Область техники, к которой относится изобретение
Данное изобретение относится к способу получения высокочистых производных D-эритро-2,2-дифторо-2-дезокси-1-оксорибозы.
Уровень техники изобретения
D-эритро-2,2-дифторо-2-дезокси-1-оксорибоза является важным промежуточным соединением, используемым в получении гемцитабина формулы (A), агента для лечения немелкоклеточного рака легкого.

Поскольку гемцитабин является эритро-энантиомером, имеющим 3-гидроксигруппу, ориентированную вниз (напротив 5-гидроксигруппы по отношению к расположению тетрагидрофуранового кольца), в получении гемцитабина важно разработать способ получения эритро-соединений 1-оксорибозы, имеющих 3-гидроксигруппу, ориентированную вниз.
Патент US № 4526988 раскрывает способ получения соединения эритро-1-оксорибозы через алкил-2,2-дифтор-3-гидрокси-3-(2,2-диалкилдиоксоран-4-ил)пропионат, смесь 3:1 3R-гидроксиэнантиомера формулы (B) и 3S-гидроксиэнантиомера формулы (B'):


где
R4 и R5 являются независимо C1-3алкилом.
Однако подобный способ включает неэкономичную стадию выделения из смеси соединений (B) и (B') только 3R-гидроксиэнантиомера формулы (B) для того, чтобы селективно получить желаемое производное эритро-1-оксорибозы, поскольку соединения (B) и (B') приводят к эритро-соединению формулы (C) и трео-соединению формулы (C') соответственно, как показано на Схемах реакции A и B.
Схема А

Схема В

Более того, способ также имеет проблему, заключающуюся в длительном времени реакции, почти четыре дня при комнатной температуре.
Между тем, патенты US № 4965374; 5223608 и 5434254 раскрывают способ получения эритро-энантиомера формулы (D), как показано на Схеме реакции C, при помощи (i) гидролиза и азеотропной перегонки 3-бензоилоксипропионового эфира формулы (Е) (смесь 3:1 3R- и 3S-энантиомеров) с получением соединения лактона формулы (F); (ii) защиты 5-гидроксигруппы соединения формулы (F) бензоильной группой с получением соединения 3,5-дибензоилокси формулы (G); и (iii) охлаждения соединения формулы (G) до -5˜10°C для осаждения только эритро-энантиомера формулы (D).
Схема С

где Bz является бензоилом.
Однако данный способ является неэкономичным по причине его относительно низкого выхода, примерно 25%, и использования дорогой и токсичной трифторуксусной кислоты в избыточном количестве в процессе гидролиза.
Более того, патент US № 5428176 и 5618951 указывает способ получения эфира 2,2-дифтор-β-силилокси-1,3-диоксолан-4-пропионовой кислоты формулы (H), имеющего высокое содержание 3R-силилгидроксиэнантиомера, взаимодействием силилацеталя 2,2-дифторкетена с производным глицеринового альдегида в таком растворителе, как 1,3-диметилпропиленмочевина (DMPU), как показано на Схеме реакции D.
Схема D

где от R6 до R9 являются алкилом; и R10 и R11 являются C1-3алкилом.
Однако данный способ также требует неэкономичного процесса колоночной хроматографии для выделения 3R-энантиомера из смеси энантиомеров.
Соответственно, авторы изобретения предприняли попытку разработать рациональный способ селективного получения соединений 1-оксорибозы, имеющих эритро-структуру, и неожиданно обнаружили оригинальный эффективный способ получения высокочистой 2,2-дифтор-2-дезокси-1-оксорибозы, имеющей эритро-структуру.
Сущность изобретения
Соответственно, целью данного изобретения является обеспечение эффективного способа селективного получения производных 2,2-дифтор-2-дезокси-1-оксорибозы, имеющих эритро-структуру.
Другой целью данного изобретения является обеспечение соединения 3R-энантиомера, которое может быть использовано в вышеупомянутом способе.
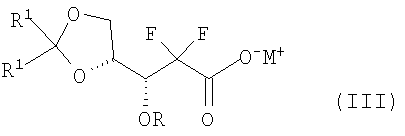
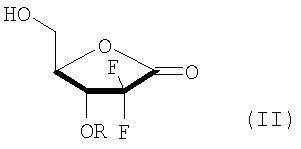
В соответствии с одним аспектом данного изобретения, обеспечен способ получения производного 2,2-дифтор-2-дезокси-1-оксорибозы формулы (I), включающий стадии (i) взаимодействия соединения формулы (V) с бифенилкарбонильным производным с получением соединения формулы (IV), имеющего 3-гидроксигруппу, защищенную бифенилкарбонильной группой; (ii) взаимодействия соединения формулы (IV) с основанием в смешанном растворителе, содержащем главным образом воду, с получением 3R-энантиомера карбоксилата формулы (III); (iii) взаимодействия соединения формулы (III) с кислотой с получением производного 5-гидрокси-1-оксорибозы формулы (II); и (iv) защиты 5-гидроксигрупы соединения формулы (II) при помощи R3:

где R является 
R1 является метилом или этилом;
R2 является C1-3алкилом;
R3 является бензоилом или 
R4 является фенилом или замещенным фенилом; и
М является аммонием (NH4), натрием или калием.
В соответствии с другим аспектом настоящего изобретения обеспечен 3R-энантиомер карбоксилата формулы (III):

где R, R1 и M имеют те же значения, что определены выше.
Подробное описание изобретения
В неочевидном способе соединения формул (IV) и (V) являются смесью 3R- и 3S-энантиомеров в данном соотношении каждый.
Способ кратко изложен в Схеме реакции I.
Схема реакции I

где R, R1, R2, R3 и M имеют те же значения, что определены выше.
В Схеме реакции I производное 2,2-дифторо-2-дезокси-1-оксорибозы формулы (I) может быть получено с высоким выходом путем защиты 3-гидроксигруппы соединения формулы (V) бифенилкарбонильной группой с получением соединения формулы (IV); гидролизом соединения формулы (IV) основанием с получением соли 3R-карбоксилата формулы (III), в которой 3R-энантиомер формулы (III) может быть выделен из образующейся смеси 3R- и 3S-энантиомеров, поскольку только 3R-энантиомер получается в виде твердого вещества; снятием защиты диоксолановой группы соединения (III) кислотой с получением производного карбоновой кислоты и переводом производного карбоновой кислоты в лактон с отгонкой воды с получением 5-гидрокси-1-оксорибозы формулы (II), имеющей эритро-структуру; и путем защиты 5-гидроксисоединения формулы (II) в соответствии с обычным способом.
Способ изобретения характеризуется тем, что возможно селективно получить 3R-энантиомер карбоксилата формулы (III) путем защиты 3-гидроксигруппы соединения формулы (V) бифенилкарбонильной группой и получением производного 1-оксорибозы формулы (I), имеющего требуемую эритро-структуру.
Поскольку по способу изобретения соединение формулы (III) может быть селективно получено в виде твердого вещества, то оно может быть легко выделено простым фильтрованием без проведения неэкономичной колоночной хроматографии или других процессов очистки. Соответственно, использование соединения формулы (II) в качестве промежуточного является одной особенностью способа изобретения, который является пригодным для производства производного 1-оксорибозы в крупных масштабах.
Соединение формулы (V), используемое в качестве исходного сырья в способе изобретения, может быть получено обычным способом, описанным в патентах US № 4526988; 4965374; 5223608 и 5434254, как показано на Схеме реакции II.
Схема реакции II
где R, R1 и R2 имеют те же значения, что определены выше.
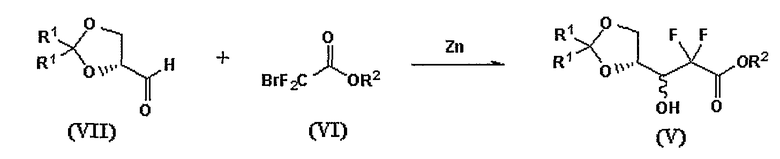
В Схеме реакции II соединение формулы (V), смесь 3:1 3R- и 3S-энантиомеров, может быть получена смешением кетонида альдегида формулы (VII) с дифторосоединением формулы (VI) и выдерживанием смеси для протекания реакции Реформатского с цинком.
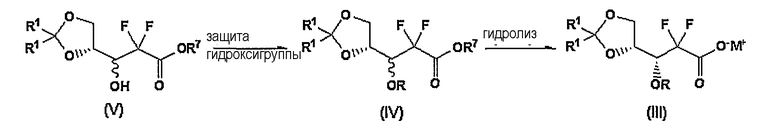
К тому же, 3R-карбоксилат формулы (III) может быть получен из соединения формулы (V), как показано на Схеме реакции III.
Схема реакции III
где R, R1, R2 и M имеют те же значения, что определены выше.
В Схеме реакции III 3R-карбоксилат формулы (III) может быть получен в виде твердого вещества; (i) защитой 3-гидроксигруппы соединения формулы (V) бифенилкарбонильной защитной группой с получением соединения формулы (IV); и (ii) гидролизом соединения формулы (IV) основанием.
В способе изобретения защитная группа, используемая на стадии (i), может быть бифенилкарбонильной группой, которая является бензоильной группой, необязательно замещенной бензольным кольцом, бензольное кольцо является замещенным одним или более заместителем, выбранным из группы, состоящей из водорода, циано, галогена, карбоалкокси, толуоила, нитро, алкокси, алкила и диалкиламино. Представительные примеры бифенилкарбонила включают такие, как 2-фенилбензоил (2-бифенилкарбонил), 4-фенилбензоил (4-бифенилкарбонил) и замещенный 2-(или 4-) фенилбензоил, предпочтительно 2-фенилбензоил и 4-фенилбензоил.
Гидрофобность, увеличивающаяся из-за двух бензольных колец бифенилкарбонильной группы, делает возможным отделить соль 3R-карбоксилата формулы (III) в виде твердого вещества, даже в воде или в содержащем воду смешанном растворителе.
С другой стороны, в случае использования в качестве защитной группы гидроксигруппы обычной бензоильной группы, невозможно получить соль 3R-карбоксилата в воде или в содержащем воду смешанном растворителе.
Напротив, в случае введения обычной защитной группы гидроксигруппы, такой как 1-нафтоил, 2-нафтоил, пивалоил или ацетил, для 3-гидроксигруппы соединения формулы (V) очень сложно селективно выделить из образующейся реакционной смеси 3R-карбоксилат формулы (III) в виде твердого вещества.
Соединение на основе бифенилкарбонила, используемое на стадии (i), может быть выбрано из группы, состоящей из бифенилкарбонила (либо замещенного бифенилкарбонила), хлорида, бромида, цианида или азида, который может быть коммерчески получен или химически синтезирован в соответствии с обычными способами.
Также основание, используемое в процессе нейтрализации на стадии (i), может быть выбрано из группы, состоящей из пиридина, триэтиламина, трибутиламина, диизопропилэтиламина и метилпиперидина, предпочтительно триэтиламин; катализатор, используемый при ацилировании, может являться 4-диметиламинопиридином либо 4-пирролидинопиридином; и ацилирование может проводиться при от -25 до 50°С.
При гидролизе на стадии (ii) основание может быть выбрано из группы, состоящей из газообразного аммиака, водного аммиака, карбоната натрия, бикарбоната натрия, гидроксида натрия, карбоната калия, бикарбоната калия, гидроксида калия и их смеси, предпочтительно бикарбонат натрия, которое может использоваться в количестве 1 эквивалента или более, предпочтительно в диапазоне от 1,5 до 5 эквивалентов по отношению к соединению формулы (IV).
Также смешанный растворитель, содержащий главным образом воду, может быть смесью воды и органического растворителя, выбранного из группы, состоящей из тетрагидрофурана, диоксана, ацетонитрила, ацетона, метилизобутилкетона, метилэтилкетона, метанола, этанола, пропанола, изопропанола, диметилацетамида, диметилформамида, диметилсульфоксида, этилацетата и их смеси, предпочтительно, смесь тетрагидрофурана и метанола; и вода может использоваться в количестве от 3 до 15 мл, предпочтительно от 5 до 11 мл, и органический растворитель от 3 до 30 мл, предпочтительно от 6 до 18 мл по отношению к 1,0 г соединения формулы (IV). Гидролиз может проводиться при от 5 до 50°С, предпочтительно от 10 до 30°С в течение от 30 мин до 2 часов.
Соединение формулы (III) может быть легко выделено из реакционной смеси, полученной на стадии (ii) удалением органического растворителя при пониженном давлении и фильтрованием полученной смеси; либо экстракцией реакционной смеси органическим растворителем и перекристаллизацией продукта из смешанного растворителя, включающего главным образом воду.
В способе изобретения 3R-карбоксилат формулы (III) может быть получен из соединения формулы (V) с высоким выходом от 60 до 70% через калиевую иди натриевую соль или примерно 40%, когда на стадии (ii) используется аммонийная соль как основание. Более того, соединение формулы (III), полученное в способе изобретения, имеет содержание 3R-карбоксилата более чем 99,7%, в то время как содержание 3S-карбоксилата менее чем 0,3% (следовательно, значение энантиомерного избытка выше 99,4%).
Соединение формулы (I), имеющее эритро-структуру, может быть получено из соединения формулы (III) высокоэнантиоселективным способом, как показано на Схеме реакции IV.
Схема реакции IV

где R, R1, R3 и M имеют те же значения, как определено выше.
В Схеме реакции IV соединение формулы (I) может быть получено путем (iii) взаимодействия соединения формулы (III) с кислотой в растворителе с получением соединения формулы (II), имеющего эритро-структуру как результат проведения каскада реакций, включающего нейтрализацию карбоксилата, удаление изоалкилиденовой защитной группы с получением диола карбоновой кислоты формулы (VIII), и образование лактона соединения формулы (VIII); и (iv) защиты 5-гидроксигруппы соединения формулы (II) защитной группой, содержащей гидрофобное бензольное кольцо.
Кислота, используемая в стадии изобретения (iii) может быть сильной кислотой, имеющей значение рКа, колеблющееся от -10,0 до 2,0, которая может быть выбрана из группы, состоящей из неорганической кислоты, такой как от 1 до 12N HCl и от 1 до 9N H2SO4, и органической кислоты, такой как метансульфоновая кислота, п-толуолсульфоновая кислота, трифторуксусная кислота и трифторметансульфоновая кислота, предпочтительно 12N HCl и трифторуксусная кислота, более предпочтительно 12N HCl; используемая в количестве в диапазоне от 1 до 2 эквивалентов, предпочтительно 1,1 до 1,5 эквивалентов по отношению к соединению формулы (III).
Между тем, продукт реакции на стадии (iii) может варьироваться таким образом, чтобы содержать 1 до 10 эквивалентов, предпочтительно 2 до 5 эквивалентов воды по отношению к соединению формулы (III), например, путем добавления водного раствора к нему, например водной неорганической кислоты, имеющей соответствующую концентрацию, либо водного растворителя, такого как 95% этанол, для того чтобы эффективно удалить изоалкилиденовую группу. Растворитель, используемый на стадии (iii), может быть выбран из группы, состоящей из ацетонитрила, тетрагидрофурана, 1,4-диоксана, этанола, метанола и изопропанола, предпочтительно, ацетонитрила.
Для удаления изоалкилиденовой группы стадия (iii) может быть проведена при температуре кипячения растворителя в течение от 4 до 8 часов; и образующаяся смесь может быть смешана с таким растворителем, как бензол и толуол, азеотропно перегнана для удаления воды из реакционной смеси с получением лактонизованного соединения формулы (II).
На стадии (iv) защитная группа может быть выбрана из группы, состоящей из бензоила, фенилбензоила и замещенного бензоила, предпочтительно 2-фенилбензоила, 4-фенилбензоила и замещенного 2-(или 4-) фенилбензоила.
Также стадия (iv) может быть проведена после выделения соединения формулы (II), полученного на стадии (iii), или проведена без процесса выделения in situ.Предпочтительным является способ in situ.
Соединение формулы (I), полученное по способу изобретения, демонстрирует высокую чистоту, примерно 99%, соединения 1-оксорибозы, имеющего требуемую эритро-структуру.
Более того, способ изобретения дает общий выход от 45 до 50%, который является улучшенным более чем на 20% по сравнению с обычными способами.
Последующие примеры предназначены для дополнительного пояснения настоящего изобретения, не ограничивая его рамки.
Получение 1: Получение 2,2-дифтор-3-гидрокси-3-(2,2-диметил-[1,3]диоксоран-4-ил)пропионата (соединение формулы (V))
Стадия 1: Получение 1,2-бис-(2,2-диметил-1,3-диоксоран-4-ил)этан-1,2-диола

100 г d-маннита смешали со 160 мл 2,2-диметоксипропана, 240 мл 1,2-диметилэтандиола и 0,1 г безводного SnCl2, смесь нагрели до получения гомогенного раствора, кипятили с обратным холодильником в течение 30 мин и добавили в нее 0,2 мл пиридина. Реакционную смесь охладили до комнатной температуры и перегнали при пониженном давлении для удаления растворителя. К остатку добавили 700 мл метилхлорида и кипятили с обратным холодильником в течение 1 часа. Образовавшуюся смесь отфильтровали через 10 г целлита при комнатной температуре и для удаления растворителя перегнали фильтрат при пониженном давлении. Остаток перекристаллизовали из 1 л гексана, отфильтровали и высушили, получив 72,4 г (выход 50%) названного соединения в виде белого твердого вещества.
ЯМР (300 МГц, CDCl3): 1,30 (с, 6H), 1,36 (с, 6H), 2,52 (д, 2H), 3,67 (т, 2H), 3,91 (м, 2H), 4,04-4,14 (м, 4H).
Температура плавления (Т. пл.): 119-121°С.
Стадия 2: Получение 2,2-диметил-[1,3]-диоксоран-4-карбальдегида

72,4 г соединения, полученного на стадии 1, растворили в 724 мл метилхлорида и добавили к нему 30 мл насыщенного гидрокарбоната натрия. Смесь охладили на водяной бане и маленькими порциями в течение 20 минут добавляли к ней 118 г метапериодата натрия, поддерживая температуру ниже 25°С. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. После того как завершение реакции было подтверждено тонкослойной хроматографией (ТСХ), к реакционной смеси добавили 36 г безводного сульфата магния и перемешивали в течение 20 мин. Образовавшуюся смесь отфильтровали и для удаления растворителя перегнали при пониженном давлении при 30°С, и остаток был дополнительно подвергнут дистилляции при атмосферном давлении при 55°С для полного удаления растворителя. Реакционный остаток перегнали при 10 торр при примерно 40°С, получив 61,6 г (выход 86%) названного соединения в виде бесцветной жидкости.
ЯМР (300 МГц, CDCl3): 1,41 (с, 3H), 1,47 (с, 3H), 4,07-4,19 (м, 2H), 4,35˜4,40 (м, 1H), 9,71 (с, 1H).
Стадия 3: Получение этил-2,2-дифтор-3-гидрокси-3-(2,2-диметил-[1,3]диоксоран-4-ил)пропионата

К 26 мл тетрагидрофурана добавили 12 г цинка, туда же добавили 0,51 мл дибромэтана, и реакционную смесь выдержали в течение 1 мин при 60°С. Туда же при 40°С добавили 0,76 мл триметилсилилхлорида и оставили смесь взаимодействовать на 10 мин. Реакционную смесь нагрели до 60°С, прибавили по каплям раствор, приготовленный из 25,5 мл этилового эфира бромдифторуксусной кислоты, 30,8 г соединения, полученного на стадии 2, и 39 мл тетрагидрофурана, и кипятили смесь с обратным холодильником в течение 30 мин. После добавления в нее 65 мл диэтилового эфира и 260 г льда, добавили 260 мл 1 N HCl и перемешивали до полного таяния льда. Водный слой трижды проэкстрагировали диэтиловым эфиром порциями по 90 мл, объединенные органические слои последовательно промыли NaCl и гидрокарбонатом натрия порциями по 65 мл, высушили над безводным сульфатом магния и отфильтровали. После удаления растворителя остаток перегнали при 10 торр, получив 28,9 г (57%) названное соединение (R:S=3:1) при 130-140°С в виде бесцветной жидкости.
ЯМР (300 МГц, CDCl3): 1,31˜1,52 (м, 9H), 2,67 (с, 1H, (R)-OH), 2,90 (д, 1H, (S)-OH), 3,7˜4,4 (м, 6H).
В следующих Примерах термин "-OCOBiPh" или "BiPhOCO-" относится к 
ВЭЖХ анализы соединений формулы (I) и (III) были проведены на колонке YMC pack pro C18 RS (4,6×150 мм, 5 мкм) с использованием в качестве элюента смеси буфера и ацетонитрила (65:35, об/об) (для соединения формулы (III)) или 80% ацетонитрила (для соединения формулы (I)). Буфер был получен смешением 7,0 г NaClO4, 1,74 г K2HPO4 и 1 л воды и добавлением H3PO4 до рН 2,75.
Пример 1: Получение этил-2,2-дифтор-3-(4-бифенилкарбонил)окси-3-(2,2-диметил-[1,3]диоксоран-4-ил)пропионата (соединение формулы (IV); R=4-бикарбонил)

К 50,0 г соединения, полученного в Получении 1, добавили 500 мл хлористого метилена, добавили к ним 42 мл триэтиламина и 51,1 г 4-бифенилкарбонилхлорида и оставили смесь при комнатной температуре на 6 часов. После добавления к ней 360 мл 1N HCl, органический слой последовательно промыли гидрокарбонатом натрия и NaCl и высушили над сульфатом магния. Остаток отфильтровали и перегнали при пониженном давлении, получив 83,7 г (выход 98%) названного соединения в виде жидкости кремового цвета.
ЯМР (300 МГц, CDCl3): 1,25˜1,74 (м, 9H), 4,11˜4,19 (м, 2H), 4,30˜4,36 (м, 2H), 4,56˜4,58 (м, 2H), 5,72˜5,83 (ддд, 1H×1/3), 5,88˜6,02 (ддд, 1H×2/3), 7,42˜7,53 (м, 3H), 7,63˜7,73 (дд, 4H), 8,15˜8,17 (д, 2H).
Пример 2: Получение 2,2-дифтор-3R-(4-бифенилкарбонил)окси-3-(2,2-диметил-[1,3]диоксоран-4-ил)пропионата калия (соединение формулы (III); R=4-бифенилкарбонил)

Способ А
К 83,8 г соединения, полученного в примере 1, добавили 1,4 л смеси тетрагидрофурана и метанола (2:3, об/об), добавили туда 107 г карбоната калия, растворенного в 750 мл воды. Смесь перемешивали в течение 30 мин и оставили при пониженном давлении для удаления органического растворителя. После фильтрации твердое вещество добавили к 100 мл эфира, перемешали, отфильтровали, промыли эфиром и высушили, получив 60,1 г (выход 70%) названного соединения в виде белого твердого вещества.
ВЭЖХ: R-изомер 99,86%, S-изомер 0,11%.
ЯМР (300 МГц, ДМСО): 1,07 (с, 3H), 1,22 (с, 3H), 3,99 (т, 1H), 4,11 (т, 1H), 4,49 (т, 1H), 5,88 (ддд, 1H), 7,38˜7,54 (м, 3H), 7,75 (д, 2H), 7,85 (д, 2H), 8,07 (д, 2H).
Способ В
К 90,4 г соединения, полученного в примере 1, добавили 600 мл смеси тетрагидрофурана и метанола (1:1, об/об), добавили туда 54,4 г карбоната калия, растворенного в 500 мл воды. Смесь перемешивали в течение 1 часа, дважды промыли гексаном порциями по 500 мл, проэкстрагировали 500 мл этилацетата и оставили при пониженном давлении для удаления растворителя. Образовавшееся твердое вещество смешали со 100 мл воды и 300 мл изо-пропилового спирта, нагревали до растворения и добавили к этому 700 мл изо-пропилового спирта. Образовавшуюся смесь выдержали при комнатной температуре в течение 2 часов для перекристаллизации твердого вещества, которое было отфильтровано, промыли изо-пропиловым спиртом и высушили, получив 62,5 г (выход 65%) названного соединения в виде белого твердого вещества.
ВЭЖХ: R-изомер 99,91%, S-изомер 0,06%.
ЯМР (300 МГц, ДМСО): 1,07 (с, 3H), 1,22 (с, 3H), 3,99 (т, 1H), 4,11 (т, 1H), 4,49 (т, 1H), 5,88 (ддд, 1H), 7,38˜7,54 (м, 3H), 7,75 (д, 2H), 7,85 (д, 2H), 8,07 (д, 2H).
Пример 3: Получение D-эритро-2-дезокси-2,2-дифторопентофураноз-1-илоз-3-(4-фенил)бензоата (соединение формулы (II); R=4-бифенилкарбонил, R3=бензоил)
Способ А: Получение выделением каждого продукта на каждой стадии
Стадия 1: Получение D-эритро-2-дезокси-2,2-дифторопентофураноз-1-илоз-3-(4-фенил)бензоата (соединение формулы (II); R=4-бифенилкарбонил)

10 г соединения, полученного в примере 2, диспергировали в 60 мл ацетонитрила, добавили туда 2,5 мл 12N HCl и кипятили смесь с обратным холодильником в течение 6 часов. К ней добавили 60 мл толуола и перегнали реакционную смесь для удаления растворителя. Этот процесс повторили дважды. К остатку добавили 100 мл эфира, отфильтровали для удаления KCl и перегнали при пониженном давлении для удаления растворителя. К образовавшемуся остатку добавили 50 мл эфира и туда добавили 100 мл гексана для того, чтобы вызвать кристаллизацию твердого вещества. Твердое вещество выделили фильтрацией (первая порция твердого вещества); и перегнали фильтрат при пониженном давлении и подвергли его второй стадии перекристаллизации, используя 20 мл эфира и 50 мл гексана, получив вторую порцию твердого вещества. Твердые вещества были объединены и высушены под вакуумом с получением 5,9 г (выход 75%) названного вещества в виде белого твердого вещества.
ЯМР (300 МГц, CDCl3): 1,8˜2,4 (шир.д с, 1H), 3,78˜4,02 (дд, 1H), 4,11˜4,13 (дд, 1H), 4,71˜4,73 (м, 1H), 5,79˜5,87 (м, 1H), 7,44˜7,54 (м, 3H), 7,64˜7,66 (д, 2H), 7,21˜7,75 (д, 2H).
Температура плавления (т. пл.): 107-111°С.
Стадия 2: Получение D-эритро-2-дезокси-2,2-дифторо-пентафураноз-1-илоз-5-бензоил-3-(4-фенил)бензоата

15,0 г соединения, полученного на стадии 1, добавили к 150 мл хлористого метилена, и при перемешивании при комнатной температуре в него добавили по каплям 6,9 мл пиридина, туда медленно добавили 7,4 мл бензоилхлорида, растворенного в 40 мл хлористого метилена, поддерживая температуру от 5 до 10°С. Реакционную смесь оставили на 7 часов при комнатной температуре, для нейтрализации пиридина в смеси туда добавили 105 мл 1N HCl и затем добавили туда воду для того, чтобы вызвать отделение органического слоя. Органический слой отделили, последовательно промыли порциями по 100 мл насыщенного гидрокарбоната натрия и NaCl, высушили над сульфатом магния и отфильтровали. Оставшийся раствор выдержали при пониженном давлении, получив твердое вещество кремового цвета. Твердое вещество перекристаллизовали из смеси эфира и гексана (5:1, об/об), получив 16,8 г названного соединения (выход: 86%).
ЯМР (300 МГц, CDCl3): 4,90˜4,75 (ддд, 2H), 5,10 (дд, 1H), 5,87 (ддд, 1H), 7,65˜7,50 (м, 5H), 7,78˜7,67 (м, 3H), 7,81 (д, 2H), 8,13 (д, 2H), 8,23 (д, 2H).
Температура плавления (т. пл.): 130-131°С.
ВЭЖХ чистота: 99,21% (трео-изомер обнаружен не был).
Способ В: получение in situ

Смешали 232 мл ацетонитрила с 38,8 г соединения, полученного в Примере 2, и 9,2 мл 12N HCl, и кипятили смесь с обратным холодильником в течение 6 часов. После добавления к ней 464 мл толуола реакционную смесь перегоняли для удаления воды и ацетонитрила до тех пор, пока температура не стала выше 100°С. Образовавшийся концентрат отфильтровали и выдержали при пониженном давлении, получив пенообразное твердое вещество. Твердое вещество растворили в 300 мл этилацетата, туда при перемешивании добавили 14 мл пиридина и добавили туда 15 мл бензоилхлорида, растворенного в 75 мл этилацетата. Смесь выдержали при комнатной температуре в течение 6 часов и для нейтрализации пиридина к ней добавили 210 мл 1N HCl. Органический слой отделили, последовательно промыли порциями по 150 мл воды, насыщенным гидрокарбонатом натрия и NaCl, высушили над сульфатом магния и выдержали при пониженном давлении, получив твердое вещество кремового цвета. Твердое вещество перекристаллизовали из смеси эфира и гексана (5:1, об/об), получив 28,4 г названного соединения (выход: 72%) в виде белого твердого вещества.
ЯМР (300 МГц, CDCl3): 4,90˜4,75 (ддд, 2H), 5,10 (дд, 1H), 5,87 (ддд, 1H), 7,65˜7,50 (м, 5H), 7,78˜7,67 (м, 3H), 7,81 (д, 2H), 8,13 (д, 2H), 8,23 (д, 2H).
Температура плавления (т.пл.): 130-131°С.
ВЭЖХ чистота: 99,05% (трео-изомер обнаружен не был).
Пример 4: Получение D-эритро-2-дезокси-2,2-дифторопентофураноз-1-илоз-3,5-ди(4-фенил)бензоата (соединение формулы (I); R и R3 = 4-бикарбонил)
Способ А: Получение выделением каждого продукта на каждой стадии
Стадия 1: Получение D-эритро-2-дезокси-2,2-дифторопентофураноз-1-илоз-3-(4-фенил)бензоата (соединение формулы (II); R = 4-бифенилкарбонил)
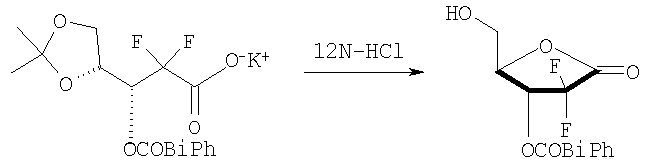
Для получения названного соединения был повторен процесс стадии 1 способа А Примера 2 с получением названного соединения (выход 75%).
Стадия 2: Получение D-эритро-2-дезокси-2,2-дифторопентофураноз-1-илоз-3,5-ди(4-фенил)бензоата

К 20 г соединения, полученного на стадии 1, добавили 300 мл хлороформа, при перемешивании при комнатной температуре туда добавили 9,5 мл пиридина и добавили 10,1 мл 4-бифенилкарбонилхлорида, растворенного в 55 мл хлороформа. Реакционную смесь оставили на 6 часов при комнатной температуре, оставшийся пиридин нейтрализовали 140 мл 1N HCl. Органический слой отделили, последовательно промыли порциями по 150 мл воды, насыщенного гидрокарбоната натрия и NaCl, высушили над сульфатом магния и выдержали при пониженном давлении, получив твердое вещество кремового цвета. Твердое вещество перекристаллизовали из смеси ацетата и гексана (3:1, об/об), получив 21,8 г названного соединения (выход: 72%) в виде белого твердого вещества.
ЯМР (300 МГц, CDCl3): 4,72˜4,79 (м, 2H), 5,03 (кв, 1H), 5,84˜5,76 (м, 1H), 7,48˜7,44 (м, 6H), 7,72˜7,60 (м, 8H), 8,15˜8,07 (м, 4H).
Температура плавления (т. пл.): 137-139°С.
ВЭЖХ чистота: 98,95% (трео-изомер обнаружен не был).
Способ В: Получение in situ

40,0 г соединения, полученного в Примере 2, добавили к 240 мл ацетонитрила, туда добавили 10 мл 12N HCl и кипятили смесь с обратным холодильником в течение 6 часов. После добавления к ней 250 мл толуола, реакционную смесь перегнали для удаления воды и ацетонитрила, охладили до комнатной температуры, отфильтровали и выдержали при пониженном давлении, получив 5-гидрокси-1-оксорибозу в качестве промежуточного соединения. Промежуточное соединение растворили в 480 мл этилацетата, туда добавили смесь 21,8 мл пиридина, 39 г 4-бифенилкарбонилхлорида и оставили взаимодействовать при комнатной температуре в течение 12 часов. Для нейтрализации оставшегося пиридина к реакционной смеси добавили 320 мл 1N HCl; органический слой отделили, последовательно промыли порциями по 160 мл воды, насыщенным гидрокарбонатом натрия и NaCl, высушили и отфильтровали. Фильтрат выдержали при пониженном давлении для удаления растворителя и остаток перекристаллизовали из смеси этилацетата и гексана (3:1, об/об), получив 31,9 г названного соединения (выход: 65%) в виде белого твердого вещества.
ЯМР (300 МГц, CDCl3): 4,72˜4,79 (м, 2H), 5,03 (кв, 1H), 5,84˜5,76 (м, 1H), 7,48˜7,44 (м, 6H), 7,72˜7,60 (м, 8H), 8,15˜8,07 (м, 4H).
Температура плавления (т. пл.): 137-139°С.
ВЭЖХ чистота: 98,33% (трео-изомер обнаружен не был).
Хотя изобретение было описано относительно конкретных вышеназванных воплощений, следует осознавать, что специалистом в данной области могут быть сделаны различные модификации и изменения, которые также попадают в рамки изобретения, как определено в прилагаемой формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1-α-ГАЛОГЕН-2,2-ДИФТОР-2-ДЕЗОКСИ-D-РИБОФУРАНОЗЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2005 |
|
RU2346948C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2'-ДЕЗОКСИ-2', 2'-ДИФТОРЦИТИДИНА | 2005 |
|
RU2360919C2 |
| АМИДНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ АГОНИСТОВ GRP119 | 2014 |
|
RU2642429C2 |
| ТРИЦИКЛИЧЕСКОЕ ТЕТРАГИДРОИЗОХИНОЛИНОВОЕ ПРОИЗВОДНОЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2021 |
|
RU2830109C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВОРИКОНАЗОЛА | 2008 |
|
RU2434009C1 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА В КАЧЕСТВЕ АГОНИСТОВ GPR119 | 2013 |
|
RU2603346C2 |
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ ЦИКЛИЧЕСКОЙ СТРУКТУРОЙ | 2018 |
|
RU2795119C2 |
| БИАРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛОК-БЕЛКОВОГО ВЗАИМОДЕЙСТВИЯ YAP/TAZ-TEAD | 2021 |
|
RU2830596C1 |
| СПОСОБ ПОЛУЧЕНИЯ КАПЕЦИТАБИНА И ИСПОЛЬЗУЕМОГО ПРИ ЭТОМ ОБОГАЩЕННОГО β-АНОМЕРОМ ТРИАЛКИЛКАРБОНАТНОГО СОЕДИНЕНИЯ | 2008 |
|
RU2439064C1 |
| ПРОИЗВОДНОЕ ОЛИГОСАХАРИДОВ | 2004 |
|
RU2290408C2 |
Предложен способ получения производного 2,2-дифтор-2-дезокси-1-оксорибозы (I), где R является 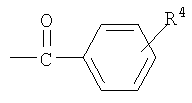 ; R1 - метил или этил; R2 - C1-3алкил; R3 - бензоил или ; R4 - фенил; и М - натрий или калий; включающий стадии: (i) взаимодействия соединения (V) с производным бифенилкарбонила с получением соединения (IV), имеющего 3-гидроксигруппу, защищенную бифенилкарбонильной группой; (ii) взаимодействия соединения (IV) с основанием (карбонат натрия или карбонат калия) в смешанном растворителе, включающем главным образом воду (смесь воды, тетрагидрофурана и метанола), с получением смеси 3R-энантиомера карбокислата (III) с 3S-энантиомером карбокислата (III) и выделения из смеси 3R-энантиомера карбокислата (III) (предложенного также в качестве нового соединения); (iii) взаимодействия соединения (III) с 12N HCl с получением производного 5-гидрокси-1-оксорибозы (II); и (iv) введения защиты R3 на 5-гидроксигруппу соединения (II). Способ изобретения характеризуется возможностью селективного получения 3R-энантиомера карбокислата (III) путем защиты 3-гидроксильной группы соединения формулы (V) бифенилкарбонильной группой и получения производного 2,2-дифтор-2-дезокси-1-оксорибозы формулы (I), имеющего требуемую эритро-структуру и являющегося важным промежуточным соединением, используемым в получении гемцитабина формулы (А), агента для лечения немелкоклеточного рака легкого. 2 н. и 4 з.п. ф-лы.
; R1 - метил или этил; R2 - C1-3алкил; R3 - бензоил или ; R4 - фенил; и М - натрий или калий; включающий стадии: (i) взаимодействия соединения (V) с производным бифенилкарбонила с получением соединения (IV), имеющего 3-гидроксигруппу, защищенную бифенилкарбонильной группой; (ii) взаимодействия соединения (IV) с основанием (карбонат натрия или карбонат калия) в смешанном растворителе, включающем главным образом воду (смесь воды, тетрагидрофурана и метанола), с получением смеси 3R-энантиомера карбокислата (III) с 3S-энантиомером карбокислата (III) и выделения из смеси 3R-энантиомера карбокислата (III) (предложенного также в качестве нового соединения); (iii) взаимодействия соединения (III) с 12N HCl с получением производного 5-гидрокси-1-оксорибозы (II); и (iv) введения защиты R3 на 5-гидроксигруппу соединения (II). Способ изобретения характеризуется возможностью селективного получения 3R-энантиомера карбокислата (III) путем защиты 3-гидроксильной группы соединения формулы (V) бифенилкарбонильной группой и получения производного 2,2-дифтор-2-дезокси-1-оксорибозы формулы (I), имеющего требуемую эритро-структуру и являющегося важным промежуточным соединением, используемым в получении гемцитабина формулы (А), агента для лечения немелкоклеточного рака легкого. 2 н. и 4 з.п. ф-лы.

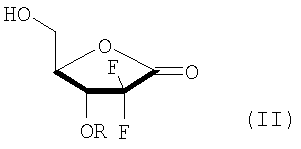



(ii) взаимодействия соединения формулы (IV) с основанием в смешанном растворителе, включающем главным образом воду, с получением смеси 3R-энантиомера карбокислата формулы (III) с 3S-энантиомером карбокислата формулы (III) и выделения из смеси 3R-энантиомера карбокислата формулы (III);
где основание представляет собой карбонат натрия или карбонат калия и смешанный растворитель, включающий главным образом воду, представляет собой смесь воды, тетрагидрофурана и метанола;
(iii) взаимодействия соединения формулы (III) с 12N HCl с получением производного 5-гидрокси-1-оксорибозы формулы (II); и
(iv) введения защиты R3 на 5-гидроксигруппу соединения формулы (II)



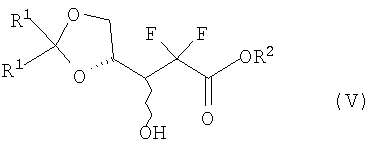
где R является 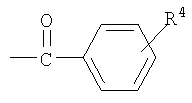 ;
;
R1 является метилом или этилом;
R2 является С1-3алкилом;
R3 является бензоилом или ;
R4 является фенилом; и
М является натрием или калием.
где R является ;
R1 является метилом или этилом;
R4 является фенилом; и
М является натрием или калием.
| US 4526988 А, 02.07.1985 | |||
| US 5945547 А, 31.08.1999 | |||
| US 4965374 А, 23.10.1990 | |||
| US 5464826 А, 07.11.1995 | |||
| US 5428176 А, 27.06.1995 | |||
| Способ получения нуклеозида или его фармацевтически приемлемых солей | 1984 |
|
SU1442076A3 |

