Изобретение относится к новым производным N-ацил-2,3-бензодиазепина общей формулы [I]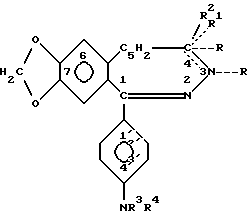
в которой R это C1-6 алифатическая ацильная группа, возможно замещенная метокси-, циано, карбоксильной, амино-, C1-4-алкиламино, ди(C1-4 алкил)амино-, пирролидино-, фталимидо- или фенильной группой, или одним или более галогеном(ами), или R это бензоил, циклопропанкарбонил, C1-5-карбамоил или фенилкарбамоильная группа, или R отсутствует, когда между N(3) и C(4) атомами существует двойная связь, R1 представляет атом водорода или R1 отсутствует, когда между N(3) и C(4) атомами существует двойная связь, R2 представляет C1-3 алкил, или R1 и R2 вместе представляют метиленовую группу, а между N(3) и C(4) атомами нет двойной связи, R3 означает атом водорода или C1-4-алифатическую ацильную группу, R4 представляет атом водорода, C1-6 алифатическую ацильную группу, возможно замещенную метокси-, циано-, карбоксильной, амино-, C1-4-алкил-амино-, ди (C1-4 алкил) амино-, пирролидино-, фталимидо или фенильной группой, или одним или более галогеном (ами), а также бензоил, пальмимитоил, циклопропанкарбонил, C1-5 алкилкарбамоил или фенилкарбамоильную группу, а пунктирные линии представляют возможно присутствующие валентные связи, при условии, что, когда оба заместителя R3 и R4 представляют атомы водорода, между N(3) и C(4) атомами нет двойной связи, а также стереоизомеры этих соединений наряду с кислыми солями присоединения (в тех случаях, когда это возможно) и фармацевтические композиции, содержащие эти соединения.
Соединения формулы (I), соответствующие настоящему изобретению, имеют ассиметрическую молекулярную структуру. Общая формула (I) относится ко всем возможным индивидуальным изомерам и их смесям.
Также в настоящем изобретении предлагается способ получения новых соединений общей формулы (I) и кислых солей присоединения на их основе.
Цель настоящего изобретения состоит в том, чтобы разработать новые соединения общей формулы (I), которые обладают значительной активностью, связанной с воздействием на центральную нервную систему (ЦНС), в частности, обладают способностью вызывать релаксацию мышц и/или противоконвульсивным действием. Единственное соединение, проявляющее такое действие, известно только среди 2,3- бензодиазепинов, а именно, это -1 -(4-аминофенил)-4-метил-7,8-метилендиокси -5Н-2,3-бензодиазепин (патент США N 4164740), также получено авторами настоящего изобретения. Однако в результате детального фармакологического скрининга было установлено, что вышеназванное соединение положительное в Ames-тесте, т.е. оно обладает мутагенным действием. Итак, конкретная цель настоящего изобретения состоит в том, чтобы новое производное бензодиазепина, которое бы сохраняло свои ценные свойства, связанные со способностью снимать мышечное напряжение и обладало противоконвульсивной активностью, но не обладало мутагенным действием.
Новые соединения общей формулы (I), в которой R, R1, R2, R3, R4 и пунктирные линии имеют значения, указанные выше, и их фармацевтически приемлемые кислые соли присоединения полностью удовлетворяют этому требованию.
Согласно настоящему изобретению соединения общей формулы (I) получают путем:
а) ацилирования соединения формулы (II)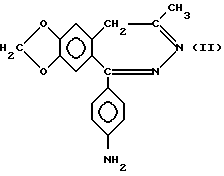
C1-6 алифатической карбоновой кислотой, возможно замещенной метокси -, циано, карбоксильной, или фенильной группой или одним или более галогеном (ами), или бензойной кислотой, циклопропанокарбоновой кислотой или пальмитиновой кислотой или производными этих кислот, и, если в этом есть необходимость, реакции нового, полученного таким образом соединения общей формулы (I), в которой R4 означает C1-6 алифатическую ацильную группу, замещенную галогеном, с C1-4-алкиламином, ди-(C1-4 алкил) амином или пирролидином с образованием соединений общей формулы (I), в которой R2, R3 и пунктирные линии имеют смысл, определенный выше, R4 представляет C1-6-алифатическую ацильную группу, возможно замещенную метокси-, циано-, карбоксильной, фенильной, C1-4- алкиламино-, ди(C1-4 алкил)амино- или пирролидиновой группой, или одним или более галогеном(ами), или представляет бензоил, циклопропанокарбонил или пальмитоил, R и R1 отсутствуют, а между N(3) и C(4) атомами наличествует двойная связь,
b) ацилирования соединения общей формулы (III)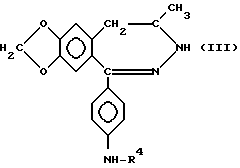
в которой R4 определен выше, C1-6 алифатической карбоновой кислотой, возможно замещенной метокси-, циано-, карбоксильной или фенильной группой или одним или более атомами галогенов, или бензойной или циклопропанкарбоновой кислотой или реакционноспособными производными на их основе, и, если в этом есть необходимость, реакция нового соединения общей формулы (I), полученной таким образом, в которой R4 представляет C1-6 алифатическую ацильную группу, замещенную галогеном, с C1-4 алкиламином, ди (C1-4 алкил) амином или пирролидином, приводящая к получению соединений общей формулы (I), в которой R1, R2, R3, R4 и пунктирные линии имеют смысл, определенный выше, R - C1-6 алифатическая ацильная группа, возможно замещенная метокси-, циано-, карбоксильной, фенильной, C1-4 алкиламино-, ди(C1-4 алкил) амино- или пирролидиновой группой или одним или более атомами галогенов, или бензоил или циклопропанкарбонил, и между N(3) и C(4) атомами отсутствует двойная связь, или
с)ацилирования соединения формулы (II) N-фталоиламинокислотой общей формулой (VI)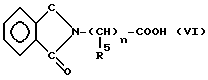
в которой R5 представляет атом водорода или C1-4- алкильную группу и n равен 1 в случае α -аминокислот, тогда же, когда R5 означает атом водорода, то n варьируется в пределах от 2 до 5, что имеет место в случае аминокислот, и если в этом есть необходимость, удаляют фталоильную группировку и получают соединения общей формулы (I), в которой R2 и пунктирные линии имеют значения, указанные выше, R3 означает атом водорода, R4 это C1-6 алифатическая ацильная группа, замещенная амино- или фталимидной группой, оба R и R1 отсутствуют, а двойная связь существует между N(3) и C(4) атомами,
d) или ацилирования соединения общей формулы (III), в которой R4, определен выше, N фталоидаминокислотой общей формулой (VI) в которой R5 представляет атом водорода или C1-4 алкильную группу и n равен 1 в случае a -аминокислот, тогда же, когда R5 представляет атом водорода, то n варьируется в пределах от 2 до 5, что имеет место в случае b-ε -аминокислот, и если в этом есть необходимость, удаляют фталоильную группировку и получают соединения общей формулы (I), в которой R1, R2 и пунктирные линии имеют значения, определенные выше, R3 означает атом водорода, R4 имеет значения, определенные выше, за исключением водорода, R представляет C1-6 алифатическую ацильную группу, замещенную амино-, или фталимидной группой, а двойная связь между N(3) и С(4) атомами отсутствует, или
e) реакции соединения формулы (II) с C1-5 алкилизоцианатом или фенилизоцианатом, приводящей к получению соединений общей формулы (I), в которой R2 и пунктирные линии имеют значения, обозначенные выше, R3 означает атом водорода, R4 представляет C1-5 - алкилкарбамоил или фенилкарбамоил, R и R1 отсутствуют, а между N(3) и C(4) атомами существует двойная связь, или
f) при взаимодействии соединения общей формулы (III), в которой R4 определен выше, с C1-5 алкил изоцианатом или фенил-изоцианатом, получают соединения общей формулы (I), в которой R1, R2 и пунктирные линии имеют значения, определенные выше, R3 означает водород, R4 имеет значения, определенные выше, за исключением водорода, R представляет C1-5 алкилкарбамоил или фенилкарбамоильную группу, двойная связь между N(3) и C(4) атомами отсутствует, или
g) селективным восстановлением нитросоединения формулы (IV)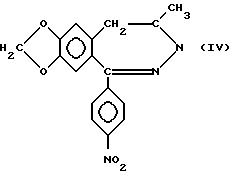
в новое соединение общей формулы (V)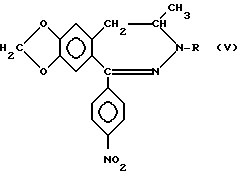
в которой R представляет водород, с последующим либо ацилированием полученного таким образом соединения формулы (V), с использованием описанных выше способов b), d) или f) и восстановлением нитрогруппы полученного таким образом нового соединения общей формулы (V), в которой R определен выше, в аминогруппу, либо восстановлением сначала нитрогруппы и затем ацилирования, соединения общей формулы (III), полученного таким образом, в котором R представляет атом водорода, с использованием одного из вышеперечисленных процессов b), d), f) или h) с получением соединений общей формулы (I), в которой R1, R3 и R4 представляют атом водорода, R2, R и пунктирные линии имеют смысл, определенный выше и между N(3) и C(4) атомами двойная связь отсутствует, или
i) ацилирование нового соединения общей формулы (I), в которой R, R1, R2 и пунктирные линии имеют смысл, определенный выше, R3 и R4 представляют атомы водорода и нет двойной связи между N(3) и C(4) атомами, C1-6 алифатической карбоновой кислотой, возможно замещенной метокси-, циано- или карбоксильной группой или одним или более атомами галогенов, или бензойной кислотой, или реакционноспособным производным на ее основе, с образованием соединений общей формулы (I), в которой R1, R2, R3 и пунктирные линии имеют смысл, определенный выше, R и R4 представляют C1-6 алифатическую ацильную группу, возможно замещенную метокси-, циано- или карбоксильной группой, или одним или более атомами галогенов или бензоильную группу, а двойная связь между N(3) и C(4) атомами отсутствует, или взаимодействие нового соединения общей формулы (I), в которой R, R1, R2 и пунктирные линии имеют смысл, определенный выше, R3 и R4 представляют атом водорода, а двойная связь между N(3) и C(4) атомами отсутствует, с C1-5 алкилизоцианатом или фенилизоцианатом с образованием соединений общей формулы (I), в которой R1, R2 и пунктирные линии имеют смысл, определенный выше, R представляет C1-6 алифатическую ацильную группу, возможно замещенную метокси-, циано- или карбоксильной группой, или одним или более атомами галогенов, или бензоил, R3 представляет атом водорода, R4 представляет C1-5 алкилкарбамоил или фенилкарбамоил, и двойная связь между N(3) и C(4) отсутствует, или
j) ацилирование нового соединения общей формулы (I) в которой R1, R2 и пунктирные линии имеют смысл, обозначенный выше, R3 и R4 представляют атомы водорода, и двойная связь между N(3) и C(4) атомами отсутствует с N -фталоиламинокислотой общей формулы (VI), в которой R5 представляет атом водорода или C1-4 алкильную группу и n равно 1, в том случае, когда это α аминокислоты, в тех случаях, когда используются b-ε аминокислоты, то R5 означает атом водорода и множитель n варьируется от 2 до 5, и, если в этом есть необходимость, удаляют фталоильную группу и получают соединения общей формулы (I), в которой R1, R2 и пунктирные линии имеют смысл, определенный выше, R представляет C1-6 алифатическую ацильную группу, возможно замещенную метокси-, циано- или карбоксильной группой или одним или более атомами галогенов, или бензоил, R3 представляет атом водорода, R4 представляет C1-6 алифатическую ацильную группу, замещенную амино или фталимидной группой, и двойная связь между N(3) и C(4) атомами отсутствует, и, если в этом есть необходимость, переводят основание общей формулы (I), полученное по любому из вышеперечисленных процессов с a) по j), в кислую соль присоединения.
В соответствии с предпочтительным вариантом осуществления способа настоящего изобретения ацилирование соединений общей формулы (I), (II), (III) и (V) может быть проведено предпочтительно с использованием подходящей карбоновой кислоты в присутствии дициклогексилкарбодиимида, в температурном интервале от 10 до 30oC в течение 1 до 25 ч.
В соответствии с другим предпочтительным вариантом настоящего изобретения соединения общей формулы (I), (II), (III) и (V) могут быть ацилированы в температурном интервале от 0 до 150oC с использованием подходящего реакционноспособного ацильного производного, например, ангидрида карбоновой кислоты, смешанного ангидрида или хлорангидрида, в отсутствии или в растворителе, обычно используемом для таких способов ацилирования, например, в хлороформе или дихлорэтане, в отсутствии или в присутствии средства, связывающего кислоту, такого, как триэтиламин. Если дополнительное ацилирование осуществляется с использованием изоцианатов, то реакцию преимущественно проводят в диметилформамиде, бензоле или дихлорметане в температурном интервале от 15 до 100oC в промежуток времени от 0,5 до 100 ч.
Селективнее восстановление соединения общей формулы (IV) в соединение общей формулы (V), в которой R представляет атом водорода может быть осуществлено с использованием неорганического или неорганического -органического комплексного соединения гидрида металла, предпочтительно с использованием борогидрида натрия, в растворителе или смеси растворителей, который имеет только низкую реакционную способность по отношению к комплексному соединению металлогидрида, используемому в реакции, или таковая способность отсутствует. В этих реакциях растворителем выбора является C1-4 спирт или пиридин. (Подобное селективное восстановление описано в патентах США N 4423044 или 4835152).
Для восстановления нитрогруппы новых соединений общей формулы (V) в амино-группу используют гидразин или гидразин-гидрат, восстановление проводят в присутствии катализатора, такого как палладий, платина или никель Ренея в C1-4 спирте, диоксане, тетрагидрофуране, бензоле, диметилформамиде, диметилацетамиде или смесях этих растворителей.
Согласно предпочтительному варианту осуществления настоящего изобретения восстановление может быть проведено в метаноле гидразином или гидразингидратом в присутствии катализатора-никеля Ренея в температурном интервале от 10 до 65oC (патент США N 4614740), но, если в этом есть необходимость, восстановление и удаление защитной фталоильной группы, описанное в способе d) может быть проведено в одном и том же реакционном сосуде.
N-фталоидаминокислоты общей формулы (IV), содержащие хиральный атом углерода, где R5 означает C1-4 алкильную группу и множитель n равен 1, могут быть получены из DL-L- и/или D-альфа -аминокислот.
Соединения формулы (I) настоящего изобретения, которые содержат основную аминогруппу, в которой R3 и R4 представляют атом водорода или R и/или R4 представляет аминоацильную группу, могут быть превращены в кислые соли присоединения в соответствии с известными методами.
Получение соединений общей формулы (II), используемых в качестве исходных материалов в процессе настоящего изобретения, описано в патенте США N 4614740, соединений общей формулы (III), в которой R4, представляет атом водорода в патенте США N 4835152, а соединений общей формулы (IV) - опубликовано во Французской патентной заявке N 8509793. Соединения общей формулы (III), в которой R4, представляет различные ацильные группы, являются новыми соединениями. Способ их получения описан здесь далее до табл. 10, или они могут быть синтезированы описанными здесь методами. Получение новых исходных материалов общей формулы (V) описано в примерах. Производные ( a-ε )-аминокислот общей формулы (VI) получены в соответствии со способами, известными из литературных данных [J. Am. Chem. Soc. 35, 1133 (1913), 41, 845 (1919), Berichte der Deutschen Chemischen Gesellschaft, 40, 498, 2649 (1907), 46,1103, 3159 (1913), 47, 3166 (1914)] или согласно известным методам, использующим реакцию фталимида калия с нужной галоидкарбоновой кислотой.
Соединения общей формулы (I), полученные по способу настоящего изобретения, обладают активностью в отношении центральной нервной системы (ЦНС), включая антиконвульсивное действие, способствуют мышечной релаксации и оказывают нейропротекторное действие, что может быть подтверждено фармакологическими испытаниями.
В сравнительном исследовании 1-(4 -аминофенил)-4-метил-7,8-метилендиокси-5Н -2,3-бензодиазепин (патент США N 4614740 далее именуемое "соединение прототипа"), имеющий подобную структуру и эффективность действия, как и соединения настоящего изобретения, использовался как соединение прототипа.
Как уже упоминалось во введении, это соединение обладает ценными фармакологическими свойствами, но как доказано, является Ames-положительным. В отличие от него соединения настоящего изобретения являются Ames -отрицательными.
Фармакологическое действие этих соединений общей формулы (I) представлено в табл. 1-8.
Наркоз-потенциирующее действие на мышах
Наркоз-потенциирующее действие было исследовано с использованием трех пероральных доз на 10 мышах. Значение ЕД50 соответствует дозе, пролонгирующей длительность общего наркоза, вызванного введением внутривенно гексобарбитала в количестве 50 мг/кг, в два раза у 50% животных в сравнении с контрольной группой животных, которым давали только разбавитель. Значения ЕД50 рассчитывались по методу Litchfield -Wilcoxon [J.Pharmacol Exp. Ther. 96, 99 (1949)] Результаты представлены в табл. 1.
Из данных, приведенных в табл.1 следует, что эффективность некоторых соединений совпадает или значительно превосходит эффективность соединения прототипа. Соединения примеров 15(16), 45, 60, 73, 98 относятся к соединениям, отличающимся особенно высокой эффективностью.
Противоконвульсивное действие на мышах
Противоконвульсивное действие соединений измеряли, используя испытание электрошоком [Swinyard: J. Pharmacol. Exp. Ther. 106, 319 (1952)] кроме того, были использованы различные химические средства, такие как пентатетразол [Goodman: J. Pharmacol. Exp. Ther, 108, 168 (1953)] стрихнин [Roskovski: J. Pharmacol. Exp. Ther. 129, 75(1960)] бемегрид, никотин и 4 аминопиридин. Тестируемые соединения перорально тремя дозами вводили самцам мышей линии CFLP, каждая доза давалась 10 мышам.
Результаты представлены в табл. 2.
Вышеприведенные данные свидетельствуют о том, что противоконвульсивное действие некоторых соединений (примеры 15, 42, 45, 46, 73, 98, 107, 108, 109 и 115) превосходит аналогичное действие соединения прототипа.
Миорелаксантная активность на мышах
Определение миорелаксантной активности было проведено в двух испытаниях. В скрининговом испытании по Randall's [J. Pharmacol. Exp. Ther. 129, 163, (1960)] 10 мышам линии CFLP на дозу, внутрибрюшинно вводили 3 дозы соединения. Результаты приведены в табл. 3.
Для определения мышечного тонуса и координации движения был использован rotarod тест. [Dunham and Mija J. Am. Pharm. Assoc. 46, 208, (1957)] Результаты, полученные с тремя выбранными соединениями высшей активности и с соединением прототипа, представлены в табл. 4.
Данные, приведенные в табл. 3 и 4 свидетельствуют о том, что некоторые соединения обладают сильной миорелаксантной активностью (соединения примеров 15, 18, 42, 45, 48, 49, 62, 73, 98 и 115).
Влияние на спинальную функцию
Было определено влияние самых активных соединений (соединение примера 15 или 16) и соединения прототипа на спинальную функцию. В табл. 5 приведены результаты, свидетельствующие о влиянии на полисинаптические сгибательные рефлексы у кошек [Farkas and Karpati. Pharm. Res. Comm. 20, S1, 141 (1988)]
Определение влияния вышеуказанных соединений на напряжения в спинальном корне у кошек было проведено на спинально иммобилизованных животных. [Farkas et al. Neuropharmacology 21. 161, (1989)] Результаты представлены в табл. 6.
Значения ЕД50, ингибирующие моносинаптический рефлекс
Соединение прототипа 2,20(1,02 -4,75)мг/кг, внутривенно.
Соединение N 15(16) 2,30(1,06 -5,01)мг/кг, внутривенно.
Значения ЕД50, ингибирующие полисинаптический рефлекс
Соединение прототипа: 0,60 ( 0,32 -1,13) мг/кг, внутривенно
Соединение N 15(16) 0,73(0,39 -1,37) мг/кг, внутривенно.
Электрофизиологические испытания
Данные по ингибирующему действию на участки напряжений, вызванные электростимуляцией in vitro срезов неокортекса у выживших крыс, суммированы в табл. 7 [Fletcher и др. Br. J. Pharmacology 95, 585 (1988)]
He-NMDA (quisqualate) антагонистическое действие определили на срезах неокортекса крыс, используя метод Harrison и Simnionds [Br. J. Pharmacol. 84, 381 (1981)] Изменения напряжения постоянного тока на срезах неокортекса крыс, вызванные перфузией quisqualate ингибировались соединением прототипа в концентрационном интервале от 10 до 50 мкМ. При определенной концентрации соединение примера 15(16) в два раза активнее соединения прототипа при ингибировании ответа на двухминутную перфузию 10 mM quisqualate. Однако обе молекулы (оба соединения) оказываются не в состоянии воздействовать на реакцию, индуцируемую NMDA. Следовательно, соединение примера 15(16) можно рассматривать как не NMDA, но quisqualate типа селективный возбуждающий аминокислотный антагонист.
Острая токсичность на крысах
Данные по острой токсичности на крысах суммированы в табл. 8.
*При токсических уровнях дозы соединения вызывали дозозависимое снижение мышечного тонуса, атаксию, адинамию и потерю установочного рефлекса. Причиной смерти служила респираторная недостаточность, развивающаяся в течение 1-2 ч после в.б. введения и в течение 10-20 ч после перорального приема.
Основываясь на вышеприведенных результатах фармакологических исследований, можно сделать вывод о том, что соединения, соответствующие настоящему изобретению формулы (I), обладают значительным противоконвульсивным действием, миорелаксантной активностью и являются возбуждающими аминокислотными антагонистами (нейропротекторами). Таким образом, они полезны в качестве терапевтических средств при лечении эпилепсии, а также различных заболеваний, связанных со спазмами скелетной мускулатуры и церебральной ишемии ("удар").
Это изобретение также относится к фармацевтическим композициям, содержащим соединения общей формулы (I) или фармацевтически приемлемым кислым солям присоединения на их основе в качестве активных ингредиентов, а также к способу получения этих композиций.
Для терапевтического использования, активные соединения согласно настоящему изобретению подходящим образом вводят в фармацевтические составы путем смешения с традиционно используемыми нетоксичными, инертными, твердыми или жидкими фармацевтическими носителями и/или дополнительными материалами, полезными для энтерального или парентерального введения. В качестве носителей можно использовать, например, воду, желатин, лактозу, крахмал, пектин, стеарат магния, стеариновую кислоту, тальк или растительные масла. В качестве добавок, например, могут быть использованы: консерванты и увлажнители, такие как эмульгаторы, диспергаторы и ароматизирующие средства, а также буферные растворы.
При использовании вышеупомянутых носителей и добавок активные ингредиенты изобретения могут быть внесены в обычные фармацевтические композиции, например, в твердые композиции (выпускаемые в виде таблеток, капсул, пилюль или суппозиториев) или жидкие композиции, такие как водные или масляные растворы, суспензии, эмульсии или сиропы), а также в виде растворов для инъекций, суспензий или эмульсий.
Для терапевтических целей ежедневная доза соединений настоящего изобретения обычно составляет от 0,2 до 1,5 мг/кг массы тела, эта доза, возможно разбивается на несколько приемов.
Основываясь на выше приведенных фактах, настоящее изобретение также обеспечивает:
способ блокирования одного или более возбуждающих аминокислотных рецепторов у млекопитающих. Этот способ включает введение млекопитающим, нуждающимся в такого рода лечении, фармацевтически эффективного количества соединений общей формулы (I),
способ лечения эпилепсии у животных. Этот способ включает введение млекопитающим, нуждающимся в такого рода лечении, противоэлептического количества соединения настоящего и соединения общей формулы (I),
способ снятия спазм (судорог) скелетной мускулатуры у млекопитающих. Этот способ включает введение млекопитающим, нуждающимся в такого рода лечении, миорелаксантного количества соединения общей формулы (I);
способ лечения церебральной ишемии ("удара") у млекопитающих. Этот способ включает, нуждающимся в такого рода лечении, введение фармацевтически эффективного количества соединения общей формулы (I).
Соединения, полученные по способу данного изобретения, идентифицированы методом элементного анализа, чистота этих соединений и их структура контролировалась и были подтверждены методами тонкослойной хроматографии, ИКС, 1H-ЯМР, 13C-ЯМР и масс -спектрометрии.
Это изобретение детально проиллюстрировано следующими далее примерами, которые не носят ограничивающий характер.
Пример 1
1-(4-диацетиламино(-3-ацетил-4 -метилен-7,8-метилендиокси-4,5-дигидро-ЗН -2,3-бензодиазепин.
2,93 г (0,01 моля) 1-(4 -аминопропенил)-4-метил-7,8-метилендиокси -5Н-2,3-бензодиазепина кипятили с обратным холодильником с 20 мл уксусного ангидрида в течение 6 ч. Этот раствор упаривали при пониженном давлении, остаток собрали двумя порциями по 20 мл безводного этанола, полученный раствор вновь упарили, и полученный остаток массой 4,55 г пропустили через хроматографическую колонку (адсорбент кизельгур 60, элюент смесь этилацетата и бензола 4:1). Сырой продукт растерли с 20 мл изопропанола, в результате получили 1,44 г (34,4% ) нужного продукта, т.пл. 240-245oC( слабое разложение), C23H21N3O5=419,445.
Пример 2
1-(4-формиламинофенил)-4-метил-7,8 -метилендиокси-5Н-2,3-бензодиазепин
3,0 г (10, 2 ммоля) 1-(4-аминофенил -4-метил-7,8-метилендиокси-5Н-2,3 -бензодиазепина растворили в 160 мл дихлорметана и сначала к раствору добавили 2,75 г (13,3 ммоля) дициклогексилкарбодиммида, затем 0,51 мл (13,3 ммоля) 100% муравьиной кислоты, и полученную реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Выпавший осадок N,N' -дициклогексилмочевины отфильтровали, фильтрат проэкстрагировали двумя порциями по 30 мл 10% водного раствора карбоната натрия, затем двумя порциями по 30 мл дистиллированной воды, органический слой высушили и упарили при пониженном давлении. Остаток растворили в этилацетате, отфильтровали и упарили при пониженном давлении. Полученный сырой продукт перекристаллизовали при пониженном давлении из 20 мл 50% этанола, в результате получили 2,93 г (83,3%) нужного продукта, т.пл. 152 -154oC (незначительное разложение), C8H15N3O3=321,342.
Примеры с 3 до 7
Соединения примеров с 3 по 7 были получены по способу, описанному в примере 2.
Пример 3
1-(4-цианоацетиламинофенил)-4-метил -7,8-метилендиокси-5Н-2,3-бензодиазепин. C20H16N4O3=360,380, т.пл. 241-243oC (разл.).
Пример 4
1-(4-метоксиацетиламинофенил)-4 -метил-7,8-метилендиокси-5Н-2,3-диазепин. C20H19N3O4=365,396, т.пл. 203-205oC.
Пример 5
1-(4-валериламинофенил)-4-метил-7,8 -метилендиокси-5Н-2,3-бензодиазепин. C22H23N3O4=377,450, т.пл. 217-219oC (разл.).
Пример 6
1-(4-фенилацетиламинофенил)-4-метил -7,8-метилендиокси-5Н-2,3-бензодиазепин. C25H21N3O3=411,467, т.пл.245-247oC (разл.).
Пример 7
1(4-циклопропанкарбониламинофенил) -4-метил-7,8-метилендиокси-5Н-2,3 -бензодиазепин. C21H19N3O3=361,407, т.пл. 260-262oC (разл.).
Пример 8
1-(ацетиламинофенил)-4-метил-7,8 -метилендиокси-5Н-2,3-бензодиазепин.
10 г (34 ммоля) 1-(4-аминофенил)-4 -метил-7,8-метилендиокси-5Н-2,3-бензодиазепина перемешивали в течение 3 ч со 100 мл уксусного ангидрида. Образовавшиеся кристаллы отфильтровывали, промыли 5 х 10 мл безводного этилового спирта и высушили, в результате получили 9,2 г сырого продукта, т.пл. 252-254oC (разл. ). Этот продукт обработали 45 мл горячего 99,5% этанола. После охлаждения кристаллы отфильтровывали, промыли 3 х 10 мл этанола и высушили, в результате получили 8,68 г, 76,1% нужного продукта, т.пл. 256- 258oC (разл.), C19H17N3O3=335,369.
Пример 9
1-(4-пропиониламинофенил)-4-метил -7,8-метилендиокси-5Н-2,3-бензодиазепин
Это соединение было получено согласно процедуре, описанной в примере 8. C20H19N3O3 349,396, т.пл. 228-230oC (разл.).
Пример 10
1-(4-пивалоиламинофенил)-4-метил -7,8-метилендионси-5Н-2,3-бензодиазепин.
1,56 мл (11,2 ммоля) триэтиламина и 1,38 мл (11,2 ммоля) пивалоилхлорида добавили к раствору 3 г (10,2 ммоля) 1 -(4-аминофенил)-4-метил-7,8-метилендиокси -5Н-2,3-бензодиазепина в 160 мл дихлорметана и реакционную смесь перемешивали при 25oC в течение 1 ч. Образовавшийся осадок отфильтровывали, промыли 3 х 5 мл дихлорметана, затем 3 х 20 мл дистиллированной воды и высушили, в результате получили 1,59 г чистого продукта, т.пл. 225-227oC (разл.). Другую порцию продукта выделили из органической фазы. Фильтрат проэкстрагировали 3 х 20 мл дистиллированной воды, затем 3 х 15 мл 4%-ным водным раствором гидроксида натрия, и в заключение 2 х 30 мл дистиллированной воды. Затем органический слой высушили и упарили при пониженном давлении. Кристаллический остаток объединили с полученными ранее 169 г продукта и суспендировали в 20 мл горячего этанола. Продукт отфильтровывали после охлаждения, промыли 3 х 3 мл этанола и высушили, в результате получили 3,38 г (87,8%) чистого продукта т.пл. 225-227oC (разл.), C22H23N3O3 377,450.
Пример 11
1-(4-бензоиламинофенил)-4-метил-7,8 -метилендиокси-5Н-2,3-бензодиазепин
1,0 мл (15 ммоля) бензоилхлорида и 2,1 мл (15 ммоля) триэтиламина добавили к раствору 4 г (13,6 ммоля)1-(4 -аминофенил)-4-метил-7,8-метилендиокси -2,3-диазепина в дихлорметане и реакционную смесь перемешивали при 25oC в течение 24 ч. Раствор проэкстрагировали 3 х 30 мл дистиллированной воды, 3 х 20 мл 4% водного раствора гидроксида натрия и, в заключение, 2 х 30 мл дистиллированной воды. Органический слой высушили, упарили при пониженном давлении, затем кристаллический осадок обработали 20 мл горячего этанола, на следующий день его отфильтровали при 0-5oC, промыли 3 х 3 мл этанола и высушили при 100oC, в результате получили 3,85 г (71,3%) чистого нужного продукта, т.пл. 246 -247oC (разл.), C24H19N3O3 397,40.
Пример 12
1-(4-пальмитоиламинофенил)-4-метил -7,8-метилендиокси-5Н-2,3- -бензодиазепин
Следуя процедуре, описанной в примере 11, с перекристаллизацией сырого продукта из 50% этанола, получили чистый нужный продукт, т.пл. 138-140oC, C33H45N3O3 531,747.
Пример 13
1-(4-фенилкарбамоиламинофенил)-4 -метил-7,8-метилендиокси-5Н-2,3 - бензодиазепин.
0,22 мл (2,04 ммоля) фенилизоцианата добавили к раствору 0,50 г (1,7 ммоля) 1-(4-аминофенил)-4-метил -7,8-метилендиокси-5Н-2,3-бензодиазепина в 4 мл диметилформамида и реакционную перемешивали при 25oC в течение часа. Затем ее разбавили 20 мл диэтилового эфира и отфильтровали при 5oC. Кристаллы промыли 2 х 25 мл диэтилового эфира и высушили при 60 100oC. 0,70 г сырого продукта с т.пл. 239-240oC (спекается при 180oC) кипятили с обратным холодильником в 15 мл этанола, потом отфильтровывали после охлаждения, промыли 3 х 1 мл этанола и высушили при 100oC, в результате получили 0,55 г нужного продукта (78,6%) т.пл. 240-241oC (разл.), C24H20N4O3X 412,456.
Пример 14
1-[4-(4- карбоксибутириламино)фенил] -4-метил-7,8 -метилендиокси-5Н-2,3-бензодиазепин.
Раствор 0,50 г (1,7 ммоля) 1-(4 -аминофенил)-4-метил- 7,8-метилендиокси -5Н-2,3-бензодиазепина в 30 мл безводного дихлорметана перемешивали с 0,18 г (1, 87 ммоля) ангидрида глутаровой кислоты при 20-25oC в течение 6 ч. На следующий день образовавшиеся кристаллы отфильтровывали при 0-5oC, промыли 3 х 2 мл дихлорметана и высушили при 60-80oC, в результате получили 0,60 г (87% ) чистого нужного продукта, т. пл. 225 -227oC (разл.), C22H21N3O5S 407,434.
Пример 15
1(4-аминофенил)-3-ацетил-4-метил -7,8-метилендиокси-5Н-2,3-бензодиазепин.
К раствору 3,58 г (12,1 ммоля) 1-(4 -аминофенил)-4-метил-7,8-метилендиокси-5Н -2,3-бензодиазепина в 100 мл хлороформа в начале добавили к 1,68 мл (12,1 ммоля) триэтиламина, затем при постоянном охлаждении льдом и перемешивании добавили 1,15 мл (12,1 ммоля) уксусного ангидрида. Перемешивание продолжали в течение 2 ч. Затем раствор проэкстрагировали 3 х 100 мл дистиллированной воды, органический слой высушили и упаривали при пониженном давлении. Кристаллический остаток перекристаллизовали из 40 мл изопропанола, в результате получили 3,50 г (85,7%) нужного продукта, т.пл. 220 -222oC. После повторной перекристаллизации, т.пл. достигла 223 -225oC. C19H19N3O3 373,385, гидрохлорид (C19H20N3O3)C 373,850, т.пл. 248-252oC (разл).
Пример 16
1-(4-аминофенил)-3-ацетил-4-метил -7,8-метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепин.
К суспензии 1,91 г (5,37 ммоля) 1 -(4-нитрофенил)-3-ацетил-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепина (продукт примера 27) в 40 мл метанола добавили около 0,2 г катализатора никель Ренея и 1,4 мл (28 ммоля) 100% гидразингидрата, после чего реакционную смесь перемешивали в течение 1 ч при 20-25oC. Исходное нитропроизводное растворили в течение 10 -20 мин. После фильтрования фильтрат упарили при пониженном давлении, белый кристаллический остаток промыли 30 мл дистиллированной воды на фильтре, затем промыли 3 х 10 мл дистиллированной воды и высушили на 100oC, в результате получили 1,50 г неочищенного продукта с т.пл. 218- 220oC. Этот неочищенный продукт растворили в 12 мл горячего изопропанола. После охлаждения смесь отфильтровали при 5oC, промыли 3 х 1 мл изопропанола и высушили при 100oC, в результате получили 1,40 г (77, 35%) белого кристаллического порошка, т.пл. 221-223oC. На основе проведенных анализов и данных спектров было установлено, что полученный продукт идентичен продукту примера 15, полученному другим способом.
Пример 17-25
Согласно способу, описанному в примере 16, были получены другие 1-(4 -аминофенил)-3-R-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепины общей формулы (I). Сведения об этих продуктах представлены в табл. 9.
Новые нитросоединения общей формулы (V), в которой R=H или ацильная группа, используемые при получении продуктов примеров с 16 по 25 могут быть получены согласно способам, описанным в примерах с 26 по 36.
Пример 26
1-(4-нитрофенил)-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазелин.
К суспензии 5,0 г (15,5 ммоля) известного 1-(4-нитрофенил)-4-метил-7,8 -метилендиокси-5Н-2,3-бензодиазепина (патент Франции N 8509793) в 380 мл этанола сначала добавили 22,5 мл (0,278 моль) концентрированной хлористоводородной кислоты при постоянном перемешивании при атом за несколько минут образовался раствор, затем в течение 30 мин частями добавляли 11,5 г (0,3 моль) борогидрида натрия. Перемешивание продолжили еще 15 мин, затем образовавшийся осадок оранжевого цвета отфильтровали и на фильтре проэкстрагировали 4 х 30 мл хлороформа. Объединенный фильтрат упарили при пониженном давлении, кристаллический остаток перенесли на фильтр с 200 мл дистиллированной воды, затем промыли 3 х 20 мл дистиллированной воды и высушили при 80-100oC, в результате получили 34,90 г (97,2%) нужного продукта, т.пл. 162-164oC, C17H15N3O4 325,331.
Пример 27
1-(4- нитрофенил)-3-ацетил-4-метил -7,8-метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепин.
2,0 г продукта примера 26 перемешали с 10 мл уксусного ангидрида при 25oC, перемешивание длилось 3 ч, затем добавили 50 мл дистиллированной воды и перемешивание продолжили в течение 1 ч. Образовавшийся желтый осадок отфильтровали, промыли 3 х 10 мл дистиллированной воды и высушили при 80 -100oC, в результате получили 2,6 г неочищенного (сырого) продукта. После перекристаллизации из 10 мл этанола получили 1,94 г (85,8%) нужного продукта, т.пл. 140-142oC, C19H17N3O5 367, 369.
Пример 28
1-(4-нитрофенил(-3-трифторацетил-4 -метил-7,8-метилен-диокси-3,4-дигидро-5Н -2,3-бензодиазепин.
К раствору 1,5 г (4,61 ммоля( часть продукта примера 26, в 30 мл безводного дихлорметана добавили 0,75 мл (5,3 ммоля) трифторуксусной кислоты и 0, 75 ммоля (триэтиламина, и реакционную смесь перемешивали при 25oC в течение 3 ч. Затем смесь проэкстрагировали 3 х 20 мл дистиллированной воды, и органический слой высушили и упарили при пониженном давлении. Кристаллический остаток обработали 15 мл горячего этанола, охладили, отфильтровали, промыли 3 х 1 мл этанола и высушили при 80-100oC, в результате получили 1,84 г (94,85%) нужного соединения в виде светло-желтого кристаллического продукта, т.пл. 165- 167oC (разл.), C19H14N3F3O5 421,339.
Пример 29
1-(4-нитрофенил)-3-пропионил-4 -метил-7,8-метилендиокси-3,4-дигидро-5Н -2,3-бензодиазепин.
Часть продукта примера 26 1,54 г (4,7 ммоля) перемешивали при 25oC в течение 3 ч с 8 мл ангидрида пропионовой кислоты, затем добавили 30 мл диэтилового эфира, и раствор оставили на ночь при 0-5oC. Образовавшийся осадок отфильтровали, промыли 3 х 8 мл диэтилового эфира и высушили, в результате получили 1,32 г (73,7%) нужного соединения в виде светло-желтого продукта, т.пл. 189-190oC, C20H19N3O5 381,396.
Пример 30
1-(4-нитрофенил)-3-валерил-4-метил -7,8-метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазелин.
К раствору 2, 5 г (7,68 ммоля) продукта примера 26 в 40 мл безводного дихлорметана добавили 4,75 г (23 ммоля) дициклогексилкарбодиимида и 2,88 г (23 ммоля( н-валериановой кислоты и реакционную смесь поддерживали при 25oC при переменном перемешивании в течение 24 ч. Затем N,N'-дициклогексилмочевину, образовавшуюся в качестве побочного продукта отфильтровали, фильтрат упарили при пониженном давлении, остаток смешали с 2 х 40 мл дистиллированной воды, декантировали и влажный продукт оставили отверждаться при 50 мл 50% этанола. Твердое соединение отфильтровывали, промыли 2 х 10 мл 50% этанола и высушили при 80oC. Сырой продукт был перекристаллизован из 24 мл ж этанола, и кристаллы высушили при 100oC, в результате получили 2,20 г (70% выход) нужного продукта в виде порошка желтого цвета, т.пл. 145-147oC, C22H23N3O5 409,450.
Пример 31
1-(4-нитрофенил)-3-пивалоил-4-метил -7,8-метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепин
Согласно процедуре, описанной в примере 28, но и с использованием пивалоилхлорида вместо ангидрида трифторуксусной кислоты получили 1,68 г (89,4%) нужного продукта, т.пл. 164 -166oC, C22H23N3O5 409,450.
Пример 32
1-(4-нитрофенил)-3-бензоил-4-метил -7,8-метилендиокси-3,4-дигидро-5Н -бензодиазепин.
Согласно процедуре, описанной в примере 31, но с использованием в качестве ацилхлорида-бензоилхлорида, получили 1,72 г (86,9% ) продукта цвета желтой охры, т.пл. 222-224oC (разл), C245H19N3O5 429,404
Пример 33
1-(4-нитрофенил)-3-фенилацетил-4 -метил-7,8-метилендиокси-3,4-дигидро-5Н -2,3-бензодиазепин.
Согласно процессу, описанному в примере 30, но с использованием 50% расчетного молярного количества дициклогексилкарбодиимида и фенилуксусной кислоты, получили светло -желтый продукт, т. пл. 193-195oC, C25H21N3O5 443,567
Примеры с 34 по 36
Следуя процедуре, описанной в примере 33, были получены продукты примеров с 34 по 36, только использовались соответствующие компоненты кислот.
Пример 34
1-(4-нитрофенил)-3 -циклопропанкарбонил-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепин, т.пл. 225-228oC, C21H19N3O5 393,407.
Пример 35
1-(4-нитрофенил)-3-цианоацетил-4 -метил-7,8-метилен-диокси-3,4-дигидро-5Н -2,3-бензодиазепин.
Т.пл. 185-188oC, C20H16N4O5 392,380
Пример 36
1-(4-нитрофенил(-3-методсиацетил-4 -метил-7,8-метилен-диокси-3,4-дигидро-5Н -2,3-бензодиазепин.
Т.пл. 187-189oC, C20H19N3O6 397,396.
Пример 37
1-(4-нитрофенил)-3-(4 -карбоксибутирил)-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепин.
Используя продукт примера 26 в качестве исходного продукта и, проводя ацилирование согласно примеру 14, используя в качестве ацилирующего средства ангидрид глутаровой кислоты, после окончательной перекристаллизации неочищенного продукта из этанола, получили нужный продукт в чистом виде. Т.пл. 148-150oC, C22H21N3O7 439,434.
Пример 38
1-(4-аминофенил)-3-фенилкарбамоил-4 -метил-7,8-метилендиокси-3,4- -дигидро-5Н -2,3-бензодиазепин.
К раствору 0,70 г (2,3 ммоля) 1-(4 -аминофенил)-4-метил-7,8-метилендиокси -3,4-дигидро-5Н-2,3-бензодиазепина в 10 мл безводного бензола добавили 0,24 мл (2,3 ммоля) фенилизоцианата и реакционную смесь нагревали с обратным холодильником в течение 1 ч. После этого раствор упарили при пониженном давлении, и аморфный остаток смешали с 20 мл горячего 50% этанола. Суспензию охладили до 0oC и отфильтровали, в результате получили 0,76 г сырого продукта, т.пл. 190-200oC. После перекристаллизации из 99,5% этанола и растирания с этилацетатом получили нужный продукт, плавящийся при 207-209oC. C24H22N4O3 414,472.
Получение исходного материала этого примера описано в патентной заявке N 198 494, Венгрия. Однако это соединение может быть также получено по новому методу согласно процедуре примера 16 с использованием соединения примера 26 в качестве исходного материала с блестящим выходом (84%). Неочищенный продукт может быть перекристаллизован из 50% этанола, т.пл. 118-120oC.
Пример 39
1-(4-диацетиламинофенил)-3-ацетил-4 -метил-7,8-метилендиокси-3,4- -дигидро-5Н -2,3-бензодиазепин.
2,0 г 1-(4-аминофенил)-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н- -2,3 -бензодиазепина нагревали с 40 мл уксусного ангидрида в течение 3 ч с обратным холодильником, затем раствор упарили до сухости при пониженном давлении. Кристаллический остаток перенесли с 25 мл дистиллированной воды на фильтр и промыли 5 х 3 мл дистиллированной воды. После высушивания получили 2,9 г неочищенного продукта триацетилпроизводного. После промывания 20 мл изопропанола и высушивании при 100oC получили 2,9 г (84, 6%) чистого нужного продукта, т.пл. 224-227oC, C23H23N3O5 421,461
Пример 40
N1-[4-(3-ацетил-4-мотил-7,8 -метилендиокси-3,4-дигидро- -5Н-2,3 -бензодиазепин-1-ил)-фенил]-N3 -метилмочевина
0,70 г продукта примера 15 растворили в бензоле, дегидратированном над гидридом кальция, добавили 0,3 мл (5 ммоля) метилизоцианата и реакционную смесь перемешивали при 50oC в течение 4 ч. Образовавшиеся после охлаждения кристаллы отфильтровали, промыли 3 х 3 мл бензола, затем растерли с 20 мл горячего бензола. Горячую смесь отфильтровали, осадок промыли 3 х 3 мл бензола и высушили, в результате получили 0,65 г (79,6%) нужного продукта, т.пл. 168-170oC (разл.), C21H22N4O4 394, 439.
Пример 41
N1-[4-(3-ацетил-4-метил-7,8 -метилендиокси-3,4-дигидро- -5Н-2,3 -бензодиазеиин-1-ил)-фенил] N3 -фенилмочевина
Следуя процедуре, описанной в примере 40, но используя фенилизоцианат вместо метилизоцианата, нагревая смесь с обратным холодильником в течение 10 ч, упаривая раствор при пониженном давлении, затем суспендируя остаток сначала в 50 мл диэтилового эфира, затем в 15 мл этилацетата, получили 0,69 г нужного продукта, т.пл. 184-186oC (разл.), C26H24N4O4 456, 510.
Пример 42
1-(4-ацетиламинофенил)-3-ацетил-4 -метил-7,8-метилен-диокси-3,4- -дигидро -5Н-2,3-бензодиазепин.
1,3 г (4,4 ммоля) 1-(4-аминофенил) -3-ацетил-4-метил-7,8-метилендиокси- 3,4 -дигидро-5Н-2,3-бензодиазепина перемешивали при 20-25oC с 5 мл уксусного ангидрида в течение 1 ч, затем раствор желтого цвета вылили в 100 г ледяной воды и перемешивали до тех пор, пока полностью не разложился избыток ангидрида. Образовавшийся осадок отфильтровали, промыли 3 х 10 мл дистиллированной воды и высушили. В результате получили 1,6 г сырого продукта. После перекристаллизации из 20 мл бензола получили 1,50 г (89,85%) нужного продукта, т. пл. 158- 160oC (разл.), C21H21N3O4 379,423.
Пример 43
1-(4-формиламинофенил)-3-формил-4 -метил-7,8-метилен-диокси-3,4-дигидро-5Н -2,3-бензодиазепин.
К 6,0 мл (0,014 моль) уксусного ангидрида по каплям при 0oC в течение 5 мин при постоянном перемешивании добавляли 3,0 мл (0,08 моль) 100% муравьиной кислоты. Перемешивание продолжалось при 50oC в течение 15 мин. Затем к полученному таким образом смешанному ангидриду добавили 1 г (3,3 ммоля) 1-(4-аминофенил-4-метил-7,8 -метилен-диокси-3,4-дигидро-5Н-2,3 -бензодиазепина. Реакционную смесь перемешивали при 25oC в течение 1,5 ч, затем ее вылили в ледяную воду, образовавшийся осадок отфильтровали, промыли 4 х 5 мл дистиллированной воды и высушили, в результате получили 0,80 г неочищенного (сырого) продукта. После кристаллизации из 3 мл этилацетата получили 0,65 г (56,2%) нужного продукта, т.пл. 193-195oC, C19H17N3O4 351,369.
Пример 44
1-(4-трифторацетиламинофенил)-3 -трифторацетил-4-метил-7,8-метилен- диокси-3,4-дигидро-5Н-2,3-бензодиазепин.
1,48 г (5 ммоля) 1-(4-аминофенил)-4 -метил-7,8-метилендиокси-3,4-дигидро-5Н -2,3-бензодиазелина растворили в 30 мл безводного хлороформа, затем добавили 2,1 мл (15 ммолей) триэтиламина, и при 20-25oC 2,12 мл (15 ммолей) ангидрида трифторуксусной кислоты, и реакционную смесь перемешивали в течение 2,5 ч, затем сначала проэкстрагировали 2 х 30 мл дистиллированной воды, а затем 20 мл, 5% хлористоводородной кислоты. Органический слой высушили над безводным сульфатом натрия, упарили при пониженном давлении, и аморфный остаток перекристаллизовали из 10 мл 70% этанола, в результате получили 1,41 г (57,9%) нужного диацилпроизводного, т.пл. 177-178oC, C21H15F6N3O4 487,363
Пример 45
1-(4-пропиониламинофенил)-3 -пропионил-4-метил-7,8-метилендиокси -3,4-дигидро-5Н-2,3-бензодиазепин.
Была повторена процедура, описанная в примере 44, за исключением того, что в данном опыте были использованы 11,2 ммоля вместе триэтиламина и ангидрида пропионовой кислоты, а кристаллический осадок перекристаллизовали из 15 мл 50% этанола, затем из 11,5 мл 99% этанола и в результате получили 2,48 г (60,9%) нужного продукта, т.пл. 152-154oC. C23H25N3O4 407,477.
Примеры с 46 по 65
Другие диацилпроизводные общей формулы (I), в которой R ацильной группе, R1 R3, R2 CH3 и R4 ацильной группе, в которой R и R4 могут быть как одинаковыми, так и различаться друг от друга, представлены в табл. 10. Эти соединения были частично получены из соединений общей формулы (III), в которой R R1 R3 H и R4 ацильной группе, а часть из этих соединений была получена из новых соединений общей формулы (1), в которой R ацильной группе, R1=R3=R4=H и R2=CH3, в соответствии с процедурами, описанными в ранее приведенных примерах.
Получение исходных веществ общей формулы (III), в которой R=R1= R3=H и R4 ацильной группе, проиллюстрировано детально ниже на производных, несущих в качестве R4 ацетильную группу:
1-(4-ацетиламинофенил)4-метил-7,8 -метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепин.
Способ А
К раствору, содержащему 6,0 г (20 ммолей) 1-(4-аминофенил)-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепина в 30 мл этилацетата добавили 1,38 мл (21 ммоля) метансульфокислоты. Кристаллический осадок отфильтровали и промыли 5 х 5 мл этилацетата. Вес сухого продукта составил 7,37 г, т. пл. он спекался выше 190oC и плавился с небольшим разложением при 210-212oC. Полученная таким образом соль метансульфокислоты в качестве исходного вещества была проацетилирована следующим образом.
7,37 г порошкообразной соли суспендировали в 110 мл уксусного ангидрида, полученную суспензию перемешивали при комнатной температуре в течение 2 ч, затем кристаллический осадок отфильтровали, промыли 5 х 10 мл этилацетата и высушили, в результате получили 6,54 г соли метансульфокислоты нужного соединения, т.пл. 240-241oC (с разложением).
Основание выделили из соли метансульфокислоты нужного соединения следующим образом: 6,54 г соли растворили в 90 мл воды, раствор осветлили на активированном угле, затем по частям добавили к нему 3,6 г гидрокарбоната натрия. Выпавший осадок отфильтровали, промыли 5 х 10 мл дистиллированной воды и высушили, в результате получили 5,54 г сырого продукта. После перекристаллизации из 130 мл изопропанола получили 3,11 г (46%) нужного продукта с т.пл. 221 -223oC и (слабое разл.), т.пл. которого возросла до 223-225oC после выварки с 15 мл бензола. C19H19N3O3 337, 385. Хлористоводородная соль этого продукта разлагалась при 262-264oC.
Способ В
После растворения при слабом нагревании 15,0 г (44,7 ммоля) 1-(4 -ацетиламинофенил)-4-метил-7,8-метилен -диокси-5Н-2,3-бензодиазепина в 150 мл пиридина добавили 10,2 г (0,269 ммоля) борогидрида натрия, и смесь перемешивали при 100oC на масляной бане в течение 5 ч. Затем реакционную смесь охладили примерно до 25oC, при непрерывном перемешивании по каплям добавили 150 мл дистиллированной воды, затем добавили смесь, содержащую 180 мл концентрированной хлористоводородной кислоты и 265 мл дистиллированной воды, добавление велось при охлаждении ледяной водой. Образовалась суспензия желтоватого цвета. Осадок отфильтровали, промыли 5 х 20 мл дистиллированной воды и высушили, получили в результате 15,2 г соли, т.пл. выше 250oC. Для выделения основания эту соль суспендировали в 150 мл 50% этанола и затем добавили по частям при перемешивании 5,7 г гидрокарбоната натрия. Полученную таким образом суспензию отфильтровали через 30 мин, промыли интенсивно 3 х 10 мл 50% этанола, 5 х 20 мл дистиллированной воды, и, наконец, 20 мл 50% этанола и высушили, получили в результате 10,95 г неочищенного продукта, т. пл. 218 220oC (слабое разложение). После выварки этого продукта с 50 мл горячего изопропанола и затем со 100 мл горячего 99,5% этанола, было получено 8,63 г (57,2%) нужного продукта, т.пл: 220-222oC (слабое разл.).
Физические характеристики других 1 -(4-ациламинофенил)-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепинов следующие:
R4-аналог т.пл.oC
пропионил 237-239
бензоил 247- 248 (разл.)
фенилацетил 213-215 (разл.)
пивалоил 132-135 (разл).
В табл. 10 даны соединения общей формулы (I) в которой R1 R3 H, R2 CH3, и R4 представляют ацильные группы.
Пример 66
1-(4-глициламинофенил)-4-метил-7,8 -метилендиокси-5Н-2,3-бензодиазепин.
К суспензии 2,89 г (5,97 ммоля) 1 -(4-фталоилглициламинофенил)-4-метил-7,8 -метилендиокси-5Н-2,3-бензодиазепина (пример 79) в 50 мл метанола добавили 0,6 мл (11,9 ммоля) 100% гидразингидрата, смесь нагревали с обратным холодильником в течение 2 ч. Реакционную смесь охладили, упарили при пониженном давлении, часть кристаллического осадка смешали с 40 мл дихлорметана, отфильтровали и побочный продукт промыли 2 х 10 мл дихлорметана. Раствор проэкстрагировали 3 х 15 мл 5% хлористоводородной кислоты, водный слой подщелочили добавлением 24 мл 10% водного раствора гидроксида натрия, образовавшийся осадок отфильтровали, промыли 3 х 10 мл дистиллированной воды и высушили при 100oC, в результате получили 1,67 г неочищенного продукта. После перекристаллизации из 73 мл этанола было получено 1,50 г (71,8%) нужного продукта, т.пл. 223-225oC. C19H18N4O4 350,385.
Примеры с 67 по 78
Другие соединения общей формулы (I), в которой R2 CH3, R3 H и некоторые их кислые соли присоединения получены согласно процедуре, описанной в примере 66, и представлены в табл. 11. Соли были получены в соответствии с известными способами. Примечание: H-Fu - гидрофумарат( H фумарат). Fu фумарат.
Продукты примеров 70-72 были получены из соответствующих исходных веществ в две стадии, первая стадия процедура примера 66, затем примера 16.
Пример 79
1-[4-(N-фталоилглициламино)фенил]-4 -метил-7,8-метиленцдокси-5Н-2,3 -бензодиазепин.
К раствору 2,0 г (6,88 ммоля) 1-(4 -аминофенил)-4-метил-7,8-метилендиокси-5Н -2,3-бензодиазепина в дихлорметане добавили 1,84 г (8,94 ммоля) дициклогексилкарбодиимида и 1,84 г (8,94 ммоля) порошкообразной фталилимидоуксусной кислоты, реакционную смесь перемешивали при 25oC в течение 8 ч, затем оставили при 0-5oC на ночь. Образовавшийся осадок отфильтровали, промыли 3 х 3 мл дихлорметана и высушили при 60-80oC, в результате получили 5 г продукта, состоящего из смеси нужного продукта и N,N'-дициклогексилмочевины побочный продукт. Очистку этой смеси осуществили путем кипячения с 210 мл этанола в течение 30 мин, затем горячую смесь отфильтровали и промыли 2 х 10 мл горячего этанола, после чего высушили при 100oC, в результате получили 2,42 г (73,3% ) нужного продукта, т. пл. 266 268oC (разл.), C27H20N4O5 480,489.
Пример 80
1-[4-(N-фталоил- γ -аминобутириламино)фенил]-4-метил-7,8 -метилендиокси-5Н-2,3-бензодиазепин.
Следуя процедуре, описанной в примере 79, но использовав g - фталимидомасляную кислоту, получили 3,8 г смеси, которую объединили с дихлорметановым маточным раствором, предварительно проэкстрагированным 2 х 40 мл 10%-ного водного раствора гидрокарбоната натрия. После упаривания при пониженном давлении остаток пропустили через хроматографическую колонку (адсорбент кизельгур 60) 0,063 2 мм(, элюент смесь этилацетат метанол 4:1. Остаток растерли с 10 мл горячего этанола, охладили, промыли 3 х 1 мл этанола и высушили, в результате получили 3,12 г (90%) нужного продукта, т.пл. 233- 235oC (разл.), C29H24N4O5 508, 543.
Пример 81
1-[4-(N-фталоил-DL-аланиламино)фенил] -4-метил-7,8 -метилендиокси-5Н-2,3-бензодиазепин.
Процесс, описанный в примере 79 был повторен, за исключением лишь того, что использовали в этот раз N-фталоил-DL -аланин (DL-2-фталимидопропионовая кислота). После того как отфильтровали легкий осадок, фильтрат упарили, остаток после упаривания смешали с 15 мл дихлорметана, тщательно профильтровали и полученный прозрачный раствор вновь упарили. Остаток очистили путем его кипячения с 60 мл этилацетата. Образование кристаллов началось уже в горячем растворе. Выпавшие кристаллы отфильтровали при 0-5oC, почти белый кристаллический порошок промыли 3 х 3 мл этилацетата и высушили при 100oC, в результате получили 2,75 г (80,95%) нужного продукта, т.пл. 243- 245, C28H22N4O5 494,516.
Пример 82
1-(4-нитрофенил)-3-глицил-4-метил -7,8-метилендиокси-3,4-дигидро-5Н- -2,3 -бензодиазенин.
Для получения этого продукта следовали процедуре, описанной в примере 66, но использовали в качестве исходного соединения продукт, полученный в примере 85, а также раствор дихлорметана экстрагировали только 3 х 20 мл дистиллированной воды, а затем органический слой упарили при пониженном давлении. Кристаллический остаток очистили, суспендировав его в 7 мл этанола, в результате получили чистый нужный продукт с выходом 86,1% т.пл. 201-203oC (разл.), C19H18N4O5 382,385.
Пример 83
1-(4-нитрофенил)-3-( g -аминобутирил)-4-метил-7,8-метилендиокси -3,4-дигидро-5Н-2,3-бензодиазепин.
Следуя процедуре, описанной в примере 82, и, используя в качестве исходного материала соединение, полученное в примере 86, был получен продукт, содержащий кристаллизационный растворитель, с выходом 89,4% т.пл. 110 -112oC (перекристаллизован из 50% этанола), C21H22N4O5 410,439
Пример 84
1-(4-нитрофенил)-3-(DL-аланил)-4 -метил-7,8-метилендиокси-3,4- -дигидро-5Н -2,3-бензодиазепин.
Следуя процедуре, описанной в примере 82, и, используя соединение, полученное согласно процедуре, описанной в примере 87 в качестве исходного материала, получили нужное соединение с т.пл. 220-221oC (разл.), C20H20O5N4 396, 412
Следуя примеру 82 и, используя в качестве исходного материала соединение, полученное в соответствии с процедурой, описанной в примере 87, получили нужный продукт, т.пл. 220-221oC (разл.), C20H20N4O5 396,412.
Примеры с 85 по 87
Новые промежуточные продукты, используемые в примерах 82-84 в качестве исходных веществ, были получены из соединения, полученного в соответствии с процедурой примера 26, согласно способу, описанному в примере 81.
Пример 85
1-(4-нитрофенил)-3-(N -фталоилглицил)-4-метил-7,8-метилендиокси -3г, 4-дигидро-5Н-2,3-бензодиазепин
Выход: 93,3% т.пл. 173-174oC (разл.), C27H20N4O7 512,489.
Пример 86
1-(4-нитрофенил)-3-(N-фталоил-g -аминобутирил)-4-метил-7,8-метилендиокси -3,4-дигидро-5Н-2,3-бензодиазепин т.пл. 218-220oC, C29H24N4O7 540,543.
Пример 87
1-(4-нитрофенил)-3-(N-фталоил-DL -аланил)-4-метил-7,8-метилендиокси-3,4 -дигидро-5Н-2,3-бензодиазепин.
Т.пл. 210-212oC, C28H22N4O7 526,516
Примеры с 88 до 94
Промежуточные соединения общей формулы (I), в которой R и/или R4, представляют(ет) C1-6 ацильную группу, замещенную фталимидной группой, необходимы для получения соединений, полученных с использованием процедур, описанных в примерах с 73 по 87, и суммированы в табл. 12. Они были получены из соединения примера 15(16) или из соединения общей формулы (III), в которой R4 водород, см. патент США N 4835152 или из 1-(4-ацетиламинофенил)-4 -метил-7,8-метилендиокси-3,4-дигидро-5Н -2,3-бензодиазепина, описанного выше (перед табл. 10) согласно процедуре примера 81.
В примере 93 следует использовать двухкратное количество фталоилглицина и дициклогексилкарбодиимида. Итак, в табл. 12 даны новые соединения общей формулы (I), в которой R и R4 представляют ацильную группу, R1=R3=H и R2= CH3.
Пример 95
1-(4-аминофенил)-3-( g аминобутирил)-4-метил-7,8-метилендиокси -3,4-дигидро-5Н-2,3-бензодиазепина гидрофумарат.
Это соединение было получено из соединения примера 83 по способу, описанному в примере 16, т.пл. 150-152oC (разл.), [C29H25N4O3]•C4H3O4 496, 531.
Пример 96
1-(4-аминофенил)-3-(4 -карбоксибутирил)-4-метил-7,8 -метилендиокси- -3,4-дигидро-5Н-2,3 -бензодиазепина гидрохлорид.
Это соединение получили из соединения примера 37 согласно способу, описанному в примере 16, т.пл. 224-226oC (разл.), [C22H24N3O5]Cl 445,915.
Пример 97
1-(4-трифторацетиламинофенил)-4 -метил-7,8-метилендиокси-5Н- -2,3 -бензодиазепин.
Это соединение получили, следуя процедуре примера 2, т.пл. 258-260oC (разл.), C19H14F3N3O3 389,339.
Пример 98
1-(4-аминофенил)-3-метилкарбамоил-4 -метил-7,8-метилендиокси-3,4-дигидро-5Н -2,3-бензодиазепин.
Это соединение было получено из 1 -(4-нитрофенил)-3-метилкарбамоил-4-метил -7,8-метилендионси-3,4-дигидро-5Н-2,3 -бензодиазепина согласно способу примера 16, т.пл. 199-201oC, C19H20N4O3 352,401.
Т.пл. гидрохлорида этого соединения равна 219-221oC (разл.), [C19H21N4O3]Cl 388,866.
Исходное нитросоединение было получено следующим образом:
1,1 мл (18,4 ммоля) метилизоцианата добавили к 3,0 г (9,22 ммоля)
1-(4-нитрофенил)-4-метил-7,8 -метилен-диокси-3,4-дигидро-5Н- -2,3 -бензодиазепина (см. пример 26) растворенного в 60 мл дихлорметана и перемешивали в течение 24 ч, полученный раствор затем упарили при пониженном давлении. Кристаллический остаток растерли с 30 мл горячего этанола при 80 -100oC, в результате получили 3,35 г (95%) нужного продукта лимонно-желтого цвета, т.пл. 238-240oC (разл.), C19H18N4O5 382,385.
Пример 99
1-(4-аминофенил)-3-(1 -пирролидиноацетил)-4-метил-7,8 -метилендиокси- -3,4-дигидро-5Н-2,3 -бензодиазепин.
Это соединение было получено из 1 -(4-нитрофенил)-3-(1-пирролидиноацетил)-4 -метил-7,8-метилендиокси-3,4-дигидро-5Н -2,3-бензодиазепина согласно способу, описанному в примере 16, т.пл. 212 -214oC, C23H26N4O3 306,493.
Исходное вещество для синтеза этого соединения было получено из 1-(4 -нитрофенил)-3-хлорацетил-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н-2,3 - бензодиазепина (см. пример 116) согласно способу, описанному в примере 102, т.пл. 189-190oC (разл.), C23H24N4O5 436,47.
Пример 100
1-(4-аминофенил)-3-(N, N -диметилглицил)-4-метил-7,8-метилендиокси - -3,4-дигидро-5Н-2,3-бензодиазепина гидрофумарат.
Это соединение было получено из 1 -(4-нитрофенил)-3-(диметилглицил)- -4 -метил-7,8-метилендиокси-3,4-цигидро-5Н -2,3-бензодиазенина согласно способу, описанному в примере 16, т.пл. 202-204oC (разл), [C21H25N4O3]•C4H3O4 496,531.
Исходное вещество было получено из 1-(4-нитрофенил)-3-хлороацетил-4-метил -7,8-метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепина согласно способу, описанному в примере 103, т.пл. 158 160oC, C21H22N4O5 410, 439.
Пример 101
1-(4-хлороацетиламинофенил)-4-метил -7,8-метилендиокси-5Н- 2,3-бензодиазепин.
Это соединение было получено согласно способу, описанному в примере 2, за исключением того, что была использована в качестве реагента хлоруксусная кислота, т.пл. 209-214oC (обугливание), C19H16ClN3O3 369,818.
Пример 102
1-[4-(1 -пирролидиноацетиламино)фенил] -4-метил -7,8-метилендиокси-5Н-2,3-бензодиазепин
К суспензии 1,5 г (406 ммоля) 1-(4 -хлороацетиламинофенил)-4-метил-7,8 -метилендиокси-5Н-2,3-бензодиазепина в 60 мл этанола добавили 0,71 мл (8,53 ммоля) пирролидина, и смесь кипятили с обратным холодильником в течение 4 ч, затем упарили ее при пониженном давлении. Остаток обработали водой, в результате получили 1,48 г неочищенного продукта, т.пл. 186-188oC. После перекристаллизации из 12 мл этанола получили 1,22 г (74,4%) нужного продукта, т.пл. 210-212oC, C23H24N4O3 404,477.
Пример 103
1-[4-(N, N-диметилглициламино)фенил] -4-метил-7,8-метилендиокси- -5Н-2,3 -бензодиазепин.
После добавления 0,66 г (8,12 ммоля) диметиламина гидрохлорида и 1,86 мл (13,4 ммоля) триэтиламина к суспензии 1,5 г (4,06 ммоля) 1,-4 -хлороацетиламинофенил-4-метил-7,8 -метилендиокси-5Н-2,3-бензодиазепина в 60 мл этанола, реакционную смесь, нагревали в течение 8 ч, затем ее упарили. Остаток растворили в 30 мл дихлорметана, промыли 20 мл 4% раствора гидроксида натрия, затем 2 х 20 мл дистиллированной воды, высушили и упарили при пониженном давлении. После обработки водой кристаллический остаток отфильтровывали, в результате получили 1,27 г сырого продукта, т.пл. 211-213oC. После перекристаллизации из 10 мл этанола получили 1,1 г нужного продукта (71,4%) т. пл. 213-215oC, C21H22N4O6 378,439.
Пример 104
1-(4-метилкарбамоиламинофенил)-4 -метил-7,8-метилендиокси-5Н- 2,3 -бензодиазепин.
0,8 мл (13,4 ммоля) метилизоцианата добавили к раствору, содержащему 1,0 г (3,41 ммоля) 1-(4-аминофенил)-4-метил -7,8-метилендиокси-5Н-2,3-бензодиазепина в III мл диметилформамида (ДМФ), затем реакционную смесь перемешивали при 25oC в течение 24 ч. После разбавления 86 мл воды, отфильтровали при 5oC и высушили в температурном интервале 60-100oC, в результате получили 1,06 г неочищенного продукта, т. пл. 204-207oC (спекание начинается при 160oC), после перекристаллизации из 5 мл этанола получили 0,85 г (71,4%) нужного продукта, т.пл. 223-224oC (разл.) C19H18N4O3 350,385.
Пример 105
1-(4-ацетиламинофенил)-3 -метилкарбамоил-4-метил-7,8 - метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепин.
Это соединение было получено из 1 -(4-аминофенил)-3-метилкарбаиоил-4-метил -7,8-метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепина с использованием способа примера 42. Неочищенный продукт был перекристаллизован из этилацетата, в результате получили продукт с выходом 71,4% т.пл. 150-152oC, C21H22N4O4 394, 439.
Пример 106
1-(4-хлорацетиламинофеиил)-3-ацетил -4-метил-7,8-метилен-диокси- -3,4-дигидро -5Н-2,3-бензодиазепин.
Это соединение было получено из 1 -(4-аминофенил)-3-ацетил-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н- -2,3 -бензодиазепина и при использовании процесса, описанного в примере 2, т.пл. 139- 140oC, C21H20ClO4N3 413,972.
Пример 107
1-[4-(N-N-диметилглициламино)фенил] -3-ацетил-4-метил-7,8-метилендиокси-3,4 -дигидро-5Н-2,3-бензодиазепин.
Это соединение было получено из продукта предыдущего примера, согласно способу, описанному в примере 103, т.пл. 206- 208oC, C23H26N4O4 422,493.
Пример 108
1-[4-(N, N-диэтилглициламино)фенил-3 -ацетил-4-метил-7,8-метилендиокси- 3,4 -дигидро-5Н-2,3-бензодиазепин.
Это соединение было получено из 1 -(4-хлорацетиламинофенил)-3-ацетил-4 -метил-7,8-метилендиокси-3,4-дигидро-5Н -2,3-бензодиазепина и диэтиламина согласно способу, описанному в примере 102, т.пл. 175-176oC, C25H30N4O4 450,547.
Пример 109
1-[4-(1-пирролидиноацетиламино)фенил-3-ацетил-4 -метил-7,8-метилен-диокси-3,4-дигидро-5Н -2,3-бензодиазепина гидрофумарат.
Это соединение было получено из 1 -(4-хлороацетиламино-фенил)-3-ацетил -4метил-7,8-метилендиокси-3,4- -дигидро -5Н-2,3-бензодиазепина согласно способу примера 2 и выделено в форме гидрофумарата, т.пл. 181-183oC (разл.), [C25H29N4O4]•C4H3O4 564,607.
Пример 110
1-(4-ацетиламинофенил)-3 -хлороацетил-4-метил-7,8-метилендиокси -3,4- -дигидро-5Н-2,3-бензодиазепин.
Это соединение было получено из соединения общей формулы (III), в которой R4 COCH3 согласно способу, описанному в примере 2 и с использованием хлоруксусной кислоты вместо муравьиной кислоты, т.пл. 138 -140oC, C21H20ClN3O4 413,972.
Пример 111
1-[4-(N, N-диэтилглициламино)фенил] -4-метил-7,8-метилендиокси- -5Н-2,3 -бензодиазепин.
Это соединение получено из 1-(4 -хлорацетиламинофенил)-4-метил-7,8 -метилендиокси-5Н-2,3-бензодиазелина согласно способу, описанному в примере 102, но с использованием вместо пирролидина диэтиламина, т.пл. 157 -158oC, C23H26N4O3 406,493.
Пример 112
1-(4-ацетиламинофенил)-3 -циклопропанокарбонил-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепин.
Это соединение было получено из 1 -(4-аминофенил)-3-циклопропанокарбоиил-4 -метил-7,8-метилендиокси-3,4-дигидро-5Н -2,3-бензодиазепина согласно способу, описанному в примере 42, т.пл. 242 -243oC, C23H23N3O4 405,461.
Пример 113
N1-4-(3-метилкарбамоил-4-метил-7,8 -метилендионси-3,4-дигидро-5Н-2,3 -бензодиазепин-1-ил)-фенил-N3 -метилмочевина.
После добавления 0,5 мл (8,5 ммоля) метилизоцианата к 0,6 г (1,7 ммоля) 1-(4 -аминофенил)-3-метилкарбамоил-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепина, растворенному в 45 мл безводного дихлорметана, реакционную смесь перемешивали при 25oC в течение 6 дней. Затем кристаллический осадок отфильтровали, промыли 3 х 2 мл дихлорметана и высушили при 60-80oC, в результате получили 0,55 г (79,7%) чистого нужного продукта, т.пл. 181 -183oC, C21H23N5O4 409,455.
Пример 114
1-(4-аминофенил)-3-н-бутилкарбамоил -4-метил-7,8-метилендиокси- -3,4-дигидро -5Н-2,3-бензодиазелин.
Это соединение было получено из 1 -(4-нитрофенил)-3-н-бутил-карбамоил-4 -метил-7,8-метилендиокси-3,4- -дигидро-5Н -2,3-бензодиазепина, т.пл. 173-175oC, C22H26N4O3 394, 482.
Исходное вещество для этого синтеза было получено так, что синтезировано исходное соединение примера 98, но вместо метилизоцианата использовали н -бутилизоцианат и реакционную смесь, перемешивали в течение 5 дней при 25oC, т.пл. 176-178oC, C22H24N4O5 424,466.
Пример 115
1-(4-глициламинофенил)-3 -метилкарбамоил-4-метил-7,8-метилендиокси - -3,4-дигидро-5Н-2,3-бензодиазепин.
Это соединение было получено из 1 -[4-(N-фталоилглициламино)-фенил-3 -метилкарбамоил-4-метил-7,8 метилендиокси-3,4-дигидро-5Н-3 -бензодиазепина согласно способу примера 66, модифицированного в примере 82, т.пл. 163- 165oC, C21H23N5O4 409,455.
Исходное вещество для этого синтеза было получено из 1-(4-аминофенил)-3 -метилкарбамоил-4-метил-7,8-метилен -диокси-3,4- -дигидро-5Н-2,3 -бензодиазепина (см. 98) согласно способу примера 79, т.пл. 270-271oC (разл.), C29H25N5O6 539,559.
Пример 116
1-(4-аминофенил)-3-(N-метилглицил) -4-метил-7,8- -метилендиокси-3,4-дигидро -5Н-2,3-бензодиазелин.
1,03 г (15,3 ммоля) гидрохлорида метиламина и 2,64 мл (18,3 ммоля) триэтиламина добавили к суспензии, содержащей 1,23 г (3,06 ммоля) 1-(4 -нитрофенил)-3-хлорацетил-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазеляна в 140 мл этанола, и реакционную смесь нагревали с обратным холодильником в течение 10 ч, затем упарили при пониженном давлении. Остаток растворили в 30 мл хлороформа, промыли 20 мл 4% раствора гидроксида натрия, затем 2 х 20 мл воды, высушили и упарили при пониженном давлении. Остаток восстановили согласно способу примера 16, и порученный продукт очистили, использовав хроматографическую колонку (адсорбент кизельгур 60, элюент смесь метанол бензол 4:1). Полученный сырой продукт растерли с 5 мл этилацетата при 25oC, в результате получили 0,60 г (63, 6%) нужного продукта, т.пл. 198-200oC (слабое разл.), C20H22N4O3 366,428.
Исходное вещество для этого синтеза было получено из 1-(4-нитрофенил)-4 -метил-7,8-метилендиокси-3,4-дигидро-5Н -2,3-бензодиазепина (см. пример 26) и хлоруксусной кислоты согласно способу, описанному в примере 33, т.пл. 189
191oC (разл.), C19H16ClN3O5 401, 818.
Пример 117
1-[4-(N-метилглициламино)фенил] -3 -ацетил-4-метил-7,8-метилендиокси-3,4 -дигидро-5Н-2,3-бензодиазепин.
1,31 г (19,5 ммоля) гидрохлорида метиламина и 3,24 мл (23,3 ммоля) триэтиламина добавили к суспензии, содержащей 1,61 г (3,89 ммоля) 1-(4 -хлорацетиламинофенил)-3-ацетил-4-метил -7,8-метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепина (см. пример 106) в 100 мл этанола и реакционную смесь нагревали с обратным холодильником в течение 10 ч, затем содержимое упарили при пониженном давлении. Остаток очистили, пропустив через хроматографическую колонку (абсорбент кизельгур 60, элюент смесь хлороформа и метанола 9:1). Сырой продукт растерли с 3 мл 50% этанола при 25oC, в результате получили 0,61 г (38,6% ) нужного продукта, т.пл. 220-222oC (слабое разл.), C22H24N4O4 498, 466.
Пример 118
Приготовление фармацевтических композиций
Таблетки или разделяемые таблетки, содержащие 25 мг 1-(4-аминофенил)-3 -ацетил-4-метил-7,8-метилендиокси-3,4 -дигидро-5Н-2,3-бензодиазепина (соединение примера 15 или 16) или 25 мг 1-(4-ацетил-аминофенил)-3-ацетил-4-метил -7,8-метилендиокси-3,4-дигидро-5Н-2,3 -беняодиазепина (соединение примера 42) или 25 мг 1-(4-аминофенил)-3 -металкарбамоил-4-метил-7,8-метилендиокси -3,4-дигидро-5Н-2,3-бензодиазепина (соединение примера 98), каждый в качестве активного ингредиента, были получены традиционными способами.
Состав одной таблетки:
Активный ингредиент 25 мг
Картофельный крахмал 43 мг
Лактоза 160 мг
Поливинилпирролидон 6 мг
Стеарат магния 1 мг
Тальк 30 мг
В) Другой предпочтительный состав таблетки
Активный ингредиент 25 мг
Лактоза 130 мг
Кукурузный крахмал 25 мг
Микрокристаллическая целлюлоза 10 мг
Желатин 4 мг
Тальк 2 мг
Стеарин 1 мг
Стеарат магния 1 мг
Пример 119
(-)-1-(4-Нитрофенил)-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепин
Раствор (S)-(-)-2-амино-3-метил-1,1 -дифенилбутан-1-ол (4,75 г) в сухом хлористом метилене охлаждают до -70oC и обрабатывают раствором борана в ТГФ (1,8 М, 9,5 мл) в течение 20 мин в атмосфере сухого азота. Раствор нагревают постепенно до 0oC и затем оставляют на ночь при 4oC. Эту смесь обрабатывают в течение 1 ч при комнатной температуре раствором 1-(4-нитрофенил)-4-метил-7,8 -метилендиокси-5Н-2,3-бензодиазепина (5,0 г) в сухом хлористом метилене (100 мл). Полученный раствор оставляют при комнатной температуре на 7 дней. Реакцию останавливают при добавлении 10% карбоната натрия (15 мл). Образовавшуюся фазу отделяют, органический слой промывают водой (2 х 50 мл), сушат над сульфатом натрия, упаривают в вакууме и получают желтое кристаллическое вещество. Этот твердый продукт суспендируют а этаноле (50 мл), фильтруют, сушат и получают 4,47 г названного соединения. Протонный магнитный резонанс, использующий в качестве реагента сдвига (Eu (hfc)3), идентифицирует продукт как смесь энантиомеров 90:10. Этот материал растворяют в горячем этилацетате (24 мл) и оставляют при комнатной температуре на ночь. Выпавший кристаллический осадок отделяют фильтрацией, промывают этилацетатом (3 х 5 мл), сушат и выделяют 2,87 г названного соединения. Протонный магнитный резонанс, использующий в качестве реагента сдвига (Eu (hfc)3), показывает, что это вещество имеет энантиомерную чистоту более 98% Температура плавления 171 172,5oC.
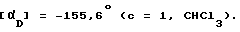
Пример 120
(+)-1-(4-Нитрофенил)-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н- -2,3 -бензодиазепин.
Названное соединение получают как описано в примере 119 с использованием (R)-(+)-2-амино-3-метил-1,1-дифенилбутан -1-ола. Температура плавления 172 - 174oC.
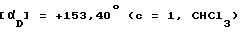
Энантиомерная чистота: > 98%
Пример 121
(-)-1-(4-Нитрофенил)-3-метилкарбамоил-4-метил-7,8-метилендиокси -3,4-дигидро-5Н-2,3-бензодиазепин.
Раствор соединения, полученного в примере 119 (4,0 г) в сухом хлористом метилене (80 мл), обрабатывают метилизоцианатом (2,18 мл). Реакционную смесь перемешивают 3 дня при комнатной температуре, упаривают в вакууме. Маслообразный остаток обрабатывают водой (60 мл), выпавший осадок отфильтровывают, сушат и получают 4,49 г названного соединения в виде желтого порошка. Это вещество используют на следующей стадии без дальнейшей очистки.
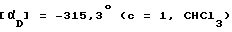
Пример 122
(+)-1-(4-Нитрофенил)-3 -метилкарбомоил-4-метил-7,8-метилендиокси - -3,4-дигидро-5Н-2,3-бензодиазепин.
Названное соединение получают из соединения примера 120 с помощью методики примера 121.
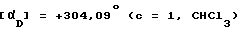
Пример 123
(-)-1-(4-Аминофенил)-3 -метилкарбамоил-4-метил-7,8-метилендиокси - -3,4-дигидро-5Н-2,3-бензодиазепин.
Суспензию соединения, полученного а примере 122 (2,42 г), и никеля Ренея (W -2, 0,5 г) в метаноле (50 мл) обрабатывают 98% гидразин гидратом (1,1 мл), перемешивают 1 ч при 25oC, отфильтровывают катализатор, фильтрат упаривают. Маслообразный остаток обрабатывают водой (50 мл). Образовавшийся осадок отфильтровывают, сушат и получают 2,06 г сырого твердого вещества, которое растворяют в этаноле (10 мл), упаривают в вакууме до небольшого объема. Остаток обрабатывают бензолом (26 мл) для инициирования кристаллизации. Кристаллический продукт отфильтровывают, сушат а вакууме (36 ч, 100oC, 80 торр) и получают 1,98 г названного соединения в виде светло -желтого порошка. Температура плавления 133 135oC.
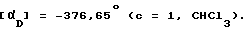
Энантиомерная чистота: > 99%
Пример 124
(+)-1-(4-Аминофенил)-3 -метилкарбамоил-4-метил-7,8-метилендиокси - -3,4-дигидро-5Н-2,3-бензодиазепин.
Названное соединение получают из соединения примера 121 с помощью методики, приведенной в примере 123. Температура, плавления 133 135oC.
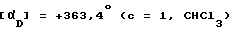 .
.
Энантиомерная чистота: > 99%
Пример 125
(-)-1-(4-Нитрофенил)-3-ацетил-4 -метил-7,8-метилен-диокси-3,4-дигидро-5Н -2,3-бензодиазепин.
Смесь соединения, полученного в примере 119 (2,34 г), и уксусного ангидрида (11,7 мл) перемешивают при комнатной температуре, через 15 мин бензодиазепин полностью растворяется и реакционную массу перемешивают 2 ч, охлаждают на бане со смесью лед-вода, обрабатывают водой (60 мл), перемешивают ночь, собирают кристаллический продукт, промывают водой (4 х 5 мл), сушат и получают 1,5 г названного соединения как бледно-желтых кристаллов. Температура плавления 173 177oC.
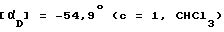 .
.
Пример 126
(+)-1-(4-Нитрофенил)-3-ацетил-4 -метил-7,8-метилен-диокси-3,4-дигидро-5Н -1,3-бензодиазепин.
Названное соединение получают из соединения примера 120 с помощью методики, приведенной в примере 125. Температура плавления 173- 177oC.
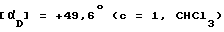 .
.
Пример 127
(+)-1-(4-Аминофенил)-3-ацетил-4 -метил-7,8-метилен-диокси-3,4-дигидро-5Н -2,3-бензодиазепин.
Суспензию соединения, полученного по методике примера 125 (2,6 г), и никеля Ренея (W 2, 0,5 г) в метаноле (52 мл) обрабатывают 98% гидразин гидратом (1,2 мл), перемешивают 1 ч при комнатной температуре, катализатор отфильтровывают, фильтрат упаривают а вакууме. Маслообразный остаток обрабатывают водой (50 мл), выпавший осадок отфильтровывают, сушат и получают 2,17 г сырого твердого вещества, которое перекристаллизовывают из этилацетата (39 мл), кристаллический продукт собирают, сушат (36 ч, 120 130oC, 80 торр) и получают 1,8 г названного соединения. Температура плавления 169-171,5oC.
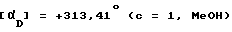
Энантиомерная чистота: > 99%
Пример 128
(-)-1-(4-Аминофенил)-3-ацетил-4 -метил-7,8-метилен-диокси-3,4-дигидро-5Н -2,3-бензодиазепин
Названное соединение получают из соединения примера 126 по методике примера 123. Температура плавления 169 -172oC. 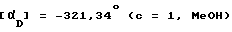 .
.
Энантиомерная чистота: > 99%
Приложение
Нейрозащитная активность
Острые и хронические нейродегенеративные болезни различного происхождения имеют схожие механизмы, приводящие к смерти клетки. Роль глутамат-регулируемой увеличенной токсичности и увеличение концентрации кальция является обычными характеристиками заболеваний и условиями подобных удару, травме мозга, обширной мозговой ишемии после остановки сердца, болезни Паркинсона и т. д. Этот факт предполагает, что ингибирование глутамат регулируемой нейротоксичности может быть успешной стратегией при лечении нейродегенеративных заболеваний.
Нейрозащитное действие соединений формулы (I) вследствие скоротечной (4 мин) закупорки обоих каротидных артерий испытывают на находящихся а сознании мышах.
Эта модель скоротечной закупорки (обширной ишемии) воспроизводит проблемы, возникающие в течение и после остановки сердца у человека.
Сохранение памяти тестировано в задаче пассивной разгрузки, выполненной 24 ч после повторной перфузии [J. Pharm. Meth. 23, 311 (1990)]
Результаты приведены в табл 8.
Защита против повреждения памяти, вызванной скоротечной двусторонней закупоркой самой артерии, на мышах, находящихся в сознании (см. табл.13)
Данные демонстрируют, что соединения перечисленные в табл. 8 в значительной степени предохраняли животных от функционального дефицита, вызванного скоротечной обширной ишемией. Цианид калия (10 мг/кг i.v.) вызывает летальный исход, который возникает обычно в течение 22 с [Meth. Find. Exptl. Clin Pharmacol 10, 349 (1988)] нейрозащитные соединения пролонгируют время выживания. Соединения формулы I испытаны этим методом для демонстрации их нейрозащитной способности.
Результаты представлены в табл. 14.
Соединения примеров 15 (16), 98 и 123 включенных в табл. 9 оказывали статистически значимое защитное действие.
АМРА введенное (2,5 нмол в 0,25 мкл ) в полосатое тело семидневных крыс вызывает тканевое поражение. [Brain Res. 526, 165 (1990)] При лечении животных соединениями формулы I (четыре раза, ежечасно), тканевое поражение уменьшалось.
Результаты показаны в табл.15.
Данные табл. 10 демонстрируют, что эффективность различных соединений формулы I превосходит эффективность известного соединения (ссылочное соединение). Одним из методов для моделирования хронических нейродегенеративных заболеваний (расстройств), связанных с допаминэргической дисфункцией является метод метамфетамин наведенной нейротоксичности.
Высокие дозы метамфетамина (2 х 20 мг/кг i.p.) уменьшают стриарный (относящийся к полосатому телу) уровень допамина, который может быть предотвращен антагонистами глутамата, например, NMDA антагонист МК 810 [Science 243, 398 (1989)]
Эксперименты выполняются с соединением примера 128 (3 х 5 мг/кг, i.p.), для демонстрации способности предотвращать потерю метамфетамин наведенного стриарного допамина и увеличение скорости (степени) обращения (обмена) стриарного допамина.
Результаты представлены в табл. 16.
Данные табл. 16 показывают, что соединение примера 128 влияет на защитное действие против допаминэргической нейротоксичности, которое сравнимо с действием NMDA антагониста МК-801 (3 х 2,5 мг/кг, i.p.). Этот результат указывает, что соединения формулы I могут быть полезны для лечения хронических нейрологических расстройств, таких как болезнь Паркинсона.
В заключение можно сказать, что положительные результаты, полученные на четырех моделях, обсуждаемых выше, служат признаком терапевтического потенциала соединений формулы I при лечении острых и хронических нейродегенеративных заболеваний.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1-[2-(ЗАМЕЩЕННЫЙ ВИНИЛ)]-3,4-ДИГИДРО-5H-2,3-БЕНЗОДИАЗЕПИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ, СПОСОБ ПОЛУЧЕНИЯ ФАРМАЦЕВТИЧЕСКОГО СОСТАВА И СПОСОБ ИНГИБИРОВАНИЯ КОНВУЛЬСИЙ. | 1996 |
|
RU2155757C2 |
| ПРОИЗВОДНЫЕ 1-[2-(ЗАМЕЩЕННЫЙ ВИНИЛ)]-5Н-2,3-БЕНЗОДИАЗЕПИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ И СПОСОБ ЕГО ПОЛУЧЕНИЯ, СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | 1996 |
|
RU2161607C2 |
| Способ получения 1-(4-аминофенил)-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина или его кислотно-аддитивных солей | 1987 |
|
SU1779251A3 |
| ПРОИЗВОДНЫЕ 2,3-БЕНЗОДИАЗЕПИНА, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ ПОЛУЧЕНИЯ И СРЕДСТВО, ОБЛАДАЮЩЕЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ АМРА-РЕЦЕПТОРОВ | 1997 |
|
RU2179557C2 |
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ДИГИДРО-2,3-БЕНЗОДИАЗЕПИНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1995 |
|
RU2151149C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ 2,3-БЕНЗОДИАЗЕПИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2243228C2 |
| Способ получения производных 1-(гидроксистирил)-5Н-2,3-бензодиазепина | 1987 |
|
SU1503681A3 |
| ТРИАЗОЛО[4,3-А][1,4]-БЕНЗОДИАЗЕПИНЫ И ТИЕНО[3,2-F]-[1,2,4]-ТРИАЗОЛО[4,3-А] [1,4]ДИАЗЕПИНЫ, В СЛУЧАЕ НАЛИЧИЯ ПО МЕНЬШЕЙ МЕРЕ ОДНОГО АСИММЕТРИЧЕСКОГО ЦЕНТРА ИХ ЭНАНТИОМЕРЫ, РАЦЕМАТЫ И ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ПРИСОЕДИНЕНИЯ КИСЛОТ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИНГИБИРУЮЩАЯ ФАКТОР АКТИВАЦИИ ТРОМБОЦИТОВ. | 1992 |
|
RU2094436C1 |
| ФИЗИЧЕСКАЯ ФОРМА (R)-7-АЦЕТИЛ-5-(4-АМИНОФЕНИЛ)-8,9-ДИГИДРО-8-МЕТИЛ-7Н-1,3- ДИОКСОЛО[4,5-H] - [2,3]-БЕНЗОДИАЗЕПИНА, СПОСОБ ЕЕ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2142465C1 |
| 3-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 3Н-2,3-БЕНЗОДИАЗЕПИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2146678C1 |
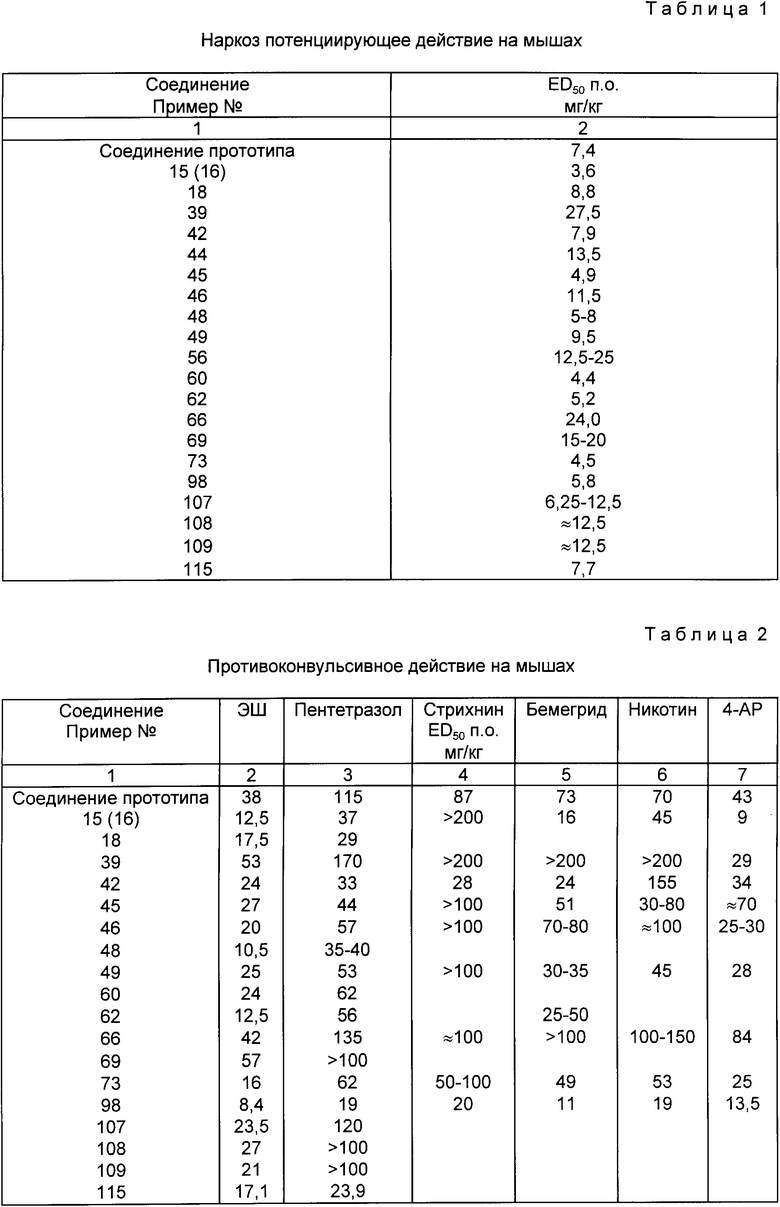
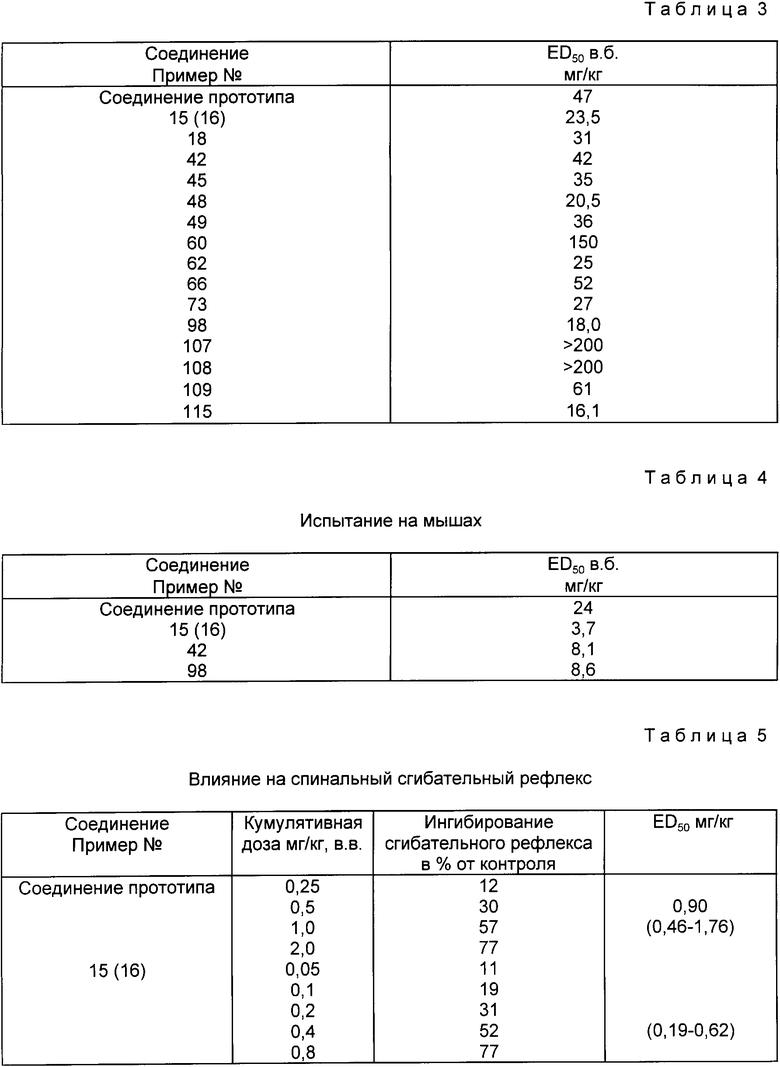
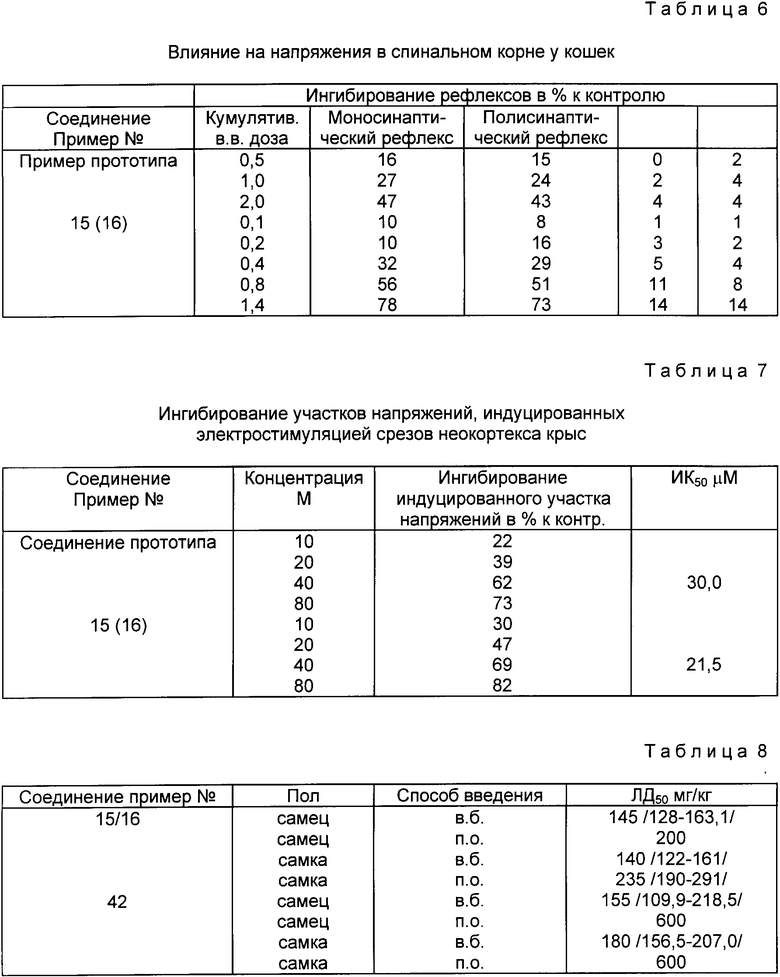
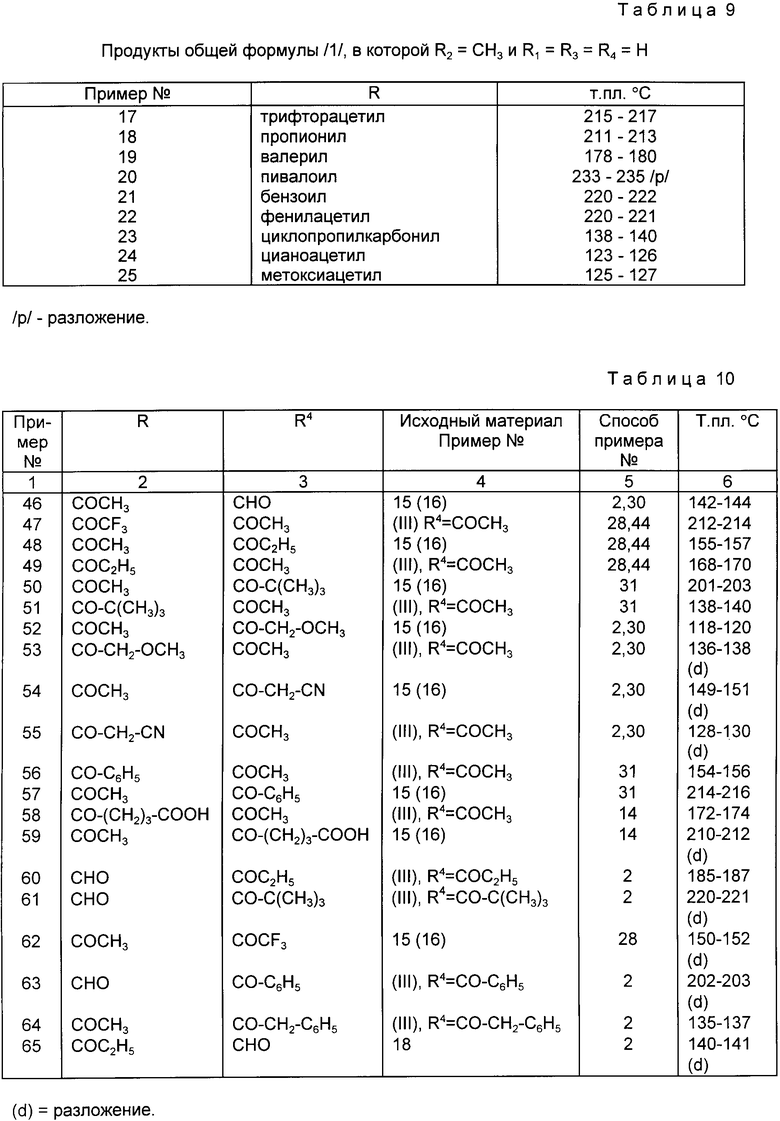
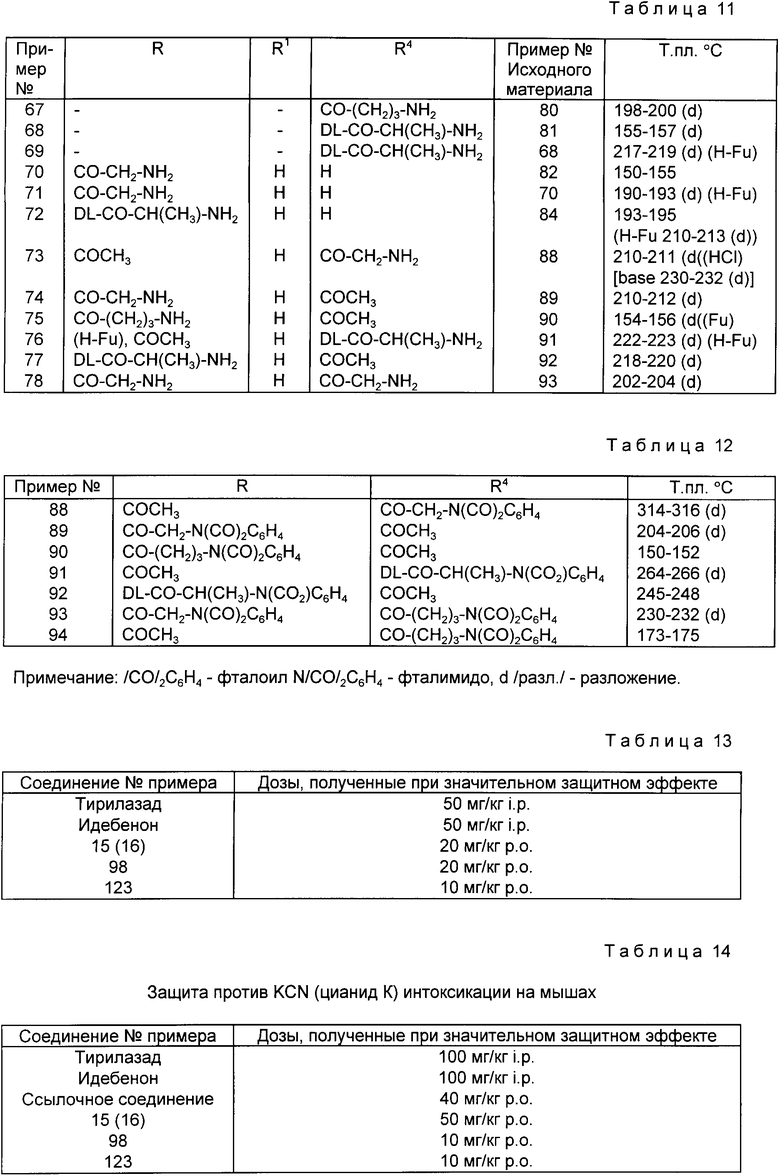
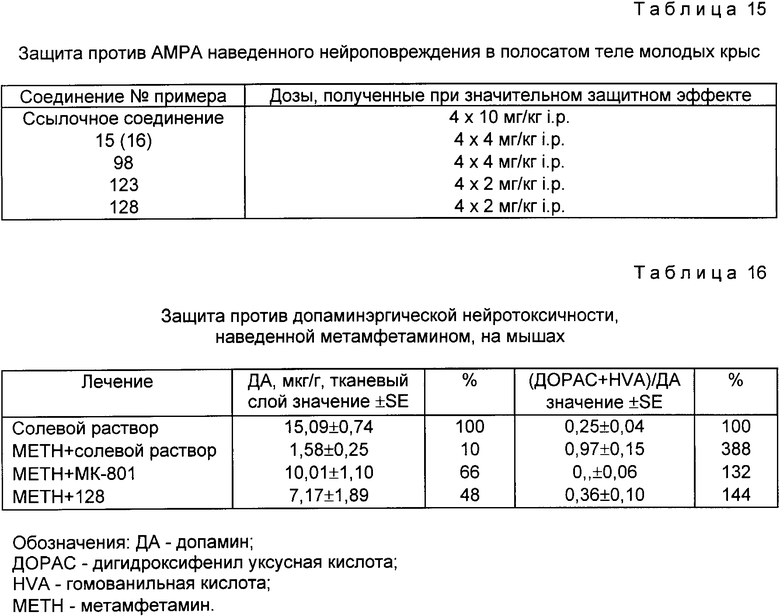
Производные N-ацил-2,3 -бензодиазепина или их стереоизомеры или кислые соли присоединения, обладающие биологической активностью, связанной с воздействием на центральную нервную систему, и фармакологически активная композиция на их основе. Сущность изобретения: производные N-ацил-2,3 -бензодиазепина общей формулы I, указанной в описании, где R - C1-6 алифатическая ацильная группа, незамещенная или многократно замещенная галоидом, C1-5 алкилкарбамоил, бензоилрадикал или R отсутствует, когда между N(3) и C(4) атомами существует двойная связь; R1 - водород или R1 отсутствует, когда между N(3) и C(4) атомами существует двойная связь; R2 - C1-3 алкил; R3 - водород или C1-4 алифатическая ацильная группа; R4 - водород, C1-4 алифатическая ацильная группа, незамещенная или замещенная амино, ди C1-4 алкиламино, пирролидино или многократно замещенная галоидом; пунктирные линии представляют двойную связь, возможно присутствующую в молекуле при условии, что когда оба радикала R3 и R4 представляют атом водорода, то двойная связь между N(3) и C(4) отсутствует. Соединения получают, например, путем ацилирования соответствующего N-незамещенного 2,3 -бензодиазепина. 2 с. и 1 з.п. ф-лы, 16 табл.
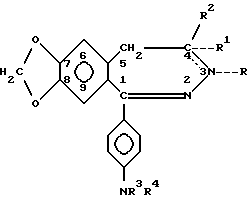
где R С1 С6-алифатическая ацильная группа, незамещенная или многократно замещенная галоидом, С1 С5-алкилкарбамоил, бензоилрадикал или R отсутствует, когда между N(3) и С(4) атомами существует двойная связь;
R1 водород или R1 отсутствует, когда между N(3) и С(4) атомами существует двойная связь;
R2 С3 С3-алкил;
R3 водород и С1 С4-алифатическая ацильная группа;
R4 водород, С1 С4-алифатическая ацильная группа, незамещенная или замещенная амино, ди-С1 С4-алкиламино, пирролидино или многократно замещенная галоидом;
пунктирные линии представляют двойную связь, возможно присутствующую в молекуле, при условии, что когда оба радикала R3 и R4 представляют атом водорода, то двойная связь между N(3) и С(4) отсутствует,
или их стереоизомеры, или кислые соли присоединения, обладающие биологической активностью, связанной с воздействием на центральную нервную систему.
1-(4-аминофенил)-3-ацетил-4-метил-7,8-метилендиокси -3,4-дигидро-5Н-2,3-бензодиазепина,
1-(4-аминофенил) -3-пропионил-4-метил-7,8-метилендиокси- 3,4-дигидро-5Н -2,3-бензодиазепина,
1-(4-ацетиламинофенил)-3-ацетил-4-метил -7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина,
1-(4-пропиониламинофенил)-3-пропионил-4-метил-7,8-метилендиокси-3,4 -дигидро-5Н-2,3-бензодиазепина,
1-(4-пропиониламинофенил) -3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина,
1-(4-ацетиламинофенил)-3-пропионил-4-метил-7,8-метилендиокси -3,4-дигидро-5Н-2,3-бензодиазепина,
1-(4-трифторацетиламинофенил) -3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина,
1-(4-глициламинофенил)-3-ацетил-4-метил-7,8-метилендиокси -3,4-дигидро-5Н-2,3-бензодиазепин дигидрохлорида,
N1-[4-/3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3 -бензодиазепин-1-ил/фенил]-N3 метилмочевины,
1-[4-(N,N-диметилглициламино)фенил]-3-ацетил -4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина,
1-[4-(N,N-диэтилглициламино)фенил]-3-ацетил-4-метил-7,8-метилен- диокси-3,4-дигидро-5Н-2,3-бензодиазепина,
1-[4-(1-пирролдиноацетиламино)фенил]-3-ацетил-4-метил-7,8 -метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина и гидрофумаратов этих соединений и
1-(4-глициламинофенил)-3-метилкарбамоил -4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3-бензодиазепина.
| DE, патент, 3209100, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| DE, патент, 3717080, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| GB, патент, 2190677, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| FR, патент, 2568252, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| US, патент, 4164740, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

