В патенте США N 5314896, на который здесь ссылаются, описывается ряд средств, действующих на центральную нервную систему. В нем также описываются фармацевтически приемлемые соли этих соединений.
Соединения, упомянутые в указанном выше патенте США, полезны в качестве допаминэргических средств. Как допаминэргические средства эти вещества полезны для лечения психозов, например шизофрении, и состояний, связанных с гиперпролактинемией, таких как галакторея, аменорея, расстройства менструации и половая дисфункция. Они также полезны в качестве антигипертензивных средств. Особенно ценным противопсихопатическим средством является (R)-(+)-1,2,3,6-тетрагидро-4-фенил-1-[(3-фенил-3-циклогексен-1- ил)метил] пиридин.
Способы, изложенные в патенте США N 5314896, являясь чрезвычайно неэффективными в отношении общего выхода и затрат времени, очень полезны для осуществления множества замещений по арильной группе при циклогексановом кольце и варьирования замещений в аминной части молекулы. В результате стадии дегидратации в ходе синтеза получали также изомеры 2,3-циклогексена, которые могли быть разделены хроматографически для биологических испытаний. Кроме того, получение соли 1,2,3,6-тетрагидро-4-фенил-1-[(3-фенил-3-циклогексен-1- ил)метил] пиридина с (R)-(-)-1,1-бинафтил-2,2'-диил-гидрофосфатом с последующей перекристаллизацией и выделением свободного основания приводило к (R)-(+)-1,2,3,6-тетрагидро-4-фенил-1-[(3-фенил-3-циклогексен- 1-ил)метил] пиридину, который оценивали биологически.
Кроме того, описан ряд других способов получения (R)-(+)-1,2,3,6- тетрагидро-4-фенил-1-[(3-фенил-3-циклогексен-1-ил)метил] пиридина и родственных соединений. Так, Johnson S.J. и др., 206-я Национальная Конференция Американского Химического Общества, Чикаго, Иллинойс, август 1993, MEDI-171, изложили несколько синтезов этого соединения. По одной методике Johnson и др. используют внутримолекулярную циклизацию по Виттигу, где исходным соединением служит раздражающее вещество и лакриматор и процедура требует разделения промежуточной рацемической ен-кислоты с потерей более 50% вещества. Кроме того, Johnson и др. и Wustrov D. и др., Tetrahedron Letters 35:61 (1994), описали методику дезоксигенирования кетона при использовании триэтилсилана и перхлората лития в диэтиловом эфире, реагентов, не подходящих для крупномасштабного синтеза. Wright J. и др., 206-я Национальная Конференция Американского Химического Общества, Чикаго, Иллинойс, август 1993, MEDI, изложили путь к желаемому соединению, требующий разделения на стереоизомеры промежуточного соединения, что приводит к более чем 50%-ной потере желаемого промежуточного соединения. Downing D.M. и др., 208-я Национальная Конференция Американского Химического Общества, Вашингтон, D.C., август 1994, MEDI-178, и Downing D. M. и др., 206-я Национальная Конференция Американского Химического Общества, Чикаго, Иллинойс, август 1993, MEDI - 173, представили различные подходы к желаемому соединению, включающие многократную хроматографию и многократное разделение стереоизомеров, которые приводят к выходу менее 50% на стадии разделения. Наконец, Wise L.D. и др., 208-я Национальная Конференция Американского Химического Общества, Вашингтон, D.C., август 1994, MEDI - 266, представили подход к желаемому соединению, не рассматривающий операцию разделения стереоизомеров для получения (R)-(+)-1,2,3,6- тетрагидро-4-фенил-1-[(3-фенил-3-циклогексен-1-ил)метил]пиридина.
Эти синтетические способы позволили установить структурные ограничения для проявления активности при биологическом использовании. Однако они не подходят для крупномасштабного способа производства, поскольку включают многократные хроматографические операции, неэффективное разделение промежуточных соединений, используют вредные реагенты и приводят к низкому общему выходу.
Объектом настоящего изобретения является улучшенный способ получения (R)-(+)-1,2,3,6-тетрагидро-4-фенил-1-[(3-фенил-3-циклогексен- 1-ил)метил] пиридина, описанного выше, при использовании нового синтеза. Далее, мы неожиданно обнаружили, что особенно ценное допаминэргическое средство, малеат (R)-(+)-1,2,3,6-тетрагидро-4-фенил- 1-[(3-фенил-3-циклогексен-1-ил)метил]пиридина, может быть получено из новых промежуточных соединений при меньшем числе стадий без хроматографии, без потерь более 50% вещества при разделении (RS)-смеси 1,2,3,6-тетрагидро-4-фенил-1-[(3-фенил-3- циклогексен-1-ил)метил] пиридина и с более высокими выходами, чем в предыдущих способах. Кроме того, настоящий способ использует недорогие исходные вещества и пригоден для крупномасштабного синтеза.
Краткое описание изобретения
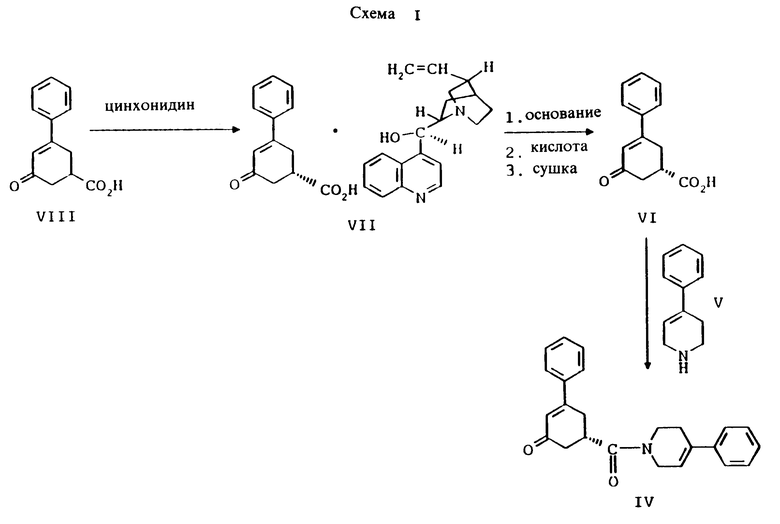
Таким образом, первым аспектом настоящего изобретения является улучшенный способ получения соединения формулы I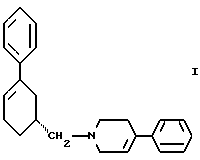
и его фармацевтически приемлемых солей, включающий
стадию (а) обработки рацемического соединения формулы VIII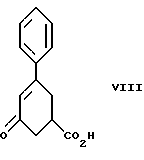
цинхонидином в растворителе с целью получения соединения формулы VII
стадию (б) обработки соединения формулы VII основанием в растворителе с целью получения после подкисления соединения формулы VI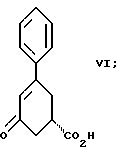
стадию (в) обработки соединения формулы VI соединением формулы V
в присутствии реагента для сочетания и растворителя с целью получения соединения формулы IV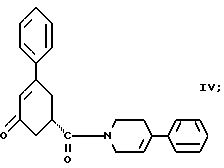
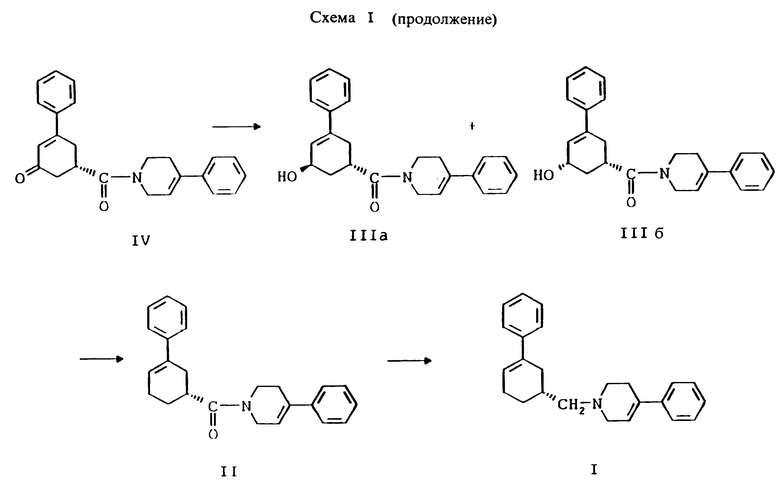
стадию (г) обработки соединения формулы IV восстанавливающим реагентом в растворителе с целью получения смеси соединений формулы IlIа и формулы IIIб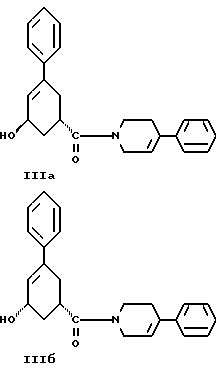
стадию (д) обработки смеси соединений формулы IlIа и формулы IIIб смесью хлористого цинка и цианборгидрида натрия в растворителе, а затем раствором карбоновой кислоты в растворителе с целью получения соединения формулы II
стадию (е) обработки соединения формулы II восстанавливающим агентом, представляющим гидрид металла, в растворителе с целью получения соединения формулы I;
стадию (ж) и, если желательно, превращение полученного соединения формулы I в соответствующую фармацевтически приемлемую соль в результате присоединения кислоты, используя общепринятые способы, и, если так требуется, превращение соответствующей фармацевтически приемлемой соли, полученной в результате присоединения кислоты, в соединение формулы I с использованием общепринятых способов.
Вторым аспектом настоящего изобретения является способ получения соединения формулы VI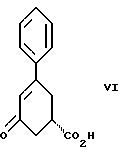
включающий
стадию (а) обработки рацемического соединения формулы VIII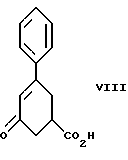
цинхонидином в растворителе с целью получения соединения формулы VII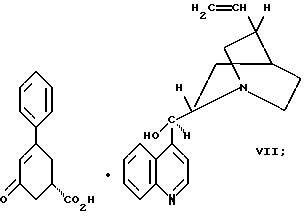
и стадию (б) обработки соединения формулы VII основанием в растворителе с целью получения после подкисления соединения формулы VI.
Третьим аспектом настоящего изобретения является способ получения соединения формулы VI
включающий обработку рацемического соединения формулы IX
в растворителе при pH около 5 с помощью α-химотрипсина с целью получения после отделения непрореагировавшего сложного эфира и подкисления соединения формулы VI.
Четвертым аспектом настоящего изобретения является способ получения соединения формулы II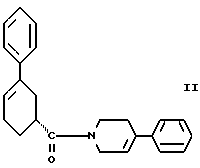
включающий
стадию (а) обработки рацемического соединения формулы VIII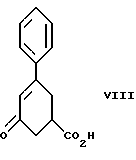
цинхонидином в растворителе с целью получения соединения формулы VII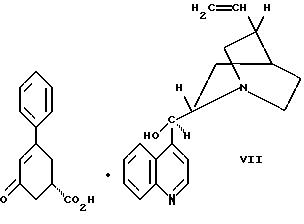
стадию (б) обработки соединения формулы VII основанием в растворителе с целью получения после подкисления соединения формулы VI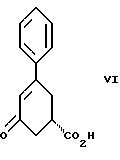
стадию (в) обработки соединения формулы VI с помощью соединения формулы V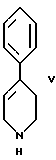
в присутствии реагента для сочетания и растворителя с целью получения соединения формулы IV
стадию (г) обработки соединения формулы IV восстанавливающим агентом в растворителе с целью получения смеси соединений формулы IlIа и формулы IIIб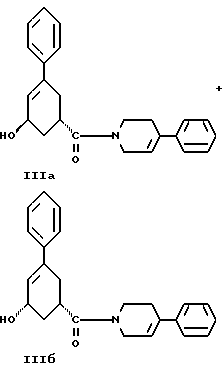
и стадию (д) обработки смеси соединений формулы IIIа и формулы IIIб смесью хлористого цинка и цианборгидрида натрия в растворителе, а затем раствором карбоновой кислоты в растворителе с целью получения соединения формулы II.
Пятым аспектом настоящего изобретения является способ получения соединений формулы IlIа и формулы IIIб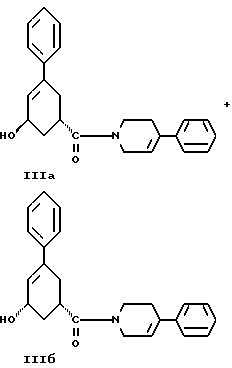
включающий
стадию (а) обработки рацемического соединения формулы VIII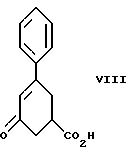
цинхонидином в растворителе с целью получения соединения формулы VII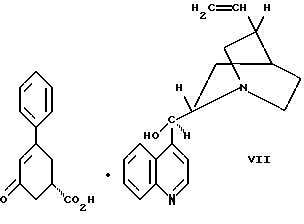
стадию (б) обработки соединения формулы VII основанием в растворителе с целью получения после подкисления соединения формулы VI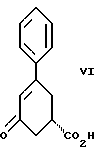
стадию (в) обработки соединения формулы VI с помощью соединения формулы V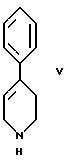
в присутствии реагента для сочетания и растворителя с целью получения соединения формулы IV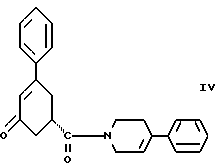
и стадию (г) обработки соединения формулы IV восстанавливающим агентом в растворителе с целью получения смеси соединений формулы IlIа и формулы IIIб.
Шестым аспектом настоящего изобретения является способ получения соединения формулы IV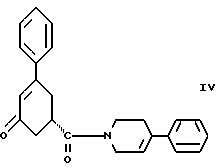
включающий
стадию (а) обработки рацемического соединения формулы VIII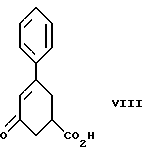
цинхонидином в растворителе с целью получения соединения формулы VII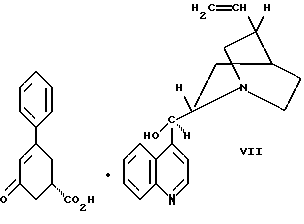
стадию (б) обработки соединения формулы VII основанием в растворителе с целью получения после подкисления соединения формулы VI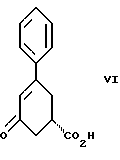
стадию (в) обработки соединения формулы VI соединением формулы V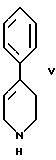
в присутствии реагента для сочетания и растворителя с целью получения соединения формулы IV.
Седьмым аспектом настоящего изобретения является новое промежуточное соединение формулы IlIа или формулы IIIб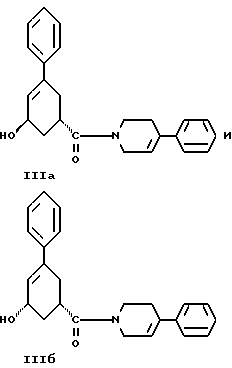
пригодное для получения соединения формулы II, которое в свою очередь пригодно для получения противопсихопатического соединения формулы I.
Восьмым аспектом настоящего изобретения является новое промежуточное соединение формулы IV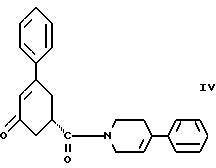
пригодное для получения соединения формулы IlIа или формулы IIIб, которое в свою очередь пригодно для получения соединения формулы II, последнее в свою очередь пригодно для получения противопсихопатического соединения формулы I.
Девятым аспектом настоящего изобретения является новое промежуточное соединение формулы VI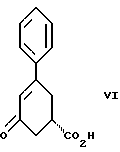
пригодное для получения соединения формулы IV, которое в свою очередь пригодно для получения соединения формулы IlIа или формулы IIIб, которое в свою очередь пригодно для получения соединения формулы II, которое в свою очередь пригодно для получения противопсихопатического соединения формулы I.
Десятым аспектом настоящего изобретения является новое промежуточное соединение формулы VII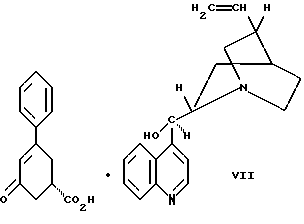
Одиннадцатым аспектом настоящего изобретения является новое промежуточное соединение формулы IX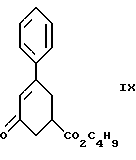
пригодное для получения соединения формулы VI, которое в свою очередь пригодно для получения соединения формулы IV, которое в свою очередь пригодно для получения соединения формулы IlIа или формулы IIIб, которое в свою очередь пригодно для получения соединения формулы II, последнее в свою очередь пригодно для получения противопсихопатического соединения формулы I.
Подробное описание изобретения
В данном изобретении термин "алкил" означает углеводородную группу с нормальной или разветвленной цепью, содержащую от 1 до 6 углеродных атомов, и включает, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, н-пентил, трет-амил, н-гексил и им подобные группы.
"Щелочной металл" означает металл группы IA периодической таблицы элементов и включает, например, литий, натрий, калий и им подобные металлы.
"Минеральная кислота" означает сильную кислоту, которая включает, например, соляную кислоту, бромистоводородную кислоту, серную кислоту и им подобные кислоты.
"Карбоновая кислота" означает органическую кислоту, которая включает, например, уксусную кислоту, пропионовую кислоту, масляную кислоту, триметилуксусную кислоту и им подобные кислоты.
"Боргидрид металла " означает восстанавливающий агент, который включает литийборгидрид, натрийборгидрид, калийборгидрид и им подобные боргидриды.
"Восстанавливающий агент, представляющий гидрид металла", означает восстанавливающий агент, который восстанавливает амиды карбоновых кислот и который включает алюмогидрид лития, бис(2-метоксиэтокси)алюмогидрид натрия и им подобные.
"Аппарат" означает реакционную систему с холодильником, позволяющую отгонять растворитель.
"Реактор" означает реакционную систему с холодильником, позволяющую возвращать растворитель непосредственно в реакционную систему.
Соединение формулы I способно далее при присоединении кислоты образовывать фармацевтически приемлемые соли. Все эти формы находятся в рамках настоящего изобретения.
Фармацевтически приемлемые соли соединения формулы I, получаемые при присоединении кислот, включают соли, образованные при использовании нетоксичных неорганических кислот, как, например, соляная, азотная, фосфорная, серная, бромистоводородная, иодистоводородная, фосфористая и им подобные кислоты, а также соли, полученные при использовании нетоксичных органических кислот, как, например, алифатические моно- и дикарбоновые кислоты, фенилзамещенные алкановые кислоты, гидроксиалкановые кислоты, алкандиовые кислоты, ароматические кислоты, алифатические и ароматические сульфокислоты и т. д.
Эти соли включают, таким образом, сульфат, пиросульфат, бисульфат, сульфит, бисульфит, нитрат, фосфат, гидрофосфат, дигидрофосфат, метафосфат, пирофосфат, хлорид, бромид, иодид, ацетат, пропионат, каприлат, изобутират, оксалат, малонат, сукцинат, суберат, себацат, фумарат, малеат, манделат, бензоат, хлорбензоат, метилбензоат, динитробензоат, фталат, бензолсульфонат, толуолсульфонат, фенилацетат, цитрат, лактат, малеат, тартрат, метансульфонат и им подобные. Также рассматриваются соли аминокислот, как, например, аргинат и ему подобные и глюконат, галактуронат (см., например, Berge S.M. и др. , "Фармацевтические соли", Journal of Pharmaceutical Science. 66:1 - 19 (1977).
Для получения солей упомянутых основных соединений с кислотами вещество в виде свободного основания подвергают взаимодействию согласно общепринятому способу с достаточным количеством соответствующей кислоты. Вещество в виде свободного основания может быть регенерировано при взаимодействии соли с основанием и выделении свободного основания общепринятым способом. Вещества в виде свободных оснований несколько отличаются от их соответствующих солей некоторыми физическими свойствами, например растворимостью в полярных растворителях, но в других отношениях соли эквивалентны соответствующим им свободным основаниям с точки зрения целей настоящего изобретения.
Как ранее показано в патенте США N 5314896, соединение формулы I полезно в качестве допаминэргического средства, как определено с помощью методологии, известной для таких исследований. Соединение формулы I обладает активностью, характерной для противопсихопатического средства.
Способ, описываемый настоящим изобретением, в своем первом аспекте является новым, улучшенным, экономичным и коммерчески выгодным способом получения противопсихопатического средства формулы I. Процесс получения согласно настоящему изобретению представлен на схеме I (см. в конце описания).
Соединение формулы VIII получают из бензоилакриловой кислоты, используя модификацию способа, описанного S. Julia и Y. Bonnet. Bull. Soc. Chim., 1354-1364 (1957).
Так, соединение формулы VIII растворяют в растворителе, например, спирте содержащем 1 - 4 атома углерода, как, например, метиловый спирт, этиловый спирт, изопропиловый спирт, н-бутиловый спирт и им подобные, при температуре примерно от 25oC до температуры кипения растворителя, и полученный раствор подвергают обработке суспензией или раствором цинхонидина в том же растворителе, чтобы получить соль с цинхонидином формулы VII. Если требуется, соль для достижения более высокой энантиомерной чистоты подвергают перекристаллизации из спирта, содержащего 1 - 4 атома углерода, такого, как, например, метиловый спирт, этиловый спирт, изопропиловый спирт, н-бутиловый спирт и им подобные. Соединение формулы VII растворяют в растворителе, например в спирте, содержащем 1 - 4 атома углерода, как, например, метиловый спирт, этиловый спирт, изопропиловый спирт, н-бутиловый спирт и им подобные, при температуре примерно от 25oC до температуры около точки кипения растворителя. Суспензию или раствор подвергают обработке водным раствором гидроокиси щелочного металла, как, например, едкого натра, едкого кали и им подобных, для образования соли щелочного металла соединения формулы VI. Спирт удаляют при вакуумной перегонке и частично заменяют водой, кристаллический цинхонидин возвращают при фильтрации. Водный фильтрат, содержащий соль щелочного металла соединения формулы VI, подвергают обработке избытком раствора минеральной кислоты, охлаждают и фильтруют, получают соединение формулы VI. R-энантиомер может быть выделен и подвергнут рацемизации в соединение из примера A при использовании стандартной методики. Соединение формулы VI подвергают сушке в вакууме при температуре примерно от 55oC до 80oC примерно от 24 до 48 часов до содержания влаги менее 0.2%.
Соединение формулы VI подвергают обработке реагентом для сочетания, например реагентом или реагентами, активирующими кислоту, как, например, карбонилдиимидазол или 1-(3-диметиламинопропил)-3- этилкарбодиимид, гидрат 1-гидроксибензотриазола и триэтиламин и им подобные. Активированную кислоту подвергают обработке соединением формулы V, 4-фенил-1,2,3,6-тетрагидропиридином или гидрохлоридом 4-фенил-1,2,3,6-тетрагидропиридина и триэтиламином в инертном растворителе, как, например, ацетонитрил, тетрагидрофуран и им подобные. Образование амида происходит при температуре примерно от 0oC до температуры около точки кипения растворителя примерно в течение 3 - 24 часов. Реакцию останавливают при прибавлении водного раствора бикарбоната, карбоната или гидроокиси щелочного металла, например бикарбоната натрия, карбоната натрия, едкого натра и им подобных. Соединение формулы IV экстрагируют инертным несмешивающимся с водой растворителем, как, например, толуол, диэтиловый эфир, трет-бутилметиловый эфир и им подобные. Раствор промывают разбавленной минеральной кислотой для удаления не вступивших в реакцию веществ основного характера. Раствор концентрируют и вместо растворителя прибавляют спирт, содержащий 1 - 4 атома углерода, как, например, метиловый спирт, этиловый спирт, изопропиловый спирт, н-бутиловый спирт и им подобные. Раствор охлаждают до температуры примерно от -5oC до 25oC и соединение формулы IV выделяют при фильтрации. Соединение формулы IV может быть высушено в вакууме, если его нужно хранить, или использовано влажным в следующей реакции.
Соединение формулы IV растворяют в растворителе, например, в спирте содержащем 1 - 4 атома углерода, как, например, метиловый спирт, этиловый спирт, изопропиловый спирт, н-бутиловый спирт и им подобные или в их водных смесях, и прибавляют восстанавливающий агент, например боргидрид металла, как, например, натрийборгидрид и ему подобные, в виде твердого вещества или раствора. Смесь перемешивают примерно от 3 до 24 часов при температуре примерно между 15oC и 35oC. Прибавляют водный раствор хлористого аммония и смесь охлаждают примерно до 0 - 5oC. Смесь соединений формул IlIа и IIIб выделяют при фильтрации и сушат в вакууме при температуре примерно от 25oC до 55oC до содержания воды менее чем 0.2%.
Соединения формул IlIа и IIIб смешивают с безводным хлористым цинком, цианборгидридом натрия и инертным растворителем, как, например, гексан или гептан и им подобные. Прибавляют раствор карбоновой кислоты, как, например, уксусная кислота, пропионовая кислота, масляная кислота, триметилуксусная кислота и им подобные, в инертном растворителе, таком как, например, гексан, гептан, тетрагидрофуран и им подобные, при температуре реакции примерно от 25oC до 35oC. Смесь перемешивают примерно от 6 до 24 часов и прибавляют водный раствор хлористого аммония. Соединение формулы II фильтруют через закрытый фильтр и твердый осадок промывают водой. Твердый осадок растворяют в растворителе, таком как, например, тетрагидрофуран, трет-бутилметиловый эфир или нагретый спирт, содержащий 1 - 4 атома углерода, как, например, метиловый спирт, этиловый спирт, изопропиловый спирт, н-бутиловый спирт и им подобные, для удаления вещества из закрытого фильтра. Растворитель удаляют при дистилляции и, если растворителем был не спирт, вместо него прибавляют спирт, содержащий 1 - 4 углеродных атома, как, например, метиловый спирт, этиловый спирт, изопропиловый спирт, н-бутиловый спирт и им подобные. Раствор охлаждают до температуры около 0 - 5oC и выделяют при фильтрации соединение формулы II. Соединение формулы II сушат в вакууме до содержания растворителя менее чем 0.1%.
Соединение формулы II суспендируют или растворяют в инертном растворителе, как, например, тетрагидрофуран, диэтиловый эфир, трет-бутилметиловый эфир и им подобные, и обрабатывают восстанавливающим реагентом, представляющим гидрид металла, как, например, алюмогидрид лития и ему подобные, в инертном растворителе, как, например, тетрагидрофуран и ему подобные, при температуре примерно от 25oC до 55oC около 3 - 4 часов. Раствор охлаждают примерно до 20 - 25oC. Прибавляют определенное количество воды в тетрагидрофуране, а затем определенное количество насыщенного водного раствора сульфата натрия. Полученную суспензию нагревают примерно до 50 - 60oC и неорганические соли удаляют при фильтрации. Инертный растворитель отгоняют в вакууме и вместо него прибавляют спирт, содержащий 1 - 4 атома углерода, как, например, метиловый спирт, этиловый спирт, изопропиловый спирт, н-бутиловый спирт и им подобные. Также может быть использован ацетонитрил. Растворитель затем отгоняют в вакууме и опять вместо него прибавляют спирт, содержащий 1 - 4 атома углерода, как, например, метиловый спирт, этиловый спирт, изопропиловый спирт, н-бутиловый спирт и им подобные. Раствор охлаждают примерно до 0 - 5oC и перемешивают, как минимум, 2 часа. Соединение формулы I выделяют при фильтрации.
Соединение формулы I растворяют в минимальном количестве спирта, содержащем 1 - 4 атома углерода, как, например, метиловый спирт, этиловый спирт, изопропиловый спирт, н-бутиловый спирт и им подобные, примерно при температуре кипения растворителя. Раствор обрабатывают фармацевтически приемлемой кислотой, как, например, суспензия или раствор малеиновой кислоты в спирте, содержащем 1 - 4 атома углерода, как, например, метиловый спирт, этиловый спирт, изопропиловый спирт, н-бутиловый спирт и им подобные. Раствор охлаждают примерно до 0 - 5oC и перемешивают, как минимум, 2 часа. Соль соединения формулы I с малеиновой кислотой выделяют при фильтрации. Соль соединения формулы I с малеиновой кислотой сушат в вакууме при температуре около 25 - 35oC в течение примерно 16 - 24 часов.
Способ по данному изобретению в своем третьем аспекте является новым, улучшенным, экономичным и коммерчески выгодным альтернативным способом получения соединения формулы VI. Устанавливают pH раствора рацемического соединение формулы IX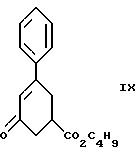
в растворителе, как, например, вода, примерно около 5 с помощью кислоты, как, например, разбавленная соляная кислота и ей подобные. Смесь затем подвергают обработке α-химотрипсином и pH поддерживают около 5 путем прибавления основания, как, например, разбавленный едкий натр и ему подобные, при температуре окружающей среды примерно 2.09 дня. Реакция заканчивается, когда расход основания составляет примерно от 90% до 100% от теоретического количества, необходимого для гидролиза 50% рацемического сложного эфира формулы IX. Предпочтительно реакцию проводят в воде при pH около 5 при использовании разбавленной соляной кислоты и разбавленного едкого натра при температуре окружающей среды примерно 2.09 дня. Не вступивший в реакцию сложный эфир экстрагируют и соль кислоты со щелочным металлом подвергают обработке кислотой, чтобы получить соединение формулы VI.
Соединение формулы IX получают из соединения формулы VIII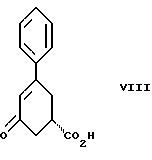
при реакции с н-бутиловым спиртом в присутствии кислоты, как, например, серная кислота и ей подобные, при температуре от комнатной до около 100oC в течение примерно 18 - 24 часов, в результате получают соединение формулы IX. Предпочтительно реакцию проводят с использованием серной кислоты при температуре около 50 - 55oC в течение примерно 22 часов.
Следующий нелимитирующий пример иллюстрирует предпочитаемый изобретателями способ получения соединения согласно изобретению.
Пример 1. Малеат (R)-(+)-1,2,3,6-тетрагидро-4-фенил-1-[(3-фенил- 3-циклогексен-1-ил)метил]пиридина
Стадия А: Получение (S)-5-оксо-3-фенил-3-циклогексенкарбоновой кислоты
Способ А: Методика с использованием цинхонидина
В аппарат емкостью 400 л загружают 50.0 кг (231.4 моля) 5-оксо-3-фенил-3-циклогексенкарбоновой кислоты (пример А, формула VIII) и 175 л изопропилового спирта и начинают перемешивание. Вещество в аппарате нагревают до 70 - 75oC, при этом вещество растворяется. В реактор загружают 50.0 кг (170 молей) цинхонидина и 275 л изопропилового спирта и начинают перемешивание. Вещество в реакторе нагревают до 50 - 75oC, чтобы получить подвижную легко транспортируемую суспензию. Суспензию/раствор передают из реактора в аппарат, содержащий раствор 5-оксо-3-фенил-3-циклогексенкарбоновой кислоты. В реактор загружают 31 л изопропилового спирта для промывки. Промывной спирт из реактора передают в аппарат. В этот момент завершается полное растворение. Содержимое аппарата охлаждают до 50 - 55oC более 1 часа. Температуру теплоносителя для аппарата устанавливают при 50oC, как минимум, на 2 часа. Температуру теплоносителя для аппарата устанавливают при 40oC, как минимум, на 1 час. Температуру теплоносителя для аппарата устанавливают при 25oC, как минимум, на 10 часов. Содержимое аппарата охлаждают до 0 - 5oC и перемешивают не менее 2 часов. Твердый продукт из аппарата отфильтровывают на центрифуге и маточный раствор направляют в реактор для регенерации вещества. Это делается для того, чтобы затем извлечь цинхонидин и (R)-5-оксо-3-фенил-3-циклогексенкарбоновую кислоту. В аппарат загружают 41 л изопропилового спирта, который используют для промывки осадка на фильтре. Промывной спирт направляют в реактор для регенерации вещества. В аппарат емкостью 200 л загружают полученный влажный осадок из центрифуги. В аппарат емкостью 200 л загружают 50 кг этилового спирта, 2В. Смесь перемешивают и нагревают до кипения около 15 минут. Перемешивание замедляют и температуру теплоносителя для аппарата устанавливают при 25oC. Перемешивание продолжается по меньшей мере в течение 8 часов. Содержимое аппарата охлаждают до 0 - 5oC и перемешивают не менее 2 часов. Твердый продукт из аппарата емкостью 200 л фильтруют на центрифуге и маточный раствор направляют в реактор для регенерации. В аппарат емкостью 200 л загружают 4 кг этилового спирта, 2В, который используется для промывки осадка. Промывной спирт направляют в реактор для регенерации. Примерно 10 г твердого вещества отбирают из центрифуги и свободную кислоту выделяют при подкислении соляной кислотой, экстрагировании (дважды) этилацетатом и концентрировании экстракта на роторном испарителе. Полученный продукт анализируют с помощью хиральной высокоэффективной жидкостной хроматографии (ВЭЖХ). Если отношение S:R не более 99:1, перекристаллизация из этилового спирта, 2В, даст возможность достичь этот уровень.
Соль с центрифуги загружают в аппарат емкостью 400 л с 67 кг метилового спирта и начинают перемешивание. В аппарат загружают раствор 3.75 кг едкого натра, 50%-ного, в 40 л воды. Смесь перемешивают около 1 часа при 20 - 25oC, в аппарат загружают 120 л воды. Спирт отгоняют под вакуумом при перемешивании при температуре 30 - 35oC до заметного уменьшения скорости дистилляции. Содержимое аппарата охлаждают до 20 - 25oC. В аппарат подают азот и дистиллят выливают. Регенерированный твердый цинхонидин из аппарата подвергают фильтрации на центрифуге. Фильтраты направляют в реактор. Регенерированный цинхонидин промывают на центрифуге 20 л воды и промывную воду направляют в реактор. Влажный регенерированный цинхонидин направляют в полочную вакуумную сушилку и сушат при 35 ± 5oC, как минимум, 24 часа при максимально возможном разрежении. Регенерированный цинхонидин может быть заново использован. Раствор натриевой соли продукта в реакторе обрабатывают при перемешивании 13 кг 37%-ной химически чистой соляной кислоты. pH суспензии проверяют с помощью индикаторной бумаги. pH должен быть от 1.5 до 2.0. Если pH выше 2.0, прибавляют дополнительно 37%-ную химически чистую соляную кислоту. Полученную суспензию в аппарате перемешивают около 1 часа и охлаждают до 5 - 10oC. (S)-5-Оксо-3-фенил-3-циклогексенкарбоновую кислоту из аппарата фильтруют на центрифуге. Фильтрат выливают. В аппарат загружают 20 л воды и ее используют для промывки осадка после центрифугирования. Промывную воду выливают. Влажную (S)-5-оксо-3-фенил-3-циклогексенкарбоновую кислоту направляют в полочную вакуумную сушилку и сушат при 65 ± 5oC, как минимум, 24 часа при максимально возможном разрежении. Получают 20 кг (S)-5-оксо-3-фенил-3-циклогексенкарбоновой кислоты в виде твердого вещества от белого до бледно-желтого цвета; т. пл. 111 - 113oC (нескорректированная).
200 МГц 1H ЯМР (дейтерохлороформ): δ 2.3 - 2.95 (м, 4H), 2.96 - 3.45 (м, 3H), 6.45 (с, 1H), 7.25 - 7.65 (м, 5H), 10.0 - 10.8 (уш. с, 1H).
Анализ с помощью хиральной ВЭЖХ: S-энантиомер 99.24%, время удерживания 26.9 минуты, R-энантиомер 0.76%, время удерживания 20.9 минуты
Условия хиральной ВЭЖХ:
Колонка: Chiralpak AD, 250 х 4.6 мм
Скорость потока: 1.0 мл/мин
Подвижная фаза: 900 гексан/100 изопропанол/1 муравьиная кислота (о/о)
Длина волны: 254 нм
Инжектируемый объем: 20 мкл
Концентрация образца: ≈ 10.0 мг/10 мл в изопропаноле
Время удерживания: S-энантиомер ≈ 25-27 минут, R-энантиомер ≈ 20-21 минут
Колонку после использования хранят в гексане/изопропаноле (900:100) (о/о).
Фильтраты из реактора для регенерации передают в аппарат. В аппарат загружают при перемешивании 11.2 кг едкого натра, 50%-ного, в 100 л воды. Растворители перемешивают и подвергают перегонке в вакууме при 30 - 35oC до тех пор, пока скорость перегонки значительно не замедлится. Содержимое аппарата охлаждают до 20 - 25oC и в аппарат подают азот. Дистиллят выливают. В аппарат загружают 50 л воды, регенерированный цинхонидин из аппарата отфильтровывают на центрифуге и маточный раствор направляют в реактор. Регенерированный цинхонидин промывают на центрифуге 20 л воды и промывную воду направляют в реактор. Влажный регенерированный цинхонидин передают в полочную вакуумную сушилку и сушат при 35 ± 5oC не менее 24 часов при максимально возможном разрежении. Фильтраты (раствор натриевой соли в основном (R)-5-оксо-3-фенил-3-циклогексенкарбоновой кислоты) из реактора передают в аппарат. В аппарат загружают при перемешивании 15 кг химически чистой 37%-ной соляной кислоты. Полученную суспензию в аппарате перемешивают около 1 часа и охлаждают до 5 - 10oC. Твердую (R)-5-оксо-3-фенил-3-циклогексенкарбоновую кислоту из аппарата фильтруют на центрифуге. Фильтрат выливают. В аппарат загружают 20 л воды, которую используют для промывки отфильтрованного осадка. Фильтрат выливают. Влажную (R)-5-оксо-3-фенил-3-циклогексенкарбоновую кислоту передают в полочную вакуумную сушилку и сушат при 80 ± 5oC, как минимум, 24 часа при максимально возможном разрежении. Вещество может быть подвергнуто рацемизации в соединение формулы VIII из примера А при использовании стандартной методики.
Способ Б: Методика с использованием α-химотрипсина
1 г н-бутилового эфира 5-оксо-3-фенил-3-циклогексенкарбоновой кислоты (пример Б) помещают в круглодонную колбу емкостью 250 мл, снабженную механической мешалкой. К эфиру прибавляют примерно 80 мл воды и смесь перемешивают. Доводят pH смеси до 5.0, используя разбавленную соляную кислоту. К этому раствору прибавляют 0.1 г (≈ 10% по весу) α-химотрипсина и смывают 20 мл воды. PH реакционной смеси поддерживают при 5 прибавлением разбавленного раствора едкого натра, осуществляя контроль с помощью лабораторного pH-метра. Реакцию проводят при температуре окружающей среды. Реакцию останавливают через 2.09 дня, например, когда расход основания составляет 90 - 100% от теоретического количества, нужного для гидролиза 50% рацемической смеси.
Отношение (R)-эфира к (S)-эфиру составляет 94:6.
Желаемую (S)-5-оксо-3-фенил-3-циклогексенкарбоновую кислоту получают при подкислении разбавленной соляной кислотой. Твердый осадок фильтруют и сушат в вакууме при 80 ± 5oC, как минимум, 24 часа при максимально возможном разрежении.
Стадия Б: Получение (S)-1,2,3,6-тетрагидро-1-[(5-оксо-3-фенил-3- циклогексенил)карбонил]-4-фенилпиридина
Способ А
В аппарат емкостью 400 л загружают 6.0 кг (27.8 моля) (S)-5-оксо-3-фенил-3-циклогексенкарбоновой кислоты, 4.1 кг (30.3 моля) гидрата 1-гидроксибензотриазола, 7.0 кг (31.6 моля) хлоргидрата 1-(3-диметиламинопропил)-3-этилкарбодиимида, 6.2 кг (33.4 моля)
хлоргидрата 4-фенил-1,2,3,6-тетрагидропиридина и 90.0 кг ацетонитрила. Смесь перемешивают и охлаждают до 5 - 10oC, используя дозирующий насос, загружают 4.6 кг (45.5 моля) триэтиламина со скоростью около 0.3 кг/минуту. Температуру загруженной смеси поддерживают при 15 - 20oC. Смесь в аппарате перемешивают, как минимум, 6 часов и дают ей нагреться до температуры окружающей среды.
В реактор емкостью 200 л загружают 60.0 л воды и 4.0 кг (47.6 моля) бикарбоната натрия и начинают перемешивание. Водный раствор бикарбоната натрия передают из реактора в аппарат. В аппарат загружают 80 л воды, а затем 90.0 л толуола. Смесь в аппарате перемешивают, как минимум, 30 минут и оставляют для разделения фаз. Нижнюю водную фазу из аппарата сливают в реактор. В аппарат загружают 50.0 кг воды. Смесь в аппарате перемешивают не менее 30 минут и оставляют для разделения фаз. Нижнюю водную фазу из аппарата сливают в реактор. В реактор загружают толуол (30.0 л), смесь в реакторе перемешивают в течение 30 минут и оставляют для разделения фаз. Нижнюю водную фазу выливают в систему для химических отходов. Толуольную фазу из реактора передают в аппарат. В реактор загружают 50 л воды и 6.8 кг (65.7 моля) химически чистой 37%-ной соляной кислоты. Разбавленную водой соляную кислоту передают из реактора в аппарат емкостью 400 л. Водный солянокислый экстракт выливают в систему для химических отходов. В реактор загружают 40.0 л воды и 4.0 кг хлористого натрия. Водный раствор хлористого натрия передают из реактора в аппарат, смесь в аппарате перемешивают в течение 20 минут и оставляют для разделения фаз. Нижнюю водную фазу выливают из аппарата в систему для химических отходов. Верхнюю толуольную фазу, содержащую продукт, из аппарата емкостью 400 л передают в аппарат емкостью 200 л (для удобства при дистилляции). Аппарат емкостью 200 л предназначается для вакуумной перегонки в реактор емкостью 200 л. Температуру теплоносителя для холодильника устанавливают при 5 - 10oC и раствор концентрируют до объема 20 ± 5 л. Температура находящегося в аппарате содержимого поддерживается ниже 60oC. Дистиллят выливают. В аппарат загружают 30 кг этилового спирта, 2В. Раствор в аппарате охлаждают до 0 ± 5oC и выдерживают, как минимум, 2 часа. Твердый продукт из аппарата фильтруют на центрифуге, направляя фильтрат в реактор. Продукт, содержащий остатки спирта и толуола, помещают в полочную вакуумную сушилку и сушат при 40 ± 5oC не менее 24 часов при максимально возможном разрежении. Фильтрат передают из реактора в аппарат. Аппарат предназначается для вакуумной перегонки в реактор. Фильтрат упаривают до 5 ± 5 л. Раствор передают в бутыль емкостью 20 л. Дистиллят выливают. Сконцентрированный фильтрат охлаждают при 0 ± 5oC в течение 24 - 48 часов. Твердый продукт фильтруют на воронке Бюхнера. Продукт, содержащий следы спирта и толуола, передают в полочную вакуумную сушилку и сушат при 40 ± 5oC не менее 24 часов при максимально возможном разрежении. Сухой продукт помещают в облицованные пластиком барабаны и хранят в сухом месте при температуре ниже 35oC.
Получают 8.4 кг первой порции продукта и 0.6 кг второй порции продукта.
Продукт, (S)-1,2,3,6-тетрагидро-1-[(5-оксо-3-фенил-3- циклогексенил)карбонил]-4-фенилпиридин, является твердым веществом, имеющим цвет от не совсем белого до бледно-желтого; т. пл. 137 - 141oC (нескорректированная).
200 МГц 1H ЯМР (дейтерохлороформ): δ 2.62 (м, 2H), 2.89 (м, 2H), 3.00 (м, 1H), 3.74 (м, 1H), 3.88 (м, 1H), 3.91 (м, 1H), 4.30 (м, 2H), 6.04 (м, 2H), 6.14 (с, 1H), 7.23 - 7.60 (м, 10H).
Способ Б
(S)-5-Оксо-3-фенил-3-циклогексенкарбоновую кислоту, 24.0 кг (111.1 моля), загружают в аппарат емкостью 200 л с 150.0 кг ацетонитрила. В отдельный реактор загружают 20.0 кг (123.5 моля) карбонилдиимидазола и 100.0 кг ацетонитрила. Раствор в реакторе охлаждают до 20 ± 5oC. Раствор карбонилдиимидазола медленно передают из реактора в аппарат. Температура содержимого аппарата поддерживается при 25 ± 5oC. В реактор загружают 50.0 кг ацетонитрила, промывают и все передают из реактора в аппарат. Температуру смеси в аппарате поддерживают при 20 - 30oC в течение 3 - 4 часов. Смесь в аппарате охлаждают до 10 - 15oC при перемешивании. В аппарат загружают 18.0 кг (112.9 моля) 4-фенил-1,2,3,6-тетрагидропиридина и поддерживают температуру в аппарате при 20 - 25oC. Шланги для передачи веществ промывают 50.0 кг ацетонитрила и промывной ацетонитрил направляют в аппарат. В аппарат загружают 12.0 кг (118.6 моля) триэтиламина, используя дозирующий насос, со скоростью около 1.0 кг/минуту. Температуру содержимого аппарата поддерживают при 20 - 25oC. (Если используют хлоргидрат 4-фенил-1,2,3,6-тетрагидропиридина, загружают 24 кг триэтиламина). Смесь в аппарате перемешивают, как минимум, 6 часов. Температура содержимого аппарата поднимается до температуры окружающей среды. В реактор загружают 500 л воды и 10 кг (119 молей) бикарбоната натрия, смесь перемешивают. Водный раствор бикарбоната натрия из реактора медленно загружают в аппарат. В аппарат загружают 500 л толуола. Смесь в аппарате перемешивают около 30 минут и оставляют для разделения фаз. Нижнюю водную фазу из аппарата передают в реактор. В аппарат загружают 200 л воды. Смесь в аппарате перемешивают не менее 15 минут и оставляют для разделения фаз. Нижнюю водную фазу из аппарата передают в реактор. В реактор загружают 100 л толуола. Смесь в реакторе перемешивают около 15 минут и оставляют для разделения фаз. Нижнюю водную фазу из реактора выливают. Верхнюю толуольную фазу из реактора передают в аппарат. В реактор загружают 300 л воды, 12 кг 37%-ной химически чистой соляной кислоты и смесь перемешивают. Разбавленную водой соляную кислоту медленно передают при перемешивании из реактора в аппарат. Смесь в аппарате перемешивают не менее 15 минут и оставляют для разделения фаз. Нижнюю водную фазу из аппарата выливают. В реактор загружают 300 л воды и 20 кг хлористого натрия. Водный раствор хлористого натрия из реактора направляют в аппарат, смесь перемешивают около 20 минут и оставляют для разделения фаз. Нижнюю водную фазу из аппарата выливают. Верхнюю толуольную фазу, содержащую продукт, из аппарата передают порциями в меньший аппарат для перегонки под вакуумом. Меньший аппарат предназначается для перегонки под вакуумом, и раствор в нем концентрируют до объема около 60 ± 5 л. Температура загруженной в аппарат реакционной смеси поддерживается ниже 65oC. Дистиллят выливают. В аппарат загружают 120 кг этилового спирта, 2В. Раствор в аппарате охлаждают до 0 ± 5oC и выдерживают не менее 2 часов. Твердый продукт из аппарата фильтруют на центрифуге. Фильтрат направляют в реактор. В аппарат загружают 50 кг этилового спирта, 2В. Смесь в аппарате перемешивают, как минимум, 10 минут и промывную жидкость направляют из аппарата на осадок в центрифуге. Фильтрат направляют в реактор. Твердый продукт из центрифуги переносят в полочную сушилку и, если нужно, сушат в вакууме. Продукт обычно используют на следующей стадии влажным. Фильтраты передают из реактора в аппарат. Содержимое аппарата подвергают перегонке в вакууме, поддерживая температуру в аппарате ниже 60oC. Содержимое подвергают перегонке до объема 50 ± 10 л. Дистиллят выливают. Раствор в аппарате охлаждают до 0 ± 5oC в течение, как минимум, 2 часов. Твердый продукт из аппарата отфильтровывают на центрифуге. Фильтрат направляют в реактор. В аппарат загружают 30 кг этилового спирта, 2В. Смесь в аппарате перемешивают не менее 10 минут и промывную жидкость направляют из аппарата на осадок в центрифуге. Фильтраты выливают. Твердый продукт из центрифуги помещают в полочную сушилку и сушат в вакууме, если необходимо. Продукт обычно используется на следующей стадии влажным.
Стадия В: Получение (S)-1,2,3,6-тетрагидро-1-[(5-гидрокси-3- фенил-3-циклогексенил)карбонил]-4-фенилпиридина
(S)-1,2,3,6-Тетрагидро-1-[(5-оксо-3-фенил-3-циклогексенил)карбонил] - 4-фенилпиридин, 8.4 кг (23.5 моля), загружают в аппарат емкостью 200 л с 30 кг этилового спирта, 2В. Смесь в аппарате охлаждают до 15 ± 5oC. В реактор загружают 1.2 кг (32.4 моля) натрийборгидрида и 30 кг этилового спирта, 2В. Содержимое реактора передают в аппарат. Температуру смеси в аппарате поддерживают при 20 ± 5oC. В реактор загружают 15 кг этилового спирта, 2В. Этиловый спирт в реакторе перемешивают не менее 3 минут и передают в аппарат. Суспензию в аппарате перемешивают, как минимум, в течение 12 часов. Температура содержимого в аппарате поднимается до 25 ± 5oC. Смесь в аппарате охлаждают до 10 ± 5oC. В реактор загружают 30 л воды и 5 кг (92.5 моля) хлористого аммония. Раствор хлористого аммония направляют из реактора в аппарат, используя дозирующий насос, со скоростью около 0.3 л/минуту. Если происходит вспенивание, уменьшают или замедляют прибавление до исчезновения пены. Раствор в аппарате охлаждают в атмосфере азота до 0 ± 5oC не менее 2 часов. Твердый продукт из аппарата отфильтровывают на центрифуге. Фильтраты выливают. Влажный продукт передают в полочную вакуумную сушилку и сушат при 40 ± 5oC не менее 24 часов при максимально возможном разрежении.
Выход (S)-1,2,3,6-тетрагидро-1-[(5-гидрокси-3-фенил-3- циклогексенил)карбонил] -4-фенилпиридина составляет 6.38 кг (первая порция продукта) и 0.3 кг (вторая порция продукта), полученный продукт является твердым веществом, имеющим цвет от белого до бледно-желтого; т. пл. 162 - 167oC (нескорректированная).
200 МГц 1H ЯМР (дейтерохлороформ): δ 1.75 (м, 2H), 2.62 (м, 2H), 3.16 (м, 1H), 3.21 (м, 2H), 3.73 (м, 2H), 3.79 (м, 1H), 4.25 (м, 2H), 4.28 (м, 1H), 6.03 (с, 1H), 6.17 (м, 1H), 7.26 - 7.42 (м, 10H).
Анализ (ВЭЖХ): 96.2% по площади пика (главный изомер) и 1.7% по площади пика (второстепенный изомер).
Условия ВЭЖХ:
Колонка: YMC-AQ, C18 5 мкм, 250 х 4.6 мм
Скорость потока: 1.5 мл/минуту
Подвижная фаза: 600 ацетонитрил/400 раствор А* (о/о)
Длина волны: 214 нм
Инжектируемый объем: 20 мкл
Концентрация образца: ≈ 5.0 мг/25 мл в подвижной фазе
* Раствор А: 5.75 г кислого однозамещенного фосфата аммония растворяют в 1 л воды с чистотой для ВЭЖХ, прибавляют 6 мл триэтиламина и доводят pH раствора до 3.0, используя 85%-ную ортофосфорную кислоту.
Стадия Г: Получение (R)-(+)-1,2,3,6-тетрагидро-4-фенил-1-[(3- фенил-3-циклогексенил)карбонил]пиридина
В скруббер загружают 750 л водопроводной воды и 300 кг едкого натра, 50%-ного. Скруббер должен быть соединен со всеми частями оборудования для удаления в целях безопасности выделяющегося в ходе процесса цианистого водорода.
В аппарат емкостью 400 л загружают 8.4 кг (23.4 моля) (S)-1,2,3,6-тетрагидро-1-[(5-гидрокси-3-фенил-3-циклогексенил)карбонил] - 4-фенилпиридина, 3.8 кг (27.9 моля) хлористого цинка, 3.5 кг (55.7 моля) цианборгидрида натрия и 70 кг гептана. Смесь в аппарате перемешивают. В реактор емкостью 80 л загружают 15 кг тетрагидрофурана и 5 кг (83.3 моля) ледяной уксусной кислоты. Температуру загруженной в аппарат смеси поддерживают при 25 ± 5oC. Раствор ледяной уксусной кислоты в тетрагидрофуране передают из реактора в аппарат. Смесь в аппарате перемешивают, как минимум, 6 часов при 25 ± 5oC. В реактор емкостью 80 л загружают 30 л воды и 5.3 кг (99 молей) хлористого аммония и начинают перемешивание.
Водный хлористый аммоний передают из реактора в аппарат с помощью дозирующего насоса со скоростью около 1 л/минуту. Температуру загруженной в аппарат смеси поддерживают при 20 ± 5oC. Смесь в аппарате перемешивают не менее 30 минут. В реактор загружают 20 л воды и 3.6 кг (34.8 моля) концентрированной 37%-ной соляной кислоты.
Разбавленную соляную кислоту передают из реактора в аппарат с помощью дозирующего насоса со скоростью около 0.5 л/минуту. Температуру загруженной в аппарат смеси поддерживают при 20 ± 5oC. Смесь в аппарате перемешивают не менее 2 часов.
Аппарат используют для перегонки под вакуумом с применением работающей под вакуумом скрубберной системы. Раствор концентрируют до объема примерно 350 ± 50 л. Температуру загруженной в аппарат смеси поддерживают ниже 50oC. Дистиллят, представляющий смесь гептана и тетрагидрофурана, выливают. В реактор загружают 200 л воды.
Внимание: Фильтрат содержит цианистый водород, и пары могут содержать цианистый водород.
Герметизированный фильтр для отделения капель жидкости испытывается перед использованием при регистрируемом манометром давлении азота 1.0545 кг/см2. Любое падение давления не должно превышать 0.0703 кг/см2 по манометру за период в 15 минут. Другими словами, фиксируют утечки и заново проверяют. Продукт из аппарата фильтруют на указанном фильтре, направляя фильтрат в реактор. В аппарат загружают 600 л тетрагидрофурана. Содержимое аппарата перемешивают и нагревают до 40 ± 5oC. Около 300 л теплого тетрагидрофуранового раствора направляют из аппарата на герметизированный фильтр. Продукт с фильтра заново суспендируют и нагревают до 40 ± 5oC, используя воду с температурой около 50oC в рубашке для теплоносителя. Содержимое перемешивают и выдерживают при этой температуре не менее 30 минут.
Фильтр Полла вставляют у входного вентиля аппарата для удаления любых мелких частиц. Теплый тетрагидрофуран с растворенным продуктом из упомянутого фильтра передают через фильтр Полла в аппарат. Остаток теплого тетрагидрофуранового раствора из первого аппарата передают на герметизированный фильтр. Содержимое с фильтра заново суспендируют и нагревают до 40 ± 5oC, используя в рубашке воду с температурой около 50oC. Смесь перемешивают и выдерживают при этой температуре не менее 30 минут. Теплый тетрагидрофуран с растворенным продуктом передают из герметизированного фильтра через фильтр Полла в аппарат. Герметизированный фильтр промывают 50 л тетрагидрофурана через распылительные форсунки. Промывной тетрагидрофуран направляют через фильтр Полла и трубопровод в аппарат. Аппарат, в котором находится раствор продукта в тетрагидрофуране, используют для перегонки в вакууме, применяя работающую под вакуумом скрубберную систему. Раствор концентрируют до объема примерно 60 ± 20 л. Температуру содержимого в аппарате поддерживают ниже 50oC. В аппарат со сконцентрированным раствором продукта загружают 150 кг абсолютного этилового спирта. В аппарате проводят перегонку под вакуумом. Раствор концентрируют до объема примерно 60 ± 20 л. Температуру содержимого аппарата поддерживают ниже 50oC. В аппарат загружают 300 кг абсолютного этилового спирта. Дистиллят, состоящий из тетрагидрофурана и этилового спирта, выливают. Смесь в аппарате перемешивают около 1 часа при 75 ± 5oC. Раствор в аппарате охлаждают в атмосфере азота до 0 ± 5oC и выдерживают не менее 2 часов. Твердый продукт из аппарата отфильтровывают на центрифуге. Фильтрат направляют в реактор емкостью 80 л. В аппарат загружают 15 кг этилового спирта, 2В. Смесь в аппарате перемешивают не менее 3 минут и промывную жидкость передают из аппарата на центрифугу, направляя при этом фильтрат в реактор емкостью 80 л. Фильтраты из реактора передают в аппарат емкостью 200 л. В аппарате проводят перегонку под вакуумом. Раствор упаривают до объема примерно 15 ± 5 л. Температуру содержимого аппарата поддерживают ниже 40 ± 5oC. Раствор передают в бутыль емкостью 20 л. Бутыль с содержимым охлаждают до 0 ± 5oC. Образовавшийся твердый осадок фильтруют на воронке Бюхнера. Влажный продукт направляют в полочную вакуумную сушилку и сушат при 40 ± 5oC не менее 24 часов при максимально возможном разрежении.
Выход (R)-(+)-1,2,3,6-тетрагидро-4-фенил-1-[(3-фенил-3- циклогексенил)карбонил] пиридина составляет 4 кг (первая порция продукта) и 0.5 кг (вторая порция продукта), полученный продукт является твердым веществом, имеющим цвет от белого до бледно-желтого; т. пл. 142 - 147oC (нескорректированная).
200 МГц 1H ЯМР (дейтерохлороформ): δ 1.91 (м, 2H), 2.37 (м, 2H), 2.52 - 2.61 (м, 1H), 3.77 (м, 2H), 3.88 (с, 1H), 4.24 (м, 2H), 5.29 (м, 2H), 6.04 (c, 1H), 6.14 (м, 2H), 7.23 - 7.42 (м, 10H).
Стадия Д: Получение (R)-(+)-1,2,3,6-тетрагидро-4-фенил-1-[(3- фенил-3-циклогексен-1-ил)метил]пиридина
Емкость с абсолютным этиловым спиртом помещают в холодное помещение для дальнейшего использования для промывок. В аппарат емкостью 200 л загружают 4.0 кг (11.64 моля) (R)-(+)-1,2,3,6-тетрагидро- 4-фенил-1-[(3-фенил-3-циклогексенил)карбонил] пиридина. В аппарат загружают 44.4 кг безводного трет-бутилметилового эфира (содержащего менее 0.02% воды).
В аппарат загружают 16.6 кг (18 молей) 1 М раствора алюмогидрида лития в тетрагидрофуране при скорости подачи от 0.1 до 0.5 кг/минуту и давлении аргона менее 0.8436 кг/см2. Температуру загруженной в аппарат смеси поддерживают при 30 - 45oC. Эта реакция успешно проходила при температуре между 0oC и температурой кипения (≈ 66oC). Для достижения лучших результатов прибавление прекращают, если температура поднимается выше 45oC. Охлаждают до температуры ниже 45oC и возобновляют прибавление.
По окончании прибавления линию подачи алюмогидрида лития промывают примерно 5 кг тетрагидрофурана. Промывной раствор прибавляют в аппарат. Смесь в аппарате перемешивают при 30 - 45oC в течение 3 - 4 часов. Смесь в аппарате охлаждают до 20 - 25oC. В стеклянной бутыли, помещенной в металлическую канистру, готовят раствор 1.27 кг воды в 4.4 кг тетрагидрофурана. Раствор 1.27 кг воды в 4.4 кг тетрагидрофурана загружают в аппарат, используя дозирующий насос, начиная со скорости 0.05 - 0.1 л в час до уменьшения вспенивания, затем прибавление всего раствора завершают при скорости 0.1 - 0.5 л в час. Готовят насыщенный раствор сернокислого натрия в воде путем смешивания 1 кг сернокислого натрия с 5 кг воды в стеклянной бутыли, помещенной в металлическую канистру. Если весь сернокислый натрий растворился, прибавляют еще сернокислый натрий, чтобы оставалось некоторое нерастворившееся количество. В аппарат загружают 4 кг насыщенного раствора сернокислого натрия с помощью дозирующего насоса со скоростью 0.1 - 0.5 л в час. Температуру содержимого аппарата поднимают до 40 - 45oC. Суспензию из аппарата, содержащую неорганические вещества, фильтруют через фильтр и фильтрат направляют в реактор емкостью 400 л. В аппарат загружают 20 л тетрагидрофурана (для промывки) и нагревают до 40 - 45oC. Промывной тетрагидрофуран направляют через фильтр в реактор емкостью 400 л. Температуру раствора продукта в реакторе поддерживают при 40 - 45oC.
Раствор продукта передают в аппарат и большую часть растворителя отгоняют под вакуумом. Прибавляют абсолютный этиловый спирт и большую часть растворителя отгоняют под вакуумом. Прибавляют абсолютный этиловый спирт и раствор охлаждают до -10oC - 0oC в течение 1 - 2 часов, (R)-(+)-1,2,3,6-тетрагидро-4-фенил-1-[(3-фенил-3- циклогексен-1-ил)метил] пиридин выделяют при центрифугировании. Отделенный при центрифугировании осадок промывают холодным абсолютным этиловым спиртом и сушат в вакууме. Хранят так же, как и малеат.
Стадия Е: Получение малеата (R)-(+)-1,2,3,6-тетрагидро-4-фенил- 1-[(3-фенил-3-циклогексен-1-ил)метил]пиридина
Раствор продукта в тетрагидрофуране после стадии Д передают в аппарат и подвергают перегонке под вакуумом. Температуру содержимого в аппарате поддерживают при 25 - 75oC. В аппарат загружают 100 л абсолютного этилового спирта и большую часть этилового спирта и тетрагидрофурана отгоняют в вакууме. Температуру содержимого в аппарате поддерживают при 25 - 75oC. В аппарат загружают 75 л абсолютного этилового спирта и смесь нагревают до слабого кипения. В реактор емкостью 200 л загружают 1.74 кг (15 молей) малеиновой кислоты и 15.8 кг абсолютного этилового спирта. Полученную суспензию перемешивают в реакторе и температуру смеси поддерживают при 60 - 65oC. Раствор продукта в этиловом спирте прибавляют к раствору малеиновой кислоты или альтернативно раствор малеиновой кислоты прибавляют к раствору продукта в этиловом спирте в реакторе емкостью 400 л и через реактор продувают аргон.
Температуру содержимого реактора поддерживают при 50 - 55oC и перемешивают в течение 1 - 2 часов. Неорганические примеси удаляют при фильтрации. В реактор вносят при необходимости в качестве затравки немного очищенного продукта.
Содержимое реактора охлаждают до 35 - 40oC и перемешивают в течение 1 - 2 часов. Содержимое реактора охлаждают до -10oC - -5oC и перемешивают в течение 1 - 2 часов. Твердый продукт отделяют на центрифуге и фильтрат направляют в реактор емкостью 400 л. В реактор загружают 15.8 кг холодного абсолютного этилового спирта, которым затем промывают осадок в центрифуге. Фильтрат направляют в реактор емкостью 400 л. Влажный продукт со следами этилового спирта передают в полочную вакуумную сушилку и сушат при 30 ± 5oC не менее 24 часов при максимально возможном разрежении.
Сухой продукт помещают в пластиковые мешки, выложенные внутри двойным полиэтиленом, находящиеся в защищенных от попадания влаги барабанах, снабженных респиратором на воздушной линии. Продукт следует хранить в сухом помещении при температуре ниже 35oC.
Вторая порция продукта:
Фильтраты из реактора передают в аппарат. Фильтраты упаривают в вакууме до тех пор, пока уровень раствора не станет ниже уровня мешалки. В аппарат загружают такое количество абсолютного этилового спирта, чтобы его было достаточно для перемешивания, и охлаждают содержимое до температуры -10 - -5oC. Дистиллят выливают. Твердый продукт из аппарата фильтруют на центрифуге и фильтрат выливают. В реактор емкостью 200 л загружают 8 кг холодного абсолютного этилового спирта. Этот промывной спирт направляют на промывку осадка в центрифуге, фильтрат выливают. Продукт со следами спирта направляют в полочную вакуумную сушилку и сушат при 30 ± 5oC не менее 24 часов при максимально возможном разрежении.
Сухой продукт помещают в пластиковые мешки, выложенные внутри двойным полиэтиленом, находящиеся в защищенных от попадания влаги барабанах. Продукт хранят при температуре ниже 35oC в сухом помещении.
Выход малеата (R)-(+)-1,2,3,6-тетрагидро-4-фенил-1-(3-фенил-3- циклогексен-1-ил)метилпиридина составляет 3.6 кг (первая порция продукта) и 0.6 кг (вторая порция продукта), полученный продукт является твердым веществом белого или не совсем белого цвета.
200 МГц 1H ЯМР (дейтерохлороформ): δ 1.65 - 1.80 (м, 1H), 1.93 - 2.05 (м, 1H), 2.05 - 2.25 (м, 4H), 2.25 - 4.20 (м, 9H), 6.0 (м, H), 6.08 (м, 1H), 6.15 (с, 2H), 7.15 - 7.45 (м, 10H); сигналы кислотных протонов малеиновой кислоты не наблюдаются.
Анализ (ВЭЖХ) (Площадь, %): ВЭЖХ: не менее 98%.
Время удерживания (мин) - Площадь, %
23.2 - 99.52
Примеси (ВЭЖХ Площадь, %): Не более 0.5% каждой
Время удерживания (мин) - Площадь, %
6.50 - 0.04
6.90 - 0.28
9.40 - 0.01
11.50 - 0.06
18.50 - 0.07
21.0 - 0.01
21.50 - 0.01
Всего примесей: 0.48%
Не более 2.0%
Условия ВЭЖХ
Колонка: YMC-AQ, C18, 5 мкм, 250 х 4.6 мм
Скорость потока: 1.5 мл/минуту
Подвижная фаза: 350 ацетонитрил/650 раствор А* (о/о)
Длина волны: 214 нм
Инжектируемый объем: 20 мкл
Концентрация образца: ≈ 5.0 мг/25 мл в подвижной фазе
* Раствор А: 5.75 г кислого однозамещенного фосфата аммония растворяют в 1 л воды с чистотой для ВЭЖХ, прибавляют 6 мл триэтиламина и доводят pH раствора до 3.0, используя 85%-ную ортофосфорную кислоту.
Титрование малеиновой кислоты: 26.00% (среднее из двух значений)
Теоретическое значение 26.05%
Удельное вращение: [α]
Отношение по данным хиральной ВЭЖХ:ВЭЖХ:
Время удерживания (мин) - Площадь, % - Отношение
12.6 - не опред.(-)
Энантиомер (граница определения 0.03%)
33.2 - 99.96%
Условия:
Колонка: Chiralcel OJ, 250 х 4.6 мм
Скорость потока: 1.0 мл/минуту
Подвижная фаза: - 700 гексан/300 изопропиловый спирт (о/о)
Длина волны: 254 нм
Инжектируемый объем: 20 мкл
Концентрация образца: ≈ 10.0 мг/25 мл в изопропиловом спирте (ультразвуковое воздействие для растворения)
Приготовление исходных веществ
Пример А. 5-Оксо-3-фенил-3-циклогексенкарбоновая кислота
В аппарат емкостью 800 л загружают 53.9 кг (306 молей) 3-бензоилакриловой кислоты. В аппарат емкостью 800 л загружают 41 кг (315 молей) этилового эфира ацетоуксусной кислоты. В аппарат загружают 150 л воды. Начинают перемешивание содержимого аппарата. В аппарат загружают 56 кг (1475 молей) 50%-ного едкого натра, а затем 20 л воды. Полученный раствор перемешивают и температуру содержимого аппарата поддерживают при 45 - 50oC в течение 16-18 часов после прибавления едкого натра. Раствор в аппарате затем кипятят в течение 20 - 24 часов (≈ 100oC). В реактор емкостью 800 л загружают 140 л воды и 90.6 кг (1100 молей) концентрированной соляной кислоты, а затем 20 л воды и начинают перемешивание. Реакционную смесь в аппарате охлаждают до 25 - 40oC. Аппарат сообщается через холодильник со скруббером, работающим под атмосферным давлением. Охлажденный раствор из аппарата (25 - 40oC) прибавляют к разбавленному раствору соляной кислоты в реакторе со скоростью около 2 - 4 л/минуту. В аппарат загружают 100 л воды, которую передают в реактор. Полученную в реакторе суспензию перемешивают при 25 - 40oC в течение 2 - 4 часов. Твердый продукт из реактора отфильтровывают на центрифуге. В реактор загружают 100 л воды и этой водой промывают осадок в центрифуге. Маточные растворы и промывную воду выливают. В аппарат загружают 100 л воды, эту воду направляют для промывки отфильтрованного осадка и фильтрат выливают. Влажный осадок продукта из центрифуги возвращают в аппарат, затем туда заливают 400 л воды и суспензию перемешивают в течение 2 - 4 часов. Твердый продукт отфильтровывают на центрифуге. Аппарат и отфильтрованный осадок промывают 200 л воды. Фильтраты выливают. Влажную 5-оксо-3-фенил-3-циклогексенкарбоновую кислоту направляют в полочную вакуумную сушилку и сушат при 80 ± 5oC не менее 24 часов при максимально возможном разрежении, получают 62.5 кг твердого вещества, имеющего цвет от не совсем белого до бледно-желтого; т. пл. 140 - 160oC (среднее значение из двух определений). Высушенный продукт хранят в барабанах, облицованных изнутри пластиком.
200 МГц 1H ЯМР (дейтерохлороформ): δ 2.3 - 2.95 (м, 4H), 2.96 - 3.45 (м, 3H), 6.45 (с, 1H), 7.25 - 7.65 (м, 5H), 10.0 - 10.8 (уш.с, 1H).
Пример Б. н-Бутиловый эфир 5-оксо-3-фенил-3-циклогексенкарбоновой кислоты
Смесь 5-оксо-3-фенил-3-циклогексенкарбоновой кислоты (пример А) (216 г) и н-бутилового спирта (500 г) подвергают обработке концентрированной серной кислотой (10 мл) при перемешивании. Смесь выдерживают при температуре окружающей среды в течение 18 часов, нагревают при 50 - 55oC в течение 1 - 2 часов и затем оставляют для охлаждения. Раствор упаривают в вакууме до остатка в виде масла. Масло выливают в избыток насыщенного раствора карбоната натрия и экстрагируют толуолом. Толуольный экстракт пропускают через слой силикагеля и упаривают при пониженном давлении при температуре 90 - 95oC. Получают продукт в виде масла; т. кип. 150 - 155oC при 0. 01 мм рт. ст. Чистота по данным ВЭЖХ составляет 96.9%, примесью в продукте является толуол.
200 МГц 1H ЯМР (дейтерохлороформ): δ 0.87 - 1.1 (т, 3H), 1.1 - 1.8 (сложный мультиплет, 4H), 2.5 - 3.2 (сложный мультиплет, 4H), 2.5 - 3.2 (сложный мультиплет, 5H), 4.0 - 4.15 (т, 2H), 6.3 - 6.5 (с, 1H), 7.2 - 7.7 (сложный мультиплет, 5H).
Описывается улучшенный пятистадийный способ получения R-(+)-1,2,3,6-тетрагидро-4-фенил-1-[(3-фенил-3-циклогексен-1-ил) метил] пиридина из 5-оксо-3-фенил-3-циклогексенкарбоновой кислоты. Кроме того, описываются способы разделения на стереоизомеры 5-оксо-3-фенил-3-циклогексенкарбоновой кислоты с использованием цинхонидина для получения (S)-5-оксо-3-фенил-3-циклогексенкарбоновой кислоты или α-химотрипсина для селективного гидролиза н-бутилового эфира 5-оксо-3-фенил-3-циклогексенкарбоновой кислоты с получением (S)-5-оксо-3-фенил-3-циклогексенкарбоновой кислоты. Описываются и другие ценные промежуточные соединения, используемые в предлагаемых способах получения. Способ использует недорогие исходные вещества и пригоден для крупномасштабного синтеза. 11 с. и 28 з.п.ф-лы.

и его фармацевтически приемлемых солей, включающий стадию (а) обработки рацемического соединения формулы VIII
цинхонидином в растворителе с целью получения соединения формулы VII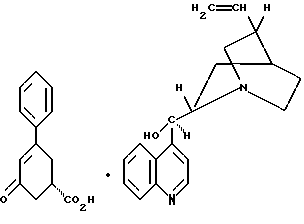
стадию (б) обработки соединения формулы VII основанием в растворителе с целью получения после подкисления соединения формулы VI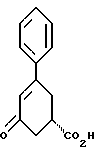
стадию (в) обработки соединения формулы VI соединением формулы V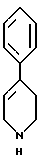
в присутствии реагента для сочетания и растворителя с целью получения соединения формулы IV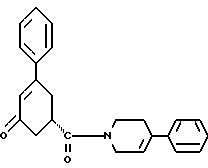
стадию (г) обработки соединения формулы IV восстанавливающим реагентом в растворителе с целью получения смеси соединений формулы IIIа и формулы IIIб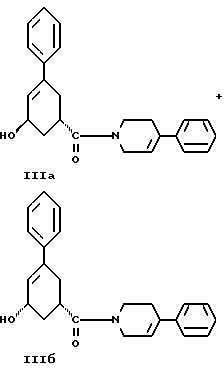
стадию (д) обработки смеси соединений формулы IIIа и формулы IIIб смесью хлористого цинка и цианборгидрида натрия в растворителе, а затем раствором карбоновой кислоты в растворителе с целью получения соединения формулы II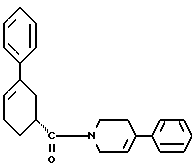
стадию (е) обработки соединения формулы II восстанавливающим агентом, представляющим гидрид металла, в растворителе с целью получения соединения формулы I; стадию (ж) и, если желательно, превращение полученного соединения формулы I в соответствующую фармацевтически приемлемую соль в результате присоединения кислоты, используя общепринятые способы, и, если так требуется, превращение соответствующей фармацевтически приемлемой соли, полученной при присоединении кислоты, в соединение формулы I при использовании общепринятых способов.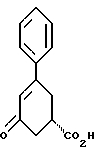
включающий стадию (а) обработки рацемического соединения формулы VIII
цинхонидином в растворителе с целью получения соединения формулы VII
и стадию (б) обработки соединения формулы VII основанием в растворителе с целью получения после подкисления соединения формулы VI.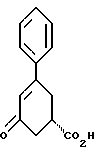
включающий обработку рацемического соединения формулы IX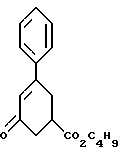
в растворителе при pH около 5 с помощью α-химотрипсина с целью получения после отделения непрореагировавшего сложного эфира и подкисления соединения формулы VI.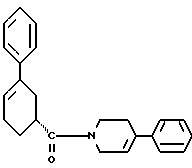
включающий стадию (а) обработки рацемического соединения формулы VIII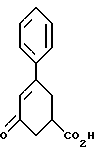
цинхонидином в растворителе с целью получения соединения формулы VII
стадию (б) обработки соединения формулы VII основанием в растворителе с целью получения после подкисления соединения формулы VI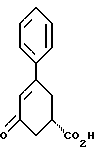
стадию (в) обработки соединения формулы VI соединением формулы V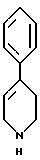
в присутствии реагента для сочетания и растворителя с целью получения соединения формулы IV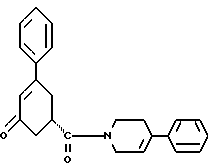
стадию (г) обработки соединения формулы IV восстанавливающим реагентом в растворителе с целью получения смеси соединений формулы IIIа и формулы IIIб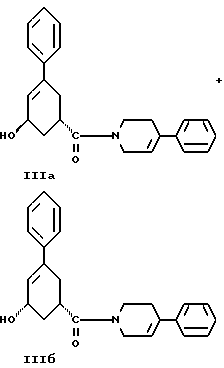
и стадию (д) обработки смеси соединений формулы IIIа и формулы IIIб смесью хлористого цинка и цианборгидрида натрия в растворителе, а затем раствором карбоновой кислоты в растворителе с целью получения соединения формулы II.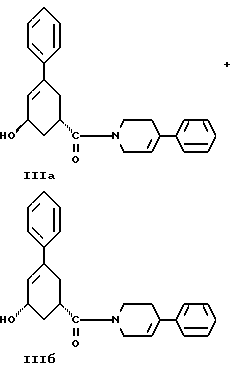
включающий стадию (а) обработки рацемического соединения формулы VIII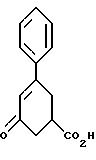
цинхонидином в растворителе с целью получения соединения формулы VII
стадию (б) обработки соединения формулы VII основанием в растворителе с целью получения после подкисления соединения формулы VI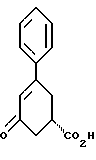
стадию (в) обработки соединения формулы VI соединением формулы V
в присутствии реагента для сочетания и растворителя с целью получения соединения формулы IV
и стадию (г) обработки соединения формулы IV восстанавливающим реагентом в растворителе с целью получения смеси соединений формулы IIIа и формулы IIIб.
включающий стадию (а) обработки рацемического соединения формулы VIII
цинхонидином в растворителе с целью получения соединения формулы VII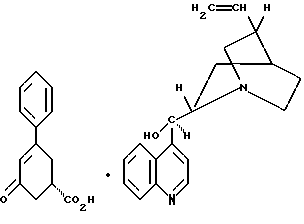
стадию (б) обработки соединения формулы VII основанием в растворителе с целью получения после подкисления соединения формулы VI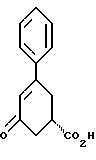
и стадию (в) обработки соединения формулы VI соединением формулы V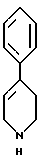
в присутствии реагента для сочетания и растворителя с целью получения соединения формулы IV.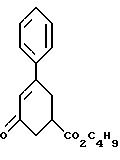
36. Соединение, выбранное из группы, состоящей из соединений формулы IIIа и формулы IIIб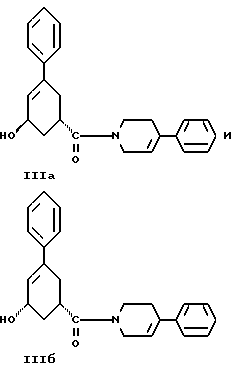
37. Соединение формулы IV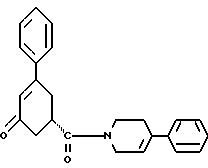
38. Соединение формулы VI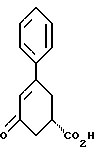
39. Соединение формулы VII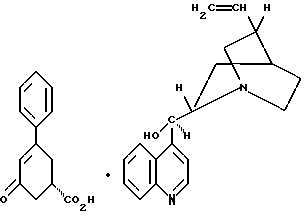
| US 5314896 A, 24.05.94 | |||
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Зажимной патрон бурового станка | 1984 |
|
SU1189982A1 |
| 1-/4-(4-Фторфенил)-4-оксибутил/ -4- (индолил-31)-1,2,3,6-тетрагидропиридин или его соли, обладающие психотропными свойствами | 1976 |
|
SU639243A1 |

