Изобретение относится к гену, который получен из Rseudomonas chlororaphis В23 и который кодирует полипептид, обладающий активностью нитрилгидратазы, гидратирующей нитрилы в амиды. Изобретение относится к рекомбинантной ДНК, содержащей ген, и трансформанту, трансформированному этой рекомбинантной ДНК. Изобретение относится, кроме того, к способу получения нитрил гидратазы, используя трансформант, и амидов, используя нитрил гидратазу.
Нитрилгидратаза или нитрилаза известна как фермент, который гидратирует нитрилы в амиды. Микроорганизмы, которые продуцируют нитрил гидратазу, включают те, которые принадлежат роду Bacillus, роду Bacteridium роду Micrococcus и роду Brcvibacterium (см. JP-B-62-21517/1989, патент США N 4001081), роду Coryhebacterium и роду Vocardia (см. JP-B-56-17918/1989, патент США 4248968), роду Pseudomonas (см.JP B-59-37951/1984, патент США 4637982), роду Rhodococcus, роду Arthrobacter и роду Microbacterium (см, JP-А-61-162193/1986, ЕП-А-0188316) и Rhodococcus rhodochroous (см. JP-А-2-470/1990, ЕП-А-0307928).
Нитрилгидратаза использовалась для гидратации нитрилов в амиды. Конструиру-ются микроорганизмы, содержащие несколько копий рекомбинантной ДНК, кодирующей нитрилгидратазу, с использованием приемов рекомбинантной ДНК. Рекомбинант продуцирует значительные уровни нитрилгидратазы по сравнению с известными приемами.
Предложен ранее ген, полученный из Rhodococcus вида N-774 (FERM ВР-1936), который также кодирует полипептид, обладающий активностью нитрилгидратазы (JP-А-2-119778/1988). Напротив, в соответствии с настоящим изобретением используют ген, полученный из Pseudomonas chlororaphis В23, описанный в патенте США N 4637982 для продуцирования нитрилгидратазы. Выделен ген, кодирующий нитрилгидратазу, вставил ген в соответствующий вектор плазмиды и трансформировал соответствующего хозяина этой рекомбинантной плазмидой, получая таким образом трансформант, продуцирующий нитрилгидратазу.
Изобретение относится к
1) гену, кодирующему полипептид, который обладает активностью нитрилгидратазы и который содержит α-субблок со следующей аминокислотной последовательностью:
MetSerThrSerIl SerThrThrAlaT
SerThrThrAlaT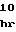 ProSerThrProG
ProSerThrProG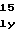 GluArgAlaTrpA
GluArgAlaTrpA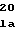 LeuPheGlnValL
LeuPheGlnValL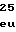 LysSerLysGluL
LysSerLysGluL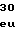 IleProGluGlyT
IleProGluGlyT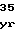 ValGluGlnLeuT
ValGluGlnLeuT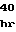 GlnLeuMetAlaH
GlnLeuMetAlaH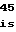 AspTrpSerProG
AspTrpSerProG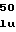 AsnGlyAlaArgV
AsnGlyAlaArgV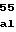 ValAlaLysAlaT
ValAlaLysAlaT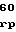 ValAspProGlnP
ValAspProGlnP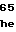 ArgAlaLeuLeuL
ArgAlaLeuLeuL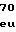 LysAspGlyThrA
LysAspGlyThrA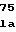 AlaCysAlaGlnP
AlaCysAlaGlnP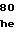 GlyTyrThrGlyP
GlyTyrThrGlyP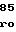 GlnGlyGluTyrI
GlnGlyGluTyrI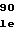 ValAlaLeuGluA
ValAlaLeuGluA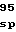 ThrProGlyVal
ThrProGlyVal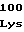 AsnValIleVal
AsnValIleVal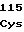 SerLeuCysSer
SerLeuCysSer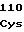 ThrAsnTrpPro
ThrAsnTrpPro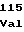 LeuGlyLeuPro
LeuGlyLeuPro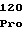 GluTrpTyrLys
GluTrpTyrLys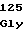 PheGluPheArg
PheGluPheArg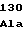 ArgLeuValArg
ArgLeuValArg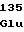 GlyArgThrVal
GlyArgThrVal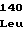 ArgGluLeuGly
ArgGluLeuGly GluLeuProSer
GluLeuProSer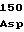 ThrValIleLys
ThrValIleLys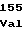 TrpAspThrSer
TrpAspThrSer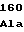 GluSerArgTyr
GluSerArgTyr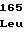 ValLeuProGln
ValLeuProGln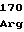 ProGluGlySer
ProGluGlySer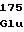 HisMetSerGlu
HisMetSerGlu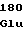 GlnLeuGlnGln
GlnLeuGlnGln ValThrLysAsp
ValThrLysAsp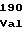 LeuIleGlyVal
LeuIleGlyVal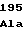 LeuProArgVal
LeuProArgVal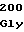
и β -субъединицу со следующей аминокислотной последовательностью
MetAspGlyPheHi AspLeuGlyGlyP
AspLeuGlyGlyP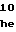 GlnGlyPheGlyL
GlnGlyPheGlyL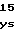 ValProHisThrI
ValProHisThrI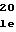 AsnSerLeuSerT
AsnSerLeuSerT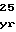 LysGlnValPheL
LysGlnValPheL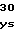 GlnAspTrpGluH
GlnAspTrpGluH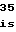 LeuAlaTyrSerL
LeuAlaTyrSerL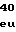 MetPheValGlyV
MetPheValGlyV AspGlnLeuLysL
AspGlnLeuLysL PheSerValAspG
PheSerValAspG ValArgHisAlaV
ValArgHisAlaV GluArgLeuAspV
GluArgLeuAspV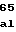 ArgGlnHisValG
ArgGlnHisValG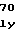 ThrGlnTyrTyrG
ThrGlnTyrTyrG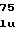 ArgTyrIleIleA
ArgTyrIleIleA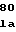 ThrAlaThrLeuL
ThrAlaThrLeuL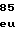 ValGluThrGlyV
ValGluThrGlyV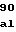 IleThrGlnAlaG
IleThrGlnAlaG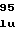 LeuAspGlnAla
LeuAspGlnAla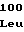 GlySerHisPhe
GlySerHisPhe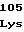 LeuAlaAsnPro
LeuAlaAsnPro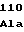 HisAlaThrGly
HisAlaThrGly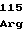 ProAlaIleThr
ProAlaIleThr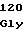 ArgProProPhe
ArgProProPhe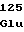 ValGlyAspArg
ValGlyAspArg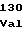 ValValArgAsp
ValValArgAsp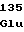 TyrValAlaGly
TyrValAlaGly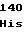 IleArgMetPro
IleArgMetPro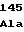 TyrValArgGly
TyrValArgGly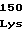 GluGlyValVal
GluGlyValVal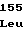 HisArgThrSer
HisArgThrSer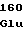 GlnTrpProPhe
GlnTrpProPhe AspAlaIleGly
AspAlaIleGly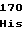 GlyAspLeuSer
GlyAspLeuSer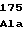 AlaHisGlnPro
AlaHisGlnPro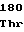 TyrHisValGlu
TyrHisValGlu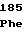 ArgValLysAsp
ArgValLysAsp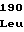 TrpGlyAspAla
TrpGlyAspAla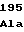 AspAspGlyTyr
AspAspGlyTyr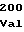 ValValAspLeu
ValValAspLeu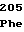 GluSerTyrLeu
GluSerTyrLeu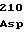 LysAlaProGly
LysAlaProGly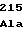 GlnAlaValAsn
GlnAlaValAsn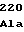
2) гену, описанному в (1), кодирующему α- и β -субблоку, содержащему кодирующую α- субблок последовательностью
15 30 45
ATGAGTACATCTATTTCCACGACTGCGACACCTTCGACACCCGGC
60 75 90
GAGAGGGCATGGGCCTTGTTTCAAGTGCTCAAGAGCAAGGAACTC
105 120 135
ATCCCAGAGGGCTATGTCGAGCAGCTCACTCAATTGATGGCCCAT
150 165 180
GACTGGAGCCCGGAGAACGGCGCTCGCGTGGTCGCCAAGGCATGG
195 210 225
GTCGATCCGCAGTTCCGGGCGCTGCTGCTCAAGGACGGAACAGCC
240 255 270
GCTTGCGCGCAGTTCGGCTACACCGGCCCACAAGGCGAATACATC
285 300 315
GTCGCCCTGGAAGATACACCGGGGGTGAAGAACGTCATCGTCTGC
330 345 360
AGCCTGTGCTCCTGCACCAACTGGCCGGTCCTCGGCCTGCCGCCC
375 390 405
GAGTGGTACAAGGGCTTTGAGTTTCGTGCGCGCCTGGTCCGGGAG
420 435 450
GGGCGCACCGTACTGCGCGAGCTGGGGACGGAGTTGCCGAGCGAC
465 480 495
ACGGTCATCAAAGTCTGGGATACCAGCGCCGAAAGCCGTTACCTG
510 525 540
GTGTTGCCGCAAAGGCCTGAAGGCTCTGAGCACATGAGTGAAGAA
555 570 585
CAGCTTCAACAGCTGGTGACCAAAGACGTGCTGATTGGCGTCGCC
600
CTGCCACGCGTTGGC
и последовательность, кодирующую β-субъединицу:
15 30 45
ATGGATGGCTTTCACGATCTCGGCGGTTTCCAAGGCTTTGGCAAA
65 75 90
GTGCCGCACACCATCAACAGCCTCAGCTACAAACAGGTTTTCAAG
105 120 135
CAGGACTGGGAACACCTGGCCTATAGCTTGATGTTTGTCGGCGTT
150 165 180
GACCAATTGAAAAAGTTCAGCGTGGACGAAGTGCGTCATGCCGTC
195 210 225
GAACGCCTGGACGTTCGCCAGCATGTCGGCACCCAGTACTACGAA
240 255 270
CGCTACATCATCGCGACCGCCACGCTGCTGGTGGAAACGGGCGTT
285 300 315
ATCACCCAGGCGGAGCTCGATCAGGCATTGGGTTCCCACTTCAAG
330 345 360
CTGGCGAACCCCGCCCATGCGACAGGTCGCCCGGCGATCACCGGC
375 390 405
AGGCCGCCCTTCGAAGTGGGCGATCGGGTTGTGGTTCGAGACGAA
420 435 450
TATGTGGCGGGGCATATCCGCATGCCGGCCTACGTGCGCGGTAAG
465 480 495
GAAGGCGTGGTCCTGCACCGCACCTCAGAGCAGTGGCCCTTCCCC
510 525 540
GACGCCATTGGCCACGGCGACTTGAGCGCAGCCCATCAGCCTACC
555 570 585
TACCACGTCGAGTTTCGCGTGAAAGATCTATGGGGTGACGCGGCA
600 615 630
GATGACGGTTACGTCGTGGTCGATCTTTTCGAAAGCTACTTGGAT
645 660
AAGGCCCCCGGTGCCCAAGCGGTGAACGCA
3) рекомбинантной ДНК, содержащей вектор, включающий ген, описанный в [1] или [2]; 4) трансформанту, трансформированному рекомбинантной ДНК, описанной в [3] ; 5) способу продуцирования нитрилгидратазы, который содержит культивирование трансформанта, и выделение нитрилгидратазы из культуры; 6) способу продуцирования амидов, который содержит гидратирование нитрилов, используя нитрилгидратазу, с целью получения амидов; 7) способу получения амидов, который содержит культивирование трансформанта и гидратирование нитрилов, используя полученную в результате культуру изолированных бактериальных клеток, обработанных им, или фиксированным их материалом, чтобы получить амиды.
Изоляция и очистка нитрилгидратазы и частичный анализ последовательности аминокислот нитрилгидратазы.
Нитрилгидратазу изолируют и подвергают очистке из Psudomonas chlororaphis В 23 и разделяют между α и β субблоками, используя ЖХВД (жидкосткую хроматографию под высоким давлением). Определяют часть аминокислотной последовательности субблоками (фиг.1).
Получение ДНК-зонда для гена нитрилгидратазы.
ДHК-зонд получают из штамма J М105/ру V К121 (FERM ВР-1937), как это описано в JP-А-2-119778/1990, благодаря высокой степени гомологичности в аминокислотной последовательности между β-субблоком нитрилгидратазы Rhodococcus вида N-744, описанным в вышеупомянутой Японской Государственной Патентной Газете, и соответствующим β-субблоком Pseudomonas chlororahis В23. Плазмиду РуVК121, содержащую ген нитрилгидратазы, полученный из Rhodococcus вида N-744, приготавливают из культуры JM105/рУVК121, ДНК рУVК121 переваривают при помощи ферментов Sph I и Sal I. Фрагмент Sph I-Sal I содержит ген нитрилгидратазы (фиг. 2-5) Rhodococcus вида N-744. ДНК-фрагмент метят радиоизотопом, чтобы получить зонд.
Обнаружение ДНК-сегмента, содержащего ген нитрилгидратазы из хромосомы Pseurdomonas chlororaphis В 23.
Хромосомную ДНК получают из культуры Pseudomonas chlororaphis В23. Хромосомную ДНК переваривают ферментами рестрикции и подвергают гибридизации с зондом, описанным в [2], используя процедуру гибридизации Саутерна (Саутерн, Э.M, J Mol. Biol, т.98, с. 503 (1975). Просеивают два ДНК-фрагменты различной длины.
Конструкция рекомбинантной плазмиды. Рекомбинантную плазмиду конструируют при помощи вставки фрагмента хромосомной ДНК, полученной в [3], в вектор плазмиды.
Трансформация и просеивание трансформанта, содержащего рекомбинантную плазмиду. Трансформанты получают с использование известной рекомбинантной плазмиды. Трансформант, содержащий рекомбинантную плазмиду, подвергают селекции с использованием зонда, описанного в [2], в соответствии с процедурой гибридизации колоний (Р.Брюс Уоллес и др. NUC/ Aci. Res, т.9, с.879 (1981)). Кроме того, присутствие гена нитрилгидратазы в рекомбинантной плазмиде подтверждают при помощи процедуры гибридизации Саутерна. Отобранные таким образом плазмиды обозначают рРСN1 и pРCN3.
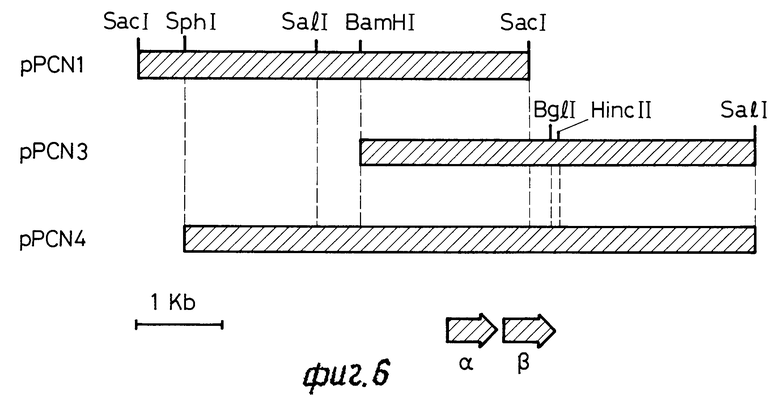
6) Изоляция и очистка ДНК-плазмиды, и конструкция карты рестрикции. ДНК плазмид рРСN1 и pPCN3, изолируют и подвергают очистке. Конструируют карту рестрикции ДНК (фиг.6), чтобы определить область, содержащую ген нитрилгидратазы.
7) Анализ ДНК-последовательности. Дополнительный сегмент вставленного ДНК-фрагмента в рРСN1 и рРСN3 расщепляют с использованием соответствующего фермента рестрикции. Вставленный ДНК-фрагмент затем используют для анализа последовательности. Нуклеотидная последовательность ДНК-фрагмента (фиг.7-10) показывает, что она содержит последовательность, полученную из аминокислотной последовательности, как это описано в (1).
8) Вставка ДНК-фрагмента в вектор экспрессии и трансформация. ДНК-фрагмент отсекают от рРСN1 и рPСN3, используют соответствующие ферменты рестрикции. Эти два фрагмента подвергают лигации и вставляют в вектор экспрессии рVС19. Эту конструкцию используют для трансформации Е.coli JM105 (Амершэм) и этот трансформант обозначают через JM105/pPCN4.
9) Продуцирование нитрилгидратазы с использованием трансформанта и превращение нитрилов в амиды. Культивируют трансформант, описанный в (8). Бактериальные клетки смешивают с нитрилами, субстратом нитрилгидратазы и получают амиды.
Pseudomonas chlororaphis В23 сдан на хранение в Исследовательский институт ферментации. Агенство по промышленной науке и технологии, и ему присвоен шифр хранения FEPM BР-187. Трансформант JM105/pPCN4 сдан на хранение в тот же институт и ему присвоен шифр хранения FERM ВR-2779.
Любой вектор, включающий вектор плазмиды (например, рАТ 153, рМР9, рНС624, рКС7, и т.д.), вектор фага (например, λ gt II (Тоиобо), Чарон 4А (Амершэм) и т,д,) можно использовать в соответствии с настоящим изобретением. Ферменты, которые можно при этом использовать включают Sph I, Sal I, Sac, Bam, HI, EcoRI, Pst I и т.д., которые производятся промышленностью (фирма Такара Шузо). Самых разнообразных хозяев можно использовать для трансформации, например, Е.coli, JM105 и TGI (но ими не исчерпывается весь список). Культурной средой для трансформанта может быть любая среда, которую в общем случае используют для этих целей.
Превращение нитрилов в амиды осуществляют с использованием нитрилгидратазы, неочищенной нитрилгидратазы, культуры трансформанта, изолированных бактериальных клеток или их обработанного материала и т.п., полученных из культуры трансформанта.
Соответствующие нитрилы настоящего изобретения включают те, которые содержат 2-4 атома углерода, также, как ацетонитрил, пропионитрил, акрилонитрил, метакрилонитрил, н-бутиронитрил и изобутилонитрил, причем акрилонитрил является предпочтительным.
На фиг.1 показана N-терминальная аминокислотная последовательность α- и β -субблоков нитрилгидратазы, полученной с использованием Pseudomonas chlororaphis B 23; на фиг.2-5 - ДНК-последовательность гена нитрилгидратазы Rhodococcus виды N-774, использованная в качестве ДН-зонда; на фиг.6 - частичные карты рестрикций рекомбинантных плазмид, pPСN1, pPCN3 и ppCN4; на фиг. 7-10 - нуклеoтидная последовательность ДНК-вставки в pPСN3, полученная из В23, и выделенная аминокислотная последовательность.
В соответствии с настоящим изобретением предлагается аминокислотная последовательность и нуклеотидная последовательность α- и β -субблоков нитрил- гидратазы, получены из Pseudomonas chlororaphis B23. Ген, кодирующий нитрилгидратазу, вставляют в вектор экспрессии, а рекомбинантный вектор используют для трансформации. Трансформант содержит несколько копий гена и продуцирует более высокие концентрации нитрилгидратазы по сравнению с известными, используемыми для этих целей, микроорганизмами.
Изобретение описано подробно в приводном ниже примере. В этом примере используют следующие сокращения.
TЕ: трис-HСl (10 мМ, рН 7,8), ЭДТК (1 мМ, рН 8,0)
ТNE: трис-HCl (50 мМ, рН 8,0), ЭДТК (1 мМ, рН 8,0), NaCl (50 мМ)
ТЕ: трис-HCl (50 мМ, рН 8,0), ЭДТК (5 мМ, рН 8,0), сахароза (35 мМ)
Среда 2 ХУТ: 1,6% Триптон, 1,0% экстракт дрожжей, 0,5% NaCl
1. Изоляция и очистка нитрилгидратазы и анализ аминокислотной последовательности части нитрилгидратазы Pseudomonas chlororaphis В23 культивировали в среде (10 г/л сахарозы, 4 г/л метакриламида, 0,5 г/л КН2РО4, 0,5 г/л К2НPO4, 0,5 г/л MgSO4.7H2O, 0,01 г/л FeSO4.7H2O, рН 7,0) при 25оС в течение 28 ч. Бактериальные клетки собирали. 100 г бактериальных клеток разрушали и фракционировали при помощи сульфата аммония. Пробу подвергали диализу и диализат подвергали центрифугированию. Верхний слой удаляли и загружали на хроматографическую колонну ДЕАЕ-Сефадцел, Октил-Сефароза КЛ-4Б, Фенил-Сефароза КЛ-4Б и Сефадекс-Г-150. Активные фракции собирали и подвергали диализу. Диализат, содержащий фермент, загружали на колонну высокоэффективной жидкостной хроматографии, используя обращенно-фазовую колонну (Сеншу Цак ВР-304-1251, Сеншу Кагаку) и получали два субблока (α и β) , N-терминальную аминокислотную последовательность α- и β -субблоков определяли с использованием анализатора аминокислотных последовательностей (фирмы Апплайед Биосистемз, Модели 470А), N-терминальные аминокислотные последовательности α- и β -субблоков приведены на фиг.1.
2. Получение ДНК-зонда для гена нитрилгидратазы E.coli JM105, содержащую pУVK121 (FERM BP-1927), культивировали в 100 мл среды 2 х УТ, содержащей 50 μ г/мл ампициллина, при 30оС в течение ночи (12 ч). Бактериальные клетки собирали и в клетки добавляли TNE. Суспензию клеток затем подвергали центрифугированию. В таблетку добавляли 8 мл STEи 10 мг лизоцима. Смесь инкубировали при 0оС в течение 5 мин, затем добавляли 4 мл 0,25 М ЭДТК. Далее, добавляли в смесь при комнатной температуре 2 мл 10% ДСН )додецил сульфата натрия пер. ) и 5 мл 5М NaCl. Полученную в результате смесь выдерживали при 0-4оС в течение 3 ч, а затем подвергали ультрацентрифугированию. В верхний слой добавляли 1/2 объема 30% ПЕГ 6000. Смесь выдерживали при 0-4оС в течение ночи (12 ч) и подвергали центрифугированию. В таблетку добавляли TNE, чтобы получить объем 7,5 мл, а затем в суспензию добавляли CSCl. Смесь подвергали центрифугированию, чтобы удалить протеины. Далее в верхний слой добавляли 300-500 мг/мл бромида этидия. Смесь переносили в пробирку для центрифугирования. Пробирку запаивали, а затем подвергали ультрацентрифугированию, ДНК экстрагировали, используя перистальтический насос. В экстракт, чтобы удалить бромид этидия, добавляли несколько больше, чем равное количество изопропилового спирта, насыщенного водой. Пробу подвергали диализу против ТЕ. Получали примерно 3 мл очищенной рУVК121.
рУVК121 ДНК переваривали ферментами Sph I и Sal I, получая в результате ДНК-фрагмент в 2,07 ко (килооснования - пер.), содержащий ген нитрилгидратазы, полученный и Rpodococcus вида N-774. Фрагмент метили радиоизотопом 32Р, чтобы получить зонд. Нуклеатидная последовательность зонда приведена фиг.2-5.
3. Получение ДНК-фрагмента, содержащего геннитрил гидратазы хромосомы. Pseudomonas chlororaphis В23 культивировали в 100 мл среды, описанной [1]. Бактериальные клетки собирали и таблетку промывали при помощи TNE. Затем таблетку суспендировали в 10 мл ТЕ. В суспензию добавляли 4 мл 0,25M ЭДТК, 10-20 мг лизозима, 10-20 мл ахромопротеазы и 10 мл 10% ДСН. Суспензию инкубировали при 37оС в течение 3 ч. В суспензию добавляли 15 мл фенола. Смесь инкубировали при комнатной температуре 60оС в течение 15 мин, а затем центрифугировали. В 15 мл верхнего слоя добавляли 0,7 мл 2,5М ацетата натрия и диэтиловый простой эфир, смесь центрифугировали. Верхний слой сбрасывали. В нижний слой добавляли два объема этанола и ДНК удаляли стеклянным стержнем. ДНК промывали в течение 5 мин при помощи смеси ТЕ : этанол 2:8, 1:9и 0:10 (о/o). ДНК затем снова суспендировали в 2-4 мл ТЕ (37оС). 10 μ л смеси РНазы А и Т1 добавляли в суспензию смесь инкубировали при 37оС. В смесь добавляли равное количество фенола, а затем подвергали центрифугированию. В 2-4 мл верхнего слоя добавляли больше, чем равное количество простого эфира. Смесь подвергали центрифугированию. После центрифугирования верхний слой сбрасывали. Нижний слой подвергали диализу относительно 2 л ТЕ, содержащего небольшое количество хлороформа, в течение ночи, а затем диализу относительно свежего ТЕ в течение 3-4 чл. Получали 4 мл неочищенной хромосомной ДНК. Переваривание ферментом хромосомной ДНК осуществляли следующим образом:
а) 2 μ л Sac I + 3 μ л реакционного буфера (10х) + 15 л хромосомной ДНК + 10 μ л ТЕ
в) 2 μ л Bam HI + 2 μ л Sal I + 3 μ л реакционного буфера (10х) + 15 μ л хромосомной ДНК + 10 μ л ТЕ.
Смесь инкубировали при 37оС в течение 1 ч, а затем подвергали электрофорезу на геле агарозы при разности потенциалов 60 В в течение 3 ч. Гибридизацию Саутерна хромосомной ДНК осуществляли с использованием зонда, описанного в (2). Обнаруживали фрагменты в примерно 4,6 ко и 4,7 ко, что говорит о сильной гибридизации. 15 μ л хромосомной ДНК переваривали при помощи Sac I Bam HI и Sal I и подвергали электрофорезу на геле агарозы, как это было описано выше. ДНК-фрагменты в 4,6 ко и 4,7 ко срезали с геля и переносили в три объема 8М NaClO4. Раствор наносили каплями на фильтровальную бумагу GF/C/Bатман/ (диаметр 6 мм). На фильтровальную бумагу добавляли десять капель (≈ 100 μ л) ТЕ, содержащего 6М NaClO4, и десять капель (≈ 100 μ л) 95% этанола. Затем бумагу сушили воздухом и помещали в 0,5 мл пробирку Эппендорфа. В пробирку добавляли 40 μ л ТЕ и все это инкубировали при 37оС в течение 30 мин. Затем пробирку центрифугировали. Получали примерно 40 μ л верхнего слоя, который содержал ДНК-фрагменты в 4,6 ко и 4,7 ко, содержащие ген нитрилгидратазы хромосомной ДНК.
4. Вставка хромосомного ДНК-фрагмента в вектор.
а) ДНК-фрагмента 4,6 ко
2 μ л Sac I, 3 μ л реакционного буфера (10х) и 10 мл ТЕ добавляли в 10 μ в ДНК pVC18. Смесь инкубировали при температуре 37оС в течение 1 ч. 2 μ л 0,25M ЭДТК добавляли в смесь, чтобы прекратить реакцию. Затем в смесь добавляли 7 μ л ТМ Тpис-HCl (pH 9) и 3 μ л БДФ (бактериальной щелочной фосфатазы). Смесь инкубировали при 65оС в течение 1 ч. Затем в смесь добавляли ТЕ, чтобы довести общий объем до 100 μ л. Смесь экстрагировали 3х равным количеством фенола. В экстракт добавляли равное количество простого эфира. Нижний слой удаляли и в нижний слой добавляли 10 μ л 3М ацетат натрия и 250 μ л этанола. Смесь инкубировали при -80оС а течение 30 мин, подвергали центрифугированию. Сушили и снова суспендировали в ТЕ.
Полученные таким образом 5 μ л ДНК Р С18 и 40 μ л ДНК-фрагмента в 4,6 ко, описанного в (3), смешивали. В смесь добавляли 6 μ л буфера лигации, 6 μ л АТФ (6 мг/мл) и 3 μ л ДНК Т4-лигазы. Смесь инкубировали при 4оС в течение ночи (12 ч), чтобы получить рекомбинантную плазмиду, содержащую ДНК-фрагмент в 4,6 ко в Сайте Sal I ч РVC18.
в) ДНК-фрагмент в 4,7 ко
рVC18 переваривали при помощи Bam HI и Sal I. ДНК-фрагмент в 4,7 ко вставляли в сайт Bam HI - Sal I в рVC18 точно так же, как это описано в (4а). Получали рекомбинантную плазмиду, содержащую ДНК-фрагмент в 4,7 ко в сайтe Bam HI - Sal I.
5. Трансформация и просеивание трансформантов.
E. coli JM105 (Амерщэм) прививали на 10 мл среды 2 х УТ и инкубировали при 37оС в течение 12 ч. После инкубирования полученную в результате культуру добавляли в свежую среду 2 х УТ до концентрации 1%, смесь инкубировали при 37оС в течение 1 ч. Культуру подвергали центрифугированию и таблетку суспендировали в 5 мл холодного 50 мМ CaCl2. Суспензию помещали в температуру 0оС на 40 мин, а затем подвергали центрифугированию. В таблетку в отдельной пробирке добавляли 0,25 мл холодного 50 мМ CaCl2 и 60 μ л каждой из рекомбинантных плазмид, полученных в (4а,в). Смесь инкубировали при 0оС в течение 40 мин, подвергали термическому удару до 42оС в течение 2 мин, помещали в температуру 0оС 5 мин и добавляли в 10 мл среды 2 х УТ. Смесь инкубировали при 37оС в течение 90 мин при встряхивании, затем центрифугировали. Таблетку суспензировали в 1 мл среды 2 х УТ и 10 μ л суспензии наносили на агарозную пластинку 2 х УТ, содержащую 50 μ г/мл ампициллина. Пластинку инкубировали при 37оС. Кологию выращивали на пластинке, затем подвергали селекции методом гибридизации колонии: Колонию переносили на нитроцеллюлозные фильтр и переваривали. ДНК фиксировали на фильтре и гибридизировали с зондом, описанным в [2]. Фильтр подвергали авторадиографии и отбирали рекомбинантную колонию. Кроме того, присутствие гена нитрилгидратазы в трансформанте подтверждали в соответствии с процедурой гибридизации Саутерна.
6) Изоляция и очистка рекомбинантной плазмиды и конструкция карты рестрикции вставленных ДНК-фрагментов.
Трансформант выращивали в 100 мл среды 2 х УТ, содержащей 50 μ г/мл ампициллина при 37оС в течение ночи (12 ч). Бактериальные клетки собирали и в клетки добавляли TNЕ. Клетки собирали снова центрифугированием и в клетки добавляли 8 мл ТЕ и 10 мг лизоцима. Смесь инкубировали при 0оС в течение 5 мин. В смесь добавляли 4 мл 0,25 м ЭДТК, 2 мл 10% ДСН (при комнатной температуре) и 5 мл 5М NaCl. Смесь инкубировали при 0-4оС в течение 3 ч и подвергали ультрацентрифугированию. В верхний слой добавляли 1/2 объема 30- ПЕН 6000. Смесь инкубировали при 0-4оС в течение ночи (12 ч) и снова центрифугировали. TNE добавляли в таблетку, чтобы довести до объема 7,5 мл. В суспензию добавляли CSCl. Суспензию центрифугировали, чтобы удалить протеины. Далее, в верхний слой добавляли 300-500 мг/мл бромида этидия и смесь переносили в пробирку для центрифугирования. Пробирку запаивали нагреванием и подвергали ультрацентрифугиро- ванию. ДНК удаляли, используя перистальтический насос. В ДНК, чтобы удалить бромид этидия, добавляли несколько больше, чем равное количество изопропилового спирта, насыщенного водой. Пробу ДНК подвергали диализу относительно ТЕ, что приводило к образованию примерно 3 мл рекомбинантной ДНК. Полученную таким образом рекомбинантную плазмиду, содержащую ДНК-фрагмент в 4,6 ко, переваривали как pPCN1 (Рекомбинантную плазмиду, содержащую ДНК-фрагмент в 4,5 ко, переваривали как pPCN3).
Эти ДНК плазмид переваривали при помощи Eco RI, Bam HI, Pst I, Sac I и Cal I. Карты рестрикции конструировали и они приведены на фиг.6.
7) Анализ ДНК-последовательности.
Расположение гена нитрилгидратазы в ДНК-фрагментах pPCN1 и pPCN3 определяли при помощи карты рестрикции и использованием процедуры гидролизации Саутерна. Основываясь на этих результатах, анализировали ДНК-фрагмент Bam HI - Hinc II при помощи процедуры Сангера (Сангер, Ф., Science, т.214, с. 1205-1210, (1981)), используют вектор фага М13. ДНК-фрагмент в 2456 ко из Pseudomonas chlororaphis В23 приведен на фиг.4.
Вся нуклеотидная последовательность, полученная из аминокислотной последовательности, определенной в [1], была обнаружена в последовательности в соответствии с описанием, приведенным выше. Анализ последовательности также обнаруживали, что этот ДНК-фрагмент содержал последовательность, кодирующую α - и β -субблоки.
8) Вставка ДНК-фрагмента Bam HI - Hinc IIв вектор экспрессии и трансформации.
Фрагмент Sph I - Bam HI в 2,2 ко в pPCN1 и фрагмент Nam HI - Sal I в 4,7 ко в pPCN3 расщепляли, оба фрагмента подвергали лигации (фиг.3). Фрагмент после лигации вставляли в сайт Sph I - Sal I вектора экспрессии pVC19 и полученную конструкцию обозначали как pPCN4.
E. coli JМ105 трансформировали при помощи pPCN4 точно так же, как это делали в (5), и трансформант обозначали через JM105/pPCN4 (FERM ВР-2779).
9) Продуцирование нитрилгидратазы, используя трансформант, и превращение нитрилов в амиды, используя нитрилгидратазу.
JM105/pPCN4 прививали в 10 мл среды 2 х УТ, содержащей 50 μ г/мл ампициллина, и инкубировали при 26,5оС в течение ночи (12 ч). 100 μ л полученной в результате культуры добавляли в 10 мл среды 2 х УТ (50 г/мл ампициллина, 50 мг/л FeSO4.7H2O, 10 мг/л MgSO4.7H2O, 1 мг/л пирролохинолинхонона). Эту смесь инкубировали при 26,5оС в течение 5 ч. В смесь добавляли IPTG до финальной концентрации I мм. Смесь инкубировали при 26,5оС в течение 10 мин. После сбора клеток клетки суспендировали в 5 мл 1/20 М фосфатного буфера (рН 7,7). В 0,1 мл суспензии добавляли 10 μ л раствора субстрата (1М акрилонитрила). Смесь инкубировали при 20оС в течение 20 мин. В качестве контрольного испытания использовали смесь, полученную при помощи аналогичной процедуры, что была oписана выше, но из E.coli JM105. Реакционную смесь испытывали на присутствие акриламида (продукта ферментной реакции) и акрилонитрила, используя ЖХВД. Акриламид, но не акрилонитрил обнаруживали в реакционной смесь JM105 pPCN4, в то время, как акрилонитрил, а не акриламид обнаруживали в реакционной смеси JM105.









Изобретение относится к биотехнологии и генетической инженерии, в частности к получению ферментов клетчатого метаболизма. Сущность изобретения состоит в том, что из Pseudomonas chlororaphis В23 получен фрагмент ДНК, который кодирует полипептид, обладающий активностью нитрилгидратазы и способный гидратировать нитрилы в амиды. Получена рекомбинантная ДНК, содержащая этот ген, и трансформант, образованный путем трансформации штамма E. coli этой рекомбинантной ДНК. Изобретение, кроме того, включает способ получения нитрилгидратазы, используя трансформант. 4 с.п. ф-лы, 10 ил.
ФРАГМЕНТ НУКЛЕИНОВОЙ КИСЛОТЫ, КОДИРУЮЩИЙ ПОЛИПЕПТИД, ОБЛАДАЮЩИЙ АКТИВНОСТЬЮ НИТРИЛГИДРАТАЗЫ, РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК рPCL 4, КОДИРУЮЩАЯ ПОЛИПЕПТИД, ОБЛАДАЮЩИЙ АКТИВНОСТЬЮ НИТРИЛГИДРАТАЗЫ, ШТАММ БАКТЕРИЙ ESCHERICHIA COLI - ПРОДУЦЕНТ ПОЛИПЕПТИДА, ОБЛАДАЮЩЕГО АКТИВНОСТЬЮ НИТРИЛГИДРАТАЗЫ, СПОСОБ ПОЛУЧЕНИЯ НИТРИЛГИДРАТАЗЫ.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| УСТРОЙСТВО для ПООПЕРАЦИОННОЙ ТРАНСПОРТИРОВКИ ПОЛЫХ РЕЗИНОВЫХ ИЗДЕЛИЙ | 0 |
|
SU204555A1 |
| Способ гальванического снятия позолоты с серебряных изделий без заметного изменения их формы | 1923 |
|
SU12A1 |
