Данное изобретение относится к производным 4-(замещенный фениламино)хиназолина, которые полезны при лечении гиперпролиферативных заболеваний, таких как злокачественные опухоли (рак) у млекопитающих.
Многие из теперешних методов лечения рака используют соединения, которые ингибируют синтез ДНК. Такие соединения являются обычно токсичными к клеткам, но их токсическое действие на быстро делящиеся опухолевые клетки может быть благоприятным. Были разработаны альтернативные подходы к противораковым агентам, механизм действия которых иной, чем ингибирование синтеза ДНК, для того, чтобы увеличить селективность действия против раковых клеток.
Известно, что клетки могут становиться раковыми за счет трансформации части ее ДНК в онкогены (т.е. гены, которые после активации ведут к образованию клеток злокачественной опухоли). Многие онкогены кодируют белки, которые являются аберрантными тирозинкиназами, способными вызывать трансформацию клеток. Альтернативно, сверхэкспрессия нормальной протоонкогенной тирозинкиназы может также приводить в результате к пролиферативным нарушениям, иногда приводящим в результате к злокачественному фенотипу.
Рецепторные тирозинкиназы представляют крупные ферменты, которые охватывают клеточную мембрану и обладают внеклеточным связывающим доменом для факторов роста, таких как эпидермальный фактор роста, трансмембранным доменом и внутриклеточной частью, которая функционирует как киназа, фосфорилируя специфические тирозиновые остатки в белках и, следовательно, оказывая влияние на пролиферацию клеток. Известно, что такие киназы являются часто аберрантно экспрессированными в обычных раковых опухолях человека, таких как рак груди, рак желудочно-кишечного тракта, такой как рак ободочной кишки, рак прямой кишки или желудка, лейкемия и рак яичников, бронхиальный или панкреатический рак. Показано было также, что рецептор фактор эпидермального роста (EGFR), который обладает активностью тирозинкиназы, мутируется и/или сверхэкспрессируется во многих раковых опухолях человека, таких как опухоли мозга, легких, сквамозных клеток, мочевого пузыря, желудка, груди, головы и шеи, азофаговые, гинекологические и тироидные опухоли.
Соответственно, признано было, что ингибиторы рецепторных тирозинкиназ являются полезными в качестве селективных ингибиторов роста раковых клеток млекопитающих. Например, эрбстатин, ингибитор тирозинкиназы, селективно аттенуирует (ослабляет) в атимических голых мышах трансплантированной рост карциномы молочной железы человека, которая экспрессирует рецепторную тирозинкиназу фактора эпидермального роста (EGFR), но не оказывает действие на рост другой карциномы, которая не экспрессирует рецептор EGF.
Показано было также, что ингибирующими свойствами в отношении тирозинкиназы обладают различные другие соединения, такие как производные стирола. Совсем недавно в пяти европейских патентных публикациях, а именно в ЕР 0566226 А1, ЕР 0602851 А1, ЕР 0635507 А1, ЕР 0635498 А1 и ЕР 0520722 А1, было описано, что некоторые производные хиназолина обладают противо-раковыми свойствами, которые являются результатом их свойств ингибирования тирозинкиназы. В публикации РСТ WO 92/20642 также раскрываются бис-моно и бициклические арильные и гетероарильные соединения в качестве ингибиторов тирозинкиназы.
Хотя описанные выше противо-раковые соединения вносят важный вклад в данную область техники, в данной отрасли техники ведется продолжающийся поиск улучшенных противораковых фармацевтических веществ.
Данное изобретение направлено на 4-замещенный фениламино/хиназолиновые производные формулы
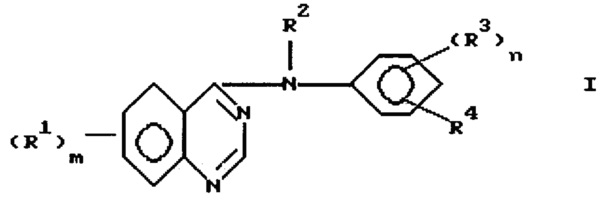
и их фармацевтически приемлемые соли и пролекарства, где:
m представляет 1, 2 или 3;
каждый R1 независимо выбран из водорода, галоида, гидрокси, амино, гидроксиамино, карбокси, (1-4)С-алкоксикарбонила, нитро, гуанидино, уреидо, карбамоила, циано, трифторметила, (R6)2 N-карбонила и фенил-W-алкила, где W выбран из одинарной связи, О, S и NH;
или каждый R1 независимо выбран из циано-(1-4)С алкила и R9, где R9 выбран из группы, состоящей из R5, R5O, (R6)2N, R7C(=O), R5ONH, А и R5Y; R5 представляет (1-4)C алкил; R6 представляет водород или R5, где символы R5 являются одинаковыми или различными; R7 представляет R5, R5O или (R6)2N; А выбран из пиперидино-, морфолино, пирролидино и 4-R6 -пиперазин-1-ила имидазол-1-ила, 4-пиридон-1-ила, карбокси-(1-4)С-алкила, фенокси, фенила, фенилсульфанила, (2-4)С алкенила, (R6)2-N-карбонил-(1-4)C алкила; и Y выбран из S, SO, SO2; алкильные фрагменты в (R6)2N являются необязательно замещенными галоидом или R9, где R9 имеет значения, определенные выше, а алкильные фрагменты в R5 и R5O являются необязательно замещенными галоидом, R6O или R9, где R6 и R9 имеют значения, определенные выше, и где получающиеся в результате группы необязательно замещены галоидом или R9 при условии, что атом азота, кислорода или серы и еще один гетероатом не могут быть присоединены к одному и тому же атому углерода, и с дополнительным условием, что не более чем три "R9" звена могут быть включены в R1; или каждый R1 независимо выбран из R5-сульфониламино, фталимидо-(1-4)С алкилсульфониламино, бензамидо, бензолсульфониламино, 3-фенилуреидо, 2-оксопирролидин-1-ила, 2, 5-диоксопирролидин-1-ила и R10-(2-4)C алканоиламино, где R10 выбран из галоида, R6O, (2-4)С-алканоилокси, R7C(=), и (R6)2N; и где указанный бензамидо, или бензолсульфониламино, или фенильный, или фенокси, или анилино, или фенилсульфанильный заместитель в R1 может необязательно нести один или два галогена, (1-4)С алкильных, циано, метансульфонильных или (1-4)С алкокси заместителя;
или любые два R1, взятые вместе с атомами углерода, к которым они присоединены, включают 5-8-членное кольцо, включающее, по крайней мере, один или два гетероатома, выбранных из кислорода, серы или азота; и где алкильные группы или алкильные части алкокси или алкиламино групп могут быть с прямой цепью, или, если они составлены, по крайней мере из трех атомов углерода, они могут быть разветвленными или циклическими;
R2 выбран из водорода и необязательно замещенного (1-6)С-алкила;
n представляет 1 или 2, и каждый R3 независимо выбран из водорода, необязательно замещенного (1-6)С-алкила, необязательно замещенного амино, галоида, гидрокси, необязательно замещенного гидрокси; R4 представляет азидо или R11-этинил, где R11 выбран из водорода, необязательно замещенного (1-6) С-алкила, где заместители выбраны из водорода, амино, гидрокси, R5O, R5NH и (R5)2N.
Более особенно данное изобретение относится к соединениям формулы I, в которой m, n, R1 и R3 имеют значения, определенные выше, и R2 представляет водород, a R4 представляет R11-этинил, где R11 выбран из водорода, необязательно замещенного (1-6)С-алкила, где заместители выбраны из водорода, амино, гидрокси, R5O, R5NH и (R5)2N, или R4 представляет азидо.
Изобретение относится также к соединениям формулы I, в которой n имеет значения, определенные выше, и m представляет 1 или 2, каждый R1 независимо выбран из водорода, гидрокси, амино, гидроксиамино, карбокси, нитро, карбамоила, уреидо;
R5, необязательно замещенного галоидом, R6O, НОС(=O), (R6)2NC(=O), А и (R6)2N;
R12O, где R12 представляет HK, и K представляет (2-4)С-алкил, необязательно замещенный галоидом, R6O, (2-4)С-алканоилокси, НОС(=O), А и (R6)2N, R6OKO, R6OKNH, CN и фенил;
R5NH, необязательно замещенного галоидом, (2-4)С-алканоилокси, R6O, R7C(=O), (R6)2N, A, R6OKO, R6OKNH, C6H5Y, CN;
(R6)2 N(C=O), R5ONH, R5S, (1-4) С-алкилсульфониламино, фталимидо-(1-4)С-алкилсульфониламино: 3-фенилуреидо, 2-оксопирролидин-1-ила, 2, 5-диоксопирролидин-1-ила, галоид-(2-4)С-алканоиламино, гидрокси-(2-4)С-алканоиламино, (2-4)С-алканоилокси-(2-4)С-алканоиламино, (1-4)С-алкокси-(2-4) С-алканоиламино, карбокси-(2-4)С-алканоиламино, (1-4) С-алкоксикарбонил-(2-4)С-алканоиламино, карбамоил-(2-4)С-алканоиламино, N-(1-4)С-алкилкарбамоил-(2-4)С-алканоиламино, N, N-ди[(1-4)С-алкил]-карбамоил-(2-4)С-алканоиламино, амино- (2-4)С-алканоиламино, (1-4)С-алкиламино-(2-4)С-алканоиламино, ди-(1-4)С-алкиламино-(2-4)С-алканоиламино, и где указанный фенил или фенокси или анилино заместитель в R1 может необязательно нести один или два галогена, (1-4)С-алкильных или (1-4)С-алкокси заместителя; или любые два R1, взятые вместе с атомами углерода, к которым они присоединены, образуют 5-8-членное кольцо, включающее, по крайней мере, один или два гетероатома, выбранных из кислорода, серы или азота; и где алкильные группы и алкильные части алкокси или алкиламино групп могут быть с прямой цепью, или, если они составлены по крайней мере, из трех атомов углерода, они могут быть разветвленными или циклическими;
каждый R3 независимо выбран из водорода, метила, этила, амино, галоида и гидрокси;
R4 представляет R11-этинил, где R11 представляет водород.
Наиболее особенно данное изобретение относится к соединениям формулы I, в которой m, n, R1, R2 и R3 имеют значения, определенные выше, и каждый R1 независимо выбран из водорода, гидрокси, амино, гидроксиамино, нитро, карбамоила, уреидо, R5, необязательно замещенного галоидом, R6O, НОС(=O), H2NC(=O);
R5O, необязательно замещенного галоидом, R6O, (2-4)С-алканоилокси НОС(=О), (R6)2N, А, фенилом; R5NH, (R5)2N, R5NH2, (R5)2NH, R5NHC(=O), (R5)2 NC(=O), R5S, фенил-(2-4)С-алкокси, и где указанный фенильный заместитель в R1 может необязательно нести один или два галоида, R5 или R5O заместителя; или любые два R1, взятые вместе с углеродами, к которым они присоединены, составляют 5-8-членное кольцо, включающее, по крайней мере, один или два гетероатома, выбранных из кислорода, серы или азота; и где алкильные группы и алкильные части алкокси или алкиламино групп могут быть с прямой цепью, или, если они составлены, по крайней мере, из трех атомов углерода, то они могут быть разветвленными или циклическими.
Данное изобретение наиболее особенно относится к соединениям формулы I, выбранным из группы, состоящей из следующих:
(6,7-диметоксихиназолин-4-ил)-(3-этинилфенил)-амин;
(6,7-диметоксихиназолин-4-ил)-[3-(3'-гидроксипропин-1-ил)фенил]-амин,
(6,7-диметоксихиназолин-4-ил)-[(3-(2'-(аминометил)-этинил)фенил]-амин;
[(3-этинилфенил)-(6-нитрохиназолин-4-ил)]-амин,
(6,7-диметоксихиназолин-4-ил)-(4-этинилфенил)-амин,
(6,7-диметоксихиназолин-4-ил)-(3-этинил-2-метилфенил)-амин,
(6-аминохиназолин-4-ил)-(3-этинилфенил)-амин,
(3-этинилфенил)-(6-метансульфониламинохиназолин-4-ил)-амин,
(3-этинилфенил)-(6,7-метилендиоксихиназолин-4-ил)-амин,
(6,7-диметоксихиназолин-4-ил)-(3-этинил-6-метилфенил)-амин,
(3-этинилфенил)-(7-нитрохиназолин-4-ил)-амин,
(3-этинилфенил)-[6-(4'-толуолсульфониламино)-хиназолин-4-ил]амин,
(3-этинилфенил)-{6-[2'-фталимидо-этан-1'-ил-сульфониламино]хиназолин-4-ил}-амин,
(3-этинилфенил)-(6-гуанидинохиназолин-4-ил)-амин,
(7-аминохиназолин-4-ил)-(3-этинилфенил)-амин,
(3-этинилфенил)-(7-метоксихиназолин-4-ил)-амин,
(6-карбометоксихиназолин-4-ил)-(3-этинилфенил)-амин,
(7-карбометоксихиназолин-4-ил)-(3-этинилфенил)-амин,
[6,7-бис(2-метоксиэтокси)хиназолин-4-ил]-(3-этинилфенил)амин,
(3-азидофенил)-(6,7-диметоксихиназолин-4-ил)-амин,
(4-азидофенил)-(6,7-диметоксихиназолин-4-ил)амин,
(3-азидо-5-хлорфенил)-(6,7-диметоксихиназолин-4-ил)амин,
(3-этинилфенил)-(6-метансульфонил-хиназолин-4-ил)-амин,
(6-этансульфанил-хиназолин-4-ил)-(3-этинилфенил)-амин,
(6,7-диметокси-хиназолин-4-ил)-(3-этинил-4-фтор-фенил)-амин,
(6,7-диметокси-хиназолин-4-ил)-(3-пропин-1-ил-фенил)-амин,
[6,7-бис-(2-метокси-этокси)-хиназолин-4-ил]-(5-этинил-2-метилфенил)-амин,
[6,7-бис-(2-метокси-этокси)-хиназолин-4-ил]-(3-этинил-4-фторфенил)-амин,
[6,7-бис-(2-хлор-этокси)-хиназолин-4-ил]-(3-этинил-фенил)-амин,
[6-(2-хлор-этокси)-7-(2-метокси-этокси)-хиназолин-4-ил]-(3-этинил-фенил)-амин,
[6,7-бис-(2-ацетокси-этокси)-хиназолин-4-ил]-(3-этинил-фенил)-амин,
2-[4-(3-этинил-фениламино)-7-(2-гидрокси-этокси)-хиназолин-6-илокси]-этанол,
[6-(2-ацетокси-этокси)-7-(2-метокси-этокси)-хиназолин-4 ил]-(3-этинил-фенил)-амин,
[7-(2-хлор-этокси)-6-(2-метокси-этокси)-хиназолин-4-ил]-(3-этинил-фенил)-амин,
[7-(2-ацетокси-этокси)-6-(2-метокси-этокси)-хиназолин-4-ил]-(3-этинил-фенил)-амин,
2-[4-(3-этинил-фениламино)-6-(2-гидрокси-этокси)-хиназолин-7-илокси]-этанол,
2-[4-(3-этинил-фениламино)-7-(2-метокси-этокси)-хиназолин-6-илокси]-этанол,
2-[4-(3-этинил-фениламино)-6-(2-метокси-этокси)-хиназолин-7-илокси]-этанол,
[6-(2-ацетокси-этокси)-7-(2-метокси-этокси)-хиназолин-4-ил]-(3-этинил-фенил)-амин,
(3-этинил-фенил)-{6-(2-метокси-этокси)-7-[2-(4-метил-пиперазин-1-ил)-этокси]-хиназолин-4-ил}-амин,
(3-этинил-фенил)-[7-(2-метокси-этокси)-6-(2-морфолин-4-ил)-этокси)-хиназолин-4-ил]-амин,
(6,7-диэтоксихиназолин-1-ил)-(3-этинилфенил)-амин,
(6,7-дибутоксихиназолин-1-ил)-(3-этинилфенил)-амин,
(6,7-диизопропоксихиназолин-1-ил)-(3-этинилфенил)-амин,
(6,7-диэтоксихиназолин-1-ил)-(3-этинил-2-метил-фенил)-амин,
[6,7-бис-(2-мстокси-этокси)-хиназолин-1-ил]-(3-этинил-2-метилфенил)-амин,
(3-этинилфенил)-[6-(2-гидрокси-этокси)-7-(2-метокси-этокси)-хиназолин-1-ил]-амин,
[6,7-бис-(2-гидрокси-этокси)-хиназолин-1-ил]-(3-этинил-2-метилфенил)-амин и
2-[4-(3-этинил-фениламино)-6-(2-метокси-этокси)-хиназолин-7-илокси]-этанол.
Еще один аспект изобретения представляет процесс получения соединения формулы
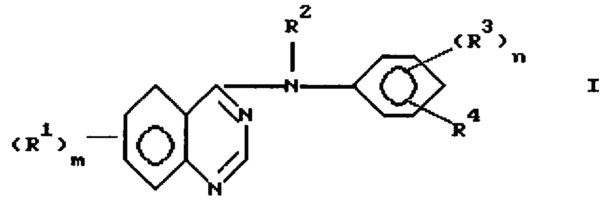
в которой
m представляет 1, 2 или 3;
каждый R1 независимо выбран из водорода, галоида, гидрокси, амино, гидроксиамино, карбокси, (1-4)С-алкоксикарбонила, нитро, гуанидино, уреидо, карбамоила, циано, трифторметила, (R6)2N-карбонила и фенил-W-алкила, где W выбран из одинарной связи, О, S и NH;
или каждый R1 независимо выбран из циано-(1-4)С-алкила и R9, где R9 выбран из группы, состоящей из R5, R5O, (R6)2N, R7C(=O), R5ONH, А и R5Y; где R5 представляет (1-4)С-алкил; R6 представляет водород или R5, где радикалы R5 являются одинаковыми или различными; R7 представляет R5, R5O или (R6)2N; А выбран из пиперидино, морфолино, пирролидино и 4-R6-пиперазин-1-ила имидазол-1-ила, 4-пиридон-1-ила, карбокси-(1-4)С-алкила, фенокси, фенила, фенилсульфанила, (2-4)С-алкенила, (R6)2-N-карбонил-(1-4)С-алкила; и Y выбран из S, SO, SO2; алкильные фрагменты в (R6)2N являются необязательно замещенными галоидом или R9, где R9 имеет значения, определенные выше, а алкильные фрагменты в R5 и R5O являются необязательно замещенными галоидом, R6O или R9, где R6 и R9 имеют значения, определенные выше, и где получающиеся в результате группы являются необязательно замещенными галоидом или R9 при условии, что атом азота, кислорода или серы и еще один гетероатом не могут быть присоединены к одному и тому же атому углерода, и при дополнительном условии, что не более чем три "R9" звена могут составлять R1;
или каждый R1 независимо выбран из R5-сульфониламино, фталимидо-(1-4)С-алкилсульфониламино, бензамидо, бензолсульфониламино, 3-фенилуреидо, 2-оксопирролидин-1-ила, 2,5-диоксопирролидин-1-ила и R10-(2-4)C-алканоиламино, где R10 выбран из галоида, R6O, (2-4)С-алканоилокси, R7C(=O) и (R6)2N; и где указанный бензамидо или бензолсульфониламино, или фенил или фенокси; или анилино или фенилсульфанильный заместитель в R1 может необязательно нести один или два галогена, (1-4)С-алкильных, циано, метансульфонильных или (1-4)С-алкокси заместителя;
или любые два R1, взятые вместе с углеродами, к которым они присоединены, составляют 5-8-членное кольцо, включающее, по крайней мере, один или два гетероатома, выбранных из кислорода, серы или азота; и где алкильные группы и алкильные части алкокси или алкиламино групп могут быть с прямой цепью, или, если они составлены из по крайней мере трех атомов углерода, они могут быть разветвленными или циклическими;
R2 выбран из водорода и необязательно замещенного (1-6) С-алкила;
n представляет 1 или 2, и каждый R3 независимо выбран из водорода, необязательно замещенного (1-6)С-алкила, необязательно замещенного амино, галоида, гидрокси, необязательно замещенного гидрокси; R4 представляет азидо или R11-этинил, где R11 выбран из водорода необязательно замещенного (1-6)С-алкила, где заместители выбраны из водорода, амино, гидрокси, R5O, R5NH и (R5)2N, который включает
а) обработку соединения формулы
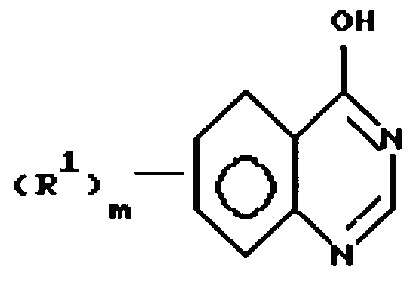
или
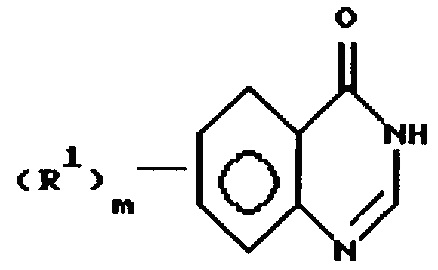
где R1 и m имеют значения, определенные выше,
с CCl4 и необязательно замещенным триарилфосфином, необязательно осажденным на инертном полимере, формулы Ar3P, где каждый Ar представляет необязательно замещенную (6-10) С-арильную группу, и каждый из заместителей независимо выбран из (1-6)С-алкила; и
b) обработку продукта стадии а) соединением формулы
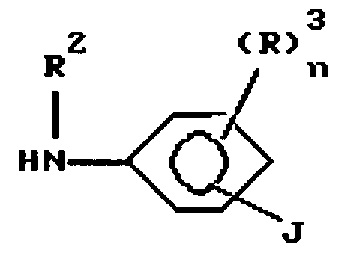
где R2, R3 и n имеют значения, определенные выше, и J представляет Y или R4, где R5 имеет значения, определенные выше, при условии, что, когда J представляет Y, тогда продукт стадии b) должен далее обрабатываться алкином.
Еще один аспект данного изобретения направлен на способ лечения гиперпролиферативных заболеваний млекопитающих путем назначения млекопитающему, страдающему от гиперпролиферативного заболевания, соединения формулы I в эффективном для лечения гиперпролиферативного заболевания количестве.
Данное изобретение также направлено на фармацевтические композиции для лечения гиперпролиферативных заболеваний млекопитающих, которые включают эффективное для лечения гиперпролиферативного заболевания количество соединения формулы I и фармацевтически приемлемый носитель.
Галоид означает хлор, бром, иод или фтор.
Под алкилом имеется в виду углеводородный фрагмент с прямой цепью, циклический или разветвленный, насыщенный или ненасыщенный, при условии, что указанный алкил должен включать три или более атомов углерода, если он является разветвленным или циклическим.
В том смысле, как оно используется здесь, выражение "реакционно-инертный растворитель" относится к растворителю, который не взаимодействует с исходными материалами, реагентами, промежуточными продуктами или продуктами каким-либо образом, который пагубно влияет на выход желаемого продукта.
Другие признаки и преимущества изобретения будут видны из описания и формулы изобретения, которые описывают изобретение.
Подробное описание изобретения
Соединения формулы I, их фармацевтически приемлемые соли и пролекарства (называемые здесь далее активные соединения) могут получаться с помощью любого процесса, известного применительно к получению химически родственных соединений.
Обычно активные соединения могут получаться из соответствующим образом замещенных хиназолинов с использованием соответственным образом замещенного амина.
Как показано на схеме (см. в конце описания), соответствующий 4-замещенный хиназолин 2, в котором X представляет подходящую вытесняемую удаляемую группу, такую как галоид, арилокси, алкилсульфинил, алкилсульфонил, такую как трифторметансульфонилокси, арилсульфинил, арилсульфонил, силокси, циано, пиразоло, триазоло или тетразоло, предпочтительно 4-хлорхиназолин, подвергается реакции с соответствующим амином или гидрохлоридом (хлоргидратом) амина 4 или 5, где R4 имеет значения, описанные выше, и Y представляет бром, иод или трифторметан-сульфонилокси, в растворителе, таком как (1-6)С-спирт, диметилформамид (ДМФ), N-метилпирролидин-2-он, хлороформ, ацетонитрил, тетрагидрофуран (ТГФ), 1-4-диоксан, пиридин или другой апротонный растворитель. Реакция может проводиться в присутствии основания, предпочтительно карбоната или гидроокиси щелочного или щелочноземельного металла, или третичного аминового основания, такого как пиридин, 2,6-лютидин, коллидин, N-метил-морфолин, триэтиламин, 4-диметиламино-пиридин или N,N-диметиланилин. Эти основания далее здесь называются подходящими основаниями. Реакционная смесь поддерживается при температуре примерно от температуры окружающей среды до температуры дефлегмации растворителя, предпочтительно примерно от 35°C до температуры дефлегмации, до тех пор, пока по существу не сможет обнаруживаться оставшийся 4-галоидхиназолин, в типичном случае примерно от 2 до 24 часов. Предпочтительно, реакция проводится в инертной атмосфере, такой как сухой азот.
Обычно реагенты объединяются в стехиометрических количествах. Когда используется аминовое основание для тех соединений, когда используется соль (обычно HCl соль) амина 4 или 5, предпочтительно использовать избыток аминового основания, обычно дополнительный эквивалент аминового основания. (Альтернативно, если аминовое основание не используется, может использоваться избыток амина 4 или 5). Для тех соединений, для которых используется пространственно (стерически) затрудненный амин 4 (такой как 2-алкил-3-этиниланилин) или очень реакционноспособный 4-галоидхиназолин; предпочитается использовать трет-бутиловый спирт или полярный апротонный растворитель, такой как ДМФ или N-метил-пирролидин-2-он, в качестве растворителя.
Альтернативно, 4-замещенный хиназолин 2, в котором X представляет гидроксил или оксо (и 2-азот гидрирован), подвергается реакции с четыреххлористым углеродом и необязательно замещенным триарилфосфином, который необязательно нанесен на инертный полимер (например, трифенилфосфин, нанесенный на полимер. Кат. Aldrich N 36645-5, который представляет 2% дивинилбензольный сшитый полистирол, содержащий 3 ммоля фосфора на грамм смолы) в растворителе, таком как четыреххлористый углерод, хлороформ, дихлорэтан, тетрагидрофуран, ацетонитрил или другой апротонный растворитель или смеси их. Реакционная смесь поддерживается при температуре примерно от равной температуре окружающей среды до температуры дефлегмации, предпочтительно, примерно от 35°C до температуры дефлегмации, в течение 2-24 часов. Данная смесь подвергается реакции с соответствующим амином или хлоргидратом амина 4 или 5 или непосредственно, или после удаления растворителя, например, с помощью вакуумного испарения, и добавления подходящего альтернативного растворителя, такого как (1-6)С-спирт, ДМФ, N-метилпирролидин-2-он, пиридин или 1-4 диоксан. Затем, реакционная смесь поддерживается при температуре примерно от температуры окружающей среды до температуры дефлегмации растворителя, предпочтительно примерно от 35°C до температуры дефлегмации, до тех пор, пока не будет достигнуто по существу полное образование продукта, обычно примерно от 2 до 24 часов. Предпочтительно реакция проводится в инертной атмосфере, такой как сухой азот.
Когда в качестве исходного материала в реакции с хиназолином 2 используется соединение 4, в котором Y представляет бром, иод или трифторметансульфонилокси, образуется соединение формулы 3, в котором R1, R2, R3 и Y имеют значения, описанные выше. Соединение 3 превращается в соединения формулы I, в которых R4 представляет R11-этинил, и R11 имеет значения, определенные выше, по реакции с подходящим палладиевым реагентом, таким как татракис(трифенилфосфин)-палладий или бис(трифенилфосфин) палладий-дихлорид, в присутствии подходящей кислоты Льюиса, такой как хлорид меди, и подходящего алкина, такого как триметилсилилацетилен, пропаргиловый спирт или 3-(N,N-диметиламино)-пропин в растворителе, таком как диэтиламин или триэтиламин. Соединения 3, в которых Y представляет NH2, могут превращаться в соединения 1, в которых R4 представляет азид, с помощью обработки соединения 3 диазотирующим агентом, таким как кислота и нитрит (например, уксусная кислота и NaNO2) с последующей обработкой получающегося в результате продукта азидом, таким как азид натрия (NaN3).
Для получения тех соединений формулы I, в которой R1 представляет амино или гидроксиамино группу, применяется восстановление соответствующего соединения формулы I, в которой R1 представляет нитро.
Восстановление может удобно осуществляться с помощью любой из многих процедур, известных для таких преобразований. Восстановление может осуществляться, например, с помощью гидрирования нитро соединения в реакционно инертном растворителе, в присутствии подходящего металлического катализатора, такого как палладий, платина или никель. Дополнительным подходящим восстанавливающим агентом является, например, активированный металл, такой как активированное железо (получаемое с помощью промывки порошка железа разбавленным раствором кислоты, такой как соляная кислота). Так, например, восстановление может осуществляться с помощью нагревания смеси нитро соединения и активированного металла с концентрированной соляной кислотой в таком растворителе, как смесь воды и спирта, например, метанола или этанола, до температуры в интервале, например, 50-150°C удобным образом при равной или близкой к 70°C. Еще один подходящий класс восстанавливающих агентов составляют дитиониты щелочного металла, такие как дитионит натрия, которые могут использоваться в (1-4)С-алкановых кислотах, (1-6)С-алканолах, воде или в смесях их.
Для получения тех соединений формулы I, в которой R2 или R3 включает первичный или вторичный аминовый фрагмент (иной, чем амино группа, предназначенная для реакции с хиназолином), такая свободная амино группа предпочтительно защищается перед описанной выше реакцией с последующим снятием защиты после описанной выше реакции с 4-(замещенным)хиназолином 2.
Могут использоваться несколько хорошо известных групп, защищающих азот. Такие группы включают (1-6) С-алкоксикарбонил, необязательно замещенный бензилоксикарбонил, арилоксикарбонил, трититл, винилоксикарбонил, О-нитрофенилсульфонил, дифенилфосфинил, п-толуол-сульфонил и бензил. Добавление защищающей азот группы может осуществляться в хлорированном углеводородном растворителе, таком как метиленхлорид или 1,2-дихлорэтан, или в простом эфирном растворителе, таком как глим, диглим или ТГФ, в присутствии или отсутствие третичного аминового основания, такого как триэтиламин, диизопропилэтиламин или пиридин, предпочтительно триэтиламин, при температуре примерно от 0°C до 50°C, предпочтительно примерно при температуре окружающей среды. Альтернативно, защитные группы удобным образом присоединяются с использованием условий Шоттен-Бауманна.
После описанной выше реакции присоединения соединений 2 и 5, защитная группа может удаляться с помощью методов деблокирования известных специалистам в данной области, таких как обработка трифторуксусной кислоты в метиленхлориде, для продуктов, защищенных трет-бутоксикарбонилом.
Что касается описания защитных групп и их использования, см. публикации T.W. Greene and P.G.H. Wuts, "Protective Groups in Organic synthesis" Второе изд., Джон Вили энд Санз, Нью Йорк, 1991.
Для получения соединений формулы I, в которой R1 или R2 представляет гидрокси, предпочитается расщепление соединения формулы I, в которой R1 или R2 представляет (1-4)С-алкокси.
Реакция расщепления может удобно осуществляться с помощью любой из многих процедур, известных для таких преобразований. Обработка защищенного производного формулы I расплавленным хлоргидратом пиридина (20-30 эквив.) при 150-175°C может применяться для реакций О-деалкилирования. Альтернативно, реакция расщепления может осуществляться, например, с помощью обработки защищенного хиназолинового производного (1-4)С-алкилсульфидом щелочного металла, таким как этантиолат натрия, или путем обработки диарилфосфидом щелочного металла, таким как дифенилфосфид лития. Реакция расщепления может также удобно осуществляться с помощью обработки защищенного производного хиназолина тригалогенидом бора или алюминия, таким как трехбромистый бор. Такие реакции предпочтительно осуществляются в присутствии реакционно-инертного растворителя при подходящей температуре.
Соединения формулы I, в которой R1 или R2 представляет (1-4)С-алкилсульфинил или (1-4)С-алкилсульфонил, предпочтительно получаются с помощью окисления соединения формулы I, в которой R1 или R2 представляет (1-4) С-алкилсульфанильную группу. Для окисления сульфанила в сульфинил и/или сульфонил подходящие окисляющие агенты хорошо известны в технике, например, перекись водорода, перкислота (такая как 3-хлорпероксибензойная или пероксиуксусная кислота), пероксисульфат щелочного металла (такой как пероксимоносульфат калия), трехокись хрома или газообразный кислород, в присутствии платины. Окисление обычно осуществляется в насколько возможно мягких условиях с использованием стехиометрического количества окисляющего агента, для того чтобы снизить риск переокисления и повреждения других функциональных групп. Обычно данная реакция осуществляется в подходящем растворителе, таком как метиленхлорид, хлороформ, ацетон, тетрагидрофуран или трет-бутилметиловый эфир, и при температуре примерно от -25 до 50°C, предпочтительно при температуре, равной или близкой к температуре окружающей среды, т.е. в интервале 15-35°C. Когда желательно соединение, несущее сульфинильную группу, следует использовать более мягкие окисляющие агенты, такие как метапериодат натрия или калия, удобным образом, в полярном растворителе, таком как уксусная кислота или этанол. Соединения формулы I, содержащие (1-4)С-алкилсульфонильную группу, могут получаться окислением соответствующего (1-4)С-алкилсульфинильного соединения, также как и соответствующего (1-4)С-алкилсульфанильного соединения.
Соединения формулы I, в которой R1 представляет необязательно замещенный (2-4)С-алканоиламино, уреидо, 3-фенилуреидо, бензамидо или сульфонамидо, могут получаться ацилированием или сульфонилированием соответствующего соединения, в котором R1 представляет амино. Подходящими ацилирующими агентами являются любые агенты, известные в технике для ацилирования амино в ациламино, например, ацилгалогениды, например, (2-4)С-алканоилхлорид или бромид, или бензоил-хлорид или -бромид, ангидриды алкановых кислот или смешанные ангидриды, например, уксусный ангидрид или смешанный ангидрид, образовавшийся по реакции алкановой кислоты и (1-4)С-алкоксикарбонилгалогенида, например (1-4)С-алкоксикарбонилхлорида, в присутствии подходящего основания. Для получения тех соединений формулы I, в которой R1 представляет уреидо или 3-фенилуреидо, подходящим ацилирующим агентом является, например, цианат, например, цианат щелочного металла, такой как цианат натрия, или изоцианат, такой как фенилизоцианат. Реакции N-сульфонилирования могут осуществляться с подходящими сульфонилгалогенидами или сульфонилангидридами в присутствии третичного аминового основания. Обычно ацилирование или сульфонилирование осуществляется в реакционно-инертном растворителе и при температуре в интервале примерно от -30 до 120°C, удобным образом, при температуре, равной или близкой к температуре окружающей среды.
Соединения формулы I, в которой R1 представляет (1-4)С-алкокси или замещенный (1-4)С-алкокси, или R1 представляет (1-4)С-алкиламино или замещенный моно-N- или ди-N,N-(1-4)С-алкиламино, получаются с помощью алкилирования, предпочтительно в присутствии подходящего основания, соответствующего соединения, в котором R1 представляет гидрокси или амино, соответственно. Подходящие алкилирующие агенты включают алкил- или замещенные алкилгалогениды, например, необязательно замещенный (1-4)С-алкилхлорид, -бромид или -иодид, в присутствии подходящего основания, в реакционно-инертном растворителе, и при температуре в пределах примерно 10-140°C, удобным образом при равной или близкой к температуре окружающей среды.
Для получения тех соединений формулы I, в которой R1 представляет амино-, окси- или циано-замещенный (1-4)С-алкильный заместитель, соответствующее соединение, в котором R1 представляет (1-4)С-алкильный заместитель, несущий группу, которая способна вытесняться или замещаться амино-, алкокси- или циано группой, подвергается реакции с соответствующим амином, спиртом или цианидом, предпочтительно в присутствии подходящего основания. Реакция предпочтительно осуществляется в реакционно-инертном растворителе или разбавителе, при температуре в интервале примерно 10-100°C, предпочтительно при равной или близкой к температуре окружающей среды.
Соединения формулы I, в которой R1 представляет карбокси заместитель или заместитель, который включает карбокси группу, получаются с помощью гидролиза соответствующего соединения, в котором R1 представляет (1-4)С-алкоксикарбонильный заместитель или заместитель, который включает (1-4)С-алкокси-карбонильную группу. Гидролиз может удобно проводиться, например, в основных условиях, например, в присутствии гидроокиси щелочного металла, такой как проиллюстрирована в сопровождающих примерах.
Соединения формулы I, в которой R1 представляет амино, (1-4)С-алкиламино, ди-[(1-4)С-алкил] амино, пирролидин-1-ил, пиперидино, морфолино, пиперазин-1-ил, 4-(1-4)С-алкилпиперазин-1-ил или (1-4)С-алкилсульфанил, могут получаться с помощью реакции, в присутствии подходящего основания, соответствующего соединения, в котором R1 - группа, замещаемая амином или тиолом, с соответствующим амином или тиолом. Реакция предпочтительно осуществляется в реакционно-инертном растворителе или разбавителе, и при температуре в интервале примерно 10-180°C, предпочтительно в интервале 100-150°C.
Соединения формулы I, в которой R1 представляет 2-оксопирролидин-1-ил или 2-оксопиперидин-1-ил, получаются с помощью циклизации, в присутствии подходящего основания, соответствующего соединения, в котором R1 представляет галоид-(2-4)С-алканоиламино группу. Реакция предпочтительно осуществляется в реакционно-инертном растворителе или разбавителе при температуре в интервале примерно 10-100°C удобным образом, при равной или близкой к температуре окружающей среды.
Для получения соединений формулы I, в которой R1 представляет карбамоил, замещенный карбамоил, алканоилокси или замещенный алканоилокси, удобным является карбамоилирование или ацилирование соответствующего соединения, в котором R1 представляет гидрокси.
Подходящие ацилирующие агенты, известные в технике для ацилирования гидроксиарильных фрагментов в алканоилоксиарильные группы, включают, например, (2-4)С-алканоилгалогениды, (2-4)С-алканоилангидриды и смешанные ангидриды, как описаны выше, и могут применяться подходящие их замещенные производные, в типичном случае в присутствии подходящего основания. Альтернативно, (2-4)С-алкановые кислоты или их подходящим образом замещенные производные могут присоединяться к соединению формулы I, в котором R1 представляет гидрокси, с помощью конденсирующего агента, такого как карбодиимид. Для получения тех соединений формулы I, в которой R1 представляет карбамоил или замещенный карбамоил, подходящими карбамоилирующими агентами являются, например, цианаты или алкил или арилизоцианаты, в типичном случае, в присутствии подходящего основания. Альтернативно, подходящие промежуточные соединения, такие как хлорформиат или карбонилимидазолильное производное соединения формулы I, в которой R1 представляет гидрокси, может получаться, например, с помощью обработки указанного производного фосгеном (или фосгеновым эквивалентом) или карбонилдиимидазолом. Получающееся в результате промежуточное соединение может затем подвергаться реакции с соответствующим амином или замещенным амином для получения желаемого карбамоильного производного.
Соединения формулы I, в которой R1 представляет аминокарбонил или замещенный аминокарбонил, могут получаться с помощью аминолиза подходящего промежуточного соединения, в котором R1 представляет карбокси.
Активирование и присоединение соединений формулы I, в которой R1 представляет карбокси, может выполняться с помощью большого разнообразия методов, известных специалистам в данной области. Подходящие способы включают активирование карбоксила, такого как галоидангидрид кислоты, азид, симметричный или смешанный ангидрид кислоты, или активного сложного эфира соответствующей реакционноспособности для сочетания с желаемым амином. Примеры таких типов промежуточных соединений и их получение и использования в реакции присоединения или сочетания с аминами, можно найти широко в литературе, например, М. Bodansky и A. Bodansky, "The Practice of Peptide Synthesis"; Springer.-Verlag, Нью Йорк, 1984. Получающиеся в результате соединения формулы I могут выделяться и очищаться с помощью стандартных способов, таких как удаление растворителя и перекристаллизация или хроматография.
Исходные материалы для описанных выше реакционных схем (например, амины, хиназолины и защищающие амин группы) являются легко доступными или могут быть легко синтезированы специалистами в данной области с использованием общепринятых методов органического синтеза. Например, получение 2,3-дигидро-1,4-бензоксазиновых производных описано в работе R.C. Elderfield, Todd, S. Gerber, Гл. 12, в "Heterocyclic Compounds" том. 6, R.C. Elderfield ред, Джон Вили энд Санз, Инк, Нью Йорк, 1957. Замещенные 2,3-дигидро-бензотиазинильные соединения описаны авторами R.C. Elderfield и Е.Е. Harris в гл. 13 тома 6 книги Elderfield "Heterocyclic Compounds".
Некоторые хиназолины формулы I могут существовать в сольватированной, а также в несольватированной формах, таких как гидратированные формы. Следует понимать, что изобретение охватывает все такие сольватированные, так же как и несольватированные формы, которые обладают активностью против гиперпролиферативных заболеваний.
Подходящей фармацевтически приемлемой солью соединения формулы I является например кислотно-аддитивная соль соответствующего соединения, которое является достаточно основным, например кислотно-аддитивная соль, например, с неорганической или органической кислотой, такой как соляная, бромистоводородная, серная, фосфорная, метансульфоновая, бензолсульфоновая, трифторуксусная, лимонная, молочная или малеиновая кислота. Подходящей фармацевтически приемлемой аддитивной солью основания соединения формулы I, которое является кислотным, является соль со щелочным металлом, например, литиевая, натриевая или калиевая соль; соль со щелочноземельным металлом, например, кальциевая или магниевая соль; аммониевая соль; или соль с органическим основанием, которое дает физиологически приемлемый катион, например, соль с метиламином, диметиламином, триметиламином, пиперидином, морфолином или трис-(2-гидрокси-этил) амином. Все такие соли находятся в сфере данного изобретения, и они могут получаться с помощью общепринятых способов. Например, они могут получаться просто путем введения в контакт кислотных или основных молекул, обычно в стехиометрическом соотношении, или в водной, или неводной, или частично водной среде, в зависимости от того, что является подходящим. Соли выделяются с помощью фильтрования; осаждения нерастворителем, предпочтительно простым эфирным или углеводородным растворителем с последующим фильтрованием и выпариванием растворителя, или, в случае водных растворов, с помощью лиофилизации.
Некоторые из соединений формулы I имеют асимметрические атомы углерода. Такие диастереомерные смеси могут разделяться на их отдельные диастереомеры на основе их физических и химических различий, по способам, известным самим по себе, например, с помощью хроматографии и/или фракционной кристаллизации. Энантиомеры могут отделяться путем превращения энантиомерных смесей в диастереомерную смесь по реакции с соответствующим оптически активным соединением (например, спиртом), разделением диастереомеров и превращением (например, гидролизом) индивидуальных диастереомеров в соответствующие чистые энантиомеры. Все такие изомеры, включая диастереомерные смеси и чистые энантиомеры, считаются частью данного изобретения.
Активные соединения данного изобретения являются сильными ингибиторами семейства erb В онкогенных и протоонкогенных белковых тирозинкиназ, таких как рецептор эпидермального фактора роста (EGFR), erb В2, HER3 или HER4, и таким образом все применимы для терапевтического использования в качестве антипролиферативных агентов (например, противораковых) для млекопитающих, особенно людей. В частности, соединения данного изобретения являются терапевтическими или профилактическими агентами для лечения разнообразных опухолей человека (ренальных, печени, почек, мочевого пузыря, груди, желудка, яичников, прямой кишки, простаты, поджелудочной железы, легких, наружных женских половых органов, щитовидной железы, гепатических карцином, саркомы, глиобластомы, различных опухолей головы и шеи), и других гиперпластических состояний, таких как доброкачественной гиперплазии кожи (например, псориаза) или простаты (например, ВРН). В дополнение к изложенному ожидается, что хиназолины настоящего изобретения могут обладать активностью против ряда лейкемий и лимфоидных злокачественных образований.
Можно также ожидать, что активные соединения полезны при лечении дополнительных нарушений, в которые вовлечены аберрантная экспрессия лиганд/рецепторных взаимодействий, активирование сигнализирующих явлений, связанных с различными белковыми тирозинкиназами, активность которых ингибируется агентами формулы I.
Такие расстройства могут включать нарушения нейронального, глиального, астроцитарного гипоталамического характера и других желез, макрофагового, эпителиального, стромального и бластоколического характера, при которых может быть вовлечена аберрантная функция, экспрессия или сигнализирование erb В тирозинкиназ. Кроме того, соединения формулы I могут быть терапевтически полезными при воспалительных ангиогенных и иммунологических расстройствах, вовлекающих как идентифицированные, так и еще неидентифицированные тирозинкиназы, которые ингибируются соединениями формулы I.
Активность ин витро активных соединений в ингибировании рецепторной тирозинкиназы (и таким образом последующего пролиферативного ответа, например, рака) может быть определена с помощью процедуры, подробно описанной ниже.
Активность активных соединений ин витро может определяться по степени ингибирования испытываемыми соединениями по отношению к контролю фосфорилирования экзогенного субстрата (например, LyS3-гастрин или поли Glu Tyr (4: 1) рэндом сополимер (J. Posner et al. J. Biol>Chem. 267 (29), 20638-47 (1992)) рецепторной киназой эпидермального фактора роста по тирозину.
Очищенный от связи растворимый рецептор EGF человека (96 нг) получается в соответствии с процедурой, описанной в работе G.N. Gill, W. Weber, Methods in Enzymology 146, 82-88 (1987) из клеток A431 (Американская коллекция типов культур, Роквилл МД) и предварительно инкубируется в микроцентрифужной пробирке с EGF (2 мкг/мл) в смеси буфер фосфорилирования + ванадат (PBV:50 мМ HEPES, рН 7.4; 125 мМ NaCl; 24 мМ MgCl2; 100 мкм ортованадат натрия) в общем объеме 10 мкл, в течение 20-30 минут при комнатной температуре. Испытываемое соединение, растворенное в диметилсульфоксиде (ДМСО), разбавляется PBV, и 10 мкл смешивается со смесью рецептор EGF/EGF, и инкубируется в течение 10-30 минут при 30°C. Реакция фосфорилирования инициируется добавлением 20 мкл смеси 33Р-АТФ/субстрат (120 мкМ LyS3-гастрин (последовательность однобуквенным кодом для аминокислот, KKKGPWEEEEEYGWLDF), 50 мМ Hepes, рН 7.4, 40 мкМ АТР, 2 МкCi -[33Р]-АТФ) к смеси рецептор EGF/EGF и инкубируется в течение 20 минут при комнатной температуре. Реакция останавливается добавлением 10 мкл стоп-раствора (0.5М ЕДТА, рН 8; 2 мМ АТФ) и 6 мкл 2 н. HCl. Пробирки центрифугируются при 14000 об./мин, 4°C, в течение 10 минут. По 35 мкл супернатанта из каждой пробирки отбирается пипеткой на 2,5 см кружок бумаги Ватман Р81, масса промывается четыре раза 5% уксусной кислотой, по 1 литру на промывку, и затем сушится на воздухе. Это приводит в результате к связыванию субстрата с бумагой с потерей свободного АТФ после промывки. Включенный [33Р] измеряется с помощью жидкостного сцинтилляционного подсчета. Включение в отсутствии субстрата (например, LyS3-гастрин) вычитается из всех величин в виде фона и процент ингибирования вычисляется относительно контрольного опыта, без присутствия испытываемого соединения.
Такие анализы, проводимые с рядом доз испытываемого соединения, позволяют определить приблизительную IC50 величину для ингибирования ин витро активности EGFR киназы. Хотя ингибирующие свойства соединений формулы I, как ожидается, изменяются в зависимости от структурных изменений, обычно активность, проявляемая под действием этих агентов, измеренная по способу, описанному выше, составляет в интервале IC50=0.0001-30 мкМ (IC50=50% ингибирующая концентрация). (Таблица 2).
Активность активных соединений ин виво может быть определена по степени ингибирования роста опухоли под действием испытываемого соединения относительно контроля. Ингибирующее действие на рост опухоли различных соединений измеряется в соответствии с методикой Corbett и др. "Tumor Induction Relationships in Development of Transplantable Cancers of the Colon in Mice for Chemotherapy Assays, with a Note on Cancinogen Structure", Cancer Res., 35, 2434-2439 (1975) и Corbett, Т.Н., и др., "A Mouse Colon-tumor Model for Experimental Therapy", Cancer Chemother. Rep. (Part 2)", 5, 169-186 (1975), с незначительными модификациями. Опухоли индуцируются в левом боку путем подкожной инъекции культивированных в 1⋅106 log фазе опухолевых клеток (клетки карциномы груди человека MDA-MB-468 или карциномы головы и шеи человека HN5), суспендированных в 0.10 мл RPM1 1640. По прохождении достаточного периода времени, чтобы опухоли стали ощутимыми (2-3 мм в диаметре), испытываемых животных (атимические мыши) подвергали лечению активным соединением (преобразованным в готовую препаративную форму растворением в ДМСО обычно в концентрации от 50 до 100 мг/мл с последующим 1:9 разбавлением солевым раствором или, альтернативно, 1:9 разбавлением 0,1% Плуроник Р105 в 0.9% солевом растворе) с помощью интраперитонального (i.p.) или орального (ро) способов назначения дважды в день (т.е. через каждые 12 часов) в течение 5 дней подряд. Для того чтобы определить противоопухолевое действие, измеряется опухоль в миллиметрах с помощью циркулей Вернье по двум диаметрам, и размер опухоли (мг) вычисляется с использованием следующей формулы: Вес опухоли = [длина ⋅ ширина]2)/2, согласно методике Geran, R.I., и др. "Protocols for Screening Chemical Agents and Natural Products Against Animal Tumors and Other Biological Systems", Третье издание, Cancer Chemother Rep., 3, 1-104 (1972). Результаты выражаются в виде процента ингибирования согласно формуле: Ингибирование (%) = (Вес опухоликонтроля - Вес опухолиопытного) / Вес опухоликонтроля ⋅ 100%.
Границы участка имплантации опухоли дают воспроизводимый эффект зависимости от дозы для разнообразных хемотерапевтических агентов, и способ измерения (диаметра опухоли) является надежным способом оценки скоростей роста опухоли.
Назначение активных соединений может проводиться любым методом, который дает возможность доставки соединений к участку действия (например, к раковым клеткам). Эти методы включают оральные пути назначения, интрадуоденальный (в двенадцатиперстную кишку), парэнтеральную инъекцию (включая внутривенную, подкожную, внутримышечную, внутрисосудистую инъекцию или вливание), топический или местный способ назначения и др.
Назначаемое количество активного соединения будет, конечно, зависеть от субъекта, подвергаемого лечению, от тяжести недуга, от способа назначения и от мнения или квалификации предписывающего врача. Однако, эффективная дозировка составляет приблизительно в интервале 0.001-100 мг/кг, предпочтительно 1-35 мг/кг в виде одной или раздельных доз. Для человека весом в среднем 70 кг это составило бы 0.05-7 г/день, предпочтительно 0.2-2,5 г/день.
Композиция может быть, например, в форме, подходящей для орального назначения, такой как таблетки, пилюли, порошки, готовые формы препарата с задержанным высвобождением лекарства; в виде растворов, суспензий; для парентеральных инъекций в виде стерильного раствора, суспензии или эмульсии; для топического или местного назначения в виде мази или крема, или для ректального назначения в виде суппозиториев или медицинских свечей. Фармацевтическая композиция может быть в форме единичных доз, подходящих для разового назначения с точной дозировкой. Фармацевтическая композиция включает обычно общепринятый фармацевтический носитель или эксципиент и соединение согласно изобретению в качестве активного ингредиента. В дополнение к указанным она может включать другие медицинские или фармацевтические агенты, носители, адьюванты и проч.
Фармацевтические композиции согласно изобретению могут содержать 0.1-95% соединения, предпочтительно 1-70%. В любом случае композиция или готовая форма препарата, предназначенная для применения, содержит активное соединение в количестве, эффективном для облегчения или снижения признаков болезни у субъекта, подлежащего лечению, т.е. симптомов гиперпролиферативных заболеваний, на протяжении курса лечения.
Примеры парентеральных форм для назначения включают растворы или суспензии активных соединений в стерильных водных растворах, например, в водных растворах пропиленгликоля или декстрозы. Такие дозированные формы, если необходимо, могут подходящим образом буферироваться.
Подходящие фармацевтические носители включают инертные разбавители или наполнители, воду и различные органические растворители. Фармацевтические композиции, если необходимо, могут содержать дополнительные ингредиенты, такие как вкусовые или ароматизирующие добавки, связующие, эксципиенты и аналогичные. Так, для орального назначения могут применяться таблетки, содержащие различные эксципиенты, такие как лимонная кислота, вместе с разнообразными дезинтегрирующими агентами, такими как крахмал, альгиновая кислота и некоторые сложные силикаты, и со связующими агентами, такими как сахароза, желатин и камедь акации. Для целей таблетирования часто полезными дополнительно являются смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции аналогичного типа могут также применяться в заполненных ими мягких или твердых желатиновых капсулах. Предпочтительные материалы для них включают лактозу или молочный сахар и полиэтиленгликоли с высоким молекулярным весом. Когда для орального назначения желательны водные суспензии или эликсиры, активное соединение в них может комбинироваться с различными подслащивающими или вкусовыми агентами, красящими веществами, или красителями, и, если необходимо, эмульгирующими агентами или суспендирующими агентами, вместе с разбавителями, такими как вода, этанол, пропиленгликоль, глицерин или их сочетания.
Способы получения разнообразных фармацевтических композиций с конкретным количеством активного соединения являются известными или являются очевидными для специалистов в данной области техники. Например, см. Remington's Pharmaceutical Sciences., Мак Паблишинг Компани, Истер, Ра., 15-е издание (1975).
Лечение гиперпролиферативных заболеваний, описанное выше, может применяться в виде единственного метода терапии или может включать в дополнение к активному соединению одно или более других противоопухолевых веществ. Такое совместное лечение может достигаться путем одновременного, последовательного, циклического или отдельного дозирования отдельных компонентов лечения.
Жидкостная хроматография высокого давления (ЖХВД), используемая в следующих ниже примерах и получениях, выполнялась в соответствии со следующим методом, если она не видоизменялась в отдельных примерах. Картриджная колонка Перкин-Элмер Пекосфер ЗХЗС (3 мм × 3 см, С18; поставляемая фирмой Перкин Элмер Корп., Норволк, СТ 06859) с предколонкой Броунлии (торговая марка) RP-8 Ньюгард (7 мкм, 3.2 мм × 15 мм, поставляемая фирмой Эпплайд Биосистемз Инк., San Tose СА 95134), которая предварительно уравновешивается при рН 4.50, в 200 мМ аммоний ацетатном буфере. Образцы элюировались с использованием линейного градиента 0-100% ацетонитрил/рН 4.50, 200 мМ ацетат аммония на протяжении 10 минут со скоростью потока 3.0 мл/мин. Хроматограммы получались в интервале 240-400 нм с использованием диодного лучевого детектора.
Следует понимать, что данное изобретение не ограничивается конкретными воплощениями, показанными и описанными здесь, и могут производиться различные изменения и модификации без отклонения от сути и объема настоящего изобретения, определенных в пунктах формулы изобретения.
Пример 1
Хлоргидрат (4-азидофенил)-(6,7-диметиоксихиназолин-4-ил)-амина
4-Хлор-6,7-диметоксихиназолин (250 мг, 1.12 ммоля) и 4-азидоанилин-гидрохлорид (200 мг, 1.11 ммоля) нагревались в условиях дефлегмации в 10 мл изопропилового спирта в течение 0.5 часа, охлаждались и фильтровались, давая твердый целевой продукт, который промывался 10 мл изопропилового спирта и сушился в вакууме, при 70°C, 392 мг (98%); т. пл. 200-205°C (разлож.).
Пример 2
Хлоргидрат (6,7-диметоксихиназолин-4-ил)-(3-этинилфенил)-амина
4-Хлор-6,7-диметоксихиназолин (250 мг, 1.12 ммоля) и 3-этиниланилин (137 мг, 1.17 ммоля) нагревались в условиях дефлегмации в 10 мл изопропилового спирта в течение 0.5 часа, охлаждались и фильтровались, давая твердый целевой продукт, который промывался 10 мл изопропилового спирта и сушился в вакууме, при 70°C, 338 мг (99%); т. пл. 269-270°C.
Пример 3
(6,7-диметоксихиназолин-4-ил)[3-(3'-гидроксипропин-1-ил)фенил]-амин
Смесь хлоргидрата (3'-бромфенил)-(6,7-диметоксихиназолин-4-ил)-амина (250 мг, 0,591 ммоля), тетракис (трифенилфосфин) палладия (100 мг), пропаргилового спирта (600 мкл), 7 мл сухого продуваемого азотом диэтиламина и иодида меди (10 мг) нагревалась в условиях дефлегмации в течение 5 часов, охлаждалась и фильтровалась, давая твердый целевой продукт, который промывался два раза 2 мл 50% диэтиламина: метанола; 136 мг. Твердое вещество перекристаллизовывалось из метанола, давая чистый целевой продукт после сушки, в вакууме, при 70°C, 73 мг (37%); т.пл. 267-268°C.
Пример 4
Хлоргидрат [(3-(2'-аминометил-этинил)фенил]-(6,7-диметокси-хиназолин-4-ил)-амина
Целевой продукт примера 3 (50 мг, 0.149 ммоля), трифенилфосфин (60 мг, 0.225 ммоля), фталимид (165 мг, 1.12 ммоля) и диэтилазодикарбоксилат (36 мкл, 0.228 ммоля) перемешивались при комнатной температуре в 3 мл сухого тетрагидрофурана в течение 16 часов. Реакционная смесь концентрировалась до твердого вещества и подвергалась флэш хроматографии на силикагеле, элюируемом смесью 15% ацетон : метиленхлорид, давая чистый твердый [3-(2'-{фталимидометил}-этинил)фенил]-(6,7-диметоксихиназолин-4-ил)амин, который превращался в его хлоргидратную соль добавлением 1 мл безводного 1 М HCl в метаноле, а затем 3 мл изопропилового спирта. Соль собиралась фильтрованием, сушилась и использовалась сразу же на следующей стадии; 15 мг. Данные 15 мг, 0.0323 ммоля, обрабатывались 0.5 мл гидразингидрата и 1 мл метанола в течение 0.5 часа. Реакционная смесь выпаривалась в вакууме, и продукт отделялся с помощью флэш хроматографии при элюировании 10% метанолом в метиленхлориде. Чистый целевой продукт выделялся после превращения его в хлоргидратную (гидрохлоридную) соль с помощью 1 мл 1 М HCl в метаноле, осаждения изопропиловым спиртом и диэтиловым эфиром и сушки в вакууме; 5.6 мг (47%), т.пл. 275°C разл.
Пример 5
Хлоргидрат (3-этинилфенил)-(6-нитрохиназолин-4-ил)-амина
4-Хлор-6-нитрохиназолин (1.06 г, 5.00 ммолей) и 3-этиниланилин (1.00 г, 5.30 ммоля) нагревались в условиях дефлегмации в 10 мл изопропилового спирта в течение 3 часов, охлаждались и после 16-часового нахождения при комнатной температуре фильтровались, давая твердый целевой продукт, который промывался 10 мл изопропилового спирта и сушился в вакууме при 70°C, 1.27 г (78%); т. пл. 255-258°C.
Пример 6
(6,7-Диметоксихиназолин-4-ил)-(4-этинилфенил)-амин
Целевой продукт получался с помощью следующей трехстадийной последовательности реакций без очистки промежуточных продуктов. 4-Хлор-6,7-диметоксихиназолин (250 мг, 1.113 ммоля) и 4-иоданилин (268 мг, 1.224 ммоля) нагревались в условиях дефлегмации в 10 мл изопропилового спирта в течение 3 часов, охлаждались до комнатной температуры и фильтровались, давая твердый хлоргидрат (4-иодфенил)-(6,7-Диметоксихиназолин-4-ил)амина, который промывался 10 мл изопропилового спирта и сушился в вакууме при 70°C, 396 мг (76%). Смесь, состоящая из хлоргидрата (4'-иодфенил)-(6,7-диметокси-хиназолин-4-ил)-амина (250 мг, 0.564 ммоля), тетракис(трифенилфосфин)палладия (50 мг), триметилсилилацетилена (160 мкл, 1.13 ммоля), 4 мл сухого продуваемого азотом диэтиламина и йодистой меди (10 мг) нагревалась в условиях дефлегмации в течение 2 часов, охлаждалась и концентрировалась в вакууме, давая остаток, который распределялся между хлороформом и 1 н. HCl. Твердый [4-(2'-{триметилсилил}этинил)фенил]-(6,7-диметоксихиназолин-4-ил)-амин, образовавшийся на поверхности раздела двух жидких фаз, фильтровался и сушился в вакууме; 170 мг (80%) [4-(2'-{Триметилсилил}-этинил)фенил]-(6,7-диметоксихиназолин-4-ил)амин (100 мг, 0.265 ммоля) и безводный карбонат калия (125 мг, 0.906 ммоля) перемешивались в 3 мл метанола и 1 мл воды при комнатной температуре в течение 2.5 часов. Реакционная смесь концентрировалась в вакууме и распределялась между 20 мл хлороформа и 20 мл 1 н. соляной кислоты. Органический слой сушился сульфатом магния, фильтровался и выпаривался в вакууме, давая целевой продукт, который растирался с диэтиловым эфиром и сушился в вакууме при 70°C; 81 мг (90%), т. пл. 239°C разл.
Пример 7
(6,7-диметоксихиназолин-4-ил)-(3-этинил-2-метилфенил)-амин
Целевой продукт получался с помощью следующей трехстадийной последовательности процедур без очистки промежуточных продуктов. Смесь, состоящая из 3-бром-2-метиланилина (1.00 г, 5.37 ммоля), тетракис(трифенилфосфин)палладия (200 мг), триметилсилилацетилена (1.053 г, 10.75 ммоля), 10 мл сухого продуваемого азотом диэтиламина и йодистой меди (910 мг) нагревалась в условиях дефлегмации в течение 16 часов, охлаждалась и концентрировалась в вакууме, давая остаток, который распределялся между хлороформом и 1 н. HCl. Органический слой промывался солевым раствором, сушился сульфатом магния и выпаривался в вакууме, давая остаток, 3-[2'-(триметилсилил)этинил]-2-метиланилин, который очищался с помощью флэш хроматографии на силикагеле, элюируемом смесью 1:1 гексаны : метиленхлорид; 200 мг (18%).
4-Хлор-6,7-диметоксихиназолин (104 мг, 0,466 ммоля) и 3-[2'-(триметилсилил)этинил]-2-метиланилин (100 мг, 0.491 ммоля) нагревались в условиях дефлегмации в 3 мл изопропилового спирта в течение 16 часов, охлаждались до комнатной температуры и фильтровались, давая остаток твердого хлоргидрата {3-[2'-(триметилсилил)этинил]-2-метилфенил}-(6,7-диметоксихиназолин-4-ил) амина, который промывался 10 мл изопропилового спирта и растирался в течение 16 часов с диэтиловым эфиром. Тонкослойная хроматография на силикагеле, элюируемом смесью 9:1 хлороформ : метанол, показал, что остаток представлял неочищенный или сырой продукт. Остаток очищался с помощью флэш хроматографии на силикагеле, элюируемом смесью 9:1 метиленхлорид; метанол, давая после концентрирования и сушки в вакууме чистый продукт, 64 мг (33%). Продукт растворялся в 3 мл метанола и обрабатывался 64 мг безводного карбоната калия при комнатной температуре в течение 3 часов. Реакционная смесь концентрировалась в вакууме и распределялась между 1 н. HCl и хлороформом. Твердый целевой продукт образовывался на поверхности раздела двух жидких фаз и фильтровался и сушился в вакууме; 40 мг (84%), т.пл. 225°C разл.
Пример 8
(6-Амино-хиназолин-4-ил)-(3-этинилфенил)-амин
(3-Этинил-фенил)-(6-нитро-хиназолин-4-ил)-амин хлоргидрат (500 мг, 1.50 ммоля) растворялся в 10 мл муравьиной кислоты и обрабатывался порционно дитионитом натрия (1.10 г, 6.28 ммоля) при комнатной температуре. Спустя 2 часа, смесь гасилась 120 мл воды и фильтровалась. Фильтрат выпаривался в вакууме до остатка, который растворялся в 100 мл смеси 1:1 метанол : хлороформ, фильтровался и выпаривался в вакууме до еще одного остатка. Данное вещество растиралось с 200 мл 5% бикарбоната натрия в течение 30 минут, фильтровалось, промывалось водой и сушилось в вакууме в течение 16 часов. Флэш хроматография на силикагеле, элюируемом этилацетатом, давала чистый (6-амино-хиназолин-4-ил)-(3-этинилфенил)-амин; 140 мг (34%); т. пл. 165°C разл.
Пример 9
(3-Этинилфенил)-(6-метансульфониламинохиназолин-4-ил)амин
Целевой продукт примера 8 (100 мг, 0.384 ммоля), пиридин (140 мкл, 1.68 ммоля) и метансульфонилхлорид (99 мкл, 1.26 ммоля) нагревались с обратным холодильником в 10 мл 1,2-дихлорэтана в течение 7 часов. Реакционная смесь охлаждалась и выпаривалась в вакууме до остатка, который растирался в 10 мл 1 н. HCl, фильтровался и сушился в вакууме, давая (3-этинилфенил)-(6-метансульфониламинохиназолин-4-ил)-амин; 102 мг (78%), т. пл. 248°C разл.
Пример 10
Хлоргидрат (3-этинилфенил)-(6,7-метилендиоксихиназолин-4-ил)-амина
4-Хлор-6,7-метилендиоксихиназолин (200 мг, 1.04 ммоля) и 3-этиниланилин (127 мг, 1.09 ммоля) нагревались в условиях дефлегмации в 5 мл изопропилового спирта в течение 16 часов, охлаждались, фильтровались, давая твердый целевой продукт, который промывался 10 мл изопропилового спирта и сушился в вакууме при 70°C, 266 мг (79%); т.пл. выше 350°C.
Пример 11
Хлоргидрат ((6,7-диметоксихиназолин-4-ил)-3-этинил-6-метилфенил)-амина
Целевой продукт получался в следующей трехстадийной последовательности процедур без очистки промежуточных продуктов. Смесь, состоящая из 4-бром-2-нитротолуола (1.50 г, 6.94 ммоля), тетракис (трифенилфосфин)палладия (750 мг), триметилсилилацетилена (3.00 мл, 21.21 моля) и йодистой меди (20 мг) в 20 мл продуваемого азотом сухого диэтиламина нагревалась в условиях дефлегмации в течение 2 часов, охлаждалась и концентрировалась в вакууме, давая остаток, который распределялся между 100 мл этилацетата и 100 мл 1 н. HCl. Органический слой промывался два раза 50 мл 1 н. HCl, а затем солевым раствором, сушился сульфатом магния и выпаривался в вакууме до остатка. Остаток растворялся в 10 мл этилацетата и разбавлялся 200 мл петролейного эфира. Твердые вещества отфильтровывались, и масло, полученное после выпаривания фильтрата в вакууме, затвердевало, давая 4-[2'-(триметилсилил)этинил]-2-нитротолуол. Данный продукт восстанавливался до амино продукта с помощью обработки порошком железа (1.76 г, 98.5 ммоля) в 30 мл метанола и 5 мл концентрированной соляной кислоте при 80°C в течение 2 часов. Охлажденная реакционная смесь фильтровалась через "целит", и фильтрат выпаривался в вакууме. Остаток распределялся между этилацетатом и 5% водным бикарбонатом натрия. Органический слой промывался солевым раствором, сушился сульфатом магния, фильтровался и выпаривался в вакууме, давая масло, 5-[2'-(триметилсилил)этинил]-2-метиланилин, который затвердевал при стоянии; 1.37 г.
Указанный выше продукт (185 мг, 0.909 ммоля) и 4-хлор-6,7-диметоксихиназолин (200 мг, 0.890 ммоля) нагревались в условиях дефлегмации в трет-бутиловом спирте в течение 16 часов. После охлаждения реакционная смесь фильтровалась, давая чистый хлоргидрат [2-метил-5-(2'-{триметилсилил}-этинил)-фенил]-(6,7-диметоксихиназолин-4-ил)-амина после промывки простым эфиром и сушки в вакууме; 326 мг (85%). Триметилсилильная группа удалялась путем растворения указанного выше продукта в 5 мл метанола и 1 мл воды и обработки карбонатом калия (320 мг). После перемешивания в течение 1 часа смесь фильтровалась и концентрировалась в вакууме. Остаток, полученный таким образом, распределялся между 100 мл метиленхлорида и 100 мл 1 н. HCl. Водный слой экстрагировался дополнительными 100 мл метиленхлорида. Охлажденные органические слои сушились сульфатом магния, фильтровались и выпаривались в вакууме до остатка, который растворялся в безводной 1 н. HCl в метаноле, концентрировался и осаждался эфиром. Твердый целевой продукт собирался фильтрованием и промывался диэтиловым эфиром, затем сушился в вакууме при 70°C; 236 мг (88%) т. пл. 266-267°C.
Пример 12
Хлоргидрат (3-этинилфенил)-(7-нитрохиназолин-4-ил)-амина
4-Хлор-7-нитрохиназолин (7.97 г, 38.0 ммолей) и 3-этиниланилин (4.54 г, 38.8 ммолей) нагревались в условиях дефлегмации в 125 мл трет-бутилового спирта в течение 3 часов, охлаждались до комнатной температуры и фильтровались, давая целевой продукт в виде твердого вещества, которое промывалось 10 мл изопропилового спирта и сушилось в вакууме при 70°C, 9.95 г (80%); т. пл. 209-210°C разл.
Пример 13
Хлоргидрат (3-этинилфенил)-[6-(4'-толуолсульфониламино)-хиназолин-4-ил]-амина
Целевой продукт примера 8 (0.201 мг, 0.771 ммоля) и 4-толуолсульфонилхлорид (0.441 мг, 2.31 ммоля) нагревались в условиях дефлегмации в 3 мл 1,2-дихлорэтана и 0.5 мл пиридина в течение 5 минут. Реакционная смесь охлаждалась до комнатной температуры, разбавлялась 75 мл этилацетата и промывалась два раза, 75 мл воды, один раз 75 мл 3% бикарбоната натрия и один раз 75 мл солевого раствора. Органический слой сушился над сульфатом магния, фильтровался и выпаривался в вакууме до остатка, который очищался с помощью хроматографии с использованием Хроматотрона (торговая марка), элюируемого этилацетатом, давая твердый целевой продукт; 86.7 мг (27%), т. пл. 220-222°C.
Пример 14
Хлоргидрат (3-этинилфенил)-{6-[2'-фталимидо-этан-1'-илсульфониламино]хиназолин-4-ил}амина.
Целевой продукт примера 8 (0.20 мг, 0.768 ммоля) и 2-фталимидо-1-этансульфонилхлорид (0.615 мг, 2.25 ммоля) нагревались в условиях дефлегмации в 2 мл 1, 2-дихлорэтана и 0.5 мл пиридина в течение 16 часов, охлаждались до комнатной температуры, разбавлялись 100 мл хлороформа и промывались 50 мл 3% водного бикарбоната натрия и 50 мл солевого раствора. Органический слой сушился сульфатом магния, фильтровался и выпаривался в вакууме до остатка, который растворялся в минимальном количестве метиленхлорида и осаждался петролейным эфиром, 188 мг. Осадок очищался с помощью хроматографии с использованием хроматоторона, элюируемого этилацетатом, давая целевой продукт в виде твердого вещества; 53.4 мг (14%), т. пл. 197-200°C.
Пример 15
Хлоргидрат (3-этинилфенил)-(6-гуанидинохиназолин-4-ил)-амина
Целевой продукт примера 8 (0.302 мг, 1.16 ммоля) и 3,5-диметилпиразол-1-карбоксамидин (0,328 мг, 2.36 ммоля) нагревались в условиях дефлегмации в 10 мл 1,2-дихлорэтана и 0,97 мл уксусной кислоты в течение 24 часов, охлаждались до комнатной температуры и фильтровались, давая сырой ацетат целевого продукта. Продукт растворялся в 35 мл метанола и обрабатывался 15 мл безводной 1 н. HCl в метаноле в течение 15 минут, а затем осаждался 75 мл диэтилового эфира. Твердый целевой продукт собирался фильтрованием и сушился в вакууме при 70°C; 91.2 мг (23%), т. пл. выше 400°C.
Пример 16
(7-Аминохиназолин-4-ил)-(3-этинилфенил)амин
Целевой продукт примера 12 (1.039 г, 3.18 ммоля) растворялся в 50 мл тетрагидрофурана, 10 мл метанола и 5 мл хлороформа при 50°C. Добавлялись первичный кислый фосфит натрия (NaH2PO2, 3.822 г, 36 ммолей) и 10% палладия на угле (0,19 г) с последующим добавлением по каплям 10 мл воды. Когда было добавлено 3 мл воды, смесь становилась заметно более гомогенной. Спустя 1 час, смесь фильтровалась через целит. Целит промывался тщательно метанолом и хлороформом. Объединенные органические растворы выпаривались в вакууме до остатка, который растирался с водой, 3% водным бикарбонатом натрия и фильтровался. Твердый целевой продукт промывался водой, затем диэтиловым эфиром и сушился в вакууме, 1.054 г (127%, влажный). Часть указанного выше продукта перекристаллизовывалась из минимального количества горячего этанола и воды, давая после удаления незначительного первого сбора примесного вещества чистый целевой продукт, (43%), т. пл. 180°C (разлож.)
Пример 17
Хлоргидрат (3-этинилфенил)-(7-метоксихиназолин-4-ил)амина
4-Хлор-7-метоксихиназолин (274 мг, 3.72 ммоля) и 3-этиниланилин (436 мг, 3.72 ммоля) нагревались с обратным холодильником в 15 мл трет-бутилового спирта в течение 3 часов, охлаждались и фильтровались, давая твердый целевой продукт, который промывался 10 мл изопропилового спирта и сушился в вакууме при 70°C, 977 мг (84%), т.пл. 229-231°C.
Пример 18
Хлоргидрат (6-карбометоксихиназолин-4-ил)-(3-этинилфенил)-амина
4-Хлор-6-карбометоксихиназолин (100 мг, 0.450 ммоля) и 3-этиниланилин-хлоргидрат (53.4 мг, 0.456 ммоля) нагревались в условиях дефлегмации в 2 мл трет-бутилового спирта в течение 2 часов, охлаждались, разбавлялись 2 мл изопропилового спирта и фильтровались, давая твердый целевой продукт, который промывался 10 мл диэтилового эфира и сушился в вакууме при 70°C, 122 мг (80%); т. пл. 232-233°C (разложение).
Пример 19
Хлоргидрат (7-карбометоксихиназолин-4-ил)-(3-этинилфенил)-амина
4-Хлор-7-карбометоксихиназолин (202 мг, 0.907 ммоля) и 3-этиниланилин (110 мг, 0.939 ммоля) нагревались в условиях дефлегмации в 4 мл трет-бутилового спирта в течение 2 часов, охлаждались, разбавлялись 4 мл изопропилового спирта и фильтровались, давая твердый целевой продукт, который промывался 10 мл диэтилового эфира и сушился в вакууме при 70°C, 248 мг (80%); т. пл. 219.5-221°C.
Пример 20
Хлоргидрат [6,7-бис-(2-метоксиэтокси)-хиназолин-4-ил]-(3-этинилфенил)-амина
3-Этиниланилин (37 мг, 0.32 ммоля) и 4-хлор-6,7-бис-(2-метоксиэтокси)-хиназолин (90 мг, 0.29 ммоля) добавлялись к изопропанолу (1.5 мл), содержащему пиридин (25 мкл, 0.32 ммоля), и смесь нагревалась в условиях дефлегмации в течение 4 часов в атмосфере сухого азота. Растворитель удалялся в вакууме, и остаток распределялся между 10% метанолом в CHCl3 и насыщенным водным бикарбонатом натрия. Органическая фаза сушилась над сульфатом натрия, фильтровалась и концентрировалась в вакууме. Остаток подвергался флэш хроматографии на силикагеле с использованием 30% ацетона в гексанах, давая 81 мг свободного основания целевого продукта в виде бледно-желтого твердого вещества. Свободное основание растворялось в минимальном объеме CHCl3, разбавлялось несколькими объемами эфира и растиралось с 1 М HCl в эфире с осаждением целевого продукта в виде его хлоргидратной соли; 90 мг, 71%; т. пл. 228-230°C.
Пример 21
(3-Азидофенил)-(6,7-диметоксихиназолин-4-ил)-амин
4-Хлор-6,7-диметоксихиназолин (5.01 г, 22.3 ммоля) добавлялся порциями на протяжении 1.5 часов к м-фенилендиамину (2.66 г, 24.6 ммоля) в дефлегмирующем изопропаноле (100 мл) в атмосфере сухого азота. После того, как добавление завершалось, смесь нагревалась при температуре дефлегмации в течение 4 часов. Смесь охлаждалась до 20°C, и осадок отфильтровывался, промывался охлажденным изопропанолом и сушился в вакууме, давая 6.97 г (93%) хлоргидрата (3-аминофенил)-(6,7-диметоксихиназолин-4-ил)-амина (αС-МС: 297 (МН+). К раствору указанного выше продукта (50 мг, 0.169 ммоля) в смеси 80% уксусная кислота/вода (2 мл) при 0°C добавлялся раствор NaNO2 (18.4 мг, 0.186 ммоля) в воде (100 мкл). После перемешивания в течение 10 минут при 0°C добавлялся раствор азида натрия (12 мг, 0.185 ммоля) в воде (100 мкл). Смесь оставлялась подогреваться до 20°C и перемешивалась в течение 1.5 часов. Реакционная смесь лиофилизовалась, и остаток распределялся между этилацетатом и насыщенным водным бикарбонатом натрия. Органическая фаза промывалась далее солевым раствором, сушилась над сульфатом натрия, фильтровалась и концентрировалась в вакууме. Перекристаллизация из смеси CHCl3/гексаны давала 36 мг целевого продукта в виде белого твердого вещества; т. пл. 110-113°C.
Пример 22
(3-Азидо-5-хлорфенил)-(6,7-диметоксихиназолин-4-ил)-амин
4-Хлор-6,7-диметоксихиназолин (200 мг, 0.89 ммоля) и 5-амино-3-хлоранилин (253 мг, 1.78 ммоля) объединялись в изо- пропаноле (3 мл) и нагревались в условиях дефлегмации в течение 17 часов в атмосфере сухого азота. После охлаждения до 20°C смесь разбавлялась метанолом (5 мл) и получающийся в результате осадок отфильтровывался и сушился в вакууме, давая 252 мг (77%) хлоргидрата (3-амино-5-хлорфенил)-(6, 7-диметокси-хиназолин-4-ил) амина (т. пл. 298-301°C, αС-МС, 331 (МН+)). Часть данного продукта (175 мг, 0.476 ммоля) растворялась в смеси 80% уксусная кислота/вода (12 мл), охлаждалась до 0°C, и добавлялся раствор нитрита натрия (NaNO2) (36 мг, 0.516 ммоля) в воде (300 мкл). Раствор перемешивался в течение 10 минут при 0°C, и добавлялся NaN3 (33 мг, 0.50 ммоля) в воде (300 мкл). Реакционная смесь оставлялась подогреваться до 20°C и перемешивалась в течение 16 часов. Получающийся в результате осадок отфильтровывался и растворялся в 10% метаноле в CHCl3, и раствор промывался насыщенным водным бикарбонатом натрия и солевым раствором, сушился над сульфатом натрия, фильтровался и концентрировался в вакууме, давая 59 мг (35%) целевого продукта в виде желтого твердого вещества; т. пл. 205-206°C.
Пример 23
Хлоргидрат (3-этинилфенил)-(6-метансульфонил-хиназолин-4-ил)-амина
6-Метансульфонил-хиназолин-4-он (200 мг, 0.89 ммоля), трифенилфосфин (566 мг, 2.15 ммоля) и четыреххлористый углерод (815 мкл, 8.92 ммоля) нагревались в условиях дефлегмации в 3 мл хлороформа в течение 3.5 часов. Растворитель выпаривался в вакууме, давая остаток. Данное вещество растворялось в 5 мл изопропилового спирта и 3-этиниланилина (156 мг, 1.33 ммоля) и нагревалось в условиях дефлегмации в течение 16 часов. Охлажденная реакционная смесь фильтровалась, промывалась минимальным количеством холодного изопропилового спирта и сушилась в вакууме при 70°C в течение 16 часов, давая чистый целевой продукт; 63 мг (20%), т. пл. 281-282°C.
Пример 24
Хлоргидрат (6-этансульфанил-хиназолин-4-ил)-(3-этинилфенил)-амина
6-Этансульфанил-хиназолин-4-он (100 мг, 0.48 ммоля), трифенилфосфин (305 мг, 1.16 ммоля) и 3 мл четыреххлористого углерода нагревались в условиях дефлегмации в течение 16 часов. Растворитель выпаривался в вакууме, давая остаток. Данный остаток растворялся в 5 мл изопропилового спирта и 3-этиниланилина (68 мг, 0.58 ммоля) и нагревался в условиях дефлегмации в течение 1 часа. Охлажденная реакционная смесь фильтровалась, промывалась минимальным количеством холодного изопропилового спирта и сушилась в вакууме при 70°C в течение 16 часов, давая чистый целевой продукт; 70 мг (42%), т. пл. 239-240°C.
Пример 25
Хлоргидрат (6,7-диметокси-хиназолин-4-ил)-(3-этинил-4-фторфенил)-амина
4-Хлор-6,7-диметоксихиназолин (500 мг, 2.23 ммоля) и 3-(2'-триметилсилилэтинил)-4-фторанилин (507 мг, 2.44 ммоля) нагревались в условиях дефлегмации в 5 мл трет-бутилового спирта в течение 16 часов, охлаждались и фильтровались, давая твердый хлоргидрат (6,7-диметокси-хиназолин-4-ил)-(3'-этинилфенил)-амина, который промывался 10 мл изопропилового спирта и сушился в вакууме при 70°C, 832 мг (83%). Данный материал подвергался реакции в 10 мл метанола и 1 капле воды, содержащих 250 мг карбоната калия, в течение 3 часов. Смесь фильтровалась, и фильтрат выпаривался в вакууме. Данный остаток растирался в течение 1 часа с 1 н. соляной кислотой, фильтровался и промывался минимальным количеством воды, затем метанолом и сушился в вакууме; 506 мг (63%), т. пл. 229°C, разлож.
3-(2'-Триметилсилил-этинил)-4-фторанилин, использованный выше, получался из 3-бром-4-фторанилина (7,0 г, 36.8 ммоля), тетракис(трифенилфосфин)палладия (1.4 г), триметилсилил-ацетилена (7.2 г, 74 ммоля) и йодистой меди (40 мг) в 140 мл продуваемого азотом сухого диэтиламина при нагревании в условиях дефлегмации в течение 16 часов. Охлажденная реакционная смесь фильтровалась через целит, и целит промывался эфиром. Объединенные фильтраты выпаривались в вакууме до остатка, который очищался с помощью флэш хроматографии на силикагеле, элюируемом 35% гексана в метиленхлориде. Фракции, содержащие чистый 3-(2'-триметилсилил-этинил)-4-фторанилин, выпаривались в вакууме до остатка, который использовался без дальнейшей очистки.
Пример 26
Хлоргидрат (6,7-диметокси-хиназолин-4-ил)-[3-(пропил-1-ил)фенил]-амина
4-Хлор-6,7-диметоксихиназолин (585 мг, 2.60 ммоля) и 3-(пропин-1-ил)-анилин (361 мг, 2.74 ммоля) нагревались в условиях дефлегмации в 5 мл трет-бутилового спирта в течение 16 часов, охлаждались и фильтровались, давая твердый (6,7-диметокси-хиназолин-4-ил)-[3-(пропин-1-ил) фенил]-амин-хлоргидрат, который промывался 5 мл изопропилового спирта и 25 мл эфира, затем сушился в вакууме при 70°C, 869 мг (94%); т. пл. 260-261°C.
3-(Пропин-1-ил)анилин, использовавшийся выше, получался из 3-бром-нитробензола в четыре стадии. 3-бром-нитробензол (5.0 г, 24.7 ммоля), тетракис(трифенилфосфин)палладий (1.0 г), триметилсилил-ацетилен (3.6 г, 37 ммолей) и йодистая медь (20 мг) в 20 мл сухого диэтиламина, продуваемого азотом, при нагревании в условиях дефлегмации в течение 16 часов. Охлажденная реакционная смесь выпаривалась в вакууме, разбавлялась 50 мл метиленхлорида и 50 мл 1 н. соляной кислоты и фильтровалась. Органический слой собирался и сушился сульфатом магния, фильтровался и выпаривался в вакууме до остатка. 3-Триметилсилилэтинилнитробензол очищался с помощью флэш хроматографии на силикагеле, элюируемом смесью 2:1 гексаны : метиленхлорид. Фракции, содержащие чистый материал, выпаривались в вакууме, давая чистый 3-триметилсилил-этинилнитро-бензол (4.6 г). 4.0 г данного вещества растворялись в 30 мл метанола и 1 капле воды, содержащей 1.16 г карбоната калия. Спустя один час, смесь выпаривалась в вакууме и разбавлялась 100 мл метиленхлорида. Органический слой промывался 100 мл 1 н. соляной кислоты, сушился сульфатом магния, фильтровался и выпаривался в вакууме до остатка (2.96 г). 790 мг данного вещества растворялось в 10 мл бензола и обрабатывалось тонко распыляемым 87% гидроксидом калия (377 мг, 5.91 ммоля), метилиодидом (2 мл) и 10 мг 18-кроун-6 (Aldrich) в условиях дефлегмации в течение 16 часов. Добавлялось дополнительно 0.5 мл метилиодида, и нагревание в условиях дефлегмации продолжалось дополнительно в течение 2 часов. Охлажденная реакционная смесь выпаривалась в вакууме до остатка, который разбавлялся 100 мл метиленхлорида и промывался 100 мл 1 н. соляной кислоты, сушился сульфатом магния, фильтровался и выпаривался в вакууме до масла. Данное масло очищалось с помощью флэш хроматографии на силикагеле, элюируемом смесью 1:1 гексаны : метиленхлорид. Фракции, содержащие чистый 3-(пропин-1-ил) нитробензол, выпаривались в вакууме до масла, которое использовалось без дальнейшей очистки; 530 мг (61%), 3-(Пропин-1-ил)-нитробензол (530 мг, 3.3 ммоля), порошок железа (400 мг, 7.27 ммоля), 3 мл концентрированной соляной кислоты и 10 мл метанола нагревались в условиях дефлегмации в течение 1 часа. Реакционная смесь фильтровалась и выпаривалась в вакууме до твердого вещества, которое распределялось между 100 мл метиленхлорида и 100 мл 1 н. гидроокиси натрия. Две фазы фильтровались, а затем органическая фаза отделялась, сушилась сульфатом магния, фильтровалась и выпаривалась в вакууме, до масла, которое использовалось непосредственно при получении целевого продукта; 321 мг (78%).
Пример 27
Хлоргидрат [6,7-Бис-(2-метокси-этокси)-хиназолин-4-ил]-(3-этинил-4-фторфенил)-амина
4-Хлор-6,7-бис-(2-метокси-этокси)-хиназолин (140 мг, 0.446 ммоля) и 3-этинил-4-фторанилин (66 мг, 0.452 ммоля) подвергались реакции в дефлегмирующем изопропаноле (3 мл) в атмосфере азота в течение 16 часов. Растворитель удалялся в вакууме, и остаток распределялся между CHCl3 и насыщенным водным бикарбонатом натрия. Органические экстракты промывались солевым раствором, сушились над сульфатом натрия, фильтровались и концентрировались в вакууме. Сырой продукт хроматографировался на двуокиси кремния с использованием смеси 40% ацетон/метиленхлорид, давая 116 мг чистого целевого продукта в виде его свободного основания. Данное масло растворялось в минимальном объеме CHCl3, разбавлялось несколькими объемами эфира и растиралось с 1М HCl в эфире с осаждением целевого продукта в виде белого твердого вещества (99 мг, 50%, т. пл. 170-190°C (разл.), αС-МС: 412 (МН+), анализ RP-18 ЖХВД RT: 4.33 мин) (R.T. = время удерживания).
Пример 28
Хлоргидрат [6,7-бис-(2-метокси-этокси)-хиназолин-4-ил]-(5-этинил-2-метил-фенил)-амина
4-Хлор-6,7-бис-(2-метокси-этокси)-хиназолин (153 мг, 0.49 ммоля), пиридин (40 мкл) и 3-этинил-6-метиланилин (71 мг, 0.54 ммоля) подвергались реакции в ДМФ (3 мл) при 110°C в атмосфере азота в течение 36 часов. Растворитель удалялся в вакууме, и остаток распределялся между хлороформом и насыщенным водным бикарбонатом натрия. Органические экстракты промывались солевым раствором, сушились над сульфатом натрия, фильтровались и концентрировались в вакууме. Сырой продукт хроматографировался на двуокиси кремния с использованием смеси 40% ацетон/метиленхлорид, давая 40 мг (19%) чистого продукта в виде его свободного основания. Данное масло растворялось в минимальном объеме хлороформа, разбавлялось несколькими объемами эфира и растиралось с 1М HCl в эфире с осаждением целевого продукта в виде белого твердого вещества (т. пл. 170-185°C (разл.), αС-МС: 408 (МН+), анализ RP18-ЖХВД RT: 3.93 мин).
Пример 29
Хлоргидрат [6,7-бис-(2-хлор-этокси)-хиназолин-4-ил]-(3-этинил-фенил)-амина
4-Хлор-6,7-бис-(2-хлорэтокси)-хиназолин (600 мг, 1.87 ммоля) и 3-этинил-анилин (219 мг, 1.87 ммоля) подвергались реакции в дефлегмирующем изопропаноле (15 мл) в атмосфере азота в течение 2.5 часов. Смесь охлаждалась до 20°C, и выпавший в осадок продукт отфильтровывался, промывался изопропанолом и эфиром и сушился в вакууме. (707 мг, 86%, т. пл. 230-240°C (разл.), αС-МС: 402 (МН+), анализ RP18-ЖХВД RT: 5.35 мин).
Пример 30
Хлоргидрат [6-(2-хлор-этокси)-7-(2-метокси-этокси)-хиназолин-4-ил]-(3-этинил-фенил)-амина
Целевой продукт получался из 4-хлор-6-(2-хлор-этокси)-7-(2-метокси-этокси)-хиназолина (399 мг, 1.26 ммоля) и 3-этинил-анилина (147 мг, 1.26 ммоля), как описано в примере 29 (515 мг, 94%, т. пл. 215-225°C (разл.), αС-МС: 398 (МН+), анализ RP18-ЖХВД RT: 4.85 мин.
Пример 31
6,7-Бис(2-ацетокси-этокси)-4-(3-этинил-фениламино)-хиназолин
Целевой продукт примера 29 (200 мг, 0.456 ммоля) обрабатывался ацетатом цезия (1.75 г, 9.12 ммоля) в ДМФ (3 мл) при 120°C в атмосфере азота в течение 16 часов. Реакционная смесь распределялась между солевым раствором и хлороформом, и органический экстракт промывался солевым раствором, сушился над сульфатом натрия, фильтровался и концентрировался в вакууме, давая масло (277 мг), которое перекристаллизовывалось из смеси метиленхлорид/гексан (184 мг, 90%, т. пл. 137-138°C, αС-МС: 450 (МН+), анализ RPIS-ЖХВД RT: 4.64 мин).
Пример 32
Хлоргидрат 2-[4-(3-этинил-фениламино)-7-(2-гидрокси-этокси)-хиназолин-6-илокси]-этанола
6,7-Бис-(2-ацетокси-этокси)-4-(3-этинил-фенил-амино)-хиназолин (199 мг, 0.443 ммоля) в метаноле (3 мл) обрабатывался 7М водной КОН (0.25 мл). Смесь перемешивалась при 20°C в течение 2 часов, перед удалением растворителя в вакууме. Твердый остаток промывался водой для удаления солей, и сушился азеотропно с помощью растворения два раза в ацетонитриле и концентрирования в вакууме, давая 116 мг целевого продукта в виде его свободного основания. Данное вещество, превращалось в его HCl соль согласно методу, использованному в примере 28 (115 мг, 65%, т. пл. 215-218°C (разл.), αС-МС: 366 (МН+), анализ RPIS-ЖХВД RT: 3.08 мин).
Пример 33
6-(2-Ацетокси-этокси)-4-(3-этинил-фениламино)-7-(2-метокси-этокси)-хиназолин
Целевой продукт примера 30 (160 мг, 0.368 ммоля) обрабатывался ацетатом цезия (707 мг, 3.68 ммоля) в ДМФ (3 мл) при 120°C в атмосфере азота в течение 16 часов. Реакционная смесь распределялась между солевым раствором и хлороформом, и органический экстракт промывался солевым раствором, сушился над сульфатом натрия, фильтровался и концентрировался в вакууме, давая остаток (285 мг), который перекристаллизовывался из смеси этилацетат/гексан (134 мг, т. пл. 84-87°C, α С-МС: 422 (МН+), анализ RP18 - ЖХВД RT: 4,38 мин).
Пример 34
Хлоргидрат [7-(2-хлор-этокси)-6-(2-метокси-этокси)-хиназолин-4-ил]-(3-этинил-фенил)амина
Данный продукт получался из 4-хлор-7-(2-хлор-этокси)-6-(2-метокси-этокси)-хиназолина (600 мг, 1.89 ммоля) и 3-этинил-анилина (147 мг, 1.26 ммоля), как описано в примере 29 (737 мг, 90%, т. пл. 225-235°C (разл.), αС-МС: 398 (МН+), анал. RP18-ЖХВД RT: 4.89 мин).
Пример 35
7-(2-Ацетокси-этокси)-4-(3-этинил-фениламино)-6-(2-метокси-этокси)-хиназолин
Целевой продукт примера 34 (160 мг, 0.368 ммоля) обрабатывался ацетатом цезия (707 мг, 3.68 ммоля) в ДМФ (3 мл) при 120°C в атмосфере азота в течение 16 часов. Реакционная смесь распределялась между солевым раствором и хлороформом, и органический экстракт промывался солевым раствором, сушился над сульфатом натрия, фильтровался и концентрировался в вакууме, давая остаток (288 мг), который перекристаллизовывался из смеси этилацетат/гексаны (134 мг, т. пл. 134-135°C, α С-МС: 422 (МН+), анализ RP-18-ЖХВД RT: 4.43 мин).
Пример 36
Хлоргидрат 2-[4-(3-этинил-фениламино)-6-(2-метокси-этокси)-хиназолин-7-ил-окси]-этанола
Целевой продукт примера 35 (149 мг, 0.354 ммоля) в метаноле (3 мл) обрабатывался 5 М водной КОН (0.25 мл). Смесь перемешивалась при 20°C в течение 30 минут перед удалением растворителя в вакууме. Твердый остаток промывался водой для удаления солей и сушился азеотропно путем растворения два раза в ацетонитриле и концентрирования в вакууме, давая 100 мг целевого продукта в виде его свободного основания. Данное вещество превращалось в его HCl соль согласно методу, использованному в примере 28 (87 мг, 59%, т.пл. 230-235°C (разлож.), αС-МС: 380 (МН+), анализ RР18-ЖХВД RT: 3.42 мин).
Пример 37
Дихлоргидрат (3-этинил-фенил)-{6-[2-метокси-этокси)-7-[2-(4-метилпиперазин-1-ил)-этокси]-хиназолин-4-ил}-амина
Целевой продукт примера 34 (110 мг, 0.253 ммоля) в ДМФ (2 мл) обрабатывался N-метил-пиперазином (281 мкл, 2.53 ммоля) при 110°C в течение 16 часов. Реакционная смесь распределялась между хлороформом и насыщенным водным бикарбонатом натрия. Органические экстракты промывались солевым раствором, сушились над сульфатом натрия, фильтровались и концентрировались в вакууме. Сырой продукт хроматографировался на двуокиси кремния с использованием 15% метанол/метиленхлорид, давая 56 мг чистого продукта в виде его свободного основания. Данное белое твердое вещество растворялось в минимальном объеме хлороформа и растиралось с 2 экв. 1 М HCl в эфире для осаждения целевого продукта в виде белого твердого вещества (65 мг, 48%, т. пл. 130-142°C (разл.), αС-МС: 462 (МН+), анализ RP18-ЖХВД RT: 3,69 мин).
Пример 38
Дихлоргидрат (3-этинил-фенил)-[7-(2-имидазол-1-ил-этокси)-6-(2-метокси-этокси)хиназолин-4-ил]-амина
Целевой продукт из примера 34 (110 мг, 0.253 ммоля) в ДМФ (2 мл) обрабатывался имидазолом (172 мг, 2.53 ммоля) при 110°C в течение 48 часов. Реакционная смесь распределялась между хлороформом и насыщенным водным бикарбонатом натрия. Органические экстракты промывались солевым раствором, сушились над сульфатом натрия, фильтровались и концентрировались в вакууме. Сырой продукт (119 мг) хроматографировался на двуокиси кремния с использованием смеси 10% метанол : метиленхлорид, давая 85 мг чистого целевого продукта в виде его свободного основания. Данное белое твердое вещество растворялось в минимальном объеме хлороформа, и растиралось с 2 экв. 1М HCl в эфире, с осаждением целевого продукта в виде белого твердого вещества (95 мг, 75%, т. пл. 220-227°C (разл.), αС-МС: 430 (МН+), анализ RP18-ЖХВД RT: 3.75 мин).
Пример 39
Дихлоргидрат (3-этинил-фенил)-[6-(2-имидазол-1-ил-этокси)-7-(2-метокси-этокси)-хиназолин-4-ил]-амина
Целевой продукт примера 30 (110 мг, 0.253 ммоля) в ДМФ (2 мл) обрабатывался имидазолом (172 мг, 2.53 ммоля) при 110°C в течение 48 часов. Реакционная смесь распределялась между хлороформом и насыщенным водным бикарбонатом натрия. Органические экстракты промывались солевым раствором, сушились над сульфатом натрия, фильтровались и концентрировались в вакууме. Сырой продукт (125 мг) хроматографировался на двуокиси кремния с использованием смеси 10% метанол/метиленхлорид, давая 86 мг чистого целевого продукта в виде его свободного основания. Данное белое твердое вещество растворялось в минимальном объеме хлороформа и растиралось с 2 экв. 1М HCl в эфире с осаждением целевого продукта в виде белой твердой дихлоридгидратной соли (95 мг, 78%, т. пл. 85-100°C (разл.), αС-МС: 430 (МН+), анализ RP18-ЖХВД RT: 4.13 мин).
Пример 40
Дихлоргидрат (3-этинил-фенил)-[7-(2-метокси-этокси)-6-(2-морфолин-4-ил-этокси)-хиназолин-4-ил]-амина
Целевой продукт из примера 30 (107 мг, 0.245 ммоля) в ДМФ (2 мл) обрабатывался морфолином (214 мкл, 2.45 ммоля) при 80°C в течение 24 часов. Реакционная смесь распределялась между хлороформом и насыщенным водным бикарбонатом натрия. Органические экстракты промывались солевым раствором, сушились над сульфатом натрия, фильтровались и концентрировались в вакууме. Сырой продукт (168 мг) хроматографировался на двуокиси кремния с использованием смеси 7.5% метанол/метиленхлорид, давая 65 мг чистого целевого продукта в виде свободного основания. Данное белое твердое вещество растворялось в минимальном объеме хлороформа и растиралось с 2 экв. 1М HCl в эфире с осаждением целевого продукта в виде белого твердого вещества (88 мг, 59%, т. пл. 115-130°C (разл.), αС-МС: 449 (МН+), анализ RP-18-ЖХВД RT: 4.00 мин).
Пример 41
Хлоргидрат 2-[4-(3-этинил-фениламино)-7-(2-метокси-этокси)-хиназолин-6-илокси]-этанола
Целевой продукт из примера 33 (149 мг, 0.354 ммоля) в метаноле (3 мл) обрабатывался 5М водной КОН (0.25 мл). Смесь перемешивалась при 20°C в течение 30 минут перед удалением растворителя в вакууме. Твердый остаток промывался водой для удаления солей и сушился азеотропно с помощью растворения два раза в ацетонитриле и концентрирования в вакууме, давая 95 мг целевого продукта в виде свободного основания. Данное вещество превращалось в его HCl соль в соответствии с методом, использованным в примере 28 (89 мг, 61%, т.пл. 190-215°C (разл.), α С-МС: 380 (МН+), анализ RP18-ЖХВД RT: 3.66 мин).
Пример 42
Хлоргидрат (6,7-диэтокси-хиназолин-4-ил)-(3-этинил-фенил)-амина
6,7-Диэтоксихиназолин-4-он (120 мг, 0.512 ммоля), трифенилфосфин (295 мг, 1.126 ммоля) в 3 мл четыреххлористого углерода нагревались в условиях дефлегмации в течение 16 часов. Реакционная смесь концентрировалась в вакууме до остатка, который разбавлялся 3 мл изопропилового спирта и 3-этиниланилином (66 мг, 0.562 ммоля) и нагревалась в условиях дефлегмации в течение 3 часов. Охлажденная реакционная смесь фильтровалась, давая твердый целевой продукт, который промывался 10 мл изопропилового спирта и сушился в вакууме при 70°C, 140 мг (75%), т. пл. 269-270°C.
Пример 43
Хлоргидрат (6,7-диэтокси-хиназолин-4-ил)-(3-этинил-2-метил-фенил)-амина
4-Хлор-6,7-диэтоксихиназолин (200 мг, 0,792 ммоля) и 3-(2'-триметилсилилэтинил)-2-метил-анилин (168 мг, 0.871 ммоля) в 4 мл третбутилового спирта нагревались в условиях дефлегмации в течение 16 часов. Охлажденная реакционная смесь разбавлялась 5 мл этилового эфира и фильтровалась, давая твердый хлоргидрат (6,7-диэтокси-хиназолин-4-ил)-(3-(2'-триметилсилил-этинил)-2-метил-фенил)-амина, который промывался 10 мл этилового эфира и сушился в вакууме при 70°C. Данное вещество десилилировалось непосредственно обработкой 2 мл метанола, содержащего 1 каплю воды и 100 мг карбоната калия, в течение 0.5 часа. Гетерогенная реакционная смесь фильтровалась через целит и выпаривалась в вакууме до остатка, который растворялся в избытке 1 н. HCl в метаноле, осаждался этиловым эфиром, фильтровался и сушился в вакууме при 70°C, давая целевой продукт; 160 мг (75%), т. пл. 258-259,5°C.
Пример 44
Хлоргидрат (3-этинил-фенил)-(6-метил-хиназолин- 4-ил)-амина
6-Метил-хиназолин-4-он (350 мг, 2,18 ммоля) добавлялся к суспензии осажденного на полимере трифенилфосфина (от Флюка, 3,63 г примерно 3 ммолей Р/г смолы; 10.9 ммоля) в смеси CCl4 (3.35 г, 21.80 ммолей) и 1,2-дихлорэтана (10 мл). Смесь нагревалась до 60°C в течение 2 часов, и затем полимер удалялся фильтрованием и промывался дихлорэтаном. Фильтрат собирался в колбу, содержащую 3-этинил-анилин (0.644 г, 2.18 ммоля) и концентрировался до 5 мл с помощью выпаривания. После 4-часового нагревания в условиях дефлегмации в атмосфере азота с последующим охлаждением до 20°C целевой продукт собирался фильтрованием (551 мг, 86%, т. пл. 256-257°C, αС-МС: 260 (МН+), анализ RP-ЖХВД RT: 4,41 мин).
Пример 45
Аммониевая соль 2-{2-[4-(3-этинил-фениламино)-6-(2-метокси-этокси)-хиназолин-7-илокси]-этилсульфанил}-пропионовой кислоты
Целевой продукт примера 34 (150 мг, 0.34 ммоля) добавлялся к раствору тиомолочной кислоты (100 мкл, 1.14 ммоля) и КОН (150 мг, 2.7 ммоля) в дегазированном ДМФ (5 мл)/вода (0.5 мл). Реакционная смесь перемешивалась при 50°C в атмосфере азота в течение 72 часов, а затем охлаждалась до комнатной температуры. Величина рН смеси доводилась примерно до 4.0 с помощью уксусной кислоты, а затем смесь распределялась между хлороформом и солевым раствором. Органические экстракты промывались солевым раствором, сушились над сульфатом натрия, фильтровались и концентрировались в вакууме. Сырой продукт очищался с помощью препаративной RP18 ЖХВД с использованием градиента 15% до 100% CH3CN/pH 4.5, 50 мМ ацетата аммония с последующей лиофилизацией соответствующих чистых фракций, давая целевой продукт (28 мг, 18%, т. пл. 95-103°C (разл.), αС-МС: 468 (МН+), анализ RP-ЖХВД RT: 3.57 мин).
Пример 46
Аммониевая соль {2-[4-(3-этинил-фениламино)-6-(2-метокси-этокси)-хиназолин-7-илокси]этилсульфанил}-уксусной кислоты
Целевой продукт получался из целевого продукта примера 34 и меркаптоуксусной кислоты согласно методу примера 45 (3%, αС-МС: 454 (МН+), анализ RP-ЖХВД RT: 3.37 мин).
Пример 47
4-(3-Этинил-фениламино)-6-(2-метокси-этокси)-хиназолин-7-ол
Данный продукт выделялся в виде более липофильного продукта (с помощью препаративной RP18 ЖХВД) по реакции, использовавшейся для получения целевого продукта примера 46 (5%, αС-МС: 336 (МН+), анализ RP-ЖХВД RT: 3,60 мин).
Пример 48
(3-Этинил-фенил)-[7-(2-метокси-этокси)-6-винилокси-хиназолин-4-ил]амин и хлоргидрат [6-(2-этокси-этокси)-7-(2-метокси-этокси)-хиназолин-4-ил]-(3-этинил-фенил)-амина
Целевой продукт примера 30 (107 мг, 0.245 ммоля) обрабатывался этилатом натрия (0.582 ммоля) в дефлегмирующем этаноле (3 мл) в течение 24 часов. Растворитель удалялся в вакууме, и продукт выделялся с помощью флэш хроматографии на двуокиси кремния с использованием смеси 10% ацетон/метиленхлорид, давая 30 мг 6-винилокси продукта (33%, т. пл. 113-114°C, αС-МС: 362 (МН+), анализ RP-ЖХВД RT: 4.84 мин), 6-(2-этокси-этокси) производное элюировалось в виде более полярного продукта (45 мг) и превращалось в его HCl соль согласно процедуре, описанной для примера 28 (43%, т.пл. 220-225°C (разл. αС-МС: 408 (МН+), анализ RP-ЖХВД RT: 4.35 мин).
Пример 49
Хлоргидрат 4-(3-этинил-фениламино)-7-(2-метокси-этокси)-хиназолин-6-ола
(3-Этинил-фенил)-[7-(2-метокси-этокси)-6-винилокси-хиназолин-4-ил]-амин (20 мг, из примера 48) гидролизовался с помощью обработки 6М соляной кислотой/метанол (30: 70, 3 мл) при 50°C в течение 5 дней. Раствор концентрировался в вакууме, и остаток распределялся между хлороформом и солевым раствором при рН около 7. Органические экстракты промывались солевым раствором, сушились над сульфатом натрия, фильтровались и концентрировались в вакууме, давая целевой продукт в виде его свободного основания (15 мг), которое превращалось в его HCl соль согласно процедуре, описанной для примера 28 (т. пл. 135-150°C (разл.), αС-МС: 336 (МН+), анализ RP-ЖХВД RT: 3.77 мин).
Пример 50
Хлоргидрат 1-{2-[4-(3-этинил-фениламино)-6-(2-метокси-этокси)-хиназолин-7-илокси]-этил}-1Н-пиридин-4-она
Гидрид натрия (30 мг 60% в минеральном масле, 0.77 ммоля) добавлялся к безводному ДМФ (2,0 мл) с последующим добавлением пирид-4-она (79 мг, 0.83 ммоля). Смесь перемешивалась в течение 40 минут при 22°C до тех пор, пока все твердые вещества не растворялись и не прекращалось выделение водорода. Добавлялись целевой продукт примера 34 (120 мг, 0.28 ммоля) и тетрабутиламмонийиодид (15 мг), и реакционная смесь перемешивалась при 22°C в течение 7 дней в атмосфере азота. Дополнительное количество пирид-4-она (79 мг) и NaH (30 мг 60%) растворялись в ДМФ (2 мл), и раствор добавлялся к реакционной смеси. После перемешивания еще в течение 4 дней смесь распределялась между хлороформом и солевым раствором. Органические экстракты сушились над сульфатом натрия, фильтровались и концентрировались в вакууме. Сырой продукт очищался с помощью флэш хроматографии на двуокиси кремния с использованием смеси 10% метанол/метиленхлорид, давая 65 мг свободного основания целевого продукта, которое превращалось в моно-хлоргидратную) соль в соответствии с процедурой, описанной для примера 28 (66 мг, т. пл. 240-248°C (разл.), αС-МС: 457 (МН+), анализ RP-ЖХВД RT: 3.23 мин).
Пример 51
Хлоргидрат 1-{2-[4-(3-этинил-фениламино)-7-(2-метокси-этокси)-хиназолин-6-илокси]-этил}-1Н-пиридин-4-она
Свободное основание данного продукта получалось из целевого продукта примера 30 и натриевой соли пирид-4-она, как описано для примера 50. Свободное основание выделялось с помощью флэш хроматографии с использованием смеси 15% метанол/хлороформ и превращалось в целевой продукт в соответствии с процедурой, описанной для примера 28 (32%, т. пл. 155-168°C (разл.), αС-МС: 457 (МН+), анализ RP-ЖХВД RT: 3.45 мин).
Пример 52
Хлоргидрат (3-этинил-фенил)-(6-метокси-хиназолин-4-ил)-амина
25 мМ раствор 6-метокси-3Н-хиназолин-4-она в 1,2-дихлорэтане добавлялся к осажденному на полимере трифенилфосфину (от Флюка, примерно 3 ммоля Р/г полимера, 2.5 мол. экв.) и четыреххлористому углероду (10 мол. экв.). Реакционная смесь нагревалась при встряхивании при 60°C в течение 21 часа, охлаждалась до 22°C, и добавлялся 30 мМ раствор 3-этиниланилина (1.5 мол. экв.) в трет-бутаноле. Получающаяся в результате смесь затем нагревалась при встряхивании при 60°C в течение 18 часов с последующим охлаждением до 22°C. Полимер отфильтровывался и промывался дважды метанолом. Метанольные промывные воды добавлялись к фильтрату, и раствор концентрировался в вакууме, давая целевой продукт (73%, αС-МС: 276 (МН+), анализ RP18-ЖХВД RT: 5.82 мин). Для этих случаев аналитическая RP18-ЖХВД система состояла из Уотерс 717 (торговая марка) автоматического пробоотборника, Уотерс 996 фотодиодного Лучевого Детектора (торговая марка) и Уотерс 600 четвертичной системы доставки растворителя и контролировалась с помощью программного обеспечения Милленниум (торговая марка). Аликвоты проб или образцов хроматографировались с использованием линейного градиента 0-100% ацетонитрил/0.2 М аммонийацетатный буфер (рН 4.5) на протяжении десяти минут при скорости потока 3 мл/мин с использованием С18 колонки Перкин-Элмер Пекосфер (торговая марка) (3 мм × 3 см).
Соединения примеров 53-94 в виде их хлоргидратных солей, указанные в таблице 1, приготавливались аналогично способу примера 52 из соответствующего 3Н-хиназолин-4-онового производного и 3-этинил-анилина.
Пример 95
Хлоргидрат (6,7-дибутокси-хиназолин-4-ил)-(3-этинил-фенил)-амина
6,7-дибутокси-хиназолин-4-он (105 мг, 0,362 ммоля), трифенил фосфин (208 мг, 0,796 ммоля) и 5 мл четыреххлористого углерода нагревались с обратным холодильником в течение 16 часов, и реакционная смесь концентрировалась в вакууме, давая остаток, который разбавлялся 3 мл изопропилового спирта и 3-этиниланилином (47 мг, 0,398 ммоля) и нагревался с обратным холодильником в течение 3 часов. Охлажденная реакционная смесь фильтровалась, давая в виде твердого вещества хлоргидрат (6,7-дибутокси-хиназолин-4-ил)-(3-этинил-фенил)-амина, который промывался 10 мл изопропилового спирта и сушился в вакууме при 70°C, в количестве 92 мг (60%); т. пл. 247-248°C.
Пример 96
Хлоргидрат (6,7-диизопропокси-хиназолин-4-ил)-(3-этинил-фенил)-амина
6,7-диизопропоксихиназолин-4-он (55 мг, 0,210 ммоля), трифенилфосфин (121 мг, 0,462 ммоля) и 3 мл четыреххлористого углерода нагревались в условиях дефлегмации в течение 16 часов, и реакционная смесь концентрировалась в вакууме до получения остатка, который разбавлялся 3 мл изопропилового спирта и 3-этиниланилина (30 мг, 0,257 ммоля) и нагревался с обратным холодильником в течение 3 часов. Охлажденная смесь упаривалась в вакууме, давая целевой продукт, который хроматографировался на силикагельной колонке при элюировании 5%-ным ацетоном в метиленхлориде, содержащем 0,25% триэтиламина. Фракции, содержащие чистый продукт, концентрировались в вакууме до получения твердого вещества, которое растворялось в 2 мл 1 н. HCl в метаноле, осаждалось этиловым эфиром, отфильтровывалось и сушилось в вакууме при 70°C, давая целевой продукт в количестве 140 мг (75%); т. пл. 241-242°C.
Пример 97
Хлоргидрат (6-Хлор-7-(2-метоксиэтилсульфанил)-хиназолин-4-ил)-(3-этинил-фенил)амина
6-хлор-7-(2-метоксиэтилсульфанил)-хиназолин-4-он (200 мг, 0,739 ммоля), трифенилфосфин (427 мг, 1,63 ммоля) и 0,7 мл четыреххлористого углерода нагревались в условиях дефлегмации в 4 мл 1,2-дихлорэтана в течение 4 часов, концентрировались в вакууме до остатка, разбавлялись 4 мл изопропилового спирта и 3-этиниланилина (129 мг, 1,104 ммоля) и нагревались с обратным холодильником в течение 16 часов. Горячая реакционная смесь фильтровалась для отделения неочищенного продукта, который подвергался колоночной хроматографии на силикагеле с элюированием 5% метанолом в хлороформе. Фракции, содержащие чистый продукт, концентрировались в вакууме, давая целевой продукт в виде твердого вещества в количестве 23 мг (8,4%); т. пл. 230-232°C.
Пример 98
(6,7-бис-[2-метоксиэтокси]-хиназолин-4-ил)-(3-этинил-2-метил-фенил)-амин
6,7-бис-[2-метоксиэтокси]-4-хлор-хиназолин (90 мг, 0,288 ммоля) и 3-(2'-триметилсилилэтинил)-2-метил-анилин (62 мг, 0,317 ммоля) нагревались с обратным холодильником в 4 мл трет-бутилового спирта в течение 16 часов. Охлажденная реакционная смесь разбавлялась 1 мл изопропилового спирта и фильтровалась, давая твердый хлоргидрат (6,7-бис-(метоксиэтокси)-хиназолин-4-ил)-(3-(2'-триметилсилилэтин-1-ил)-2-метил-фенил)-амина, который промывался 10 мл этилового эфира и сушился в вакууме при 70°C; 70 мг. 51 мг данного вещества десилилировался путем обработки в 3 мл метанола, содержащего 1 каплю воды и 50 мг карбоната калия, в течение получаса при комнатной температуре. Гетерогенная реакционная смесь фильтровалась через целит и упаривалась в вакууме до остатка, который сушился в вакууме при 70°C, давая целевой продукт в виде сухой пены в количестве 38 мг (75%); т. пл. 232°C.
Пример 99
Хлоргидрат (6,7-бис-[2-метоксиэтокси]-хиназолин-4-ил)-(3-этинил-5-фтор-фенил)-амина
6,7-бис-(2-метоксиэтокси)-4-хлор-хиназолин (90 мг, 0,288 ммоля) и 3-(2'-триэтилсилилэтинил)-5-фтор-анилина (69 мг, 0,317 ммоля) в 3 мл третбутилового спирта нагревались в условиях дефлегмации в течение 5 часов. Охлажденная реакционная смесь разбавлялась 2 мл изопропилового спирта и фильтровалась, давая твердый хлоргидрат (6,7-бис-метоксиэтоксихиназолин-4-ил)-(3-(2'-триметилсилил-этинил)-5'-фтор-фенил)-амина, который промывался 10 мл этилового спирта и сушился в вакууме при 70°C; 131 мг. Все это вещество десилилировалось путем растворения в 3 мл метанола, содержащего 1 каплю воды и 35 мг карбоната калия, в течение получаса при комнатной температуре. Величина рН реакционной смеси доводилась до 2,5 водным раствором 1 н. соляной кислоты, и смесь фильтровалась. Твердое вещество сушилось в вакууме, давая 92 мг целевого продукта (78%). Т. пл. 249-250°C.
Пример 100
(7-Пропилсульфанил-хиназолин-4-ил)-(3-этинил-фенил)-амина.
Хлоргидрат 7-пропилсульфанил-хиназолин-4-он (300 мг, 1,36 ммоля), трифенилфосфин (785 мг, 2,99 ммоля), 1,31 мл четыреххлористого углерода и 5 мл хлороформа нагревались с обратным холодильником в течение 16 часов, и реакционная смесь концентрировалась в вакууме до остатка, который разбавлялся 5 мл изопропилового спирта и 3-этиниланилина (175 мг, 1,49 ммоля) и нагревался в условиях дефлегмации в течение 3 часов. Охлажденная реакционная смесь концентрировалась в вакууме, и остаток очищался колоночной хроматографией на силикагеле с элюированием 10% метанолом в хлороформе. Фракции, содержащие чистый целевой продукт, в виде свободного амина, концентрировались в вакууме, давая твердое вещество, которое добавлялось к 3 мл 1 н. HCl в метаноле. Данный раствор упаривался в вакууме до остатка, который растирался с 4 мл горячего изопропилового спирта, охлаждался и фильтровался. Полученное таким образом твердое вещество сушилось в вакууме при 70°C, давая 239 мг чистого целевого продукта (55%), т. пл. 229-230°C.
Пример 101
Хлоргидрат [7-(2-метоксиэтилсульфанил)-хиназолин-4-ил]-(3-этинил-фенил)-амина
Тем же способом, что и в примере 42, хлоргидрат [7-(2-метоксиэтилсульфанил)-хиназолин-4-ил]-(3-этинил-фенил)-амина получался из 7-(2-метоксиэтилсульфанил)-хиназолин-4-она (200 мг, 0,847 ммоля), трифенилфосфина (533 мг, 2,03 ммоля) и 3 мл четыреххлористого углерода с выходом 74%; 233 мг, т. пл. 208-209°C.
Пример 102
Хлоргидрат (7-хлор-6-нитро-хиназолин-4-ил)-(3-этинил-фенил)-амина
7-хлор-6-нитро-хиназолин-4-он (1,002 г, 4,44 ммоля), оксихлорид фосфора (11,5 г, 7,51 ммоля) и пентахлорид фосфора (1,62 г, 7,74 ммоля) нагревались с обратным холодильником в течение 2 часов, и реакционная смесь концентрировалась в вакууме до остатка, который растирался с толуолом, а затем снова с хлороформом и сушился в вакууме, давая неочищенный 4,7-дихлор-6-нитро-хиназолин. Последний растворялся в 35 мл изопропилового спирта и 3-этиниланилина (639 мг, 5,45 ммоля) и нагревался в условиях дефлегмации в течение 3 часов. Охлажденная реакционная смесь фильтровалась, давая целевое соединение в виде твердого вещества, которое промывалось 10 мл изопропилового спирта и сушилось в вакууме при 70°C, 1,055 г (66%); т. пл. 230,8-232,6°C.
Пример 103
Хлоргидрат (6-амино-7-хлор-хиназолин-4-ил)-(3-этинил-фенил)-амина
Хлоргидрат (7-хлор-6-нитро-хиназолин-4-ил)-(3-этинил-фенил)-амина (166 мг, 0,295 ммоля) и дитионит натрия (207 мг, 1,19 ммоля) перемешивались в 1,5 мл муравьиной кислоты в течение 4 часов при комнатной температуре. 45 мл метанола добавлялись к реакционной смеси, которая оставлялась на 16 часов при комнатной температуре. Полученный таким образом осадок отфильтровывался, растирался с 3%-ным бикарбонатом натрия в течение получаса и отфильтровывался. Твердое вещество растворялось в 20 мл 1 н. HCl в метаноле и осаждалось 200 мл этилового эфира. Данное вещество фильтровалось и сушилось в вакууме при 70°C, давая 72 мг целевого продукта (83%); т. пл. 260-265°C.
Пример 104
(3-этинил-фенил)-(7-метокси-6-нитро-хиназолин-4-ил)-амин
Хлоргидрат (7-хлор-6-нитро-хиназолин-4-ил)-(3-этинил-фенил)-амина (100 мг, 0,306 ммоля) и сухой метилат натрия (120 мг, 2,22 ммоля) перемешивались в 2 мл сухого 2-метил-пирролидин-1-она в течение 8 часов при 30°C. К охлажденной реакционной смеси добавлялось 0,93 мл 3 н. HCl и 1 мл воды. Смесь разбавлялась 60 мл воды и экстрагировалась дважды 60 мл этилацетата. Объединенные органические слои трижды промывались 50 мл воды и 50 мл солевого раствора, сушились сульфатом магния, фильтровались и упаривались в вакууме, давая 80 мг целевого продукта в виде твердого вещества (82%); т. пл. 213-218°C разл.
Пример 105
Аммониевая соль {2-[4-(3-этинил-фениламино)-7-(2-метокси-этокси)-хиназолин-6-илокси]-этилсульфанил}-уксусной кислоты
Данное вещество получалось из целевого продукта примера 30 и меркаптоуксусной кислоты при 22°C в течение 10 дней в соответствии со способом, приведенным в примере 45 (16%, т. пл. 98-113°C (разл.); αС-МС: 454 (МН+); анал. RP-ЖХВД, 3,24 мин).
Получение 1
6,7-Бис(2-метокси-этокси)-хиназолин
К этил-3,4-дигидроксибензоату (36,4 г, 0.200 моля), карбонату калия (60.8 г, 0.44 моля) и тетрабутиламмонийиодиду (750 мг) в дегазированном ацетоне (400 мл) добавлялся 2-бромэтилметиловый эфир (69.5 г, 47 мл). Смесь перемешивалась в атмосфере азота при нагревании с обратным холодильником в течение 64 часов. К смеси добавлялся эфир (600 мл), и после перемешивания в течение 30 минут при 20°C осажденные соли удалялись фильтрованием. Фильтрат концентрировался в вакууме, и остаток растирался с гексаном (500 мл) в течение 30 минут, и белый твердый этил 3,4-бис(2-метокси-этокси) бензоат отфильтровывался и сушился в вакууме (55,5 г, 93%, т. пл. 50-51°C). Часть данного продукта (45.7 г, 0.158 моля) в уксусной кислоте (150 мл) обрабатывалась по каплям концентрированной азотной кислотой (40 мл) при 5°C, и раствор перемешивался в течение 24 часов перед выливанием в холодную воду (1,6 л). Смесь экстрагировалась этилацетатом (1.1 л) и органическая фаза промывалась три раза 200 мл воды и солевым раствором и сушилась над сульфатом натрия, фильтровалась и концентрировалась в вакууме, давая этил 4,5-бис(2-метокси-этокси)-2-нитробензоат (54.3 г) в виде коричневого масла. Данный нитро продукт (52.0 г, 0.15 моля) растворялся в этаноле (1000 мл), содержащем 1 экв. HCl (получаемого в этаноле путем предварительного добавления 11 мл ацетилхлорида), PtO2⋅H2O (1.0 г) добавлялся, и смесь гидрировалась под давлением водорода 45 фунт./кв. дюйм (3.164 кг/см2) в течение 6 часов. Катализатор удалялся фильтрованием через целит, и фильтрат концентрировался в вакууме до густой суспензии, которая разбавлялась эфиром (400 мл). Твердая белая хлоргидратная соль этил 2-амино-4,5-бис-(2-метокси-этокси)бензоата отфильтровывалась и сушилась в вакууме (44.7 г, 88%). Часть данного вещества (42 г, 0.12 моля) и формиат аммония (7,6 г, 0.12 моля) растворялись в формамиде (63 мл), и перемешанная смесь нагревалась до 160-165°C в атмосфере азота в течение 3 часов. Добавлялась вода (200 мл), и после охлаждения осажденный сырой целевой продукт выделялся фильтрованием, промывался холодной водой и сушился в вакууме. Фильтрат экстрагировался пять раз хлороформом, и объединенные органические экстракты промывались солевым раствором, сушились над сульфатом натрия и концентрировались в вакууме. Остаток и осадок сырого хиназолина объединялись, растирались в горячем ацетонитриле (250 мл) в течение 30 минут, охлаждались до 20°C и обрабатывались эфиром (250 мл). После охлаждения до 4°C белое твердое вещество отфильтровывалось и сушилось в вакууме (30.4 г, 86%, GC-MC m/z 294 (М+)).
Получение 2
4-Хлор-6,7-бис-(2-метокси-этокси)-хиназолин
К 6,7-бис(2-метокси-этокси)-хиназолину (500 мг, 1.7 ммоля) из получения 1 в хлороформе (10 мл), содержащем одну каплю ДМФ, добавлялся оксалилхлорид (490 мкл, 5.6 ммоля) в виде нескольких порций на протяжении 5 минут. После того, как пенообразование прекращалось, раствор нагревался с обратным холодильником в течение 1,5 часов. Растворитель удалялся в вакууме, и остаток растворялся в 1,2-дихлорэтане (20 мл) и промывался два раза 80 мл насыщенного водного карбоната натрия. Органическая фаза сушилась над сульфатом натрия и концентрировалась в вакууме, давая твердый целевой продукт (520 мг, 92%, т. пл. 108-109°C).
Получение 3
4-Хлор-6, 7-бис-(2-хлор-этокси)-хиназолин, 4-хлор-6-(2-хлор-этокси)-7-(2-метокси-этокси)-хиназолин, 4-хлор-6,7-бис-(2-метокси-этокси)-хиназолин и 4-хлор-7-(2-хлор-этокси)-6-(2-метокси-этокси)-хиназолин 6,7-Бис (2-метокси-этокси) хиназолин (5.4 г, 18.3 ммоля) из получения 1 и пиридин (3.0 мл, 37 ммолей) нагревались в дефлегмирующем POCl3 (22 мл) в атмосфере сухого азота в течение 2.5 часов. После концентрирования смеси в вакууме при 60°C остаток растворялся в хлороформе (150 мл) и осторожно добавлялся порциями при перемешивании к холодному насыщенному водному бикарбонату натрия (100 мл). Смесь перемешивалась в течение 10 минут после завершения добавления, и органическая фаза отделялась, промывалась солевым раствором, сушилась над сульфатом натрия и концентрировалась в вакууме. Остаток подвергался флэш хроматографии на двуокиси кремния с использованием градиента 20-60% этилацетат/гексаны, давая 3.41 г 4-хлор-6,7-бис-(2-метокси-этокси)-хиназолина, 234 мг 4-хлор-6-(2-хлор-этокси)-7-(2-метокси-этокси)-хиназолина, 532 мг 4-хлор-7-(2-хлор-этокси)-6-(2-метокси-этокси)-хиназолина и 330 мг 4-хлор-6,7-бис(2-хлор-этокси)-хиназолина.
Результаты испытаний соединений представлены в таблице 2.
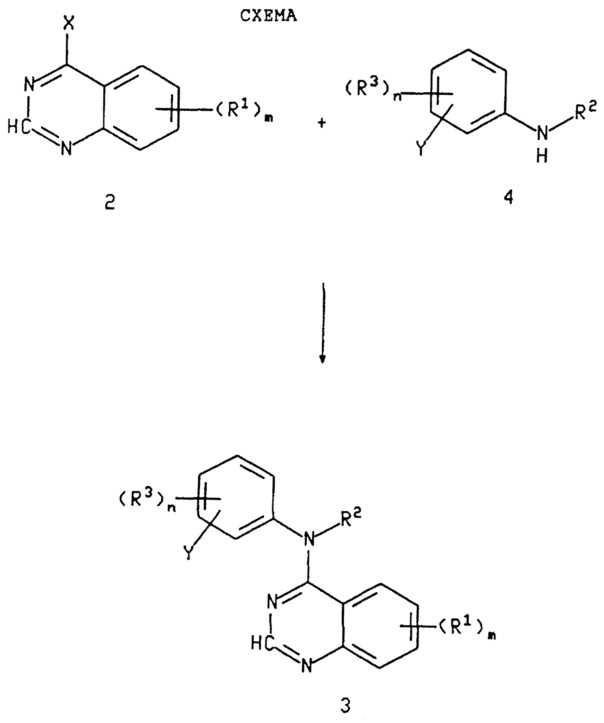
СХЕМА (ПРОДОЛЖЕНИЕ)
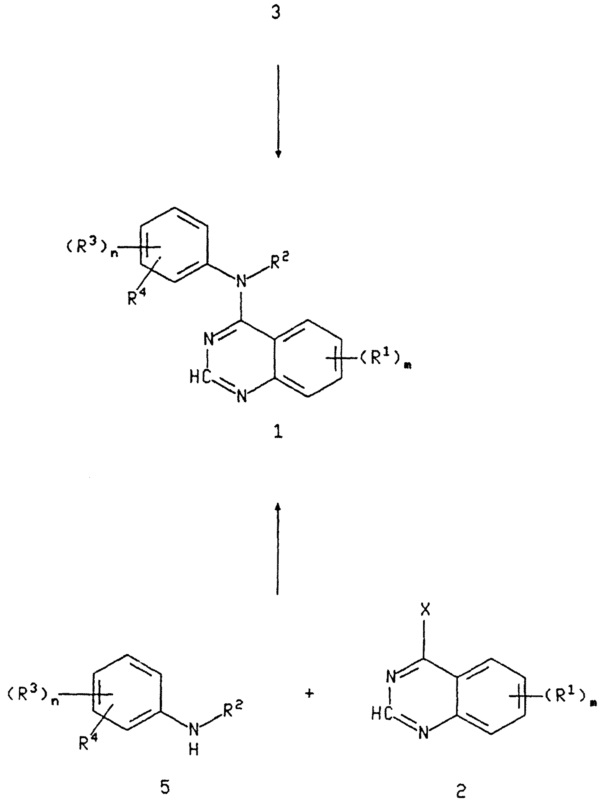








Изобретение относится к производным 4-(замещенного фениламино)хиназолина, имеющего приведенную ниже формулу X, в которой m представляет 2, n представляет 1, каждый R1 выбран из группы, состоящей из R5O, замещенного (1-4)С алкокси; R2 представляет водород; группа R3 выбрана из водорода; R4 представляет (2-4)С алкинил; R5 представляет (1-4)С алкил; или к их фармацевтически приемлемым солям. Указанные соединения ингибируют рецепторную тирозинкиназу и могут быть полезны при лечении пролиферативных заболеваний, таких как злокачественные опухоли у млекопитающих. Изобретение также относится к фармацевтической композиции, содержащей эти соединения. 2 н.п. ф-лы, 2 ил., 2 табл., 105 пр.
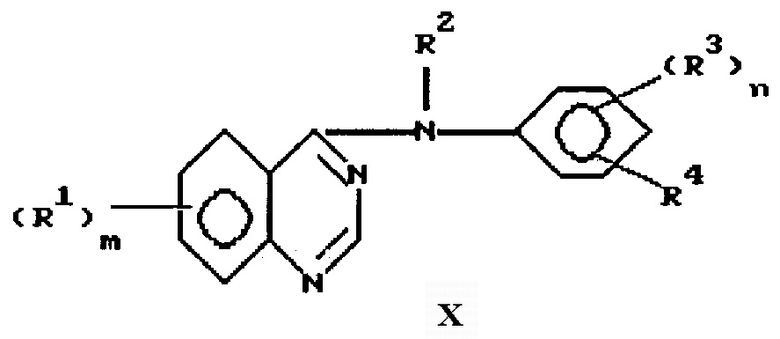
1. Производные 4-(замещенного фениламино)хиназолина формулы
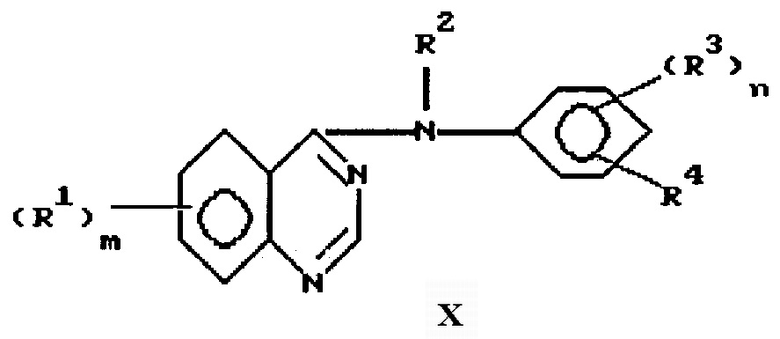
в которой m представляет 2,
n представляет 1,
каждый R1 выбран из группы, состоящей из R5O, замещенного (1-4)С алкокси;
R2 представляет водород;
группа R3 выбрана из водорода;
R4 представляет (2-4)С алкинил;
R5 представляет (1-4)С алкил;
или их фармацевтически приемлемые соли.
2. Фармацевтическая композиция, обладающая антипролиферативной активностью и содержащая активный агент и фармацевтически приемлемый носитель, отличающаяся тем, что в качестве активного агента она содержит терапевтически эффективное количество соединения по п. 1.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Устройство для передачи сигналов времени и кодовой информации о текущем времени в составе телевизионных сигналов | 1975 |
|
SU520722A1 |
| Магнитный запоминающий элемент с неразрушающим считыванием информации | 1975 |
|
SU566266A1 |
| 0 |
|
SU400048A1 | |
| Способ получения производных 2-метил-4-диалкиламиноалкиламинохиназолина | 1973 |
|
SU466233A1 |

