Изобретение касается композиций статистических сополимеров пропена, содержащих в качестве сомономера один или более α -олефинов C4-C10, и способа для получения указанных композиций.
Сополимерные композиции, описанные ниже, пригодны для получения пленок, способных свариваться при нагревании, которые можно использовать сами по себе благодаря их механическим и физическим свойствам, или при получении многослойных пленок, получаемых, например, путем совместной экструзии с полипропиленом. Кроме того, указанные композиции могут также быть использованы в пищевой промышленности благодаря низкому содержанию в них компонента, растворимого в ксилоле при комнатной температуре.
Использование композиций сополимеров пропена с олефином, главным образом, с этиленом и/или 1-бутеном или их смесей с другими полимерами олефинов для получения способных свариваться материалов известно в технике.
В пат. США N 4481336, кл. 220-295 описаны композиции, состоящие из двух фракций сополимера пропена с этиленом и/или пропена с бутеном, причем указанные композиции обладали способностью к сварке, прозрачностью и антиадгезионными свойствами. В указанном патенте отмечается, что содержание бутена в сополимерной фракции, которая присутствует в композиции в меньших количествах, должно быть больше 25 мас. % по отношению к общему количеству мономеров, а количество пропена должно быть менее 75 мас. %. Если количества бутена и пропена будут выходить за эти пределы (нижний и верхний соответственно), то полученная композиция будет плохо свариваться. Более того, пленка, полученная на основе указанной композиции, не должна быть ориентированной, иначе в процессе тепловой сварки часть свариваемой пленки может дать усадку.
В пат. США N 4822840, кл. 925-323 описаны сополимерные композиции, состоящие из статистических сополимеров пропена с этиленом и с α -олефинами. Основным компонентом является пропен, а этилен присутствует в количествах от 2,0 до 3,5 мас.%.
В итальянской заявке N 21601 A/90 описаны композиции, состоящие из сополимеров пропена с α -олефином и предпочтительно сополимера этилена с пропеном в качестве второго компонента сополимера.
Однако композиции, известные к настоящему времени, имеют характеристики, которые неадекватны для некоторых областей применения, для которых предназначены указанные композиции. Недостатки указанных композиций - это недостаточно низкая температура сварки, слишком высокая растворимость в ксилоле при 25oC и низкая кристалличность.
Специалистам в данной области хорошо известно, что для того чтобы получить композиции с низкой температурой сварки, они должны иметь низкую температуру плавления.
Было найдено, что новая композиция имеет низкую растворимость в ксилоле и более низкую температуру сварки, но высокую температуру плавления.
Особое преимущество сополимерных композиций изобретения состоит в том, что их можно использовать для получения слоистых одноосно и двухосно ориентированных пленок, имеющих низкую температуру сварки.
Кроме того, преимуществом сополимерных композиций изобретения является их жесткость. Указанные композиции имеют значительно более высокую жесткость, чем композиции, известные в технике.
Цель изобретения - частично кристаллические композиции, содержащие (по весу)
А) 20-60% статистического сополимера пропена с α -олефином С4-С10 (фракция A), содержащего от 1 до 10% α -олефина C4-C10;
В) 40-80% статистического сополимера пропена с α -олефином C4-C10 (фракция B), содержащего от 15 до 40% α -олефина C4-C10, одинакового или отличающегося от α -олефина, присутствующего во фракции A, где мас. % фракции B (% B) по отношению ко всей композиции и мас. % α -олефина C4-C10 во фракции B (C
% B C
Предпочтительными частично кристаллическими полиолефиновыми композициями являются те, в которых фракция A составляет 25-50 мас. % и фракция B - 50-75 мас. %.
Предпочтительными α -олефинами C4-C10 являются 1-бутен, 1-пентен, 1-гексен и 4-метил-1-пентен. Особенно предпочтителен 1-бутен.
Предпочтительное процентное содержание α -олефинов C4-C10 во фракции A составляет от 3 до 8 мас. %, а во фракции B предпочтительное содержание составляет от 20 до 30 мас. %.
Композиции изобретения имеют следующие свойства: температура плавления от 135 до 150oC; фракция, растворимая в ксилоле при 25oC, составляет менее 20%, предпочтительно менее 16%; модуль упругости при изгибе выше 700 МПа; температура сварки от 90o до 105oC.
Композиции, предложенные в изобретении, предпочтительно получают путем последовательной полимеризации мономеров в присутствии стереоспецифических катализаторов Циклера - Натта, нанесенных на дигалогениды магния в активной форме. Указанные катализаторы содержат как существенный элемент твердый компонент катализатора, состоящий из соединения титана, имеющего по крайней мере одну связь титан-галоген, и электронодонорного соединения, причем оба они нанесены на дигалогениды магния в активной форме. Дигалогенид магния присутствует предпочтительно в виде сферических частиц, имеющих узкое распределение по размеру.
Катализаторы, использованные в изобретении, характеризуются тем, что на них образуется полипропилен, имеющий показатель изотактичности выше 90, предпочтительно выше 95. Катализаторы, которые имеют вышеупомянутые характеристики, хорошо известны в патентной литературе. Особенно пригодны для применения катализаторы, описанные в пат. США N 4339054 и в европейской пат. N 45977. Другие примеры подходящих катализаторов описаны в пат. США N 4472524 и N 4473660.
Твердые компоненты катализатора, используемые при получении указанного катализатора, содержат в качестве электронодонора соединения, выбранные из простых эфиров, кетонов, лактонов, соединений, содержащих атомы N, P и/или S, а также эфиров моно- и дикарбоновых кислот. В частности, для этого пригодны эфиры фталевой кислоты, такие, как диизобутил-, диоктил- и дифенилфталат, и эфиры малоновой кислоты, так же, как диизобутил- и диэтилмалонат; арил- и алкилпивалаты; алкил-, циклоалкил- и арилмалеаты; алкил- и арикарбонаты, такие как диизобутилкарбонат, этилфенил - карбонат и дифенилкарбонат; эфиры янтарной кислоты, такие как моно- и диэтилсукцинат.
Другими подходящими электронодонорами являются простые 1,3-диэфиры формулы (I)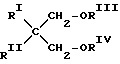
где RI и RII - одинаковые или различные алкильные C1-C18, циклоалкильные C3-C18 или арильные C6-C18 радикалы; RIII и RIV - одинаковые или различные алкильные радикалы, содержащие от 1 до 4 атомов углерода.
Простые эфиры такого типа описаны в европейском пат. N 361493.
Примерами простых эфиров формулы (I) являются 2-метил-2-изопропил-1,3-диметоксипропан, 2,3-диизобутил-1,3-диметоксипропан и 2-изопропил-2-циклопентил-1,3-диметоксипропан.
Описанные компоненты катализатора получают различными способами. Один из них состоит в измельчении в мельнице дигалогенида магния (безводного, содержащего менее 1% воды) совместно с соединением титана и с электронодонорным соединением в условиях, которые вызывают активацию дигалогенида магния; после этого измельченный продукт обрабатывают один или более раз избытком TiCl4 при температуре от 80o до 135oC и затем промывают несколько раз углеводородами (например, гексаном) до тех пор, пока в промывной воде не перестанут обнаруживаться ионы хлора.
По другому способу безводный дигалогенид магния вначале предварительно активируют с использованием известных методов, затем он реагирует с избытком TiCl4, содержащим растворенное в нем электронодонорное соединение. Процесс проводят при температуре от 80o до 135oC. Обработка TiCl4 может быть повторена. Затем твердый продукт промывают гексаном или другими углеводородными растворителями, чтобы удалить все следы непрореагировавшего TiCl4.
Другой способ основан на реакции между аддуктом MgCl2•nROH (особенно в форме сферических частиц), где n - обычно число от 1 до 3, а ROH - этанол, бутанол или изобутанол, и избытком TiCl4, содержащим растворенное электронодонорное соединение. Температура реакции обычно находится в пределах от 80 до 120oC. Твердый осадок затем отделяют и проводят его реакцию один или несколько раз с TiCl4, затем промывают углеводородным растворителем для того, чтобы удалить все следы непрореагировавшего TiCl4. Альтернативный способ состоит в том, что алкоголяты и хлоралкоголяты магния (последние получают, в частности, согласно методу, описанному в пат. США N 4220554) реагируют с избытком TiCl4, содержащим растворенное в нем электродонорное соединение, при тех же условиях реакции, которые были описаны выше.
Обычно содержание соединения титана в твердом компоненте катализатора (выраженное как содержание Ti) составляет от 0,5 до 10 мас.%, а количество электронодонорного соединения, которое остается фиксированным на твердом компоненте катализатора (внутренний донор), обычно находится в пределах от 5 до 20 мол.% по отношению к дигалогениду магния.
Соединения титана, которые могут быть использовано для получения твердого компонента катализатора (а), представляют собой галогениды и галогено-алкоголята титана. Предпочтительным соединением является четырехлористый титан.
Удовлетворительные результаты могут быть получены также с тригалогенидами титана, в частности, с TiCl3HR, TiCl3ARA и галогеноалкоголятами титана, такими как TiCl3OR, где R - фенильный радикал.
Реакции, показанные выше, приводят к образованию дигалогенида магния в активной форме. Кроме этих реакций, в технике известны другие реакции, которые приводят к образованию активированных галогенидов магния, исходя из других соединений магния, отличающихся от галогенидов, например таких, как карбоксилаты магния.
На активную форму дигалогенидов магния в твердых компонентах катализатора указывает тот факт, что в рентгенограмме компонента катализатора полуширина пика наиболее интенсивного рефлекса по крайней мере на 30% больше, чем полуширина рефлекса, который имеется на рентгенограмме неактивированного дигалогенида магния, или тот факт, что наиболее интенсивный рефлекс на рентгенограмме неактивированного дигалогенида магния (имеющего величину удельной поверхности менее 3 м2/г) отсутствует, но на его месте появляется гало с максимумом интенсивности, сдвинутым относительно положения наиболее интенсивного рефлекса на рентгенограмме неактивированного дигалогенида магния.
Наиболее активными формами являются те, у которых на рентгенограмме появляется вышеупомянутое гало.
Среди галогенидов магния предпочтительным соединением является хлорид. В случае наиболее активных форм хлорида магния на рентгенограмме компонента катализатора видно гало вместо рефлекса, который на рентгенограмме неактивированного хлорида магния появляется на расстоянии 2,56 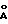 .
.
Все алюминийалкильные соединения, используемые в качестве сокатализаторов, представляют собой триалкилы алюминия, такие как триэтилалюминий, триизобутиолалюминий, три-н-бутилалюминий, а также линейные или циклические алюминийалкилы, содержащие два или более атомов Al, связанных через атомы 0 или N, или через группы SO2, SO3 и SO4.
Некоторые примеры этих соединений приведены ниже:
(C2H5)2Al-O-Al(C2H5)2,
(C2H5)2Al-N(C6H5)-Al (C2H5)2,
(C2H5)2Al-SO2-Al (C2H5)2,
CH3/[(CH3)Al-O/]n-Al(CH3)2,
-[(CH3)Al-O]n,
где n - число от 1 до 20.
Можно также использовать соединения общей формулы AlR2H и AlR2OR1, где R - алкильный радикал, имеющий от 1 до 6 атомов углерода, а R1 - арильный радикал, замещенный в одном или нескольких положениях.
Алюминийалкильные соединения обычно присутствуют в катализаторе в таких количествах, что отношение Al/Ti лежит в пределах от 1 до 1000.
Электронодонорные соединения, которые могут использоваться в качестве внешних доноров (добавляемых вместе с алюминийорганическим соединением), включают эфиры ароматических кислот (такие как алкилбензоаты), гетероциклические соединения (такие как 2,2,6,6-тетраметилпиперидин и 2,6-диизопропилпиперидин) и особенно соединения кремния, содержащие по крайней мере одну связь Si-OR (где R - углеводородный радикал). Примерами соединений кремния является (t-C4H9)2 Si(OCH3)2, (C6H11)2 Si(OCH3)2 и (C6H5)2•Si(OCH3)2. Простые 1,3-диэфиры формулы (I) также могут с успехом использоваться в качестве внешних доноров. В том случае, когда внутренним донором является один из простых 1,3-диэфиров формулы (I), внешний донор можно не принять.
Полимеризацию проводят по крайней мере в две стадии, в ходе которых получают фракции A и B изобретения. Эти фракции образуются в отдельных и последовательных стадиях. На каждой стадии процесс проводят в присутствии полученного полимера и катализатора, использованного на предыдущей стадии. Порядок, в котором получают фракции A и B, не имеет значения, однако предпочтительно получать сначала фракцию A и затем фракцию B.
Процесс полимеризации можно проводить в жидкой фазе, в присутствии или в отсутствии инертного растворителя, или в газовой фазе, или с использованием смешанных жидкой и газовой фаз. Предпочтительным является процесс, который проводится в газовой фазе.
Продолжительность и температура полимеризации во время полимеризационных стадий не критичны; при любой скорости предпочтительный интервал температур составляет от 20o до 100oC.
Регулирование молекулярной массы осуществляется путем использования известных регуляторов, предпочтительно водорода.
Процессу полимеризации может предшествовать процесс форполимеризации, в котором катализатор контактирует с малыми количествами олефина.
Следующие примеры иллюстрируют, но не ограничивают методы получения и характеристики композиции, предложенной в изобретении.
Получение твердого компонента катализатора.
Твердый компонент катализатора получают из аддукта MgCl2 3C2H5OH следующим образом: в инертной атмосфере вводят в колбу, погруженную в баню с температурой 120oC, при перемешивании 28,4 г безводного MgCl2, 49,5 г чистого безводного этанола, 100 мл вазелинового масла ROL OB/30, 100 мл силиконового масла (вязкость 350 сСт) и перемешивают до тех пор, пока MgCl2 не растворится. При этом образуется аддукт MgCl2 с этанолом в смеси с маслами. Затем горячую реакционную смесь переносят в инертной атмосфере в сосуд емкостью 1500 мл с рубашкой для нагрева, содержащий 150 мл вазелинового масла и 150 мл силиконового масла. Температуру смеси поддерживают на уровне 120oC и содержимое сосуда перемешивают при помощи мешалки Ультра Терракс Т-45 N фирмы "Джанке унд Кункель К.Г. Ика Верке". Перемешивание продолжают 3 мин при 3000 об/мин. Затем смесь выгружают в сосуд емкостью 2 л, содержащий 1000 мл безводного н-гептана, продолжают перемешивание и охлаждают таким образом, чтобы температура не превышала 0oC. Полученные таким образом микросферы MgCl2 • 3C2H5OH выделяют путем фильтрования и сушат под вакуумом при комнатной температуре. Затем от полученного аддукта отщепляют спирт путем постепенного повышения температуры от 50o до 100oC в токе азота до тех пор, пока содержание спирта не снизится до 1,5 молей на моль MgCl2. Аддукт после частичного отщепления спирта имеет удельную поверхность 9,1 м2/г и кажущуюся плотность 0,564 г/см3.
25 г указанного аддукта добавляют к 625 мл TiCl4 при перемешивании при 0oC. Затем смесь нагревают до 100oC в течение одного часа. Когда температура достигает 40oC, добавляют диизобутилфталат при мольном отношении магний/диизобутилфталат, равном 8.
Содержимое реактора нагревают до 100oC в течение двух часов, затем дают твердым частицам отстояться, и горючую жидкость декантируют при помощи сифона. Осадок промывают 6 раз 200 мл безводного гексана при 60oC и затем три раза при комнатной температуре. Твердый осадок после высушивания под вакуумом имеет следующие характеристики:
пористость - 0,261 см3/г;
удельная поверхность - 66,5 м2/г;
объемная плотность - 0,55 г/см3.
Примеры 1, 2, 3, 4 и сравнительные примеры 1с и 2с.
Общие методики.
Опыты по полимеризации проводили в автоклаве из нержавеющей стали емкостью 22 л, снабженном магнитной винтовой мешалкой, вращающейся со скоростью 90 об/мин.
Газовую фазу анализировали непрерывно при помощи газового хроматографа, чтобы определить концентрацию 1-бутена, пропилена и водорода. Во время полимеризации 1-бутен, пропилен и водород подавались в автоклав через регуляторы-расходомеры, чтобы поддерживать постоянную концентрацию в газовой фазе.
Реакцию проводили при постоянной температуре и давлении, если это не оговорено специально.
Периодический процесс проводили в две стадии, обе в газовой фазе: на первой стадии проводили сополимеризацию пропилена с 1-бутеном, чтобы получить фракцию A, на второй стадии проводили сополимеризацию тех же сополимеров, чтобы получить фракцию B.
1-ая стадия. При комнатной температуре вводили в автоклав по порядку
а) такие количества пропилена, 1-бутена и водорода, чтобы получить желаемый состав газовой фазы и желаемое давление;
б) каталитическую систему, состоящую из твердого компонента (около 1 г), полученного, как описано выше, и из смеси 25 мл 25%-ного раствора триэтилалюминия (ТЭА) в гексане с таким количеством дициклопентилдиметоксисилана (ДЦПМС), которое обеспечивает молярное отношение ТЭА/ДЦПМС = 8. Каталитическую систему инжектировали в реактор давлением пропилена. Затем температуру доводили до рабочего уровня (примерно за 10 мин) и реакцию продолжали в течение желаемого времени. Непрореагировавшие мономеры удаляли путем дегазации при 60oC при атмосферном давлении, и образец сополимера отбирали через кран, расположенный в дне автоклава, и отправляли на анализ. Операция продолжалась около 10 мин.
2-я стадия. В реактор вновь вводили по порядку пропилен, 1-бутен и водород в таком соотношении и количестве, чтобы получить желаемый состав и давление газовой фазы, и затем реакцию продолжали в течение требуемого времени, которое менялось в зависимости от реакционноспособности каталитической системы и требуемого процентного содержания фракции B.
После окончания полимеризации полимер выгружали со дна автоклава, стабилизировали пентаэритритил-тетракис 3-(3,5-дитрет-бутил-4-гидроксифенил)пропаноатом (0,2 мас.%) и 2,6-дитрет-бутил-п-крезолом (ВНТ) (0,2 мас.%) и сушили в сушильном шкафу в токе азота при 60oC.
Массовый процент фракции A и B (% A и % B соответственно) по отношению к полученной композиции, массовый процент бутена, содержащегося во фракции B, ( (C
% A = ClF/ClA
(C
J • V • B = [J, V • F - (% A J • V •A/100)] 100/% B,
где ClF и ClA - это соответственно содержание хлора (из остатков катализатора) в полученной целевой композиции и во фракции A;
- (C
- J • V • F и J • V • A - это соответственно характеристическая вязкость целевой композиции и фракции A.
Все проведенные опыты и относительные условия их проведения приведены в табл. 1 и 2, а в табл. 3 показаны данные по выходу полимеров при полимеризации и характеристики полученных композиций.
Для того чтобы охарактеризовать композиции, описанные в примерах, были использованы следующие аналитические методы:
- содержание 1-бутена определяли методом ИК-спектроскопии;
- температуру плавления определяли методом ДСК;
- фракцию, растворимую в ксилоле, определяли путем растворения образца полимера в ксилоле при 125oC и последующего охлаждения его до комнатной температуры. Растворимую и нерастворимую фракцию разделяли фильтрованием;
- индекс расплава определяли по методу ASTMD 1238, условие L;
- характеристическую вязкость определяли в тетрагидронафталине при 135oC;
- мутность определяли по методу ASTMD 1003 на образцах толщиной 1 мм;
- температуру начала сварки определяли следующим образом: получали пленки толщиной 50 мкм путем экструзии композиций, полученных в примерах, при температуре около 200oC. Каждую полученную таким образом пленку накладывали на пленку из полипропилена, имеющего показатель изотактичности 97 (в кипящем н-гептане), индекс расплава 4,5 г/10 мин и толщина указанной пленки составляла 560 мкм. Наложенные друг на друга пленки склеивали в прессе при 200oC и нагрузке 9000 кг.
Указанную нагрузку поддерживали в течение 5 мин. Полученный склеенные пленки затем вытягивали в шесть раз по длине и ширине при помощи вытяжного устройства для пленок ТМ Long, получая в результате пленки толщиной около 20 мкм. Из указанных пленок вырезали образцы размером 5•10 см. Величины, характеризующие способность к сварке, получали путем приложения нагрузки 200 г к сваренным образцам. Для каждого измерения два описанных выше двуслойных образца накладывались друг на друга свариваемыми слоями, полученными из композиций, описанных в примерах, так что эти слои соприкасались между собой. Затем указанные наложенные друг на друга образцы сваривали в продольном направлении через 5 см при помощи комбинированного лабораторного сварочного устройства Ксентинел, модель 12-12 AS. Время сварки 5 с, давление 1,2 ат и ширина сварного шва 2,5 см. Температура сварки возрастала на 2oC для каждого следующего измеряемого образца. Сваренные образцы затем разрезали, чтобы получить полоски размером 2,5•10 см, несваренные концы которых присоединялись к динамометру, и определялась минимальная температура сварки, при которой сварной шов не разрушался при нагрузке 200 г. Эта температура для наложенных друг на друга образцов и представляет собой температуру начала сварки (Seal initiation temperature - SJT).
Композиция частично кристаллических полиолефинов содержит (по весу):
А) 20-60% статистического сополимера пропена с α - олефином С4-С10 (фракция A), содержащего от 1 до 10% α -олефина C4-C10;
В) 40-80% статистического сополимера пропена с α - олефином C4-C10 (фракция B), содержащего от 15 до 40% α -олефина C4-C10, одинакового или отличающегося от олефина, присутствующего во фракции A, где мас.% фракции B (% B) по отношению ко всей композиции и мас.% α -олефина C4-C10 во фракции B ( (C
% B • C
2 с. и 8 з.п. ф-лы, 3 табл.
% B•C
2. Композиция по п.1, отличающаяся тем, что альфа-олефины C4 - C1 0 во фракциях A и B одни и те же.
| US, патент, 4481336, кл | |||
| Ветряный много клапанный двигатель | 1921 |
|
SU220A1 |
| US, патент, 4822840, кл | |||
| Телескоп | 1920 |
|
SU525A1 |
| EP, патент, 045977, кл | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |

