Изобретение относится к новым химическим соединениям, конкретно пиразолидам циклопропанкарбоновых кислот формулы I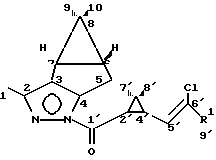
где Ia R1 Cl
Ib R1 CF3
в качестве промежуточных соединений при получении пиретроидов.
Задачей изобретения является усовершенствование способа получения пиретроидов в части выбора промежуточных соединений, которое позволит получать оптически активные пиретроидные соединения (с чистотой не менее 90 95%) по более простой и технологичной схеме.
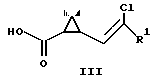
Описываемые соединения I использованы в найденных условиях для получения 1R-цис кислот (II) чистотой 90 95% и сложных эфиров 1R-цис кислот и низших спиртов, из которых получают оптически активные пиретроидные соединения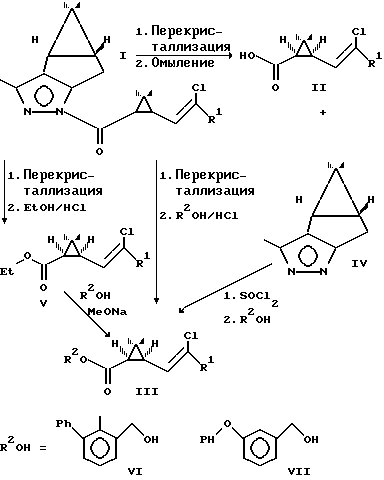
Ранее пиратроиды получали по схеме [1]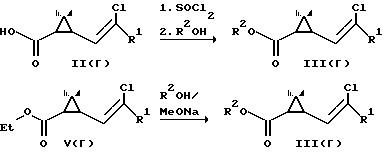
где индекс (r) обозначает рацемическая смесь соответствующих соединений.
Рацемические кислоты II(r) переводили в хлорангидриды действием хлористого тионила и полученные хлорангидриды использовали для ацилирования алкогольных компонент R2OH. При этом получали пиретроидные соединения III(r) в виде смеси энантиомеров.
Рацемические этиловые эфиры V(r) пиретроидных кислот переалкилировали пиретроидными алкогольными компонентами в присутствии оснований (метилата натрия) и получали при этом пиретроидные соединения III(r) в виде смеси энантиомеров.
Представленная схема имеет ряд недостатков.
Пиретроид III(r) содержит 50% балластного 15-изомера, имеющего активность на 2 3 порядка ниже соответствующей величины для 1R-изомера. С учетом указанного факта активность препарата снижается в 2 раза, и для поражения членистоногих требуются в 2 раза большие нормы пиретроида по сравнению с чистым 1R-изомером. Это, в свою очередь, неизбежно приводит к накоплению токсичных веществ -остатков пиретроидов и их метаболитов в природной среде. Кроме этого в 2 раза возрастает расход пиретроидных алкогольных компонент на производство препарата одинаковой удельной активности по сравнению с 1R-препаратом. Известен также антагонизм изомеров пиретроидов, когда присутствие 1S-изомера уменьшает активность 1R-изомера [2]
Возможно также получение пиретроидов по схеме [3]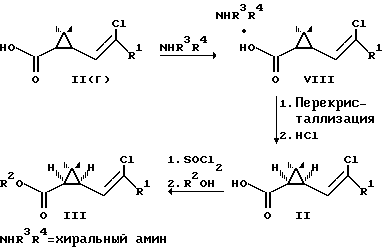
По этой схеме рацемические кислоты II(r) обрабатывают оптически активным амином с образованием смеси диастереомерных солей аминов, которые разделяют перекристаллизацией и выделяют диастереомерную соль, содержащую 1R-кислоту. Действием минеральной кислоты полученную соль разлагают и выделяют оптически активную кислоту, которую переводят в хлорангидрид и получают оптически активный пиретроид III.
Недостатком данного способа является неустойчивость применяемых аминов к действию кислорода воздуха, а также, как правило, высокая растворимость свободных аминов в воде. Указанные факты затрудняют регенерацию дорогостоящих оптически активных аминов в целях их многократного использования.
Возможно также получения пиретроидов по схеме [4]
По этой схеме рацемические кислоты II(r) через хлорангидриды переводят в смесь диастереомерных эфиров оптически активного ментола IX. Разделение диастереомеров осуществляют перекристаллизацией с отделением менее растворимого изомера. Щелочным гидролизом полученного диастереомера выделяют оптически активную кислоту II, которую используют в виде хлорангидрида в синтезе оптически активного пиретроида.
Недостатками данного способа является высокая стоимость D-(+)-ментола X, используемого для разделения рацемических смесей 1R-кислот, устойчивость сложных эфиров ментола к гидролизу. В связи с последним фактом гидролиз осуществляют автоклавированием в метанольных растворах щелочи при 120oC в течение 6 ч, что подразумевает потребность в сложном технологическом оборудовании и значительных энергетических затратах. Кроме этого, длительная обработка соединений, содержащих галогенвинильный фрагмент, основаниями при повышенной температуре может приводить к элиминированию хлористого водорода и появлению примеси ацетиленовых соединений [5] что снижает качество выделяемой циклопропанкарбоновой кислоты II.
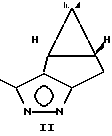
Новые промежуточные соединения получают по схеме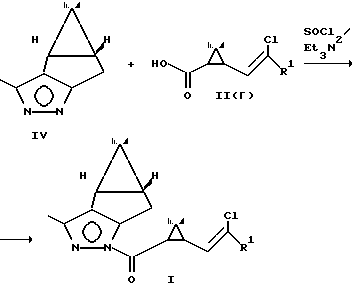
Раствор пиразола IV и рацемической циклопропанкарбоновой кислоты II(r) в эфире в присутствии триэтиламина обрабатывают хлористым тионилом и выделяют пиразолид I с выходом 93% в расчете на кислоту.
Использование заявляемых соединений I позволяет разделить рацемические циклопропанкарбоновые кислоты и получить 1R-цис кислоты чистотой 90-95% путем гидролиза диастереомерных соединений II в водно-спиртовых растворах щелочи; использование заявляемых соединений I после разделения диастереомеров позволяет получать алкоголизом в среде низших спиртов в присутствии сухого хлористого водорода оптически активные сложные эфиры циклопропановых кислот V, являющиеся промежуточными соединениями при получении пиретроидов.
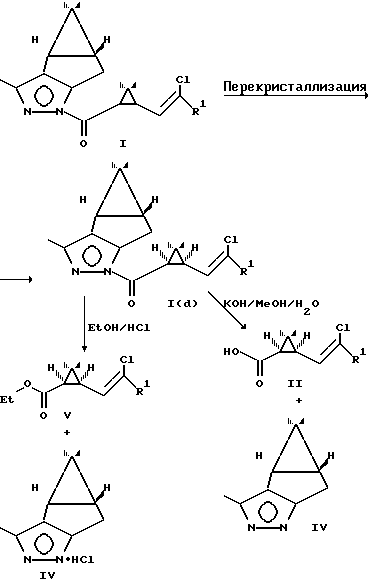
где индекс (d) здесь и далее по тексту обозначает единственный диастереомер, а именно (2'R, 4'S).
Использование заявляемых соединений I после разделения диастереомеров позволяет получать алкоголизом в среде пиретроидных алкогольных компонент в присутствии сухого хлористого водорода оптически активные пиретроидные препараты перметрин III(a) и бифентрин III(b) чистотой 90-95%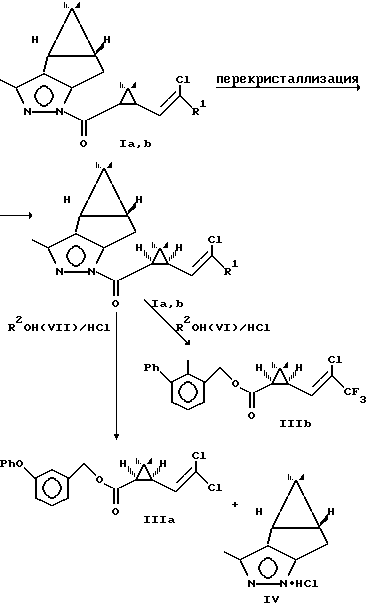
Таким образом, предлагаемая новая общая схема синтеза пиретроидов через соединения I имеют вид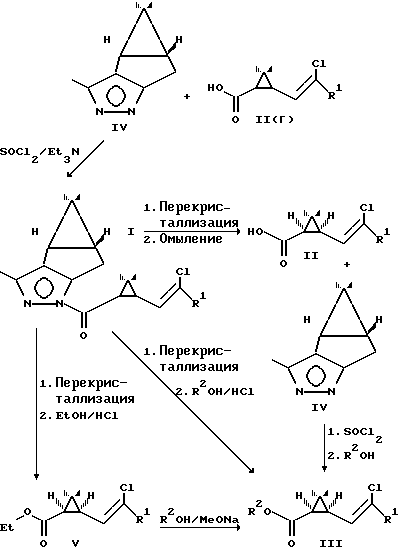
Преимущества пиразолидов I по сравнению с известными соединениями заключаются в следующем.
1. Большая доступность промежуточного соединения I. Пиразол IV, используемый в синтезе пиразолидов I, является доступным соединением. Пиразол IV синтезируют из (+)-3 карена, являющегося крупнотоннажным продуктом химической промышленности, с использованием недорогих реагентов с выходом 76% [6-7]
2. Технологические преимущества при получении промежуточных соединений. Химическая стабильность пиразола IV по отношению к большинству окислителей значительно выше, чем у известных аминов, используемых для расщепления рацемических кислот. Наряду с этим пиразол IV обладает весьма незначительной растворимостью в воде, что также выгодно отличает соединение IV от большинства аминов расщепляющих агентов. Химическая стабильность пиразола IV и его незначительная растворимость в воде облегчает его регенерацию в целях многократного использования. Получение промежуточных соединений I осуществляется в одну стадию с выходом 93% при обработке смеси пиразола IV и циклопропанкарбоновой кислоты хлористым тионилом в присутствии триэтиламина (примеры 1, 2) в отличие от двухстадийного синтеза сложных эфиров ментола IX.
3. Преимущества при получении изомерных полупродуктов пиретроидов. Использование пиразолидов циклопропанкарбоновых кислот Ia, b позволяет после перекристаллизации и омыления получать 1R-цис кислоты чистотой 90-95% (примеры 5, 6). Гидролиз пиразолидов I(d) осуществляется в значительно более мягких условиях, чем гидролиз известных сложных эфиров ментола IХ и не требует сложного технологического оборудования. Алкоголиз пиразолидов I(d) в среде низших спиртов также легко осуществим и позволяет в мягких условиях получать соответствующие оптически активные сложные эфиры 1R-цис циклопропанкарбоновых кислот Va,b чистотой 90-95% (примеры 7, 8), являющиеся промежуточными соединениями при получении пиретроидов. Как в результате гидролиза, так и в результате алкоголиза пиразолидов I(d) удается регенерировать пиразол IV, используемый в синтезе пиразолидов.
4. Преимущества при получении пиретроидных препаратов (примеры 9, 10). Алкоголиз пиразолидов I(d) в среде пиретроидных алкогольных компонент - спиртов VI и VII осуществляется с высоким выходом и одновременно регенерацией расщепляющего агента пиразола. При этом вместо трех стадий 1) омыления диастереомерного соединения с получением циклопропанкарбоновой кислоты; 2) получения хлорангидрида кислоты; 3) ацилирования пиретроидного алкогольного компонента и получения пиретроида используется единственная стадия: алкоголиза пиразолидов I(d).
Изобретение демонстрируется следующими примерами.
Пример 1. Получение смеси диастереомеров пиразолида цис-Z-цигалотриновой кислоты Ib. К раствору 24,3 г рацемической цис-Z-цигалотриновой кислоты IIb(r) и 17 г пиразола IV в 100 мл бензола и 11 мл триэтиламина при 10oC прибавляют при перемешивании 6,54 г хлористого тионила. Смесь перемешивают при комнатной температуре в течение 3 ч, промывают 1М раствором HCl (50 мл), 0,5 М раствором Na2CO3 (30 мл), высушивают безводным сульфатом натрия и после упаривания получают 36,0 г (93%) смеси диастереомеров пиразолида цис-Z-цигалотриновой кислоты Ib.
Пример 2. Получение смеси диастереомеров пиразолида цис-перметриновой кислоты Ia. К раствору 20,9 г рацемической цис-перметриновой кислоты IIa(r) и 17 г пиразола IV в 100 мл бензола и 11 мл триэтиламина при 10oC прибавляют при перемешивании 6,54 г хлористого тионила. Смесь перемешивают при комнатной температуре в течение 3 ч, промывают 1 М раствором HCl (50 мл), 0,5 М раствором Na2CO3 (30 мл), высушивают безводным сульфатом натрия и поле упаривания получают 33,0 г (93,5%) смеси диастереомеров пиразолида цис-перметриновой кислоты Ia.
Пример 3. Получение диастереомера пиразолида 1R-цис-Z-цигалотриновой кислоты Ib (d), 100 мл бензольного раствора, содержащего 36,0 г диастереомерной смеси пиразолида цис-Z-цигалотриновой кислоты Ib, полученного согласно примеру 1, после стадии высушивания безводным сульфатом натрия, разбавляют 100 мл метанола и охлаждают до -5oC и отделяют 16,7 г (92,7%) светло-желтого вязкого масла, содержащего согласно данным спектров ЯМР не менее 90% диастереомера пиразолида 1R-цис-цигалотриновой кислоты Ib (d). Полученный продукт имеет следующие характеристики:
[α]
ЯМР 1H (200 Мгц, δ м.д.): 2.21 с (3H-1); 2.733 ддд ( a 5); 3.013 ддд ( b H 5); 1.75 м (H 5, H -6); 1.07 с (3H 9); 0.673 с (3H - 10); 3.424 д (H 2); 2.358 дд (H 4'); 6.921 дкв (H 5'); 1.325 с (3H - 7'); 1.370 с (3H 8'). ЯМР 13C ' d м. д. 50.32 Мгц) 12.8 кв. 148.19 с. 131.20 с; 152.27 т; 27.13 т; 34.91 д; 25.63 д; 21.89 с; 26.19 кв; 13.59 кв; 167.86 с; 32.83 д; 30.87 с; 31.13 д; 129.94 д; 121.69 с; 28.33 кв; 14.66 кв; 120.36 с.
Пример 4. Получение пиразолида 1-R-цис-перметриновой кислоты Ia(d). 100 мл бензольного раствора, содержащего 33,0 г диастереомерной смеси пиразолида цис-перметриновой кислоты Ia, полученного согласно примеру 2, после стадии высушивания безводным сульфатом натрия, разбавляют 100 мл метанола, охлаждают до -5oC и отделяют 15,2 г (92,0%) светло-желтых кристаллов, содержащих согласно данным спектров ЯМР не менее 95% диастереомера пиразолида 1R-цис-цигалотриновой кислоты.
Полученный продукт имеет следующие характеристики.
[α]
ЯМР 1H (200 МГц, CHCl3, δ m.); 2719 c (3H 1); 2.775 ддд ( a 5-); 3.00 ддд ( b 5); 1.74 м (H 6, H 7); 1.06 м (3H 9); 0.67 с (3H 10); 3.280 д (H 2'); 2.192 (H 4'); 6.304 д (H 5'); 1.288 с (3H 7'); 1.313 с (3H 8') ЯМР 13C ( d м.д. 50.32 Мгц); 12.78 кв; 147.86 с; 130.92 с; 152.15 т; 27.14 т; 34.89 д; 25.64 д; 217.5 с; 26.21 кв; 13.59 кв; 168.11 с; 30.08 с; 29.89 с; 34.77 д; 124.68 д; 120.65 с; 28.34 кв; 14.68 кв.
Пример 5. Получение 1-R-цис-Z-цигалотриновой кислоты IIb. К раствору 3,86 г пиразолида 1-R-цис-Z-цигалотриновой кислоты Ib(d) в 50 мл метанола прибавляют раствор 0,7 г KOH в 10 мл метанола и через 5 мин при перемешивании добавляют 50 мл воды. Полученную смесь кипятят в течение 3 ч при перемешивании. Метанол упаривают при пониженном давлении, а охлажденную смесь разбавляют водой (100 мл) и экстрагируют хлористым метиленом (2x30 мл). После высушивании безводным сульфатом натрия и упаривания растворителя при пониженном давлении получают 1,38 г (85.2%) пиразола IV в виде желтоватых кристаллов. Водную фазу подкисляют концентрированной соляной кислотой (5 мл), насыщают хлористым натрием и экстрагируют хлороформом (2x30 мл). После высушивания безводным сульфатом натрия и упаривания растворителя при пониженном давлении получают 2,21 г (91%) 1-R-цис-Z-цигалотриновой кислоты IIb в виде кристаллов белого цвета.
[α]
Пример 6. Получение 1-R-цис-перметриновой кислоты IIa. К раствору 3,5 г пиразолида 1-R-цис-перметриновой кислоты Ia(d) в 50 мл метанола прибавляют раствор 0,7 г KOH в 10 мл метанола и через 5 мин при перемешивании добавляют 50 мл воды. Полученную смесь кипятят в течение 3 ч при перемешивании. Метанол упаривают при пониженном давлении, а охлажденную смесь разбавляют водой (100 мл) и экстрагируют хлористым метиленом (2x30 мл). После высушивания безводным сульфатом натрия и упаривания растворителя при пониженном давлении получают 1,35 г (83,3%) пиразола IV в виде желтоватых кристаллов. Водную фазу подкисляют концентрированной соляной кислотой (5 мл), насыщают хлористым натрием и экстрагируют хлороформом (2•30 мл). После высушивания безводным сульфатом натрия и упаривания растворителя при пониженном давлении получают 1,95 г (93%) 1-R-цис-перметриновой кислоты IIa в виде кристаллов белого цвета.
[α]
Пример 7. Получение этилового эфира 1-R-цис-Z-цигалотриновой кислоты Vb. В раствор 3.86 г пиразолида 1-R-цис-Z-цигалотриновой кислоты IIb(d) в 50 мл этанола вдувают сухой газообразный хлористый водород в течение 1,5 ч. Реакционную смесь упаривают в вакууме, разбавляют 150 мл 1M соляной кислоты и экстрагируют диэтиловым эфиром (2x30 мл). Органический экстракт высушивают безводным сульфатом натрия и после упаривания растворителя получают 2,30 г (85,3% ) этилового эфира 1-R-цис-Z-цигалотриновой кислоты Vb в виде слабо окрашенного масла. К кислой водной фарез прибавляют 30 мл концентрированного водного аммиака и полученную смесь экстрагируют хлороформом (2x20 мл). Органический экстракт высушивают безводным сульфатом натрия и после упаривания растворителя получают 1,35 г (83,3%) пиразола IV в виде желтоватых кристаллов.
Пример 8. Получение этилового эфира 1-R-цис-перметриновый кислоты Va. В раствор 3,5 г пиразолина 1-R-цис-перметриновой кислоты Ia(d) в 50 мл этанола вдувают сухой газообразный хлористый водород в течение 1,5 ч. Реакционную смесь упаривают в вакууме, разбавляют 150 мл 1M соляной кислоты и экстрагируют диэтиловым эфиром (2x30 мл). Органический экстракт высушивают безводным сульфатом натрия и после упаривания растворителя получают 2,05 г (86,4%) этилового эфира 1-R-цис-перметриновой кислоты Va в виде слабо окрашенного масла. К кислой водной фазе прибавляют 30 мл концентрированного водного аммиака и полученную смесь экстрагируют хлороформом (2x20 мл). Органический экстракт высушивают безводным сульфатом натрия и после упаривания растворителя получают 1,4 г (87,5%) пиразола IV в виде желтоватых кристаллов.
Пример 9. Получение 1R-цис-бифентрина IIIb. В раствор 3,9 г пиразолида 1-R-цис-Z-цигалотриновой кислоты Ib(d) и 3,0 г 2-метил-3-фенилбензилового спирта VI в 15 мл хлороформа вдувают сухой хлористый водород в течение 1,5 ч. Реакционную смесь упаривают при пониженном давлении, к остатку прибавляют 1M соляную кислоту (100 мл) и экстрагируют бензолом (2x75 мл). Органические экстракты объединяют промывают насыщенным раствором NaCl (100 мл), высушивают безводным сульфатом натрия и упаривают при пониженном давлении. Полученный сырой продукт очищают хроматографией на силикагеле (элюент-пентан-бензо 10:1) и получают после упаривания 3,28 г (75%) 1-R-цис бифентрина IIIb в виде кристаллов желтоватого цвета с температурой плавления 70-71oC. К кислой водной фазе после экстракции прибавляют 1 мл концентрированного водного аммиака и полученную смесь экстрагируют хлороформом (2x20 мл). Органический экстракт высушивают безводным сульфатом натрия и после упаривания растворителя получают 1,34 г (83%) пиразола IV в виде желтоватых кристаллов.
Пример 10. Получение 1R-цис-перметрина IIIa. В раствор 3,5 г пиразолида 1-R-цис-перметриновой кислоты Ia(d) и 3,0 г 3-феноксибензилового спирта VII в 15 мл хлороформа вдувают сухой хлористый водород в течение 1,5 ч. Реакционную смесь упаривают при пониженном давлении, к остатку прибавляют 1 M соляную кислоту (100 мл) и экстрагируют бензолом (2x75 мл). Органический экстракты объединяют промывают насыщенным раствором NaCl (100 мл), высушивают безводным сульфатом натрия и упаривают при пониженном давлении. Полученный сырой продукт очищают хроматографией на силикагеле (элюент-пентан-бензол 10:1) и получают после упаривания 3,20 г (76%) 1-R-цис перметрина IIIa в виде белой кристаллической массы с температурой плавления 64-65oC.
К кислой водной фазе после экстракции прибавляют 30 мл концентрированного водного аммиака и полученную смесь экстрагируют хлороформом (2x20 мл). Органический экстракт высушивают безводным сульфатом натрия и после упаривания растворителя получают 1,34 г (83%) пиразола IV в виде желтоватых кристаллов.
Пиразолиды циклопропанкарбоновых кислот формулы I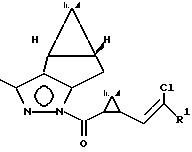
где R' = Cl, CF3, используют в качестве промежуточных соединений при получении оптически активных пиретроидных соединений. Соединения I получают с выходом 93% из пиразола II и рацемической циклопропанкарбоновой кислоты III обработкой хлористым тионилом в присутствии триэтиламина в среде эфира.
Преимущества соединений I: большая доступность; технологические преимущества при их получении и при дальнейшей их переработке в оптически активные пиретроиды.
Пиразолиды циклопропанкарбоновых килот в виде смеси диастереомеров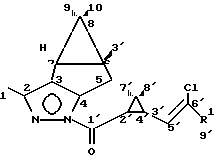
где R1 хлор или R1 CF3,
в качестве промежуточных соединений при получении пиретроидов.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Актуальные направления синтеза перметрина | |||
| Промышленность по производству минеральных удобрений | |||
| Серия: Химические средства защиты растений | |||
| НИИТЭХИМ | |||
| Кузнечная нефтяная печь с форсункой | 1917 |
|
SU1987A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Naumann K., Synthetic Pyrethroid insecticides: Structure and Properties in Chemistry of plant proteetion, Vol 4, Synthetic Pyrethoid insecticides, Springer-Verlag: Heidelberg, 1990 | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| СПОСОБ КОНТАКТНОЙ СУШКИ МИКРООРГАНИЗМОВ | 1992 |
|
RU2008589C1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Прибор для определения рентабельности судового рейса | 1928 |
|
SU22972A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Марч Дж | |||
| Органическая химия | |||
| - М.: Мир, 1988, т.4, с.60 | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Tkachev A.V., Rukavishnikov A.K | |||
| Mendeleev Commun, 1992, s | |||
| Вага для выталкивания костылей из шпал | 1920 |
|
SU161A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Popov S.A., etal, Tetrahedron, Asymmetry, Vol 5, N 3, 1994, s.479 - 489. | |||

