Изобретение относится к новым производным 1-[2-(замещенный винил)]-3,4-дигидро-5H-2,3-бензодиазепина, способу его получения, фармацевтическому составу, применению указанных производных бензодиазепина для лечения заболеваний и приготовления фармацевтических составов, пригодных при лечении заболеваний.
Как известно, в литературе описаны производные 3,4-дигидро- 5H-2,3-бензодиазепина, содержащие в качестве заместителей атом водорода, фенил, нафтил, замещенный фенил, фурил или тиенил в 1 положении основной цепи молекулы (HU 168760, HU 198494, HU 206719; заявка на патент Венгрии N 8398/90; Chem. Ber. 95, 2012, 1962; Helv. Chem. Acta 59, 2786, 1976; Synthesis, 1973, 159; Synthesis, 1977, 1; Acta Chim. Hung. 83, 115, 1974; Rec. Trav. Chim. 84, 661, 1965; J. Chem. Soc. Chem. Comm. 1972, 823; Il. Farmaco-Ed. Sc. 40, 942, 1985; Chem. Pharm. Bull, 30, 3764, 1982.
Известное соединение 1-(4-аминофенил)-4-метил-7,8-метилендиокси-5H-2,3-бензодиазепин (которое имеет также название GY-КI-52466) представляет собой антагонист не-NMDA-глутаминовой кислоты, проявляющий спазмолитическую и противоишемическую активность, однако длительность его действия довольно коротка, что является недостатком с точки зрения возможности его терапевтического применения.
Задачей изобретения является получение новых производных 2,3-бензодиазепина, сравнимых с известными бензодиазепинами по действию на центральную нервную систему, однако превосходящих их по длительности воздействия.
Было обнаружено, что соединения в соответствии с изобретением характеризуются следующими признаками.
В соответствии с настоящим изобретением получены новые производные 1-[2-(замещенный винил)]-3,4-дигидро-5H-2,3-бензодиазепина обшей формулы (I)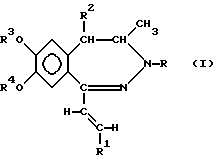
где R представляет собой водород или C1-4 алканоил,
R1 представляет собой фенил, который может содержать от 1 до 3 одинаковых или различных заместителей, выбранных из группы, включающей галоген, нитро-, амино-, C1-4 алкиламино-, ди-(C1-4 алкил)-амино-, C1-4 алканоиламиногруппу, C1-4 алкил, C1-4 алкоксигруппу, метилендиоксил и гидроксил, либо
нафтил, который может содержать заместитель, выбранный из группы, включающей гидроксил, C1-4 алкил и C1-4 алкокси,
R2 представляет собой водород или C1-4 алкил,
R3 и R4 независимо друг от друга представляют собой C1-4 алкил, или
R3 и R4 вместе образуют метилен,
их стереоизомеры и фармацевтически приемлемые соли кислотного присоединения.
Предпочтительными соединениями общей формулы (I) являются такие соединения, в которых R представляет собой C1-4 алканоил, R1 представляет собой фенил или нафтил, содержащий в качестве заместителя C1-4 алканоиламиногруппу или C1-4 алкоксигруппу, R2 представляет собой водород или этил, а R3 и R4 независимо представляют собой C1-4 алкил.
В частности, предпочтительными соединениями в соответствии с изобретением являются следующие производные:
1-(4-ацетиламиностирил)-3-ацетил-4-метил-7,8- диметокси-3,4-дигидро-5Н-2,3-бензодиазепин,
1-[2-(1-нафтил)-винил] -4-метил-7,8-диметокси-3,4-дигидро-5Н -2,3-бензодиазепин,
1-(2,3-диметоксистирил)-3-ацетил-4-метил-7,8-диметокси-3,4-дигидро- 5Н-2,3-бензодиазепин,
их стереоизомеры и фармацевтически приемлемые соли кислотного присоединения.
Употребляемый в описании и формуле изобретения термин "низшие" относится к группам или соединениям с числом атомов углерода от 1 до 4. Термин "алкил" относится к линейным или разветвленным группам, имеющим такое же число атомов углерода, например, таким как метил, этил, н-пропил и т.п. Термин "алкоксигруппа" относится к эфирным группам, содержащим алкилы с линейной или разветвленной цепью, например, таким как метокси-, этокси-, изопропоксигруппа и др. Термин "алканоиламино-" относится к линейным или разветвленным алифатическим карбоксильным кислым амидным группам (например, термин "атом галогена" включает все четыре атома галогена, т.е. фтор, хлор, йод и бром).
Фармацевтически приемлемые соли кислотного присоединения соединений общей формулы (I) могут быть образованы с неорганическими кислотами (например, галоговодородами, такими как хлористый или бромистый водород, серной, фосфорной или пергалоидными кислотами, такими как хлорная кислота), органическими карбоновыми кислотами (например, фумаровой, уксусной, пропионовой, гликолевой, малеиновой, гидроксималеиновой, аскорбиновой, лимонной, яблочной, салициловой, молочной, коричной, бензойной, фенилуксусной, пара-аминобензойной, пара-гидроксибензойной, пара-аминосалициловой кислотой и др.), алкилсульфоновыми кислотами (например, метансульфокислотой, этансульфокислотой) или арилсульфоновыми кислотами (например, пара-толуолсульфоновой, пара-бромфенилсульфоновой, нафтилсульфоновой, сульфанильной кислотами).
В изобретении предложен также способ получения производных 1-[2-(замещенный винил)] -3,4-дигидро-5H-2,3-бензодиазепина общей формулы (I), включающий
а) восстановление 5H-2,3-бензодиазепина общей формулы (II)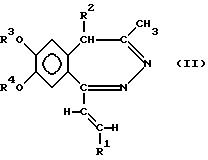
где R1, R2, R3 и R4 как указано выше, комплексным гидридом металла и/или комплексом борана и при необходимости последующее ацилирование полученного таким образом соединения общей формулы (I), где R - водород, a R1, R2, R3 и R4 такие же, как указано выше, или
б) восстановление соединения общей формулы (I), где R1 представляет собой нитрофенил, который может содержать 1 или 2 одинаковых или различных заместителя, выбранных из группы, включающей галоген, нитро-, амино-, метилендиокси-, гидрокси-, C1-4 алкил- и C1-4 алкоксигруппу, a R, R2, R3 и R4 такие же, как указано выше, гидратом гидразина в присутствии катализатора с получением 1-[2-(замещенный винил)]-3,4-дигидро-5H-2,3-бензодиазепина общей формулы (I), где R представляет собой аминофенил, (C1-4 алкил)аминофенил, ди-(C1-4 алкил)-аминофенил или (C1-4 алканоил)-аминофенил, при этом указанные группы могут иметь один или два одинаковых или различных заместителя, выбранных из группы, включающей галоген, нитро-, амино-, метилендиокси-, C1-4 алкил- и C1-4 алкоксигруппу, a R, R2, R3 и R4 - такие же, как указано выше, и при необходимости ацилирование или алкилирование полученного таким образом аминосоединения,
а также при желании растворение полученного таким образом соединения общей формулы (I) или превращение полученного основания общей формулы (I) в фармацевтически приемлемую соль кислотного присоединения.
В соответствии с изобретением вариант (а) способа включает восстановление комплексным гидридом металла и/или комплексом борана 5-H-2,3-бензодиазепина общей формулы (I) и при необходимости ацилирование полученного таким образом соединения общей формулы (I), содержащее в качестве R водород. Для селективного восстановления соединений общей формулы (I) можно использовать следующие восстановительные агенты: борогидрид натрия, гидрид лития-алюминия, боран и комплексы борана. Восстановление предпочтительно проводят в растворителе. В качестве растворителей можно использовать воду, низшие спирты, низшие карбоксильные кислоты, растворители эфирного типа, ароматические углеводороды, хлорированные алифатические углеводороды, пиридин или их смеси. Растворитель или смесь растворителей, которую используют в данном случае, зависит от выбранного восстановительного агента.
Восстановление проводят при температуре от 0 до 100oC с использованием предпочтительно от 1,1 до 25 молярных эквивалентов восстановительного агента.
В соответствии с изобретением предпочтительный пример осуществления варианта (а) способа включает добавление от 1,5 до 2,0 эквивалентов эфирата боротрифторида к раствору или суспензии производного 5H-2,3-бензодиазепина общей формулы (II) в сухом дихлорметане при температуре от 10 до 15oC, добавление к раствору полученного таким образом комплекса 1,1 эквивалента комплекса борана-триметиламина и перемешивание полученной реакционной смеси при 25oC в течение от 0,5 до 4 ч. Органическую фазу затем обрабатывают карбонатом натрия, промывают водой, сушат, выпаривают, кристаллизуют требуемый продукт, фильтруют и при необходимости подвергают перекристаллизации из подходящего растворителя, например низшего спирта, или суспендируют в подходящем растворителе.
В соответствии с изобретением другой предпочтительный пример осуществления варианта (а) способа включает растворение или суспендирование соединения общей формулы (II) в сухом тетрагидрофуране, охлаждение до температуры от 0 до 5oC, добавление 1 моль-эквивалента гидрида лития-алюминия и перемешивание реакционной смеси при комнатной температуре в течение 2 ч. Комплекс затем подвергают разложению и выпаривают органическую фазу. Из остатка получают требуемый 3,4-дигидро-5H-2,3-бензодиазепин либо на хроматографической колонке, либо путем кристаллизации и при желании превращают его в соответствующее ацилпроизводное.
В соответствии с изобретением другой пример осуществления варианта (а) способа включает растворение или суспендирование в метаноле исходного основания общей формулы (II), добавление избытка концентрированной хлористо-водородной или уксусной кислоты и введение борогидрида натрия к полученному таким образом хлориду или ацетату. После обработки реакционной смеси требуемое 3,4-дигидросоединение получают путем кристаллизации и при желании превращают его в соответствующее ацилпроизводное.
Ацилирование можно осуществить известными в литературе методами, предпочтительно, реакцией с галогенидами карбоновых кислот или их ангидридами.
В соответствии с изобретением в примере выполнения варианта (б) способа получают производные 1-[2-(замещенный винил)]-3,4-дигидро -5H-2,3-бензодиазепина общей формулы (I), включающий бензодиазепин общей формулы (I), где R представляет собой аминофенил, моно- или ди-(C1-4 алкил)- аминофенил, который может содержать 1 или 2 одинаковых или различных заместителя, выбранных из группы, включающей галоген, нитро-, метилендиокси-, гидрокси-, C1-4 алкил- и C1-4 алкоксигруппу, путем восстановления соответствующего производного нитрофенила общей формулы (I) гидратом гидразина в присутствии катализатора и при необходимости ацилирования или алкилирования полученного аминосоединения. Для восстановления нитрогруппы используют метод селективного восстановления, который не приводит к насыщению винильной группы. Описание способа восстановления таких соединений в литературе неизвестно. Было обнаружено, что использование гидразингидрата в присутствии катализатора может быть применено для селективного восстановления соединений подобного типа. Известно, что гидразингидрат в присутствии катализатора использовали для превращения в аминосоединения только таких нитросоединений, которые не содержали других групп, способных к восстановлению [Chem. Rev. 65, 52, 1965, J. Am. Chem. Soc. 75, 4334, 1953, Chem. Lett. 1975, 259].
Восстановление предпочтительно осуществляют в присутствии органического растворителя. Предпочтительно можно использовать следующие растворители или их смеси: низшие спирты, диоксан, тетрагидрофуран, бензол, хлороформ, дихлорметан, диметилформамид, диметилсульфоксид и пиридин. Предпочтительно проводить реакцию в избытке 90-100%-ного гидрата гидразина. В качестве катализатора предпочтительно использовать палладий на костяном угле, платину или никель Рэнея. Реакцию проводят при температуре от 0oC до точки кипения растворителя, предпочтительно от +10 до +100oC.
В соответствии с изобретением предпочтительный пример выполнения варианта (б) способа включает суспендирование в метаноле производного 1-нитростирил-5Н-2,3-бензодиазепина общей формулы (II) и осуществление его взаимодействия с 2-4, предпочтительно 3 эквивалентами 98-100%-ного гидразингидрата в присутствии в качестве катализатора никеля Рэнея при комнатной температуре в течение 1-2 ч. Сырой продукт отделяют от реакционной смеси известными методами. Если полученное соединение труднорастворимо в метаноле, так что происходит частичное его отделение, предпочтительно промыть катализатор несколько раз растворителем, в результате чего продукт легко будет растворен (например, хлороформом). Сырой продукт при необходимости очищают путем перекристаллизации или растирания в порошок в растворителе. В качестве растворителя можно использовать спирт, воду или их смеси.
Если желательно провести алкилирование, его осуществляют известными методами, предпочтительно с алкилгалогенидами в индифферентном растворителе в присутствии агента, связывающего кислоту, при температуре от комнатной до точки кипения растворителя. Предпочтительно можно использовать следующие растворители: алифатические спирты, кетоны, нитрилы, тетрагидрофуран, диоксан, диметилформамид или диметилсульфоксид. В качестве агента, связывающего кислоту, предпочтительно используют карбонат или гидрокарбонат щелочного металла или 1-2 эквивалента низшего третичного амина.
Полученные, как указано выше, производные аминостирилбензодиазепина при необходимости ацилируют. Ацилирование проводят с помощью 1-2 эквивалентов галогенангидрида кислоты или ангидрида кислоты. Реакцию предпочтительно проводят в присутствии агента, связывающего кислоту, предпочтительно низшего алифатического третичного амина или в пиридине. Предпочтительно проводить реакцию в растворителе (например, в алифатическом кетоне, нитриле, тетрагидрофуране, диоксане, пиридине), однако реакцию можно провести также без растворителя, в избытке реагента.
Соединения общей формулы (II), использованные в качестве исходных веществ, являются новыми и могут быть получены по способу, описанному в литературе [HU 195788]. Температуры их плавления указаны ниже.
Новые соединения общей формулы (I) в соответствии с изобретением обладают ценными фармацевтическими свойствами, в частности активностью по отношению к центральной нервной системе. Эти соединения образуют сильные связи с центрами, специфичными по отношению к гомофталазинам (2,3-бензодиазепинам) [FEBS Letters 308(2), 215-217, 1992], в результате чего можно предположить, что, принимая во внимание аналогичную абсорбцию и метаболизм 2,3-бензодиазепинов, они будут проявлять значительную активность in vivo по отношению к центральной нервной системе.
Величины Ki, определенные с использованием 5 нМ 3H-гиризопама [1-(3-хлорфенил)-4-метил-7,8-диметокси -5H-2,3- бензодиазепина] представлены в таблице 1. Величину Ki рассчитывали по следующей формуле: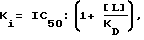
где KD - константа диссоциации меченого комплекса лиганд-рецептор, [L] - концентрация меченого лиганда и IC50 половина максимально ингибирующей концентрации испытуемого образца.
Новые соединения в соответствии с изобретением значительно снижают спонтанную двигательную активность (СДА) мышей после внутрибрюшинного или орального применения.
Ингибирующее действие на СДА, а также величина кратковременного токсического эффекта (умершие/обраб. указаны в скобках) новых соединений приведены в таблице II.
Эксперименты проводили по методу Ирвина [Psychopharmacology, 13, 222, 1968.]
В противоположность известным веществам, молекулы которых имеют аналогичную химическую структуру, соединения в соответствии с изобретением характеризуются значительным потенцированием стереотипии, вызванной амфетамином, проявляя, таким образом, вероятную активность в качестве антидепрессанта. Потенцирование стереотипии, индуцированной введением амфетамина оценивали по шкале Constall'a и Naylor'а [Eur. J. Pharmacol. 18, 95, 1972.]. Результаты представлены в таблице III.
Соединения общей формулы (I) проявляют также умеренную антиконвульсивную активность. Антиконвульсивная активность была измерена на мышах по методу Goodman'a [J. Pharm. Exp. Ther., 106, 319, 1952]. Конвульсии, вызванные введением 50 мг/кг i.v. пентетразола, ингибировали на 30-40% и 50-55% после i. p. применения 30 мг/кг соединений, полученных в примерах 10, 11, 13, 19, 28, 35, 40 и примерах 34 и 38 соответственно.
Действие соединений на glutamatergic передачу исследовали на срезе гиппокампа по методу Tarnawa [Acta Physiol. Hung., 79, 163, 1992]. Были получены срезы гиппокампа мозга крысы толщиной 400 мкм, которые находились в камере мембранного типа в условиях, имитирующих физиологические. Была проведена коллатеральная стимуляция по Schaffer'y и измерены потенциалы полей пирамидальных клеток области CA1 гиппокампа. В качестве нейротрансмиттера в этом процессе участвует глутамат, действующий главным образом посредством АМРА рецепторов. Известный антагонист АМРА, соединение, называемое GYKI-52466, ингибирует с концентрационной зависимостью потенциалы полей CA1. Полученные результаты представлены в таблице IV.
Соединения в примерах 39 и 41 обладают по меньшей мере такой же активностью, как молекулы GYKI-52466. Однако в случае применения последнего соединения ингибирование, наблюдаемое после промывки, длившейся 30 мин, оказалось значительно ниже, чем ингибирование, измеренное после инкубационного периода, длившегося 60 мин, в то время как в случае соединений из примеров 41 и 49 30-минутная промывка не привела к снижению эффекта. Эти результаты показывают, что длительность воздействия последних соединений превосходит длительность воздействия GYKI-52466. Следующие три испытуемые образца показали, что ингибирование, наблюдаемое после промывки, оказалось даже выше ингибирования, измеренного после инкубационного периода.
Ингибирование потенциалов полей гиппокампа подтверждает возможность терапевтического применения новых соединений в качестве антиишемических и нейропротективных средств [Eur. J. Neurosci., 4 (suppl.), 1068, 1991]. Таким образом, результаты проведенных нами экспериментов показали, что длительность воздействия новых соединений превосходит длительность воздействия известных веществ аналогичного действия.
Предлагаемое изобретение включает также фармацевтичесие составы, содержащие в качестве активного компонента соединение общей формулы (I) или его фармацевтически приемлемую соль в смеси с подходящим инертным твердым или жидким фармацевтическим наполнителем.
Фармацевтический состав в соответствии с настоящим изобретением может быть получен известными методами путем смешения активного компонента с подходящим инертным твердым или жидким наполнителем и перевода смеси в галеновую форму.
Фармацевтический состав в соответствии с настоящим изобретением можно применять орально (например, в виде таблеток, гранул, гранул с покрытием, драже, твердых или мягких желатиновых капсул, растворов, эмульсий или суспензий), парентерально (например, в виде раствора для инъекций) или ректально (например, в виде суппозитория).
В качестве наполнителя для приготовления таблеток, таблеток с покрытием, драже и твердых желатиновых капсул можно использовать, например, лактозу, кукурузный и картофельный крахмал, тальк, карбонат магния, стеарат магния, карбонат кальция, стеариновую кислоту или ее соли и т.п. В качестве наполнителя мягких желатиновых капсул можно использовать, например, растительные масла, жиры, воска или многоатомные спирты подходящей консистенции. В качестве наполнителя для растворов и сиропов можно использовать, например, воду, многоатомные спирты (полиэтиленгликоль), сахарозу или глюкозу. Растворы для инъекций могут содержать, например, воду, спирты, многоатомные спирты, глицерол или растительные масла. Суппозитории можно приготовить с помощью, например, масел, восков, жиров или многоатомных спиртов подходящей консистенции.
В дополнение к этому фармацевтические составы могут содержать вспомогательные компоненты, обычно применяемые в фармацевтической промышленности, например смачивающие агенты, подслащающие и ароматизирующие вещества, соли, вызывающие изменение осмотического давления, буферы и т.п. Фармацевтические составы могут содержать также другие активные компоненты.
Суточная доза соединений общей формулы (I) может изменяться в широком интервале в зависимости от нескольких факторов, например активности активного компонента, состояния и возраста пациента, тяжести заболевания и т.п. Предпочтительно оральная доза составляет от 0,1 до 500 мг/сутки. Следует подчеркнуть, что вышеуказанная доза носит справочный характер и применять состав можно только в дозах, назначенных врачом-терапевтом.
Настоящее изобретение включает также использование соединений общей формулы (I) или его фармацевтически приемлемых солей кислотного присоединения для получения фармацевтических составов, воздействующих, в частности, на центральную нервную систему.
Настоящее изобретение включает также способ лечения расстройств центральной нервной системы, включающий назначение пациенту эффективного количества соединения общей формулы (I) или его фармацевтически приемлемой соли кислотного присоединения.
Более подробно изобретение описано в последующих примерах его реализации, которые, однако, не ограничивают область изобретения.
Новые соединения, полученные в соответствии с изобретением, были исследованы методами элементного анализа, ИК-спектроскопии, H-NMR (ядерного магнитного резонанса) и масс-спектроскопии. Протоны насыщенных связей находятся исключительно в транс-положениях.
Пример 1
1-(3,4-диметоксистирил)-4-метил-7,8-метилендиокси- 3,4-дигидро-5H-2,3-бензодиазепин
К раствору 2,04 г (5,6 ммолей) 1-(3,4-диметоксистирил)-4-метил-7,8- метилендиокси-2,3-бензопирила в 40 мл безводного дихлорметана добавили 1,0 мл (8,4 моля) эфирата бортрифторида при охлаждении проточной водой, а затем 0,45 г (6,16 моль) комплекса триметиламина борана. Реакционную смесь перемешивали при температуре 25oC в течение получаса и затем добавили 30 мл 10%-ного водного раствора карбоната натрия по каплям при охлаждении проточной водой, после чего смесь перемешивали в течение 1 ч. Органическую фазу отделили, промыли четыре раза порциями дистиллированной воды по 30 мл, высушили и подвергли выпариванию. Кристаллический осадок суспендировали в 10 мл этанола, отфильтровали, промыли три раза, порциями по 1 мл, этанолом и высушили при 80-100oC. Было получено 1,76 г требуемого продукта. Тпл.: 166-168oC.
Для очистки сырой продукт кипятили в 10 мл этанола, охлаждали, фильтровали, промывали тремя порциями по 1 мл этанола и сушили. В результате было получено 1,69 г (82,4%) конечного продукта. Тпл: 168-170oC.
Другие соединения общей формулы (I), где R представляет собой водород, полученные по способу, описанному в примере 1, представлены в таблице V.
Пример 16
1-(2,3-диметоксистирил)-4-метил-7,8- метилендиокси-3,4-дигидро-5H-2,3-бензодиазепин гидрохлорид
1,5 г (4,12 ммолей) 1-(2,3-диметоксистирил)-4-метил-7,8- метилендиокси-5H-2,3-бензодиазепина восстанавливали, как указано в примере 1, затем остаток после выпаривания растворили в этилацетате и к раствору добавили 10 мл 10%-ного этилацетата, насыщенного газообразным хлоридом водорода. Отделенный продукт отфильтровали, промыли три раза порциями по 5 мл этилацетата и высушили при 80-100oC. Было получено 0,76 г (45,8%) требуемого продукта. Тпл: 193-195oC (разл.).
Пример 17
1-(2,4-диметоксистирил)-4-метил-7,8-метилендиокси-3,4-дигидpo- 5H-2,3-бензодиазепин
К суспензии 1,1 г (3,0 ммолей) 1-(2,4-диметоксистирил)-4-метил- 7,8-метилендиокси-5H-2,3-бензодиазепина в 15 мл безводного тетрагидрофурана охладили до температуры от 0 до 5oC и добавили 0,114 г (3,0 моля) гидрида лития-алюминия. Реакционную смесь перемешивали при температуре 25oC в течение 2 ч, снова охладили до температуры от 0 до 5oC и подвергли разложению путем добавления 0,36 мл 10%-ного водного раствора тартарата калия-натрия. Затем смесь перемешивали в течение 1 ч при 25oC, осадок отфильтровали, фильтрат высушили и выпарили при пониженном давлении. Сырой продукт подвергли перекристаллизации из 10 мл этанола, отфильтровали, промыли три раза порциями по 1 мл этанола и высушили при 80-100oC. Было получено 0,84 г (76%) требуемого продукта. Тпл.: 176-178oC.
Пример 18
1-(2,4-диметоксистирил) -4-метил-7,8-диметокси- 3,4-дигидро-5H-2,3-бензодиазепин
В качестве исходного вещества использовали 1-(2,4-диметоксистирил)-4- метил-7,8-диметокси-5H-2,3-бензодиазепина, которое подвергли операциям, описанным в примере 17, с тем отличием, что сырой продукт, полученный после выпаривания, очищали на хроматографической колонке, в качестве адсорбента использовали Kieselgel 60 с частицами размером 0,063-2 мм, элюент: бензол-метанол-NH4OH (8:2:0,2). После выпаривания фракций был получен конечный продукт в кристаллической форме. Выход 53%. Тпл.: 118-120oC.
Пример 19
1-(2,4-диметоксистирил)-3-ацетил -4-метил-7,8- метилендиокси-3,4-дигидро -5H-2,3-бензодиазепин
Сырой продукт, полученный после выпаривания соединения в примере 17, растворили в 7 мл хлороформа, добавили 0,7 мл уксусного ангидрида и полученную смесь кипятили в течение 2 ч. Затем смесь охладили до комнатной температуры, добавили 10 мл воды и добавили гидрокарбонат натрия до достижения pH 7 - 8. Органическую фазу отделили, водную фазу экстрагировали три раза хлороформом, порциями по 5 мл, экстракты объединили и промыли дважды дистиллированной водой порциями по 10 мл, высушили и выпарили при пониженном давлении. Остаток после выпаривания подвергли перекристаллизации из этанола. Было получено 0,8 г (65%) конечного продукта. Тпл.: 185-187oC.
В примерах 20-23 были получены соединения по способу, описанному в примере 19.
Пример 20
1-стирил-3-ацетил-4-метил-7,8-диметокси-3,4-дигидро-5Н-2,3-бензодиазепин
Выход: 50,0%. Тпл.: 118-120oC.
Пример 21
1-(2,3-диметоксистирил)-3-ацетил-4-метил-7,8-диметокси-3,4-дигидро-5Н- 2,3-бензодиазепин
Выход 56,0%. Тпл.: 85-87oC.
Пример 22
1-(2,4-диметоксистирил)-3-ацетил-4-метил-7,8-диметокси-3,4-дигидро-5Н- 2,3-бензодиазепин
Выход: 58,0%. Тпл.: 72-74oC (эт. сп.).
Пример 23
1-(2,3-диметоксистирил)-3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро- 5Н-2,3-бензодиазепин
Выход: 60,0%. Тпл.: 125-128oC (эт. сп.).
Пример 24
1-(4-нитростирил)-4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3- бензодиазепин
К суспензии 3,6 г (10,3 ммолей) 1-(4-нитростирил)-4-метил- 7,8-метилендиокси-5Н-2,3-бензодиазепина в 130 мл метанола добавили 17,7 мл (0,218 молей) концентрированной соляной кислоты при перемешивании. К полученному раствору через несколько минут добавили 9,8 г (0,259 моль) гидроборида натрия порциями в течение 30 мин, и смесь продолжали перемешивать еще 30 мин. Затем к полученной суспензии оранжевого цвета добавили 150 мл дистиллированной воды по каплям, отфильтровали сырой продукт, промыли четыре раза дистиллированной водой порциями по 20 мл и высушили при температуре от 80 до 100oC. Было получено 3,37 г требуемого продукта. Для очистки сырой продукт кипятили в 17 мл этанола, охлаждали, фильтровали, промывали и сушили. В результате было получено 2,67 г (73,8%) конечного продукта. Тпл.: 175-177oC (разл.).
Соединения в примерах 25-29 могут быть получены по способу, описанному в примере 24.
Пример 25
1-(4-нитростирил) -4-метил-7,8-диметокси-3,4-дигидро-5Н-2,3- бензодиазепин
Выход: 65,0%. Тпл.: 173-175oC (разл.) (эт. сп.).
Пример 26
1-(4-нитростирил) -4-метил-5-этил-7,8-диметокси-3,4-дигидро-5Н-2,3- бензодиазепин
Выход: 64,5%. Тпл.: 168-169oC (разл.) (эт.сп.).
Пример 27
1-стирил-4-метил-5-этил-7,8-метилендиокси-3,4-дигидро-5Н-2,3- бензодиазепин
Выполняют операции, указанные в примере 24, за исключением того, что после добавления борогидрида натрия и завершения реакции смесь выпаривают и сырой продукт, осажденный водой, подвергают перекристаллизации из этанола. Выход: 40,0%. Тпл.: 153-154oC.
Пример 28
1-(3,4-дихлорстирил) -4-метил-7,8-диметокси-3,4-дигидро-5Н-2,3- бензодиазепин
Выполняют операции, как указано в примере 24, и затем реакционную смесь обрабатывают, как указано в примере 27. Выход: 54,0%. Тпл.: 132-133oC (эт. сп.).
Пример 29
1-(3-хлорстирил) -4-метил-7,8-метилендиокси-3,4- дигидро-5Н-2,3-бензодиазепин
Выполняют операции, как указано в примере 24, и затем реакционную смесь обрабатывают, как указано в примере 27. Выход: 40,0%. Тпл.: 114-117oC (эт. сп.).
Пример 30
1-(4-нитростирил)-3-ацетил-4-метил-7,8-диметокси-3,4-дигидро-5Н-2,3- бензодиазепин
2,6 г (7,07 моля) соединения, полученного в примере 25, перемешивали в 13 мл уксусного ангидрида в течение 1 ч при 25oC, добавили 50 мл дистиллированной воды и перемешивали смесь еще 1 ч. Отделенный желтый осадок отфильтровали, промыли трижды дистиллированной водой порциями по 15 мл и высушили при температуре от 80 до 100oC. Было получено 2,68 г сырого продукта, который подвергли перекристаллизации из 13 мл горячего этанола. Было получено 2,62 г (90,6%) конечного продукта в чистом виде. Тпл.: 182-184oC.
В примерах 31-32 были получены соединения по способу, описанному в примере 30.
Пример 31
1-(4-нитростирил)-3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро- 5Н-2,3-бензодиазепин
Выход: 91,0%. Тпл.: 188-190oC (эт. сп.).
Пример 32
1-(4-нитростирил)-3-ацетил-4-метил-5-этил-7,8-диметокси-3,4-дигидро- 5Н-2,3-бензодиазепин
Выход: 88,0%. Тпл.: 184-185oC.
Пример 33
1-(4-аминостирил)-4-метил-7,8-диметокси-3,4-дигидро-5H-2,3-бензодиазепин
6,95 г (18,9 ммолей) 1-(4-нитростирил)-4-метил-7,8-диметокси-3,4- дигидро-5Н-2,3-бензодиазепина, полученного в соответствии с примером 25, суспендировали в 170 мл метанола, добавили 0,7 г сухого (что соответствует 1,4 г влажного) катализатора никель Рэнея и 3,3 мл (66 молей) 100%-ного гидрата гидразина, после чего перемешивали реакционную смесь в течение 1 ч. Был получен раствор, температура которого вначале поднялась до 40-45oC. Катализатор отфильтровали, промыли три раза метанолом порциями по 15 мл, фильтрат выпарили в вакууме, сырой продукт перенесли на фильтр в 80 мл воды, промыли три раза водой порциями по 15 мл и высушили. Было получено 5,46 г требуемого продукта. Для очистки сырой продукт подвергли перекристаллизации из 25 мл 50%-ного этанола. В результате было получено 4,21 г (66,0%) конечного продукта. Тпл.: 152-154oC.
Соединения в примерах 34-38 могут быть получены по способу, описанному в примере 33.
Пример 34
1-(4-аминостирил) -4-метил-7,8-метилендиокси-3,4-дигидро-5Н-2,3- бензодиазепин
Выход: 80,0%. Тпл.: 159-161oC (50% эт. сп.).
Пример 35
1-(4-аминостирил)-4-метил-5-этил-7,8-диметокси-3,4-дигидро-5Н-2,3- бензодиазепин
Выход: 75,5%. Тпл.: 155-158oC (50% эп. сп.).
Пример 36
1-(4-аминостирил)-3-ацетил-4-метил-7,8-диметокси-3,4-дигидро-5Н- 2,3-бензодиазепин
Выполняют операции, указанные в примере 33, за исключением того, что вследствие плохой растворимости исходного вещества и конечного продукта в качестве растворителя использовали смесь дихлорметана и метанола 2:1. Выход: 81,4%. Тпл.: 253-255oC (разл.) (эт.сп.).
Пример 37
1-(4-аминостирил)-3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро- 5Н-2,3-бензодиазепин
Выход: 68,9%. Тпл.: 233-234oC (разл.) (эт. сп.).
Пример 38
1-(4-аминостирил) -3-ацетил-4-метил-5-этил -7,8-диметокси-3,4-дигидро- 5Н-2,3-бензодиазепин
Выход: 77,1%. Тпл.: 104-106oC (эт.сп.).
Пример 39
1-(4-ацетиламиностирил)-3-ацетил-4-метил-7,8-диметокси-3,4-дигидpo- 5H-2,3-бензодиазепин
1,2 г (3,56 ммолей) 1-(4-аминостирил)-4-метил-7,8-диметокси-3,4- дигидро-5Н-2,3-бензодиазепина, полученного в соответствии с примером 33, суспендировали в 6 мл уксусного ангидрида. Суспензию перемешивали в течение 1 ч при комнатной температуре. За это время исходное вещество растворилось, началось отделение конечного продукта и загущение реакционной смеси. Отделенный продукт отфильтровали, промыли три раза диэтиловым эфиром порциями по 15 мл и высушили при температуре от 80 до 100oC. Было получено 1,07 г (71,3%) конечного продукта. Тпл: 243-246oC (разл.).
Используя в качестве исходного вещества соединение, полученное в примере 36, и выполняя операции примера 39, можно получить конечное вещество с выходом 78%.
Соединения в примерах 40-41 могут быть получены по способу, описанному в примере 39.
Пример 40
1-(4-ацетиламиностирил) -3-ацетил-4-метил-7,8-метилендиокси-3,4-дигидро- 5Н-2,3-бензодиазепин
Выход: 91,0%. Тпл.: 252-255oC (разл.).
Пример 41
1-(4-ацетиламиностирил)-3-ацетил-4-метил-5-этил-7,8-метилендиокси-3,4- дигидро-5Н-2,3-бензодиазепин
Выход: 73,5%. Тпл.: 137-140oC (эт. сп.).
Пример 42
1-[2-(1-нафтил)-винил] -4-метил-7,8-диметокси-3,4-дигидро-5Н-2,3- бензодиазепин
К раствору 5,35 г (14,3 ммолей) 1-[2-(1-нафтил)-винил]-4-метил-7,8- диметокси-5Н-2,3-бензодиазепина в 30 мл ледяной уксусной кислоты добавили раствор 1,76 г (46,3 ммолей) борогидрида натрия в 10 мл воды по каплям при 50oC при перемешивании и затем перемешивали реакционную смесь еще 2 ч. Продукт затем размешали в 250 мл воды, добавили раствор гидроксида аммония для получения щелочной реакции, отфильтровали полученный желтый осадок, промыли водой и подвергли перекристаллизации из изопропанола. В результате было получено 3,75 г (70,5%) конечного продукта.
Тпл.: 148-152oC.
В таблице VI представлены новые исходные вещества для получения соединений в соответствии с приведенными примерами.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1-[2-(ЗАМЕЩЕННЫЙ ВИНИЛ)]-5Н-2,3-БЕНЗОДИАЗЕПИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ И СПОСОБ ЕГО ПОЛУЧЕНИЯ, СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | 1996 |
|
RU2161607C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 2,3-БЕНЗОДИАЗЕПИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2243228C2 |
| ПРОИЗВОДНЫЕ N-АЦИЛ-2,3-БЕНЗОДИАЗЕПИНА, ИЛИ ИХ СТЕРЕОИЗОМЕРЫ, ИЛИ КИСЛЫЕ СОЛИ ПРИСОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ БИОЛОГИЧЕСКОЙ АКТИВНОСТЬЮ, СВЯЗАННОЙ С ВОЗДЕЙСТВИЕМ НА ЦЕНТРАЛЬНУЮ НЕРВНУЮ СИСТЕМУ, И ФАРМАКОЛОГИЧЕСКИ АКТИВНАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1991 |
|
RU2102387C1 |
| 3-ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 3Н-2,3-БЕНЗОДИАЗЕПИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2146678C1 |
| ДИФФУЗИОННО-ОСМОТИЧЕСКАЯ ФАРМАЦЕВТИЧЕСКАЯ СИСТЕМА С РЕГУЛИРУЕМЫМ ВЫДЕЛЕНИЕМ ЛЕКАРСТВА (ВАРИАНТЫ) И СПОСОБ ЕЕ ИЗГОТОВЛЕНИЯ (ВАРИАНТЫ) | 1994 |
|
RU2133605C1 |
| ПРОИЗВОДНОЕ 2-(1,2,4-ТРИАЗОЛ-1-ИЛ)-1,3,4-ТИАДИАЗОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1998 |
|
RU2180903C2 |
| Способ получения производных 1-(гидроксистирил)-5Н-2,3-бензодиазепина | 1987 |
|
SU1503681A3 |
| ПРОИЗВОДНЫЕ 5-(ЗАМЕЩЕННЫЙ АМИНО)-1,2,4-ТРИАЗОЛ-[1,5А]-ПИРИМИДИНА, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ КАРДИОТОНИЧЕСКИМ И КОРОНАРОРАСШИРЯЮЩИМ ДЕЙСТВИЕМ, И СПОСОБ КАРДИОТОНИЧЕСКОГО И/ИЛИ КОРОНАРОРАСШИРЯЮЩЕГОСЯ ВОЗДЕЙСТВИЯ НА СЕРДЦЕ | 1992 |
|
RU2097382C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ДИГИДРО-2,3-БЕНЗОДИАЗЕПИНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1995 |
|
RU2151149C1 |
| Способ получения производных 5Н-2,3-бензодиазепина или их солей присоединения кислот | 1979 |
|
SU1402258A3 |






Описываются новые производные 1-[2-(замещенный винил)]-3,4-дигидро-5Н-2,3-бензодиазепина общей формулы I, где R представляет собой водород или C1-4 алканоил; R1 представляет собой фенил, который может содержать от 1 до 3 одинаковых или различных заместителей, выбранных из группы, включающей галоген, нитро-, амино-, ди-(C1-4 алкил)-амино-, C1-4 алканоиламиногруппу, C1-4 алкил, C1-4 алкоксигруппу, метилендиоксил и гидроксил, либо нафтил; R2 представляет собой водород или C1-4 алкил; R3 и R4 независимо друг от друга представляют собой C1-4 алкил или R3 и R4 вместе образуют метилен, и их фармацевтически приемлемые соли, полученные присоединением кислоты. Новые соединения образуют сильные связи с центрами, специфичными по отношению к гомофталазинам, следовательно, будут проявлять значительную активность in vivo по отношению к центральной нервной системе. Описывается также способ их получения, фармацевтический состав и способ его получения, а также способ ингибирования конвульсий. 7 c. и 5 з.п. ф-лы, 6 табл.
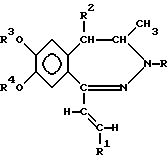

где R представляет собой водород или C1-4 алканоил;
R1 представляет собой фенил, который может содержать от 1 до 3 одинаковых или различных заместителей, выбранных из группы, включающей галоген, нитро-, амино-, ди(C1-4 алкил)-амино-, C1-4 алканоиламиногруппу, C1-4 алкил, C1-4 алкоксигруппу, метилендиоксил и гидроксил, либо нафтил;
R2 представляет собой водород или C1-4 алкил;
R3 и R4 независимо друг от друга представляют собой C1-4 алкил или
R3 и R4 вместе образуют метилен,
и их фармацевтически приемлемые соли, полученные присоединением кислоты.
1-(4-ацетиламиностирил)-3-ацетил-4-метил-7,8-диметокси-3,4-дигидро-5H-2,3-бензодиазепин,
1-[2-(1-нафтил)-винил] -4-метил-7,8-диметокси-3,4-дигидро-5H-2,3-бензодиазепин,
1-(2,3-диметоксистирил)-3-ацетил-4-метил-7,8-диметокси-3,4-дигидро-5H-2,3-бензодиазепин,
и их фармацевтически приемлемые соли, полученные присоединением кислоты.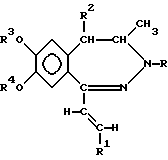
где R, R1, R2, R3 и R4 такие же, как указано в п.1,
включающий восстановление 5H-2,3-бензодиазепина общей формулы II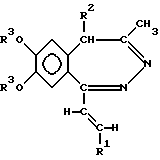
где R, R1, R2, R3 и R4 - такие же, как указано выше,
комплексным гидридом металла и/или комплексом борана и, при необходимости, последующее ацилирование полученного таким образом соединения общей формулы I, где R - водород, а R1, R2, R3 и R4 такие же, как указано выше, а также, при желании, растворение полученного таким образом соединения общей формулы I или превращение полученного основания общей формулы I в фармацевтически приемлемую соль, полученную присоединением кислоты.
1-(4-ацетиламиностирил)-3-ацетил-4-метил-7,8-диметокси-3,4-дигидро-5H-2,3-бензодиазепин,
1-[2-(1-нафтил)-винил] -4-метил-7,8-диметокси-3,4-дигидро-5H-2,3-бензодиазепин,
1-(2,3-диметоксистирил)-3-ацетил-4-метил-7,8-диметокси-3,4-дигидро-5H-2,3-бензодиазепин,
или их фармацевтически приемлемые соли, полученные присоединением кислоты.
| Способ получения производных 3,4-дигидро-5 @ -2,3-бензодиазепина | 1982 |
|
SU1151206A3 |
| Способ получения производных 1-(гидроксистирил)-5Н-2,3-бензодиазепина | 1987 |
|
SU1503681A3 |
| ВСЕСОЮЗНАЯ I | 0 |
|
SU362013A1 |
| Вещество,обладающее транквилизирующей снотворной и противосудорожной активностью | 1974 |
|
SU484873A1 |
| 1-(Гидразинокарбонил)алкил-1,2-дигидро-3Н-1,4-бенздиазепин-2-оны, обладающие транквилизирующими и противосудорожными свойствами | 1980 |
|
SU953819A1 |
| US 4734412, 1988 | |||
| СПОСОБ УПРАВЛЕНИЯ РАБОТОЙ ДВИГАТЕЛЯ ВНУТРЕННЕГО СГОРАНИЯ И ДВИГАТЕЛЬ ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2004 |
|
RU2264545C1 |
