[Область техники]
[1] Настоящее изобретение относится к способу получения пирролопиридинового производного, проявляющего противовирусную активность.
[Уровень техники]
[2] Синдром приобретенного иммунодефицита (СПИД) возникает в результате инфицирования вирусом иммунодефицита человека (ВИЧ). Для лечения СПИДа, в соответствии с механизмом действия ВИЧ, разработаны ингибиторы ферментов. В соответствии со способом действия, ингибиторы ферментов подразделяют на нуклеозидные ингибиторы обратной транскриптазы (NRTI), ингибиторы протеазы (PI), ингибиторы слияния и ингибиторы интегразы. Поскольку для ингибиторов обратной транскриптазы, ингибиторов протеазы и ингибиторов слияния характерны такие недостатки как побочные эффекты, межлекарственные взаимодействия, лекарственная устойчивость и так далее, то активно продолжается разработка ингибиторов интегразы.
[3] По механизму их действия ингибиторы интегразы делят на ингибиторы каталитического сайта и ингибиторы некаталитического сайта. Иллюстративным примером ингибитора каталитического сайта интегразы является ралтегравир. Механизм ингибирования не каталитического сайта интегразы описан авторами Zeger Debyser et al. (Frauke Christ, Zeger Debyser at al., Nature Chemical Biology, 2010, том 6, 442), и в его отношении не было успешно разработано ни одно лекарственное средство.
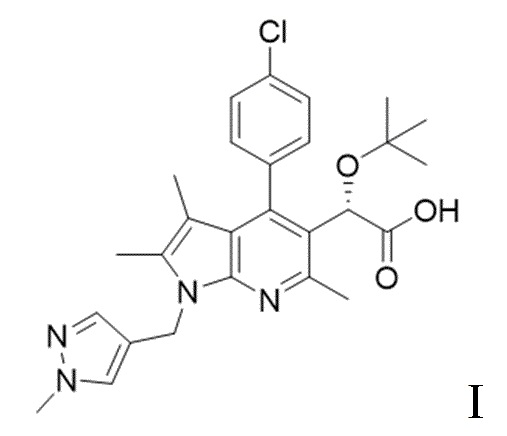
[4] Однако также было обнаружено, что ралтегравир, ингибитор каталитического сайта интегразы, проявляет лекарственную устойчивость. В случае прекращения приема лекарственного средства при ВИЧ принимаемое лекарственное средство перестает быть эффективным, поскольку происходит реактивация латентного ВИЧ и развивается лекарственная устойчивость, и с учетом этого предпринимаются попытки разработки ингибиторов не каталитического сайта интегразы в качестве лекарственного средства, способного решить проблему развития лекарственной устойчивости. В частности, в качестве ингибитора некаталитического сайта интегразы известно пирролопиридиновое производное, представленное следующей химической формулой I:
[5] [Химическая формула I]
[6] 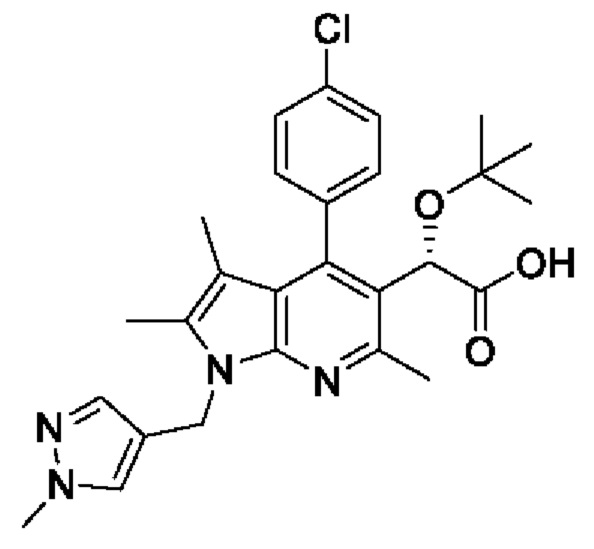
[7] В международной публикации WO 2013/073875 отсутствует непосредственное упоминание соединения химической формулы I, но описан способ получения производных, охватывающих соединение химической формулы I. Однако поскольку для получения производных, подобных соединению химической формулы I, необходимо множество стадий (в общей сложности 16 стадий), то вышеуказанный способ получения не подходит для массового производства.
[8] Кроме того, в международной публикации WO 2018/174320 представлено непосредственное описание соединения химической формулы I и способа его получения. Однако поскольку способ его получения был осуществлен в соответствии со способом получения, описанным в международной публикации WO 2013/073875, и для всех стадий требуется очистка с использованием колонки, то такой способ получения не пригоден для массового производства.
[9] Таким образом, необходимо разработать новый способ получения, обеспечивающий возможность получения пирролопиридинового производного химической формулы I с высоким выходом и высокой чистотой, путем усовершенствования вышеуказанного неэффективного способа получения.
[Сущность изобретения]
[Техническая проблема]
[10] Задача настоящего изобретения заключается в разработке способа получения пирролопиридинового производного, обеспечивающего возможность получения пирролопиридинового производного с высокой чистотой и высоким выходом, со снижением тем самым производственных затрат и обеспечением эффективных технологических стадий, пригодных для массового производства.
[11] Кроме того, другая задача настоящего изобретения заключается в обеспечении нового промежуточного соединения, применяемого в вышеуказанном способе получения.
[Техническое решение]
[12] Технический результат настоящего изобретения заключается в обеспечении способа получения пирролопиридинового производного формулы I с высокой чистотой и высоким выходом. В одном общем аспекте настоящего изобретения предложен способ получения пирролопиридинового производного, представленного следующей химической формулой I:
[13] [Химическая формула I]
[14] 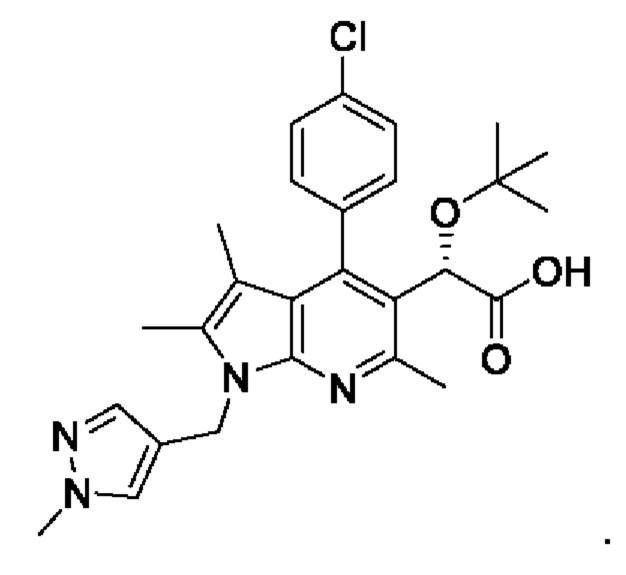
[15] В частности, способ получения согласно настоящему изобретению может включать следующие стадии (S-1) - (S-5):
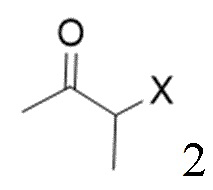
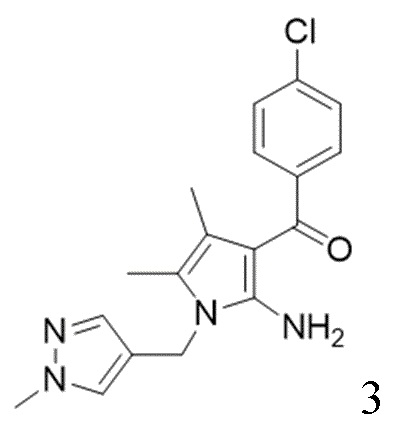
[16] (S-1) первую стадию получения соединения, представленного следующей химической формулой 3, посредством циклизации соединения, представленного следующей химической формулой 1, или его соли, и бутанонового производного, представленного следующей химической формулой 2;
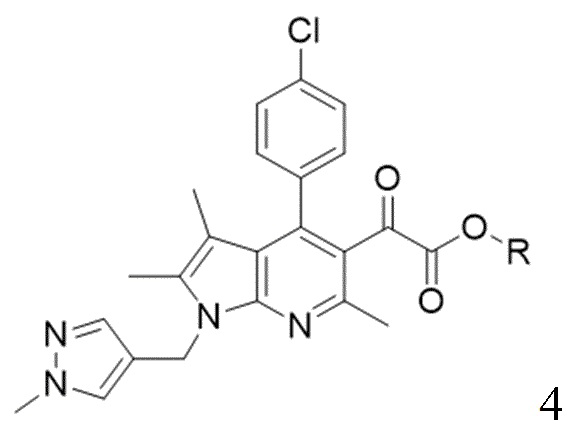
[17] (S-2) вторую стадию получения соединения, представленного следующей химической формулой 4, посредством циклизации соединения, представленного химической формулой 3, и ацетопируватного производного;
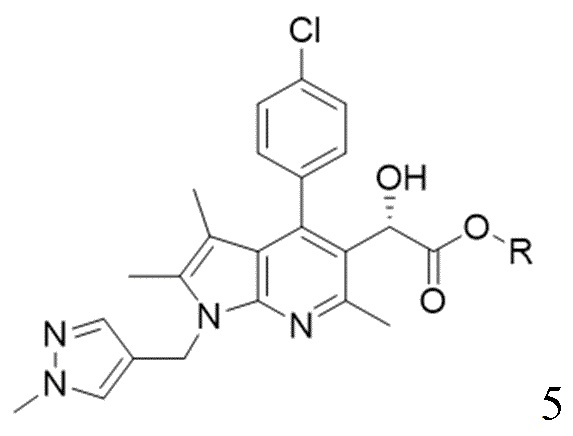
[18] (S-3) третью стадию получения соединения, представленного следующей химической формулой 5, из соединения, представленного химической формулой 4, посредством хирального восстановления;
[19] (S-4) четвертую стадию получения соединения, представленного следующей химической формулой 6, из соединения, представленного химической формулой 5, посредством алкилирования; и
[20] (S-5) пятую стадию получения соединения, представленного следующей химической формулой I, из соединения, представленного химической формулой 6, посредством гидролиза:
[21] [Химическая формула 1]
[22] 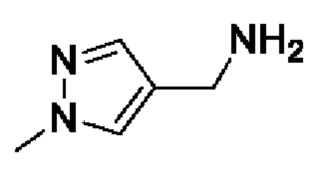
[23] [Химическая формула 2]
[24] 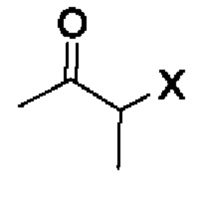
[25] [Химическая формула 3]
[26] 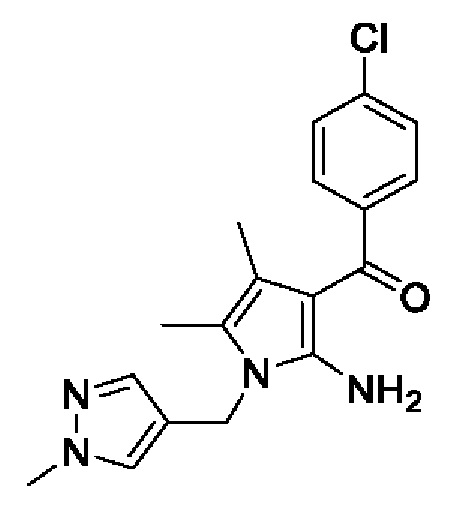
[27] [Химическая формула 4]
[28] 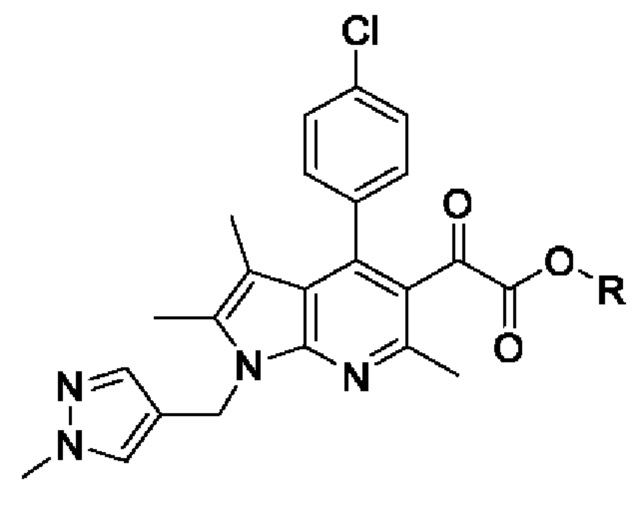
[29] [Химическая формула 5]
[30] 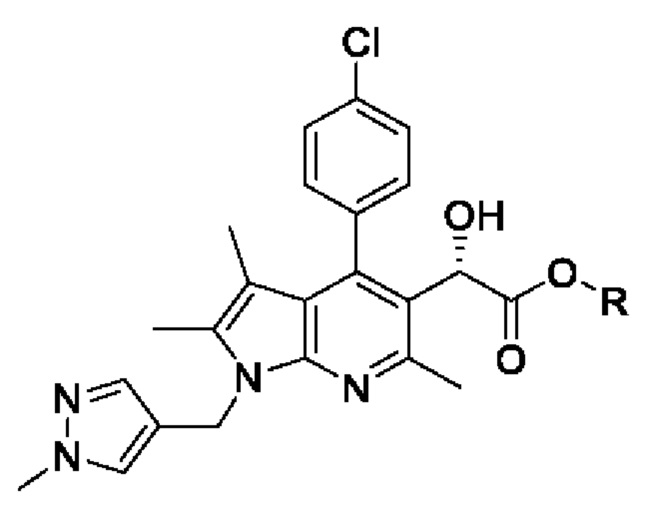
[31] [Химическая формула 6]
[32] 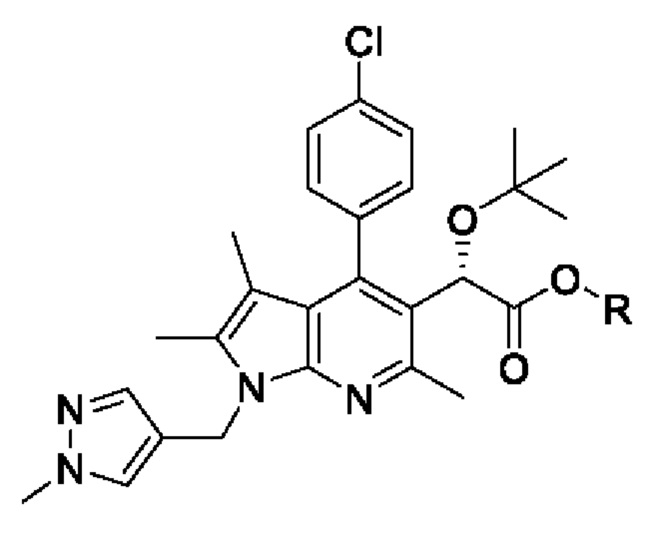
[33] [Химическая формула I]
[34] 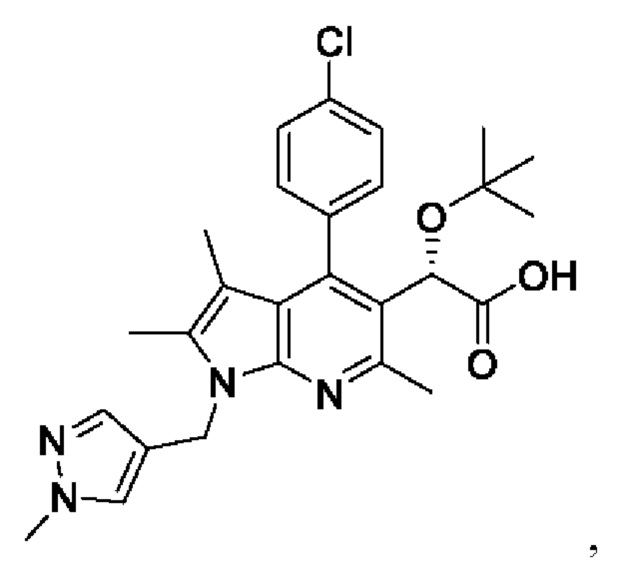
[35] где в представленной выше химической формуле X представляет собой С1, Br или I, и R представляет собой С1-4алкил.
[36] В соответствии со способами получения, описанными в международной публикации WO 2013/073875 и международной публикации WO 2018/174320, соединение, представленное химической формулой I, изображенной выше, получали, используя в общей сложности 16 стадий, что не подходит для массового производства. Однако способ получения согласно настоящему изобретению обеспечивает возможность получения соединения, представленного химической формулой I, с использованием в общей сложности лишь 5 стадий (S-1) - (S-5), и его можно применять для массового производства.
[37] Кроме того, все соединения, представленные химическими формулами 3-6, полученные на стадиях (S-1) - (S-4), являются подходящими промежуточными соединениями для получения пирролопиридинового производного, представленного химической формулой I.
[38] Далее каждая из стадий (S-1) - (S-5) описана по отдельности.
[39] Стадия (S-1)
[40] В настоящем изобретении стадия (S-1) заключается в получении производного пиррольного кольца, представленного химической формулой 3, посредством циклизации пиразольного производного, представленного химической формулой 1, или его соли, и бутанонового производного, представленного химической формулой 2 (схема реакции 1):
[41] [Схема реакции 1]
[42] 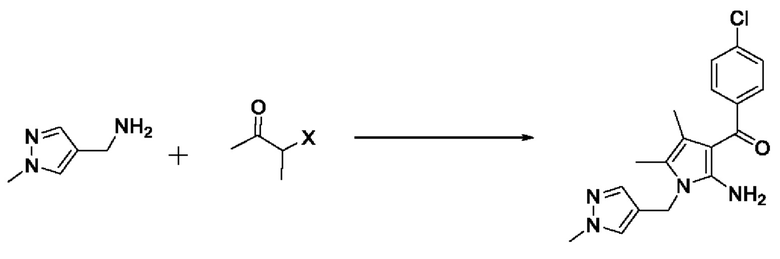
[43] на изображенной выше схеме реакции X представляет собой Cl, Br или I, и R представляет собой С1-4алкил.
[44] В соответствии с одним из вариантов реализации настоящего изобретения, указанную реакцию можно проводить посредством циклизации гидрохлорида (1-метил-1Н-пиразол-4-ил)метанамина вместе с бутаноновым производным (например, 3-хлорбутан-2-оном) и 4-хлорбензоилацетонитрилом.
[45] В вышеуказанной реакции может быть использован органический растворитель, обычно используемый для реакции циклизации. Например, растворитель может представлять собой метанол, этанол, изопропиловый спирт, трет-бутанол, тетрагидрофуран, 1,4-диоксан, ацетон или их смесь. В частности, может быть использован, без ограничения, этанол.
[46] Кроме того, реакцию можно проводить, без ограничения, при 30-60°С, более конкретно при 35-45°С.
[47] После реакции дополнительно могут быть выполнены, без ограничения, одна или более стадий разделения или очистки продукта. Например, в примере согласно настоящему изобретению продукт с высокой степенью чистоты получали посредством перемешивания продукта реакции в органическом растворителе, толуоле.
[48] Стадия (S-2)
[49] Согласно настоящему изобретению, на стадии (S-2) получают пирролопиридиновое производное, представленное химической формулой 4, посредством циклизации с использованием пиррольного производного, представленного химической формулой 3, в качестве исходного вещества (схема реакции 2):
[50] [Схема реакции 2]
[51] 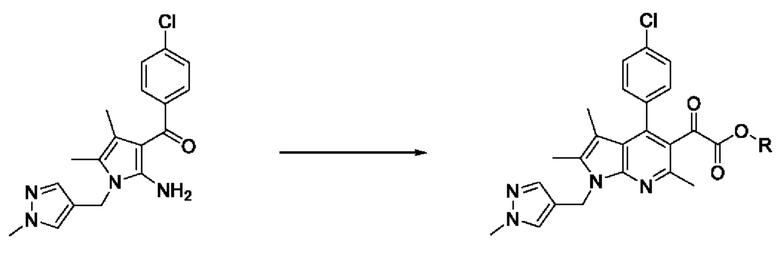
[52] на изображенной выше схеме реакции R представляет собой С1-4алкил.
[53] В соответствии с одним из вариантов реализации настоящего изобретения, в указанной реакции соединение, представленное химической формулой 4, может быть получено посредством взаимодействия с этилацетопируватом. В данном случае R представляет собой этил.
[54] Кислота, используемая в реакции, может представлять собой соляную кислоту, ацетилхлорид, серную кислоту, п-толуолсульфоновую кислоту, метансульфоновую кислоту или их смесь. В частности, может быть использована, без ограничения, соляная кислота.
[55] Кроме того, в вышеуказанной реакции может быть использован органический растворитель, обычно используемый для реакции циклизации. В данном случае в качестве органического растворителя можно использовать этанол, 1,4-диоксан, ацетонитрил, диметилсульфоксид, диметилформамид, диметилацетамид или их смесь. В частности, может быть использован, без ограничения, ацетонитрил.
[56] Кроме того, реакцию можно проводить, без ограничения, при 40-80°С, более конкретно, при 60-65°С.
[57] Стадия (S-3)
[58] Согласно настоящему изобретению на стадии (S-3) получают пирролопиридиновое производное, имеющее структуру хирального спирта, представленную химической формулой 5, посредством восстановления кетоновой группы пирролопиридинового производного, представленного химической формулой 4, методом хирального восстановления (схема реакции 3):
[59] [Схема реакции 3]
[60] 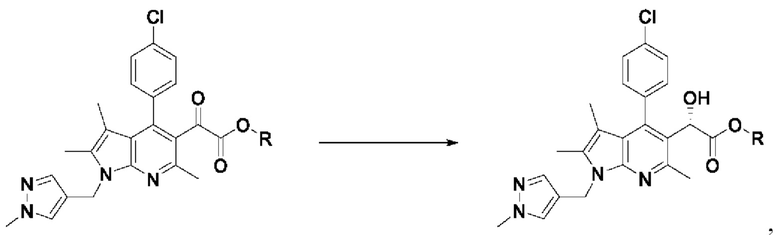
[61] где на изображенной выше схеме реакции R представляет собой С1-4алкил.
[62] В соответствии с одним из вариантов реализации настоящего изобретения, хиральное восстановление можно осуществлять с использованием комбинации (R)-(+)-2-метил-CBS-оксазаборолидина и катехолборана
[63] Кроме того, в соответствии с другим вариантом реализации настоящего изобретения, хиральное восстановление можно проводить с использованием комбинации димера дихлорида (пентаметилциклопентадиенил)родия (III) и (1S,2S)-N-(п-толуолсульфонил)-1,2-дифенилэтандиамина.
[64] В вышеуказанной реакции может быть использован органический растворитель, обычно используемый для хирального восстановления. Например, органический растворитель может представлять собой, без ограничения, толуол, ацетонитрил или их смесь.
[65] Кроме того, реакцию можно проводить, без ограничения, при температуре от -10 до 10°С, более конкретно при температуре от -5 до 5°С.
[66] Стадия (S-4)
[67] Согласно настоящему изобретению, на стадии (S-4) получают пирролопиридиновое производное, представленное химической формулой 6, посредством алкилирования хиральной спиртовой группы пирролопиридинового производного, представленного химической формулой 5 (схема реакции 4):
[68] [Схема реакции 4]
[69] 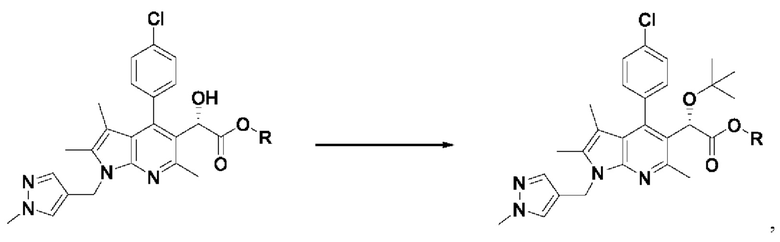
[70] где на изображенной выше схеме реакции R представляет собой С1-4алкил.
[71] В соответствии с одним из вариантов реализации настоящего изобретения, алкилирование можно осуществлять с использованием трет-бутилацетата. В данном случае реакцию можно проводить, без ограничения, в присутствии хлорной кислоты.
[72] В соответствии с другим вариантом реализации настоящего изобретения, алкилирование можно проводить с использованием изобутена. В данном случае реакцию также можно проводить, без ограничения, в присутствии хлорной кислоты.
[73] В вышеуказанной реакции может быть использован органический растворитель, обычно используемый для алкилирования. Например, в качестве органического растворителя можно использовать раствор галогенсодержащего соединения и, в частности, дихлорметановый раствор. Однако примеры органического растворителя не ограничены им.
[74] Кроме того, реакцию можно проводить, без ограничения, при температуре от -5 до 30°С, более конкретно при температуре от -5 до 20°С.
[75] Стадия (S-5)
[76] Согласно настоящему изобретению, на стадии (S-5) получают карбоновую кислоту, представленную химической формулой I, посредством гидролиза сложноэфирного соединения, представленного химической формулой 6 (схема реакции 5):
[77] [Схема реакции 5]
[78] 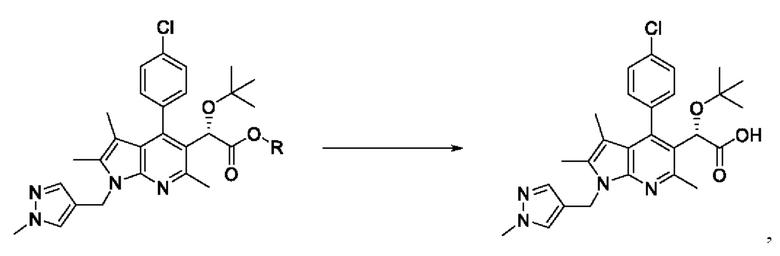
[79] где на изображенной выше схеме реакции R представляет собой С1-4алкил.
[80] В соответствии с одним из вариантов реализации настоящего изобретения, гидролиз может представлять собой щелочной гидролиз. Основание, используемое для реакции, может представлять собой гидроксид лития, гидроксид натрия или гидроксид калия, и может представлять собой, в частности, гидроксид натрия. Однако примеры основания не ограничены ими.
[81] В вышеуказанной реакции может быть использован органический растворитель, обычно используемый для гидролиза. В данном случае в качестве органического растворителя можно использовать тетрагидрофуран, метанол, этанол, изопропиловый спирт, водный раствор или их смесь. В частности, может быть использована, без ограничения, смесь тетрагидрофурана и метанола. [82] Кроме того, реакцию можно проводить, без ограничения, при температуре от 20 до 80°С, более конкретно, при температуре от 40 до 50°С.
[Полезные эффекты]
[83] Способ получения согласно настоящему изобретению обеспечивает возможность уменьшения количества реакционных стадий благодаря разработке эффективного способа получения пирролопиридинового производного с высокой чистотой и высоким выходом, что значительно снижает стоимость производства для обеспечения экономичности, что подходит для массового производства.
[Наилучший вариант реализации]
[84] Далее представлены предпочтительные примеры для облегчения понимания настоящего изобретения. Однако следующие примеры приведены лишь для облегчения понимания настоящего изобретения, и настоящее изобретение не ограничено представленными примерами.
[85] Примеры
[86] В примерах настоящего изобретения пирролопиридиновое производное, представленное химической формулой I, получали в соответствии с изображенной ниже схемой реакции I:
[87] [Схема реакции I]
[88] 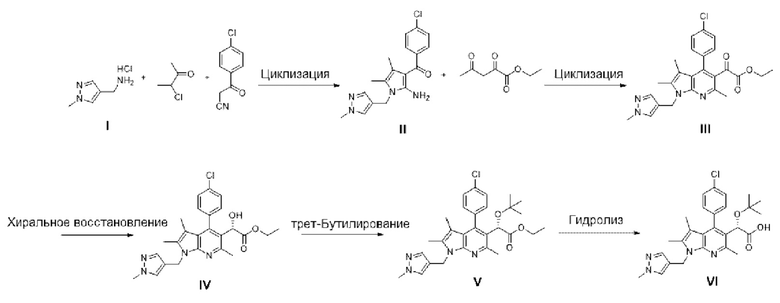
[89] Пример 1: Получение (2-амино-4,5-диметил-1-((1-метил-1Н-пиразол-4-ил)метил)-1Н-пиррол-3-ил)(4-хлорфенил)метанона
[90 ]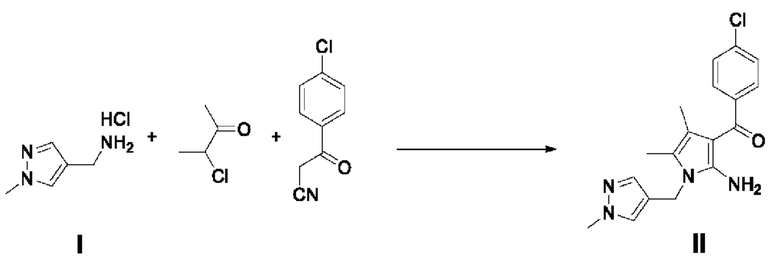
[91] Гидрохлорид (1-метил-1Н-пиразол-4-ил)метанамина (10 г, 67,76 ммоль) разбавляли 135 мл этанола, затем добавляли диизопропилэтиламин (29,5 мл, 169,37 ммоль) и 4-хлорбензоилацетонитрил (13,38 г, 74,52 ммоль) и перемешивали реакционный раствор, повышая температуру до 35-40°С. Медленно, по каплям добавляли 3-хлорбутан-2-он (10,3 мл 101,62 ммоль) в течение 30 минут и перемешивали смесь в течение 3 часов, поддерживая температуру от 35 до 40°С. После завершения реакции охлаждали смесь до 10°С и по каплям добавляли в нее 400 мл воды, после чего перемешивали в течение 30 минут. Отфильтровывали выпавшие в осадок кристаллы и разбавляли полученное твердое вещество 200 мл толуола, после чего перемешивали при 40°С в течение 30 минут. Медленно охлаждали смесь до 15°С и перемешивали в течение 30 минут, затем фильтровали и сушили при пониженном давлении с получением требуемого продукта (17,98 г, 77%).
[92] 1Н-ЯМР 400 Гц (ДМСО-d6): 7,58 (с, 1H), 7,45-7,43 (м, 2Н), 7,35-7,33 (м, 3Н), 7,18 (с, 2Н), 4,80 (с, 2Н), 3,78 (с, 3Н), 1,99 (с, 3Н), 1,36 (с, 3Н);
[93] ЖХМС: m/z 343,1 [М+1].
[94] Пример 2: Получение этил-2-(4-(4-хлорфенил)-2,3,6-триметил-1-((1-метил-1Н-пиразол-4-ил)метил)-1Н-пирроло[2,3-b]пиридин-5-ил)-2-оксоацетата
[95] 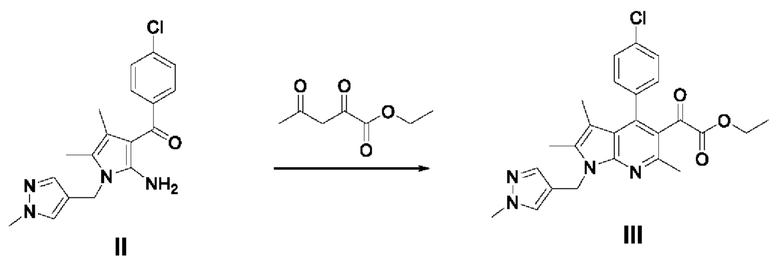
[96] (2-Амино-4,5-диметил-1-((1-метил-1Н-пиразол-4-ил)метил)-1Н-пиррол-3-ил)(4-хлорфенил)метанон (17,6 г, 51,34 ммоль), полученный в примере 1, разбавляли 103 мл ацетонитрила и затем добавляли этилацетопируват (10,81 мл, 77,00 ммоль) в атмосфере азота. Перемешивая реакционный раствор, к нему по каплям добавляли 4 М раствор соляной кислоты в диоксане (38,5 мл, 154,01 ммоль), после чего перемешивали при температуре от 62 до 65°С в течение 20 часов или более и подтверждали завершение реакции с помощью ВЭЖХ. Концентрировали реакционный раствор и разбавляли 528 мл этилацетата и 352 мл насыщенного водного раствора гидрокарбоната натрия при 0°С, затем перемешивали в течение 10 минут и выделяли органический слой при комнатной температуре. Отделенный органический слой дважды промывали 352 мл насыщенного водного раствора гидрокарбоната натрия, обезвоживали безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (этилацетат : гексан = 1:1) с получением требуемого продукта (10,2 г, 42,7%).
[97] 1H-ЯМР 400 Гц (CDCl3): 7,43-7,37 (м, 3Н), 7,26-7,22 (м, 3Н), 5,34 (с, 2Н), 3,86-3,83 (м, 5Н), 2,69 (с, 3Н), 2,32 (с, 3Н), 1,63 (с, 3Н), 1,12 (т, J=7,2 Гц, 3Н);
[98] ЖХМС: m/z 465,1 [М+1].
[99] Пример 3-1: Получение этил-(S)-2-(4-(4-хлорфенил)-2,3,6-триметил-1-((1-метил-1Н-пиразол-4-ил)метил)-1Н-пирроло[2,3-b]пиридин-5-ил)-2-гидроксиацетата
[100] 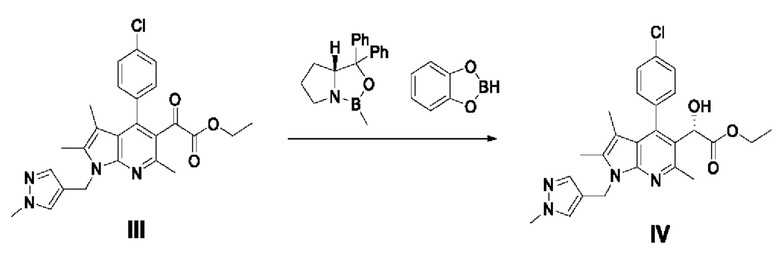
[101] Этил-2-(4-(4-хлорфенил)-2,3,6-триметил-1-((1 -метил-1H-пиразол-4-ил)метил)-1Н-пирроло[2,3-b]пиридин-5-ил)-2-оксоацетат (6,67 г, 14,35 ммоль), полученный в примере 2, разбавляли в 48 мл толуола, а затем добавляли (R)-(+)-2-метил-CBS-оксазаборолин (1 М раствор в толуоле, 5,74 мл, 5,74 ммоль) в атмосфере азота. Реакционную смесь охлаждали до -50°С, затем медленно, по каплям добавляли катехолборан (1 М раствор в тетрагидрофуране, 43,04 мл, 43,04 ммоль) в течение 25 минут и перемешивали при температуре от -10 до -5°С в течение 8 часов. После завершения реакции по каплям добавляли 267 мл гептана и перемешивали. Полученное твердое вещество перемешивали в течение 10 минут и затем фильтровали. Полученное твердое вещество растворяли в 100 мл метанола, перемешивали в течение 15 минут и концентрировали. Концентрированный остаток растворяли в 200 мл этилацетата и охлаждали до 5-10°С, и по каплям добавляли 66 мл водного раствора карбоната натрия, после чего перемешивали в течение 30 минут. Отделенный водный слой экстрагировали, используя 176 мл этилацетата. Органический слой обезвоживали безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с получением требуемого продукта (2,2 г, 32,8%, э.и.: 97%).
[102] 1H-ЯМР 400 Гц (ДМСО-d6): 7,55-7,52 (м, 3Н), 7,33-7,28 (м, 3Н), 5,70 (д, J=3,6 Гц, 1H), 5,31-5,18 (м, 2Н), 4,98 (д, J=3,6 Гц, 1H), 4,08-3,97 (м, 2Н), 3,74 (с, 3Н), 2,56 (с, 3Н), 2,29 (с, 3Н), 1,42 (с, 3Н), 1,10 (т, J=6,8 Гц, 3Н);
[103] ЖХ-МС: m/z 467,1 [М+1].
[104] Пример 3-2: Получение этил-(S)-2-(4-(4-хлорфенил)-2,3,6-триметил-1-((1-метил-1Н-пиразол-4-ил)метил)-1Н-пирроло[2,3-b]пиридин-5-ил)-2-гидроксиацетата (IV)
[105] 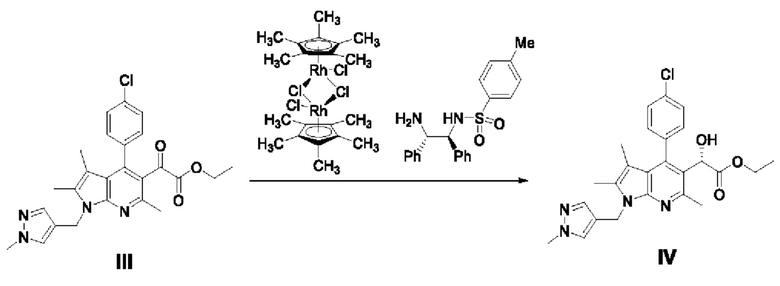
[106] Этил-2-(4-(4-хлорфенил)-2,3,6-триметил-1-((1-метил-1H-пиразол-4-ил)метил)-1Н-пирроло[2,3-b]пиридин-5-ил)-2-оксоацетат (11,71 г, 25,19 ммоль), полученный в примере 2, разбавляли в 84 мл ацетонитрила и добавляли триэтиламин (7,02 мл, 50,4 ммоль) в атмосфере азота, после чего перемешивали в течение 10 минут. После снижения температуры реакции до -5°С или менее медленно, по каплям добавляли муравьиную кислоту (2,85 мл, 75,6 ммоль).
[107] В другой реактор добавляли димер дихлорида (пентаметилциклопентадиенил)родия (III) (0,74 г, 1,2 ммоль), (1S,2S)-N-(п-толуолсульфонил)-1,2-дифенилэтандиамин (1,1 г, 3,0 ммоль) и 27 мл ацетонитрила и перемешивали в течение около 10 минут или более. Поддерживая внутреннюю температуру от 0 до 5°С, добавляли триэтиламин (1,76 мл, 12,6 ммоль) и перемешивали в течение 1 часа, а затем по каплям добавляли к реакционному раствору. Реакцию проводили в течение 60 часов при внутренней температуре от -5 до 5°С. После завершения реакции добавляли 210 мл этилацетата и 180 мл очищенной воды и экстрагировали органический слой, и дважды промывали очищенной водой. Отделенный органический слой сушили над безводным сульфатом натрия и фильтровали с помощью угольного фильтра, и концентрировали фильтрат при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с получением требуемого продукта (9,64 г, 82%, э.и.: 99%).
[108] 1H-ЯМР 400 Гц (ДМСО-d6): 7,55-7,52 (м, 3Н), 7,33-7,28 (м, 3Н), 5,70 (д, J=3,6 Гц, 1H), 5,31-5,18 (м, 2Н), 4,98 (д, J=3,6 Гц, 1H), 4,08-3,97 (м, 2Н), 3,74 (с, 3Н), 2,56 (с, 3Н), 2,29 (с, 3Н), 1,42 (с, 3Н), 1,10 (т, J=6,8 Гц, 3Н);
[109] ЖХ-МС: m/z 467,1 [М+1].
[110] Пример 4-1: Получение этил-(S)-2-(трет-бутокси)-2-(4-(4-хлорфенил)-2,3,6-триметил-1-((1-метил-1Н-пиразол-4-ил)метил)-1Н-пирроло[2,3-b]пиридин-5-ил)ацетата (V)
[111] 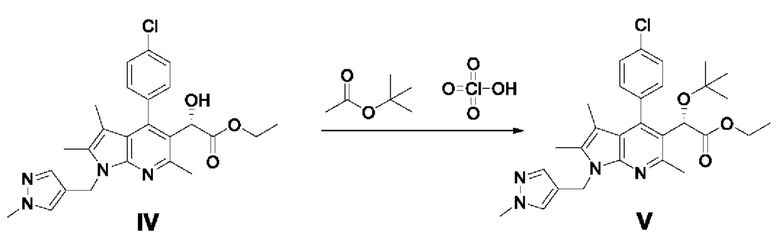
[112] Этил-(S)-2-(4-(4-хлорфенил)-2,3,6-триметил-1-((1 -метил-1 Н-пиразол-4-ил)метил)-1Н-пирроло[2,3-b]пиридин-5-ил)-2-гидроксиацетат (16 г, 34,26 ммоль), полученный в примере 3-1, разбавляли в 68,5 мл дихлорметана и добавляли 456 мл трет-бутилацетата в атмосфере азота. Реакционный раствор охлаждали до 0-5°С, медленно по каплям добавляли 70% хлорную кислоту (11,8 мл, 137,05 ммоль) в течение 1 часа, затем постепенно повышали температуру и перемешивали при 20°С в течение 4 часов. Реакционный раствор охлаждали до 0-5°С и разбавляли 480 мл дихлорметана и 960 мл насыщенного водного раствора карбоната натрия, после чего перемешивали в течение 20 минут для отделения органического слоя. Отделенный органический слой промывали, используя 240 мл воды, обезвоживали безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с получением требуемого продукта (12,4 г, 69%, э.и.: 99%).
[113] 1H-ЯМР 400 Гц (ДМСО-d6): 7,61-7,53 (м, 3Н), 7,42 (дд, J=8,2 Гц, J=2 Гц, 1H), 7,34 (с, 1H), 7,30 (дд, J=8,2 Гц, J=2 Гц, 1H), 5,28-5,18 (м, 2Н), 4,98 (с, 1H), 4,10-4,02 (м, 2Н), 3,74 (с, 3Н), 2,62 (с, 3Н), 2,29 (с, 3Н), 1,41 (с, 3Н), 0,91 (с, 9Н);
[114] ЖХ-МС: m/z 523,2 [М+1].
[115] Пример 4-2: Получение этил-(S)-2-(трет-бутокси)-2-(4-(4-хлорфенил)-2,3,6-триметил-1-((1-метил-1Н-пиразол-4-ил)метил)-1H-пирроло[2,3-b]пиридин-5-ил)ацетата (V)
[116] 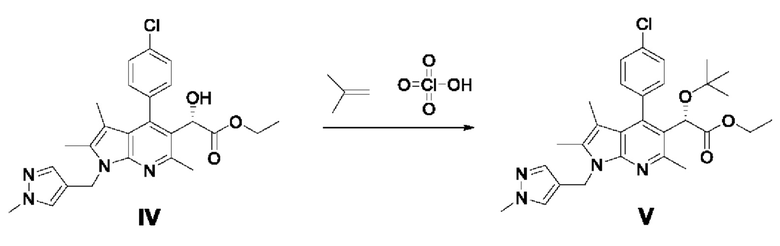
[117] Этил-(S)-2-(4-(4-хлорфенил)-2,3,6-триметил-1-((1 -метил-1 Н-пиразол-4-ил)метил)-1Н-пирроло[2,3-b]пиридин-5-ил)-2-гидроксиацетат (7 г, 14,99 ммоль), полученный в примере 3-1, разбавляли 8% раствором дихлорметана в изобутене (158,1 мл, 300 ммоль). Реакционную смесь охлаждали до температуры от -5 до 0°С и по каплям добавляли 70% хлорную кислоту (4,51 мл, 52,5 ммоль), после чего перемешивали при температуре от 5 до 10°С в течение 24 часов. После завершения реакции к реакционному раствору медленно добавляли 1 н. водный раствор гидроксида натрия (55,5 мл, 57,7 ммоль) при температуре от -5 до 0°С, после чего перемешивали при комнатной температуре в течение 10 минут. Органический слой промывали 50 мл воды, обезвоживали безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле с получением требуемого продукта (6,46 г, 82%, э.и.:99%).
[118] 1H-ЯМР 400 Гц (ДМСО-d6): 7,61-7,53 (м, 3Н), 7,42 (дд, J=8,2 Гц, J=2 Гц, 1H), 7,34 (с, 1H), 7,30 (дд, J=8,2 Гц, J=2 Гц, 1H), 5,28-5,18 (м, 2Н), 4,98 (с, 1H), 4,10-4,02 (м, 2Н), 3,74 (с, 3Н), 2,62 (с, 3Н), 2,29 (с, 3Н), 1,41 (с, 3Н), 0,91 (с, 9Н);
[119] ЖХ-МС: m/z 523,2 [М+1].
[120] Пример 5: Получение (S)-2-(трет-бутокси)-2-(4-(4-хлорфенил)-2,3,6-триметил-1-((1-метил-1Н-пиразол-4-ил)метил)-1Н-пирроло[2,3-b]пиридин-5-ил)уксусной кислоты
[121] 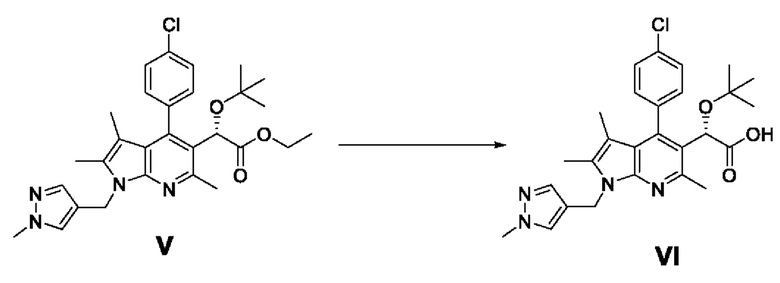
[122] Этил-(S)-2-(трет-бутокси)-2-(4-(4-хлорфенил)-2,3,6-триметил-1-((1-метил-1Н-пиразол-4-ил)метил)-1Н-пирроло [2,3-b]пиридин-5-ил)ацетат (13,5 г, 25,8 ммоль), полученный в примере 4-1, разбавляли в 108 мл тетрагидрофурана и 27 мл метанола. Добавляли гидроксид натрия (3,1 г, 77,4 ммоль) и перемешивали полученную смесь при 40-45°С в течение 4 часов. После завершения реакции концентрировали смесь при пониженном давлении и добавляли к ней 120 мл дихлорметана и 60 мл очищенной воды. Полученную смесь охлаждали до 0-5°С и доводили значение рН до 4,5-5,0 с помощью 2 н. водного раствора соляной кислоты. Отделяли органический слой, промывали 60 мл очищенной воды, обезвоживали безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. К полученному остатку добавляли 30 мл ацетонитрила и перемешивали полученную смесь в течение 3 часов при охлаждении до 5-10°С. Выпавшие в осадок кристаллы отфильтровывали и сушили при пониженном давлении с получением требуемого продукта (9,25 г, 72%, э.и.: 99%) в виде белого твердого вещества.
[123] 1H-ЯМР 400 Гц (ДМСО-d6): 7,60 (дд, J=8,4 Гц, J=2,4 Гц, 1H), 7,56-7,53 (м, 2Н), 7,46 (дд, J=8 Гц, J=2,4 Гц, 1H), 7,35 (с, 1H), 7,30 (дд, J=8,4 Гц, J=2,4 Гц, 1H), 5,28-5,19 (м, 2Н), 4,93 (с, 1Н), 3,74 (с, 3Н), 2,63 (с, 3Н), 2,29 (с, 3Н), 1,42 (с, 3Н), 0,89 (с, 9Н);
[124] ЖХ-МС: m/z 495,2 [М+1].
[125] Были подробно описаны некоторые элементы настоящего изобретения. Специалистам в данной области техники понятно, что изложенное выше конкретное описание относится лишь к предпочтительным вариантам реализации, и объем настоящего изобретения не ограничен ими. Таким образом, объем настоящего изобретения определяется прилагаемой формулой изобретения и его эквивалентами.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИРРОЛОПИРИДИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2021 |
|
RU2815649C1 |
| НОВОЕ ПИРРОЛОПИРИДИНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2017 |
|
RU2733723C1 |
| ИЗОИНДОЛИНОНОВЫЕ ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ MDM2-P53, ОБЛАДАЮЩИЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2794333C1 |
| ИЗОИНДОЛИНОНОВЫЕ ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ MDM2-P53, ОБЛАДАЮЩИЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2797295C1 |
| ПИРРОЛОПИРИМИДИНОВОЕ СОЕДИНЕНИЕ | 2015 |
|
RU2701206C2 |
| Соединение-ингибитор мультикиназ и его кристаллическая форма и применение | 2017 |
|
RU2723985C1 |
| ИНГИБИТОРЫ КАНАЛОВ С ТРАНЗИТОРНЫМ ПОТЕНЦИАЛОМ НА ОСНОВЕ ОКСАДИАЗОЛОВ | 2019 |
|
RU2818244C2 |
| ПИРАЗОЛБЕНЗОДИАЗЕПИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ CDK2, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ СОДЕРЖАЩАЯ | 2000 |
|
RU2249593C2 |
| НОВЫЕ АМИНОАЛКИЛБЕНЗОТИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2679891C2 |
| АГЕНТЫ, ИНГИБИРУЮЩИЕ Р38 КИНАЗУ | 2010 |
|
RU2532376C2 |
Изобретение относится к способу получения пирролопиридинового производного формулы I, включающему: (S-1) получение соединения формулы 3 посредством циклизации соединения формулы 1, бутанового производного формулы 2 и 4-хлорбензоилацетонитрила при температуре 30-60°С; (S-2) получение соединения формулы 4 посредством циклизации соединения формулы 3 и ацетопируватного производного при температуре 40-80°С; (S-3) получение соединения формулы 5 из соединения формулы 4 посредством хирального восстановления при температуре -10-10°С; (S-4) получение соединения формулы 6 из соединения формулы 5 посредством алкилирования при температуре -5-30°С; (S-5) получение соединения формулы I из соединения формулы 6 посредством гидролиза при температуре 20-80°С; где Х представляет собой Cl, Br или I и R представляет собой С1-4алкил. Также изобретение относится к соединениям формулы 3, 4 и 5, где R представляет собой С1-4алкил. Технический результат - способ получения пирролопиридинового производного формулы I с высокой чистотой и высоким выходом. 4 н. и 9 з.п. ф-лы, 7 пр.
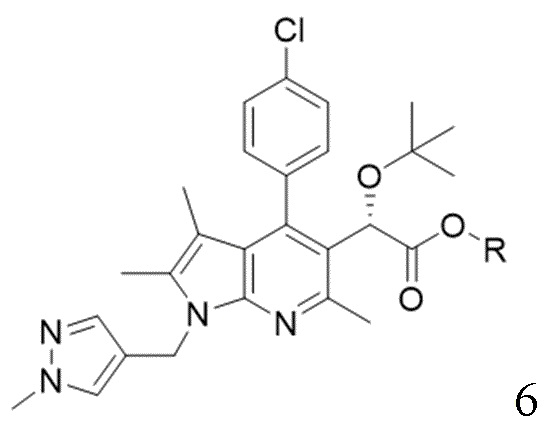
1. Способ получения пирролопиридинового производного, включающий:
(S-1) первую стадию получения соединения, представленного следующей химической формулой 3, посредством циклизации соединения, представленного следующей химической формулой 1, или его соли, бутанонового производного, представленного следующей химической формулой 2, и 4-хлорбензоилацетонитрила при температуре от 30 до 60°С;
(S-2) вторую стадию получения соединения, представленного следующей химической формулой 4, посредством циклизации соединения, представленного химической формулой 3, и ацетопируватного производного при температуре от 40 до 80°С;
(S-3) третью стадию получения соединения, представленного следующей химической формулой 5, из соединения, представленного химической формулой 4, посредством хирального восстановления при температуре от -10 до 10°С;
(S-4) четвертую стадию получения соединения, представленного следующей химической формулой 6, из соединения, представленного химической формулой 5, посредством алкилирования при температуре от -5 до 30°С; и
(S-5) пятую стадию получения соединения, представленного следующей химической формулой I, из соединения, представленного химической формулой 6, посредством гидролиза при температуре от 20 до 80°С:
[Химическая формула 1]
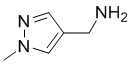 ,
,
[Химическая формула 2]
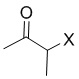 ,
,
[Химическая формула 3]
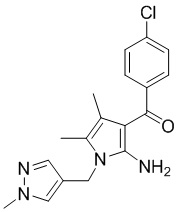 ,
,
[Химическая формула 4]
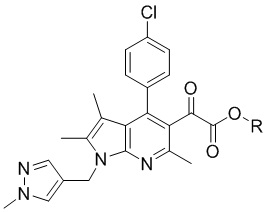 ,
,
[Химическая формула 5]
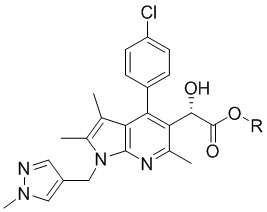 ,
,
[Химическая формула 6]
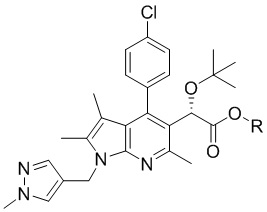 ,
,
[Химическая формула I]
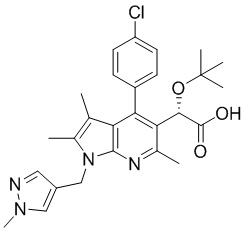 ,
,
где в представленных выше химических формулах:
X представляет собой Cl, Br или I и
R представляет собой C1-4алкил.
2. Способ по п. 1, отличающийся тем, что соль соединения, представленного химической формулой 1, представляет собой гидрохлорид.
3. Способ по п. 1, отличающийся тем, что X представляет собой Cl.
4. Способ по п. 1, отличающийся тем, что стадия (S-1) дополнительно включает процесс разделения или очистки выпавшего в осадок соединения, представленного химической формулой 3.
5. Способ по п. 1, отличающийся тем, что стадия (S-2) представляет собой реакцию с этилацетопируватом.
6. Способ по п. 1, отличающийся тем, что стадия (S-3) представляет собой реакцию с (R)-(+)-2-метил-CBS-оксазаборолидином и катехолбораном.
7. Способ по п. 1, отличающийся тем, что стадия (S-3) представляет собой реакцию с димером дихлорида (пентаметилциклопентадиенил)родия (III) и (1S,2S)-N-(п-толуолсульфонил)-1,2-дифенилэтандиамином.
8. Способ по п. 1, отличающийся тем, что стадия (S-4) представляет собой реакцию с трет-бутилацетатом или изобутеном.
9. Способ по п. 8, отличающийся тем, что стадию (S-4) проводят в присутствии хлорной кислоты.
10. Способ по п. 1, отличающийся тем, что стадия (S-5) представляет собой щелочной гидролиз.
11. Соединение, представленное следующей химической формулой 3, или его соль:
[Химическая формула 3]
.
12. Соединение, представленное следующей химической формулой 4, или его соль:
[Химическая формула 4]
,
где в приведенной выше химической формуле R представляет собой C1-4алкил.
13. Соединение, представленное следующей химической формулой 5, или его соль:
[Химическая формула 5]
,
где в приведенной выше химической формуле R представляет собой C1-4алкил.
| US 20140249162 A1, 04.09.2014 | |||
| KR 1020190141152 A, 23.12.2019 | |||
| WO 2013012649 A1, 24.01.2013 | |||
| KR 1020150141275 A, 18.12.2015 | |||
| Прибор для наметки центра | 1927 |
|
SU14957A1 |

