Настоящее изобретение касается новых замещенных имидазолов, которые могут найти применение в фармацевтических составах и использованы как самостоятельно, так и в сочетании с другими лекарствами, в особенности с диуретиками и нестероидными противовоспалительными препаратами (НСПВП), а также фармацевтической композиции на их основе.
Соединения, отвечающие настоящему изобретению, подавляют действие гормона ангиотензина II (AII) и потому полезны для лечения гипертонии, вызванной назначением ангиотензина. Энзим ренин воздействует на ангиотензиноген α2-глобулин в плазме крови, вызывая образование ангиотензина I, который затем превращается в AII под действием энзима, специфического по отношению к ангиотензину. Гормон AII сильнейший стимулятор повышения давления в сосудах, который, как известно, является главной причиной повышения давления крови как у животных разных биологических видов, включая крыс и собак, так и у человека. Соединения, отвечающие настоящему изобретению, подавляют активность AII на соответствующих рецепторах клеток-мишеней и тем самым предотвращают повышение кровяного давления, вызванного взаимодействием указанного гормона с его рецепторами. В результате назначения соединения согласно настоящему изобретению упомянутым животным (относящимся к млекопитающим), болеющим гипертонией, вызванной AII, кровяное давление в организме этих животных удается снизить. Соединения, отвечающие настоящему изобретению, кроме того, полезны при лечении сердечной недостаточности, обусловленной закупоркой сосудов. Назначение соединения, отвечающего настоящему изобретению, в сочетании с диуретиком, таким как фуросемид или гидрохлортиазид, в качестве самостоятельного этапа лечения (когда на первом этапе назначают диуретик) или же в виде физической смеси указанных препаратов усиливает антигипертензивный эффект предложенного соединения. Назначение соединения, отвечающего настоящему изобретению, вместе с нестероидным противовоспалительным препаратом (НСПВП) может предотвратить почечную недостаточность, нередко возникшую вследствие назначения НСПВП.
Выложенная Европейская патентная заявка N 0253310, опубликованная 20 января 1988 года, раскрывает, что определенным образом замещенные имидазолы блокируют рецепторы AII и, следовательно, применимы для смягчения протекания гипертонии, вызванной ангиотензином, а также для лечения сердечной недостаточности, обусловленной закупоркой сосудов. Указанные имидазолы имеют формулу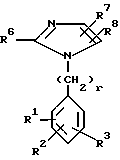
Имидазолы настоящего изобретения отличаются от аналогичных соединений, описанных в упомянутой выше заявке EPA 0253310, группами R7 и R8 в положениях 4 и 5 кольца имидазола. В заявке EPA 0253310 радикалы R7 и R8 определены следующим образом:
R7 означает H, F, Cl, Br, I, NO2, CF3 или CN;
R8 означает H, CN, алкил с 1-10 атомами углерода, алкенил с 3-10 атомами углерода или их фторзамещенные производные; фенилалкенил, алифатическая часть которого содержит 2-6 атомов углерода;
-(CH2)m имидазол-1-ил; -(CH2)m - 1,2,3-тризолил, возможно замещенный одной или двумя группами, выбранными из таких групп, как CO2CH3 или алкил с 1-4 атомами углерода; -(CH2)m тетразолил.
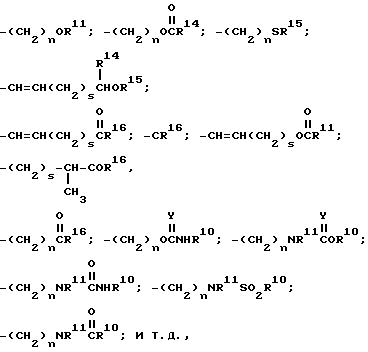
группы R10, R11, R14, R15, R16 и Y - те же, что определены ниже для соединений настоящего изобретения.
Пэлз и сотр. (Pals et al. Circulation Research, 1971, v.29, p.673) описывают присоединение к эндогенному гормону AII, сжимающему сосуды, остатка сарколизина (в положении 1) и аланина (в положении 8) с получением октапептида, блокирующего действие AII на кровяное давление у крыс, подвергнутых пункции спинного мозга. Было установлено, что данный (Sar1, Ala8) аналог AII, который вначале обозначили как P-113, а затем назвали саралазином (Saralasin), является наиболее эффективным средством борьбы с вредным воздействием AII, хотя сам по себе указанный аналог подобно большинству так называемых пептидных антагонистов AII тоже вызывает нежелательные побочные эффекты. Было показано, что саралазин снижает артериальное давление у млекопитающих и человека в тех случаях, когда (повышенное) кровяное давление зависит от циркуляции AII в крови [см. вышеуказанную работу Pals et al. а также монографию: Стритен и Андерсон. Руководство по гипертонии, т.5: "Клиническая фармакология лекарственных средств против гипертонии" под ред. Дойла; Научное изд-во "Эльзевир", 1984, стр.246 (Streeten and Anderson. Handbook of Hypertension, v.5: "Clinical Pharmacology of Antihypertensive Drugs", ed. A.S.Doyle; Elsevier Science Publishers, B.V. 1984, p.236)] Однако благодаря своей активной природе, саралазин, вообще говоря, в тех случаях, когда за уровень кровяного давления ответственен не AII, сам играет роль стимулятора повышения давления. Так как саралазин представляет собой пептид, то его фармакологическое действие является сравнительно кратковременным и проявляется только после назначения путем инъекций (parenterally), тогда как действие препарата, вводимого стоматически (orally), то есть через полость рта, неэффективно. В связи с тем, что терапевтическое применение пептидных блокеров гормона AII существенно ограничено из-за их неэффективности при введении стоматически, а также из-за непродолжительного времени действия, они пригодны в качестве фармацевтического стандарта при проведении исследований.
Некоторые известные непептидные антигипертензивные агенты подавляют энзим, называемый энзимом превращения ангиотензина (ЭПА), ответственный за превращение ангиотензина I в AII. Следовательно, такие агенты можно назвать ингибиторами ЭПА или же ингибиторами энзима превращения (ИЭП). Имеются в продаже такие ИЭП, как каптоприл и эналаприл.
Клинические испытания показывают, что в 40% случаев организм, подверженный гипертонии, невосприимчив к лечению препаратами типа ИЭП. Но стоит лишь совместно с ИЭП назначить прием диуретика, такого как фуросемид или гидрохлортиазид, как происходит эффективная нормализация кровяного давления в большинстве случаев заболеваний. Лечение диуретиком переводит патологическое состояние регулирования кровяного давления, не зависящее от ренина, в нормальное состояние, регулируемое ренином. Хотя имидазолы, предложенные в настоящем изобретении, действуют по другому механизму, а именно скорее благодаря блокированию рецептора AII, нежели посредством подавления энзима, ответственного за превращение ангиотензина, все же оба эти процесса связаны с каскадом ренин-ангиотензин. Фирма Merch Co поставляет на рынок препарат под фирменным названием "Вазеретик" (Vaseretic), представляющий собой сочетание малеата эналаприла (препарат типа ИЭП) и гидрохлортиазида, являющегося диуретиком. О применении диуретиков совместно с ИЭП для лечения гипертонии, когда сначала назначают диуретик, а на следующей стадии ИЭП или просто назначают смесь этих препаратов, сообщалось в публикациях Китона и Кемпбелла (Keeton T. K. Campbell W.B. Pharmacol. Rev. 1981, v.31, p.81), а также Вейнберга (Weinberg M.H. Medical Clinics N.America, 1987, v.71, p.979). Кроме того, описано использование диуретиков в сочетании с саралазином для усиления противогипертонического эффекта.
Данн (см. Dunn M.J. Hospital Practice, 1984, v.19, р.99) писал о том, что нестероидные противовоспалительные препараты (НСПВП) вызывают почечную недостаточность у пациентов с почечной подперфузией и высоким уровнем содержания AII в плазме крови. Назначение отвечающего настоящему изобретению соединения, блокирующего AII, в сочетании с НСПВП (либо путем последовательного приема на разных стадиях лечения, либо в виде физической смеси) способно предотвратить упомянутую почечную недостаточность. Было показано, что саралазин подавляет почечный эффект сжатия сосудов у собак, вызванный индометацином и меклофенаматом, см. работы Сато и сотр. (Satoh et al. Circ. Res. 1975, v.36/37, suppl.I, pp.1-89), а также Блесингема и сотр. (Blessingham et al. Am. Journ. Physiol. 1980, v.239, p.F360). Было показано, что каптоприл (препарат типа ИПЭ) противодействует вызванному индометаксином и имеющему почечный характер сжатию сосудов у собак, сопровождающемуся негипотоническим кровоизлиянием; см. работу Вонга и сотр. (Wong et al. J. Pharmacol. Exp. Ther. 1980, v.219, p.104).
Согласно настоящему изобретению предложены новые замещенные имидазолы, обладающие свойствами антагонистов по отношению к гормону ангиотензину II и применимые в качестве средств против гипертонии у животных, а также фармацевтическая композиция на их основе.
Замещенные имидазолы, предложенные согласно настоящему изобретению, объединяются общей формулой I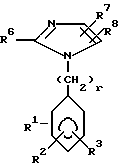
где R1 означает радикал формулы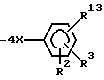
R2 означает H;
R3 означает H;
R6 означает алкил с 2-6 атомами углерода;
R7 означает винил, низший циклоалкилиденил; фенилалкинил, в котором алкинильная часть имеет 2-6 атомов углерода; 2- или 3-фурил, бифенилил, феноксифенил, фенилтио, пиридилтио или 3-меркаптофенилтио; метилтио;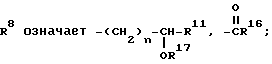
R11 означает водород;
R13 означает 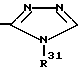 или -COOH;
или -COOH;
R16 означает водород;
R17 означает водород;
R31 означает водород;
X означает углерод-углеродную одинарную связь;
n имеет значение от 1 до 10;
r имеет значение от 1 до 2;
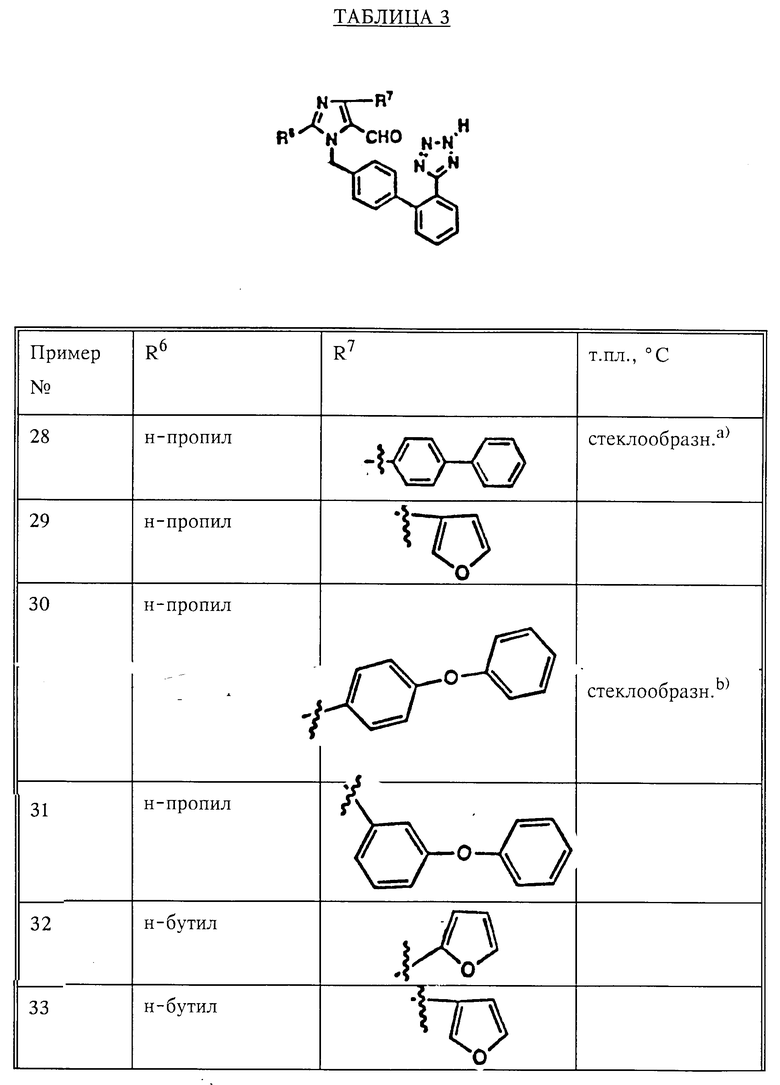
при условии: R1 не находится в орто-положении; R13 находится в орто- или мета-положении, или их фармацевтически приемлемые соли, обладающие антигипертензивной активностью.
По всему тексту необходимо иметь в виду, что если упоминается алкильный заместитель, то подразумевается нормальное строение алкила, а именно "бутил" означает "н-бутил", если не оговорено иное.
Фармацевтически приемлемые соли включают как соли металлов (неорганические соли), так и органические соли. Перечень солей дан в ремингтоновском фармацевтическом справочнике (Remington's Pharmaceutical Scinces, 1985, 17th ed. p.1418). Любому специалисту в данной области хорошо известно, что подходящий вид соли выбирают с учетом ее физической и химической стабильности, текучести, гигроскопичности и растворимости. По причинам, указанным выше, предпочтительными являются соли калия, натрия, кальция и аммония.
Объектом настоящего изобретения также является фармацевтическая композиция, включающая подходящий фармацевтический носитель и соединение формулы I в эффективном количестве. Фармкомпозицию можно использовать для лечения животных, больных гипертонией и сердечной недостаточностью, вызванной закупоркой сосудов. Фармацевтические композиции могут дополнительно содержать один или более лекарственных препаратов, таких как диуретик или нестероидный противовоспалительный препарат (НСПВП). Способ предотвращения почечной недостаточности у животных, обусловленной назначением НСПВП, включает назначение соединения формулы I на одном из этапов лечения или же в виде физической смеси с НСПВП. Соединения, отвечающие настоящему изобретению, можно также использовать в качестве диагностических агентов при исследовании системы ренин-ангиотензин.
Синтез.
Новые соединения формулы I можно получить с использованием реакций и технологий, описанных в настоящем разделе. Эти реакции проводят в среде растворителя, подходящего для используемых реагентов и веществ, а именно пригодного для осуществления необходимых химических превращений. Специалистам в области органического синтеза хорошо известно, что функциональные группы имидазола и других частей молекулы соединения, должны соответствовать намечаемым химическим превращениям. Так же, как и при осуществлении других процессов синтеза, нередко приходится принимать решения о том, какие необходимы защитные группы, условия снятия защиты, приемы активирования позиции на кольце бензила, способной к "сшиванию" с азотом в ядре имидазола. Не все соединения формулы I, упоминаемые в тексте данного раздела и относящиеся к заданному классу, можно получить всеми способами, приведенными для этого класса. Защитные группы должны быть совместимыми с условиями реакции. Ограничение, налагаемое на замещающие группы в отношении их совместимости с условиями проведения реакции, совершенно очевидно для любого специалиста в данной области и может потребовать применения одного из альтернативных способов, также описанных в данном разделе.
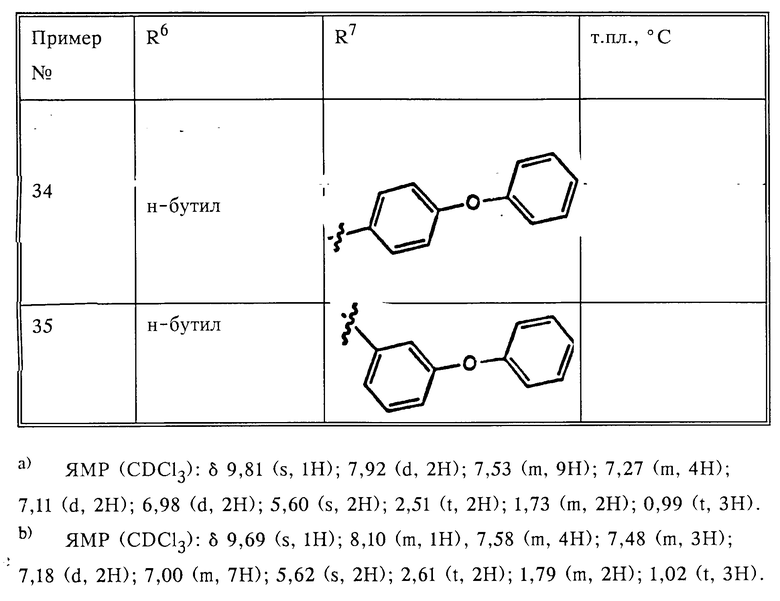
На схеме 1 показаны возможные способы получения предлагаемых соединений I.
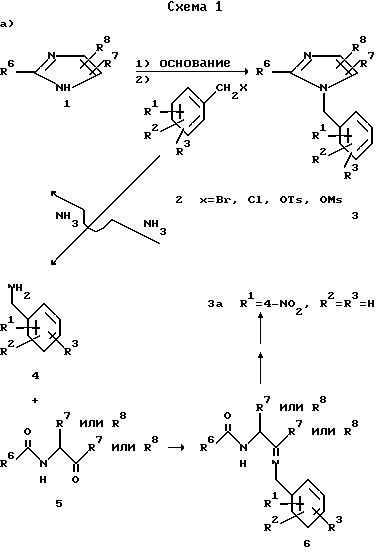
Как видно из схемы 1, соединения формулы 3 легко получить прямым алкилированием имидазола 1 защищенным соответствующим образом бензилгалидом, тозилатом или мезилатом 2 в присутствии основания: см. путь а). Предпочтительно получать металлическую соль имидазола реакцией имидазола 1 с акцептором протонов, таким как MH, где M литий, натрий или калий, в растворителе, таком как диметилформамид (ДМФ), или же проведением реакции имидазола формулы 1 с алкоксидом металла формулы MOR, где R -метил, этил, трет.-бутил и т. п. в спиртовом растворителе (этиловом или трет.-бутиловом спирте) или в диполярном апротонном растворителе, таком как диметилформамид. Соль имидазола растворяли в инертном апротонном растворителе, таком как ДМФ, и обрабатывали подходящим алкилирующим агентом 2. В другом случае имидазол 1 удавалось алкилировать бензилгалидом формулы 2, где X бром или хлор, в присутствии основания, такого как карбонат натрия, карбонат калия, триэтиламин или пиридин. Реакцию проводят в инертном растворителе, таком как ДМФ или диметилсульфоксид (ДМСО), при температурах от 20°С до температуры кипения растворителя в течение 1-10 ч. Например, 4-нитробензильное промежуточное соединение формулы За, где R1 представляет собой 4-NO2, а R2 и R3 водород, можно получить прямым алкилированием имидазола 1 4-нитробензилгалидом, тозилатом или мезилатом в присутствии основания.
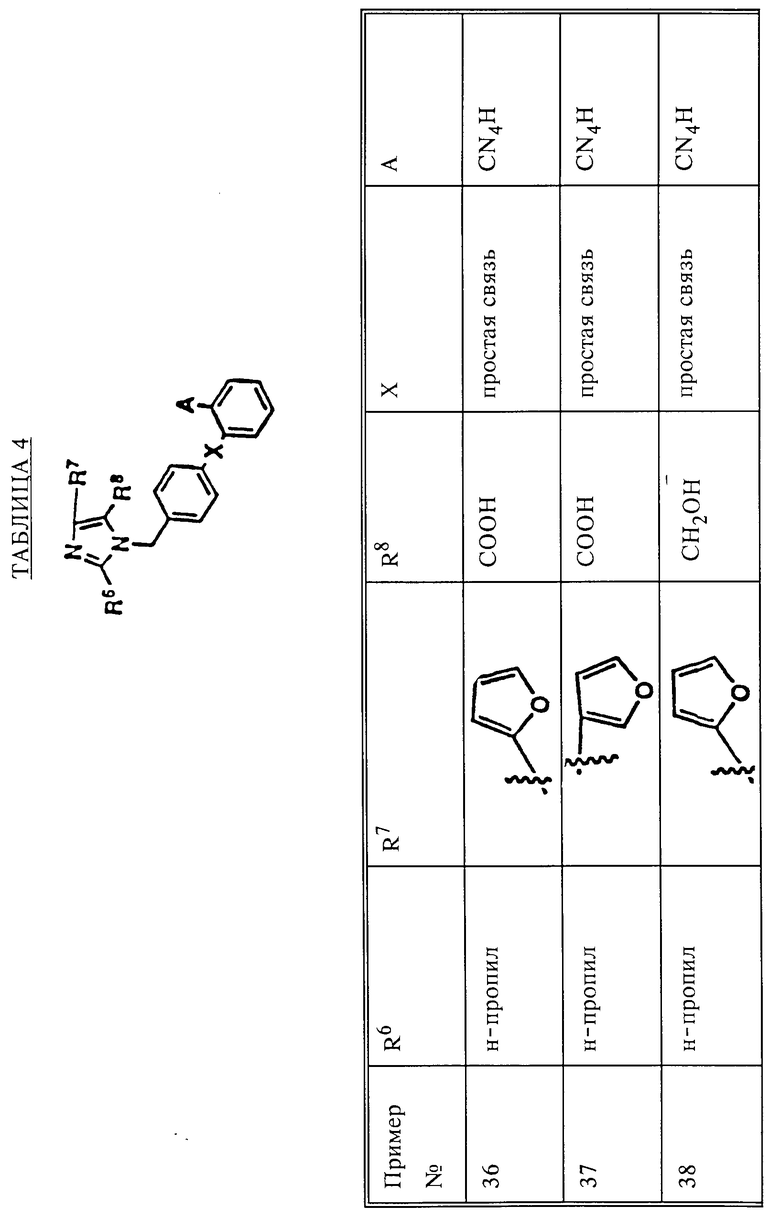
Поскольку R7 и R8 различны, то получают смеси двух продуктов (3b и 3c) алкилирования, являющихся региоизомерами, в которых R7 и R8 взаимозаменимы. Если R8 представляет собой CHO, то алкилирование протекает таким образом, что группа бензила оказывается присоединенной преимущественно к ближайшему атому азота. Такие изомеры обладают различными физическими и биологическими свойствами. Их можно разделить и изолировать один от другого с использованием обычных методик разделения, таких как хроматография и/или кристаллизация.
Из всех изученных опытов обнаружено, что первым вымывается изомер данной пары, обладающий большей биологической активностью по сравнению с изомером, вымываемым позже.
Согласно другому варианту любое производное 4 бензиламина с соответствующими функциональными группами можно превратить в имин 6 обработкой ациламинокетоном 5 в среде инертного растворителя, такого как бензол, толуол и т. п. в присутствии каталитического количества пара-толуолсульфокислоты или молекулярных сит [см. Работу Энгеля и Стеглиха (Engel N. Steglich W. - Liebigs Inn. Chem. 1978, p.1916)] или же в присутствии глинозема [см. работу Тексье-Буле (Texier-Boulet F. Synthesis, 1985, p.679)] Получающийся имин 6 можно циклизовать в N-бензилимидазол формулы 3 пентахлоридом фосфора PCl5, оксихлоридом фосфора POCl3 или трифенилфосфином PPh3 в дихлорэтане в присутствии основания, такого как триэтиламин (см. Указанную выше работу Энгеля и Стеглиха).
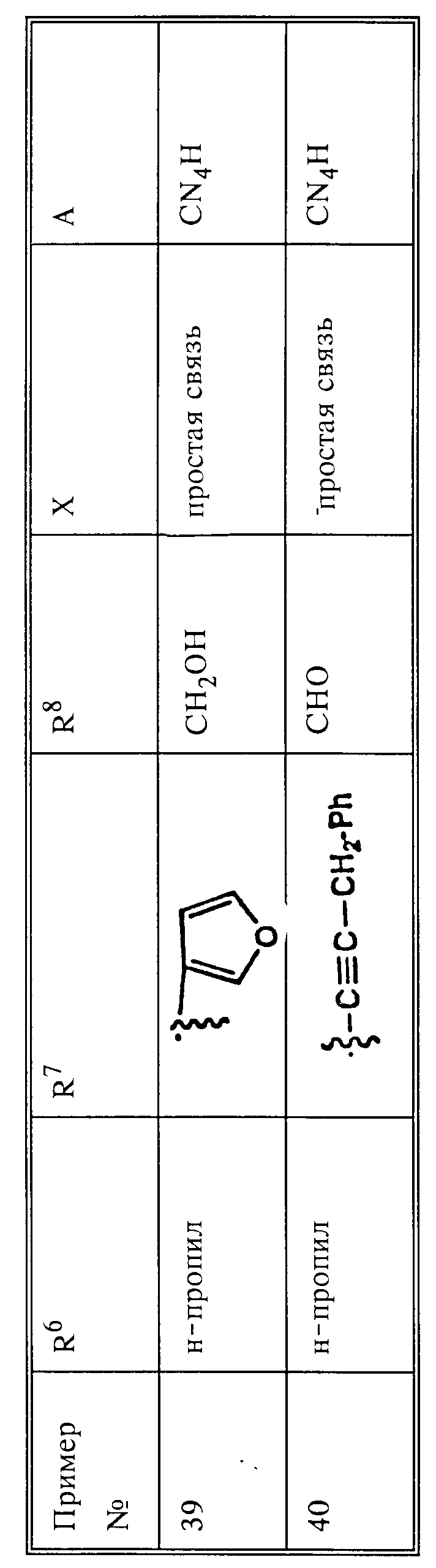
Ациламинокетон 5 легко получить из аминокислот по реакции Дакина-Уэста (см. Dakin H.D. West R. J. Biol. Chem. 1928, v.78, p.95, 745) или согласно той или иной ее модификации [см. работы Стеглиха и Гофле (Steglich W. Hofle G. -Angew. Chem. Int. Ed. Engl. 1969, v.8, p.981; Hofle G. Steglich W. Vorbruggen H. Angew. Chem. Int. Ed. Ed. Engl. 1978, v.17, p.569; Steglich W. Hofle G. Ber. 1969, v.102, p.883)] или же посредством избирательного восстановления ацилцианидов [см. работу Пфальца и Анвара (Pfaltz A. Anwars S. Fet. Lett. 1984, p.2977)] или из α-гало-, a-тозил- или a-мезилкетонов путем проведения соответствующих реакций замещения, хорошо известных специалисту в данной области.
Бензиламины 4 с функциональными группами можно синтезировать из соответствующего бензилгалида, тозилата или мезилата 2 посредством замещения азотным нуклеофилом по методике, хорошо известной специалисту в данной области. Такое замещение можно осуществить с использованием иона азида, аммиака или аниона фталимида и т.п. в нейтральном растворителе, таком как диметилформамид, диметилсульфоксид и т.п. или же в условиях фазового переноса. Бензилгалид можно синтезировать по одной из многочисленных методик бензилгалогенирования, хорошо известных специалисту в данной области, например бромированием производных толуола N-бромсукцинимидом в инертном растворителе, таком как четыреххлористый углерод, в присутствии инициатора образования радикалов, такого как бензилпероксид, при температуре, повышаемой до начала отгонки растворителя.
Самые разнообразные производные толуола можно синтезировать путем электрофильного замещения. Сюда относятся нитрование, сульфирование, фосфорилирование, алкилирование по Фриделю-Крафтсу, галогенирование и другие аналогичные реакции, известные специалисту в данной области [см. справочник Olah G. A. "Реакции Фриделя-Крафтса и связанные с ними реакции" (Friedel-Grafts and Related Reactions, v.1-5, New York Intersciense, 1965)]
Другой путь синтеза функционально замещенных бензилгалидов лежит через хлорметилирование соответствующего промежуточного соединения, превращаемого затем в ароматическое. Следовательно, подходящим образом замещенное кольцо бензола можно подвергнуть хлорметилированию, например, формальдегидом и хлористоводородной кислотой HCl при наличии или при отсутствии инертного растворителя, такого как хлороформ, четыреххлористый углерод, легкий петролейный эфир или уксусная кислота. В качестве катализатора или конденсирующего агента можно добавить одну из кислот Льюиса, такую как хлорид цинка ZnCl2, или же минеральную кислоту, например фосфорную [см. работу Fuson R.C. McKeever c.H. Org. Reactions, 1942, I, p.63]
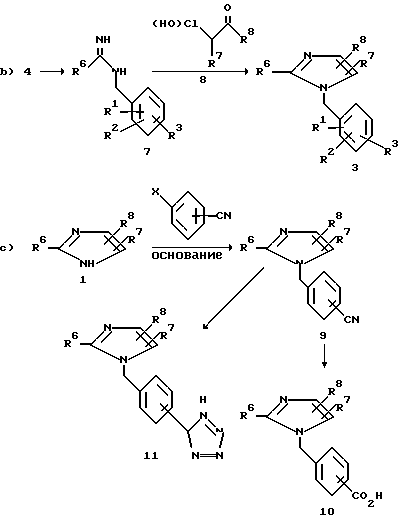
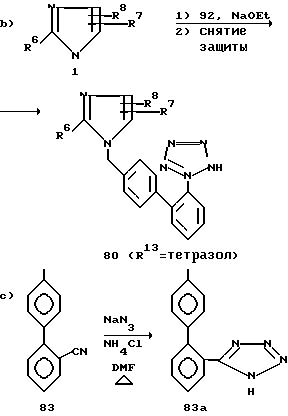
Согласно другому варианту N-бензилимидазолы 3 можно также приготовить, как показано на схеме 1b), через образование R6-замещенного амидина 7 и соответствующим образом замещенного бензиламина 4, который в свою очередь вступает в реакцию с a-галокетоном, a-гидроксикетоном 8, a-галоальдегидом или a-гидроксиальдегидом (см. работу Kunckell F. Ber. 1901, v.34, p.637).
Как показано на схеме 1a), имидазол 1 можно алкилировать различными бензильными производными. Сюда относятся соединения со скрытой кислотной функциональностью, такие как орто-, мета- и пара-цианобензилгалиды, мезилаты или тозилаты. Как показано на схеме 1c), нитрилы формулы 9 можно гидролизовать до карболовых кислот формулы 10 воздействием сильной кислоты или щелочи. Предпочтительны обработка смесью концентрированного водного раствора соляной кислоты с ледяной уксусной кислотой (объемное соотношение 1:1) при температурах отгонки растворителя в течение 2-96 ч или же обработка 1н. раствором гидроксида натрия в спиртовом растворе, таком как этиловый спирт или этиленгликоль, в течение 2-96 ч при температурах от 20oC до температуры отгонки растворителя. Если присутствует и другая нитрильная группа, то она также будет гидролизована. Нитрильная группа также может быть гидролизована в два этапа, когда на первом этапе осуществляют смешивание с серной кислотой с образованием амида, а на следующем проводят гидролиз гидроксидом натрия или минеральной кислотой с образованием карбоновой кислоты 10.
Нитрилы 9 можно превратить в соответствующее производное 11 тетразола различными методами с использованием гидразойной кислоты. Так, нитрил можно подвергнуть нагреванию с азидом натрия и хлоридом аммония в диметилформамиде при температурах от 30oC до температуры кипения растворителя в течение 1-10 сут (см. работу Hurwitz J.P. Tomson A.J. J. Org. Chem. 1961, v.26, p.3392). Предпочтительно, когда тетразол получают путем 1,3-диполярного циклоприсоединения триалкилолова или азидов триалкилолова к подходящим образом замещенному нитрилу, как это подробно описано на схеме 6.
Исходные имидазольные соединения 1 легко синтезировать по любой из многочисленных стандартных методик. Например, можно циклизовать ациламинокетон 5 с аммиаком или эквивалентными ему веществами (см. работу Davidson D. et al. J. Org. Chem. 1937, v.2, p.319) в соответствующий имидазол, как это показано на схеме 1. Соответствующий оксазол также можно превратить в имидазол 1, как правило, воздействием аммиака или аминов (см. работу Cornforth J.W. Cornforth R.H. J. Chem. Soc. 1947, 96).
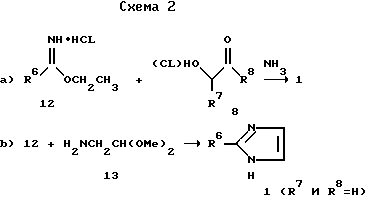
На схеме 2 представлен ряд альтернативных методик получения имидазола 1. Нумерацию и общую формулу см. схему 1. Уравнение a), приведенное на схеме 2, показывает, что проведение реакции подходящего R6-замещенного сложного эфира 12 имидата с подходящим образом замещенными a-гидрокси- или a-галокетоном, или же альдегидом 8 в аммиаке дает имидазол формулы 1 (см. работу Dziuron P. Schumack W. Archiv. Pharmaz. 1974, p.307, 470).
Исходные имидазольные соединения 1, где R7 и R8 оба представляют собой водород, можно получить согласно уравнению b) проведением реакции подходящего R6-замещенного сложного эфира имидата с a-аминоацетальдегиддиметилацеталем 13 (см. работу Grimett M.R. Adv. Heterocyclic Chem. 1970, v.12, p.103).
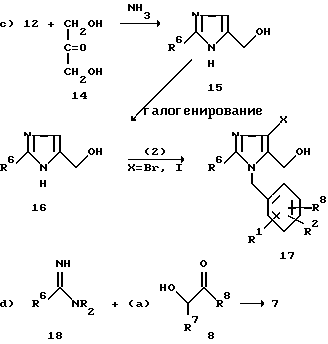
Как показывает уравнение 2c), имидазол 15 (где R7 водород, а R8 группа CH2OH) можно получить обработкой сложного эфира 12 имидата 1,3-дигидроксиацетоном 14 в аммиаке по известной методике (см. Archive der Pharmacie, 1974, p.307, 470). Галогенирование имидазола 15 или же любого имидазола, в котором R7 и R8 представляют собой водород, лучше всего осуществлять проведением реакции с одним или двумя эквивалентами N-галосукцинимида в полярном растворителе, таком как диоксан или 2-метоксиэтанол при температуре, лежащей в пределах 40-100oC, в течение 1-10 ч. Реакция галогенированного имидазола 16 с бензилгалидом 2, осуществляемая так, как это показано на схеме 1, дает соответствующий бензилимидазол 17, где R7 галоген, а R8 радикал CH2OH. Такая операция описана в патенте США 4355040. По другому варианту имидазол 17 можно получить по методике, описанной в патенте США 4207324.
Соединения формулы 17, кроме того, можно получить обработкой исходного имидазольного соединения 1, где R7 и R8 оба представляют собой водород, подходящим бензилгалидом с последующей функционализацией R7 и R8 с использованием формальдегида, как это описали Godefroie E.F. et al. Recueil, 1972, v.91, p.1383, с последующим галогенированием (см. выше).
Уравнение 2d) показывает, что имидазолы 1 можно также получить проведением реакции R6-замещенных амидинов 18 с a-гидрокси- или a-галокетоном или альдегидом 8, как это описал Kunckel F. Ber. 1901, v.34, p.637.
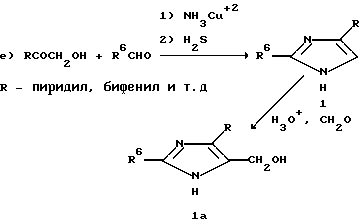
Соединения формулы 1, где R8 радикал CH2OH, можно также получить, как это показывает уравнение e). А именно, имидазолы 1 были получены, как это описал Reiter L.A. (J. Org. Chem. 1987, v.52, p.2714). Гидроксиметилирование соединения I дают гидроксиметилимидазолы Ia, как это описали Кемпе и сотр. в патенте США 4278801.
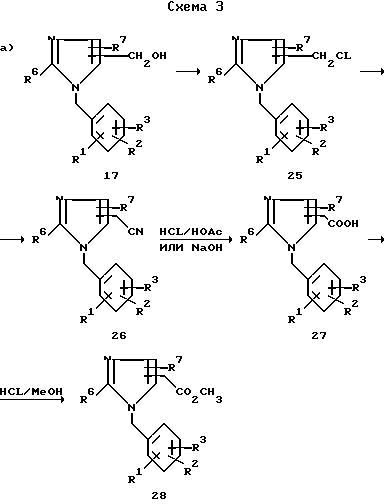
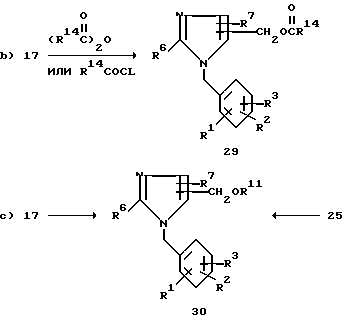
Как показывает на схеме 3 путь a) для бензилимидазолов 17, у которых R8 означает радикал CH2OH, гидроксиметильные группы можно легко превратить в соответствующий галид, мезилат или тозилат по одной из методик, множество которых хорошо известно специалисту в данной области. Предпочтительно превращать спирт 17 в хлорид 25 тионилхлоридом в инертном растворителе при температуре от 20oC до температуры отгонки растворителя.
Хлорид 25 может быть замещен различными нуклеофилами по одной из методик проведения реакции нуклеофильного замещения, хорошо известных специалисту в данной области. Так, для образования цианометилпроизводных 26 можно взять избыток цианида натрия в диметилсульфоксиде при температуре 20-100oC.
Нитрил 26 можно гидролизовать до производного уксусной кислоты 27 по одной из известных методик. Эти методики включают те, что были описаны ранее для гидролиза нитрилов формулы 9. Примеры желательных кислот и оснований для проведения данного гидролиза включают минеральные кислоты, такие как серная, соляная, а также смеси каждой из них с 30-50% уксусной кислоты (в тех случаях, когда возникает проблема растворимости), и гидроксиды щелочных металлов, такие как гидроксид натрия или калия. Реакцию гидролиза проводят при нагревании до температур 50-160oC в течение 2-48 ч. Карбоновую кислоту 27 можно этерифицировать по одной из известных методик, не затрагивая остальные части молекулы. Предпочтительно проводить реакцию в течение 2-48 ч при кипении раствора, содержащего соляную кислоту и метиловый спирт, до получения сложного эфира 28.
Сложный эфир 28 можно гидролизовать до карбоновой кислоты 27, например, после того, как будут тщательно проработаны группы R1, R2 и R3. При этом можно использовать различные методики, как кислотные, так и основные. Например, соединение 28 перемешивают с 0,5н. раствором гидроксида калия в метиловом спирте, а если основание растворимо, его перемешивают в 1н. растворе гидроксида натрия при температуре от 20oC до температуры перегонки в течение 1-48 ч.
Гидроксиметильное производное 17 можно ацилировать до соединения 29 с использованием той или иной методики. Как показывает путь b), ацилирование можно провести с использованием 1-3 эквивалентов ацилгалида или ангидрида в растворителе, таком как простой диэтиловый эфир, тетрагидрофуран, метиленхлорид и т.п. в присутствии основания, такого как пиридин или триэтиламин. По другому варианту соединение 17 можно ацилировать проведением реакции с карбоновой кислотой и дициклогексилкарбодиимидом (ДЦК) в присутствии каталитического количества 4-(N,N-диметиламино)пиридина (ДМАП) по методике, которую описал Хасснер (Hassner A. Tet. Lett. 1978, v.46, p.4475). Предпочтительная методика заключается в обработке соединения 17 раствором ангидрида карбоновой кислоты в пиридине желательно в присутствии каталитического количества ДМАП при температурах 20-100oC в течение 2-48 ч.
Как показывает путь c), простой эфир 30 можно получить из спирта 17 различными методами, такими как обработка соединения 17 в растворителе, таком как диметилформамид или диметилсульфоксид, трет.-бутоксидом калия, гидридом натрия и т. п. с последующей обработкой радикалом R11L при 25oC в течение 1-20 ч при условии, что L представляет собой галоген, тозилат или мезилат.
Согласно другому варианту обработка соединения 17 одним-пятью эквивалентами тионилхлорида в хлороформе при 25oC в течение 2-6 ч с последующей обработкой промежуточного соединения 25 одним-тремя эквивалентами радикала MOR где M натрий или калий, при 25oC в течение 2-10 ч или в растворителе формулы R11OH, или же в полярном растворителе, таком как диметилформамид и т. п. также дает в виде конечного продукта простой эфир 30.
Простой эфир 30 можно также получить нагреванием соединения 17 при 60-160oC в течение 3-15 ч в среде соединения R11OH, содержащей также неорганическую кислоту соляную или серную.
В различных процедурах синтеза необязательно, чтобы группы R1, R2 и R3 оставались теми же самыми от исходного соединения до конечных продуктов, напротив их нередко меняют, проводя известные реакции на промежуточных этапах, как это показано на схемах ниже. Все превращения, показанные на схемах, могут быть также выполнены и на терминальном ароматическом кольце, например на бифенильном кольце.
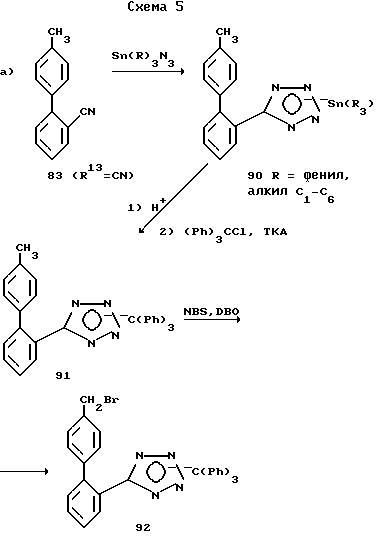
Соединения структурной формулы I, где X связь углерод-углерод, имеющие обозначение 80, можно синтезировать, как показано на схеме 4.
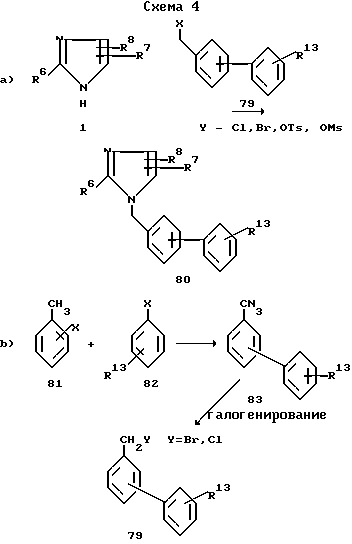
Уравнение а) показывает, что бифенильные соединения 80 можно получить алкилированием имидазола 1 подходящим галометилбифенильным соединением 79 по общей методике, приведенной в описании схемы 1.
Необходимые галометилбифенильные промежуточные соединения 79 получают ульмановским (Ullman) связыванием соединения 81 и 82, как это описано в "Organic Reactions", 1944, v.2, p.6, с целью получения промежуточных соединений 83, которые в свою очередь галогенируют. Галогенирование может сопровождаться нагреванием соединения 83 при кипении инертного растворителя, такого как четыреххлористый углерод, в течение 1-6 ч в присутствии N-галосукцинимида и инициатора реакции, такого как азобисизобутиронитрил (уравнение b).
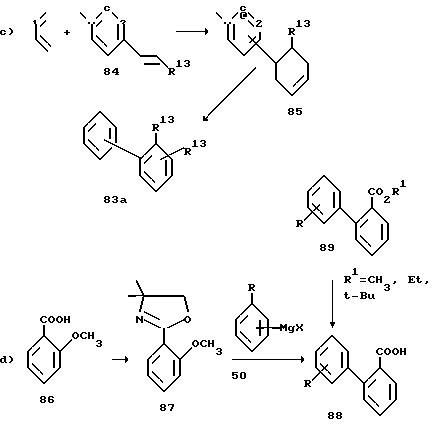
Уравнение c) показывает, что производные промежуточного соединения 83, у которых R13 находится в положении 2' (соединения 83a), можно также получить способом, описанным в J. Org. Chem. 1976, v.41, p.1320, то есть методом Дильса-Альдера (Diels-Alder) присоединения 1,3-бутадиена к стиролу 84 с последующей ароматизацией промежуточного соединения 85.
По другому варианту замещенные бифенильные соединения 83, у которых R13 -COOH, а также их сложные эфиры 89 можно получить методом, представленным уравнением d), который в качестве ключевых промежуточных соединений включает оксазолиновые соединения (см. работу Meyers A.J. Michelich E.D. J. Am. Chem. Soc. 1975, v.97, p.7383).
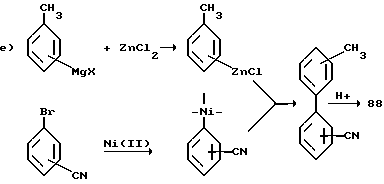
Далее уравнение e) показывает, что катализируемое никелем поперечное связывание арилцинкового гадила галобензонитрилом дает бифенилнитрил, который, в свою очередь, можно гидролизовать одним из стандартных методов и получить кислоту 88.
Замещенные бифенильные тетразолы 83, у которых R13 означает
можно получить из нитрильных соединений, у которых R13 означает CN, по методикам, описанным на схеме 1 для уравнения c) и на схеме 5 для уравнения c).
Однако предпочтительные способы получения тетразолов описаны на схеме 5 для уравнений a) и b). Соединения 90 можно получить 1,3-диполярным циклоприсоединением азидов триалкилолова или трифенилолова к подходящим образом замещенному нитрилу 83, как это представлено уравнением a). "Алкил" означает нормальный алкил с 1-6 атомами углерода, а также циклогексил.
Пример такой технологии описали Козима и сотр. (Kozima S. et al. - Organometallic Chemistry", 1971, p.337). Необходимые азиды триалкил- или триарилолова синтезируют из имеющихся в продаже хлоридов триалкил- и триарилолова и азида натрия. Группу триалкил- или триарилолова удаляют кислотным или основным гидролизом, причем тетразол можно защитить группой тритила проведением реакции с тритилхлоридом и триэтиламином с целью получения соединения 91. Описанное выше бромирование N-бромсукцинимидом и дибензоилпероксидом дает соединение 92. Алкилирование соединения 1 подходящим образом замещенным бензилгалидом в условиях, описанных выше, с последующим снятием защитной группы тритила путем гидролиза дает соединение 80, у которого R тетразол. Для защиты тетразольной части молекулы вместо группы тритила можно использовать другие защитные группы, такие как пара-нитробензил и 1-этоксиэтил. Эти группы, так же как и группу тритила, можно вводить и снимать по методике, описанной в руководстве Грина "Защитные группы в органическом синтезе" (Greene, "Protective Groups in Organic Synthesis", Wiley-Interscience, 1980).
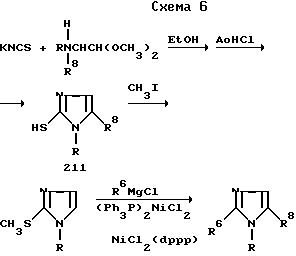
Различные 2-замещенные имидазолы можно получить проведением реакции защищенного 2-триметилсилилимидазола с подходящим электрофилом, который затем можно удалить по желанию. Соответствующую методику описали Пинкертон и Теймс (Pinkerton F.H. Thames S.F. J. Het. Chem. 1972, v.9, p.67).
По другому варианту группы R можно ввести катализируемым никелем поперечным связыванием реагентов Гриньяра (Grignard) с 2-метилтиоимидазолами (схема 6), как это описали Венкерт и Феррейра; Венкерт и сотр. Сугимара и Такэй (Wenkert E. Ferreira T.W. J. Soc. Chem. Commun. 1982, p.840; Wenkert E. et al. J. Chem. Soc. Chem. Commun. 1979, p.637; Sugimura H. Takei H. Bull. Chem. Soc. Jap. 1985, v.58, p.664). 2-Метилтиоимидазолы можно получить по методике, описанной в Германском патенте 2618370 и приведенных в нем ссылках.
Реакцией тиопиридилового эфира (229) с реактивом Гриньяра получают кетон (230)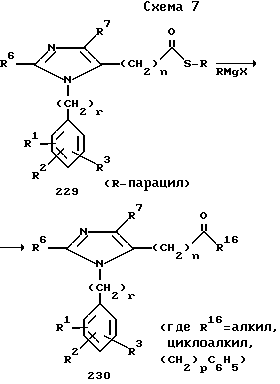
Как показано на схеме 8, если к имидазолу в положении 4- и/или 5- присоединен альдегид (см. соединение 231), то реакция этого соединения с органометаллическим реагентом, таким как реагент Гриньяра, или же с реагентом, представляющим собой алкил- или ариллитий, дает спирт 232, который можно, в свою очередь, перевести в те или иные функциональные производные, хорошо известные специалисту в данной области.
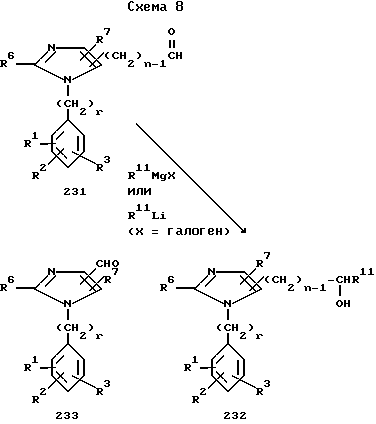
Альдегид 233, в свою очередь, можно синтезировать из соответствующего спирта 17 по различными методикам, хорошо известным специалистам в данной области, включая реакции окисления с использованием хлорхромата пиридия (XXII), церийамоний нитрата церия и аммония, а также окислительной реакцией Сверна (Swern).
Аналогично этому неалкилированное гидроксиметилимидазольное производное 1, у которого R8 радикал CH2OH, можно перевести в альдегид.
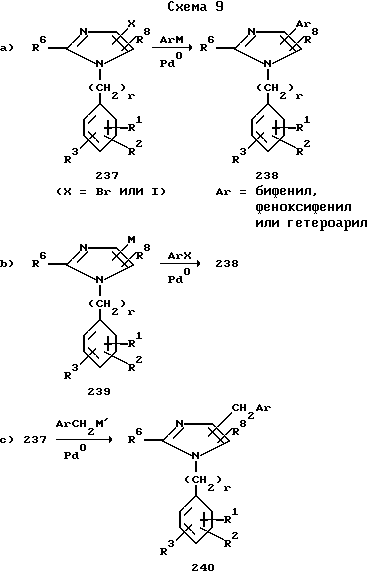
Соединения 238, у которых Ar пара-бифенилил, пара-феноксифенил или группа гетероарила, охарактеризованные при определении радикала R, можно получить связыванием арилметаллического производного ArM, где M ZnBr, Me3Zn, B(OH)2 и т.п. с галоимидазолом 237 в присутствии катализатора из переходного металла, такого как палладий, никель, платина, цирконий и т.п. (схема 9a). По другому варианту имидазол металлическое производное 239 можно связать с арилгалидом и получить таким образом соединение 238 (схема 9b).
Арилметильные производные 240 можно получить с использованием катализируемого переходным металлом связывания соединения 237 с арилметилметаллическим соединением ArCH2M', где M' ZnBr и т.п. как это показано на схеме 9c.
Соединения 241 можно получить, как показано на схеме 9d, посредством связывания алкенил- или алкинилметаллического производного AM или соответствующего алкена или алкина AH с соединением 237.
Аналогично этому неалкилированные имидазолы 1, у которых R7 бром или йод, можно подвергнуть реакциям связывания, представленным на схемах 9a-d, относительно катализируемых переходным металлом реакций связывания, см. руководство Гека "Палладиевые реагенты в органическом синтезе" (Heck R.C. "Palladium Reagents in Organic Synthesis", Ch.6,7,8; New York: Acad. Press), а также приведенные в нем ссылки на иные источники.
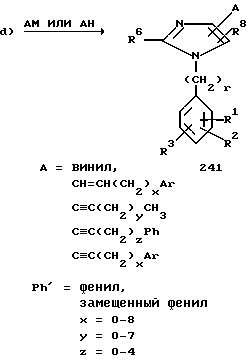
Соединения формулы I, у которых R7 -алкинильная или замещенная алкинильная группа или замещенная алкенильная группа, причем двойная или тройная связь углерод-углерод не примыкает к кольцу имидазола, когда, например, R7 представляет собой (CH2)4CH=CH(CH2)vAr, где v не равно нулю, можно получить с использованием различных приемов наращивания цепи, а также реакций связывания цепей (эти реакции хорошо известны специалисту в данной области), включая реакции, представленные на схемах 3 и 9.
Соединения формулы I, где R7 винил или арилалкенил, а R8 представляет собой CH2OH, альдегид или COOH, можно получить, как показано на схеме 10.
2-Алкилимидазол-4,5-дикарбоновые кислоты 242, полученные методом, который разработали Фаргер и Паймен (Fargher R.G. Pymen F.L. J. Chem. Soc. 1919, v. 115, p.217), можно перевести в их соответствующие сложные диэфиры 243 простой отгонкой спиртового растворителя в присутствии кислоты, такой как соляная, или одним из множества иных способов, хорошо известных специалисту в данной области.
Далее сложный эфир 243 можно превратить в его металлическую соль проведением реакции с метоксидом, этоксидом или гидридом натрия или другим основанием в подходящем растворителе, таком как диметилформамид, в присутствии акцептора кислоты, такого как карбонат калия или натрия. Полученную соль далее алкилируют подходящим образом замещенным бензилпроизводным 2 с целью получения бензилимидазола 244. Вышеуказанную последовательность реакций алкилирования можно также осуществить, подвергнув нагреванию или отгонке растворителя бензилгалид (тозилат или мезилат) 2 с имидазолом 243 в среде растворителя, такого как диметилформамид, в присутствии поглотителя кислоты, такого как карбонат калия или натрия.
Сложный диэфир 244 можно восстановить гидридом лития и алюминия в инертном растворителе, таком как тетрагидрофуран, до соответствующего диалкоголя 245. Избирательное окисление диалкоголя 245 диоксидом марганца в инертном растворителе, таком как тетрагидрофуран, дает в качестве продукта главным образом альдегид 247 с небольшим количеством диальдегида 246. Кристаллизационное либо хроматографическое разделение соединений 246 и 247 с последующим проведением реакции Виттига соединения 247 с подходящим образом замещенным арилалкилидентрифенилфосфораном в инертном растворителе, таком как тетрагидрофуран, дает в качестве продукта 4-арилалкенил-5-гидроксиметилимидазол 248. Дальнейшее окисление соединения 248 периодатом, предложенное Десс-Мартином (Dess-Martin), см. J.Org. Chem. 1983, v.48, p.4155, а также диоксидом марганца, хлорохроматом пиридиния, манганатом бария или иными окислителями, хорошо известными специалисту в данной области, в инертном растворителе, таком как тетрагидрофуран или метиленхлорид, с последующим снятием (в случае необходимости) защитных групп с любого из радикалов R1, R2 или R3, если желательно получить 4-арилалкенил-имидазол-5-карбоксальдегид 249.
Окисление соединения 249, например, сочетанием диоксида марганца с ионом цианида, см. работу Коури и сотр. (Corey E.J. et al. J. Am. Chem. Soc. 1968, v.90, p.5616), а также Сэма и сотр. (Sam D.J. et al. J. Am. Chem. Soc. 1972, v. 94, p.4024), дает в качестве продукта 4-арилалкенилимидазол-5-карбоновую кислоту 250.
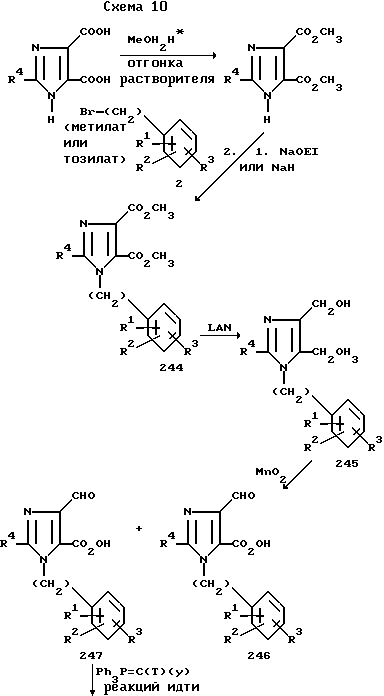
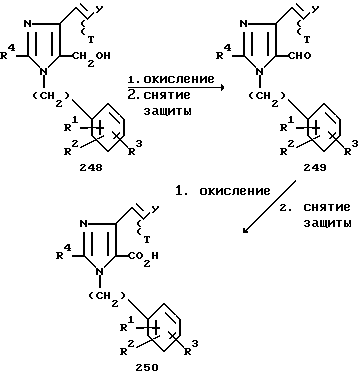
где Т и Н, y=H, (CH2)x -арил или Т и у вместе с прилегающими атомами образуют цикл с 3-8 атомами углерода и их стереоизомеры Z и E вокруг двойной связи в соединениях 248, 249, 250.
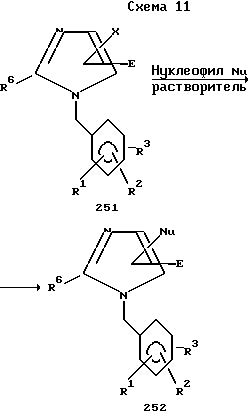
Имидазолы, представленные формулой 251, где X хлор, бром или йод, а E - электроноотнимающая группа: сложноэфирная, кето-, нитро-, алкилсульфонил и т. п. можно подвергнуть реакции нуклеофильного ароматического замещения, см. работу Шуберта и Саймона (Schubert H. Simon H. Jumar A. Z. Chem. 1968, pp. 62-63), где отщепляемая группа X замещена нуклеофилом, таким как сера, углерод или азот, для получения аддуктов 252 (схема 11).
Эту реакцию можно провести в спиртовом растворителе, таком как метиловый спирт, или в негидроксильном растворителе, таком как диметилсульфоксид, при температуре от комнатной до температуры отгонки растворителя. Иногда нуклеофил приходится превращать в его анион, чтобы сделать это соединение более нуклеофильным. Например, тиофенол кипятят в присутствии метоксида натрия и галоимидазола. Другие нуклеофилы также включают арилтиолы, гетероарилтиолы.
Соединения, отвечающие настоящему изобретению, и методы их получения будут более понятны при рассмотрении нижеследующих примеров, которые, однако, не ограничивают данное изобретение. В этих примерах, если не оговорено иное, все температуры указаны в градусах стоградусной шкалы (Цельсия), а доли и проценты в массовых единицах.
Пример 1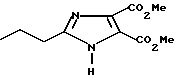
Часть А. Получение 2-н-про пил-4,5-дикарбометоксиимидазола.
2-н-пропилимидазол-4,5-дикарбоновую кислоту, полученную по методике Фаргера и Паймена (Fargher R.G. Pymen F.L. J. Chem. Soc. 1919, v.115, p. 217), с температурой плавления 257oC (dec.), взятую в количестве 17,14 г (86,6 мМ, 1 экв), 400 мл метилового спирта и 38,1 мл (534 мМ, 6 экв) ацетилхлорида перемешивали при соблюдении мер предосторожности (добавление ацетилхлорида к метиловому спирту характеризуется очень сильной экзотермичностью). Смесь подвергали кипячению растворителя в течение ночи. Далее растворитель удаляли вакуумированием, после чего добавляли 100 мл воды и 10н раствор гидроксида натрия до тех пор, пока величина pH не достигала 7. Водную смесь экстрагировали тремя порциями этилацетата, после чего органические слои объединяли, обезвоживали при помощи сульфата магния, растворитель удаляли в вакууме с тем, чтобы получить 12 г продукта в виде твердого белого вещества.
Перекристаллизация из смеси гексана с этилацетатом дала 11,41 г продукта в виде белого твердого вещества (выход 58% ) с температурой плавления 162,0-164,5oC.
Характеристики ЯМР (CDCl3): δ 3,95 (s, 6Н); 2,78 (t, 2Н); 1,83 (t из t, 2Н, J=7,7 Гц); 0,97 (t, 3Н, J=7 Гц).
ИК-спектр (для неразбавленного соединения): 1735 см-1.
Анализ формулы C10H14N2O4•(H2O): C 52,06; H 6,28; N 12,14.
Найдено: C 52,06; H 6,17; N 12,49.
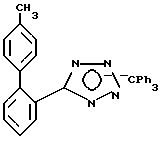
Часть B. Получение 4-ме тил-2-[N-трифенилметил(1н-тетра зол-5-ил)]бифенила.
Смешивали 4-метилбифенил-2-нитрил (его получение описано в Европейской патентной заявке 0253310, опубликованной 20.01.1988) в количестве 10 г (51,7 мМ, 1 экв), хлорид три-н-бутилолова в количестве 14 мл (51,7 мМ, 1 экв), азид натрия в количестве 3,4 г (51,7 мМ, 1 экв) и ксилол в количестве 50 мл. Реакционную смесь подвергали отгонке растворителя в течение 64 ч, после чего охлаждали до комнатной температуры. Затем добавляли 10н NaOH в количестве 6,10 мл (0,061 мМ, 1,2 экв) и тритилхлорид в количестве 14,99 г (53,8 мМ, 1,04 экв), затем эти вещества перемешивали в течение 24 ч, после чего в смесь добавляли 39 мл воды и 100 мл гептана. Получившуюся смесь перемешивали при 0oC в течение 1,5 ч. Затем образовавшиеся твердые вещества отфильтровывали, дважды промывали порциями воды по 55 мл и один раз смесью (3:2) гептана с толуолом, взятой в количестве 55 мл, после чего на протяжении ночи проводили обезвоживание под высоким вакуумом до получения 19,97 г продукта в виде светло-желтого порошка с температурой плавления 148-155oC (разл.). Далее из этих твердых веществ готовили смесь в 75 мл этилацетата, который фильтровали до получения 15 г продукта в виде светло-желтого порошка с температурой плавления 164,0-165,5oC (разл.).
Характеристики ЯМР (CDCl3): δ 7,91 (d, 1Н, J=9 Гц); 7,53-7,18 (m, 13Н); 7,02-6,84 (m, 9Н); 2,25 (s, 3Н).
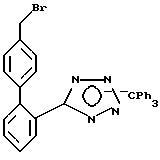
Часть C. Получение 4-бром метил-2-[N-трифенил метил-(1н-тетра зол-5-ил)] бифенила, сравнительный опыт.
Смешивали 4-ме тил-2-[N-трифенилметил-(1н-тетра зол-5-ил)] бифенил в количестве 52,07 г (109 мМ, 1 экв), 19,4 г N-бромсукцинимида (109 мМ, 1 экв), 1 г бензоилпероксида, а также 300 мл тетрахлорида углерода. Смесь подвергали кипячению в течение 2,5 ч. Далее реагенты охлаждали до комнатной температуры и отфильтровывали сукцинимид. Затем фильтрат концентрировали, а осадок растирали в порошок с простым эфиром и получали в качестве продукта первого съема 36 г вещества с температурой плавления 129,5-133,0oC (разл.).
Характеристики ЯМР (CDCl3): δ 4,37 (CH2Br).
Полученное вещество пригодно для дальнейших превращений.
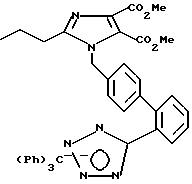
Часть D. Получение 4,5-дикарбометокси-2-н-про пил-1-[(2'-(N-трифенилметил-(1н-тетра зол-5-ил))бифенил-4-ил)метил]имидазола.
При комнатной температуре к раствору 10 г 4,5-дикарбметок си-2-н-пропилимидазола (44,2 мМ, 1 экв) в диметилформамиде добавляли 1,06 г гидрида натрия (44,2 мМ, 1 экв). При этом наблюдалось вспенивание и выделение газа. Температуру повышали до 60oC на 15 мин для полного растворения гидрида натрия. Выделение газа прекращалось, после чего смесь охлаждали до комнатной температуры. К этой смеси добавляли 24,64 г 4-бромме тил-2-[N-трифенилметил(1н-тетра зол-5-ил)] бифенила (44,2 мМ, 1 экв). Через 24 ч растворитель удаляли в вакууме, а остаток подвергали тонкослойной хроматографии в смеси гексана с этилацетатом (при их соотношении 75:25) до 100% этилацетата на силикагеле и в результате получали 15,78 г (выход 51%) белого стекловидного вещества, пригодного для дальнейших превращений.
Перекристаллизацией из этилового спирта была получена проба для анализа в виде белых кристаллов с температурой плавления 124,0-125,5oC.
Характеристики ЯМР (CDCl3): δ 7,91 (d на d, 1Н, J=3,9 Гц); 7,59-7,20 (m, 12Н); 7,09 (d, 2Н, J=9 Гц); 6,94 (m, 6Н); 6,76 (d, 2Н, J=9 Гц); 5,30 (s, 2Н); 3,89 (s, 3Н); 2,50 (t, 2Н, J=7 Гц); 1,67 (t из t, 2Н, J=7,7 Гц); 0,85 (t, 3Н, J=7 Гц).
ИК-спектр (для неразбавл.): 1718 см-1.
Анализ, вычисленный для формулы C43H38N6O4: C 73,49; H 5,45; N 11,96.
Найдено: C 73,23; H 5,48; N 12,22.
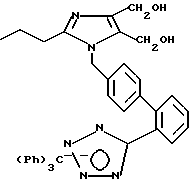
Часть E. Получение 4,5-дигидроксиметил-2-н-про пил-1-[(2'-[N-трифенилметил(1н-тетра зол-5-ил)]бифенил-4-ил)метил]имидазола.
4,5-Дикарбометокси-2-н-про пил-1-[(2'-[N-трифенил метил-(1н-тетразол-5-ил)би фенил-4-ил)метил]имидазола в количестве 9,88 г (14,1 мМ, 1 экв) растворяли в минимальном количестве тетрагидрофурана. В этот раствор медленно по каплям вводили 1М раствор алюмогидрида лития в тетрагидрофуране в количестве 15,48 мл (15,48 мМ, 1,1 экв). Смесь перемешивали в течение ночи при комнатной температуре, после чего быстро охлаждали по методике Стейнхардта (см. руководство Fieser Fieser, v.1, p.584) следующим образом. К реакционной смеси сначала осторожно добавляли 0,66 мл воды, затем 0,66 мл 15% NaOH, а затем еще 1,97 мл воды. После перемешивания в течение 72 ч получался весь тонкодисперсный осадок, который медленно отфильтровывали на цеолитном фильтре марки CeliteTM. Фильтрат обезвоживали при помощи сульфата магния, а растворитель удаляли в вакууме. В результате получали 8,83 г желтого стекловидного некристаллизующегося вещества. Это промежуточное вещество было пригодно для последующих преобразований.
Характеристики ЯМР (диметилсульфоксид-d6): δ 7,82 (d, 1Н, J=9 Гц); 7,68-7,28 (m, 12Н); 7,05 (d, 2Н, J=9 Гц); 6,87 (d, 6Н, J=9 Гц); 5,16 (s, 2Н); 4,94 (t, 1Н, J=7 Гц); 4,66 (t, 1Н, J=7 Гц); 4,37 (d, 2Н, J=7 Гц); 4,32 (d, 2Н, J=7 Гц); 2,34 (t, 2Н, J=7 Гц); 1,52 (t на q, 2Н, J=8,7 Гц); 0,77 (t, 3Н, J=7 Гц).
ИК-спектр (для неразбавл. ): 3300 уш. 3061, 1027, 1006, 909, 732, 699 см-1.
Анализ, вычисленный для формулы C41H38N6O2•H2O: C 74,07; H 6,06; N 12,64.
Найдено: C 74,06; H 5,95; N 11,86.
Часть F. Получение 5-гидрокси метил-2-н-про пил-1-[(2'-(N-трифенилметил(1Н-тетра зол-5-ил))бифенил-4-ил)метил] имида зол-4-карбоксальдегида и 2-н-про пил-1-[(2'-(N-трифенилметил(1Н-тетра зол-5-ил))бифенил-4-ил)метил] имида зол-4,5-дикарбоксальдегида.
4,5-Дигидроксиметил-2-н-про пил-1-[2'-(N-трифенилметил-(1Н-тетра зол-5-ил))бифенил-4-ил)метил] имидазола в количестве 8,56 г (13,2 мМ, 1 экв) растворяли в минимальном количестве тетрагидрофурана и добавляли в смесь, содержащую 11,14 г диоксида марганца (128,1 мМ, 9,7 экв) в 100 мл тетрагидрофурана и имеющую комнатную температуру. Спустя 24 ч содержимое реакционного сосуда фильтровали через цеолитовый фильтр марки CeliteTM. Остаток на фильтре промывали тетрагидрофураном, а растворитель из фильтрата удаляли в вакууме. Остаток подвергали тонкослойной хроматографии в смеси гексана с этилацетатом при их соотношении 1:1 до 100% этилацетата на силикагеле до получения диальдегида, вымываемого первым, а также 1,25 г дубильного стекловидного вещества (выход 15%).
Характеристики ЯМР (диметилсульфоксид-d6): d 10,27 (s, 1Н); 10,17 (s, 1Н); 7,81 (d, 1Н, J=7 Гц); 7,68 (m, 2Н); 7,50-7,23 (m, 10Н); 7,09 (d, 2Н, J= 9 Гц); 6,96 (d, 2Н, J=9 Гц); 6,86 (m, 6Н); 5,59 (s, 2Н); 2,52 (t, 2Н, J=7 Гц); 1,58 (t на q, 2Н, J=7,7 Гц); 0,77 (t, 3Н, J=7 Гц).
ИК-спектр (для неразбавл.): 1697, 1672 см-1.
Анализ для формулы C41H34N6O2: C 76,62; H 5,33; N 13,07.
Найдено: C 76,46; H 5,54; N 12,94.
Продуктом последующего вымывания был 4-гидроксиметилимида зол-5-карбоксальдегид, представляющий собой светло-желтое твердое вещество с температурой плавления 164,5-166,0oC.
Характеристики ЯМР (диметилсульфоксид-d6): d 9,86 (s, 1Н); 7,80 (d, 1Н, J= 9 Гц); 7,63 (t, 1Н, J=9 Гц); 7,53 (t, 1Н, J=7 Гц); 7,50-7,25 (m, 10Н); 7,07 (d, 2Н, J=9 Гц); 6,97-6,80 (m, 8Н); 5,47 (t, 1Н, J=7 Гц); 5,29 (s, 2Н); 4,63 (d, 2Н, J= 7 Гц); 2,37 (t, 2Н, J=7 Гц); 1,49 (t на q, 2Н, J=7,7 Гц); 0,73 (t, 3Н, J=7 Гц).
ИК-спектр (Nujol): 1668 см-1.
Анализ для формулы C41H36N6O2•(H2O)0,1: C 76,16; H 5,64; N 12,84.
Найдено: C 76,02; H 5,36; N 12,84.
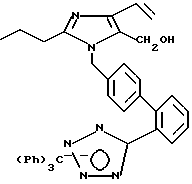
Часть G. Получение 5-гидрокси метил-2-н-пропил-1-[(2'-(N-трифенил метил-(1Н-тетразол-5-ил))би фенил-4-ил)метил]-4-винилимидазола.
2,5М Раствор н-бутиллития в тетрагидрофуране, взятый в количестве 1,70 мл (4,3 мМ, 2,1 экв), добавляли по каплям к суспензии бромида метилтрифенилфосфония, взятого к количестве 1,53 г (4,3 мМ, 2,1 экв), в 50 мл тетрагидрофурана при 0oC в газовой среде азота.
Суспензия превращалась в темно-желтый раствор, к которому далее добавляли 1,31 г 5-гидрокси метил-2-н-пропил-1-[(2-(N-трифенил метил-(1Н-тетразол-5-ил))би фенил-4-ил)метил] имида зол-4-карбоксальдегида (2 мМ, 1 экв), растворенного в минимальном количестве тетрагидрофурана. Полученный мутный светло-желтый раствор перемешивали в течение ночи при комнатной температуре. Далее этот раствор разбавляли этилацетатом и промывали тремя порциями воды. Органический слой обезвоживали при помощи сульфата магния, растворитель удаляли в вакууме, а остаток подвергали тонкослойной хроматографии над силикагелем в смеси гексана с этилацетатом при соотношении этих компонентов 1:1 до получения 620 мг (выход 48%) белого стекловидного вещества.
Характеристики ЯМР (диметилсульфоксид-d6): δ 7,79 (d, 1Н, J=7 Гц); 7,62 (t, 1Н, J=7 Гц); 7,55 (t, 1Н, J=7 Гц); 7,45 (d, 1Н, J=7 Гц); 7,41-7,18 (m, 9Н); 7,06 (d, 2Н, J=9 Гц); 6,95-6,80 (m, 8Н); 6,80-6,55 (m, 1Н); 5,73 (d из d, 1Н, J=17,3 Гц); 5,17 (s, 2Н); 5,10 (t, 2Н, J=7 Гц); 5,05 (d из d, 1Н, J= 12,3 Гц); 4,28 (d, 2Н, J=7 Гц); 2,37 (t, 2Н, J=7 Гц); 1,50 (t на q, 2Н, J= 7,7 Гц); 0,78 (t, 3Н, J=7 Гц).
ИК-спектр (для неразбавл.): 1029, 1006, 909, 733, 698 см-1.
Анализ для формулы C42H38N6O•H2O: C 76,34; H 6,10; N 12,72.
Найдено: C 76,49; H 5,88; N 12,52.
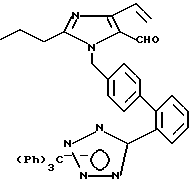
Часть H. Получение 2-н-про пил-1-[(2-(N-трифенилметил-(1Н-тетра зол-5-ил))бифенил-4-ил)ме тил]-4-винилимидазол-5-карбоксальдегида.
Готовили смесь 470 мг 5-гидроксиметил-2-н-про пил-1-[(2-(N-трифенилметил(1Н-тетра зол-5-ил))бифенил-4-ил)ме тил] -4-винилимидазола (0,73 мМ, 1 экв), 341 мг периодата Na (Dess-Martin) (0,80 мМ, 1,1 экв), полученного, как указано в J. Org. Chem. 1983, v.48, p.4155, и 10 мл метиленхлорида, которую перемешивали в течение ночи в среде азота. Растворитель удаляли в вакууме, а остаток подвергали тонкослойной хроматографии в смеси гексана с этилацетатом при соотношении этих компонентов 3:2 над силикагелем до получения в качестве продукта 310 мг (выход 66%) белого стекловидного вещества.
Характеристики ЯМР (диметилсульфоксид-d6): δ 9,91 (s, 1Н); 7,80 (d, 1Н, J= 7 Гц); 7,61 (t, 1Н, J=7 Гц); 7,54 (t, 1Н, J=7 Гц); 7,48-7,22 (m, 10Н); 7,20 (d, 1Н, J=9 Гц); 7,06 (d, 2Н, J=9 Гц); 7,00-6,75 (m, 8Н); 6,15 (d на d, 1Н, J=17,3 Гц); 5,52 (s, 2Н), 5,47 (d на d, 1Н, J=12,3 Гц); 2,49 (t, 2Н, J=7 Гц); 1,57 (t на q, 2Н, J=7,7 Гц); 0,79 (t, 3Н, J=7 Гц).
ИК-спектр (для неразбавл.): 1658 см-1.
Анализ, вычисленный для формулы C42H36N6O•(H2O)0,50: C 77,63; H 5,74; N 12,93.
Найдено: C 77,53; H 5,73; N 12,64.
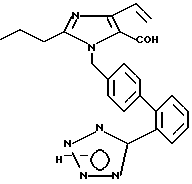
Часть I. Получение 2-н-про пил-1-[2'-(1Н-тетразол-5-ил))би фенил-4-ил)метил]-4-винилимида зол-5-карбоксальдегида.
Готовили смесь 330 мг 2-н-про пил-1-[(2'-(N-трифенил метил-(1Н-тетразол-5-ил))би фенил-4-ил)метил]-4-винилимида зол-5-карбоксальдегида, 1,65 мл трифторуксусной кислоты, 1,65 мл воды и 1,65 мл тетрагидрофурана, которую перемешивали при комнатной температуре. Спустя 8 ч, эту смесь нейтрализовали до pH 7 10н. раствором NaOH, после чего растворители удаляли под вакуумом. Остаток подвергали тонкослойной хроматографии в смеси гексана с этилацетатом при соотношении этих компонентов 1:1 до 100% этилового спирта до получения 270 мг продукта в виде белого стекловидного вещества.
Характеристики ЯМР (диметилсульфоксид-d6): δ 9,92 (s, 1Н); 7,65-7,50 (m, 1Н); 7,50-7,12 (m, 3Н); 7,09 (d, 2Н, J=9 Гц); 6,89 (d, 2Н, J=9 Гц); 6,11 (d на d, 1Н, J=17,3 Гц); 5,55 (s, 2Н); 5,45 (d на d, 1Н, J=12,3 Гц); 2,63 (t, 2Н, J=7 Гц); 1,64 (t на q, 2Н, J=7,7 Гц); 0,90 (t, 3Н, J=7 Гц).
ИК-спектр (Nujol): 1680 см-1.
Пример 2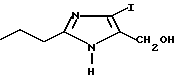
Часть A. Получение 5-гидрокси метил-4-йод-2-н-пропилимидазола.
При 45oC в течение 2 ч перемешивали раствор 31,5 г 4(5)-гидрокси метил-2-н-пропилимидазола и 50,06 г N-йодосукцинимида в смеси 560 мл 1,4-диоксана и 480 мл 2-метоксиэтанола. Затем растворители удаляли под вакуумом. Образовавшиеся твердые вещества промывали дистиллированной водой, а затем высушивали для получения 54,6 г продукта в виде желтого твердого вещества с температурой плавления 169-170oC.
Характеристики ЯМР (диметилсульфоксид-d6): δ 12,06 (уш.s, 1Н); 5,08 (t, 1Н); 4,27 (d, 2Н); 2,50 (t, 2Н); 1,59 (двойн.d, 2Н); 0,84 (t, 3Н).
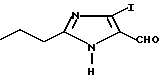
Часть B. Получение 4-йод-2-н-пропилимида зол-5-карбоксальдегида.
К раствору 35,8 г 5-гидрокси метил-4-йод-2-н-пропилимидазола в 325 мл ледяной уксусной кислоты добавляли по каплям при 20oC в течение 1 ч 290 мл 1н водного раствора церийаммонийнитрата. Полученную смесь перемешивали при 20oC в течение 1 ч. Далее прореагировавшую смесь разбавляли водой, доводили до величины pH 5-6 водным раствором гидроксида натрия и экстрагировали хлороформом. Объединенные органические фазы промывали водой и рассолом, высушивали над безводным сульфатом натрия, фильтровали и концентрировали. Получившийся сырой твердый продукт подвергали перекристаллизации из 1-хлорбутана до получения 29,9 г продукта в виде желтого твердого вещества с температурой плавления 141-142oC.
Характеристики ЯМР (CDCl3): δ 11,51 (уш.s, 1Н); 9,43 (s, 1Н); 2,81 (t, 2Н); 1,81 (двойн. 2Н); 0,97 (t, 3Н).
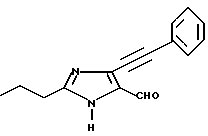
Часть C. Получение 3-н-про пил-4-(фенилэтинил)имида зол-5-карбоксальдегида.
В среде азота нагревали до 70oC раствор, содержащий 2,64 г 4-йод-2-н-пропилимида зол-5-карбоксальдегида (0,01М), 25 мл обезвоженного диметилформамида, 2,5 мл триэтиламина, 1 г (0,001426М) хлорида бис(трифенилфосфин)палладия и 5 г (фенилэтинил)трибутилолова (0,017М). Реакционную смесь перемешивали в течение 120 ч, после чего охлаждали. Осадок отфильтровывали и промывали метиленхлоридом, а фильтрат выпаривали при пониженном давлении. Остаток растворяли в 200 мл метиленхлорида и экстрагировали тремя порциями 10% HCl по 100 мл. Величину pH водного слоя регулировали до 10 50%-ным раствором гидроксида натрия, после чего этот слой экстрагировали тремя порциями метиленхлорида по 100 мл. Органическую фазу высушивали над сульфатом натрия и выпаривали при пониженном давлении. В результате получали 0,56 г 3-н-про пил-4-(фенилэтинил)имида зол-5-карбоксальдегида (0,0023М, выход 23%).
Характеристики ЯМР (CDCl3): δ 9,89 (s, 1Н); 8,22 (s, 1Н), 7,93 (m, 3Н); 7,53 (m, 2Н), 2,87 (t, 2Н); 1,87 (m, 2Н); 1,03 (t, 3Н).
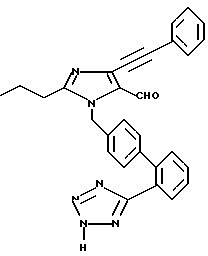
Часть D. Получение 4-фенил этинил-3-н-пропил-1-[(2'-(1Н-тетра зол-5-ил)бифенил-4-ил)метил]имида зол-5-карбоксальдегида.
3-н-пропил-4-(фенилэтинил)имида зол-5-карбоксальдегид алкилировали 4-бромметил-2'-[N-трифенил метил-(1Н-тетразол-5-ил)] бифенилом по методике, описанной в примере 1, части D и I, с целью получения продукта, название которого приведено выше.
Характеристики ЯМР (CDCl3): δ 9,88 (s, 1Н); 8,03 (m, 1Н); 7,57-7,27 (m, 8Н); 7,17 (m, 2Н); 7,01 (m, 2Н); 5,55 (s, 2Н); 2,61 (t, 2Н); 1,75 (m, 2Н); 0,99 (t, 3Н).
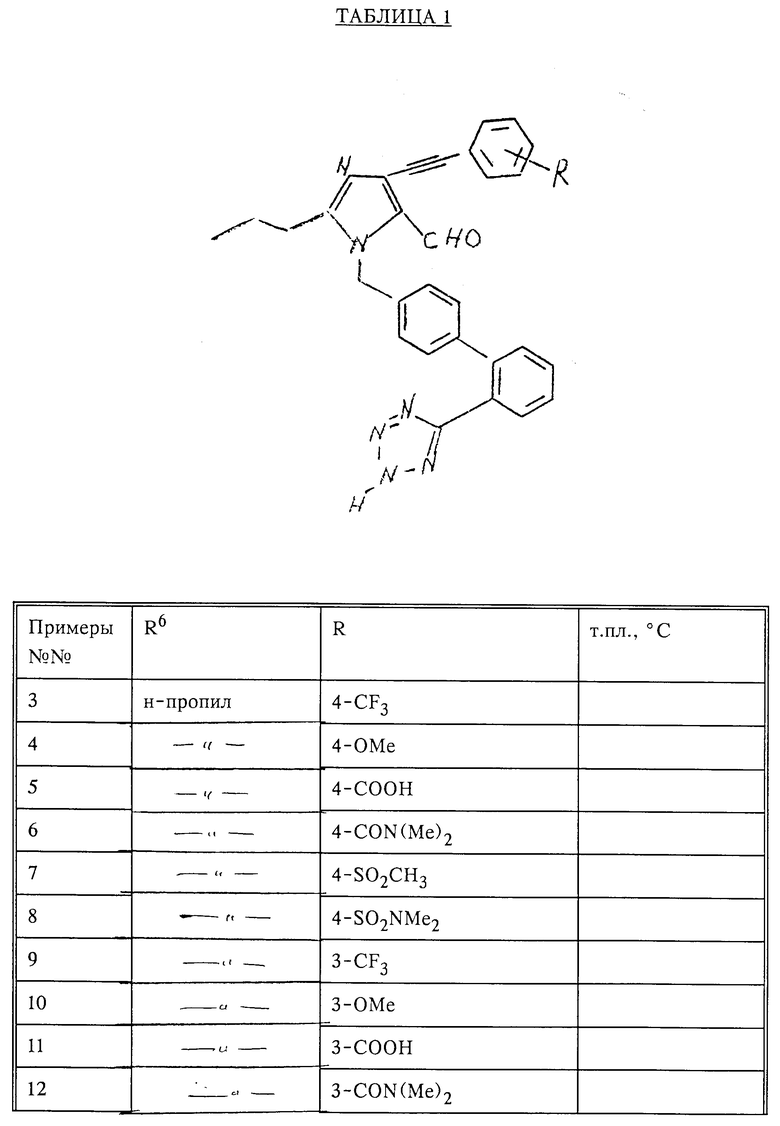
В табл. 1 приведены соединения общей формулы, указанной ниже, которые могут быть получены по методике примера 2.
Пример 27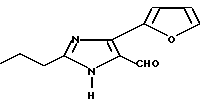
Часть A. Получение 4-(фу ран-2-ил)-2-н-пропилимида зол-5-карбоксальдегида.
В среде азота при комнатной температуре перемешивали раствор, содержащий 2,64 г 4-йод-3-н-пропилимида зол-5-карбоксальдегида (0,01М), 60 мл толуола и 0,33 г (0,00029М) тетракистрифенилфосфина палладия (O), в который постепенно вводили раствор 2,34 г фуран-2-илборной кислоты (0,0174М) в 50 мл этилового спирта. Реакция продолжалась в условиях перемешивания в течение 5 мин, после чего постепенно добавляли 12 мл 2М раствора карбоната натрия. По окончании введения этого раствора реакционную смесь подвергали отгонке растворителя в течение 8 ч, а затем охлаждали. Продукты реакции фильтровали, фильтрат выпаривали при пониженном давлении, а образовавшийся остаток растворяли в 300 мл метиленхлорида, промывали двумя порциями насыщенного раствора хлорида натрия по 100 мл, затем двумя порциями дистиллированной воды по 1200 мл и, наконец, двумя порциями 10% HCl по 300 мл. Солянокислый слой переводили в щелочной (pH 10) 50% раствором гидроксида натрия. Полученный основный водный слой экстрагировали тремя порциями метиленхлорида по 300 мл. Метиленхлоридный слой обезвоживали над сульфатом натрия и выпаривали при пониженном давлении. Выход 5-карбоксальдегида составил 0,34 г (0,00156М).
Характеристики ЯМР (CDCl3): δ 10,14 (s, 1Н); 7,54 (s, 1Н); 7,00 (d, 1Н); 6,55 (m, 1Н); 2,79 (t, 2Н); 1,80 (m, 2Н); 1,02 (t, 3Н).
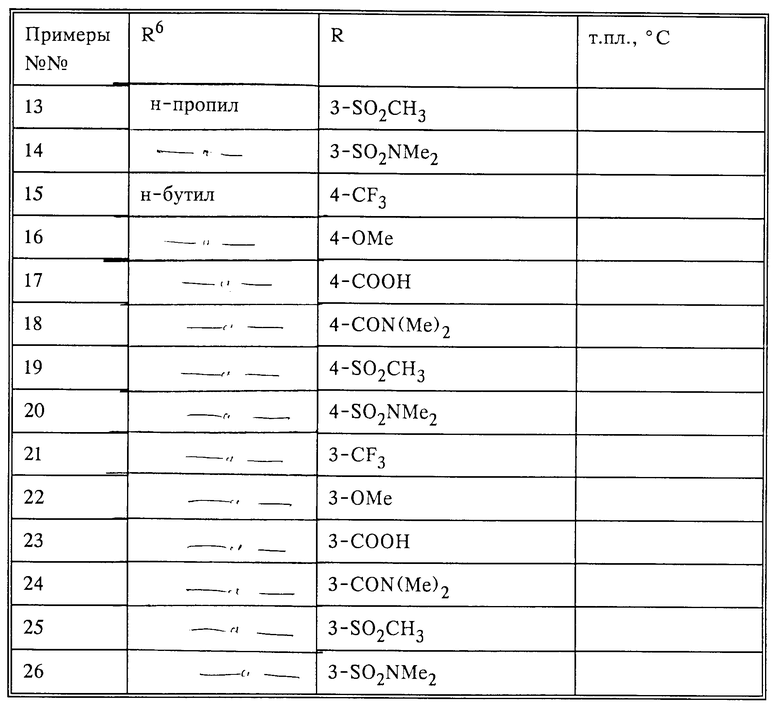
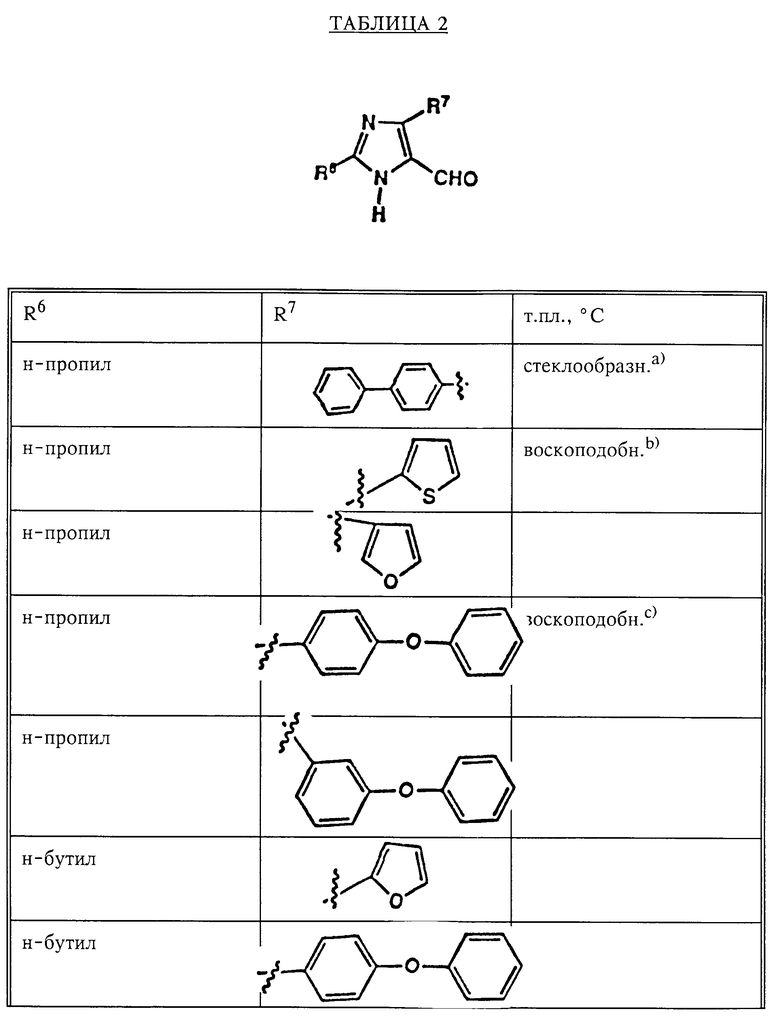
По методикам, описанным в примере 27, часть A, были получены (или могут быть получены) следующие соединения (см. табл.2).
Часть B. Получение 4-(фу ран-2-ил)-2-н-пропил-1-[2'-(1Н-тетра зол-5-ил)бифенил-4-ил)метил]имида зол-5-карбоксальдегида.
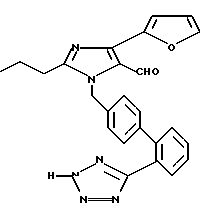
4-(Фуран-2-ил)-2-н-пропилимида зол-5-карбоксальдегид был превращен в продукт, указанный в заголовке, с использованием методик, описанных в примере 1, части D и I. Температура плавления этого продукта 129oC (разл.).
Характеристики ЯМР (CDCl3): δ 10,15 (s, 1Н); 7,95 (d, 1Н); 7,55 (m, 2Н); 7,38 (m, 2Н); 7,10 (d, 2Н); 6,98 (d, 2Н); 6,85 (d, 1Н), 6,45 (m, 1Н); 5,55 (s, 2Н); 2,55 (t, 2Н); 1,70 (m, 2Н); 0,91 (t, 3Н).
Соединения, указанные в примерах табл.3, можно получить по методикам, описанным в примере 27, с использованием подходящих исходных веществ.
Соединения, указанные в табл.4, можно синтезировать по методикам, описанным в примерах 1, 2 или 27, с использованием бифенильных исходных веществ, описанных в настоящей заявке, или другими методами, хорошо известными специалисту в данной области.
Пример 41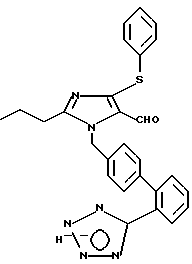
Получение 2-н-бутил-4-фенил тио-1-[(2'-(1Н-тетразол-5-ил)би фенил-4-ил)метил]имида зол-5-карбоксальдегида.
К свежеприготовленному раствору метоксида натрия в метиловом спирте, содержащему 205 мг натрия (8,9М, 10 экв) и 40 мл метилового спирта, добавили 590 мг 2-н-бу тил-4-хлор-1-[(2'-н-трифенил метил-(1Н-тетразол-5- ил)бифенил-4-ил)метил] -имидазол-5-карбоксальдегида (0,89 мМ, 1 экв), полученного, как это описано в Европейской патентной заявке 89100144.8, опубликованной 7.19.89, и 0,91 мл тиофенола (8,9 мМ, 10 экв). Из полученной смеси отгоняли растворитель в течение ночи в среде азота. Далее растворители удаляли в вакууме, а остаток растворяли в 50 мл воды. Величину pH регулировали до 10-12 10н раствором NaOH. Образующееся каучукоподобное твердое вещество, представляющее собой соединение, содержащее защитную группу тротила, растворяли добавлением 50 мл этилового эфира. Затем проводили разделение слоев и экстрагировали водный слой двумя порциями этилового эфира по 50 мл. Далее водный слой экстрагировали шестью порциями этилацетата по 50 мл. Этилацетатные слои собирали вместе, обезвоживали при помощи сульфата магния, а растворитель удаляли в вакууме. В результате получали остаток, который вновь переводили в раствор добавлением 50 мл воды. Величину pH регулировали до 1 концентрированной соляной кислотой. При этом выпадал каучукоподобный осадок, содержащий искомый продукт, который растворяли в 50 мл этилацетата. Далее проводили разделение слоев и водный слой экстрагировали двумя порциями этилацетата по 50 мл. Этилацетатные слои собирали вместе, обезвоживали при помощи сульфата магния, а растворитель удаляли в вакууме. В результате получали 200 мг белого стекловидного вещества. Кристаллизация из горячего н-бутилхлорида дала 142 мг белого твердого вещества с температурой плавления 143,5-145,5oC.
Характеристики ЯМР (диметилсульфоксид-d6): δ 9,82 (s, 1Н); 7,80-7,61 (m, 2Н); 7,58 (d, 1Н, J=8 Гц); 7,52 (d, 1Н, J=8 Гц); 7,45-7,20 (m, 5Н); 7,09 (d, 1Н, J=8 Гц); 7,03 (d, 2Н, J=8 Гц); 5,62 (s, 2Н); 2,64 (t, 2Н, J=7 Гц); 1,50 (t на t, 1Н, J=7,7 Гц); 1,25 (t на q, 2Н, J=7,7 Гц); 0,80 (t, 3Н, J=7 Гц).
Анализ, вычисленный для формулы C28N26N6OS•(H2О)0,4: C 67,02; H 5,38; N 16,75; S 6,39.
Найдено: C 66,90; H 5,20; N 16,75; S 6,00.
Соединения примеров 42-49 в табл.5 можно синтезировать с использованием методик, описанных в примере 41 и других примерах настоящей заявки, а также в Европейском патенте 89100144.8, опубликованном 19.07.89, или иных методов, хорошо известных специалисту в данной области.
Пример 50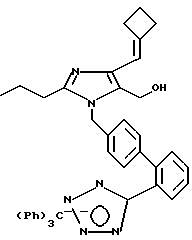
Часть A. Получение 2-н-про пил-4-циклобутилиденил-5-гидрокси метил-1-[(2'-(N-трифенил метил-1Н-тетразол-5-ил)би фенил-4-ил)метил]имидазола.
Готовили суспензию 7,42 г бромида (α-бром-н-бутил)трифенилфосфония (0,0155 мМ, 2 экв) в 125 мл тетрагидрофурана. К суспензии при комнатной температуре добавляли 41,4 мл 0,75М раствора гексаметилдисилазана калия (0,031 мМ, 4 экв). Происходило окрашивание смеси в кроваво-красный цвет. Спустя 0,5 ч, добавляли 5 г 2-н-пропил-5-гидрокси метил-1-[(2'-(N-трифенил метил-(1Н-тетразол-5-ил))би фенил-4-илметил] имида зол-4-карбоксальдегида (7,75 мМ, 1 экв) в виде смеси суспензии в тетрагидрофуране. С течением времени суспензия становилась желто-оранжевой. Через 24 ч реакцию завершали добавлением небольшого количества метилового спирта с целью быстрого охлаждения, после чего добавляли этилацетат и воду. Затем осуществляли разделение слоев и промывали органический слой сначала двумя порциями воды, а затем одной порцией рассола. Далее органический слой обезвоживали при помощи сульфата магния и растворитель удаляли в вакууме, а остаток подвергали тонкослойной хроматографии в смеси пентана и этилацетата (при соотношении этих компонентов 60:40) до 100% этилацетата. В результате получили 4,12 г (выход 78%) белого вещества с температурой плавления 181,5-182,5oC.
Характеристики ЯМР (диметилсульфоксид-d6): d 7,78 (m, 1Н); 7,61 (t, 1Н, J= 7 Гц); 7,54 (t, 1Н, J=7 Гц); 7,48-7,20 (m, 10Н); 7,03 (d, 2Н, J=7 Гц); 6,96-6,70 (m, 8Н); 5,99 (s, 1Н); 5,13 (s, 2Н); 4,97 (t, 1Н, J=7 Гц); 4,21 (d, 2Н, J=7 Гц); 3,05 (m, 2Н); 2,79 (m, 2Н); 2,31 (t, 2Н, J=7 Гц); 1,98 (m, 2Н); 1,48 (t на q, 2Н, J=7,7 Гц); 0,77 (t, 3Н, J=7 Гц).
Анализ для формулы C45H42N6O•(H2O)0,75: для C.H.N.
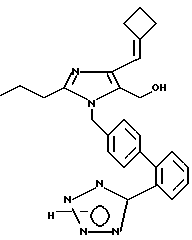
Часть B. Получение 2-н-про пил-4-циклобутилиденил-5-гидрокси метил-1-[2-(1Н-тетразол-5-ил)би фенил-4-ил)метил]имидазола.
Готовили смесь 1 г 2-н-про пил-4-циклобутилиденил-5-гидрокси метил-1-[2'-(N-трифенил метил-(1Н-тетразол-5-ил))би фенил-4-ил)метил]-имидазола, 25 мл метилового спирта и 15 мл тетрагидрофурана, которую перемешивали и кипятили в течение 24 ч. Растворители удаляли в вакууме, а остаток сразу же подвергали тонкослойной хроматографии в смеси пентана с этилацетатом (при соотношении этих компонентов 1:1) до 100% изопропилового спирта и постепенно до 100% этилового спирта до получения 320 мг светло-желтого стекловидного вещества.
Характеристики ЯМР (диметилсульфоксид-d6): δ 7,59 (d, 1Н, J=7 Гц); 7,54 (t, 1Н, J=7 Гц); 7,46 (t, 1Н, J=7 Гц); 7,42 (d, 1Н, J=7 Гц); 7,06 (d, 2Н, J= 7 Гц); 6,90 (d, 2Н, J=7 Гц); 5,97 (s, 1Н); 5,17 (s, 2Н); 4,31 (s, 2Н); 3,04 (m, 2Н); 2,77 (m, 2Н), 2,42 (t, 2Н, J=7 Гц); 1,97 (m, 2Н); 1,53 (t на q, 2Н, J=7,7 Гц); 0,86 (t, 3Н, J=7 Гц).
Анализ для формулы C26H28N6O: на C, H, N.
Полезность изобретения.
Гормон ангиотензин II (AII) вызывает различные биологические реакции, например сжатие сосудов, благодаря стимулированию имеющихся на клеточных мембранах рецепторов этого гормона. С целью идентификации соединений, как антагонистов AII, способных взаимодействовать с рецептором AII, первоначально использовали лиганд-рецепторное связывание по методике, которую описали Глоссман и сотр. (Glossman et al. J. Biol. Chem. 1974, v.249, p.825), с некоторыми изменениями. Реакционная смесь содержала надпочечные корковые микросомы (источник рецепторов AII) в Tris-буферном растворе, содержащем 2 нМ 3H-AII в присутствии потенциального антагониста AII или без него. Смесь подвергали созреванию в течение 1 ч при комнатной температуре, причем сразу же по окончании реакции осуществляли ускоренное фильтрование и промывку через стекломикроволокнистый фильтр. Гормон 3H-AII, связанный с рецептором, уловленный на фильтре, количественно определяли подсчетом сцинтилляций (вспышек). Ингибирующая концентрация IC50 потенциального антагониста к гормону AII, характеризующая 50%-ное замещение всех специфически связанных группировок 3H-AII, является мерой сродства указанных соединений по отношению к рецептору AII. Соединения, отвечающие настоящему изобретению, испытанные методом связывания, показали значения IC50 порядка 10-5М или менее (см. табл.6).
Потенциальное антигипертензивное действие соединений, отвечающих настоящему изобретению, можно продемонстрировать путем назначения данных соединений бодрствующим крысам, подверженным гипертонии, посредством перевязывания у них левой почечной артерии, см. работу Конджано и сотр. (Cangiano et al. J. Pharmacol. Exp. Ther. 1979, v.208, p.310). Такая операция повышает давление крови вследствие увеличения выработки репина, сопровождающейся повышением уровней содержания гормона AII. Соединения назначали стоматически при дозах 30 мг/кг или вводили через трубочку, вставленную в яремную вену, при дозировке 3 мг/кг. При этом непрерывно проводили прямое измерение артериального давления крови при помощи трубочки, вставленной в сонную артерию, причем для записи данных применяли датчик давления и многопозиционный самописец. Уровни давления крови до эксперимента сравнивали с соответствующими уровнями до обработки для установления противогипертонического действия испытанных соединений. Некоторые соединения, отвечающие настоящему изобретению, показали внутривенную активность при дозе 3 мг/кг, а некоторые -активность при назначении стоматически при дозе 30 мг/кг (табл. 6).
Формы применения.
Соединения, отвечающие настоящему изобретению, могут быть назначены для лечения гипертонии в соответствии с настоящим изобретением любым путем, если обеспечивается контактирование активного ингредиента с зоной воздействия в организме теплокровного животного. Например, назначение может быть реализовано путем инъекций, а именно подкожно, внутривенно, внутримышечно или введением в брюшную полость. В порядке дополнения или исключения прием препарата можно назначать также стоматически (через полость рта).
Прием препаратов можно назначать любым общепринятым методом, пригодным для применения в сочетании с другими лекарственными препаратами, представляющими собой как самостоятельные лекарства, так и сочетания лекарств. Препараты можно назначать в чистом виде, но, как правило, вместе с фармацевтическим носителем, выбранным на основе того или иного метода назначения в соответствии с обычной фармацевтической практикой.
Что касается объекта применения всего сказанного выше, то таковым является теплокровное животное, относящееся к тем представителям животного мира, которые обладают гомеостатическим механизмом; сюда относятся млекопитающие и птицы.
Назначаемая дозировка будет зависеть от возраста, состояния здоровья и массы излечиваемого животного с учетом частоты назначения природы желательного воздействия. Обычно суточная доза активного ингредиента составляет 1-500 мг/сут. Как правило, для достижения желательных результатов достаточно 10-100 мг/сут (один прием или несколько раз в сутки). Эти дозировки эффективны как для лечения гипертонии, так и при лечении сердечной недостаточности, связанной с закупоркой сосудов, а именно для снижения кровяного давления, а также для корректирования нагрузки на сердце путем раскупоривания сосудов.
Активный ингредиент можно назначать стоматически в виде твердых дозировок, таких как капсулы, таблетки или порошки, или в виде жидких дозировок, таких как эликсиры, микстуры и суспензии. Этот ингредиент можно также вводить и путем инъекций в виде стерильных жидких дозировок.
Желатиновые капсулы содержат активный ингредиент и порошковые носители, такие как лактоза, крахмал, производные целлюлозы, стеарат магния, стеариновая кислота и т.п. Аналогичные наполнители можно использовать и при изготовлении спрессованных таблеток. Таблетки и капсулы можно изготовить в виде медленно действующих форм, чтобы обеспечить непрерывное поступление в кровь лекарства в течение времени, измеряемого в часах. Прессованные таблетки могут быть покрыты слоем сахара или пленкой, чтобы замаскировать неприятный вкус и защитить таблетку от воздействия атмосферных факторов. Или же таблетки могут иметь внутренние пленочные перегородки, обеспечивающие избирательное разделение таблетки на части в пищеварительном тракте.
Жидкие дозировки для назначения стоматически могут содержать окрашивающие или ароматизирующие добавки с целью большей привлекательности для излечиваемого животного.
Для приготовления инъекционных и тому подобных растворов, вводимых "извне", пригодны такие носители, как вода, подходящее масло, рассол, водорастворимая декстроза (глюкоза) и соответствующие растворы сахаров, а также гликоли, такие как пропиленгликоль или полиэтиленгликоль. Растворы для назначения путем инъекции предпочтительно содержат водорастворимую соль активного ингредиента, подходящие стабилизирующие агенты, а в случае необходимости буферные вещества. Подходящими стабилизирующими агентами являются противоокислительные агенты, такие как бисульфит или сульфит натрия, или же аскорбиновая кислота, взятые в отдельности или в сочетаниях. Кроме того, применяют лимонную кислоту и ее соли, а также этилендиаминтетраацетат натрия. Растворы для введения путем инъекции могут содержать сохраняющие добавки, такие как хлорид бензалкония, метил- или пропилпарабен, хлоробутанол.
Подходящие фармацевтические носители описаны в "Ремингтоновском фармацевтическом справочнике" под ред. Осола ("Remington's Pharmaceutical Sciences", ed. Osol A.), являющемся общепринятым руководством в данной области.
Используемые фармацевтические дозировки для назначения соединений, отвечающих настоящему изобретению, приведены ниже.
Капсулы.
Большое число капсульных упаковок готовят путем наполнения стандартных двухчастных капсул из твердой желатины, помещая в каждую часть по 100 мг порошкового активного ингредиента, 150 мг лактозы, 50 мг целлюлозы и 6 мг стеарата магния.
Капсулы из мягкой желатины.
Готовили смесь активного ингредиента с легко усваиваемым маслом, таким как соевое, хлопковое или оливковое, которую при помощи специального приспособления инжектировали в желатину, получая мягкие желатиновые капсулы, содержащие по 100 мл активного ингредиента. Эти капсулы мыли и высушивали.
Таблетки.
Множество видов таблеток готовят по обычной технологии, когда дозировка состоит из 100 мг активного ингредиента, 0,2 мг коллоидного диоксида кремния, 5 мг стеарата магния, 275 мг микрокристаллической целлюлозы, 11 мг крахмала и 98,8 мг лактозы. Для улучшения вкусовых качеств и облегчения усвоения можно использовать подходящие покрытия таблеток.
Жидкости для инъекций.
Состав для назначения путем инъекций готовят перемешиванием 1,5% (мас. доли) активного ингредиента в 10% (об. доли) в водном растворе пропиленгликоля. При доведении объема до метки пользуются специальной водой для инъекций. Готовый раствор стерилизуют.
Суспензии.
Для назначения стоматически (через полость рта) готовят такую водную суспензию, в каждых 5 мл которой содержится 100 мг тонко диспергированного активного ингредиента, 100 мг натриевой соли карбоксиметилцеллюлозы, 5 мг бензоата натрия, 1 г сорбитолового раствора, U.S.P. а также 0,025 мл ванилина.
Те же виды дозировок, что перечислены выше, можно использовать при назначении соединений, отвечающих настоящему изобретению, на каких-либо этапах лечения в сочетании с другими лекарственными препаратами. Если два лекарства назначаются в их физическом сочетании, то вид дозировки и способ назначения необходимо выбрать с учетом совместимости обоих лекарств. Подходящие дозировки, разновидности дозировочных форм и способы назначения указаны в табл.7 и 8.
При использовании вместе с препаратом НСПВП дозы блокеров AII обычно те же, что и при самостоятельном применении блокера гормона AII: 1-500 мг/сут, обычно 10-100 мг/сут в один или несколько приемов. При использовании совместно с диуретиками начальная доза блокера AII может быть меньше: 1-100 мг/сут, а в случае более активных соединений она составляет 1-10 мг/сут.
Возможно, что соединения, отвечающие настоящему изобретению, будут также полезны и при лечении хронической почечной недостаточности.
| название | год | авторы | номер документа |
|---|---|---|---|
| АРАЛКИЛЗАМЕЩЕННЫЕ ИМИДАЗОЛЫ, ОБЛАДАЮЩИЕ ПРОТИВОГИПЕРТОНИЧЕСКИМ ДЕЙСТВИЕМ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ, ОБЛАДАЮЩИЙ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ АНГИОТЕНЗИНА II | 1990 |
|
RU2067581C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА | 1992 |
|
RU2017733C1 |
| ПРОИЗВОДНЫЕ ТЕТРАЗОЛА | 1992 |
|
RU2091376C1 |
| ЗАМЕЩЕННЫЕ 1,2,3-ТРИАЗОЛЫ | 1992 |
|
RU2076102C1 |
| Способ получения производных имидазола или их фармацевтически приемлемых солей | 1987 |
|
SU1694062A3 |
| ПРОИЗВОДНЫЕ ОКСАЗОЛИДИНОНА, ФУНГИЦИДНАЯ КОМПОЗИЦИЯ И СПОСОБ БОРЬБЫ С ГРИБКОВЫМИ ЗАБОЛЕВАНИЯМИ | 1990 |
|
RU2092051C1 |
| ПРОИЗВОДНЫЕ СЕМИКАРБАЗОНОВ, СПОСОБ УНИЧТОЖЕНИЯ АРТРОПОДОВ, АРТРОПОДИЦИДНАЯ КОМПОЗИЦИЯ | 1989 |
|
RU2067092C1 |
| Способ получения производных имидазола | 1989 |
|
SU1814646A3 |
| ФТОРОАЛКОКСИАМИНОТРИАЗИНЫ | 1992 |
|
RU2047607C1 |
| Способ получения азолов | 1989 |
|
SU1709907A3 |
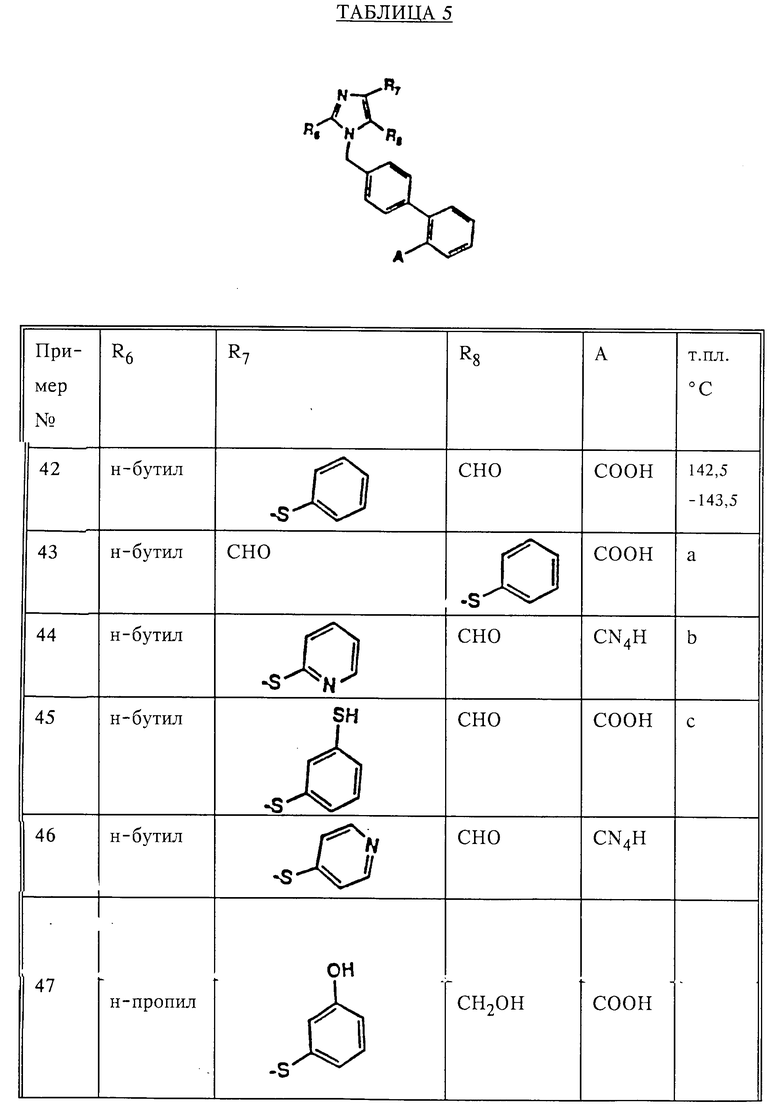
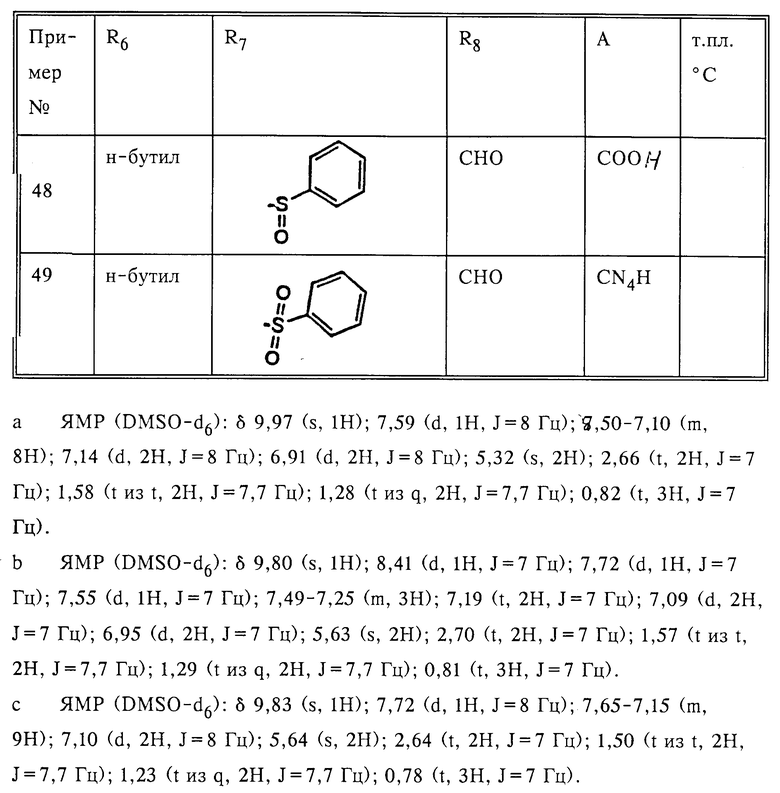



Замещенные амидазолы формулы I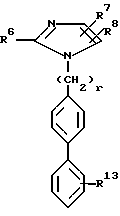
в которой R6 - алкил C2-C6; R7 - винил, низкий циклоалкилиденил; фенилалкинил, в котором алкинильная часть содержит 2-6 атомов углерода; 2- или 3-фурил; бифенилил; феноксифенил; фенилтио; пиридилтио или 3-меркаптофенилтио, метилтио; R8 означает -(CH2)n-CH2OH или -C(O)H; R13 - тегидразолил или -COOH; n = 1-10, r = 1-2; или их фармацевтически приемлемые соли. Соединение I и их указанные соли обладают антигипертензивной активностью. Описана фармацевтическая композиция, содержащая соединение I или его указанные соли. 2 с. и 2 з.п. ф-лы, 8 табл.
где R1 радикал формулы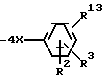
R2 H;
R3 H;
R6 C2- C6-алкил
R7 винил; низший циклоалкилиденил; фенилалкинил, в котором алкинильная часть содержит 2 6 атомов углерода; 2- или 3-фурил; бифенилил; феноксифенил; фенилтио; пиридилтио или 3-меркаптофенилтио; метилтио;
R8 -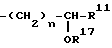
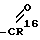
R1 1 H;
R1 3 -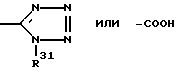
R1 6 H;
R1 7 H;
R3 1 H;
X углерод-углеродная одинарная связь;
n 1 10;
r 1 2,
при условии, что R1 не находится в ортоположении; R1 3 находится в орто- или метаположении,
или их фармацевтически приемлемые соли.
| 0 |
|
SU253310A1 | |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
