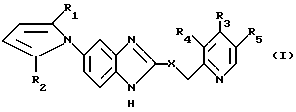
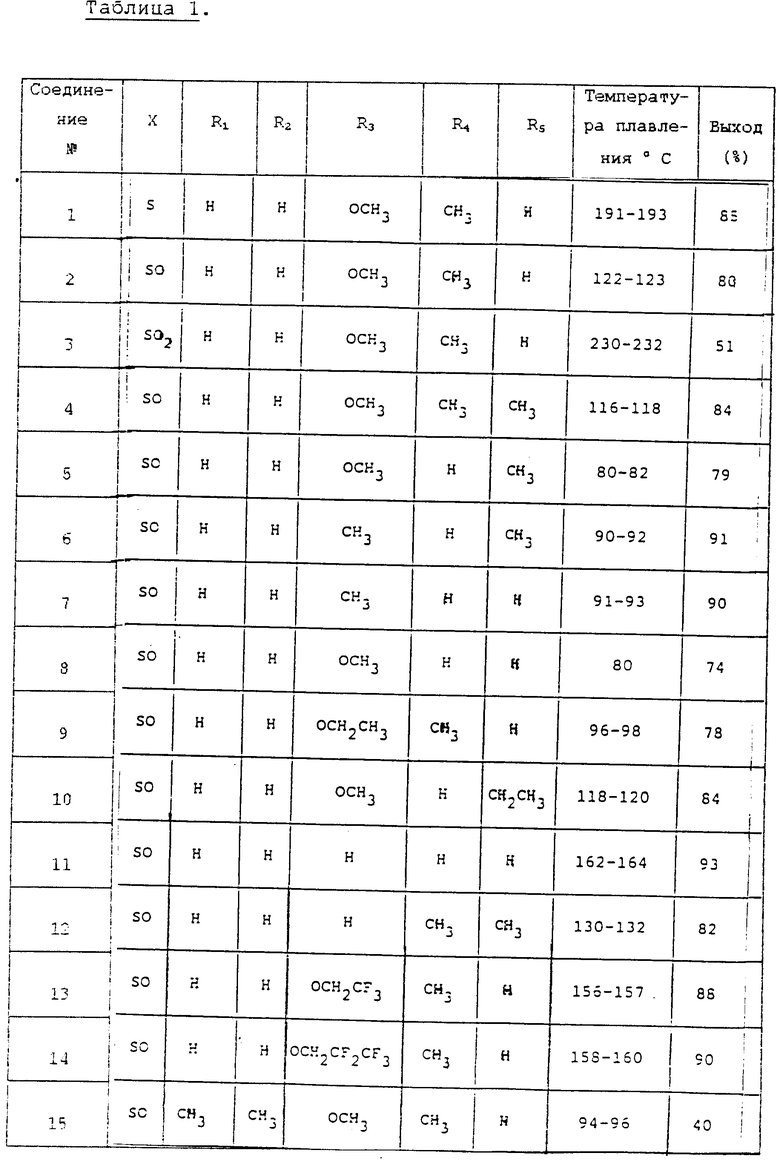
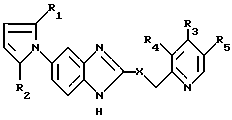
Изобретение относится к новым производным 5-пирролил-2 -пиридилметилсульфинилбензимидазола. Более конкертно, настоящее изобретение относится к новым производным 5-пирролил- 2-пиридилметилсульфинилбензимидазола, представленным следующей общей формулой: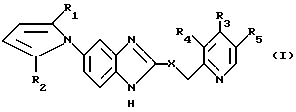
и их солям, в которой: X представляет S, SO или SO2,
R1 и R2, независимо друг от друга представляют водород или C1-C8-алкил,
R3 представляет водород, C1-C8-алкил, -ОR6,
где: R6 представляет C1-C4-алкил, C2-C5-фторалкил;
R4 и R5 независимо друг от друга представляют водород или C1-C5-алкил.
Настоящее изобретение также относится к способу получения соединения формулы (I), которое определено выше, и к применению соединения формулы (I) в качестве средства для профилактики и лечения язв желудка и двенадцатиперстной кишки.
Язвы желудка и двенадцатиперстной кишки представляют желудочно-кишечное заболевание, вызванное различными факторами, например, нервно-психическим напряжением, пищевыми привычками, потреблением раздражающей пищи и т.п. Непосредственная причина пептических язв представляет опасность для желудочной оболочки из-за избыточной секреции желудочной кислоты. Поэтому лечебные средства, которые обычно используют для лечения язв, включают, например, антацидные средства для нейтрализации желудочной кислоты, антипептические средства, средства для защиты слизистой желудочной оболочки, противохолинергические средства для ингибирования желудочной секреции, парасимпатолитические средства, антагонисты H2-рецептора и т.п. В настоящее время, с тех пор как было обнаружено, что антацидные средства и противоязвенные средства, действующие на центральную нервную систему, обеспечивают лишь неудовлетворительное лечебное воздействие и могут вызвать нежелательные действия, когда их принимают в течение длительного времени, применение антагонистов H2-рецептора в качестве средства для лечения язв желудка и двенадцатиперстной кишки возросло.
Кроме того, недавно был разработан и наглядно показан в качестве эффективного противоязвенного средства 5-метокси-2-[(4-метокси-3,5-диметил-2-пиридинил)метил]сульфинил]-1н-бензимидазол (общепринятое название: Омепразол), который обладает превосходным действием по сравнению с традиционными антагонистами H2-рецептора, например, циметидином, фамотидином, ранитидином и т. п. Смотри: описания к патентам США N 4255431, 4337257, 4508905, 4758579, патенту Великобритании N 2134523, Европейским патентам N 0005129 и 0268956. Поэтому Омепразол широко используют в различных видах лекарственных препаратов с точки зрения механизма действия, который является противоположным по отношению к общепринятым антагонистам H2-рецептора, Омепразол блокирует протонное прокачивание H+, K+-ATP-азы, присутствующей в удачной оболочке, для ингибирования желудочной секреции. Кроме того, омепразол обладает также преимуществом, состоящим в более продолжительном действии по сравнению с традиционными противоязвенными средствами.
Для создания новых противоязвенных средств настоящие изобретатели работали в течение долгого периода времени. В результате чего было синтезировано новое соединение, имеющее общую формулу (1), которая определена выше, и затем установлено, что соединение формулы (1) обладает превосходным противоязвенным воздействием по сравнению с омепразолом.
Цель настоящего изобретения обеспечение новых производных 5- пирролил - 2- пиридилметилсульфинилбензимидазола, имеющих следующую общую формулу: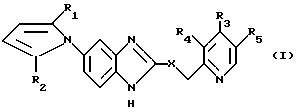
и их солей, в которой значения R1-R5 даны выше, а также способа их получения.
Предпочтительные соединения формулы (1) в соответствии с настоящим изобретением включают такие, в которых:
X представляет S, SO или SO2;
R1 и R2 независимо друг от друга представляют водород или метил;
R3 представляет водород, метил, метокси или этокси группу, 2,2,2-трифторэтокси или 3,3,3,2,2-пентафторпропокси;
R4 водород или метил;
R5 водород, метил или этил.
C2-C5-фторалкил может включать 2,2,2-трифторэтил, 2,2,3,3,3-пентафторпропил, 2,2,3,3-тетрафторпропил, 1-(трифторметил)-2,2,2-трифторэтил, 2,2,3,3,4,4,4-гептафторбутил, 2,2,3,3,4,4,5,5-октафторпентил и т.п.
Более предпочтительные соединения формулы (I) в соответствии с настоящим изобретением включают, например, такие, в которых:
X представляет SO,
R1 и R2 представляют водород,
R3 представляет метокси или этокси,
R4 и R5 независимо представляют водород, метил или этил.
Соединения настоящего изобретения формулы (I):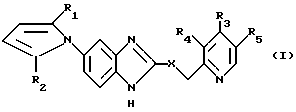
в которой X, R1-R5 имеют значения, указанные выше, и их соли могут быть получены реакцией соединения общей формулы (II):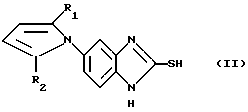
в которой R1 и R2 определены так, как они описаны ранее, с соединением, имеющим общую формулу (III):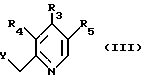
в которой R3, R4 и R5 определены так, как они описаны ранее;
Y представляет галоген, этерифицированную гидрокси или ацилокси группу
в органическом растворителе в присутствии основания, или реакцией соединения общей формулы (IV):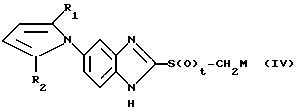
в которой R1 и R2 определены так, как они описаны ранее, t означает 1 или 2, М представляет щелочной металл, с соединением общей формулы (V):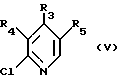
в которой R3, R4 и R5 определены так, как они описаны ранее, или реакцией соединения общей формулы (VI):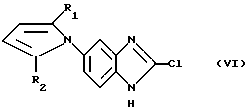
в которой R1 и R2 определены так, как описаны ранее, с соединением общей формулы (VII):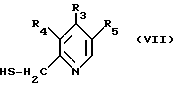
в которой R3, R4 и R5 определены так, как они описаны ранее, или реакцией соединения общей формулы (VIII):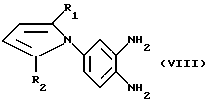
в которой R1 и R2 определены так, как они описаны ранее, с соединением общей формулы (IX):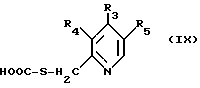
в которой R3, R4 и R5 определены так, как они описаны ранее, в полярном растворителе в присутствии серной кислоты.
Получение соединений формулы (I) реакцией соединения формулы (II) с соединением формулы (III) в органическом растворителе в присутствии основания показано на следующей реакционной схеме (А):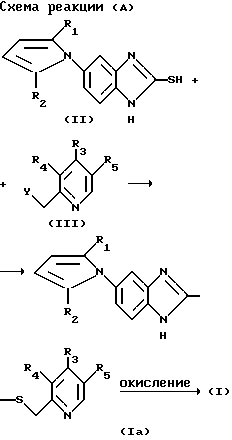
В вышеприведенной реакционной схеме X, R1, R2, R3, R4 и R5 определены как и в соединении формулы (1), приведенной выше, и Y представляет галоген, этерифицированную гидрокси и ацилокси группу.
В этой реакции используемый растворитель может включать обычный органический растворитель, например, низший алканол, такой как метанол, этанол и т. д. ацетон, простой эфир, тетрагидрофуран, метиленхлорид, ацетонитрил, диметилсульфоксид или диметилформамид, к которому не обязательно может быть добавлена вода. Температура реакции обычно составляет в диапазоне 0 - 150oC, предпочтительно в диапазоне 50 100oC.
В качестве основания для этой реакции могут быть использованы гидроксиды, карбонаты или гидриды щелочного металла, или третичные амины, примеры основания включают гидроксид натрия, гидроксид калия, карбонат калия, карбонат кальция, метоксид натрия, бикарбонат натрия, гидрид калия, гидрид натрия, пиридин, триэтиламин, этилдиизопропиламин или т.п.
Соединение формулы (I) в соответствии с настоящим изобретением может быть получено путем окисления соединения формулы (Iа) соответствующим количеством окислителя, как показано в вышеприведенной реакционной схеме (А). В этом случае полученное соединение формулы (I) может быть или сульфоксидным (-SO-) соединением или сульфоновым (-SO2-) соединением в зависимости от вида и количества используемого окислителя.
Окислитель, который используют для этой цели, включает: m-хлорпероксибензойную кислоту, пероксид водорода, пероксиуксусную кислоту, трифторпероксиуксусную кислоту, 3,5-динитропероксибензойную кислоту, пероксималеиновую кислоту, пента-оксид ванадия, азотную кислоту, озон, тетраоксид диазота, иодоксобензол, N-галосукцинимид, 1-хлорбензотриазол, третбутилгипохлорид, диазабицикло[2,2,2] -октан, метапериодат натрия, диоксид селена, диоксид марганца, хромовую кислоту, цериевый нитрат аммония, бром, хлор, сульфурилхлорид и т.п.
Реакцию можно предпочтительно осуществлять в инертном растворителе, например, ароматическом углеводороде, таком как бензол или толуол; хлорированном углеводороде, таком как хлороформ или метиленхлорид; или ацетоне.
В этом случае температура реакции обычно находится в диапазоне от -70oC до температуры кипения используемого растворителя, предпочтительно в диапазоне от -50oC до -20oC.
Получение соединений формулы (I) в соответствии с настоящим изобретением взаимодействием соединения формулы (IV) с соединением формулы (V) показано на следующей реакционной схеме (В):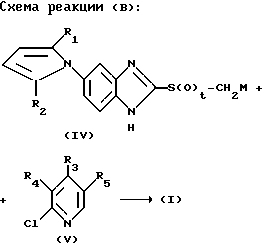
В вышеприведенной реакционной схеме: X, R1, R2, R3, R4 и R5 определены так, как они определены в соединении формулы (I), приведенной выше; t означает 1 или 2 и М представляет щелочной металл.
Эту реакцию можно предпочтительно осуществить в обычном инертном растворителе, который упоминался выше.
Кроме этого, реакцию обычно осуществляют при температуре 0 120oC, предпочтительно при температуре кипения используемого здесь растворителя. Соединение формулы (V), которое используют в качестве исходного материала в способе в соответствии с реакционной схемой (В) для получения соединения формулы (I) настоящего изобретения, может быть получено путем взаимодействия промежуточного соединения пиридин-N-оксида с обычным хлорирующим агентом, например, оксихлоридом фосфора, пентахлоридом фосфора и т.п.
Альтернативно соединение формулы (I) в соответствии с настоящим изобретением может быть также получено путем взаимодействия соединения формулы (VI) с соединением формулы (VII), как показано на следующей реакционной схеме (С):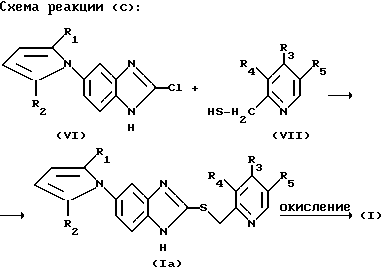
В вышеприведенной реакционной схеме X, R1, R2, R3, R4 и R5 определены так, как они определены в соединении формулы (I), приведенной выше.
В этой реакции реакционные условия по существу идентичны реакционным условиям реакции в соответствии с реакционной схемой (А) для получения соединения формулы (I) настоящего изобретения.
Кроме этого, соединение формулы (Iа), полученное в соответствии с вышеприведенным способом, может быть окислено при тех же самых условиях, как и в реакционной схеме (А), приведенной выше, для получения соединения формулы (I) в соответствии с настоящим изобретением.
Кроме этого, соединение формулы (I) в соответствии с настоящим изобретением может быть получено путем взаимодействия соединения формулы (VIII) с соединением формулы (IX) в полярном растворителе в присутствии сильной кислоты, как показано на следующей реакционной схеме (D):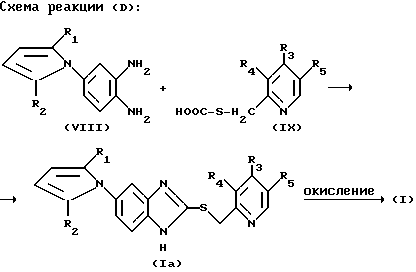
В вышеприведенной реакционной схеме X, R1, R2, R3, R4 и R5 определены так, как они определены в соединении формулы (I), приведенной выше.
В этой реакции полярный растворитель может также содержать воду.
Реакцию в соответствии с реакционной схемой (D) можно осуществить при температуре кипения используемого здесь растворителя.
Соединение формулы (I), которое может быть здесь получено в соответствии с реакционными схемами (С) и (D), может быть окислено в соответствии с той же самой методикой, как и в реакционной схеме (А) для получения соединения формулы (I) настоящего изобретения.
Исходные материалы, используемые в вышеупоминаемых процессах в соответствии с настоящим изобретением, в настоящее время известны и могут быть получены в соответствии с известными методами.
Соединение формулы (I), полученное вышеупомянутыми способами в соответствии с настоящим изобретением, можно отделить и очистить в соответствии с общепринятыми рабочими методиками или можно превратить его в фармацевтически приемлемую солевую форму в соответствии с общепринятыми способами.
Соединение формулы (I) в соответствии с настоящим изобретением может быть использовано для профилактики и лечения язв желудка и двенадцатиперстной кишки. Соединение формулы (I) имеет химическую структуру, подобную химической структуре известного производственного средства омепразола, и, поэтому показывает механизм фармакологического действия омепразола. Кроме того, как было продемонстрировано испытаниями in vitro, фармакологическая эффективность соединения формулы (I) в соответствии с настоящим изобретением приблизительно в 7 раз выше фармакологической эффективности омепразола. Кроме того, испытания in vivo на животных также показали, что соединение формулы (1) обладает сильным фармакологическим воздействием, в 2,5-3 раза превышающим фармакологическое воздействие омепразола. Кроме того, в соответствии с фармакологическими испытаниями на токсичность было установлено, что соединение формулы (I) в соответствии с настоящим изобретением не обладает острой токсичностью или токсичностью в отношении центральной нервной системы.
Поэтому новые соединения формулы (I) в соответствии с настоящим изобретением являются превосходным противоязвенным средством, которое обладает превосходным фармакологическим действием, значительно лучшим, чем фармакологическое действие известного противоязвенного средства, и также продолжительным периодом действия.
Соединение формулы (I) в соответствии с настоящим изобретением может быть введено или перорально или парентерально. Предпочтительным способом применения является пероральный прием.
Соединение формулы (I) в соответствии с настоящим изобретением можно вводить само по себе или в форме его фармацевтически приемлемой соли. Подходящие примеры таких солей соединения формулы (I) включают кислую соль присоединения и соль щелочного металла. В качестве соли щелочного металла могут быть упомянуты натриевая соль, магниевая соль, кальциевая соль или алкиламиновая соль. В качестве кислоты, которая может образовывать кислую соль присоединения соединения формулы (1), могут быть упомянуты следующие соединения:
серная кислота, сульфоновая кислота, фосфорная кислота, азотная кислота, перхлорная кислота, муравьиная кислота, уксусная кислота, пропионовая кислота, янтарная кислота, глюконовая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, аскорбиновая кислота, малеиновая кислота, оксималеиновая кислота, пировиноградная кислота, фенилуксусная кислота, бензойная кислота, р-аминобензойная кислота, р-оксибензойная кислота, салициловая кислота, р-аминосалициловая кислота, метансульфоновая кислота, этансульфоновая кислота, оксиэтансульфоновая кислота, этиленсульфоновая кислота, толуолсульфоновая кислота, нафтилсульфоновая кислота, сульфаниловая кислота, камфорсульфоновая кислота, хинная кислота, о-метиленминдальная кислота, протонная бензолсульфоновая кислота, метионин, триптофан, лизин, аргинин, пикриновая кислота или d-o-толилвинная кислота.
Соединение формулы (I) в соответствии с настоящим изобретением можно применять в соответствующей фармацевтически приемлемой композиции, которую получают путем использования фармацевтически приемлемой добавки и соответствующего носителя при использовании методов, хорошо известных специалистам в родственной области. Хотя такая композиция включает различные фармацевтически приемлемые препаративные формы, например, капсулы, таблетки, покрытые сахаром, сиропы или инъекции, предпочтительными для введения являются капсулы, покрытые растворимой в кишечнике оболочкой, или таблетки.
Соединения этого изобретения можно использовать в смеси с обычными наполнителями, т.е. фармацевтически приемлемыми органическими или неорганическими носителями, подходящими для парентерального, тонкокишечного (например орального) введения, которые не разрушают при взаимодействии активные соединения. Подходящие фармацевтически приемлемые носители включают, но при этом не ограничиваются, воду, солевые растворы, спирты, гуммиарабик, растительные масла, бензиловые спирты, полиэтиленгликоли, желатин, карбогидраты, например, лактозу, амилозу или крахмал, стеарат магния, тальк, кремниевую кислоту, вязкий парафин, душистое масло, моноглицериды и диглицериды жирной кислоты, эфиры жирных кислот и пентаэритрола, гидроксиметилцеллюлозу, поливинилпирролидон и т.д.
Фармацевтические препараты можно стерилизовать и по желанию смешать со вспомогательными веществами, например, консервантами, стабилизаторами, эмульгаторами, солями для оказания влияния на осмотическое давление, буферами, красителями, веществами, улучшающими вкус, и/или ароматическими веществами, и т.п. которые не разрушают при взаимодействии активные вещества. Их можно также сочетать с другими активными агентами, например, витаминами.
Для парентерального применения в особенности подходящими являются впрыскиваемые стерильные растворы, предпочтительно масляные или водные растворы, а также суспензии, эмульсии или имплантанты, включая суппозитории. Подходящими унифицированными дозами являются ампулы.
Для энтерального применения в особенности подходящими являются таблетки, драже, жидкости, капли или капсулы. Когда применяют подслащенный наполнитель, можно использовать сироп, эликсир или т.п.
Доза соединений в соответствии с этим изобретением обычно составляет 1 - 1000 мг/день, предпочтительно 3 100 мг/день, когда его вводят взрослым пациентам для профилактики и лечения язв желудка и двенадцатиперстной кишки. Как понятно специалистам в данной области, дозу можно определить с использованием общепринятых рассмотрений, например, путем обычного сравнения дифференциальных активностей обсуждаемых соединений и известного средства, например, посредством соответствующего общепринятого фармакологического протокола.
Настоящее изобретение будет более конкретно проиллюстрировано посредством следующих примеров. Однако, понятно, что настоящее изобретение не ограничивается этими примерами.
Пример 1. Получение 2-[[(4-метокси-3-метил)-2-пиридинил]метилтио]-5-(1н-пиррол-1-ил)-1н-бензимидазола (Соединение 1). 2 г (9,3 ммоля) 5-(1н-пиррол-1-ил)-2-меркаптобензимидазола растворили в растворе 0,74 г (2 экв. веса) гидроксида натрия в 100 мл метанола при комнатной температуре. К полученному раствору добавили 1,9 г (1 экв. вес) 4-метокси-3-метил-2-хлор-метилпиридингидрохлорида и затем смесь оставили для взаимодействия в течение 3 ч при температуре 50 60oC, после чего отфильтровали для удаления осажденного неорганического материала. Под пониженным давлением удалили растворитель и из простого эфира для получения 2,7 г (85%) желательного соединения выкристаллизовали остаток.
Температура плавления: 191 193oC.
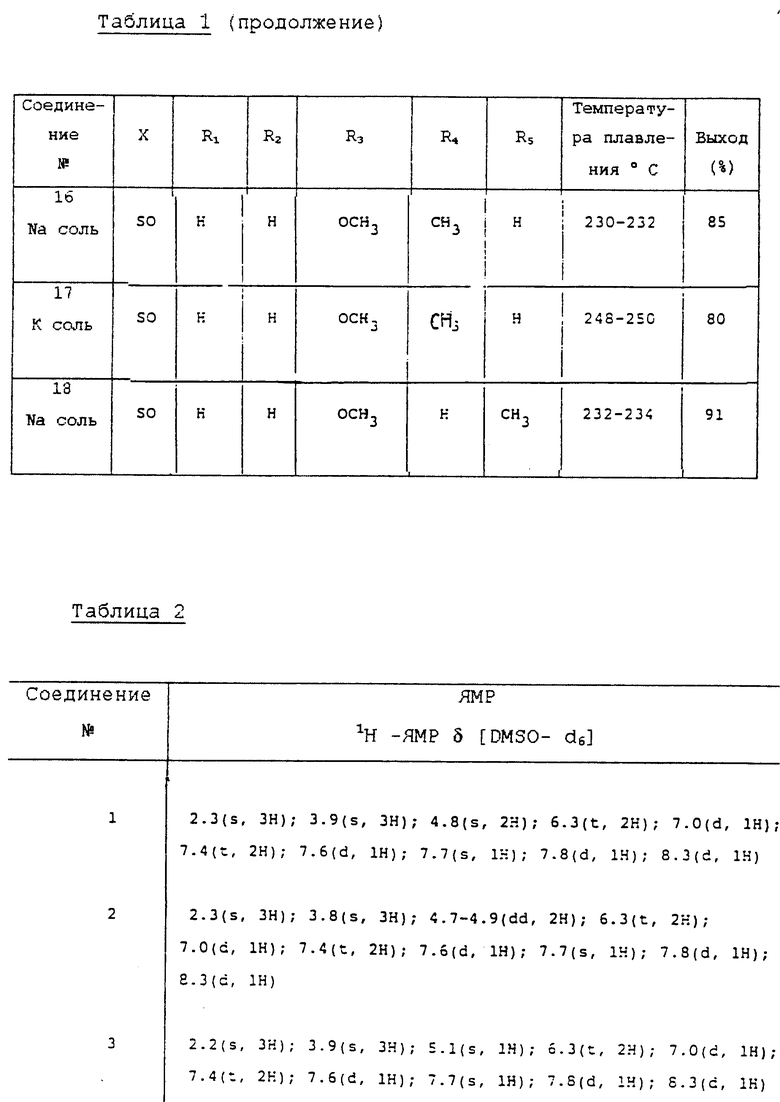
1H-ЯМР δ [DMSO-d6] 2,3 (s, 3H), 3,9 (s, 3H), 4,8 (s, 2H), 6,3 (t, 2H), 7,0 (d, 1H), 7,4 (t, 2H), 7,6 (d, 1H), 7,7 (s, 1H), 7,8 (d, 1H), 8,3 (d, 1H).
Пример 2. Получение 2-[[(4-метокси-3-метил)-2-пиридинил]метилсульфинил] -5-(1н-пиррол-1-ил)-1н-бензимидазола (Соединение 2). 6,7 г (19 ммолей) соединения, полученного в примере 1, растворили в 150 мл хлороформа и затем охладили до температуры -40oC. К нему по каплям медленно добавили m-хлорпероксибензойную кислоту (1 экв. вес), растворенную в хлороформе, и затем смесь перемешали в течение 20 мин при -40oC. Реакционную смесь разбавили хлороформом и промыли бикарбонатом натрия и насыщенным солевым раствором. Раствор хлороформа сушили сульфатом натрия. После удаления растворителя под пониженным давлением полученный сырой продукт растворили в этилацетате и затем кристаллизовали простым эфиром до получения 5,6 (80%) желательного соединения.
Температура плавления: 122 123oC.
1H-ЯМР d [DMSO-d6] 2,3 (s, 3H), 3,8 (s, 3H), 4,7-4,9 (s, 2H), 6,3 (t, 2H), 7,0 (d, 1H), 7,4 (t, 2H), 7,6 (d, 1H), 7,7 (s, 1H), 7,8 (d, 1H), 8,3 (d, 1H).
Соединения 3 12, перечисленные в последующих таблицах 1 и 2, могут быть получены в соответствии с той же самой методикой, которая приведена в примере 2.
Пример 3. Получение 2-[[(4-(2,2,2-трифторэтокси)-3-метил)-2-пиридинил] -5-(1н-пиррол-1-ил)-1н-бензимидазола (Соединение 13). 2 г (9,3 ммоля) 5-(1н-пиррол-1-ил)-2-меркаптобензимидазола растворили в растворе 0,74 г (2 экв. веса) гидроксида натрия в 100 мл метанола. К полученному раствору добавили 2,6 г (1 экв. вес) 4-(2,2,2-трифторэтокси)-3-метил-2-хлорметилпиридингидрохлорида и затем смесь оставили для взаимодействия в течение 3 ч при температуре 50 60oC. После удаления растворителя под пониженным давлением полученный продукт растворили в 150 мл хлороформа и затем охладили до -40oC. К этому реакционному раствору медленно по каплям добавили m-хлорпероксибензойную кислоту (1 экв. вес), растворенную в хлороформе, и затем смесь перемешали в течение 20 мин при -40oC, разбавили хлороформом и промыли бикарбонатом натрия и насыщенным солевым раствором. Раствор хлороформа сушили сульфатом натрия. После удаления растворителя под пониженным давлением образованный сырой продукт растворили в этилацетате и затем выкристаллизовали из простого эфира до получения 3,6 г (8,8%) желательного соединения.
Температура плавления: 156 157oC.
1H-ЯМР d [DMSO-d6] 2,2 (s, 3H), 4,3 (q, 2H), 4,6-4,9 (dd, 2H), 6,3 (t, 2H), 6,6 (d, 1H), 7,1 (t, 2H), 7,3 (d, 1H), 7,4 (s, 1H), 7,7 (d, 1H), 8,3 (d, 1H).
Пример 4. Получение 2-[[(4-(2,2,3,3,3-пентафторпропокси)-3-метил)-2-пиридинил] метилсульфинил] -5-(1н-пиррол-1-ил)-1н-бензимидазола (Соединение 14). 2 г (9,3 ммоля) 5-(1н-пиррол-1-ил)-2-меркаптобензимидазола растворили в растворе 0,74 г (2 экв. веса) гидроксида натрия в 100 мл метанола. К полученному раствору добавили 3 г (1 экв. вес) 4-(2,2,3,3,3-пентафторпропокси)-3-метил-2-хлорметилпиридингидрохлорида, и затем смесь оставили для взаимодействия в течение 3 ч при температуре 50 60oC. После удаления растворителя под пониженным давлением полученный продукт растворили в 150 мл хлороформа и затем охладили до -40oC. К этому реакционному раствору медленно по каплям добавили m-хлорпероксибензойную кислоту (1 экв. вес), растворенную в хлороформе, и затем смесь перемешали в течение 20 мин при -40oC, разбавили хлороформом и промыли бикарбонатом натрия и насыщенным солевым раствором. Раствор хлороформа сушили сульфатом натрия. После удаления растворителя под пониженным давлением полученный сырой продукт растворили в этилацетате и затем кристаллизовали из эфира до получения 4 г (90%) желательного соединения.
Температура плавления: 158 160oC.
1H-ЯМР d [DMSO-d6] 2,2 (s, 3H), 4,7-4,9 (dd, 2H), 5,0 (t, 2H), 6,3 (t, 2H), 7,1 (d, 1H), 7,4 (t, 2H), 7,6 (d, 1H), 7,7 (s, 1H), 7,8 (d, 1H), 8,3 (d, 1H).
Пример 5. Получение 2-[[(4-метокси-3-метил)-2-пиридинил]-метилсульфинил] -5-(1н-2,5-диметилпиррол-1-ил)-1н-бензимидазола (Соединение 15). 2 г (8,2 ммоля) 5-(2,5-диметилпиррол-1-ил)-2-меркаптобензимидазола растворили в растворе 0,66 г (2 экв. веса) гидроксида натрия в 100 мл метанола. К полученному раствору добавили 1,7 г (1 экв. вес) 4-метокси-3-метил-2-хлорметилпиридингидрохлорида, и затем смесь оставили для взаимодействия в течение 3 ч при температуре 50 - 60oC. После удаления растворителя под пониженным давлением полученный продукт растворили в 150 мл хлороформа и затем охладили до -40oC. В этому реакционному раствору медленно по каплям добавили m-хлорпероксибензойную кислоту (1 экв. вес), растворенную в хлороформе, и затем смесь перемешали в течение 20 мин при -40oC. После удаления растворителя под пониженным давлением остаток подвергли силикагельной хроматографии при использовании в качестве элюента этилацетата, при этом получили 1,4 г (10%) желательного соединения.
Температура плавления: 94 96oC.
1H-ЯМР d [DMSO-d6] 1,9 (s, 6H), 2,1 (s, 3H), 3,8 (s, 3H), 4,5-4,8 (dd, 2H), 5,8 (d, 2H), 4,9 (d, 1H), 7,2 (d, 2H), 7,4 (s, 1H), 7,6 (d, 1H), 8,2 (d, 1H).
Пример 6. Получение натриевой соли 2-[[(4-метокси-3-метил)-2-пиридинил] метилсульфинил]-5-(1н-пиррол-1-ил)-1н-бензимидазола. 1 г (2,7 ммоля) соединения, полученного в примере 2, растворили в 15 мл метиленхлорида и затем к нему добавили 0,1 г (2,7 ммоля) гидроксида натрия, растворенного в 10 мл воды. Смесь энергично перемешали. Водный слой отделили, промыли несколько раз метиленхлоридом и затем лиофилизовали до получения 0,9 г (85%) желательного соединения.
Температура плавления: 230 232oC.
1H-ЯМР d [DMSO-d6] 2,0 (s, 3H), 3,9 (s, 3H), 4,5-4,9 (dd, 2H), 6,4 (t, 2H), 6,9 (d, 1H), 7,4 (t, 2H), 7,5 (d, 1H), 7,7 (s, 1H), 7,8 (d, 1H), 8,2 (d, 1H).
Пример 7. Получение 2-[[(4-метокси-3-метил)-2-пиридинил]метилсульфинил] -5-(1н-пиррол-1-ил)-1н-бензимидазола (Соединение 2). 2 г (7,9 ммоля) 2-(литийметилсульфинил)-5-(1н-пиррол-1-ил) бензимидазола растворили в 100 мл бензола и затем к нему добавили 1,25 г (1 экв. вес) 2-хлор-(4-метокси-3-метил)пиридина. Реакционную смесь нагрели в колбе с обратным холодильником в течение 2 ч и отфильтровали для удаления хлорида лития. После удаления растворителя под пониженным давлением полученный сырой продукт растворили в этилацетате и затем кристаллизовали из простого эфира до получения 2,1 г (84%) желательного соединения.
Температура плавления: 102 123oC.
1H-ЯМР d [DMSO-d6] 2,3 (s, 3H), 3,8 (s, 3H), 4,7-4,9 (dd, 2H), 6,3 (t, 2H), 7,0 (d, 1H), 7,4 (t, 2H), 7,6 (d, 1H), 7,7 (s, 1H), 7,8 (d, 1H), 8,3 (d, 1H).
Пример 8. Получение 2-[[(4-метокси-3-метил)-2-пиридинил]метилтио]-5-(1н-пиррол-1-ил)-1н-бензимидазола (Соединение 1). 2 г (9,2 ммоля) 4-метокси-3-метил-2-тиометилпиридина растворили в растворе 0,4 г (1 экв. вес) гидроксида натрия в 100 мл этанола. К полученному раствору добавили 2 г (1 экв. вес) 2-хлор-5-(1н-пиррол-1-ил)бензимидазола, и затем реакционную смесь нагревали в колбе с обратным холодильником в течение 2 ч. После удаления растворителя под пониженным давлением полученный продукт кристаллизовали из эфира до получения 2,7 г (85%) желательного соединения.
Температура плавления: 191 193oC.
1H-ЯМР d [DMSO-d6] 2,3 (s, 3H), 3,9 (s, 3H), 4,8 (dd, 2H), 6,3 (t, 2H), 7,0 (d, 1H), 7,4 (t, 2H), 7,6 (d, 1H), 7,7 (s, 1H), 7,8 (d, 1H), 8,3 (d, 1H).
Пример 9. Получение 2-[[(4-метокси-3-метил)-2-пиридинил]метилтио]-5-(1н-пиррол-1-ил)-1н-бензимидазола (Соединение 1). 17,3 г (0,1 ммоля) 2-[[(4-метокси-3-метил)-2-пиридинил] метилтио] муравьиной кислоты и 21,3 г (1 экв. вес) о-[5-(1н-пиррол-1-ил)]фенилендиамина нагревали в колбе с обратным холодильником в 100 мл 4N HCl в течение 40 мин. Реакционную смесь охладили и затем нейтрализовали аммиачной водой. Раствор обработали активированным углем и затем экстрагировали этилацетатом. Растворитель удалили под пониженным давлением, и остаток кристаллизовали из простого эфира до получения 8,8 г (25%) желательного соединения.
Температура плавления: 191 193oC.
1H-ЯМР d [DMSO-d6] 2,3 (s, 3H), 3,9 (s, 3H), 4,8 (dd, 2H), 6,3 (t, 2H), 7,0 (d, 1H), 7,4 (t, 2H), 7,6 (d, 1H), 7,7 (s, 1H), 7,8 (d, 1H), 8,3 (d, 1H).
Физико-химические свойства соединений, полученных в соответствии, по существу, с той же самой методикой, как и соединения в вышеописанных примерах, описаны в табл. 1 и 2.
Противоязвенное действие соединения формулы (1), которое определено выше, в соответствии с настоящим изобретением было проиллюстрировано посредством различных экспериментов, включая ингибирование ферментативной активности, влияние на ингибирование секреции желудочной кислоты и кислотности, ED50 и т.п. Методы испытаний и результаты следуют далее.
Испытание 1: Ингибирование ферментативной активности. Ингибирование H+/K+-ATP-азы соединениями формулы (1) согласно настоящему изобретению демонстрировали испытаниями in vitro.
В этом испытании в качестве контрольного соединения использовали омепразол [5-метокси-2-[[(4-метокси-3,5-диметил-2-пиридинил)метил] сульфинил]-1н-бензимидазол]
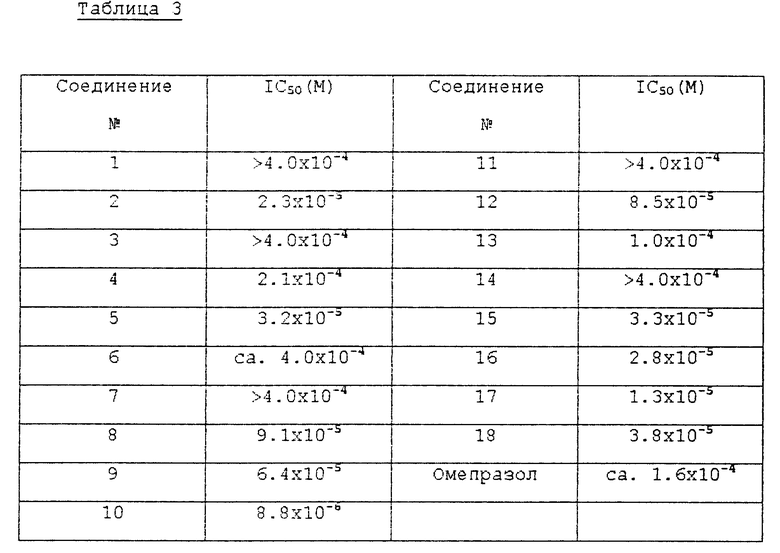
У кролика удалили желудочную слизистую оболочку и затем центрифугировали 77,000 г, используя ультрацентрифугу для отделения микросомной фракции, которую использовали в качестве источника фермента H+/K+-ATP-азы для этого испытания. В течение 5 мин при 37oC предварительно инкубировали пробой (соединением в соответствии с настоящим изобретением) 60 μг H+/K+-ATP-азы и затем к ней добавили 4 мМ ATP в качестве питательной среды и 4 мМ Mg++, 20 мМ K+ в качестве коферментов. Затем при использовании спектрофотометра при 660 нм определили количество полученного таким образом неорганического фосфора и превратили его в белок. Концентрацию соединения, которая ингибирует активность фермента на 50% т.е. IC50, вычислили из значений процентной концентрации для ингибирования активности фермента, которые получили от 3 5 пробирок, содержащих различную концентрацию испытуемого соединения, согласно методу Litchfield-Wilcoxon (Литчфилда-Вилкоксона). Результаты описаны в табл. 3.
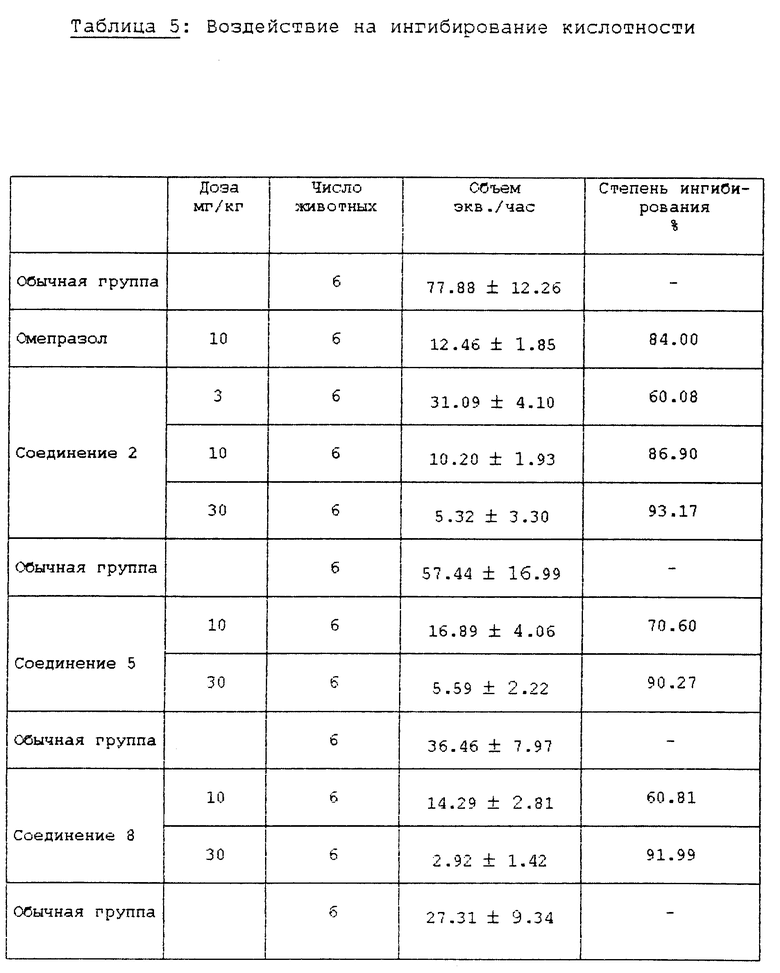
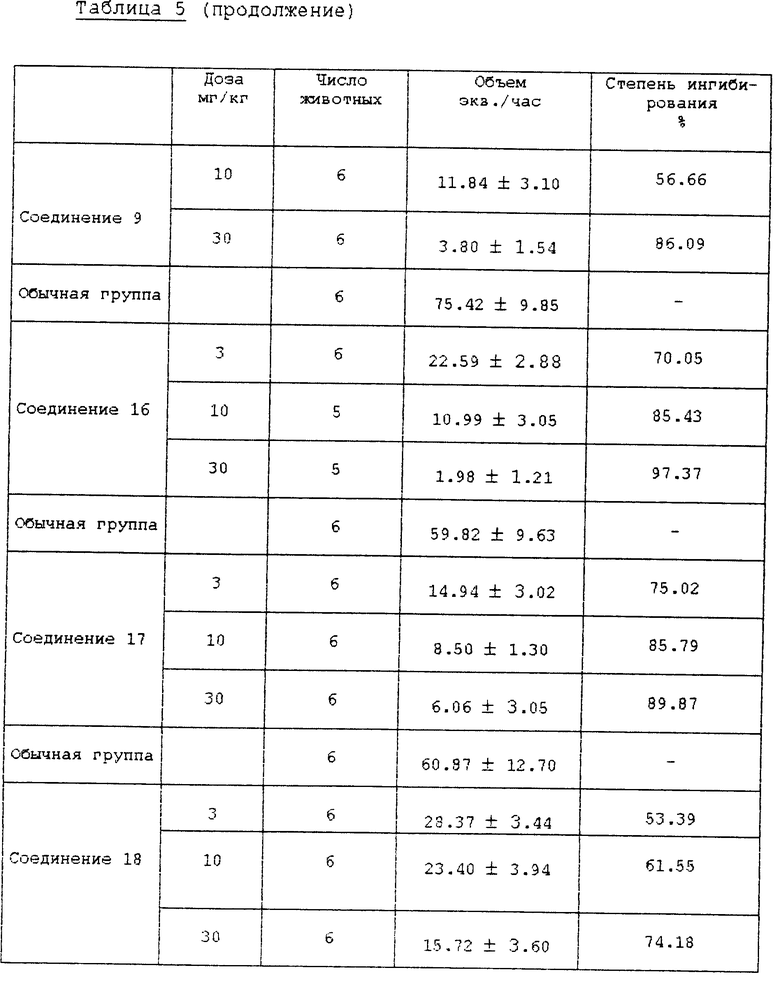
Испытание 2. Воздействие на ингибирование секреции желудочного сока и кислотности (in vivo). В качестве второго испытания in vivo, проводили испытания ингибирования секреции желудочного сока и кислотности на крысах, используя метод Shay (Шея), и результаты сравнивали с обычной контрольной группой и группой омепразола. Конкретные методы испытаний следуют ниже.
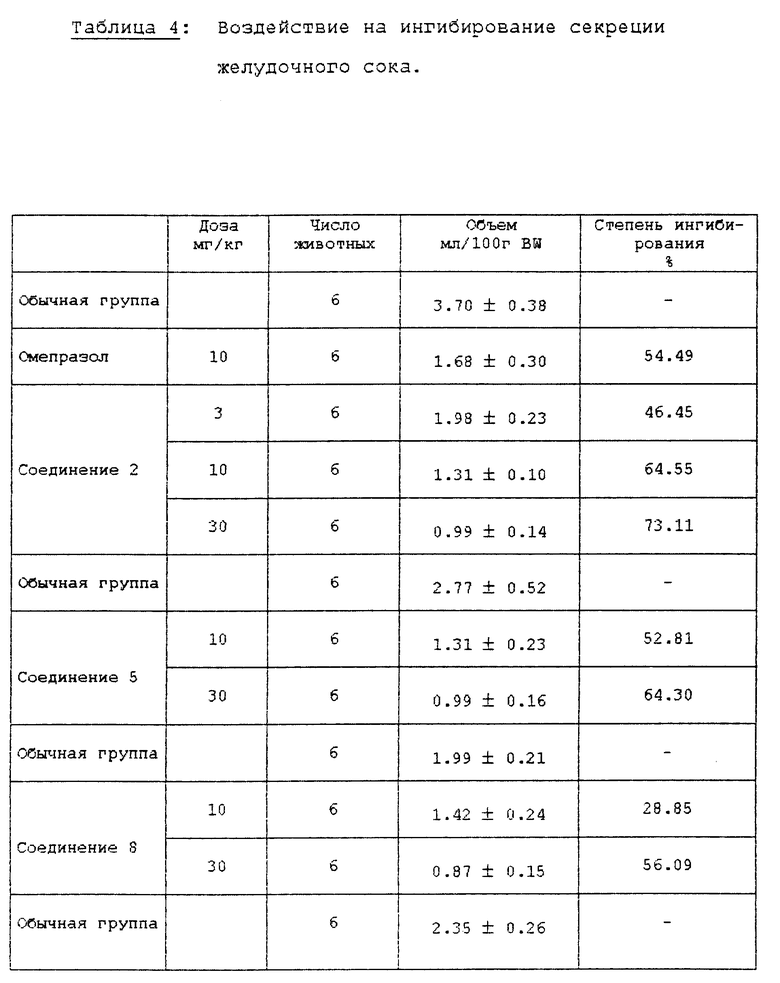
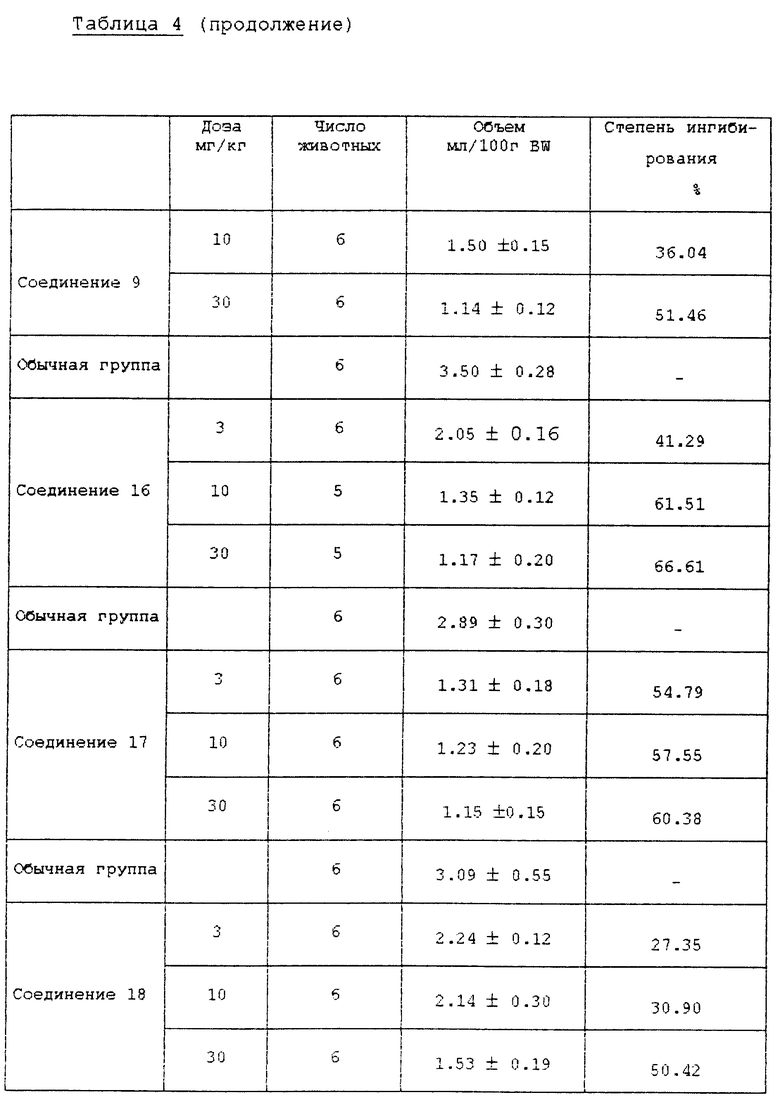
Самцы крыс SD (200±20 г) голодали в течение 24 ч, за исключением того, что им давали воду, затем их анестезировали простым эфиром. Рассекли брюшную полость и затем лигировали пилорус (суженую часть желудка). Испытуемые соединения суспендировали или растворяли в 5% СМС (карбоксиметилцеллюлозе) и впрыскивали в двенадцатиперстную кишку. После того, как раствор всосался в брюшную полость, крыс выдерживали в течение 5 ч и затем умертвили простым эфиром. У крыс удалили желудок и собрали желудочный сок. Желудочный сок центрифугировали при 10000хg в течение 10 мин при 4oC для удаления осадка. Количество желудочного сока и кислотность определили при использовании 0,02 N NaOH для создания pH 7,0 в конечной точке анализа и затем вычислили общий объем кислоты. Результаты описаны в табл. 4 и 5.
Как можно видеть из вышеприведенных результатов испытаний, среди соединений формулы (1) в соответствии с настоящим изобретением соединения 2, 4, 5, 8 10, 12, 13, 15 18 показывают активность ингибирования фермента, подобную или превосходящую активность известного противоязвенного средства омепразола, а соединения 2, 5, 8, 9, 16 и 17 показывают хорошее воздействие на ингибирование секреции желудочного сока и понижение кислотности. В частности, установили, что соединения 2 и 5 среди соединений формулы (1) в соответствии с настоящим изобретением показывают сильное ингибирование секреции желудочного сока и эффект снижения высокой кислотности даже при более низкой дозе, чем доза известного противоязвенного средства, и ED50 соединения 2 для воздействия на ингибирование секреции желудочного сока составила 3,6 мг/кг, а для воздействия на ингибирование кислотности она составила 1,6 мг/кг.
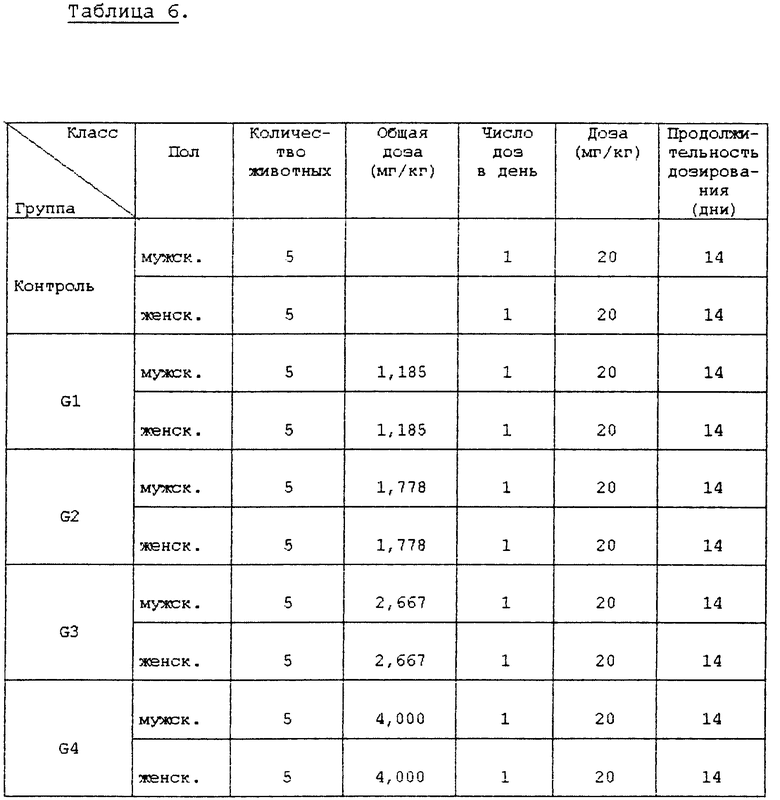
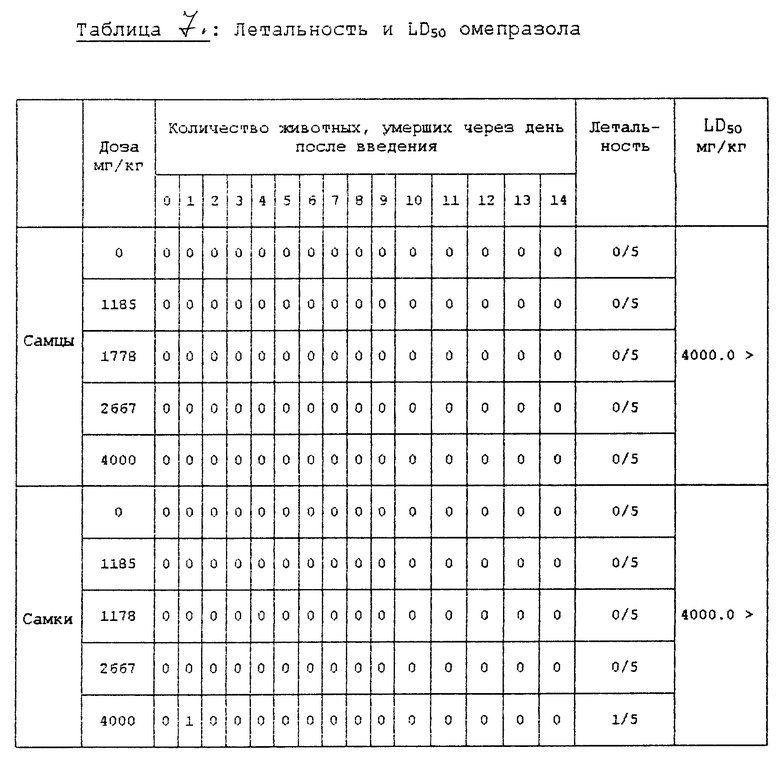
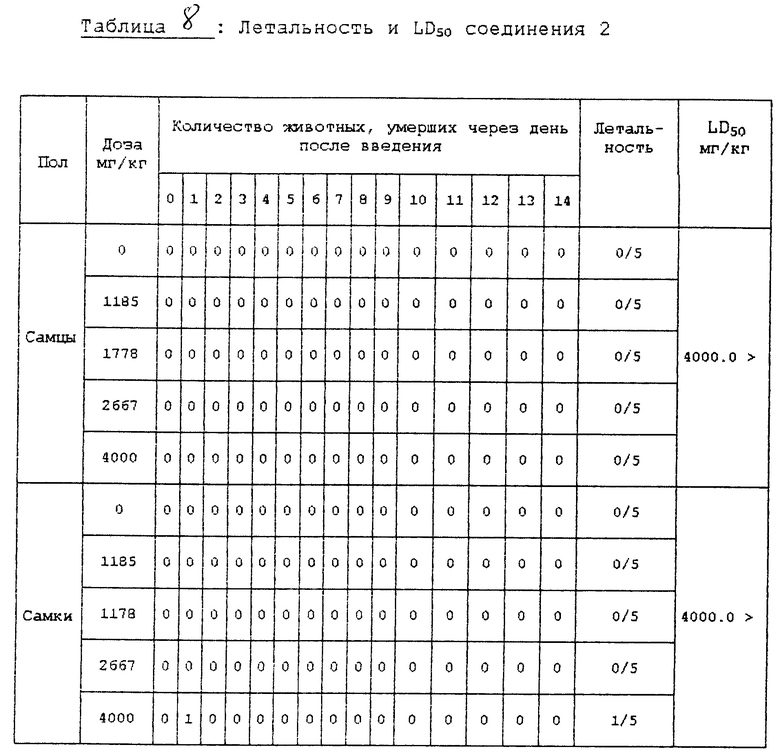
Испытание 3. Испытание острой токсичности. В клетке для разведения животных в течение одной недели предварительно вскормили пятинедельных мышей (самцов и самок) и затем беспорядочно выбрали и использовали в этом испытании животных, которые набрали вес. Количество испытуемых соединений, вводимых подопытным животным, установили на основе максимальной дозы 4000 мг/кг при обычном соотношении, равном 1,5.
Испытуемые соединения в порошковой форме суспендировали в 0,5% метилцеллюлозе и ввели перорально при использовании 1 мл шприца. Другие конкретные условия введения описаны в нижеприведенной табл. 6.
В вышеприведенном испытании контрольная группа получила только 0,5% метилцеллюлозы.
Клинические симптомы и смерть подопытных животных, вызванную испытуемыми соединениями, наблюдали тотчас же после введения испытуемых соединений и во время всего периода испытаний, при этом изменения массы тела регистрировали три раза: т. е. в день введения, через неделю после введения и в последний день испытаний.
После завершения испытаний всех подопытных животных умертвили простым эфиром и наблюдали изменения во внутренних и наружных органах, которые появились благодаря испытуемым соединениям. Результаты испытаний представлены в табл. 7 и 8.
Из результатов этого испытания, которое показано выше, можно установить, что соединение 2 в соответствии с настоящим изобретением является очень безопасным соединением, которое имеет значение LD50, равное 4000 мг/кг или более, оно не влияет на обычные изменения массы тела подопытных животных и, кроме того, не оказывает влияния на внутренние и наружные органы подопытных животных.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРРОЛОПИРИДИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2815649C1 |
| НОВЫЕ СОЕДИНЕНИЯ, ИХ ИЗОМЕР ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ В КАЧЕСТВЕ АНТАГОНИСТА ВАНИЛОИДНОГО РЕЦЕПТОРА И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2007 |
|
RU2448108C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИРАЗОЛОПИРИМИДИНОНА ДЛЯ ЛЕЧЕНИЯ ИМПОТЕНЦИИ | 2001 |
|
RU2236410C2 |
| СПОСОБ ПОЛУЧЕНИЯ КЛОПИДОГРЕЛА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В СПОСОБЕ | 2005 |
|
RU2357970C1 |
| НОВОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ ЗАМЕЩЕННОЕ ПРОИЗВОДНОЕ ПИРИМИДИНА, ПРОЯВЛЯЮЩЕЕ ИНГИБИРУЮЩЕЕ ДЕЙСТВИЕ НА РОСТ РАКОВЫХ КЛЕТОК, И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕГО | 2020 |
|
RU2834201C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПИРРОЛОПИРИДИНОВОГО ПРОИЗВОДНОГО | 2021 |
|
RU2830589C1 |
| НОВОЕ ПИРИМИДИНОВОЕ ПРОИЗВОДНОЕ, ОБЛАДАЮЩЕЕ ЭФФЕКТОМ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК, И СОДЕРЖАЩАЯ ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2018 |
|
RU2744168C1 |
| ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ОСНОВЕ ДЕРИВАТОВ ПИРРОЛИНА ДЛЯ ПРЕДОТВРАЩЕНИЯ ОМЕРТВЕНИЯ КЛЕТКИ, ПОРАЖЕННОЙ ОПУХОЛЬЮ, СПОСОБ ЕГО ПРОИЗВОДСТВА И СПОСОБ ИСПОЛЬЗОВАНИЯ | 2007 |
|
RU2473551C2 |
| Новые соединения пиридопиримидинона для модулирования каталитической активности гистонлизиндеметилаз (KDMS) | 2015 |
|
RU2684396C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО D-ЭРИТРО-2,2-ДИФТОРО-2-ДЕЗОКСИ-1-ОКСОРИБОЗЫ | 2005 |
|
RU2337917C1 |


Использование: в качестве противоязвенного препарата. 2-{[(4-метокси-3-метил)-2-пиридинил] метилтио} -5-(1H-пиррол-1-ил)-1H-бензимидазол, выход 85%, т. пл. 191 - 193oC; натриевая соль 2-{[(4-метокси-3-метил)-2-пиридинил] метилсульфинил}-5-(1H-пиррол-1-1ил)-1H-бензимидазол, выход 85%, т. пл. 230 - 232oC. 10 з.п. ф-лы, 8 табл.
где X S, SO или SO2;
R1 и R2 независимо друг от друга водород или С1 - С8-алкил;
R3 водород, С1 С8-алкил, -OR6, где R6 С1 С4-алкил, С2 С5-фторалкил;
R4 и R5 независимо друг от друга водород или С1 - С5-алкил,
или их соли.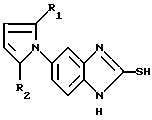
где R1 и R2 имеют указанные в п. 1 значения,
подвергают реакции с соединением формулы III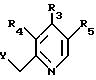
где R3, R4 и R5 имеют указанные в п. 1 значения;
Y галоген, этерифицированная гидрокси- или ацилоксигруппа,
в органическом растворителе в присутствии основания.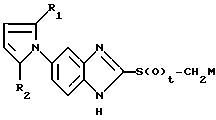
где R1 и R2 имеют указанные в п. 1 значения;
t 1 или 2;
M щелочной металл,
подвергают реакции с соединением формулы V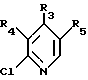
где R3, R4 и R5 имеют указанные в п. 1 значения.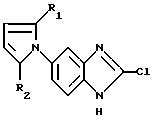
где R1 и R2 имеют указанные в п. 1 значения,
подвергают реакции с соединением формулы VII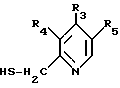
где R3, R4 и R5 имеют указанные в п. 1 значения.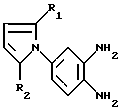
где R1 и R2 имеют указанные в п. 1 значения,
подвергают реакции с соединением формулы IX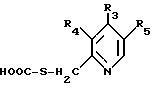
где R3, R4 и R5 имеют указанные в п. 1 значения,
в полярном растворителе в присутствии сильной кислоты.
| US, 4255431, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
| US, 4337257, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
| EP, 5129, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, 268956, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
