Изобретение относится к новым производным трет.-бутилфенолов, конкретно к 4-(ω-метоксиалкил)-2-трет.-бутилфенолам формулы: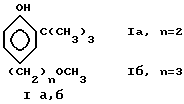
в качестве промежуточных соединений в синтезе метопролола и его аналогов, которые являются известными лекарственными средствами профилактики аритмий и инфаркта миокарда.
Соединения 1а,б в литературе не описаны.
Задачей изобретения является усовершенствование способа получения метопролола и его аналогов, которое позволит получать целевые продукты с высокой степенью чистоты и по более простой и технологичной схеме.
Описываемые соединения 1 использованы в найденных условиях для получения 4-(ω-метоксиалкил) фенолов формулы: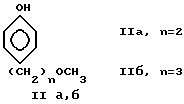
из которых синтезируются метопролол и его аналоги по схеме I: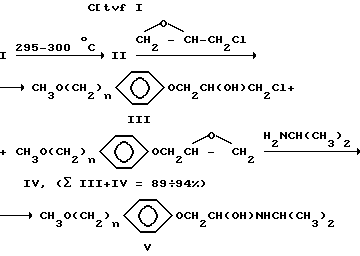
согласно которой соединения 1 подвергают термолизу при 290 300oC и полученные с выходом 70 77% 4-(ω -метоксиалкил)фенолы (II) алкилируют эпихлоргидрином при 100oC. После упаривания эпихлоргидрина, взятого в избытке, получают смесь (в сумме 90 94%) 3-[4-( w-метоксиалкил)фенокси]-1-хлорпропанола-2 (III) и 1,2-эпокси-3-[4-(w -метоксиалкил)фенокси]пропана (IV), которую, не разделяя, нагревают с изопропиламином и получают метопролол (Vа) с выходом 80% или его аналоги. Затем в комплексе с винной кислотой готовят тартрат метопролола, который является действующим началом лекарственной формы.
Ранее метопролол (Vа) получали по схеме 2, согласно которой фенол (II) синтезировали из нитропроизводного (VI) [1]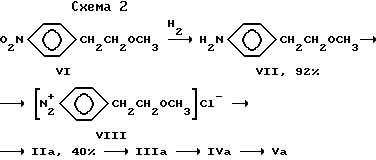
Восстановление VI водородом приводит к амину (VII). Этот амин диазотируют и полученное диазосоединение (VIII) гидролизуют, что приводит к 4-(2-метоксиэтил)фенолу IIа с выходом 40% [1] Последующие стадии, приводящие к метопрололу, по химизму аналогичны предложенной ранее схеме I [2,3]
Основным недостатком схемы 2 является низкий выход 4-(2-метоксиэтил)фенола.
Известное промежуточное нитросоединение (VI) производят по двум различным схемам. Реализован метод получения его из 2-фенилэтанола [1]
который метилировали диметилсульфатом и полученный метиловый эфир 2-фенилэтанола нитровали, получая нитропродукт VI с выходом 55% К недостаткам этой схемы следует отнести невысокий выход целевого продукта VI, а также трудности, связанные с выделением п-изомера из смеси образующихся о-, м- и п-изомеров.
Известен другой подход к синтезу нитропродукта VI взаимодействием 4-бромнитробензола и метилвинилового эфира с последующим восстановлением водородом продукта конденсации [2,4,5] Процесс осуществляют по схеме: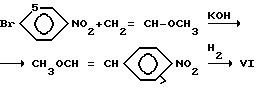
Основным недостатком схемы является отсутствие в России производств обоих компонентов, причем синтез метилвинилового эфира технологически опасен [6] Кроме того, наличие стадии бромирования требует регенерации брома, а стадия нитрования тщательной очистки от изомеров.
В рассмотренных примерах синтеза метопролола потери продуктов, связанные с введением гидроксильной группы в ароматический фрагмент промежуточных соединений, достигают 75%
Новые промежуточные соединения 1, заведомо содержащие в своей структуре гидроксильную группу, получают по схеме:
согласно которой 4-(ω -хлоралкил)-2,6-ди-трет.-бутилфенолы (IX), полученные из 2,6-ди-трет. -бутилфенола [7] нагревают с метилатом натрия или с раствором гидроокиси натрия в метаноле, получая 4-(w-метоксиалкил)-2,6-ди-трет. -бутилфенолы (X) c выходом 75 85% (пример 1). Термолиз этих соединений при 280 290oC приводит к соединениям 1 (пример 2), дальнейшее нагревание которых при 290 300oC дает 4-метоксиалкилфенолы (пример 3) с выходом 70 77% Технологически превращение соединений X в метоксиалкилфенолы II осуществляют в одном сосуде (пример 4).
Получаемые таким образом соединения II обладают высокой степенью чистоты. Так, технический 4-(2-метоксиэтил)фенол (II,а) с содержанием основного вещества (СОВ) 97% плавится в интервале 35 37oC, а очищенный кристаллизацией из толуола имеет т. пл. 41 43oC. В литературе это соединение известно как масло [8] или как твердое вещество с т.пл. 27 29 oC [9] Эта разница в чистоте продуктов является естественным следствием выбранной нами схемы синтеза, в которой отсутствуют процессы, приводящие к образованию изомеров.
Использование более чистых соединений II в синтезе метопролола привело к более чистому метопрололу с т.пл. 47.0 48.5oC (описан как масло [3,8]) и тартрату, полученному на его основе, с т.пл. 117 118oC (ср. с т.пл. 114 -116oC) [10]
Новые промежуточные соединения I в синтезе метопролола и его аналогов по сравнению с известным промежуточным продуктом VI имеют следующие преимущества:
в более технологичном способе получения самих промежуточных соединений из доступного 2,6-ди-трет.-бутилфенола;
в более высокой чистоте промежуточного продукта II,а, а также самого метопролола, получаемого с меньшими затратами;
в большей легкости и простоте получения 4-метоксиалкилфенолов, представляющих самостоятельный интерес [II]
в возможности получения аналогов метопролола и лекарственных форм на их основе.
Пример 1. Получение 4-(2-метоксиэтил)-2,6-ди-трет.-бутилфенола (X,а).
В автоклав Вишневского вместимостью 1,5 л загружают 322 г 4-(2-хлорэтил)-2,6-ди-трет. -бутилфенола (IX, а) с СОВ 94% и 70 г метилата натрия в 1 л метанола. Реакционную массу нагревают при перемешивании 6 ч при 100oC, охлаждают до комнатной температуры и содержимое нейтрализуют соляной кислотой. Осадок хлористого натрия отфильтровывают и фильтрат упаривают. Остаток 310 г перегоняют в вакууме, собирая фракцию, выкипающую в интервале 140 150oC/2 3 мм рт.ст. Получают 250 г соединения X,а с т.пл. 52 54oC (СОВ 97%).
Пример 2. Получение 4-(2-метоксиэтил)-2-трет.-бутилфенола (I,а).
В колбе с дефлегматором нагревают 264 г (1 моль) 4-(2-метоксиэтил)-2,6-ди-трет. бутилфенола (X, а) в течении 8 ч при температуре 285 295 oC. Получают 214 г бесцветного масла, содержащего по данным ГЖХ 95% продукта I, а, который растворяют в толуоле, промывают 2%-ным раствором щелочи, затем водой, упаривают растворитель, остаток перегоняют в вакууме, собирая фракцию с т.кип. 150 160oC/2 -3 мм рт.ст. Получают продукт I,а в виде масла. Найдено: M/z 208.1460. C13H20O3. Вычислено: M/z 208,1463. Спектр ПМР (CCL, d м.д. от ТМС): 1.33 (с. 9H, C4H9); 2.74 (т. J=7 Гц, 2H, -CH2Ar); 3.33 (с. 3H, CH3O-); 3.54 (т. J=7 Гц, 2Н, -CH2O-); 5.95 (с. IH, -OH); 6.31 (д. Jорто=8 Гц), 6.72 (м. Jорто8 Гц, Jмета=2 Гц), 6.94 (д. Jмета=2 Гц) группа сигналов протонов ароматического кольца, 3Н.
Пример 3. Получение 4-(2-метоксиэтил)фенола (II,а) из соединения I,а.
В перегонную колбу с дефлегматором помещают 191 г соединения 1,а (СОВ 95% ) и нагревают в токе азота при температуре реакционной массы 295 - 300oC в течение 36 ч. Затем выдержку продолжают еще 12 ч с отбором продуктов, выкипающих не выше 270oC. Получают 120 г масла, содержащего 75% соединения 11, а. Его растворяют в толуоле и встряхивают с 1 л 4%-ного раствора щелочи. Толуольный раствор отделяют и упаривают, получая соединение 1,а (СОВ 90%).
Водный слой подкисляют соляной кислотой, экстрагируют толуолом и экстракт упаривают. Остаток (95 г масла) перегоняют под вакуумом, собирая фракцию с т. кип. 130 135 oC/2 3 мм рт.ст. Получают 75 г соединения 11,а (СОВ 97%). Выход 60%
Пример 4. Получение 4-(2-метоксиэтил)фенола (11,а) из соединений X,а и I,а.
В куб ректификационной колонны со стеклянной насадкой помещают 215 г (0,815 моль) X,а (СОВ 97%) и 30 г (0,144 моль) I,а и нагревают в слабом токе азота при 290 300 oC в течение 30 ч. Затем рубашку ректификационной колонны нагревают до 280oC и начинают отбирать фракцию, выкипающую при 271 273 oC. Флегмовое число 6. Отгонку продолжают в течение 18 ч. Получают 110 г соединения II,а (СОВ 92%). Вакууммной перегонкой кубового остатка получают дополнительно 12 г продукта II,а (СОВ 80%). Отгоны объединяют и перегоняют под вакуумом, собирая фракцию с т.кип. 131 135oC/2 3 мм рт.ст. Получают 110 г II, а (СОВ 97% ), т.пл. 33 35oC. После кристаллизации из толуола т.пл. 41 43oC.
Из кубового остатка вакуумной перегонкой выделяют 12,5 г I,а (фракция с т.кип. 140 160oC/2 3 мм рт.ст.).
Выход II,а, считая на вступившие в реакцию продукты, составляет 77%
Пример 5. Получение метопролола (V,а).
Смесь 141 г (0,9 моль) 4-(2-метоксиэтил)фенола (II,а), 570 мл эпихлоридгидрина и 1,5 мл пиперидина нагревают при перемешивании 4 ч при 100oC. Затем из реакционной массы отгоняют эпихлоргидрин до содержания его в реакционной массе не более 5% Получают 223 г масла, содержащего 12% 1,2 -эпокси-3-[4-(2-метоксиэтил)фенокси] -пропана и 79% 3-[4-(2-метоксиэтил)фенокси]-1-хлорпропанола-2. Эту смесь помещают во вращающийся автоклав вместе с 300 мл изопропиламина и 350 мл изопропанола и выдерживают 12 ч при 100oC. Затем отгоняют смесь изопропиламина и растворителя, остаток растворяют в эфире. Эфирный раствор промывают 10%-ным раствором щелочи, затем водой и сушат сульфатом магния. После упаривания растворителя получают 176,7 г твердого метопролола (V,а) с т.пл. 39 -46oC, после кристаллизации из гексана с добавкой хлористого метилена т.пл. 47,0 - 48,5oC.
В изопропаноле смешивают 154 г метопролола (V,а) с т.пл. 39 46 oC и 38,8 г винной кислоты и нагревают, перемешивая при 50oC в течение 0,5 ч, охлаждают до комнатной температуры и выдерживают 12 ч. Выпавший осадок отфильтровывают и сушат на воздухе до постоянного веса. Получают 143,6 г тартрата метопролола с т.пл. 116 118oC (по данным [10] т.пл. 114 - 116oC).
Пример 6. Получение 4-(3-метоксипропил)-2,6-ди-трет.-бутилфенола (X,б).
В стальной вращающийся автоклав вместимостью 0,2 л помещают 60 г (0,264 моль) 4-(3-хлорпропил)-2,6-ди-трет.-бутилфенола (IX,б) и 13,5 г (0,25 моль) метилата натрия в 70 мл метанола, выдерживают при вращении автоклава 8 ч при 135oC. После охлаждения до комнатной температуры реакционную массу нейтрализуют соляной кислотой, выпавший хлористый натрий отфильтровывают и растворитель упаривают. Получают 57 г 4-(3-метоксипропил)-2,6-ди-трет.-бутилфенола (СОВ 86% ), который очищают, перегоняя в вакууме. Собирают фракцию с т.кип. 155 160oC/4 мм рт.ст. и получают 40 г продукта X,б в виде масла (СОВ 97%).
Пример 7. Получение 4-(3-метоксипропил)-2-трет.-бутилфенола (I,б) и 4-(3-метоксипропил)фенола (II,б).
В куб ректификационной колонны загружают 211 г (0,75 моль) 4-(3-метоксипропил)-2,6-ди-трет.-бутилфенола и выдерживают в токе азота при 295 -300oC (температура реакционной массы). По окончании выдержки реакционную массу охлаждают, растворяют в 150 мл толуола и энергично встряхивают с раствором 30 г щелочи в 0,5 л воды. Из толуольного слоя выделяют соединение I,б, а из водного -II,б.
Остаток 30 г, полученный после упаривания толуольного слоя, перегоняют в вакууме, собирая фракцию с т.кип. 165 170 oC/5 мм рт.ст. Получают 18 г 4-(3-метоксипропил)-2-трет. -бутил-фенола (I, б) (СОВ 98% ). Найдено: M/z 222,1622. C14H22O2. Вычислено: M/z 222,1619. Спектр ПМР (CCl4, d м.д. от ТМС): 1,34 (с. 9Н, C4H9 -трет.); 1,81 (м. 2Н, -CH2-); 2,53 (т. J=7 Гц, 2H, -CH2Ar); 3,31 (с. 3H, CH3O); 3,38 (т. J=7 Гц, 2Н, -CH2O-); 6,05 (с. IH, OH); 6,40 (д. Jорто8 Гц), 6,68 (м. Jорто=8 Гц, Jмета=2 Гц), 6,90 (д. Jмета=2 Гц) группа сигналов протонов ароматического кольца, 3H. УФ спектр (C2 H5OH, lmax (lg ε нм): 279,5 (3,2).
Для выделения соединения II,б водный слой подкисляют соляной кислотой до pH 5,0 и продукт II,б экстрагируют толуолом. После упаривания растворителя получают 95 г 4-(3-метоксипропил) фенола (СОВ 95%), который очищают перегонкой в вакууме, собирая фракцию с т.кип. 140 150oC/2 3 мм рт.ст. Получают 90 г продукта II,б в виде масла с выходом 73% Спектр ПМР (CCl4, d м.д. от ТМС): 1,82 (м, 2H, -CH2-); 2,58 (т. J=7 Гц, 2Н, ArCH2-); 3,34 (с. 3Н, CCH3); 3,39 (т. J=7 Гц, 2Н, -CH2-); 4,93 (с.IH, -OH); 6,70 и 6,90 (система AA'BB', 4Н, Ar-H).
Пример 8. Промежуточное образование соединения I,б в синтезе 4-(3-метоксипропил) фенола (II,б).
Термолиз образца 4-(3-метоксипропил)-2,6-ди-трет.-бутилфенола (X,б) при температуре 295 300oC в условиях примера 7 через 14 ч дал продукт, содержащий по данных ГЖХ 2% исходного соединения X,б, 19% соединения II,б и 74% 4-(3-метоксипропил)-2-трет.-бутилфенола (I,б).
Пример 9. Получение 1-изопропиламино-3-[4-(3-метоксипропил)-фенокси]пропанола-2 (V,б).
Смесь 15,2 г (0,1 моль) 4-(2-метоксипропил)фенола, 65 мл эпихлоргидрина и 0,17 мл пиперидина нагревают при перемешивании 4 ч при 100oC. Затем из реакционной массы отгоняют эпихлоргидрин до содержания его в реакционной массе не более 5% Получают 29,3 г масла, содержащего 24% 1,2-эпокси-3-[4-(2-метоксипропил)фенокси] -пропана и 59% 3-[4-(2-метоксипропил)фенокси]-1-хлорпропанола-2. Эту смесь помещают во вращающийся автоклав вместе с 35 мл изопропиламина и 45 мл изопропанола и выдерживают 12 ч при 100oC. Затем отгоняют смесь изопропиламина и растворителя, остаток растворяют в эфире. Эфирный раствор промывают 10% -ным раствором щелочи, затем водой и сушат сульфатом магния. После упаривания растворителя получают 19,7 г продукта V,б с т. пл. 40 50oC, после кристаллизации из гексана с добавкой дихлорэтана т. пл. 53 54 oC. Найдено? C 67,54; H 9,26; N 5,04. Вычислено, C 67,39; H 9,42; N 5,23. Спектр ПМР (CCl4, d м. д. от ТМС); 1,05 (д. 6Н); 1,75 (м. 2Н); 2,55-2,77 (м. 2Н); 3,23 (с. 3Н); 3,77-3,88 (м.3Н); 6,73-6,97 (система AA'BB', 4H).
Пример 10. Получение тартрата V,б.
Смесь 2,0 г продукта V,б и 0,5 г винной кислоты в 15 мл изопропанола нагревают, перемешивая, 15 мин при 50 60 oC. Полученный раствор охлаждают до комнатной температуры и выдерживают в течение 6 ч. Выпавший осадок отфильтровывают, промывают изопропанолом и сушат на воздухе до постоянного веса. Получают 1,9 г (80,5%) тартрата продукта V,б с т.пл. 106 - 107 oC. Найдено, C 61,18; H 8,62; N 3,86. Вычислено, C 60,88; H 8,22; N 3,93.
Литература.
1. Synthesis and analytical study of 4-(2-methoxyethyl)phenol Tyrazol// Rutkowska-Olma E. Sazala W. Kulawinek J.M.// Barwniki Srodki Pomocnicze -1988 32 (3) p. 55-61 // Chem. Abstr. 1989 111- 57149v.
2. Preparation of salts of (±)-1-isopropylamino-3-[4-(2-methoxyethyl)phenoxy] -2-propanol, especially the tartrate of (±)-1-isopropylamino-3-[4-(2-methoxyethyl)phenoxy] -2-propanol// Zjawiony J. Kaczur-Kaczynski E. at al.//Pol. PL 158.497 (Cl. C 07 C 217/32)// Chem. Abstr. 1993 119 203128w.
3. Phenoxy-hydroxypropylamines, metod and pharmaceutiical preparations for treating cardiovascular diseases // A.E. Brandstrom, P.A.E. Carlsson at al. // Pat. US 3928601 (US Cl. 424/300, 424/330. Int Cl2 A 61 K 31/27) - 1975.
4. Preparation of 2-methoxyethenyl-nitrobenzenes// Hallberg A. Holm B. Westfelt L. // Swed. 447, 989 (Cl. C 07 C 43/164)// Chem. Abstr. 1987 107 236227h.
5. A new route to 4-(2-methoxyethyl)phenol via palladiumcatalysed arylation of methyl vinyl ether// Hallberg A. Westfelt L. Anderssonlarl M.// Synth. Commun. 1985 15 (13) -p. 1131-1136.
6. Органикум. Практикум по органической химии, т.I. М. Мир, с. 356-357.
7. Способ получения 4-галоидалкил-2,6-ди-трет.-бутилфенолов// Патент РФ (кл. C 07 C 39/24, 37/62), N 1376511, 1985.
8. Synthesis of cabon-14-labelled metoprolol //Chaudhuri N.K. Ball T.J. // J. Labelled Comp. Radiopharm, 1981, 18 (9) p. 1273-1281.
9. The evaluation of a biogenetically based approach the synthesis of octahydro-1H-benzofuro[3,2-e] isoguinolines//Ajao J.F. Bird C.W. Chauhan Y. -P.// Tetrahedron 1985 41 (6-7) p. 1367-1372.
10. Патент СССР, N 1170968, кл. C 07 C 93/06, 1985.
11. Synthesis and physical properties of alkoxymethylene substituted phenyl cyclohexanecarboxylates// Kitamura Teruo, Mukoh Akio at al.// Mol. Cryst. Liq. Cryst. 1985 130 (3-4) p. 231-243.
Изобретение относится к 4-метоксиалкил-2-трет.-бутилфенолы (1) (алкил = этил-, пропил-), которые получают обработкой 4-хлоралкил-2,6-ди-трет.-бутилфенолов метилатом натрия (или раствором гидроокиси натрия в метаноле) при нагревании с последующим термолизом образующихся 4-метоксиалкил-2,6-ди-трет. -бутилфенолов при 280 - 290oC. Преимущества соединений 1: более технологичный способ получения 1, большая чистота последующих промежуточных соединений и метопролола, возможность получения аналогов метопролола.
4-Метоксиалкил-2-трет.бутилфенолы формулы I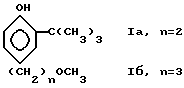
в качестве промежуточных соединений в синтезе метопролола и его аналогов.

