1148560 .J
Изобретение относится к способу изохинолин-3-карбоновой кислоты получения нового замещенного ацило- общей формулы (I) вого производного 1,2,3.,4-тетрагйдро
,
СНг 1
COOR низший aлкилi низший алкнл, водород-, X и Y - низший алкоксил, воДОРРД-, фенил; водород, низший алкил или бензил или к его фармацевтически приемлемым солям, обладающим биологической активностью. Известна реакция образования пептидной связи взаимодействием, карбоксильной группы с N-защищенной амино-группой в среде органического растворителя с использованием дегидратирующего агента 1J. Цель изобретения - синтез новых соединений, обладающих ценными физиологическими свойствами. Поставленная цель достигается, тем, что согласно способу получения замещенного ацилового производного 1,2,3,Д-тeтpaгидpoизoxинoлин-3-кapбoнoвoй кислоты указанной общей формулы (I), основанному на реакции образования пептидной связи, соединения формулы (II где X и Y имеют приведенные значен R.J - низший алкил о вый или бензи ловый сложный эфир, подвергают взаимодействию с N-заме щенной аминокислотой формулы 2 - CHj - Ш-КН СН-СООН coceti где Аг, Rj, , R имеют указанные зна чения, и вьзделением целевого проду та или с последующим удалением бло
COOR
кирующей группы и выделением целевого продукта в свободном виде или в виде фармацевтически приемлемой соли. .Пример 1. 2-(2{(1-(этоксикарбонил)-3-фенилпропил)-амино -1-оксопропил)-1,2,3,4-тетрагидро-6,7-диметокси-З-изохинолинкарбоновая кислота, гидрохлорид, гидрат (S,S,S). 0,0079 моль гидрохлорида 2-(2- {(1-(зтоксикарбонил)-3-фенилпропил) -амино -1 -оксопропип) -1,2,3,4-тетрагидро-6,7-диметокси-З-изохинолинкар-боновой кислоты, фенилметилового эфира (S S S) растворенного в too мл тетрагидрофурана, подвергают каталитическому дебензилированию водородом под низким давлением в присутствии 0,5 г 20%-го палладия на угле. Затем катализатор фильтруют и продукт осаждают в виде относительно негигроскопического твердого вещества путём добавления 10-кратного количества дизтилового эфира (3,7 г 58%-й выход) с т.пл, 120-140 С, тонкослойная хроматографическая обработка (смесь 20% метанола с хлороформом на .двуокиси кремния): одно пятно,Rf - 0,50,7, (альфа) 2 +31,6 (0,05% этанола) . Найдено,%: С 58,59, Н N 5,06. C.Hj.N.O. Вычислено,%: С 58,63; Н 6,74; N 5,07. . Некристаллический диэфиргидрохлоридный исходньй материал получают обработкой 5,54 г (0,0079 моль) малеатной соли (полученной по примеру 5) с избытком насьлценного раствора бикарбоната натрия, экстракщгонной обработкой свободного основания смесью 50% диэтштового эфира с этилацетатом, обраб(т1(ой этого
раствора избытком хлористого водорода и концентрированием смеси при пониженном давлении.
Пример 2. 2-(2-{(1-(этоксикарбонил)-3 фенш1пропид)-амино t 5 -оксопропил)-1,2,3,Д-тетрагидро-3-изохинрлинкарбоновая кислота, гидрохлррид, гидрат, ().
Процедура А. Дебензилирование.
2- (2- { (1 - (Этоксикарбонил) -3 -фенил-10 пропил)-амино -1-оксопррпил)- 1,2, 3,4-тетрагидро-З-изохинолинкарбоновую кислоту, фенилметиловый эфир, малеат (S,), полученный в соответствии с примером 6, подвергают t5 каталитическому дебеизилированию по примеру 1, в результате чего с достижением 56%-го выхода получают продукт с т.пл. 105-120С, тонко-:слойная хроматографическая обработ- 20 ка (20% метанола в смеси с хлороформом на двуокиси кремния); одно пятно, R ,6, (альфа) д «10,9° (1,03% этанола).
Найдечо,%: С 6t,00, Н 6,37/ 25 N 5,59..
C Hj N Oy-HCIH O
Вычислено,%: С 60,90; Н 6,75, N 5,58.
Процедура В. Отщепление 1,1-дн- JQ метилэтилового эфира. К 11,6 г (0,023 моль) 2-(2-{(1-этоксйкарбонил)-3-фенилпропил)-а:ь1ино -1-оксопррпил)-1,2,3,4-тетрагидро-3 изрхинолинкарбоновой 35 кислоты, 1,t-диметилэтилового сложного эфира (), полученного по примеру 7, добавляют 100 г трифторуксуснРй кислрты. Далее смесь перемешивают при комнатной температуре 40 до образования раствора, а затем еще 1 ч. Большую часть трифторуксусной кислоты удаляют в роторном испарителе, а оставшиеся следы удаляют последовательньм добавлени- 5 ем и удалением в роторном испарителе 2-х порций по 50 мл тетрагидрофурана. Остаточный маслоподобный продукт растворяют приблизительно в 400 мл сухого диэтилрвого эфира 50 и гидрохлорид осаждают добавлением
раствора 1,0 г (избытка) сухого диэтиловогр эфира. После фильтроваШ и промывки сухим диэтилрвым эфиром фильтровальный пирог растворяют 55 приблизительно в 250 мл воды. Этот
раствор фильтруют через пелит и вы сушивают вьмораживанием с получением 10,0 г ( выход) продукта в виде неполного гидрата с температурой плавления 113-120С.
Найдено,%: С 61,51; Н 6,49; N 5,70.
C25.H N20j-HCI-3/4
Вычислено,% с 61,55; Н 6,70; N 5,74.. .
Пример 3. 2-(2-f(1-Kap6oKси-3-фенилпропил)-аминоJ-1-оксопропил)-1,2,3,4-тетрагидро-6,7-д1шетокси-3-изохинолинкарбоноваякислота, гидрохлорид, гидрат (
Раствор 0,553 г (0,001 моль) 2-(2-(1-(этoкcикapбoншI)-3-фeнилпpoпил)-aминo5 -1-oкcoпpoш л)-t , 2 , 3 , 4-тетрагидро-6,7-диметокси-3-изохинолинкарбоновой кислоты, .гидрохлорида, гидрата (S.), полученного по примеру 1, в 4 мл (0,004 моль) 1 Н. раствора гидрата окиси натрия и 4 мл метанола вьщерживают в спокойном состоянии при комнатной температуре в течение 20 ч. Затем этот реакционный раствор добавляют к 5 мл 1 н. соляной кислоты при пониженном давлении. Остаточное количество воды удаляют двумя последовательными добавлениями и удалениями при пониженном давлении 25-миллилитровых порций этанола. Органическую порцию остатка растворяют в 0,5 мл метанола. Далее добавляют 30 мл хлороформа и раствор высушивают над сульфатом натрия, обрабатывают древесным углем, фильтруют и концентрируют с получением 0,45 г продукта.Этот аморфньй продукт растворяют в 20 мл тетрагидрофурана и затем добавляют 100 МП диэтилового эфира, в результате чего в рсадок выпадает 0,4 г практически белого твердого вещества с т.пл. 145-170 С (80%-й выхрд), тонкослойная хроматографическая обработка (смесь 20% метанола с хлороформом, на двуокиси кремния)i
RI - 0,1 (альфа)j5 +37,8 (1,09% метанола).
Найдено,%: С 57,17 г; Н 6,10 N 5,51
CjsHjoNjQ -HCI-HjO
Вычислено,%:. С 67,10 Н 6,34; N 5,34.
П р и м е р 4. 2-2{(1-карбокси-3-фенилпрошш)-аминоу-1-оксопропил)-1,2,3,4-тетрагидро-З-иэохиSнолинкарбоновая кислота, гидрохлорид, полугидрат (). 2(2- {(1-(этоксикарбонил)-3-фенилпропил)-амино V-1-olcconpoпил)-1,2,3,4-тетрагидро-З-иэохинолинкарбоновую кислоту, гидрохлорид, гидрат () обрабатывают в соответствии с примером 3, в результате чего с добавлением 39%-го вькода получают продукт с т.пл. 140-170 С, (альфа)23 +14,5 (1,08% метанола). Найдено,%: С 60,68; Н 6,04; N 5,89J а 7,04. C23HigN205-HCe- 1/2 Н,0 Вычислено,%: С 60,59; Н 5,97; N 6,15; се 7,77. Пример 5. 2-(2-(1-(этоксикарбонил)-3-фенилпропил)-амино1-1-оксопропил)-1,2,3,4-тетрагидро-6, 7-диметокси- З-изохинолинкарбоно вая кислота, фенилметиловьй эфир, малеат (). Перемешиваемьй раствор 5,0 г (0,0158 моль) этил-альфа-(1-карбок сиэтил)-амино)-бенэолбутаноатгидро хлорида (), полученного по примеру 8, в 200 мл метиленхлорида последовательно обрабатьшают 1,60 (0,0158 моль) триэтиламина, 2,14 г (0,0158 моль) 1-оксибензотриазола, 5,16 г (0,0158 моль) 1,2,3,4-тетра гидро-6,7-диметокси-З-изохинолинкарбоновой кислоты, свободным осно ванием фенилметилового сложного эф ра (S-форма), полученного по приме ру 9, а затем 3,26 г (0,0158 моль) дициклогексилкарбодиимида в 10 мл метилендихлорида. Постепенно .вьде.ляют дициклогексилмочевину. Далее смесь отстаивают в течение ночи при комнатной температуре, добавляют 300 мл гексана и отфильтровывают мочевину. Фильтрат промывают 250 мл насыщенного раствора бикарбоната натрия, высушивают над сульфатом натрия и концентрируют с удалением растворителя. Вязкий остаток растирают, в 50 мл диэтилового эфира и фильтруют с целью удаления нерастворимьж компонентов. Фильтрат концентрируют с полу чением 9,2 г (99%-й выход) сырого основания. Получение малеатной соли, Раст вор 9,0 г (0,015 моль) вышеуказан ного сырого основания в 50 мл этил 0 ацетата обрабатывают теплым (при 40°.С) раствором 1,86 г (0,016 моль) малеиновой кислоты в 50 мл этилацетата. В результате вьщеляют белые кристаллы в количестве 7,2 г (65%-й выход) с т.пл. 139-14ГС, тонкослойная хр.оматографи еская обработка основания (полученного обработкой соли бикарбонатом натрия с последующей экстракционной обработкой этилацетатом) свидетельствует о наличии одного пятна, R/ - 0,7 (этилацетат, на двуокиси кремния). В результате перекристаллизации из этиленацетата получают чистый продукт с той же температурой плавления, (альфа) +3,4 (1,05% этанола) . Найдено,%: С 64,48; Н 6,30; N 3,99. ,N20yC H404 Вычислено,%: С 64,74-, Н 6,29) N 3,98. Пример 6. 2-(2-{(1-этоксикарбонил)-3-фенилпропил)-амин 1-оксопропил)-1,2,3,4-теграгидро-3-изохинолинкарбоновая кислота, феннлметиловый эфир, малеат (S.,). Проводят реакцию сочетания этил-альфа(1-карбоксиметил)-амино)-бензолбутаноатгидрохлорида (), полученного по примеру 8, с 1,2,3,4-тетрагидро-З-изохинолинкарбоновойкислотой, свободным основанием фенштметилового сложного эфира (S-форма), подученного по примеру 10, в соответствии с примером 5, в результате чего с достижением 61%-ного выхода получают продукт с т.пл. 151-153 С (после перекристаллизации из этилацетата) , (альфа) 5 11,7 (1,0% метанола). НайденоД: С Г5,58-, Н 6,09; N 4,25 C3 HjjNj05-C4H404 Вычислено,%: С 67,07; Н 6,25; N 4,35. Пример 7. 2-(2-(1-этoкcикapбoнил)-3-фeнилпpoпил)-aминo J-1-оксопррпил)-1,2,3,4-тетрагидро-3-йзохинолинкарбоновая кислота, 1,1-диметилэтш1овый сложный эфир (SiS,S). Смесь 8,38 г (0,03 моль) этил-альфа-(1-кар6оксиэтил)-амино)-бензолбутаноата (свободной аминокислоты) (SS), полученной по примеру 8, 8,09 г (0,03 моль) 1,2,3,4-тетрагидро-3-изохинолинкарбоновой кислоты, гидрохлорида 1,1-диметилэтилового сложного эфира (S-форма), полученного по примеру 11, 4,05 г (0,03 моль) 1-окси-бензотриазола с 250 мл тетрагидрофурана охлаждают в бане со льдом до 3-5°С. При перемешивании добавляют J,04 г (0,03 моль) триэтиламина, а затем в течение 20 мин по каплям раствор 6,92 г (0,0335 моль) дициклогексш карбрдиимида в 30 мл тетрагидрофурана. Реакционную смесь перемешивают при 3-5fC 1 ч. Далее без ледяной бани реакционнзто смесь перемешивапи еще 3 ч. Вьщеливщуюся смесь триэтиламингидрохлорида с дициклогексилмочевиной удаляют фильтрованием и промыванием тетра гидрофураном. Фильтрат выпаривают в роторном испарителе с целью удаления всех летучих компонентов. Образовавшуюся смолоподобную массу растворяют приблизительно в 300 мл этилацетата. После фильтрования через цеолит этилацетатный раствор подвергают экстракционной.обработке двумя 100-миллнлитровыми порциями насыщенного раствора бикарбоната натрия, одной 75-миллштитровой порцией 2 н. раствора лимонной кислоты одной ЮО-миллилитровой порцией насщенного раствора бикарбоната натрия и одной 100-мшшилитровой порцией насыщенного раствора хлорида натрия. После сушки над безводным сульфатом магния и фильтрования этилацетат удаляют в роторном испартеле с получением 16., 9 г светло-коричневого смолоподобного материала, который растворяют в 350 мп кипящего гёксана и декантировали через цеолит. Гексановый раствор охлаждаю на льду, вносят в него затравку для кристаллизации и перемешивают до достижения интенсивной кристаллизации. Затем продукт фильтруют, прот мывают холодным гексаном и высушивают с получением 11,6 г (78%-й выход) вещества с т.пл. 68,, (альфа) -12,2 (2% метанола). ,Т.пл. чистого продукта 71-72 С,, (альфа)5 -12,6° (2% метанола). Т.пл. малеатной соли 127,5-128, (альфа) J, +46,4 (2% метанола).
Пример 8. Этил-альфа-(t-кapбoкcиэтшI)-aминo)-бeнзoлбyтaнoатгидрохлорид (SyS).
Раствор 2,0 г третбутилаланииа (S-форма) и 3,78 г этил-2-бром-4-фенилбутаноата в 25 мл диметилформанида обрабатывают 1,8 мл триэтиламина и раствор вьщерживают при 70С 18 ч. Растворитель удаляют при пониженном давлении и остаток смешивают с водой с последующей экстракционной обработкой диэтилоBbw эфиром. Органический слой промывают водой и высушивают над сульфатом магния. Концентрирование с удалением растворителя при пониженном давлении позволяет получить маслоподобный третбутиловый сложный эфир промежуточного продукта, который, как это установлено газо-жидкостной хроматографической обработкой, достаточно чистый для последующего использования.Раствор 143,7 г этого третбутилового сложного эфира в 630 мп трифторуксусной кислоты перемешивают при комнатной температуре 1 ч. Растворитель удаляют при пониженном давлении и остаток растворяют в диэтиловом эфире с последующим повторным выпариванием. Далее эту операцию повторяют. Затем эфирньй раствор обрабатывают добавлением по каплям раствора газообразного хлористого водорода в диэтиловом эфире до прекращения выпадения осадка. Твердый продукт, собранный фильтрованием, представляет собой смесь диастереоизомеров с т.пл. 152-165С, (альфа)) +3,6 (1% метанола).
С целью вьделения предпочтительного S S-изомера суспензию 10,0 г указанной смеси в 200 мл метиленхлорида перемешивают при комнатной температуре 5 мин с последующим фильтрованием. Твердый продукт промывают дополнительным количеством, метиленхлорида и наконец диэтиловьм эфиром. Т.пл. одного из изомеров 202-208 0 (с разложением), (альфа) -29, (1% метанола) г причем этот изомер представлял собой диастереоизомер, обладающий R,S-KOHфигурацией (символом S отмечена часть молекулы, полученная ив L-anaнина). Предпочтительный S S-диастереоизомер вьделяют из фильтрата после концентрирования и растирания остатка в диэтиловом зфире. Т.пл. этого вещества 137-139 С, (альфа) +31,3 (1% метанола).
Свободную аминокислоту (S 8-фор мы) получают обработкой водного раствора гидрохлорида насыщенным раствором ацетата натрия. Полученный продукт фильтруют,интенсивно промывают холодной водой и перекристаллизовьшают из этилацетата. Т.пл. продукта 149-1SlC (альфа)1 +29, (U 0,1 и. соляной кислоты) .
Пример 9.1,2,3,4-тетрагидро-6,7-диметокси-З-изохинолинкарбоновая кислота, фенилметиловый сложный, гидрохлорйд (S-форма).
Смесь 1,2,3,4-тетрагидро-6,7-диметокси-З-изохинолинкарбоновойкислоты, гидрохлорида (S-формы) с 600 мл бензилового спирта насыщают газообразным хлористым водородом. Температуру смеси повыщают до . Затем эту смесь перемешивают при комнатной температуре 3 дня., Далее фильтруют относительно небольшое количество твердого вещества и фильтрат обрабатывают приблизительно 2 л диэтилового эфира, в результате чего с достижением 83%-го выхода получают 37,5 г сырого продукта. Последний очищают обработкой избыточным количеством насьщ ённого раствора бикарбоната натрия, экстрагированием основания этилацетатом и осаждением гидрохлоридной соли газообразным хлористым водородом. В результате перекристаллизации из смеси метанола с диэтиловым эфиром получают чистый продукт с Т.пл. 255-260С, (альфа)р -81, (1,0% метанола), тонкослойная хроматографическая обработка (20% метанола в, смеси с хлороформом на двуокиси кремния) - одно пятноj Rр 0,8.
Найдено,%: С 62,54i Н 5,99; N 4,00.
Ci,H2iN04-HCe
Вычислено,%: С 62,72; Н 6,10; N 3,85.
Пример 1,0. 1,2,3,4-тетрагидро-3-изохинолинкарбоновая кислота, фенипметиловьй сложный эфир, гидрохлорид (S-форма) 750. мл бензилового спирта обрабатывают 150 г технической полифосфорной кислоты с последующим нагревом смеси до 90с и перемешиванием при этой температуре до образования гомогенной смеси. Далее добавляют в
смесь 165,2 г твердой 1,2,3,4-тетра-. гидро-3-изохинолинкарбоновой кислоты (S-формы). Затем смесь перемешивают 4 ч при 95-105°С с последующей
вьщержкой в спокойном состояний при комнатной температуре 18 ч. Далее добавляют 18,5 г газообразного хлористого водорода в 2,5 л безводного диэтилового эфира и продукт
0 постепенно отделяют с охлаждением в течение ночи. В результате фильтрования получают сырой гвдрохлорид бензйл-1,2,3,4-тетрагидро-З-изохинолинкарбоксилата. Последний
5 двукратной перекристаллизацией из этанола с получением продукта с т,ш1. 190,5-19iCj (альфа), -83,3 (1% смеси метанола с 1 н. соляной кислотой в соотношении 1:t).
0 Приме р 11. 1,2,3,4-тетрагидро-3-изохинолинкарбоновая кислота, 1,1-диметиловый сложный эфир, гидрохлорид (S-форма).
Это соединение получают пропусканием 447 г изобутилена через охлажденный До раствор 63,5 г 1,2,3, 4-тетрагидро-З-изохинолинкарбоновой кислоты S-формы) в 650 мл сухого
диоксана и 65 мл концентрированной серной кислоты (в токе азота). Затем реакционный сосуд закрывают и его содержимое встряхивают в течение 17 ч при комнатной температуре. Далее реакционный сосуд открывают и смесь выливают в 25 л холодного 2 н. раствора гидрата окиси натрия. Образовавшийся продукт экстрагируют диэтиловым эфиром. Эфирный раствор промывают водой, вькушивают и сконцентровьшают до остаточного объема приблизительно 500 мл. Последний обрабатываютизбытком 6 Hi изопропанолового раствора соляной кислоты с целью осаждения продукта, который отделяют фильтрование. Образец, очищенный перекристаллизацией из смеси этанола с диэтиловь м эфиром, имеет т.пЛ. 190-192С (с разложекием).-, (альфа) -88,7(2%
метанола).
Пример 12. 1000 таблеток, каждая из которых, содержит по 100 мг 2-((этоксикарбонил)-З-фенилпропил)-3-фенилпропил)-амино -1-оксопропил)-1,2,3,4-тетрагидро-6,7-диметокси-3-изохинолинкарбоновойкислоты, гидрохлорида, гидрата (S S; S) , формуют с использованием следующих компонентов, г; 2-(2-(1-(Этоксикарбонил)-З-фенилпропил)-aMHHoj-1-OKco-npoпйл)-1,2,3,4-тетрагидро-6,7-диметокси-3-изохинолинкарбоновая кислота, гидрохлорид, гидрат, (S:,) 100 Кукурузный крахмал 50 Желатин Ависел (микрокристаллическая целлюлоза)25 Стеарат магния2,5 2-(2-(1-(этоксикарбонил)-3-фенш1пропил).-амино -1-оксопропил)-1,2,3,4-тетрагидро-6,7-диметокси) -3-изохинолинкарбоновую кислоту, гидрохлорид, гидрат () и куку рузный крахмал смешивают с водным раствором желатина. Затем эту смес сушат и измельчают до тонкодисперс ного порошка. Далее с гранулнрова Ависел. нием подмешивают продукт после чего стеарат магния. После зт.ого массу прессуют в таблет-маши формуя, таким образом, 1000 таблеток, каждая из которых содержит по 100 мг активнодействующих компонен тов. Пример 13. 1000 таблеток, каждая из которых включает в себя по 200 мг 2-(2-(1-(этоксикарбоНИЛ)-З-фенилпропил)-амино -1-оксопропил)-1,2,3,4-тетрагидро-6,7-ди метокси-3-изохинолинкарбоновой кислоты, гидрохлорида гидрата ( формуют с использованием следующих компонентов, г: 2-(2-(1-(Этоксикарбонил)-3-фенилпропил)-амино - 1 -оксопропил) -1,2,3,4-тетрагидро-6,7-диметокси-3-изохинолинкарбоновая кислота,гидрохлорид, гидрат, () 200 Лактоза100 Продукт Ависел 150 Кукурузный крахмал 50 Стеарат магния5 2-(2-(1-(этoкcикapбoннл)-3-фeш лпpoпил)-амино j -1-оксопропил) -1,2,3,4-тетрагидро-6,7-диметокси-3-изохинолинкарбоновую кислоту, гидрохлорид, гидрат () лакто зу и продукт Ависел смешивают. 012 . затем перемешивают с кукурузньи Крахмалом. Далее добавляют в смесь стеарат магния. Сухую смесь прессуют в таблет-машине, в результате чего формуют 1000 таблеток весом по 505 мг каждая, причем каждая из этих таблеток включает в себя по 200 мг активноденствующего компонента. После этого таблетки покрывают раствором продукта Метосел Е 15 (метилцеллюлрза), который включает красочный лак, содержащий желтый краситель № 6. Пример 14. Двухэлементные желатиновые капсулы № 1, каядая из которых включает в себя по 250 мг 2-(2-(1-(этоксикарбонил)-3-фенил)-амино -1-оксопропш1)-1,2,3,4-тетрагидро-6,7 диметокси-3-изохинолинокарбоновой кислоты, гидрохлорида, падрата () заполняют смесью следующих компонентов, мг: 2-(2- (1-Этоксикарбонил ) -З-фенилпропил) -аминоj-1-эксопропил)-1,2,3,4-тетраг1одро-6,7-диметокси-З-изохинолинкарбоновая.кислота, гидрохлорид, гидрат (5,8., S)250 Стеарат магния7 Лактоза USP193 Пример 15. Раствор для инъекций содержит следующие компоненты:2-(2-(1-Этоксикарбонил)-З-фенилпропил)-амино J-1-оксопропил)-1,2,3,4-тетрагидро-6,7-диметокси-З-изохинолинкарбоновая кислота, гидрохлорид, гидрат (, S) 500 г Метилпарабен5 г Пропилпарабен . 1 г Хпористый натрий25 г Вода для инъекций в количестве до 5 л Активнодействующий компонент, онсерванты и хлористый натрий расторяют в 3 л воды для инъекций,посе чего общий объем раствора довоят водой до 5 л. Затем раствор ильтруют через стерильный фильтр и асептических условиях фасуют по терилизованньм пробиркам, после его закрывают их стерилизованными езииовьвш пробками-. Каждая пробирка одержит по 5 мл раствора с концекграцией активнодействующего компонента по 100 мл на миллилитр раств ра для инъекций. Под действием фермента ренина на ангиотензиноген псевдоглобулин в плазме крови образуется декапептидный ангиотензин I. Под действие ангиотензинпревращающего фермента (АПФ) происходит конверсия ангиотензина I в октапептидный ангиотен ЗИН II. Последний является активны прессорным веществом, который служит агентом, вызывающим у млекопитающих, например у крыс и собак, различные виды гипертонии. Соединения предлагаемого изобретения влияют на протекание процессов последовательности ренин-ангиотенз 1-ангиотензин II вследствие подавления активности фермента, который вызывает конверсию ангиотензина I, и снижения количества или полного подавления образования такого прес сорного вещества, как ангиотензин II, в результате чего такие соединения могут быть использованы для ослабления или лечения гиперто нии. Дпя снижения кровяного давления рекомендуется вводить предлагаемую композицию в организм в вид однократной дозы или от двух до четьфех дробных дозировок в расчете на ежедневную от О,1 до
/ CHoCH,CH-NHCH
// ьI
COOR
сн
СО OR 00 мг/кг, предпочтительнее от 1 до 50 мг/кг/день. Предлагаемые лекарственные средства вводят в организм перорально или путем подкожных, внутримышечных, внутривенных и внутрибрюшинных инъекций. Испытание ин витро. АПФ. Ингибирующее действие на АПФ определяют путем проведения эксперимента с сывороточным АПФ морской свинки в присутствии и отсутствии испытываемого соединения. Перед добавлением меченого субстрата ЗН-гиппурилглицилглиуина АПФ сьгаоротки морской свинки испытуемое соединение подвергают предварительной инкубации в течение 10 мин. После 60-минутной инкубации при реакцию прекращают добавлением 0,1 и. соляной кислоты. АПФ расщепляют гиппурил-глициловую связь с образованием депиптидглицил-глицина и ЗН-гиппуровой кислоты. Далее ЗН-гиппуровую кислоту экстрагируют этилацетатом и АПФ данной пробы рассчитывают как количество вьщелившейся ЗН-гиппуровой кислоты. В таблице даны ациловые производные 1,2,3,4-тетрагидроизохинолия-З-карбоновые кислоты (S S S-конфигурации) и их ингибирующая активность при действии на АПФ, определенная ин витро.
Продолжение таблицы







Способ получения замещенного ацилового производного 1,2,3,4-тетрагидроизохинолин-3-карбоновой кислоты общей формулы 1 COOR RI О 1 II СН-с-1 где X и Y имеют указанные значения; Rj - низший алкиловый или бензиловый сложный эфир, подвергают.взаимодействию с N-замещенной амино кислотой формулы (III) Ar-CHj - СНг - СН- Ш-СН-СООН COORj где Аг, R, R., имеют указанные значения, с вьщелением целевого продукта или с последукяцйм удалением блокирующей группы и выделеяием целевого продукта в свободном виде или в виде фармацевтически приемлемой соли.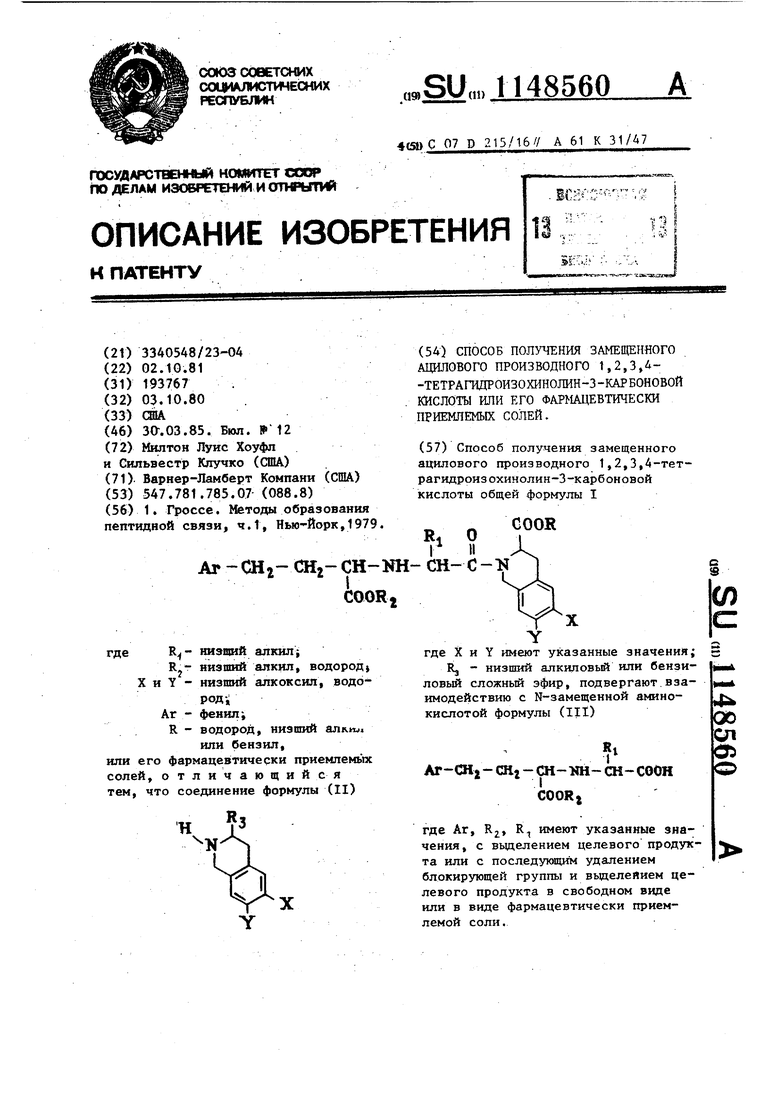
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Гроссе | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| или бензил, или его фармацев тически приемпемьпс солей, отличающийся тем, что соединение формулы (II) COO (54) СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННОГО АЦИЛОВОГО ПРОИЗВОДНОГО t,2,3,4-ТЕТРАГИДРОИЗОХИНОЛИН-3-КАРБОНОВОЙ КИСЛОТЫ или ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ. | |||
