Изобретение относится к способу получения (+) (2R)-эндо-норборнеола и (-)-(2S)-эндо-норборнела и последующего их превращения, соответственно, в фармацевтические средства, 5-(3-[(2S)-эндо-норборнеола и последующего их превращения, соответственно, в фармацевтические средства, 5-(3-[(2S)-экзо-бицикло [2.2.1] гепти-2-илокси] - 4-метоксифенил)-3,4,5,6-тетрагидропирамидин-2 (1H)-он формулы: ,
,
и его энантиомер, 5-(3- [(2R.) -экзо-бицикло [2.2.1.]гепт-2- илокси]- 4- метоксифенил)-3,4,5,6-тетрагидропиримидин-2 (1Н)-он, формулы: .
.
Настоящее изобретение также относится к этим конкретным оптически активным фармацевтическим средствам самим по себе, а также к промежуточным соединениям (формулы которых с (III) по (VI) приведены ниже), используемым при их синтезе.
(2R) -Эндо-Норборнеол альтерна тивно называют (2R) - эндобицикло - [2.2.1. ] гептан-2-олом или (1S, 2R, 4R) -бицикло [2.2.1.] гептан-2-олом. Аналогично, энантиомерный (2, S)-эндо-норборнеол альтернативно называют (2S)-эндо-бицикло [2.2.1. ] гептан-2-олом или (1R, 2S, 4S)-бицикло [2.2.1.] гептан-2-олом. Аналогично, производные замещающие группы (2S)-экзо-бицикло [2.2.1. ] гепт-2-ила и (2R)-экзо-бицикло [2.2.1.] гепт-2-ила альтернативно называют, соответственно, (1S, 2S, 4R)-бицикло [2.2.1.] гепт-2-илом и (1R, 2S, 4S)-бицикло [2.2.1.] гепт-2-илом.
Настоящие соединения формул (I) и (II) представляют собой особенно ценные виды соединений, подробно описанные Саккомано с сотр. в опубликованной Международной патентной заявке WO87/06576, обладающие полезностью как антидепрессанты. Несмотря на то, что в этой ссылке конкретно описан рацемический 5- (3- [(2S)-экзо-бицикло [2.2.1.] гепт-2-илокси]-4-метоксифенил)-3,4,5,6- тетрагидропиримидин-2-(1H)-он, там нет конкретного раскрытия настоящих оптически активных его изомеров или какого-либо конкретного способа их получения. Как дальше описано здесь, а также в параллельно поданной Международной патентной заявке N PCT/US89/00000 [Досье N 7641/PH], настоящие соединения формул (I) и (II) также являются особенно ценными для лечения астмы и некоторых кожных заболеваний.
До настоящего времени оптически активные (2R) - и (2S)- эндо-норборнеолы получали разделением рацемического экзонорборнеолового сложного эфира гемифталевой кислоты о помощью оптически активных фенэтиламинов, гидролизом до оптически активных экзонорборнеолов, окислением CrO3 до оптически активных норборнанонов и наконец восстановлением Li(S-Вu)3BH до нужных эндо-изомеров. Ирвин с сотр. Irwin et al., T. Am Chem. Soc V. 98, pp. 8476-8481 (1979) Согласно вышеуказанной ссылке энантиомерное обогащение эндо-норборнеолов осуществляли путем неполного восстановления рацемического 2-норборнанона при катализе спиртовой дегидрогеназой из печени лошади, в то время как неполное окисление рацемического экзо-норборнеола, катализируемого тем де самым ферментом, давало обогащенные энантиомерами экзо-норборнеолы, (-) -Экзо-норборнеол также получен из норборнена путем асимметрического гидроборирования, Brown, et. al. T. Org. Chem, V.47, pp. 5065-5069 (1982).
До сих пор некоторые хиральные спирты разделяют с помощью трансэстерификации, катализируемой свиной панкреатической липазой в почти безводном органическом растворителе. Kirchner et. at, T. Am. Chem. Soc. V. 107, pp. 7072-7076 (1985). Например, 47% превращение рацемического 2-октанола и 2,2,2- трихлороэтилбутирата давало (R.)-2- октилбузират высокой оптической чистоты. Однако, когда этот способ применяли к рацемическому экзонорборнеолу, то как восстановленный спирт, так и полученный сложный эфир бутират оставались по существу рацемическими, даже несмотря на то, что трансэстерификация явно происходила под действием фермента (показано по отсутствию реакции без фермента).
Несмотря на тот факт, что катализируемая липазой трансэстерификация экзо-норборнеола не дает полезного способа оптического разделения экзо-изомеров, авторы настойчиво продолжали работы и открыли, что этот способ является простым и эффективным способом оптического разделения указанного эндо-норборнеола. Таким образом, в одном из своих аспектов настоящее изобретение относится к способу получения оптически активных эндо-бицикло [2.2.1.] гептан-2-олов(эндо-норборнеолов), который содержит стадии:
(а) частичной трансэстерификации между рацемическим эндобицикло [2.2.1.] гептан-2-олом и 2,2,2-трихлороэтилбутиратом в по существу безводном реакционно инертном органическом растворителе в присутствии панкреатической липазы, полученной от млекопитающих,
(Ь) выделение из полученной смеси непрореагировавшего (-)- (2S)-эндо-бицикло [2.2.1.] гептан-2-ола [(2S)-эндонорборнеола/ формулы: ,
,
и (2R)-эндо-бицикло [2.2.1.]гепт-2-ил бутирата, сложного эфира, который гидролизуют, чтобы получить энантиомерный (+)-(2R)- эндо-бицикло [2.2.1.] гептан-2-ол [(2R)-эндонорборнеол] формулы: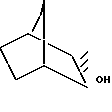 .
.
Используемый здесь выше и повсюду здесь в тексте термин "реакционно инертный растворитель" относится к растворителю, который не взаимодействует с исходными веществами, реагентами, промежуточными соединениями или продуктами реакции неким образом, который вредно влияет на выход нужного продукта или продуктов.
В предпочтительных вариантах настоящего способа фермент происходит от свиньи, растворителем является простой эфир, а полученные оптически активные (2R) - и (2S)-эндо-бицикло [2.2.1.] гептан-2-олы, далее контактируют с З-гидрокси-4-метоксибензальдегидом в присутствии трифенилфосфина и диэтилазадикарбоксилата, чтобы получить, соответственно, оптически активные альдегиды, 3 -(2S)-экзо-бицикло [2.2.1.] гепт-2-илокси)-4- метоксибензальдегид формулы: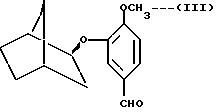 ,
,
и 3-(2R)-экзо-бицикло[2.2.1. ]гепт-2-илокси)-4-метоксибензальдегид формулы: .
.
Эти соединения в свою очередь затем превращают в соединения формулы (I) и (II) посредством следующих стадий и промежуточных веществ:
(а) реакция оптически активного 3-(экзо-бицикло[2.2.1.]гепт- -2-илокси)-4- метоксибензальдегида (III) или (IV) с не менее 2 моль-эквивалентами 2-цианоуксусной кислоты в пиридине в присутствии каталитического количества пиперидина при температуре в пределах примерно 25-100oC,чтобы получить оптически активный 3-(3- экзо-бицикло [2.2.1. ] гепт-2-илокси)-4-метоксифенил)пентандинитрил формулы (V) и (VI),приведенных ниже, где V представляет собой CN,
(Ь) обычная гидратация указанного пентандинитрила, чтобы получить оптически активный 3-(3-(экзо-бицикло [2.2.1. ]гепт-2-илокси) -4-метоксифенил)глютарамид формулы (V) или (VI), приведенные ниже, где V представляет собой CONH2, и
(с) циклизация указанного глютарамида под действием молярного избытка тетраацетата свинца в пиридине, чтобы получить оптически активный 5-(3-(экзо-бицикло[2.2.1. ] гепт-2-илокси)-4- метоксифенил)-3,4,5,6,-тетрагидроксипиримидин-2-(1H)-он формулы (I) или (II),приведенные выше.
Настоящее изобретение также относится к недоступным до настоящего времени оптически активным видам соединения формул (I) и (II), приведенных выше, и к оптически активным промежуточным соединениям формул: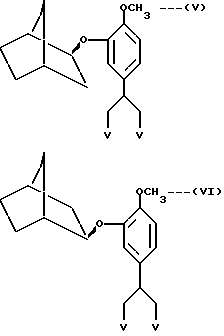 ,
,
где V представляет собой - CN или - CONH2.
Различные аспекты настоящего изобретения легко осуществить. Соответственно, рацемический эндо-норборнеол и 2,2,2-трихлорэтилбутирал, сложный эфир, по существу в моль-эквивалентных количествах растворяют в реакционно инертном органическом растворителе. Предпочтительными растворителями в данном случае являются простые эфиры, такие, как сам эфир, диизопропиловый эфир, тетрагидрофуран или диоксан, которые по существу являются безводными и в то же время легко растворяют указанные реагенты. Панкреатическую липазу, полученную от млекопитающих (предпочтительно свиная панкреатическая липаза, которую легко получить из коммерческих источников), добавляют по частям в виде сухого порошка в нужных количествах, чтобы поддержать разумную скорость трансэстерификации. Температуру, являющуюся очень критичной, обычно поддерживают в пределах примерно 15-40oC. Если температура слишком низка, скорость будет слишком низкой, в то время как слишком высокие температуры будут быстро дезактивировать фермент. За реакцией следят аналитическими методами (удобно с помощью протонного магнитного резонанса, с помощью которого легко различать исходные материалы и полученный сложный эфир) и заканчивают ее, когда реакция завершается на 40-50% с тем, чтобы обеспечить максимальную оптическую чистоту выделенных (2S)-эндо-норборнеола и (2R)-эндо-норборнилбутирата. Эти продукты разделяют известными способами, удобно с помощью хроматографических способов, а сложный эфир гидролизуют обычными способами, хорошо известными в данной области, чтобы получить (2R)-эндо-норборнеол.
Согласно настоящему изобретению полученные оптически активные (2R)- и (2S)-эндо-норборнеолы превращают, соответственно, в альдегиды вышеуказанных формул (III) и (IV ) по способу, описанному в цитированной ссылке WO89/06576 и кратко изложенному выше.
Затем, согласно настоящему изобретению, альдегиды вышеуказанных формул (III) и (IV) конденсируют с не менее, чем с двумя мольэквивалентными эквивалентными цианоуксусной кислоты, чтобы получить динитрил формулы (V) или (VI), где V представляет собой CN. Удобно эту конденсацию осуществлять в пиридине, который также частично выполняет роль основного катализатора, в присутствии вторичного амина, предпочтительно незатрудненного, такого, как пиперидин или пирролидин, в количестве, являющемся, как правило, молярным избытком. Хотя начальные стадии реакции можно осуществить при окружающей температуре, реакцию (включая декарбоксилирование) лучше всего осуществлять при нагревании при температуре в пределах примерно 80-110oC.
Динитрилы формулы (V) или (VI) затем гидратируют в бис-амиды формулы (V) или (VI), где V представляет собой CONH2, известными способами. Удобно это осуществлять путем контактирования динитрила в реакционно инертном водном органическом растворителе (например, 2:1 ацетон:H2О) с полумолярным количеством H2O2 в присутствии избытка Na2CO3 при температуре в пределах примерно 0-30oC.
Наконец, бис-амиды формулы(VI) циклизуют, чтобы получить оптически активный 5-(3- экзо-бицикло[2.2.1. ] гепт-2-илокси)-4- метоксифенил)-3,4,5,6-тетрагидропиримидин-2(1H)-он формулы (I) или (II). Это лучше всего осуществить с помощью молярного избытка тетраацетата свинца в избытке пиридина в качестве растворителя. Температура не является критической, причем, как правило, температуры в пределах 0-60oC оказываются удовлетворительными. Удобно использовать температуры окружающей среды, чтобы избежать расходов на нагревание и охлаждение.
Как указано на с.25 цитированной выше патентной заявки WO87/06576, 5-(3-(экзо-бицикло[2.2.1.] гепт-2-илокси)-4- метокси-фенил)-3,4,5,6,-тетрагидропиримидин-2(1H)-он обладает in vitro активностью ингибировать фосфодиэстеразы, полученные из коры головного мозга крыс. Более уместной для полезности этого средства при лечении астмы является его активность ингибировать фосфодиэстеразы, полученные из легкого морской свинки, как подробно описано в примере 1, где настоящие оптически активные соединения формул (I) и (II) проявляют одинаковую активность. Полезность для лечения астмы далее подтверждается способностью настоящих соединений ингибировать in vivo эозинофильную миграцию в сенсибилизированной легочной ткани в морских свинках, которым ввели антигены, как подробно описано в примере 2. Полезность настоящих соединений для лечения псориаза и дерматита, вызванных повышенной контактной чувствительностью, подтверждается способностью настоящих соединений ингибировать in vivo отек кожи в морских свинках, сенсибилизированных к овальбумину, как подробно описано в примере 3.
В системном лечении астмы или воспалительных заболеваниях кожи с помощью соединения формулы (I) или (II), либо с помощью соответствующего рацемического соединения, дозировка обычно составляет примерно от 0,01 до 2 мг/кг/день (0,5-100 мг/день при типичном весе человека 50 кг) в виде единственной или раздельных доз, безотносительно от пути введения. Конечно, в зависимости от конкретного соединения и от конкретной природы индивидуальной болезни могут быть предписаны дозировки вне указанных пределов по указанию лечащего врача. При лечении астмы, как правило, предпочтительны внутриназальные средства (капли или аэрозоль), ингаляции аэрозоля через рот и обычное оральное введение. Однако, если больной не может глотать, либо иным образом нарушено оральное поглощение, предпочтительным системным путем введения будет парентеральный путь (внутримышечный, внутривенный). При лечении воспалительных заболеваний кожи предпочтительным путем введения является оральный или местный. При лечении воспалительных заболеваний дыхательных путей предпочтительным путем введения является внутриназальный или оральный.
Соединения по настоящему изобретению обычно вводят в виде фармацевтических композиций, содержащих одно из указанных соединений вместе с фармацевтически допустимым носителем или разбавителем. Такие композиции обычно включают в рецептуру известным способом, используя твердые или жидкие носители или разбавители, соответствующие виду нужного введения: для орального введения в форме таблеток, твердых или мягких желатиновых капсул, суспензий, гранул, порошков и т.п., для парентерального введения в форме растворов или суспензий для инъекции и т.п., для местного введения в форме растворов, лосьонов, мазей, кремов и т.п., обычно содержащих примерно от 0,1 до 1%(вес/объем) активного ингредиента, и для внутриназального или ингаляторного введения обычно в виде раствора, содержащего от 0,1 до 1% (вес/объем).
Настоящее изобретение проиллюстрировано следующими примерами, но оно не ограничено его подробностями.
Пример 1. Ингибирование легочной фосфодиэстеразы (PDEIY).
Легочную ткань от морских свинок поместили в гомогенизационный буферный раствор (20 миллимоль/л Бистрис, 5 миллимоль/л 2-меркапто-этанола, 2 миллимоль/л бензамидина, 2 миллимоль/л ЭДТА, 50 миллимоль/л ацетата натрия, pH 6,5) при концентрации 10 мл/мг ткани. Ткань гомогенизировали с помощью гомогенизатора тканей Tekmar на полной скорости в течение 10 с. Фенилметилсульфонилфторид (ФМСФ, 50 миллимоль/л в 2-пропаноле) добавляли к буферу непосредственно перед гомогенизацией, чтобы получить конечную концентрацию ФМСФ 50 микромоль/л. Гомогенат центрифугировали при 12000 g в течение 10 мин при 4oC. Поверхностный раствор отфильтровали через марлю и стекловату, а затем подали в колонку 17 х 1,5 см DEAE-Sepharose CL-6В, предварительно уравновешенную гомогенизационным буфером при 4oC. Использовали скорость потока 1 мл/мин. После пропускания поверхностного раствора через колонку, колонку промывали объемом гомогенизационного буфера, по меньшей мере вдвое превышающим объем поверхностного раствора. Фосфодиэстеразу промывали линейным градиентом ацетата натрия 0,05-01 моль/л. Собрали сто фракций по 5 мл. Фракции сохранили на основании проявляемой ими специфической фосфодиэстеразной (PDEIV) активности, определяемой с помощью гидролиза с [3H] сАМР и в сравнении со способностью известной PDEIV.
Получение испытываемых соединений. Соединения растворяли в ДМСО в концентрации 10-2 моль/л, а затем разбавляли водой 1oC25 (4х10 моль/л соединения, 4% ДМСО). Затем осуществляли последовательные разбавления в 4% ДМСО, чтобы получить нужные концентрации. Конечная концентрация ДМСО в пробирках составляла 1%.
Следующие вещества добавили в перечисленном порядке в стеклянные пробирки 12 х 75 мм, в трех экземплярах, при 0oC, (все концентрации приведены как конечные концентрации в пробирке):
25 микролитров соединения или ДМСО (1%, для контроля и пустой опыт)
25 микролитров аналитического буфера (50 миллимоль/л Трис, 10 миллимоль/л MgCl2, pH 7,5)
25 микролитров /3H/-сАМР (1 микромоль/л)
25 микролитров фосфодиэстеразного фермента PDEIV (для пустого опыта фермент предварительно инкубировали в бане с кипящей водой в течение 10 мин)
Реакционные пробирки встряхивали и поместили в водяную баню (37oC) на 10 мин, после этого времени реакцию останавливали, помещая пробирки в баню с кипящей водой на 2 минуты. Промывочный буфер (0,5 мл,0,1 моль/л HEPES/0,1 моль/л NaCl, pH 8,5) добавляли в каждую пробирку на бане со с льдом. Содержимое каждой пробирки наносили на колонку Аffi-Gel60l (гель с боронатным средством, объем слоя 1,2 мл), предварительно уравновешенной промывочным буфером. [3H] сАМР промывали 2 х 6 мл промывочным буфером, а затем [3H] 5'АМР элюировали 6 мл 0,25 моль/л уксусной кислоты. После вихревого перемешивания 1 мл элюата добавили к 3 мл жидкости для атомной сцинциляции в подходящей ампуле, перемешали и подсчитали [3H].
Процент ингибирования определяли по формуле: .
.
Концентрацию ингибирования IC50 определяют как концентрацию соединения, которая ингибирует 50% специфического гидролиза [3H] сАМР в [3H] 5'АМР.
В этом тесте показано, что рацемический 5-(3-(экзо-бицикло/ [2.2.1.] гепт-2-илокси)-4-метоксифенил)-3,4,5,6, -тетрагидропиримидин-2(1Н)-он имеет IC50 равную 0,5 микромоль/л. В этом испытании по существу такую же степень активности наблюдали для каждого из двух соответствующих оптически активных энантиомерных соединений.
Пример 2. Ингибирование эозинофильной миграции в сенсибилизированную легочную ткань с введением антигеном в морских свинках.
Нормальных морских свинок Hartiey (300-350 граммов), полученных из Charles River Laboratories содержали в течение 5-7 дней перед сенсибилизированием. Затем морских свинок сенсибилизировали 0,5 мг/кг анти-ОА IgGl или солевым раствором в качестве контроля. Через 48-72 ч морским свинкам дали перорально группам по шесть животных каждого из соединений вплоть до 32 мг/кг, используя 2% Твин 80 в качестве носителя. Через 1-1,5 ч животным внутрибрюшинно ввели 5 мг/кг пиридамина. Через тридцать минут после введения пириламина животных подвергали воздействию в течение 10 мин 0,1 % овальбуминно аэрозоля, за которым следовал 15-минутный период распада облака в Tri-R Airborne Infection Apparatus (Поток сжатого воздуха= 20л/мин, поток основного воздуха= 8,4 л/мин) Морских свинок вынули из устройства и содержали в клетках в течение 18 ч перед умерщвлением и последующей процедурой промывания легких.
Морских свинок убили с помощью 3 мл уретана (0,5 г/мл), и трахею отделили от окружающей ткани. Хирургическую нить завязали вокруг трахеи и сделали надрез в трахее примерно 1-2 см от тимусной железы. Тупую иглу 15G для питания длиной 1 см вставили в трахею и затянули нить, чтобы закрепить иглу на месте. Тремя порциями по 10 мл солевого раствора промывали легкие пять раз. Примерно 20-25 мл жидкости выделили и поместили в 50 мл коническую пробирку на льду. Определенную часть (0,475 мл) промывочной жидкости поместили в полистирольную пробирку, содержащую 0,025 мл 2% моющего средства Тритон Х-100 (в двух экз).
Определенную часть образца с Тритоном разбавили 1 мл /0,1% Тритонового буфера (pH 7,0). К определенной части(0,025 мл) разбавленного образца добавили дополнительное количество 0,125 мл PBS/0,1% тритонового буфера. Колориметрическую реакцию инициировали путем добавления 0,300 мл 0,9 мг/мл о-фенилендиаминдигидрохлорида(OPD) в 50 миллимоль/л трис буфера /0,1% Тритона (pH 8,0) плюс 1 микролитр/мл перекиси водорода. После 5-минутного инкубирования добавили 0,250 мл 4н. раствора серной кислоты, чтобы остановить реакцию. Оптическую плотность смеси измеряли на длине волны 490 нм, при вычитании фоновой оптической плотности (пустая пробирка).
Повторные показания оптической плотности усредняли, чтобы получить единственное значение для каждого животного. Среднее значение оптической плотности и + /- стандартную ошибку рассчитывали, используя шесть полученных значений для каждой группы животных. Специфическую EPD реакцию из-за введенного антигена рассчитывали по формуле:
1000 х [Среднее значение оптическ. плотн. (сенсиб, введен антиген) - среднее значение оптич. плотн (несенсиб. введен антиген)]
Процент ингибирования специфической ЕРО реакции из-за предварительной лекарственной обработки рассчитывали по формуле: .
.
В этом испытании рацемический 5-(3-экзо-бицикло[2.2.I.] -гепт-2-илокси(-4-метокси-3,4,5,6,-тетрагидропиримидин-2(1H)-он проявил эффективную дозу ED50 равную 10 мг/кг.
Пример 3. Ингибирование отека кажи у морских свинок, сенсибилизированных к овальбумину
Четырех морских свинок (Hartley, самцы, 350-400 г) сенсибилизировали антиальбуминным IgGl антителом. Двум морским свинкам орально ввели испытываемое соединение при дозировке 32 мг/кг, а двум остальным морским свинкам ввели носитель (2% Твин-80). Через один час после введения каждой морской свинке внутривенно ввели 1 мл Evan Blue (7 мг/мл), а затем через их кожу им ввели подкожно 0,1 мл овальбумина (0,1% ) или PBS. Через двадцать минут после введения кожу удаляли, и визуально обследовали место отечной кожи (круглые голубые пятна в местах введения антигена).
Введение овальбумина приводило к образованию отечности на коже в месте введения овальбумина, в то время как введение PBS давало меньший отек кожи. Как интенсивность, так и площадь голубых пятен в местах введения антигена были значительно меньше у двух морских свинок, которым ввели рацемический 5- (3- экзо-бицикло [2.2.1.]гепт-2-илокси)-4-метоксифенил)-3,4,5,6, -тетрагидропиримидин-2(1Н)он, по сравнению с отеком у животных, которым ввели носитель.
Этот результат наглядно показывает, что это соединение является эффективным против отека кожи у морских свинок, вызванного антигеном.
Пример 4. (+) -(2R)- и (-)-(2S)-эндо-Норботэнеол
[(2R)- и (2S)-эндо-Бицикло [2.2.1.] гептан-2-ол/
dl -Эндонорборнеол (5,0 г, 44,6 миллимоль) и трихлороэтилбутират (5,1 г, 23,2 миллимоль) растворили в 40 мл диэтилового эфира. Добавили молекулярных сит 4А (4г), и смесь перемешивали при комнатной температуре. Порционно добавляли свиную панкреатическую липазу (Сигма, тип II, неочищена) в количествах 0,5 г, 1,0 г, 1,0 г, 1,0 г и 0,5 г в моменты времени, соответственно 0, 20, 43, 50 и 67 ч. Ход реакции контролировали с помощью протонного магнитного резонанса, и примерно при 50% превращении (92 ч) реакционную смесь отфильтровывали через диатомовую землю и выпарили под вакуумом без нагревания. (Спирт возгонялся легко) Неочищенный остаток подвергали флэш-хроматографии с программируемым градиентом элюента 2-25% эфир/гексан, чтобы выделить 2,9 г (15,9 миллимоль) (2R) - эндонорборнилбутирата в виде неочищенного масла и 1,8 г (16 миллимоль) (2S)-эендонорборнеола в виде белого твердого вещества, [альфа]D = - 2,03oC, е.е. 87,2% ( на основании протонного магнитного резонанса полученного сложного эфира (S)- альфаметокси-альфа-(трифторометил)фенилуксусной кислоты (МТРА)). Ввиду того, что удельное вращение так мало, величины е.е., определенные протонным магнитным резонансом, являются намного более надежной мерой оптической чистоты.
Выделенный эндонорборнилбутират (2,3 г , 12,6 миллимоль), К2СО3 (2,5 г, 18,0 миллимоль) и метанол (65 мл) перемешивали при комнатной температуре в течение 64 ч перед распределением между диэтиловым эфиром и водой. Органическую часть промывали водой и солевым раствором, сушили (Na2SO4), отфильтровывали и сконцентрировали под вакуумом, получив 1,3 г (11,6 миллимоль, выход 91,9%), (2R)-эндонорборнеола,
[альфа] D = + 2,7oC, е.е. 87,6% (на основании протонного магнитного резонанса сложного эфира МТРА).
Эти стадии способа были повторены при завершении сложноэфирного обмена только до 44%, в результате получили (2S)-эндонорборнеол низкой оптической чистоты с более, чем 90% выходом, [альфа]D = - 0,88, е.е. 71,4 (на основании протонного магнитного резонанса, как указано выше), и (2R) - эндонорборнеол более высокой оптической чистоты с выходом 56,4, е.е. свыше 95% ( на основании протонного магнитного резонанса, как указано выше)
Пример 5. 3-[(2S)-экзо-Бицикло [2.2.1.] гепт-2-илокси/-4-метоксибен-зальдегид
Диэтилазодикарбоксилат (28,5 г, 27,7 мл, 0,141 моль) и три-(фенилфосфин (36,9г, 0,141 моль) растворили в 200 мл тетрагидрофурана. К этому раствору добавили (+) - (2R)-эндо-норборнеол (7,9г, 0,0705 молью в 100 мл тетрагидрофурана, а затем З-гидрокси-4- метоксибензальдегид (изованиллин, 21,4 г, 0,141 моль) в 100 мл тетрагидрофурана. Полученную смесь нагревали с обратным холодильником в течение двух дней, затем охладили, разбавили 1,5 литрами эфира, промывали последовательно половинными объемами воды (2х), 0,5 н. раствора NaOH (2х), воды и рассола, сушили (NaOH), отгоняли из нее легкие фракции, и остаток хроматографировали на силикагеле, который градиентно элюировали этилацетатом от 0 до 10%, получив 8,5 г названного выше продукта, 8,5 г (49%), [альфа]D = + 24,5o (дейтерохлороформ)
Таким же способом (-)- (2S)-эндо-норборнеол превратили в 3-[(2R)-экзо-бицикло [2.2.1. ]гепт-2- илокси]-4-метоксибензальдегид, который имел аналогичные физические свойства за исключением знака вращения.
Пример 6. (3-(3-[(2S)-экзо-Бицикло[2.2.1. ] гепт-2илокси]метоксифенил) пентандинитрил
Названный продукт предшествующего примера (8,5 г, 0,0346 моль) растворили в 250 мл пиридина. Добавили цианоуксусную кислоту (14,6 г,0,171 моль) и пиперидин (5 мл), и смесь перемешивали при комнатной температуре в течение 4 часов, а затем при 60oC в течение 2 ч и наконец при 100oC в течение 24 ч. Растворитель удалили отгонкой под вакуумом, и остаток растворили в 250 мл этилацетата, промывали насыщенным NaHCO3, а затем водой, повторно отогнали легкие фракции и провели кристаллизацию из изопропилового спирта/изопропилового эфира, получив 5,84 г(54%) вышеназванного продукта, т.пл. 121-123oC, [альфа]D = + 17,8o(дейтерохлороформ)
Таким же способом энантиомерный продукт предшествующего примера превратили в 3-(3-[(2R)-экзо-бицикло[2.2.1.]гепт2-илокси]-4- метоксифенил)пентандинитрил, имеющий идентичные физические свойства за исключением знака вращения.
Пример 7. 3-(3- [(2S)-экзо-Бицикло[2.2.1.]гепт-2-илокси/-4-метокси- фенил)глутарамид
К названному продукту предшествующего примера (5,82г, 0,0188 моль) в 150 мл смеси 2:1 ацетона:H2О по объему добавили 5 мл 10% раствора Na2CO3, после чего по каплям добавили 30% H2О2 (8 мл, 0,094 моль), поддерживая температуру 0-5o. После перемешивания в течение 16 ч при комнатной температуре смесь вылили в воду (300 мл) и этилацетат (500 мл), и смесь перемешивали в течение 1 ч, чтобы растворить все твердые вещества.
Органический слой отделили; промыли H2O и солевым раствором, высушили и отгоняли легкие фракции, чтобы закристаллизовать остаток, который подвергали флэш-хроматографии на силикагеле, используя смесь 15:1 CH2Cl2, CH3ОН в качестве элюанта, чтобы получить 3,7 г вышеназванного продукта, т. пл 198,5-199,5oC, ИК-спектр (KBr)см-1: 3335, 3177, 2952, 1674, 1631, 1516, 1406, 1256, 1142, 1003, 809, 685, 641 см-1.
Таким же способом энантиомерный продукт предшествующего примера превратили в 3-(3-/(2R)-экзо-бицикло [2.2.1.]гепт-2-илокси/-4- метоксифенил)глутарамид, имеющий такие же физические свойства за исключением знака вращения.
Пример 8. 5-(3-[(2S)-экзо-Бицикло[2.2.1. ]гепт-2-илокси/-4- метоксифенил)-3,4,5,6,-тетрагидропиримидин-2(1H)-он
К названному продукту предшествующего примера (3,7 г, 0,0107 моль), который был растворен в 250 мл пиридина, добавили тетрацетат свинца (10,92г, 0,0246 моль) в 250 мл пиридина. После перемешивания в течение 30 ч реакционную смесь отогнали под вакуумом, и маслянистый остаток растворили в 100 мл CH2Cl2, промыли H2O и солевым раствором, высушили (Na2SO4), отогнали из него легкие фракции, и полученные твердые вещества растирали с эфиром, чтобы получить вышеназванный продукт в виде белого твердого вещества 1,21 г, т. пл 202-203oC, [альфа]D = + 14,45o(дейтерохлороформ)
Таким же способом энантиомерный продукт предшествующего примера превратили в 5-(3-[2R)-экзо-бицикло[2.2.1. ] гепт-2-илокси] -4- метоксифенил)-3,4,5,6-тетрагидропиримидин-2(1H)-он, имеющий такие же физические свойства за исключением знака вращения.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения производных 2-имидазолидинона | 1988 |
|
SU1653542A3 |
| Способ получения производных гексагидропиримидинона | 1987 |
|
SU1681725A3 |
| 2,5-ДИАЗАБИЦИКЛО [2.2.1] ГЕПТАНОВЫЕ СОЕДИНЕНИЯ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1990 |
|
RU2024524C1 |
| ПРОИЗВОДНЫЕ 2-ПИПЕРИДИНО-1-АЛКАНОЛА | 1991 |
|
RU2029769C1 |
| Способ получения 5-[3-(бицикло[2.2.1]-гепт-2-илокси)-4-метоксифенил]гексагидро-2-пиримидинона | 1988 |
|
SU1646488A3 |
| ПРОИЗВОДНЫЕ БЕНЗОКСАЗОЛОНА, ОБЛАДАЮЩИЕ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2024512C1 |
| ПРОИЗВОДНЫЕ БИЦИКЛО[2,2,1]ГЕПТ-7-ИЛАМИНА И ИХ ПРИМЕНЕНИЯ | 2006 |
|
RU2442771C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 2-(АМИНОМЕТИЛ)-3-ФЕНИЛ-БИЦИКЛО[2.2.1]ГЕПТАНОВ | 2009 |
|
RU2405766C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ БЕНЗОКСАЗОЛОНА | 1990 |
|
RU2036913C1 |
| ИНГИБИТОР ДИПЕПТИДИЛПЕПТИДАЗЫ-4 ДЛЯ ЛЕЧЕНИЯ САХАРНОГО ДИАБЕТА 2-ГО ТИПА, СОЕДИНЕНИЯ (ВАРИАНТЫ) | 2018 |
|
RU2712097C1 |
Использование: биотехнология. Сущность изобретения: оптически активные (2S)-эндобицикло [2.2.1] гептан-2-ол или (2R)-эндо-бицикло [2.2.1] гептан-2ол получают путем трансэтерификации рацемического эндобицикло [2.2.1] гептан-2-ола и 2,2,2-трихлорэтилбутирата в реакционно инертном по существу безводном органическом растворителе в присутствии панкреатической липазы млекопитающих, 5-(3-[(2R)-экзо-бицикло [2.2.1] гепт-2-илокси-4-метоксифенил) -3,4,5,6-тетрагидропиринидин-2(1H)-он получают взаимодействием (2S)-эндо-бицикло [2.2.1] -гептан-2-ола с по меньшей мере одним моль-эквивалентом 3-гидрокси-4-метоксибензальдегида в присутствии одного моль-эквивалента каждого из веществ: трифенилфосфина и диэтилазодикарбоксилата в реакционно инертном растворителе при 50-100oC, образующийся 3-(3- [(2R)-экзо-бицикло-[2.2.1] гепт-2-илокси]-4метоксибензальдегид) обрабатывают двумя моль-эквивалентами цианоуксусной кислоты в пиридине в присутствии каталитического количества пиперидина, образующийся 3-(3-(2R)-экзобицикло [2.2.1] гепт-2-илокси] -4-метоксифенил) пентандинитрил подвергают гидратации, с последующей циклизацией образующегося 3-(3- [(2R)-экзо-бицикло [2.2.1] гепт-2-илокси]-4метоксифенил)глутарамида в присутствии молярного избытка тетраацетата свинца в пиридине. 5 с. и 6 з.п. ф-лы.
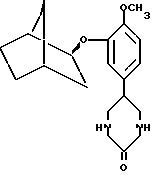
и формулу II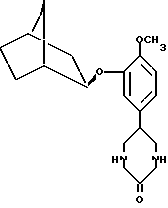
6. Соединение по п.5, имеющее формулу I
7. Соединение по п.5, имеющее формулу II.
и формулы VI
где Y -CH- или -CONH2-группа.
| J.Am | |||
| Chem Soc | |||
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| WO, 87/06576, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| US, патент, 4261995, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |

