Изобретение относится к цефалоспоринацетилгидролазному гену, белку, содержащему аминокислотную последовательность, кодируемую указанным геном и представляющему собой мультимер, предпочтительно тетрамер или октамер, а также к методу получения указанного белка.
Цефалоспорины, такие как цефалоспорин С и 7-аминоцефалоспорановая кислота (далее обозначаемая как 7-АСА), до настоящего времени получали элиминированием ацетогрупп, связанных с гидроксиметильной группой в ее 3-позиции (далее наз. деацетилирование) с целью получения деацетилированных соединений, таких как деацетилцефалоспорин С или деацетил 7-АСА, которые используют как исходный материал для синтеза различных цефалоспориновых антибиотиков, включая те, которые уже пущены в продажу.
Для деацетилирования цефалоспоринов существуют химические и ферментативные методы. Из этих методов последний считается предпочтительным, так как может осуществляться при примерно нейтральных pH и мягкой температуре и частично потому, что протекает без побочных реакций. Описано несколько ферментативных методов (например, патент Японии N 108, 790/1984, 132, 194/1974 и 67, 489/1986 и патент США N 3304236).
Заявители установили, что штамм Bacillus subtilis, выделенный из почвы, производит цефалоспоринацетилгидролазу и эффективно продуцирует деацетилцефалоспорины деацетилированием цефалоспоринов. Это открытие привело исследователей к мысли, что микроорганизмы, приобретающие способность производить цефалоспоринацетилгидролазу с помощью метода рекомбинирования ДНК, обладали бы большими преимуществами для промышленного получения деацетилцефалоспоринов. Такой микроорганизм, который производил бы только цефалоспоринацетилгидролизу, до сих пор не известен.
Изобретатели провели обширные исследования и добились успеха в выделении ДНК-фрагмента, несущего цефалоспоринацетилгидролазный ген из штамма B. subtilis, выделенного из почвы, а также в клонировании указанного ДНК фрагмента в E. coli. Они также установили, что штамм E. coli, трансформированный плазмидным вектором, в который был включен указанный ДНК-фрагмент, производит значительное количество цефалоспоринацетилгидролазы. Изобретение было сделано на основе этих открытий.
Таким образом, изобретение включает в себя цефалоспоринацетилгидролазный ген, белок, имеющий аминокислотную последовательность, кодируемую указанным геном, и метод получения указанного фермента с помощью рекомбинантных ДНК.
Недавно выделенный штамм B. subtilis SHS 0133, из которого в соответствии с изобретением получают цефалоспоринацетилгидролазный ген, был депонирован в Fermentation Research Institute, Agency of Industrial seience and Technology, 1-3, Higashi 1 chome, Tsukubashi, Ibakari-ken, 305, Japan under Budapest Treaty с номером FERM ВР-2755 (дата: 1990, февраль 15).
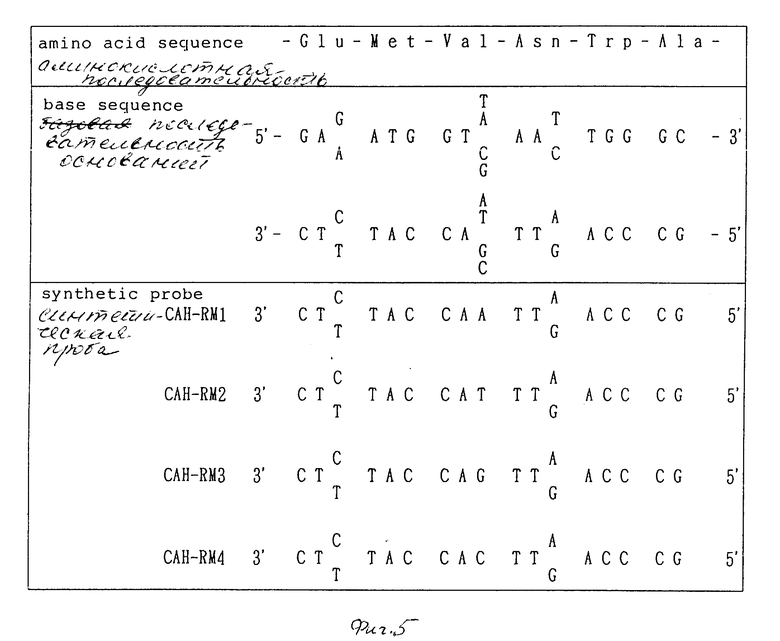
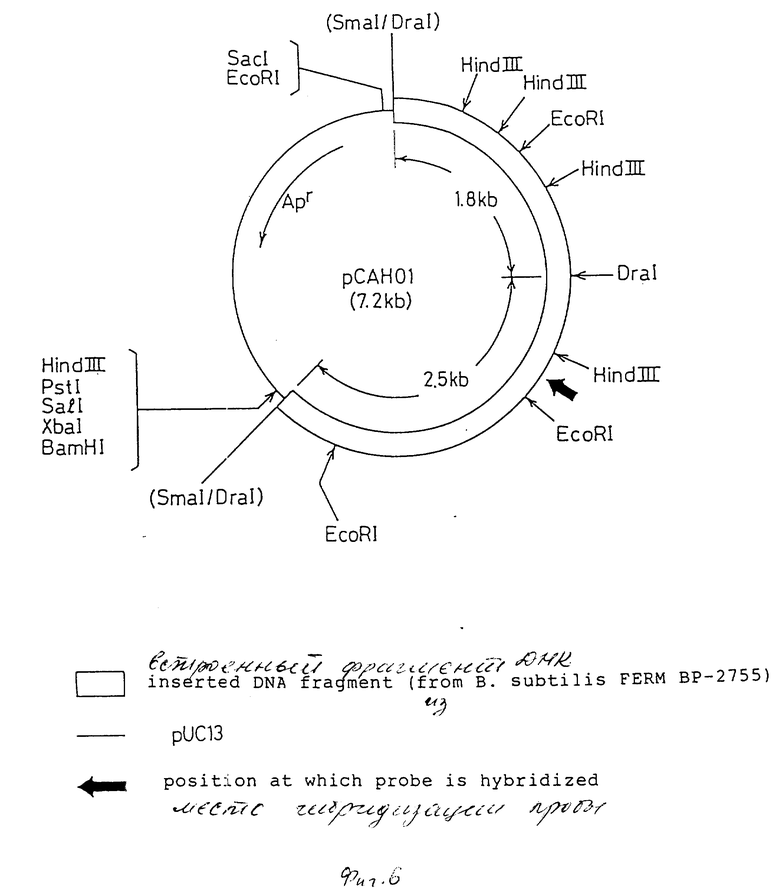
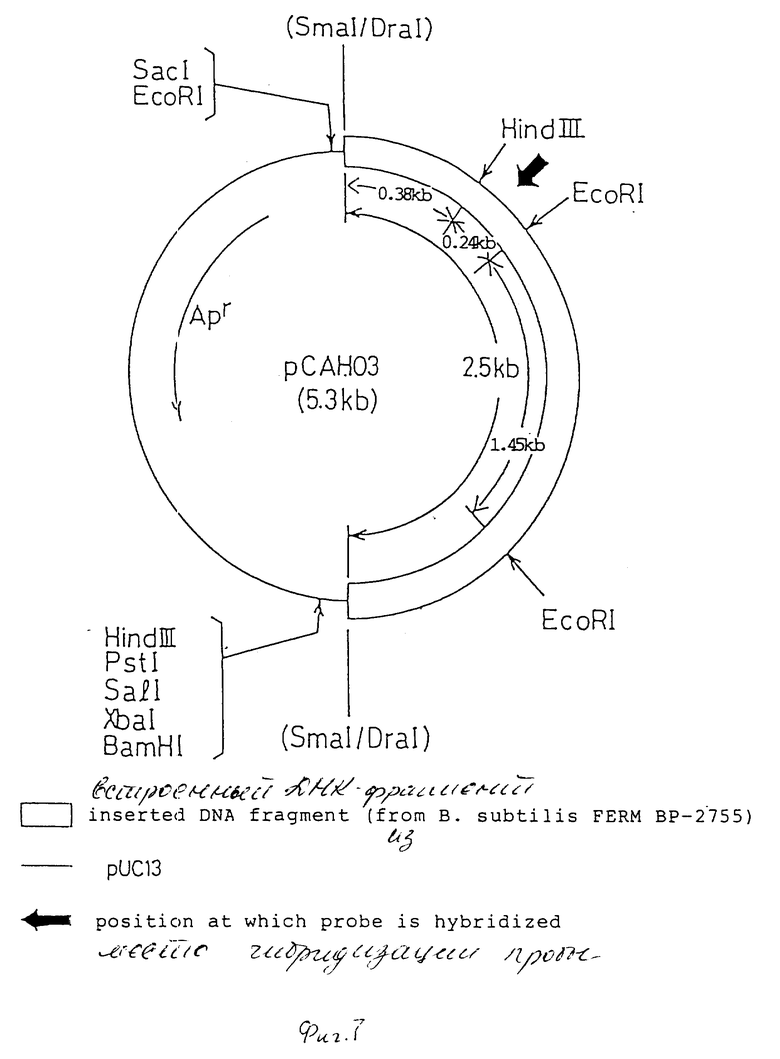
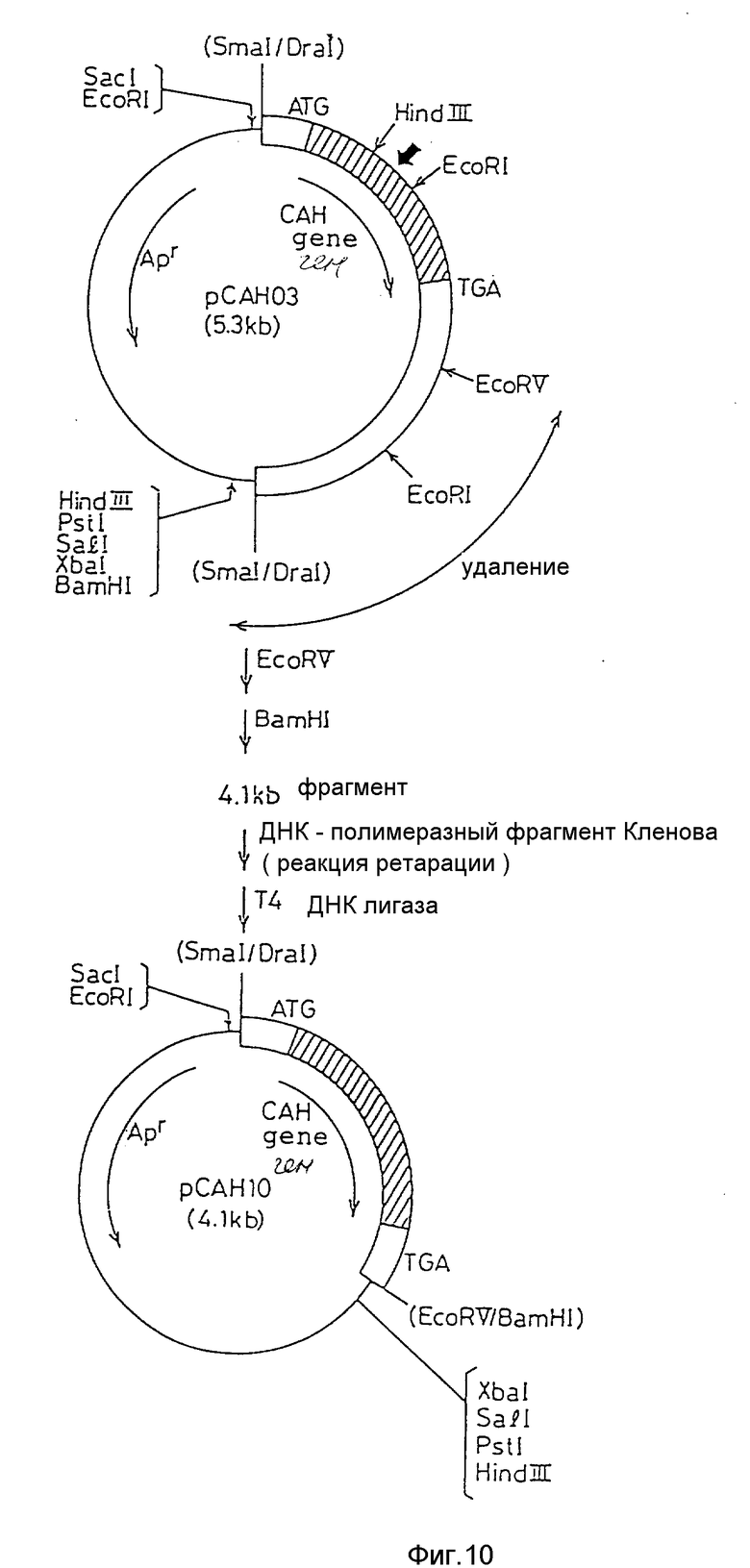
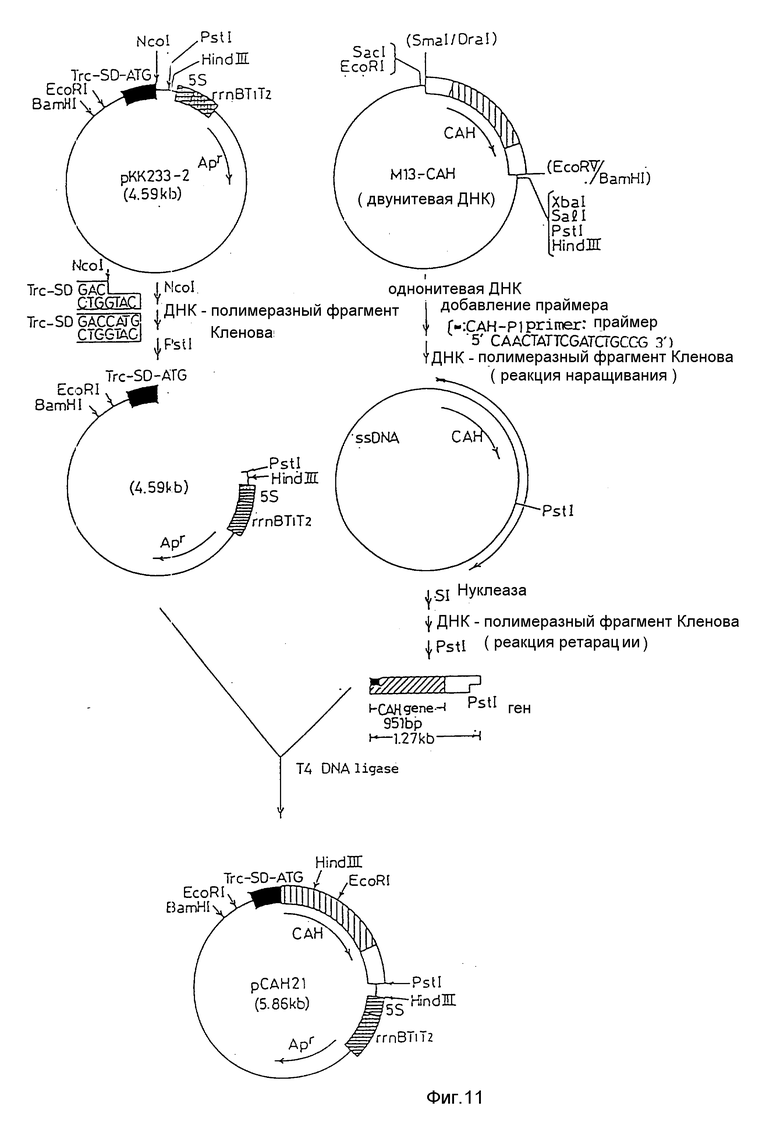
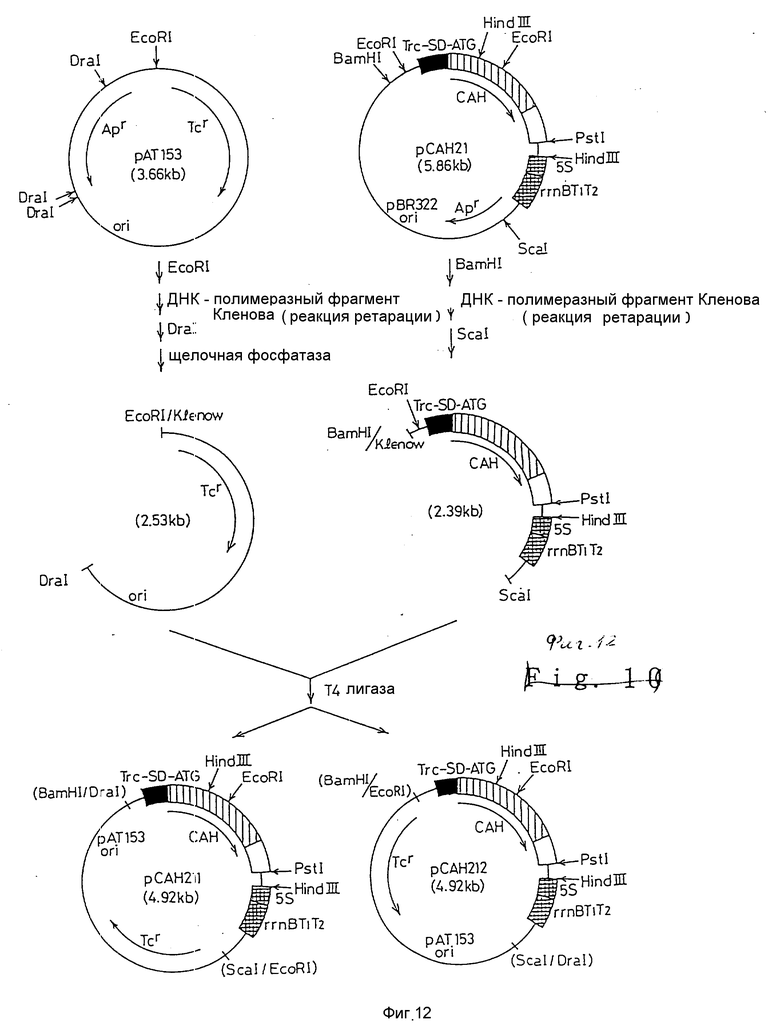
На фиг. 1 дано изображение аминокислотной последовательности цефалоспоринацетилгидролазы; на фиг. 2 и 3 - изображение основной последовательности цефалоспоринацетилгидролазного гена (нижний ряд) и аминокислотной последовательности, установленной на ее основе (верхний ряд); на фиг. 4 - изображение частично установленной аминокислотной последовательности (34 аминокислотных остатка) цефалоспоринацетилгидролазы; на фиг. 5 - изображение четырех синтезированных проб, включая все генетически возможные последовательности ДНК оснований, установленных на основе аминокислотной последовательности, приведенной на фиг. 4; на фиг. 6 - изображение карты рестриктазного расщепления рекомбинантной плазмиды pCAHOI и позиции, с которой гибридизируется ДНК проба; на фиг. 7 - изображение карты рестриктазного расщепления рекомбинантной плазмиды pCOH03; на фиг. 8 и 9 - изображение ДНК последовательности клонированного цефалоспоринацетилгидролазного гена и примыкающих областей; на фиг. 10 - изображение строения миниатюризированной плазмиды pCAHIO, которая соответствует плазмиде pCOH03, но в которой удален участок ниже цефалоспоринацетилгидролазного гена; на фиг. 11 - изображение строения экспрессионной плазмиды рСАН21, использованной в E. coli; на фиг. 12 - изображение строения экспрессионных плазмид рСАН211 и рСАН212.
Характеристики B. subtilis SHS 0133.
1. C+C мольный процент (мол.%) хромосомной ДНК: 43,4.
2. Морфологические характеристики.
Штамм является грамм-положительным, с короткими палочковидными бактериями размером 0,7 - 0,8 х 1,8 - 2,6 мкм и перитрихиозным. Этот штамм хорошо растет в аэробных условия и слабо растет в природной среде, содержащей глюкозу в анаэробных условиях. Спора размером 5,8 х 1,3 - 1,7 мкм формируется в центральной части клетки.
3. Культуральные характеристики.
(1) Культура на агаровой пластинке с мясным экстрактом (при З0oC в течение 7 дн).
Образование колоний: 24 ч после прививки
Форма: нерегулярная
Поверхность: волнообразная
Высота: выпуклая
Цвет: кремовый
Блеск: тусклый
Оптические свойства: непрозрачный
(2) Косая агаровая культура на мясном экстракте (при 30oC, в течение 7 дн).
Рост: хороший
Форма: нитевидная
Поверхность: бороздчатая
Цвет: кремовый
Блеск: тусклый
Оптические свойства: непрозрачный
Консистенция: маслянистая
(3) Бульонная культура на мясном экстракте (при 30oC в течение 7 дн).
Рост на поверхности: образуется толстая пленка
Мутность: легкая
Запах: легкий
Осадок: видимый
Количество осадка: скудный
(4) Культура с мясным экстрактом на насеченном желатине (при 24oC в течение ЗО дн).
Рост: растет однородно вдоль насеченных линий
Линия прокола: нитевидная
Выделение влаги: снижается на 7 днь при 24oC с образованием 0,5 мм слоев жидкости (стратообразование)
(5) Лакмусовая молочная культура:
Лакмус уменьшается. Казеин быстро усваивается без коагуляции.
4. Биохимические характеристики.
(1) Уменьшение нитратов: положительное
(2) Денитрификация: положительная
(3) MR тест: отрицательный
(4) YF тест: положительный
(5) Образование индола: отрицательное
(6) Образование H2: бумага с ацетатом свинца, отрицательное
ТSI: отрицательное
Krigler отрицательное
(7) Гидролиз крахмала: положительный
(8) Утилизация цитратов:
Koser - Положительное
Christensen - Положительное
(9) Утилизация неорганических источников азота:
Нитраты - Положительное
Аммиак - Положительное
(10) Образование пигмента: образуется коричневый пигмент
(11) Тест на мочевину: положительный
(12) Тест на оксидазу: положительный
(13) Тест на каталазу: положительный
(14) pH: область роста: 1,5 - 8,8
Оптимум - 6,0 - 8,8
(15) Температура: область роста: 16,5-50,5С
Оптимум - 26,0-36,0 С
(16) OF тест:
D - глюкоза: получение кислот ферментативно без образования газов
Лактоза: получение кислот ферментативно без образования газов
(17) Образование кислот и газа из сахара:
i) образуются кислоты и не образуется газ из D -арабинозы, D -ксилозы, D -манозы, D -фруктозы, мальтозы, сахарозы, трегалозы, D -маннитол, глицерол и крахмал;
ii) ни кислоты, ни газы нe образуются из D -галактозы, лактозы, D -сорбитола и инозитола.
(18) Нитритные требования: нет
(19) Деградация пектина: положительная
(20) Деградация гиппуровой кислоты: отрицательная
(21) Образование левана (сахарозы, рафинозы): положительное
(22) Аргинин гидролаза: отрицательная
(23) Лецитиназа: отрицательная
(24) Рост в анаэробных условиях: слабо растет в природной среде, содержащей D-глюкозу.
Исходя из приведенных выше результатов штамм идентифицировали как штамм B. subtilis на основании Bergey's Manual of Sistematie Bactereology, vol. 11, 1986, и обозначили как Bacillus subtilis SHS 0133.
Изобретение включает в себя процесс получения цефалоспоринацетилгидролазы с использованием рекомбинантного микроорганизма, трансформированного с помощью рекомбинантной ДНК, полученной при введении в вектор, используемый в E. coli последовательности ДНК, кодирующей аминокислотную последовательность, изображенную на фиг. 1, предпочтительно последовательности ДНК, изображенной на фиг. 2 и 3. Этот метод может быть реализован посредством 7 последовательных шагов, которые представляют историю развития изобретения, хотя при этом могут быть использованы более простые методики, потому что изобретением предлагается последовательность ДНК, кодирующая цефалоспоринацетилгидролазу.
(1) Bac. subtilis (FERM BP-2755) культивируют в подходящей среде и получают цефалоспоринацетилгидролазу, полученную цефалоспоринацетилгидролазу выделяют из среды и очищают. Очищенный фермент переваривается подходящей протеазой, результирующие пептидные фрагменты выделяют, а затем аминокислотную последовательность одного из пептидных фрагментов определяют на основании N-конца.
(2) Возможные последовательности ДНК, соответствующие части определенной аминокислотной последовательности, выводили и химически синтезировали олигонуклеотиды, имеющие установленную последовательность оснований, вводя в 5'-конец олигонуклеотидов 32p. Меченные олигонуклеотиды использовали как пробы для клонирования генов.
(3) Хромосомальные ДНК экстрагировали, очищали от B.subtilis, обрабатывали различными рестриктазами, подвергали электрофорезу на агарозном геле и переносили ДНК-фрагменты на нитроцеллюлозную мембрану. Затем проводили гибридизацию, используя нитроцеллюлазную мембрану и 32p - меченные пробы, полученные как было описано в шаге (2). Для того, чтобы выбрать ДНК фрагменты, проявляющие гомологичность к пробам, из ДНК фрагментов выбирали наибольшие по сравнению с размером желаемого гена, исходя из молекулярного веса белка цефалоспоринацетилгидролазы, вырезали релевантную область агарозного геля, содержащего ДНК фрагмент, и экстрагировали ДНК.
(4) ДНК фрагмент из шага (3) вставляли в клонирующий вектор E. coli и результирующую плазмиду переносили в клетки. Полученные трансформанты помещали в агаровую среду для образования колоний и проводили гибридизацию клетки с использованием 32р - меченных проб. Колонии E. coli, показавшие гомологичность с пробой, отбирали и выделяли.
(5) Из отобранных клеток экстрагировали рекомбинантную плазмидную ДНК и составляли рестрикционную карту фрагментов. Определяли последовательность оснований во фрагментах, кодирующих цефалоспоринацетилгидролазу B. subtilis. Аминокислотную последовательность, вычисленную из определенной ДНК последовательности, затем сравнивали с частично определенной ранее аминокислотной последовательностью, молекулярным весом, данными аминокислотного анализа концов и аминокислотного состава цефалоспоринацетилгидролазы, а затем определяли структуру гена цефалоспоринацетилгидролазы.
(6) ДНК-фрагмент, содержащий цефалоспоринацетилгидролазный ген, модифицировали и вводили его в экспрессионный вектор для E. coli таким образом, что структурный ген был оперативно связан с регуляторными структурами рекомбинантной плазмиды.
(7) Рекомбинантная экспрессионная плазмида, полученная выше, была введена в клетки E. coli с получением нового штамма E. coli, производящего цефалоспоринацетилгидролазу.
Описанная выше методика может быть известна из литературных данных и легко осуществлена в соответствии с экспериментальным протоколом, раскрытым в стандартных учебниках, таких как "Молекулярное клонирование", Maniatis etal. , Cold Spring Harbor Laboratory. Все использованные материалы, такие как ферменты и реагенты, являются коммерчески доступными и могут быть использованы в соответствии с прилагаемыми инструкциями.
Клетками E. coli, используемыми в качестве хозяйских, может быть штамм E. coli K-12 и его производные, такие как HB101, DHI, С600, JM103, JM105 и JM109. В качестве вектора E. coli, используемого в клонировании, могут применяться такие плазмиды, как pUС13, рВРЭ22 и рАТ153, а также фаг-вектор λgt10.
Вышеописанные хозяева и векторы являются коммерчески доступными и легко получаемыми.
В описанном выше шаге (1) аминокислотная последовательность белков является известной (например, коммерчески доступная автоматизированная аминокислотная последовательность может быть использована). В вышеописанном шаге (2) синтез олигомеров можно проводить, используя коммерчески доступный ДНК-синтезатор, соответствующий прилагаемому протоколу. В соответствии с шагом (5) определение последовательности ДНК можно осуществлять в соответствии с методом Sanger и др. Proc. Natl Acad. Sci. USA. 74, 5463-5467 (1977), где используется известная векторная система М13.
В представленном выше шаге (6) структура плазмиды, необходимая для того, чтобы осуществить эффективную экспрессию гена в E. coli, может быть проведена с помощью вставки ДНК фрагмента, содержащего желаемый цефалоспоринацетилгидролазный ген в экспрессионный вектор (рКК223-3, рBS, рDР540, pPL - лямбда и т.д.), содержащий промотор (Lас,Тас, Тчс, Тrp, РL и т.д.), функциональный в хозяйской клетке и последовательности Shine Dalgarno (SD) или в АТG - вектор (рКК233-2 и т.д.), который дополнительно содержит трансляционный инициатор - кодон АТG. Введение экспрессионной плазмиды в подходящего хозяина (например, такого, как штамм E. coli J. H103, JM103, JM109, HB101 и С600) приводит к получению микроорганизма, экспрессирующего цефалоспоринацетилгидролазу.
Экспрессированная цефалоспоринацетилгидролаза может быть очищена с помощью подходящих методов очистки, например с помощью комбинированного центрифугирования, колоночной хроматографии и им подобным.
Изобретение иллюстрируется далее следующими примерами, но не ограничивается ими.
Пример. Отделение и очистка цефалоспоринацетилгидролазы и определение частичной аминокислотной последовательности
(1) Отделение и очистка цефалоспоринацетилгидролазы
Среду (20 л), состоящую из 2,5% глюкозы, 0,75% экстракта пшеницы, 1,0% смеси аминокислот, 0,3% KH2PO4 и 0,8 частей на моль MnSO4/4H2O pH 7,0, помещали в 30 л ферментер. После стерилизации B. subtilis (FERM ВР-2755), который был прекультирован в среде, состоящей из 0,5% глюкозы, 0,75% экстракта пшеницы, 0,5% смеси аминокислот и 0,02% KH2PO4, pH 7,0 вливали в среду так, чтобы получился 6%-ный прививочный материал. После 48 ч культивирования при 28oC к культуре добавляли активированный уголь в количестве 1% от жидкости и перемешивали в течение 2 ч. Последующей фильтрацией получали фермент в составе фильтрата. К раствору фермента-сырца добавляли 0,7% DEAE Sephadex А-50 (Pharmacia) и смесь доводили до pH 8,0, используя 2N NAOH, а затем перемешивали в течение часа. После фильтрации с помощью DEAE Sephadex А-50, на котором адсорбировалась активная цефалоспоринацетилгидролаза, адсорбент промывали 50 мм Трис-HCl буфером (pH 8,0) (4 л), содержащим 0,1 М NaCl, и затем активный компонент смывали тем же буфером, содержащим 0,5 М NaCl. После концентрирования и обессоливания ультрафильтрационным методом активный компонент адсорбировали на колонке, заполненной DEAE Sepharose CL-6B (Pharmacia), предварительно уравновешенный тем же буфером. Колонку промывали тем же буфером и последовательно буфером, содержащим 0,15 М NaCl, а активный компонент затем смывали Трис-буфером, содержащим 0,2 М NaCl. После концентрирования и обессоливания ультрафильтрацией проводили очистку с помощью жидкостной хроматографии высокого разрешения (далее обозначаемой как HPLC). В качестве колонки использовали DEAE Toypearlpak 650М (Tosoh). Активный компонент элюировали при помощи возрастающего градиента концентраций соли. Таким образом, исходная концентрация 0,15 М NaCI последовательно возрастала до 0,5 М. Фракции, содержащие смыв активного компонента, собирали, концентрировали ультрафильтрацией и затем очищали с помощью HPIC и колонок с молекулярными ситами. Как колонку использовали TSK -G3000 (Tosoh), а 0,2 М фосфатный буфер (pH 7,0) как подвижную фазу. Элюат активной фракции собирали, конценрировали ультрафильтрацией и затем очищали посредством HPLC, используя колонку с обратной фазой. В качестве колонки использовали Microbondapak C18(Waters), а смыв проводили методом элюции в градиенте концентрации, где концентрация ацетонитрила последовательно возрастала. Таким образом, исходная водная система, содержащая 0,1% трифторуксусной кислоты, а конечная концентрация ацетонитрила возрастала до 98%. Фракции, содержащие элюированную цефалоспоринацетилгидролазу, концентрировали при пониженном давлении при 50oC и осадок растворяли в 0,5 М Триc-HCl буфере (pH 8,0), содержащем 6 М гуанидингидрохлорид. К раствору добавляли EDTA (этилендиаминтетрауксусную кислоту) до получения концентрации 2 мМ и 200-кратное по отношению к ферменту мольное количество 2-меркаптоэтанола и превращение проводили при 37oC в течение 4 ч в атмосфере азота. Затем к цефалоспоринацетилгидролазе добавляли 190-кратное мольное количество иодоацетата натрия и проводили реакцию при 37oC в течение 10 мин в темноте, чтобы снизить карбоксиметилирование. Затем осуществляли очистку с помощью HPIC, используя колонку с обращенной фазой в соответствии с описанным выше методом. Результирующие фракции цефалоспоринацетилгидролазы анализировали с помощью SDS-полиакриламидного гель электрофореза в соответствии с методикой Lacmmli, Nature, 227, 680-685 (1970). Наличие одной полосы, определенное в позиции молекулярного веса 35 KD, предполагает, что очистка прошла гомогенно. Дополнительно, тот факт, что очищенная цефалоспоринацетилгидролаза, растворенная в 0,1 М Трис-HCl буфере (pH 8,0), содержащем 7 М мочевины, взаимодействовала при 10-кратном разбавлении с 0,1 М фосфатным буфером (pH 7,0), и что активный компонент смывался с помощью колоночной хроматографии на молекулярных ситах с молекулярным весом 280 KD предполагает, что цефалоспоринацетилгидролаза в природной форме является октамером, содержащим гомогенные субъединицы.
С другой стороны молекулярный вес, определенный по методике Hedrick и др. Arch Biochem Biophys 126, 155-164 (1968) был около 150 KD, и предполагается, что тетрамер также является активным.
Анализы N-конца очищенной цефалоспоринацетилгидролазы деградацией по Edman и гидролизу гидразина показали, что N-конец должен быть метионином, а C-конец должен быть глицином.
(2) Определение частичной аминокислотной последовательности цефалоспоринацетилгидролазы
Очищенную цефалоспоринацетилгидролазу (1 мг) растворяли в 0,1 М Трис-HCl буфере (pH 8,8) (1,0 мл), содержащем 2 М мочевины, и затем к раствору добавляли лизил-эндопептидазу (0,01 мг) и составляли смесь для проведения реакции при 37oC в течение 4 ч. Реакционную смесь разделяли методом HPIC. В качестве колонки использовали Microbondapak C18 (Waters) и элюирование проводили линейным градиентом концентрации ацетонитрила. Таким образом, начиная с водной системы, содержащей 0,1% трифторуксусной кислоты, концентрация ацетонитрила возрастала до конечной концентрации 98%. Определение проводили при длине волны 214 нм. На основании разделенных пиков определяемые фракции, имеющие большое время задерживания, собирали и снова очищали, используя такие же обращенные колонки, а затем анализировали аминокислотную последовательность белков, используя газофазовое автоматизированное (Applied Biosys tems) определение аминокислотной последовательности амино-концов 34 аминокислотных фрагментов белка, как показано на фиг. 4.
2. Синтез ДНК проб и меченных 5-терминусов
Аминокислотную последовательность, подчеркнутую на фиг. 4 и описанную выше в шаге (1), синтезировали, а также синтезировали олигонуклеотиды, соответствующие всем генетически возможным последовательностям ДНК, вычисленным из аминокислотной последовательности. Как показано на фиг. 5, были синтезированы 4 группы олигонуклеотидов, которые были обозначены как ДНК пробы CAH-RMI, САН-RM2, САН-RM3 и САН-RM4. Синтез олигонуклеотидов проводили, используя автоматический ДНК синтезатор ZEON-GENET А-11 (Nihon Zeon).
5'-конец результирующих ДНК проб метили Т4 -полинуклеотидкиназой и ( γ32 - Р/АТР в соответствии с методикой Wallace и др. Nucleic Acids Res 6, 8543-3557 (1979).
3. Экстракция и очистка хромосомной ДНК из B. subtilis и гибридизация по Southern
Клетки В. subtilis обрабатывали лизоцимом, после чего N-лауроилсаркозинатом натрия, а затем хромосомную ДНК экстрагировали и очищали из результирующего лизата методом ультрацентрифугирования в градиенте плотности Cs - Cl - этидиум бромид по методике Harris-Warrick и др. Proc. Natl. Acad. Sci. USA, 72, 2207-211 (1975). ДНК в подходящих условиях переваривались рестрикционными ферментами (каждый около 10 единиц), и фрагменты подвергались электрофорезу на 0,8% агарозном геле. После анализа гель подвергали гибридизации Southern в соответствии с методом Southern и др. J. Mol. Biol. 98. 503-517 (1975). Гель обрабатывали раствором 1,5 М NaCl, содержащим 0,5 N NAOH при комнатной температуре в течение 1 ч для денатурирования белка, и затем нейтрализовали 1 М Трис-HCl буфером (pH 7,0), содержащим 1,5 М NaCl, при комнатной температуре в течение 1 ч. Затем ДНК с геля переносили на нитроцеллюлозную мембрану. Используя ДНК, перенесенную на нитроцеллюлозную мембрану, проводили гибридизацию с меченной пробой. Гибридизацию проводили при 38oC в течение ночи, используя 4-кратную концентрацию SSC (1 x SS С; 0,15 М NaCl, 0,015 М цитрат натрия, pH 7,0), 10-кратную концентрацию раствора (1 x раствора Denhardt: 0.02% Ficoll, 0,02% поливинилпирролидон, 0,02% бычий сывороточный альбумин), и примерно 1 х 106 сpm/мл меченных проб. Мембрану несколько раз промывали 4-кратной концентрацией SSC при комнатной температуре и затем подвергали аторадиографии. Дополнительно температура промывки мембраны постепенно повышалась, и каждый раз мембрану снова подвергали авторадиографии. Установили, что несколько фрагментов ДНК показывают положительный сигнал даже при высокой температуре промывки, такой как 48 и 53oC, и что размеры гибридизованых цепочек отличаются друг от друга в зависимости от использованной рестриктазы. ДНК-фрагменты, показавшие положительный сигнал (2,5-3 кв DraI - фрагмент и 4-4,5 kb HindIII - фрагмент) были признаны предпочтительными для генного клонирования благодаря их размеру, большему, чем желаемый ген.
4. Клонирование цефалоспоринацетилгидролазного гена
1. Структура генной библиотеки
К хромосомной ДНК (12 мг) из B. subtilis, экстрагированной и очищенной как описано в шаге 3, добавляли рестриктазу DraI (примерно 120 единиц) и инкубировали при 37oC в течение 90 мин. Затем реагенты экстрагировали равным объемом фенола и этанола, который добавляли к результирующей водной среде для преципитации ДНК. Осадок ДНК растворяли в ТЕ буфере (10 мМ Трис-HCl, 1 мМ EDTA, pH 8,0) (60 мл), результирующий раствор подвергали электрофорезу в 0,8% агарозном геле и область геля, соответствующая размеру 2-4 кв, вырезали. ДНК-фрагменты элюировали, извлекали из геля, используя коммерческий кит (Bio 101, CEIIECIEAII), и растворяли в ТЕ буфере (30 мл).
С другой стороны в качестве вектора для создания генной библиотеки использовали pUc13, pUC13 (10 мкг) перемешивали с рестрикционным ферментом SmaI (примерно 100 единиц), инкубировали при 37oC в течение 140 мин и затем обрабатывали 10 мин при 65oC. Алкалинфосфат (ВАР) (примерно 20 единиц) добавляли к смеси и в дальнейшем реакцию проводили при - 65oC в течение 80 мин. После этого проводили экстракцию фенолом, а затем преципитацию ДНК этанолом в соответствии с описанной выше методикой и растворение полученной ДНК в ТЕ буфере (50 мл).
Раствор, содержащий фрагмент хромосомной ДНК B. subtilis (7,5 мл) и раствор, содержащий SmaI - BAP - обработанный фрагмент вектора рUC13 (2,5 мл), объединяли и смесь инкубировали с Т4 ДНК-лигазой при 6oC в течение 20 ч для объединения фрагментов с получением рекомбинантной ДНК. Результирующую рекомбинантную ДНК использовали для трансформации штамма E. coli в соответствии с методикой Hanahan и др. J. Mol. Biol. 166, 557-580 (1983). Далее колонии, образующиеся на нитроцеллюлозной мембране, помещали на L -бульон (1% бактотриптон, 0,5% экстракт дрожжей, 0,5% NaCl (pH 7,3)) в агаровую среду, содержащую 40 мг/мл ампициллина. Эти колонии образовали генную библиотеку В. subtilis.
2. Отбор положительного по цефалоспоринацетилгидролаэе клона метолом гибридизации колоний
Колонии, образовавшиеся на нитроцеллюлозной мембране в описанном выше шаге (1), переносили на другую нитроцеллюлозную мембрану. Реплику помещали в агаровую среду на L-бульоне, содержащем 40 мг/мл ампициллина, и инкубировали при 37oC в течение 3 ч. Затем мембрану переносили на агаровую среду на L-бульоне, содержащем 250 мг/мл хлорамфенинола, и инкубировали при 37oC в течение ночи. Гибридизацию колонии проводили в соответствии с методикой Grunstein и др.
Proc. Natl. Acad. Sci USA 72, 3961-3965 (1975). Мембрану обрабатывали 0,5 N NaOH (в течение 10 - 15 мин), чтобы осуществить лизис колонии и денатурацию ДНК. Последовательно мембрану нейтрализовали 1 М Триc-HCl буфером (pH 7,2) в течение 5-10 мин и далее обрабатывали 1 М Триc-HCI буфером (pH 8,0), содержащем 1,5 М NaCI в течение 10-15 мин. После того, как мембрану выдерживали при пониженном давлении при 80oC в течение 2 ч, чтобы иммобилизовать ДНК на мембране, иммобилизованную ДНК гибридизовали меченной пробой САН-PM2. Реакцию проводили в растворе, содержащем 4-кратную концентрацию SSC, 10-кратную концентрацию раствора Denhardt и примерно 2 х 106 cpm /мл меченной пробы при 38oC в течение 16 ч. После этого мембрану промывали 3-4 раза 4-кратно концентрированным SSC при комнатной температуре, затем промывали при 38oC в течение 2 мин и подвергали авторадиографии (в условиях -80oC, 3 ч). Для того, чтобы исследовать корреляцию между температурой отмывки и интенсивностью сигнала при авторадиографии, в дальнейшем температуру промывки поднимали с определенным интервалом и в каждом случае проводили авторадиографию. Отмывку проводили при 43, 48 и 53oC, каждый раз в течение 2 мин.
В результате было установлено, что 3 из примерно 30000 колоний показали положительный сигнал даже при высокой температуре отмывки. Эти колонии были культивированы L-бульоне (5 мл), содержащем 40 мг/мл ампициллина, при 37oC в течение ночи, и, таким образом, были получены рекомбинантные плазмиды (метод Birnboim и др. Nucleic. Acids. Res. 7. 1513-1523 (1979)). Эти плазмиды были расщеплены и фрагментированы с помощью таких рестриктаз, как EcoRI, Hind III и pVU II, имеющих сайты расщепления в векторе (рUC13). Затем проводили электрофорез на агарозном геле и гибридизацию по Southern с меченной пробой. В результате было установлено, что две или три рекомбинантных плазмиды содержали рестрикционные фрагменты с вставкой гибридизованной с ДНК-пробой. Размеры гибридизующихся фрагментов различались у двух положительных плазмид, поэтому показавшие отличие друг от друга ДНК-фрагменты повторно вставляли в вектор. Соответственно, были получены рекомбинантные плазмиды, обозначенные как pCAHOI и рСАНО2.
5. Илентификация положительного клона и определение основной последовательности
(1) Идентификация положительного клона
В случае попыток получения рестрикционной карты для рекомбинантных плазмид pCAHOI и рСАНО2 после расщепления их различными ограничивающими ферментами было установлено, что плазмида pCAHOI имеет два различных усвоенных фрагмента и плазмида рСАНО2 имеет по крайней мере три различных усвоенных фрагмента, вставленных в сайт SmaI вектора, и что ДНК-проба гибридизуется только с одним из этих фрагментов. В последующих методиках использовали рекомбинантную плазмиду pCAHOI, рестриктная карта плазмиды pCAHOI изображена на фиг. 6 вместе с ДНК гибридизованными пробами.
Поскольку плазмида pCAHOI содержит два экзогенных фрагмента, производных из хромосомной ДНК В. subtilis, (1,8 и 2,5 кв), и только фрагмент 2,5 кв гибридизуется с ДНК пробой, фрагмент 1,8 кв DraI был удален из плазмиды. DraI - PstI - фрагмент (2,6 кв), содержащий фрагмент 2,5 кв, удаляли из плазмиды pCAHOI и вставляли в SmaI - PstI сайт векторной плазмиды рUС13 для того, чтобы получить миниатюризованную рекомбинантную плазмиду pCAH03 (5,3 кв). Рестриктная карта рекомбинантной плазмиды рCAH03 приведена на фиг. 7.
(2) Определение последовательности оснований
0,24 кв. EcoRI - HindIII - фрагмент, с которым была гибридизована ДНК проба, был выделен из рекомбинантной плазмиды pCAH03, и его ДНК - последовательность была определена в соответствии с методикой Sanger и др. Proc. Natl. Acad. Sci. USA 74, 5463-5467 (1977). В результате последовательность, соответствующая частичной аминокислотной последовательности цефалоспоринацетилгидролазы, полученной в описанном выше шаге (1), была найдена во фрагменте EcoRI - HindIII, обнаруживая, что этот фрагмент содержит часть цефалоспоринацетилгидролазного гена. С помощью последовательного определения последовательности оснований между DraI и HindIII сайтами (0,38 кв), а также между сайтами EcoRI и EcoRI (1,45 кв) на основе карты рестрикции плазмиды pCAH03 была установлена последовательность оснований примерно 2 кв между DraI и EcoRI местами, как показано на фиг. 8 и 9. Это подтверждало существование последовательности оснований для кодируемого белка, состоящего из 318 аминокислотных остатков, содержащих начальный метионин, кодируемый трансляционным инициирующим коленом АТG. Дополнительно предполагаемый белок, кодированный описанной выше последовательностью оснований, показал хорошее совпадение с цефалоспоринацетилгидролазой в отношении ее молекулярного веса, амино-терминальных и карбокси-терминальных аминокислот, а также аминокислот, составляющих лизилэндопептидазный фрагмент, поэтому можно верить, что этот белок должен быть цефалоспоринацетилгидролазой.
Таким образом, было доказано, что структурный ген цефалоспоринацетилгидролазы содержался полностью во фрагменте генома В., содержащемся в плазмиде pCAH03.
6. Структура экспрессионной плазмиды
Клонированный фрагмент ДНК из В. subtilis, содержащийся в плазмиде pCAH03, включает область, иррелевантную фенотипичному выражению гена, и эта область была определена в соответствии с методикой, изображенной на фиг. 10.
Для того чтобы управлять уровнем экспрессии гетерологичного гена в Е. coli, очень эффективным является конструирование экспрессионной плазмиды, в которой желаемый структурный ген включен непосредственно после последовательности, состоящей из промотера, имеющего высокую эффективность экспрессии, и трансляционного инициирующего колена АТG.
Таким образом, для того чтобы получить большое количество цефалоспоринацетилгидролазы в E. coli, конструировали плазмиду экспрессии, используя вектор, содержащий промотор, АТG -кодон и кодирующую последовательность в соответствии с методом, изображенным на фиг. 11. Цефалоспоринацетилгидролазный структурный ген для этой цели получен следующим образом.
Область, содержащую целый желаемый ген, клонировали в вектор М13mp, чтобы получить однонитевую ДНК. В соответствии с так называемым методом расширения праймера, использующего в качестве праймера олигонулкотиды, специфичные для нескольких аминокислот, включая метионин на N -конце последовательности цефалоспоринацетилгидролазе, был получен ДНК-фрагмент, в котором только участок цефалоспоринацетилгидролазного гена является двунитевым.
Фиг. 12 показывает метод конструирования модифицированной экспрессионной плазмиды, дающей большее число копий по сравнению с экспрессионной плазмидой, полученной по методу, показанному на фиг. 11, и которая несет тетрациклиновый (Tcr) маркер вместо ампициллинового (Арr) маркера.
В дальнейшем каждый шаг описан более детально.
(1) Структура миниатюризированной плазмиды
Для того чтобы укоротить нижележащую область цефалоспоринацетилгидролазного структурного гена (сокращенно обозначаемого как CАН на фиг. 10), плазмиду pCAH03 расщепляли с помощью EcoRV и BamHI и получали примерно 4,1 кв ДНК-фрагмент, который очищали и затем 3' - конец от расщепления Bam HI заполняли, используя фрагмент ДНК полимеразы Klenow (фиг. 10). После дальнейшей очистки проводили лигирование, используя Т4 ДНК лигазу, с получением миниатюризированной плазмиды pCAHIO, содержащей цефалоспоринацетилгидролазный ген.
(2) Получение однонитевой ДНК для конструирования экспрессионной плазмиды
Миниатюризированную плазмиду pCAHIO, полученную в описанном выше шаге, расщепляли с помощью SacI и SalI и полученный (примерно 1,5 кв) фрагмент очистили и назвали SacI-SalI фрагментом. С другой стороны, репликативную форму фага М13 р11 ДНК расщепляли с помощью SacI и SalI, добавляли вышеуказанный фрагмент SacI-SalI и лигировали их с помощью Т4 ДНК лигазы с образованием M13-CАН ДНК. Далее получали единично-скрученную ДНК соответственно методике Messing Methods Enzymology 101, 20-78 (1983).
(3) Расщепление праймера
Область цефалоспоринацетилгидролазного структурного гена была получена при элиминировании некодирующей белок области ДНК, полученной из В. subtilis, которая локализовалась вверх от инициирующего кодона (ATG), в соответствии с методикой Goeddel и др. Nucleic. Acids Res, 8, 4057-4074 (1980). Олигонуклеотид, содержащий последовательность оснований, соответствующую второму аминокислотному глютамину-седьмому аминокислотному пролину аминоконца цефалоспоринацетилгидролазы, был синтезирован в качестве праймера для использования при расширении праймера и обозначен как CAH-PI. Праймер CAH-PI (3 рмоль) добавляли к однонитевой ДНК (7,5 мг) фага MI3-CАН, полученного в описанном выше шаге (2), и смесь нагревали до 60oC течение 20 мин, а затем оставляли при комнатной температуре. Последовательно к смеси были добавлены dATP, dCTP, dGTP и dТТР (каждый по 0,25 мм), а также фрагмент ДНК полимеразы Klenow (2 единицы), после чего проводили расширение праймера при 37oC в течение 2 ч в реакционной смеси (20 мл): 7 мМ Трис-НСI буфера (pH 7,5), 7 мМ MgCl2, 0,1 мМ EDTA, 20 мМ NaCI. После реакции проводили экстракцию фенолом и осаждение этанолом. ДНК растворяли в небольшом количестве дистиллированной воды, к которой добавляли SI-нуклеазу (4 единицы) и инкубировали при 37oC в течение 30 мин в реакционной смеси (40 мл): 30 мМ ацетата натрия (pH 4,), 100 мМ NaCI и 1 мМ ZnSO4 для усваивания оставшейся однонитевой ДНК. Раствор, содержащий двунитевой ДНК-фрагмент, получали при последовательном проведении экстракции фенолом, осаждения этанолом и обработки фрагментом ДНК полимеразы Klenow в соответствии с приведенной выше методикой.
(4) Структура экспрессионной плазмиды
Вектор pKK233-2 (c устойчивостью к ампициллину), использованный в настоящем примере для конструирования экспрессионной плазмиды, является коммерческим (Pharmacia). Вектор является членом АТG - векторов, которые содержат Trc промотор и могут быть расщеплены непосредственно после кодона инициирования (ATG) с помощью NcoI. Как изображено на фиг. 11, ДНК-фрагмент, полученный в описанном выше шаге (3), который содержит цефалоспоринацетилгидролазный ген, был расщеплен с помощью PstI и ДНК-фрагмент размером 1,27 кв отделен и очищен. С другой стороны, рКК2ЗЗ-2, содержащую Trс промотор, расщепляли с помощью NcoI и обрабатывали ДНК полимеразным фрагментом. Результирующий фрагмент затем расщепляли с помощью PstI для получения примерно 4,6 кв фрагмента ДНК. Далее, вышеупомянутые фрагменты смешивали и связывали друг с другом с помощью Т4 ДНК-лигазы; результирующую смесь использовали для переноса в E. coli JМ103 и формировали клоны на L - бульонной агаровой среде, содержащей 40 мг/мл ампициллина. Эти клоны были культивированы на L-бульоне в течение ночи, затем клетки собирали, плазмидную ДНК экстрагировали из клеток и определяли последовательность, расположенную вслед за местом соединения АТG вектора и фрагмента, содержащего цефалоспоринацетилгидролазный ген. В результате была получена экспрессионная плазмида, в которой структурный ген цефалоспоринацетилгидролазы включает аминоконцевой кодон метионина, вставленный непосредственно после АТG кодона; она обозначена как рCАН21. Штамм E. coli, включающий экспрессионную плазмиду, обозначали как E. coli JМ103/рCАН21.
Репликативная система экспрессионной плазмиды рCАН21 является производной из рВR322. Для того чтобы увеличить количество копий этой плазмиды и повысить уровень экспрессии в E. coli, ее область начала репликации (ori) была изменена таким образом, что стала производной от рАТ153.
Одновременно заменяли маркер устойчивости к ампциллину (Арr) маркером устойчивости к тетрациклину (Тcr). Метод модификации плазмиды изображен на фиг. 12. Плазмида рCАН21 была первоначально расщеплена с помощью BamHI ДНК и обработана фрагментом полимеразы Klenow. Затем результирующий фрагмент расщепляли ScaI с получением ДНК-фрагмента примерно 2,4 кв, содержащего Тrс промотор, цефалоспоринацетилгидролазный ген и Т1Т2-терминатор 5S рибосомальной РНК (5Srrn BT1T2). В качестве векторной плазмиды была использована коммерчески доступная плазмида рАТ153. Плазмида рАТ153 была ращеплена с помощью EcoRI, ДНК обработана фрагментом полимеразы Klenow и затем защеплена с помощью DraI. Для того чтобы предотвратить самолигирование вектора, была проведена обработка алкалинфосфатазой; примерно 2,5 кв ДНК-фрагмент, содержащий репликативную область и ген тетрациклин - резистентности, был получен из рАТ153. Затем два вышеупомянутых фрагмента были смешаны и лигированы с помощью Т4 ДНК-лигазы, а результирующая смесь использована для переноса в E. coli JМ103. Затем были выбраны колонии, образовавшиеся на L-бульонной агаровой среде, содержащей 20 мг/мл тетрациклина. После этого колонии были культивированы в L-бульоне в течение ночи, клетки собраны, а плазмидная ДНК экстрагирована из клеток и проанализирована с помощью рестриктазного расщепления. В результате были получены две рекомбинантные плазмиды, отличающиеся по ориентации связывания. Одна плазмида, в которой ориентация цефалоспоринацетилгидролазного гена, была идентична с ориентацией гена тетрациклин-резистентности, была названа рCАН211, а другая плазмида, в которой эти гены были обратно ориентированы, была обозначена как рCАН212. Штаммы E. coli, включающие эти рекомбинантные экспрессионные плазмиды, были обозначены как E. coli JMIO3/рCАН211 и E. coli JМ103/рCАН212 соответственно.
7. Экспрессия цефалоспоринацетилгидролазного гена в E. coli
(1) Экспрессия цефалоспоринацетилгидролазного гена E. coli JМ103/рCАН211 или E. coli JМ103/рCАН212 была проведена на дважды концентрированном L-бульоне (50 мл), содержащем 20 мг/мл тетрациклина (в 0,5 л колбе), при 37oC в течение 24 ч с встряхиванием. Аликвота (0,5 мл) смеси была центрифугирована для того, чтобы собрать клетки. Клетки были суспендированы в 0,1 М фосфатном буфере (pH 7,0) (0,5 мл) и разрушены ультразвуком. Супернатант, полученный при центрифугировании, использовали как образец раствора, содержащий желаемый фермент. С другой стороны, супернатант, полученный при центрифугировании культуральной жидкости В. subtilis, полученный в шаге (1), был использован как фермент для сравнения. Цефалоспоринацетилгидролаза воздействует не только на цефалоспорин С и 7-АСА, но также на р-нитрофенилацетат (далее сокращенно называемый pNPA) с образованием окрашенной субстанции р-нитрофенол (далее сокращенно называемый pNP). pNp является спектрофотометрически определяемым, поэтому метод, в котором в NPA используется в качестве субстрата, применяется в качестве простого метода определения активности цефалоспоринацетилгидролазы. Реакцию проводили в растворе (3 мл), содержащем 0,02% pNPA, 0,1 М фосфатного буфера (pH 6,8) и вышеупомянутый раствор фермента при 30oC. Активность фермента определяли измерением абсорбции при 400 нм, используя спектрофотомер. Количество фермента, из которого получают 1 ммоль pNP в минуту в условиях pH 68 и З0oC, было определено как одна единица. В результате было установлено, что активность фермента в культуральной жидкости E. coli JМ103/рCАН211 и E. coli JМ103/рCАН212 была 9,9 и 12,4 ед/мл соответственно. С другой стороны, активность фермента В. subtilis была 0,36 ед/мл.
Кроме того, была сконструирована плазмида, в которой Тrc промотор и SD-АТG последовательность экспрессионной плазмиды рСАН211 были замещены Тrр промотером и SD-АТG последовательностью, производной от триптофанового оперона E. coli. После того как плазмида была введена в E. coli М109, результирующие трансформанты были культивированы аналогично тому, как было описано выше. Было получено 75,5 ед активного фермента на мл культуральной жидкости.
Полученное количество цефалоспоринацетилгидролазы можно было увеличить выращиванием E. coli, содержащей эти экспрессионные плазмиды, в подходящих условиях, используя большое количество аппаратов для культивирования, таких, например, как вибрационный ферментер.
(2) Деацетилирование цефалоспорина С и 7АСА.
Деацетилирование с помощью раствора фермента из E. coli JMIO3/pCAH212 проводили, используя цефалоспорин С или 7АСА в качестве субстрата. Реакцию проводили при 37oC в течение 40 мин после добавления раствора фермента (0,2 мл) к 0,1 М фосфатному буферу (pH 7,0) (1,0 мл), содержащему 10 мМ субстрата, и заканчивали добавлением 0,2 М ацетатного буфера (pH 4,0) (1,2 мл). Результирующий раствор подвергали HPLC и измеряли деацетилцефалоспорин С или деацитил-7ACA.
Для заполнения колонки использовали Космосил 5С8 (тест Nacalai), а промывку проводили с помощью метода элюции в концентрационном градиенте, где концентрация метанола последовательно возрастала. Таким образом, используя раствор, содержащий 20 мМ NaH2PO4 и 5 мМ тетра-n-бутиламмоний гидрохлорид (ТВАН), концентрация метанола возрастала до 20%. Определение деацетилированного продукта проводили при 254 нм. Количество фермента, производящего 1 ммоль продукта в минуту при pH 7,0 и З7oC, было определено как одна единица (ед. ). В результате было установлено, что активность фермента культуральной жидкости E. coli JМ103/рСАН212 была 7,4 ед/мл как для цефалоспорина С, так и для 7АСА.
(3) Структура рекомбинантной цефалоспоринацетилгидролазы
Определение молекулярного веса активной формы и субъединицы цефалоспоринацетилгидролазы, полученной в E. coli, дало одинаковые результаты, такие же, что были получены в описанном выше шаге (1), указывая на то, что рекомбинант цефалоспоринацетилгидролазы также существует в форме октамера, аналогичного природной форме.
Более того, рекомбинантную цефалоспоринацетилгидролазу очищали с помощью НРLС, используя риверсивную колонку. Анализ концов деградацией по Edman и гидразиновым гидролизом показал, что амино- и карбокси-концы фермента представлены метионином и глицином соответственно и идентичны с природной формой. Дополнительное определение аминотерминальной последовательности с использованием автоматизированного секвенатора показало, что аминокислотная последовательность 25 аминокислот была полностью идентична с последовательностью, определенной из структурного гена (фиг. 2 и 3).
Как детально описано в приведенных выше примерах, изобретение подтверждает, что эффективное производство цефалоспоринацетилгидролазы возможно при клонировании гена, кодирующего цефалоспоринацетилгидролазу, полученного из В. subtilis, конструировании рекомбинантной плазмиды, содержащей названный ген в подходящем векторе и трансформации им E. coli. Это позволяет предположить широкое применение этого фермента. ДНК-фрагмент, содержащий клонированный цефалоспоринацетилгидролазный ген, обеспечивает возможность использования функции цефалоспоринацетилгидролазы.





Использование: в биотехнологии при получении полипептидов с активностью цефалоспоринацетилгидролазы. Сущность изобретения: при получении полипептида, обладающего активностью цефалоспорингидролазы, ведут культивирование клеток E. coli, трансформированных рекомбинантной молекулой ДНК, содержащей ДНК-фрагмент, полученный из генома Bacillus subtilis SHS 0133, продуцирующий указанный полипептид. 3 с. и 2 з.п. ф-лы, 12 ил.
[H Y]n,
где n 4 или 8;
Y аминокислотная последовательность с мол.м. примерно 35 кД,
кодируемый геном цефалоспоринацетилгидролазы, происходящим от B.subtilis SHS 0133 (Ferm ВР-2755) и имеющим нуклеотидную последовательность I, приведенную на с.
или ее аналогом, содержащим аминокислотные замены, делецил или вставки, не влияющие на каталитическую активность.
| Applied microbiology, 30, N 3, 1975, p | |||
| СТАНОК ДЛЯ ИЗГОТОВЛЕНИЯ ГАЛЕЙ | 1923 |
|
SU413A1 |
