол-5-ил)этенсульфонамида (2,0 г), 1-метил-4-пиперидона (1,62 г) и гидроксида калия (0,7 г) в ТД (20 мл) перемешивали при комнатной температуре в течение 22 ч. Реакционную смесь концентрировали, и остаток очищали хроматографически на колонке из силикагеля. Элюировали смесью дихлорметан/этанол/аммиак (25:10:1) и получили масло, которое твердело при стоянии с превращением в коричневое твердое вещество. Растирание с эфиром привело к получению продукта в виде белого порошка (1,85 г, 64% от теории).
ЯМР: - 1,94 δ (2H) m; 2,10 δ (2H) m; 2,24 δ (3H) s; 2,46 δ (2H) m; 2,55 δ (2H + ДМСО-d5) m; 4,69 δ (1H) широкий s; 6,93 δ (1H) d, J = 15,4 Гц; 7,00 δ (1H) широкий s; 7,25 δ (1H) d, J = 1,7 Гц; 7,42 δ (1H) d, J = 8,5 Гц; 7,45 δ (1H) d, J = 15,4 Гц; 7,49 δ (1H) d из d, J = 8,5, 1,7 Гц; 11,12 δ (1H) широкий s.
Пример 1
(E)-N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H- индол-5-ил]этенсульфонамид
Смесь N-метилэтенсульфонамида (460 г), промежуточного соединения 1 (1 кг), ацетата палладия (15,4 г), три-δ-толилфосфина (315 г), триэтиламина (960 мл) и целита (400 г) в ДМФ (5 л) нагревали при 100-108o в атмосфере азота в течение 2,5 ч. Суспензию охлаждали до 5o, фильтровали и осадок на фильтре промывали ДМФ (2 л). Порцию (3,7 л) фильтрата перемешивали с водой (250 мл) и циклогексаном (3,0 л) в течение 10 мин. Фазы разделили и ДМФ-слой вновь экстрагировали циклогексаном (1х3,0 л, 1х1,5 л). ДМФ-раствор нагревали до 90o и добавляли в течение более 40 мин воду (2 л). Смесь охлаждали до 10o в течении 3 ч и затем выдерживали при 5o в течение 14 ч. Образовавшийся твердый остаток отфильтровали, промыли холодной (10o) смесью ДМФ/вода (2:1) (2х500 мл) и сушили в вакууме при 50o в течение 18 ч с получением желтого порошка (323,2 г, 60% от теории). Т.пл. 203-205oC (разложение).
ЯМР: - 2,33  (3H) s; 2,55
(3H) s; 2,55 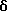 m; 2,57 δ широкий s; 2,60 δ m (7H + ДМСО-d5); 3,10 δ (2H) m; 6,30 δ (1H) m; 7,00 δ (1H) широкий резонанс; 7,05 δ (1H) d, J = 15,7 Гц; 7,45 δ (1H) d, J = 8,4 Гц; 7,47 δ (1H) широкий s; 7,51 δ (1H) d, J = 15,7 Гц; 7,54 δ (1H) d из d, J = 8,4 Гц, 1,6 Гц; 8,17 δ (1H) широкий s; 11,38 δ (1H) широкий s.
m; 2,57 δ широкий s; 2,60 δ m (7H + ДМСО-d5); 3,10 δ (2H) m; 6,30 δ (1H) m; 7,00 δ (1H) широкий резонанс; 7,05 δ (1H) d, J = 15,7 Гц; 7,45 δ (1H) d, J = 8,4 Гц; 7,47 δ (1H) широкий s; 7,51 δ (1H) d, J = 15,7 Гц; 7,54 δ (1H) d из d, J = 8,4 Гц, 1,6 Гц; 8,17 δ (1H) широкий s; 11,38 δ (1H) широкий s.
Пример 2
(E)-N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H- индол-5-ил]этенсульфонамид
Смесь N-метилэтенсульфонамида (460 г), промежуточного соединения 1 (1 кг), ацетата палладия (15,4 г), три- -толилфосфина (315 г), триэтиламина (960 мл) и целита (400 г) в ДМФ (5 л) нагревали при 100-108o в атмосфере азота в течение 2,5 ч. Затем суспензию охлаждали до 5o, фильтровали и осадок на фильтре промывали ДМФ (2 л). Затем по каплям добавляли воду (2,5 л) к охлажденной до 4o порции (3,7 л) фильтрата. Полученную суспензию охлаждали до 5o, выдерживали в течение 45 мин и фильтровали. Осадок на фильтре промывали холодной смесь. ДМФ/вода (7:5) (1 л), затем холодным ТД (1 л). Осадок перемешивали с этилацетатом (2,76 л) при комнатной температуре в течение 1 ч, затем фильтровали. Осадок на фильтре промывали этилацетатом (500 мл), и собранный твердый продукт сушили в течение ночи в вакууме при 45o (394,6 кг, 69,3% от теории). Т.пл. 203 - 205oC (разлож.).
-толилфосфина (315 г), триэтиламина (960 мл) и целита (400 г) в ДМФ (5 л) нагревали при 100-108o в атмосфере азота в течение 2,5 ч. Затем суспензию охлаждали до 5o, фильтровали и осадок на фильтре промывали ДМФ (2 л). Затем по каплям добавляли воду (2,5 л) к охлажденной до 4o порции (3,7 л) фильтрата. Полученную суспензию охлаждали до 5o, выдерживали в течение 45 мин и фильтровали. Осадок на фильтре промывали холодной смесь. ДМФ/вода (7:5) (1 л), затем холодным ТД (1 л). Осадок перемешивали с этилацетатом (2,76 л) при комнатной температуре в течение 1 ч, затем фильтровали. Осадок на фильтре промывали этилацетатом (500 мл), и собранный твердый продукт сушили в течение ночи в вакууме при 45o (394,6 кг, 69,3% от теории). Т.пл. 203 - 205oC (разлож.).
ЯМР: - 2,32 δ (3H) s; 2,55 δ m; 2,56 δ d (J = 4,8 Гц); 2,60 δ m (7H + ДМСО-d5); 3,09 δ (2H) m; 6,29 δ (1H) m; 6,99 δ (1H) q, J = 4,8 Гц; 7,04 δ (1H) d, J = 15,7 Гц; 7,44 δ (1H) d , J = 8,4 Гц; 7,46 δ (1H) широкий s; 7,51 δ (1H) d, J = 15,7 Гц; 7,54 δ (1H) d из d, J = 8,4 Гц, 1,6 Гц; 8,17 δ (1H) широкий s; 11,38 δ (1H) широкий s.
Пример 3
(E)-N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H- индол-5-ил]этенсульфонамид
Смесь N-метилэтенсульфонамида (15,74 г), промежуточного соединения 1 (30,02 г), ацетата палладия (2,08 г), три- -толилфосфина (7,21 г) триэтиламина (28,7 мл) в ДМФ (90 мл) нагревали при 110-115oC в течение 4 ч. Смесь профильтровали, пока она еще была горячей, через высокоскоростной фильтр. Полученный фильтрат охладили до 0-5o, и в течение 30 мин добавляли ледяную воду (300 мл). Смесь перемешивали при температуре 0-5o в течение 1 ч 15 мин, затем оставляли при 5o в течение ночи. Образовавшийся твердый продукт собирали фильтрованием, промывали водой (90 мл) и отсасывали насухо в течение 20 мин. Твердый желтый продукт перемешивали с этилацетатом (120 мл) при комнатной температуре в течение 3 ч. Этот продукт отфильтровали, промыли этилацетатом (30 мл) и сушили в вакууме при 55o в течение ночи (28,06 г, 82% от теории). Т.пл. 203 - 205oC (разлож.).
-толилфосфина (7,21 г) триэтиламина (28,7 мл) в ДМФ (90 мл) нагревали при 110-115oC в течение 4 ч. Смесь профильтровали, пока она еще была горячей, через высокоскоростной фильтр. Полученный фильтрат охладили до 0-5o, и в течение 30 мин добавляли ледяную воду (300 мл). Смесь перемешивали при температуре 0-5o в течение 1 ч 15 мин, затем оставляли при 5o в течение ночи. Образовавшийся твердый продукт собирали фильтрованием, промывали водой (90 мл) и отсасывали насухо в течение 20 мин. Твердый желтый продукт перемешивали с этилацетатом (120 мл) при комнатной температуре в течение 3 ч. Этот продукт отфильтровали, промыли этилацетатом (30 мл) и сушили в вакууме при 55o в течение ночи (28,06 г, 82% от теории). Т.пл. 203 - 205oC (разлож.).
ЯМР: - 2,31 δ (3H) s; 2,55 δ m; 2,56 δ d (J = 4,8 Гц); 2,60 δ m (7H + ДМСО-d5); 3,08 δ (2H) m; 6,28 δ (1H) m; 6,98 δ (1H) q, J = 4,8 Гц; 7,03 δ (1H) d, J = 15,7 Гц; 7,43 δ (1H) d, J = 8,4 Гц; 7,45 δ (1H) d, J = 1,9 Гц; 7,49 δ (1H) d, J = 15,7 Гц; 7,52 δ (1H) d из d, J = 8,4 Гц, 1,6 Гц; 8,16 δ (1H) широкий s; 11,38 δ (1H) широкий s.
Пример 4
Гидрохлорид (E)-N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H- индол-5-ил]этенсульфонамида
Смесь N-метилэтенсульфонамида (320 г), промежуточного соединения 1 (700 г), ацетата палладия (10,5 г), три- -толилфосфина (140 г), триэтиламина (670 мл) и целита (280 г) в ДМФ (3,5 л) нагревали при 85o в течение 4 ч. Смесь профильтровали, пока она еще была горячей, и осадок на фильтре промывали ДМФ (700 мл). Фильтрат охлаждали до 15-20o и добавляли по каплям воду (8,4 л). Эту смесь затем выдерживали при 8o, фильтровали, продукт промывали водой (2,1 л), затем сушили в вакууме в течение ночи при 40o. Неочищенный продукт размешивали в этилацетате (2,1 л) при 21o в течение 3 ч. Суспензию фильтровали, и твердый остаток промывали этилацетатом (700 мл). Влажный твердый остаток суспендировали в ДМФ (2,1 л), охлаждали до 15o и добавляли к суспензии концентрированную соляную кислоту (210 мл) при температуре < 25o в течение 30 мин. Затем по каплям в течение 30 мин добавляли этилацетат (1,4 л). Еще через 30 мин аналогичным способом ввели еще ДМФ (5,6 л) за 1 ч. Продукт отфильтровали, промыли этилацетатом (1,4 л), затем 2-пропан-1-олом (700 мл), и сушили в вакууме при 45o в течение ночи (791,5 г, 89,5% от теории).
-толилфосфина (140 г), триэтиламина (670 мл) и целита (280 г) в ДМФ (3,5 л) нагревали при 85o в течение 4 ч. Смесь профильтровали, пока она еще была горячей, и осадок на фильтре промывали ДМФ (700 мл). Фильтрат охлаждали до 15-20o и добавляли по каплям воду (8,4 л). Эту смесь затем выдерживали при 8o, фильтровали, продукт промывали водой (2,1 л), затем сушили в вакууме в течение ночи при 40o. Неочищенный продукт размешивали в этилацетате (2,1 л) при 21o в течение 3 ч. Суспензию фильтровали, и твердый остаток промывали этилацетатом (700 мл). Влажный твердый остаток суспендировали в ДМФ (2,1 л), охлаждали до 15o и добавляли к суспензии концентрированную соляную кислоту (210 мл) при температуре < 25o в течение 30 мин. Затем по каплям в течение 30 мин добавляли этилацетат (1,4 л). Еще через 30 мин аналогичным способом ввели еще ДМФ (5,6 л) за 1 ч. Продукт отфильтровали, промыли этилацетатом (1,4 л), затем 2-пропан-1-олом (700 мл), и сушили в вакууме при 45o в течение ночи (791,5 г, 89,5% от теории).
ЯМР: - 2,55 δ (3H) d, J = 5,0 Гц; 2,82 δ (2H) широкий m; 2,89 δ (3H) s; 3,30 δ (1H) широкий m; 3,58 δ (1H) широкий m; 3,79 δ (1H) широкий m; 3,98 δ (1H) широкий m; 6,34 δ (1H) m; 7,05 δ (1H) q, J = 5,0 Гц; 7,07 δ (1H) d, J = 15,4 Гц; 7,47 δ (1H) d, J = 8,5 Гц; 7,50 δ (1H) d, J = 15,4 Гц; 7,58 δ (1H) d из d, J = 8,5 Гц, 1,3 Гц; 7,62 δ (1H) d, J = 2,6 Гц; 8,22 δ (1H) широкий s; 10,7 δ (1H) широкий резонанс; 11,68 δ (1H) широкий s.
Пример 5
(E)-N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H- индол-5-ил]этенсульфонамид
Перемешанную смесь промежуточного соединения 2 (30,08 г), N-метилэтенсульфонамида (14,84 г), ацетата палладия (1,97 г), три- -толилфосфина (6,81 г) и триэтиламина (27 мл) в ДМФ (90 мл) нагревали при 110 - 115o в течение 4 ч. Горячую (90o) смесь фильтровали и осадок промывали ДМФ (30 мл). К фильтрату по каплям добавили воду (300 мл), затем охладили смесь до 5o и выдержали в течение 30 мин. Эту суспензию профильтровали, осадок на фильтре промыли водой (3х30 мл) и досуха отсасывали в течение 1,5 ч. Влажный осадок размешивали с этилацетатом (120 мл) в течение 3 ч, отфильтровали, и полученный на фильтре осадок промыли этилацетатом (30 мл). Этот продукт сушили в вакууме при 55o в течение 18 ч (27,52 ч, 85% от теории). Т.пл. 203 - 205oC.
-толилфосфина (6,81 г) и триэтиламина (27 мл) в ДМФ (90 мл) нагревали при 110 - 115o в течение 4 ч. Горячую (90o) смесь фильтровали и осадок промывали ДМФ (30 мл). К фильтрату по каплям добавили воду (300 мл), затем охладили смесь до 5o и выдержали в течение 30 мин. Эту суспензию профильтровали, осадок на фильтре промыли водой (3х30 мл) и досуха отсасывали в течение 1,5 ч. Влажный осадок размешивали с этилацетатом (120 мл) в течение 3 ч, отфильтровали, и полученный на фильтре осадок промыли этилацетатом (30 мл). Этот продукт сушили в вакууме при 55o в течение 18 ч (27,52 ч, 85% от теории). Т.пл. 203 - 205oC.
ЯМР: - 2,31 δ (3H) s; 2,55 δ m; 2,56 δ d, J = 4,8 Гц; 2,60  m, (7H + ДМСО-d5); 3,08 δ (2H) m; 6,28 δ (1H) m; 6,98 δ (1H) q, J = 4,8 Гц; 7,03 δ (1H) d, J = 15,7 Гц; 7,43 δ (1H) d, J = 8,4 Гц; 7,45 δ (1H) d, J = 1,9 Гц; 7,50 δ (1H) d, J = 15,7 Гц; 7,53 δ (1H) d из d, J = 8,4 Гц, 1,6 Гц; 8,16 δ (1H) широкий s; 11,39 δ (1H) широкий s.
m, (7H + ДМСО-d5); 3,08 δ (2H) m; 6,28 δ (1H) m; 6,98 δ (1H) q, J = 4,8 Гц; 7,03 δ (1H) d, J = 15,7 Гц; 7,43 δ (1H) d, J = 8,4 Гц; 7,45 δ (1H) d, J = 1,9 Гц; 7,50 δ (1H) d, J = 15,7 Гц; 7,53 δ (1H) d из d, J = 8,4 Гц, 1,6 Гц; 8,16 δ (1H) широкий s; 11,39 δ (1H) широкий s.
Пример 6
(E)-N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H- индол-5-ил]этенсульфонамид
Смесь N-метилэтенсульфонамида (115 г), промежуточного соединения 1 (250 г), ацетата палладия (3,85 г), три- -толилфосфина (77,5 г), триэтиламина (240 мл) и целита (100 г) в ДМФ (1,25 л) нагревали при 100 - 110o в атмосфере в течение 2 ч. Суспензию охладили до 20o, отфильтровали и осадок на фильтре промыли ДМФ (500 мл). Объединенные промывные воды и фильтрат перемешивали с водой (125 мл) и циклогексаном (1,5 л). Эти фазы разделили и слой с ДМФ вновь экстрагировали циклогексаном (1х1,5 л, 1х0,75 л). Раствор ДМФ обработали триэтиламином (125 мл). Затем добавляли воду (1 л) при температуре ≤ 35o в течение 20 мин. Образовавшуюся суспензию охладили примерно за 30 мин до 5o и так выдержали в течение 1,5 ч. Твердую фазу отфильтровали, промыли смесью ДМФ/вода (2:1) (2х250 мл), затем водой (125 мл), и сушили в вакууме при 50o в течение 18 ч с получением желтого порошка (194,3 г, 69% от теории). Т.пл. 203 - 205oC (разлож.).
-толилфосфина (77,5 г), триэтиламина (240 мл) и целита (100 г) в ДМФ (1,25 л) нагревали при 100 - 110o в атмосфере в течение 2 ч. Суспензию охладили до 20o, отфильтровали и осадок на фильтре промыли ДМФ (500 мл). Объединенные промывные воды и фильтрат перемешивали с водой (125 мл) и циклогексаном (1,5 л). Эти фазы разделили и слой с ДМФ вновь экстрагировали циклогексаном (1х1,5 л, 1х0,75 л). Раствор ДМФ обработали триэтиламином (125 мл). Затем добавляли воду (1 л) при температуре ≤ 35o в течение 20 мин. Образовавшуюся суспензию охладили примерно за 30 мин до 5o и так выдержали в течение 1,5 ч. Твердую фазу отфильтровали, промыли смесью ДМФ/вода (2:1) (2х250 мл), затем водой (125 мл), и сушили в вакууме при 50o в течение 18 ч с получением желтого порошка (194,3 г, 69% от теории). Т.пл. 203 - 205oC (разлож.).
ЯМР: - 2,33 δ (3H) s; 2,5 δ m, 2,56 δ широкий s, 2,61 δ m (7H + ДМСО-d5); 3,10 δ (2H) m; 6,30 δ (1H) m; 7,08 (1H) широкий резонанс, 7,04 δ (1H) d, J = 15,7 Гц; 7,44 δ (1H) d, J = 8,4 Гц; 7,46 δ (1H) s, 7,47 δ (1H) d, J = 15,7 Гц; 7,54 δ (1H) d из d, J = 8,4 Гц, 1,6 Гц; 8,18 δ (1H) широкий s; 11,4 δ (1H) широкий s.
Пример 7
(E)-N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H- индол-5-ил]этенсульфонамид
Раствор промежуточного соединения 1 (100 г) и N-метилэтенсульфонамида (46 г) в смеси ДМФ (300 мл) и 5 н. соляной кислоты (70 мл) добавляли в течение 0,75 ч к перемешанной смеси три- -толилфосфина (31,3 г), ацетата палладия (1,54 г), целита (40 г) и триэтиламина (114 мл) в ДМФ (200 мл) при 100o в атмосфере азота. Реакционную смесь перемешивали еще 4 ч при 100o, охлаждали до комнатной температуры, фильтровали и осадок на фильтре промывали ДМФ (2х100 мл). Объединенные промывные жидкости и фильтрат перемешивали с водой (50 мл) и циклогексаном (600 мл). Фазы разделяли и слой с ДМФ снова экстрагировали циклогексаном (1х600 мл, 1х300). Раствор в ДМФ обработали триэтиламином (48 мл). Затем к смеси в течение 15 мин добавляли воду (400 мл) при температуре ≤ 35o. Образовавшуюся суспензию охладили до 5o и выдерживали в течение 1 ч. Затем твердое вещество отфильтровали, промыли смесью ДМФ/вода (2: 1) (2х100 мл) и сушили в вакууме при 50o в течение ночи. Получили желтый порошок (76,9 г, 68% от теории). Т.пл. 203 - 205oC (разлож.).
-толилфосфина (31,3 г), ацетата палладия (1,54 г), целита (40 г) и триэтиламина (114 мл) в ДМФ (200 мл) при 100o в атмосфере азота. Реакционную смесь перемешивали еще 4 ч при 100o, охлаждали до комнатной температуры, фильтровали и осадок на фильтре промывали ДМФ (2х100 мл). Объединенные промывные жидкости и фильтрат перемешивали с водой (50 мл) и циклогексаном (600 мл). Фазы разделяли и слой с ДМФ снова экстрагировали циклогексаном (1х600 мл, 1х300). Раствор в ДМФ обработали триэтиламином (48 мл). Затем к смеси в течение 15 мин добавляли воду (400 мл) при температуре ≤ 35o. Образовавшуюся суспензию охладили до 5o и выдерживали в течение 1 ч. Затем твердое вещество отфильтровали, промыли смесью ДМФ/вода (2: 1) (2х100 мл) и сушили в вакууме при 50o в течение ночи. Получили желтый порошок (76,9 г, 68% от теории). Т.пл. 203 - 205oC (разлож.).
ЯМР: - 2,32 δ (3H) s; 2,55 δ m, 2,56 δ d (J = 4,8 Гц), 2,60 δ m (7H + ДМСО-d5); 3,09 δ (2H) m; 6,28 δ (1H) m; 6,98 δ (1H) q, J = 4,8 Гц; 7,03 δ (1H) d, J = 15,7 Гц; 7,43 δ (1H) d, J = 8,4 Гц; 7,45 δ (1H) s; 7,47 δ (1H) d, J = 15,7 Гц; 7,53 δ (1H) d из d, J = 8,4 Гц, 1,6 Гц; 8,17 δ (1H) широкий s; 11,4 δ (1H) широкий s.
Пример 8
(E)-N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H- индол-5-ил]этенсульфонамид
Смесь N-метилэтенсульфонамида (43,08 г), промежуточного соединения 2 (100 г), ацетата палладия (1,45 г), три- -толилфосфина (29,5 г), триэтиламина (90 мл) и целита (40 г)
-толилфосфина (29,5 г), триэтиламина (90 мл) и целита (40 г)
в ДМФ (200 мл) нагревали при температуре 100 - 110o в атмосфере азота в течение 4 ч. Затем суспензию охладили до 20o, профильтровали, и осадок на фильтре промыли ДМФ (200 мл). Промывную жидкость и фильтрат объединили и перемешали с водой (50 мл) и циклогексаном (600 мл). Фазы разделили, и слой ДМФ вновь экстрагировали циклогексаном (1х600 мл, 1х300 мл). Раствор с ДМФ обработали триэтиламином (45 мл). Затем при температуре ≤ 35o добавляли воду (400 мл) в течение 15 мин. Эту суспензию постепенно охлаждали до 5o в течение 1,5 ч, а затем выдерживали 1 ч. Твердый остаток отфильтровали, промыли смесью ДМФ/вода (2: 1) (2х100 мл), затем водой (50 мл), и сушили в вакууме при 50o в течение 18 ч. Получили желтый порошок (76,7 г, 67% от теории). Т. пл. 203 - 205oC (разлож.).
ЯМР: - 2,33 δ (3H) s; 2,55 δ m, 2,57 δ s, 2,62 δ m, (7H + ДМСО-d5); 3,10 δ (2H) m; 6,30 δ (1H) m; 7,0 δ (1H) широкий резонанс, 7,05 δ (1H) d, J = 15,7; 7,44 δ (1H) d, J = 8,4 Гц; 7,46 δ (1H) s; 7,48 δ (1H) d, J = 15,7 Гц; 7,55 δ (1H) d, J = 8,4 Гц; 8,18 δ (1H) широкий s; 11,4 δ (1H) широкий s.
Пример 9
(E)-N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H- индол-5-ил]этенсульфонамид
Смесь промежуточного соединения 3 (3,93 г), 1-метил-4-пиперидона (3,42 г) и гидроксида калия (1,41 г) в ТД (35 мл) кипятили с обратным холодильником в течение 17 ч. Эту суспензию охладили до комнатной температуры и профильтровали. Осадок на фильтре промыли ТД (5 мл), затем водой (10 мл) и снова ТД (5 мл), затем сушили в вакууме. Этот неочищенный продукт растирали с водой (30 мл), фильтровали, осадок на фильтре промывали водой (10 мл) и сушили в вакууме при 50o; получили бледно-желтое твердое вещество (2,30 г, 42% от теории). Т.пл. 203 - 205oC (разлож.).
ЯМР: - 2,35 δ (3H) s; 2,57 δ (2H + ДМСО-d5); 2,60 δ (3H) s; 2,64 δ (2H) m; 3,12 (2H) m; 6,32 δ (1H) m; 7,07 δ (1H) d, J = 15,7 Гц; 7,47 δ (1H) d, J = 8,4 Гц; 7,49 δ (1H) s; 7,53 δ (1H) d, J = 15,7 Гц; 7,56 δ (1H) d из d, J = 8,4, 1,6 Гц; 8,20 δ (1H) s.
Пример 10
(E)-N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил] -1H- индол-5-ил]этенсульфонамид
Смесь промежуточного соединения 4 (1,0 г) и гидроксида калия (90 мг) в ТД (15 мл) кипятили с обратным холодильником в течение 20 ч. Этот раствор охладили до комнатной температуры и образовавшийся желтый осадок отфильтровали, промыли водой (3х5 мл), затем ТД (2х2 мл) и высушили в вакууме; получили желтый порошок (0,16 г, 17% от теории). Т.пл. 203 - 205oC (разлож.).
ЯМР: - 2,35 δ (3H) s; 2,57 δ (2H + ДМСО-d5) m; 2,59 δ (3H) s; 2,64 δ (2H) m; 3,12 δ (2H) m; 6,22 δ (1H) m; 7,02 δ (1H) широкий резонанс; 7,07 δ (1H) d, J = 15,7 Гц; 7,47 δ (1H) d, J = 8,4 Гц; 7,49 δ (1H) s; 7,50 δ (1H) d, J = 15,7 Гц; 7,57 δ (1H) d из d, J = 8,4, 1,6 Гц; 8,20 δ (1H) s.
Пример 11
Гидрохлорид N-метил-3-(1-метил-4-пиперидинил)-1H- индол-5-этансульфонамида
Смесь (E)-N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил]-1H- индол-5-ил]этенсульфонамида (10 кг) и 10%-ного оксида палладия на угле (10 кг, паста 50%-ной влажности, добавленная в два приема) в смеси ДМФ (50 л), воды (20 л) и 2 н. соляной кислоты (15 л) гидрогенизировали при атмосферном давлении в течение 18,5 ч. Затем катализатор удалили фильтрованием. Осадок на фильтре промыли водой (20 л). Фильтрат сконцентрировали в вакууме до объема приблизительно 30 л и охладили до 20o. Затем постепенно в течение одного часа добавляли этилацетат (70 л) и образовавшуюся суспензию охладили до 5o и выдержали при этой температуре в течение 30 мин. Продукт собрали фильтрованием, промыли этилацетатом (20 л) и сушили в вакууме при 40 - 50o в течение ночи (9,34 кг, 81,6% от теории). Порцию (2,0 кг) твердого продукта перекристаллизовали из горячей воды (6,0 л) и получили белое кристаллическое вещество (1,40 кг, 70% от теории).
ЯМР: - 2,1 δ (4H) m; 2,64 δ (3H) d, J = 4,9 Гц; 2,78 δ (3H) s; 3,04 δ m; 3,11 δ широкий m, (5H); 3,33 δ (2H) m; 3,47 δ (2H) m; 7,02 δ (2H) m; 7,14 δ (1H) широкий s; 7,31 δ (1H) d, J = 8,2 Гц; 7,60 δ (1H) широкий s; 10,75 δ (1H) широкий резонанс; 10,9 δ (1H) широкий s.
Пример 12
Гидрохлорид N-метил-3-(1-метил-4-пиперидинил)-1H- индол-5-этансульфонамида
Смесь гидрохлорида (E)-N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H- индол-5-ил]этенсульфонамида (500 г) и 10%-ного оксида палладия на угле (паста 50%-ной влажности, 700 г, добавленная в три порции) в смеси ДМФ (3 л), воды (3 л) и метанола (1,5 л) гидрогенизировали при атмосферном давлении в течение 24 ч. Суспензию фильтровали и осадок на фильтре промыли водой (500 мл). Фильтрат сконцентрировали до объема приблизительно 2 л отгонкой в вакууме. Затем добавляли этилацетат (5 л) в течение примерно 10 мин и смесь охладили до 5o. Твердый продукт отфильтровали, промыли этилацетатом (1 л) и сушили в вакууме при 45o в течение ночи (453 г, 90,1% от теории). Перекристаллизацией из горячей воды (1,36 л) получили белые кристаллы (324,0 г, 71,2% от теории).
ЯМР: - 2,1 δ (4H) m; 2,64 δ (3H) d, J = 4,9 Гц; 2,79 δ (3H) s; 3,04  m; 3,11 δ широкий m, (5H); 3,33 δ (2H) m; 3,47 δ (2H) m; 7,02 δ (2H) m; 7,14 δ (1H) широкий s; 7,31 δ (1H) d, J = 8,2 Гц; 7,60 δ (1H) широкий s; 10,65 δ (1H) широкий резонанс; 10,9 δ (1H) широкий s.
m; 3,11 δ широкий m, (5H); 3,33 δ (2H) m; 3,47 δ (2H) m; 7,02 δ (2H) m; 7,14 δ (1H) широкий s; 7,31 δ (1H) d, J = 8,2 Гц; 7,60 δ (1H) широкий s; 10,65 δ (1H) широкий резонанс; 10,9 δ (1H) широкий s.
Пример 13
Гидрохлорид N-метил-3-(1-метил-4-пиперидинил)-1H- индол-5-этансульфонамида
Раствор (E)-N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H- индол-5-ил]-этенсульфонамида (25 г) в воде (244,5 мл), содержащей метансульфоновую кислоту (5,5 мл), гидрогенизировали при атмосферном давлении в присутствии 10%-ного оксида палладия на угле (25 г, паста 50%-ной влажности, добавленная в два приема). Через 18 ч поглощение водорода прекратилось и катализатор удалили фильтрованием. Фильтрат упарили в вакууме до объема приблизительно 50 мл и добавили концентрированную соляную кислоту (10 мл). Выпаривание смеси продолжили, и основную часть воды удалили азеотропной перегородкой с ТД (3х100 мл). Получившаяся в результате суспензия (~100 мл) была выдержана при 5o в течение 1,5 ч, затем профильтрована, и остаток на фильтре промыт диизопропиловым эфиром (2х100 мл). Полученное вещество бледно-желтого цвета (18,5 г, 66% от теории) сушили в вакууме при 55o в течение 20 ч. Перекристаллизацией части продукта (15 г) из воды получили беловатые кристаллы (13,2 г, 88% от теории).
ЯМР: - 2,1 δ (4H) m; 2,64 δ (3H) d, J = 4,9 Гц; 2,79 δ (3H) s; 3,04 δ m; 3,11 δ широкий m, (5H); 3,33 δ (2H) m; 3,47 δ (2H) m; 7,0 (2H) m; 7,13 (1H) широкий s; 7,31 δ (1H) d, J = 8,2 Гц; 7,60 δ (1H) широкий s; 10,6 δ (1H) широкий резонанс; 10,9 δ (1H) широкий s.
Пример 14
Гидрохлорид N-метил-3-(1-метил-4-пиперидинил)-1H- индол-5-этансульфонамида
Смесь (E)-N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H- индол-5-ил]этенсульфонамида (10 кг) и 10%-ного оксида палладия на угле (10 кг, паста 50%-ной влажности, добавленная в
два приема) в смеси ДМФ (50 л), воды (35 л) и 2 н. соляной кислоты (15,75 л) гидрогенизировали при атмосферном давлении в течение 20,5 ч. Затем катализатор удалили фильтрованием. Осадок на фильтре промыли водой (40 л). Фильтрат сконцентрировали в вакууме до объема приблизительно 30 л и охладили до 18o. Затем в течение 1 ч добавляли этилацетат (70 л), а образовавшуюся суспензию охладили до 5o и так выдержали в течение 1 ч. Затем собрали продукт фильтрованием, осадок на фильтре промыли водой (40 л). Фильтрат сконцентрировали в вакууме до объема примерно 30 л и охладили до температуры 18o. Добавили этилацетат (70 л) в течение 1 ч, а образовавшуюся суспензию охладили до 5o и так выдержали в течение 1 ч. Продукт собрали фильтрованием, промыли этилацетатом (20 л) и сушили в вакууме при 40-50o в течение ночи (8,87 кг, 79,0% от теории). Порцию (0,2 кг) этого продукта перекристаллизовали из горячей смеси ТД/вода (4:1) (1 л) и получили тонкие беловатые кристаллы (0,143 кг, 71,7% от теории).
ЯМР: - 2,1 δ (4H) m; 2,64 δ (3H) d, J = 4,9 Гц; 2,78 δ (3H) s; 3,04 δ m; 3,11 δ широкий m, (5H); 3,33 δ (2H) m; 3,47 δ (2H) m; 7,0 (2H) m; 7,13 (1H) широкий s; 7,31 δ (1H) d, J = 8,2 Гц; 7,58 δ (1H) широкий s; 10,5 (1H) широкий резонанс; 10,9 (1H) широкий s.
Пример 15
Гидрохлорид N-метил-3-(1-метил-4-пиперидинил)-1H- индол-5-этансульфонамида
Смесь (E)-N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H- индол-5-ил] этенсульфонамида (8,6 г) и 10%-ного оксида палладия на угле (2,58 кг, паста 50%-ной влажности) в смеси ДМФ (86 л) и 2 н. соляной кислоты (12,3 кг) гидрогенизировали при атмосферном давлении в течение 21 ч. Затем катализатор удалили фильтрованием, осадок на фильтре промыли смесью ДМФ/вода (1: 1) (30 л). Объединенные промывные жидкости и фильтрат затем обработали обесцвечивающим углем (0,86 кг) при температуре 75 - 80o в течение 2 ч. Затем суспензию профильтровали, остаток на фильтре промыли смесью ДМФ/вода (2х30 л). Для дальнейшей обработки можно объединить фильтраты из двух опытов по гидрогенизированию. Таким образом, объединенные фильтраты обрабатывали 3 М соляной кислотой (1,72 д), затем концентрировали в вакууме до объема приблизительно 52 л. Добавили этилацетат (120 л), образовавшуюся в результате суспензию охладили до 3o и выдержали так в течение 1 ч. Продукт собрали фильтрованием, промыли этилацетатом (2х26 л) и сушили в вакууме при 40-50o в течение ночи (15,96 кг, 83% от теории). Перекристаллизацией из горячей смеси ТД/вода (7:1; 1,206 л) получили тонкие беловатые кристаллы (13,02 кг, 84% от теории).
ЯМР: - 2,1 δ (4H) m; 2,64 δ (3H) d, J = 4,9 Гц; 2,78 δ (3H) s; 3,04 δ m; 3,11 δ широкий m, (5H); 3,33 δ (2H) m; 3,47 δ (2H) m; 7,02 (2H) m; 7,12 (1H) широкий s; 7,31 δ (1H) d, J = 8,2 Гц; 7,62 δ (1H) широкий s; 10,9 δ (1H) широкий s.
Пример 16
Гидрохлорид N-метил-3-(1-метил-4-пиперидинил)-1H- индол-5-этансульфонамида
Смесь (E)-N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1H- индол-5-ил] этенсульфонамида (100 г) и 10%-ного оксида палладия на угле (30 г, паста 50%-ной влажности) в смеси ДМФ (1 л) и 2 н. соляной кислоты (150 мл) гидрогенизировали при атмосферном давлении в течение 48 ч. Затем удалили фильтрованием катализатор, а фильтр с осадком промыли смесью ДМФ/вода (1:1; 200 мл). Затем объединенные промывные жидкости и фильтрат обработали обесцвечивающим углем (10 г) при 75 - 80o в течение 2 ч. Суспензию профильтровали, осадок промыли смесью ДМФ/вода (1:1, 400 мл) и добавили 2 М соляную кислоту (10 мл). Фильтрат концентрировали в вакууме до объема приблизительно 300 мл, и охладили до 30o. Постепенно, в течение 30 мин добавляли этилацетат (700 мл), а образовавшуюся в результате суспензию охладили до 0 - 5o и выдержали в течение 30 мин. Продукт отделили фильтрованием в вакууме и промыли этилацетатом (150 мл), затем ТД (2х150 мл). Влажный от ТД осадок перекристаллизовали из горячей смеси ТД/вода (7:1) (1,315 л), и получили тонкие беловатые кристаллы (60,1 г, 54% от теории).
ЯМР: - 2,1 δ (4H) m; 2,62 δ (3H) d, J = 4,9 Гц; 2,78 δ (3H) s; 3,02 δ m; 3,10 δ широкий m, (5H); 3,31 δ (2H) m; 3,47 δ (2H) m; 6,98 (2H) m; 7,11 (1H) широкий s; 7,29 δ (1H) d, J = 8,2 Гц; 7,58 δ (1H) широкий s; 10,5 (1H) широкий резонанс; 10,9 (1H) широкий s.
| название | год | авторы | номер документа |
|---|---|---|---|
| 5-ГЕТЕРОЦИКЛО-1,5-БЕНЗОДИАЗЕПИНЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1995 |
|
RU2152939C1 |
| ПРОИЗВОДНЫЕ 3-(5-ТЕТРАЗОЛИЛБЕНЗИЛ)АМИНОПИПЕРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1994 |
|
RU2136675C1 |
| ПРОИЗВОДНЫЕ ЛАКТАМА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ЯВЛЯЮЩАЯСЯ АНТАГОНИСТОМ 5-ОКСИТРАПТАМИНА /5-НТ/ НА 5- НТ -РЕЦЕПТОРАХ ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СОЛОВАТЫ | 1992 |
|
RU2067980C1 |
| ПРОИЗВОДНЫЕ 1,5-БЕНЗОДИАЗЕПИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЙ ИХ ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ | 1994 |
|
RU2135486C1 |
| ПРОИЗВОДНЫЕ ЛАКТАМА ИЛИ ИХ ФИЗИОЛОГИЧЕСКИЕ ПРИЕМЛЕМЫЕ СОЛИ ИЛИ СОЛЬВАТЫ | 1992 |
|
RU2081117C1 |
| ПРОИЗВОДНЫЕ АКРИДИНА, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ ОПУХОЛЕЙ | 1992 |
|
RU2119482C1 |
| ПРОИЗВОДНЫЕ 2,6-ДИАМИНОПУРИН-β-D-РИБОФУРАНУРОНАМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМСОСТАВ ДЛЯ ПОДАВЛЕНИЯ АКТИВНОСТИ ЛЕЙКОЦИТОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, СПОСОБ ПОДАВЛЕНИЯ АКТИВНОСТИ ЛЕЙКОЦИТОВ | 1994 |
|
RU2129561C1 |
| СПОСОБ ЛЕЧЕНИЯ | 2012 |
|
RU2621148C2 |
| СОЕДИНЕНИЯ ГЕТЕРОАРИЛА В КАЧЕСТВЕ ИНГИБИТОРОВ ТКБ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2742122C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 1-[2-(БЕНЗИМИДАЗОЛ-1-ИЛ)ХИНОЛИН-8-ИЛ]ПИПЕРИДИН-4-ИЛАМИНА | 2004 |
|
RU2323214C2 |
Описывается способ получения соединения формулы I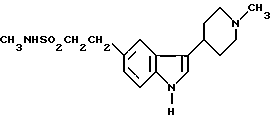
его соли, который включает восстановление соединения формулы II
в присутствии водорода, катализатора из благородного металла и растворителя. Предложено также новое промежуточное соединение формулы II и способы его получения. Технический результат - создание способа, обеспечивающего хороший выход и высокую степень чистоты целевого продукта. 6 с. и 7 з.п.ф-лы.
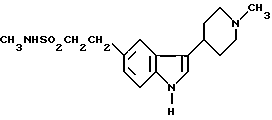
или его физиологически приемлемой соли, отличающийся тем, что восстановлению подвергают соединение формулы II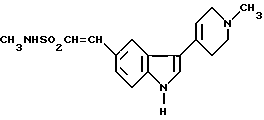
в присутствии водорода, катализатора из благородного металла и растворителя.
или его соль, где R1 представляет собой гидрокси-группу, и R2 представляет собой водород, или R1 и R2 вместе образуют двойную связь, Х представляет собой уходящий атом, такой как атом галогена, например атом брома, или уходящую группу, например трифлатную (CF3SO3) группу, конденсируют с N-метил-винилсульфонамидом формулы IV
CH2=CHSO2NZCH3
где Z представляет собой водород или аминозащитную группу, и возможно, если необходимо и/или желательно, снимают защиту с полученного таким образом защищенного производного.
дегидратируют в присутствии кислоты или основания.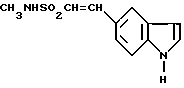
подвергают взаимодействию с соединением формулы VI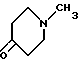
13. Способ получения N-метил-2-[3-(1,2,3,6-тетрагидро-1-метил-4-пиридинил)-1Н-индол-5-ил] этенсульфонамида, отличающийся тем, что дегидратируют соединение формулы XVI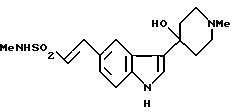
| УСТРОЙСТВО для ОПРЕДЕЛЕНИЯ ПОЛОЖЕНИЯ ПОВЕРХНОСТЕЙ | 0 |
|
SU303507A1 |
| EP 0354777 A, 1990 | |||
| Способ получения производных индолилпиперидина или их солей | 1989 |
|
SU1804460A3 |

