Изобретение относится к производным β -аминокислот, которые являются изостерами дипептидного звена Gly - Asp. Соединения изобретения представляют собой псевдопептиды, обладающие противотромбовой активностью. В частности соединения замечательны с той точки зрения, что они ингибируют связывание фибриногена с рецептором фибриногена кровяных пластинок (тромбоцитов) (гликопротеином GP IIb/IIIa).
Решающей стадией при образовании тромба является поперечное сшивание кровяных пластинок молекулами фибриногена. Требованием для этого является активация тромбоцитов веществами, обладающими сродством к рецептору, такими, как тромбин или аденозиндифосфат (ADP). Эта активация вызывает реструктурирование оболочки клетки, вследствие чего GP IIb/IIIa выступает в активной форме.
GPIIb/IIIa принадлежит к группе рецепторов адгезии, известных как интегрины (integrins). Другими лигандами, помимо фибриногена, являются фибронектин, витронектин и фактор Виллибранда (von Willebrand factor). Эти лиганды играют важную роль в гемостатических процессах, так как они вызывают адгезию и агрегацию тромбоцитов. Специфическое терапевтическое ингибирование этих взаимодействий может оказать влияние на решающую стадию образования тромбов. Связывание фибриногена и других лигандов осуществляется пептидной последовательностью Arg-Gly-Asp (RGD) (Ruos-lahti E., Pierschbacher M., Cell 1986, 44, 517-18).
Фибриноген обладает еще одной пептидной последовательностью (His - His - Leu - Gly - Gly - Ala - Lys - Gln - Ala - Gly - Asi - Val), имеющей сродство к рецептору фибриногена, на C-конце гамма-цепи. Немногочисленные синтетические пептиды, которые содержат такие последовательности, могут ингибировать связывание фибриногена, фибронектина, витронектина и фактора Виллибранда с GPIIb/IIIa, и таким образом могут ингибировать агрегацию тромбоцитов (Plon et al. Proc. Natl. Acad. Sii. USA 1985, 82, 8057-61; Riggeri et al. Proc. Natl. Acad. Sсi. USA 1986, 5708-12; Ginsberg et al. J. Biol. Chem. 1985, 260, 3931-36; Gartner et al. J. Biol. Chem. 1987, 260, 11, 891-94).
Изобретение предлагает псевдопептидные аналоги Arg - Gly - Asp, в которых звено Gly - Asp замещено, производными β -аминокислот, и в которых Arg в большинстве случаев замещен бензамидинкарбоновой кислотой. Новые псевдопептиды ингибируют агрегацию тромбоцитов и образование тромбов.
Изобретение относится к псевдопептидам формулы I в свободном состоянии и в виде соли.
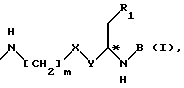 ,
,
в которой
R1 представляет собой группу формулы -COOH, -COOM или -COO(C1-C4)алкил, предпочтительно, COOH, и
в которых
M представляет собой атом щелочного или щелочноземельного металла, предпочтительно, Li,
X представляет собой -CH2-, -CH=, -CO-, -C*HOH-, или -C*HO((C1-C4)алкил)-, предпочтительно, -CH2-,
или также предпочтительно, когда X и R1 вместе являются группой 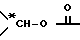 ,
,
Y представляет собой -(CH2)m-, =CH- или -NH-, предпочтительно, -(CH2)m-, и равен 1 или 2, предпочтительно 2,
A представляет собой или группу формулы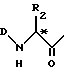 ,
,
в которой
D является водородом, защитной группой Z или α -аминокислотой, связанной через свою карбонильную группу,
R2 представляет собой группу формулы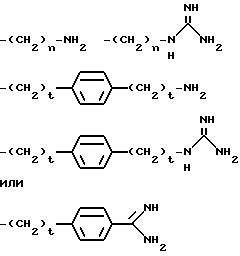
в которых
n равен 3 или 4, или t равен 0 или 1, или A представляет собой группу формулы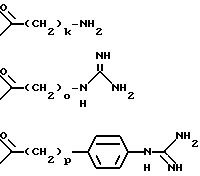
или предпочтительно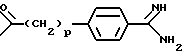
в которых
k равен 3, 4, 5 или 6;
o равен 3, 4 или 5, и
p равен 0, 1 или 2, предпочтительно 1,
B представляет собой или группу формулы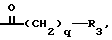 ,
,
в которой
q представляет собой 1 или 2, предпочтительно 1, и
R3 представляет собой (C1-C4)алкил, (CH3)2CH-, трет.-бутил, 1-адамантил, приметилсилил, 1-нафтил, фенил, 3-индолил или (C1-C4)алкоксифенил, предпочтительно (CH3)2CH, (C1-C4)алкоксифенил (например, п-метоксифенил), или 1-адамантил, особенно (CH3)2CH, или
B представляет собой группу формулы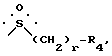 ,
,
в которой
r равен 0, 1 или 2, предпочтительно 0, и
R4 представляет собой (C1-C4)алкил, 2-пропил, трет-бутил, фенил, п-(C1-C4)алкоксифенил, 1-нафтил, толил, метилсульфонил или тризил, предпочтительно метилсульфонил, или
B представляет собой группу формулы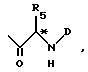 ,
,
в которой
D представляет собой водород или защитную группу Z,
R5 представляет собой фенил-(CH2)t-, индол-3-ил-(CH2)t-, нафт-1-ил-(CH2)t-, аламант-1-ил-(CH2)t-, про-2-ил-(CH2)t-, триметилсилил-(CH2)t- или трет-бутил-(CH2)t-, в которых t имеет значения, установленные выше, или
B представляет собой α -аминокислоту, связанную через ее карбонильную группу.
Каждый асимметричный C-атом, отмеченный в формулах звездочкой (*), может быть или в R или в S конфигурации.
Предпочтительно, чтобы (C1-C4)алкил в формулах представлял бы собой метил, а (C1-C4)алкоксил - метоксил. Предпочтительно, чтобы защитная группа Z представляла собой бензилоксикарбонильную или трет-бутилоксикарбонильную группу. Когда D и/или B обозначают α -аминокислоту, это может быть α -аминокислота, встречающаяся в природе, или α -аминокислота, в природе не встречающаяся.
В описании, если отсутствуют специальные указания, термины, такие как "соединения формулы I", относятся как к соединениям в форме соли, так и к соединениям в свободной форме.
Предпочтительными соединениями являются соединения формулы I'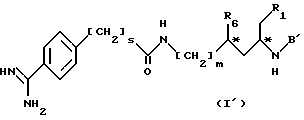
в которой
R1 и m имеют значения, установленные выше,
S равен 0 и 1, R6 = H, OH или O(C1-C4)алкилу, или
R1 и R6 вместе являются группой -O-CO,
B' является или группой формулы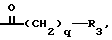 ,
,
в которой
R3 и q имеют значения, установленные выше, или
B представляет собой группу формулы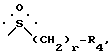 ,
,
в которой
R4 и r имеют значения, установленные выше, или
B представляет собой группу формулы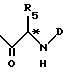 ,
,
в которой
R5 и D имеют значения, установленные выше.
Особенно предпочтительными соединениями являются соединения, имеющие формулу I''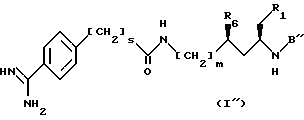
в которой
R1, R6, m и S имеют значения, установленные выше, или R1 и R6 вместе образуют -O-CO-группу, и
B'' представляет собой группу формулы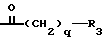
в которой
q и R3 имеют значения, установленные выше.
Наиболее предпочтительными соединениями формулы I являются соединения следующих далее формул II, III, IV, V, VI, VII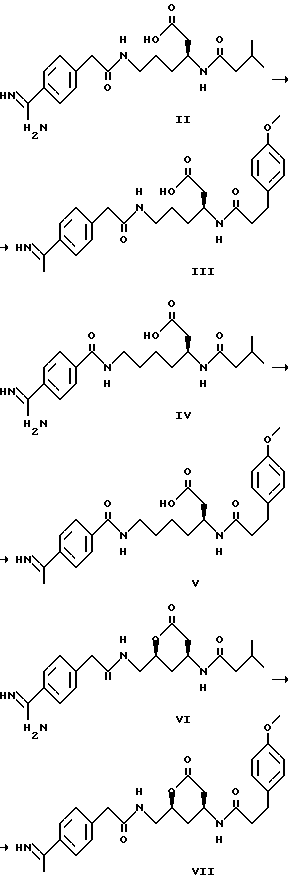
Эти соединения являются соединениями примеров 2, 3, 6, 9, 8 и 7 соответственно. Соединения формулы I такого типа, который включает соединения формул со II по V, приведенных выше, могут быть синтезированы с использованием методик, аналогичных следующей далее схеме синтеза I.
Схема синтеза I.
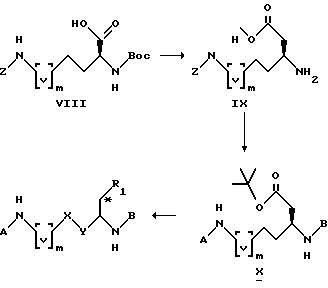
В приведенных выше формулах [v]m-являются группой [CH2]m, и m, A, B, X, Y и R1 имеют значения, установленные выше.
В соответствии со схемой синтеза I, соединения формулы I, в которых X = Y = CH2, R1 = COOH и m = 1 или 2, получают из соединения X путем обработки трифторуксусной кислотой при комнатной температуре. Соединение X получают из сложного эфира IX с защитной группой Z путем соединения аминогруппы с изовалериановой кислотой или п-метоксифенилпропановой кислотой (в зависимости от радикала B) при добавлении DCC или HOBT, последующим гидролизом метилового эфира с LiOH, превращением кислоты в трет-бутиловый эфир с помощью трет-бутил-2,2,2-трихлорацетимидата, удалением защитной группы Z в условиях восстановления в системе H2/Pd-C, и последующим соединением с подходящим радикалом A [например, A означает (п-амидинофенил)-(CH2)S-COOH, в которой S имеет установленное выше значение] , вновь добавляя DCC и HOBT. Метиловый эфир IX получают из аминокислоты VIII, защищенной группой Z = Boc, удлиняя цепь карбоновой кислоты по методу диазометан/Ag2O /Helv. Chem. Act. 58, 969 (1975)/, и удаляя Boc-группу трифторуксусной кислотой при комнатной температуре.
Кроме того, соединения формулы I такого типа, который включает соединения приведенных выше формул VI и VII, могут быть получены с использованием методик, аналогичных следующей далее схеме синтеза 2.
Схема синтеза 2.
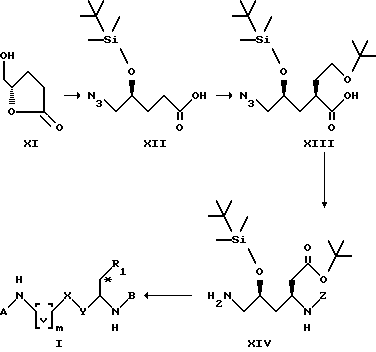
В приведенных выше формулах [v]m представляет собой [CH2]m, и m, A, B, X, Y и R1 имеют значения, установленные выше.
В соответствии со схемой синтеза 2, соединения формулы I, в которых m равен 1, X и R1 вместе являются группой HC*-O-CO- и Y представляет собой группу CH2-, могут быть получены из соединения XIV путем соединения с подходящим радикалом A (аналогично синтезу по схеме I), удалением защитной группы в условиях восстановления в системе H2/Pd-C, с последующим соединением с подходящим радикалом B (аналогично синтезу по схеме I), и удаляя потом защитную группу трифторуксусной кислотой при комнатной температуре. Соединение XIV получают из соединения XIII путем превращения карбоновой кислоты в Z-защищенный амин, используя дифенилфосфорилазид и триэтиламин в присутствии бензилового спирта, и восстанавливая азидогруппу трифенилфосфином в тетрагидрофуране. Карбоновую кислоту XIII получают из соединения XII посредством энантиоселективного введения кислотной боковой цепи в 3 стадии:
a) соединение с оксазолидиноном Эванса с целью введения дополнительной хиральной группы (J. Am. Chem. Soc. 1990, 112, 4011);
b) образование амид-енолята с Li-гексаметилдисилазидом и реакция с трет-бутиловым эфиром бромуксусной кислоты;
c) удаление дополнительной хиральной группы пергидроксидов Li. Азидокарбоновую кислоту XII получают из лактона XI, вводя метилсульфонильную группу в спирт при обработке спирта метансульфонилхлоридом, осуществляя азидирование с азидом натрия, раскрывая лактон раствором каустика в этаноле и осуществляя взаимодействие с трет-бутилдиметилсилилхлоридом в присутствии имидазола с целью введения защитной спиртовой группы.
Соединение формулы I ингибируют связывание фибриногена с рецептором фибриногена кровяных пластинок (гликопротеином GP IIb/IIIa). В результате такого свойства, соединения предотвращают агрегацию тромбоцитов человека и образование сгустков, и могут, соответственно использоваться для предотвращения и лечения тромбозов, апоплексии, сердечного инфаркта, воспаления, артериосклероза и опухолей. Другими терапевтическими областями применения являются остеопороз, острая закупорка (acute reocclusion) после РТСА и добавление во время тромболиза.
Аббревиатуры, использованные в описании:
Z - бензоилоксикарбонил;
BOC - трет-бутилоксикарбонил;
DCC дициклогексилкарбодиимид;
HOBT гидроксибензотриазол;
DMF - диметилформамид;
THF - тетрагидрофуран;
TFA - трифторуксусная кислота;
EtOAc - этилацетат;
RP - обращенная фаза;
Pd/C (10%) - катализатор - палладированный уголь, содержащий 10% палладия;
LiOH - гидроксид лития;
PTCA - чрескожная просветная (percutanions luminal) операция на коронарных сосудах
TBDMS - трет-бутилдиметилсилилхлорсилан;
MeOH - метанол;
LiHMDS - гексаметилдисилазид лития;
DPPA - дифенилфосфорилазид;
EtOH - этанол;
NMM - N-метилморфолин;
Пример 1 (S)-3-(N-тозиламино)-6-[N-(п-амидинофенилацетил)-амино] -гексановая кислота
A) Трифторацетат метилового эфира (S)-3-амино-6-[N-(бензилоксикарбонил)амино]гексановой кислоты
0,69 мл изобутилового эфира хлормуравьиной кислоты добавляют по каплям в раствор 1,95 г Boc-Orn(Z)-OH и 0,74 мл NEt3 в 12 мл THF при -15oC. После 30-минутной выдержки при -15oC, осажденный гидрохлорид отфильтровывают, и фильтрат смешивают при -15oC с 40 мл (8 ммоль) эфирного раствора диазометана. Реакционную смесь перемешивают в течение 4 ч. при 0oC, и оставляют стоять еще в течение 16 ч при 4oC. Добавляют воду, и раствор экстрагируют эфиром. Эфирную фазу промывают насыщенным водным раствором бикарбоната, осушают над сульфатом натрия и концентрируют упариванием. Полученный остаток растворяют в 15 мл метанола, смешивают с 246 мг оксида серебра (I) и нагревают с обратным холодильником в течение 12 ч. Твердое вещество удаляют фильтрацией, метанол выпаривают, и остаток хроматографируют на силикагеле (этилацетат/гексан 1:1). Выделенный таким образом продукт представляет собой метиловый эфир (S)-3-N-Boc-амино-6-N-Z-аминогексановой кислоты. Масс-спектр: 395 (M + H)+.
Полученный продукт смешивают при 0oC с 10 мл метиленхлорида и 10 мл трифторуксусной кислоты, перемешивают в течение 1 ч при комнатной температуре, растворитель удаляют отгонкой в вакууме, и остаток сушат в течение 24 ч в высоком вакууме. Получают трифторацетат метилового эфира (S)-3-амино-6-[N-(бензилоксикарбонил)амино] гексановой кислоты в виде бесцветного твердого вещества.
B) Метиловый эфир (S)-3-N-тозиламино-6-N-Z-аминогексановой кислоты
Трифторацетат со стадии A (1,3 г) растворяют в 5 мл DMF вместе с 0,75 мл триэтиламина, и затем добавляют 468 мг тозилхлорида. Через 2 ч добавляют воду, и выполняют экстракцию эфиром. Эфирную фазу промывают водой, осушают над сульфатом натрия, эфир удаляют испарением, и остаток хроматографируют на силикагеле (этилацетат/гексан 1:1). Выделенный таким образом продукт представляет собой метиловый эфир (S)-3-N-тозиламино-6-N-Z- аминогексановой кислоты; масс-спектр: 449 (M + H)+.
C) (S)-3-(N-тозиламино)-6-[N-(п-аминофенилацетил)-амино)] гексановая кислота
Продукт, полученный в соответствии со стадией (B), растворяют в 20 мл метанола и гидрируют в присутствии 0,3 г Pd/C (10%) и 1,34 мл IN соляной кислоты. Когда реакция заканчивается, катализатор удаляют фильтрованием, метанол выпаривают, и остаток сушат в высоком вакууме. Получают 500 мг хлоргидрата метилового эфира (S)-3-(N-тозиламино)-6- аминогексановой кислоты в виде бесцветной пены. 0,5 г гидрохлорида, 373 мг N-Boc-п-амидинофенилуксусной кислоты (полученной Boc-илированием п-амидинофенилуксусной кислоты (Pharmazie 29, 256-262)), 0,19 мл триэтиламина и 199 мг HOBT растворяют в 10 мл DMF, и смешивают с 276 мг DCC. После 16-часовой выдержки при комнатной температуре осажденное твердое вещество удаляют фильтрацией, DMF выпаривают, и остаток растворяют в этилацетате. Этилацетатную фазу промывают водой, осушают над сульфатом натрия, концентрируют упариванием, и остаток хроматографируют на силикагеле (этилацетат). Полученный продукт смешивают с раствором 0,2 мл анизола в 5 мл TFA, перемешивают в течение 2 ч при комнатной температуре, и по каплям добавляют в 300 мл эфира. Осажденное твердое вещество отфильтровывают, растворяют в смеси 2 мл метанола и 1 мл воды, и смешивают с 57 мг LiOH-H2O. После 5-часовой выдержки при комнатной температуре метанол выпаривают, и водный раствор нейтрализуют 0,13 мл TFA. Осажденное твердое вещество отфильтровывают, сушат и перекристаллизовывают из смеси метанол-эфир. Получают продукт - трифторацетат (S)-3-(N-тозиламино)-6-[N-(п- амидинофенилацетил)-амино] гексановой кислоты в виде белого твердого вещества, масс-спектр: 461 (M + H)+. Свободное соединение получают из трифторацетата известным способом.
Пример 2. (S)-3-[N-(3-метилбутирил)амино]-6-[N-(п-амидиноамино] гексановая кислота
A) Метиловый эфир (S)-3-[N-(3-метилбутирил)амино]-6-N-Z- аминогексановой кислоты
Трифторацетат со стадии A примера 1 (1,3 г), 250 мг изовалериановой кислоты, 0,34 мл триэтиламина и 363 мг HOBT растворяют в 5 мл DMF и смешивают с 505 мг DCC. Через 16 ч выпавший осадок удаляют фильтрацией, DMF выпаривают, и остаток растворяют в этилацетате. Этилацетатную фазу промывают водным насыщенным раствором бикарбоната и водой, осушают над сульфатом натрия, и растворитель удаляют в вакууме. Остаток очищают перекристаллизацией из смеси этилацетат : эфир, и получают продукт - метиловый эфир (S)-3-[N-(3-метилбутирил)-амино] -6-N-Z-аминогексановой кислоты - в виде бесцветных кристаллов. Масс-спектр: 379 (M + H)+.
B) (S)-3-[N-(3-метилбутирил)амино] -6-[N-(п-амидинофенилацетил)- амино] гексановая кислота
Продукт со стадии (A) примера 2 (560 мг) растворяют в 20 мл метанола и гидрируют в присутствии 0,3 г Pd/C (10%) и 1,41 мл IN соляной кислоты. После действий, описанных в стадии (C) примера 1, получают хлоргидрат метилового эфира (S)-3-[N-(3-метилбутирил)амино] - 6-аминогексановой кислоты в виде бесцветного масла. Хлоргидрат (400 мл) и 392 мг N-Boc-п-амидинофенилуксусной кислоты вводят во взаимодействие с 0,2 мл триэтиламина, 259 мг HOBT и 290 мг DCC, и действуют, как описано в стадии (C) примера 1. После расщепления Boc-группы с помощью TFA и гидролиза метилового эфира с LiOH-H2O, аналогично стадии (C) примера 1, полученный сырой продукт перекристаллизовывают из метанола. Продукт - трифторацетат (S)-3-[N-(3-метилбутирил)амино]-6-[N-(п-амидинофенилацетил) амино] гексановой кислоты - получают в виде белого порошка; масс-спектр: 391 (M + H)+. Свободное соединение получают из трифторацетата известным способом.
Пример 3 (S)-3-N-[3-(п-метоксифенил)пропионил] амино-6- [N-(п-амидинофенилацетил)амино]гексановая кислота
A) Метиловый эфир (S)-3-N-[3-(п-метоксифенил)пропионил]- амино-6-N-Z-аминогексановой кислоты
Вводят во взаимодействие 0,7 г трифторацетата со стадии (A) примера 1 и 256 мг 3-(п-метокси)пропионовой кислоты с 0,21 мл триэтиламина, 256 мг HOBT и 313 мг DCC, и выполняют действия, описанные в стадии A) примера 2. Сырой продукт перекристаллизовывают из эфира. Получают продукт - метиловый эфир (S)-3-N-[3-(п-метоксифенил)пропионил]амино-6-N-Z-аминогексановой кислоты - в виде белого кристаллического вещества; масс-спектр: 457 (M + H)+.
B) (S)-3-N-[3-(п-метоксифенил)пропионил] амино-6-[N-(п- амидинофенилацетил)амино]гексановая кислота
Метиловый эфир (440 мг) со стадии (A) примера 3 гидрируют в соответствии со стадией (C) примера 1, и полученный хлоргидрат вводят во взаимодействие и обрабатывают со 164 мг N-Boc-п-амидинофенилуксусной кислоты, 0,13 мл триэтиламина, 164 мг HOBT и 200 мг DCC, как это описано в стадии (C) примера 1. С помощью TFA и LiOH-H2О разрушают защитную группу продукта, аналогично стадии (C) примера 1, и сырой продукт перекристаллизовывают в системе метанол/вода. Продукт - трифторацетат (S)-3-N-[3-(п-метоксифенил)пропионил]амино-6-[N-(п-амидинофенилацетил)- амино] гексановой кислоты - получают в виде белого порошка. Масс-спектр: 583 (M + H)+.
Свободное соединение получают из трифторацетата известным способом.
Пример 4. (S)-3-[N-(адамант-1-илацетил)амино] -6-[N-(п- амидинофенилацетил)амино]гексановая кислота
A) Метиловый эфир (S)-3-[N-(адамант-1-илацетил)амино]-6-N-Z- аминогексановой кислоты
Трифторацетат (0,7 г) со стадии (A) примера 1 вводят во взаимодействие с 246 мг адамант-1-илуксусной кислоты, 0,21 мл триэтиламина, 246 мг HOBT и 331 мг DCC и обрабатывают, как описано в стадии A) примера 2. Сырой продукт хроматографируют на силикагеле (этилацетат), и выделяют метиловый эфир (S)-3-[N-(адамант-1-илацетил)амино]-6-N-Z-аминогексановой кислоты в виде бесцветного масла; масс-спектр: 471 (M + H)+.
B) трет-Бутиловый эфир (S)-3-[N-(адамант-1-илацетил)-амино-6-N-Z-аминогексановой кислоты
Метиловый эфир (460 мг) со стадии A) примера 4 растворяют в 4 мл MeOH и 2 мл воды, и смешивают с 84 мг LiOH-H2O. После 4-часовой выдержки при комнатной температуре, смесь нейтрализуют IN соляной кислотой и экстрагируют этилацетатом. Этилацетатную фазу осушают над сульфатом натрия, и растворитель удаляют в вакууме. Получают 400 мг сырого продукта, который растворяют в 2 мл THF, и смешивают с раствором 479 мг трет-бутил-2,2,2-трихлорацетимидата в 2,5 мл циклогексана. После добавления 0,069 мл трифторэфирата бора, смесь перемешивают в течение 3 ч при комнатной температуре. Реакционную смесь смешивают с 5%-ным водным раствором бикарбоната, экстрагируют этилацетатом, этилацетатную фазу промывают насыщенным водным раствором хлорида натрия, осушают над сульфатом натрия, и раствор сгущают выпариванием. Остаток растворяют в смеси метиленхлорид/гексан (1/1), не растворившийся трихлорацетамид отфильтровывают, и растворитель выпаривают. Полученный в виде масла сырой продукт хроматографируют на силикагеле [этилацетат/гексан (1/1)], и получают трет-бутиловый эфир (S)-3-[N-(адамант-1-илацетил)амино]-6-N-Z-аминогексановой кислоты в виде бесцветного масла. Масс-спектр: 513 (M + H)+.
C) (S)-3-[N-(адамант-1-илацетил)амино] -6-[N-(п- амидинофенилацетил)амино]гексановая кислота
Трет-Бутиловый эфир (225 мг) со стадии (B) примера 4 растворяют в 20 мл этанола, и гидрируют в присутствии 0,1 г Pd/C (10 %) и 0,028 мл уксусной кислоты, как описано в стадии (C) примера 1. Полученный сырой продукт растворяют в 5 мл DMP, и вводят во взаимодействие со 119 мг N-Boc-п-амидинофенилуксусной кислоты, 0,061 мл триэтиламина, 73 мг HOBT и 90 мг DCC, аналогично стадии (C) примера 1. Полученный сырой продукт после обработки, хроматографируют на силикагеле (этилацетат), и чистый продукт смешивают с раствором 5 мл TFA в 0,2 мл анизола.
После 3-часовой выдержки при комнатной температуре, реакционную смесь добавляют по каплям в 300 мл эфира, и осажденное твердое вещество отфильтровывают. После высушивания в высоком вакууме, получают трифторацетат (S)-3-[N-(адамант-1-илацетил)амино] -6-[N-(п-амидинофенилацетил) амино]гексановой кислоты в виде белого порошка; масс-спектр: 505 (M + H)+.
Свободное соединение получают из трифторацетата известным способом.
Пример 5 (S)-3-[N-(3-адамант-1-илпропионил)амино]-6-[N-(п- амидинофенилацетил)амино]гексановая кислота
A) Метиловый эфир (S)-3-[N-(3-адамант-1-илпропионил)амино]-6-N-Z-гексановой кислоты
Метиловый эфир (S)-3-[N-(3-адамант-1-илпропионил)амино]-6-N-Z- аминогексановой кислоты получают взаимодействием 0,85 г трифторацетата со стадии (A) примера 1 с 423 мг 3-адамант-илпропионовой кислоты, 0,28 мл триэтиламина, 337 мл HOBT и 418 мг DCC, с последующей очисткой методом хроматографии (силикагель, этилацетат), аналогично стадии (A) примера 2; масс-спектр: 485 (M + H)+.
B) трет-Бутиловый эфир (S)-3-[N-(3-адамант-1-илпропионил)амино]-6-N-Z-аминогексановой кислоты
Метиловый эфир (500 мг) со стадии (A) примера 5 подвергают гидролизу, как описано в стадии (B) примера 4, со 173 мг LiOH-H2O, и вводят во взаимодействие с 575 мг трет-бутил-2,2,2-трихлорацетимидата и 0,06 мл трифторэфирата бора. После выполнения действий, описанных в стадии (B) примера 1, выделяют трет-бутиловый эфир (S)-3-[N-(3-адамант-1-илпропионил)-амино]-6-N-Z- аминогексановой кислоты; масс-спектр: 527 (M + H)+.
C) (S)-3-[N-(3-адамант-1-илпропионил)амино]-6-[N-(п- амидинофенилацетил)амино]гексановая кислота
Продукт (510 мг) по стадии (B) примера 5 гидрируют в соответствии со стадией (C) примера 4, соединяют с 430 мг N-Boc-п-амидинофенилуксусной кислоты, и после очистки методом хроматографии (силикагель/этилацетат), в полученном продукте разрушают защитную группу 5 мл смеси TFA/анизол (95/5). Последующим осаждением из эфира получают трифторацетат (S)-3-[N-(3-адамант-1-илпропионил)амино] -6-[N-(п- амидинофенилацетил)амино] -гексановой кислоты в виде белого порошка, аналогично стадии (C) примера 5; масс-спектр: 497 (M + H)+.
Свободное соединение получают из трифторацетата известным способом.
Пример 6 (S)-3-[N-(3-метилбутирил)амино]-7-[N-(п-амидинобензоил) амино] -гептановая кислота
A) Метиловый эфир (S)-3-амино-6-N-Z-амино-гептановой кислоты
4,04 г Boc-Lys (Z)-OH, 1,38 мл изобутилового эфира хлоругольной кислоты, 1,44 мл триэтиламина и 80 мл (16 ммоль) эфирного раствора диазометана вводят во взаимодействие, затем, в дальнейшее взаимодействие с 490 мг оксида серебра (I) и действуют аналогично описанному в стадии (A) примера 1, затем сырой продукт очищают хроматографическим способом (силикагель, этилацетат/гексан = 1/1), и получают метиловый эфир (S)-3-N-Boc-амино-6-N-Z-аминогептановой кислоты. Масс-спектр: 409 (M + H)+. Метиловый эфир растворяют в 15 мл метиленхлорида и смешивают с 15 мл TFA. Через 1 ч TFA и растворитель удаляют в вакууме, и остаток высушивают в глубоком вакууме, чтобы получить в качестве продукта трифторацетат метилового эфира (S)-3-амино-6-N-Z-аминогептановой кислоты.
B) Метиловый эфир (S)-3-[N-(3-метилбутирил)амино]-7-N-Z- аминогептановой кислоты
Трифторацетат (4,1 г) со стадии (A) примера 6 вводят во взаимодействие с 1,0 г изовалериановой кислоты, 1,36 мл триэтиламина, 1,65 г HOBT и 2,02 г DCC, как описано в стадии (A) примера 2, действуя аналогичным образом. Полученный сырой продукт перекристаллизовывают из эфира, и получают метиловый эфир (S)-3-[N-(3-метилбутирил)амино] -7-N-Z-аминогептановой кислоты; масс-спектр: 393 (M + H)+.
C) трет-Бутиловый эфир (S)-3-[N-(3-метилбутирил)амино] -7- N-Z-аминогептановой кислоты
Как описано в стадии (B) примера 4, 3,25 г метилового эфира со стадии (B) примера 6 омыляют с 1,36 г LiOH-H2O, и вводят во взаимодействие с 4,04 г трет-бутил-2,2,2-трихлорацетамида и 0,42 мл трифторэфирата бора, получают продукт - трет-бутиловый эфир (S)-3-[N-(3-метилбутирил)амино]-7-N-Z-аминогептановой кислоты; масс-спектр: 435 (M + H)+.
D) (S)-3-[N-(3-метилбутирил-амино-7-[N-(п-аминобензоил) амино]гептановая кислота
После гидрирования 2,2 г продукта со стадии (C) примера 6, аналогично стадии (C) примера 4, продукт затем вводят во взаимодействие с 1,34 г N-Boc-п-амидинобензойной кислоты (получают аналогично N-Boc-п-амидинофенилуксусной кислоте), 0,7 мл триэтиламина, 0,852 г HOBT и 1,04 г DCC, как на стадии (C) примера 4, и затем отщепляют защитную группу 25 мл смеси TFA/анизол (95/5), и в результате всех действий получают трифторацетат (S)-3-[N-(3-метилбутирил)амино] -7-[N-(п-амидинобензоил)амино] гептановой кислоты в виде белого порошка; масс-спектр: 391 (M + H)+.
Свободное соединение получают из трифторацетата известным способом.
Пример 7 δ -Лактон (3S,5S)-6-(4-амидинофенилацетиламино)-3- (4-метоксифенилпропиониламино)гексановой кислоты
A) γ -Лактон (4S)-4-окси-5-0-метилсульфонилвалериановой кислоты
γ -Лактон (4S)-4,5-диоксивалериановой кислоты (16,3 г, 140 ммоль) и триэтиламин (21,52 мл, 154 ммоль) растворяют в метиленхлориде, и раствор охлаждают до -30oC. Затем при перемешивании добавляют по каплям метансульфонилхлорид (12,0 мл, 154 ммоль). Раствор затем перемешивают в течение 15 мин при -30oC и нагревают до 18oC в течение 1,5 ч. Полученную суспензию добавляют в 0,5N HCl и несколько раз экстрагируют эфиром. Объединенные органические фазы промывают насыщенным водным раствором NaHCO3 и насыщенным водным раствором NaCl. После высушивания над Na2SO4 и концентрирования в роторном испарителе, получают γ -лактон (4S)-4-окси-5-0-метилсульфонилвалериановой кислоты в виде масла, которое сразу же используют на следующей стадии реакции.
B) γ -Лактон (4S)-4-окси-5-азидовалериановой кислоты
Мезилат со стадии (A) примера 7 (25,0 г, 128,7 ммоль) растворяют в DMSO, раствор при комнатной температуре смешивают с азидом натрия (16,74 г, 257,4 ммоль), и перемешивают в течение 1,5 ч при 100oC. Суспензию коричневого цвета охлаждают, и отгоняют в вакууме DMSO, фильтруют через гифло (Hyflo) и концентрируют в вакууме. Вакуумная перегонка (0,16 мбар) дает названное в заголовке соединение в виде бесцветного масла, αD = +79,9o (c = 2,2 в CHCl3).
C) (4S)-5-азидо-4-{ [(1,1-диметилэтил)-диметилсилил] -окси}- пентановая кислота
γ -Лактон (4S)-4-окси-5-азидовалериановой кислоты (12,28 г, 87,01 ммоль) со стадии (B) примера 7 растворяют в 435 мл этанола, и затем, при перемешивании при комнатной температуре, добавляют 43,5 мл водного 2N NaOH. После выстаивания в течение 1/2 ч при комнатной температуре, раствор концентрируют в роторном испарителе и сушат в высоком вакууме. Полученный остаток (16,94 г) смешивают со 174 мл DMF, имидазолом (29,63 г, 435,05 ммоль) и TBDMS-Cl (48,3 г, 313,24 ммоль), и перемешивают в течение 19 ч при комнатной температуре. Полученную суспензию добавляют в смесь льда с 1N водным раствором NaHSO4, и несколько раз экстрагируют эфиром. Объединенные органические фазы сушат над Na2SO4 и концентрируют в вакууме. Остаток растворяют в 435 мл метанола и при перемешивании смешивают с 18,44 г Na2CO3 (в 87,0 мл H2O) при комнатной температуре, и затем энергично перемешивают в течение 1/2 ч при комнатной температуре. Полученную суспензию смешивают с 435 мл H2O, и дважды экстрагируют гексаном. Объединенные гексановые фазы промывают смесью метанол/H2O (1:1), и объединенные водные фазы подкисляют NaHSO4. После экстрагирования гексаном и промывания насыщенным водным раствором NaCl, органическую фазу сушат над Na2SO4 и концентрируют в роторном испарителе. После тонкослойной хроматографии и 1Н-ЯМР, убеждаются, что полученный продукт - сырая (4S)-5-азидо-4-{ [(1,1-диметилэтил)диметилсилил]окси}-пентановая кислота - является практически чистой, и ее используют в дальнейшем без дополнительной очистки. Масс-спектр: 274 (M + H)+.
D) [3(4S), 4R]-3-[5-азидо-4-{[(1,1-диметилэтил)диметилсилил]- окси}-1-оксопентил]-4-(фенилметил)-2-оксазолидинон
Оптически активное производное пентановой кислоты со стадии (C) примера 7 (19,79 г, 72,37 ммоль), растворяют в 405 мл сухого THF, охлаждают до -78oC, смешивают с 13,5 мл триэтиламина (97 ммоль) и затем с 10,4 мл (84,7 ммоль) пивалоилхлорида. После 5-минутной выдержки при -78oC смесь нагревают в течение часа до 0oC, и вновь охлаждают до -78oC. В другом реакционном сосуде растворяют (4R)-4-фенилметил-2-оксазолидонона (15,65 г, 88,3 ммоль) в 405 мл безводного THF, охлаждают до -78oC, смешивают с п-бутиллитием (56,1 мл, 1,6 М раствора), и перемешивают в течение 0,25 ч при -78oC. Полученный таким образом азаенолят смешивают при -78oC, используя давление иглы (pressure needle), со смешанным ангидридом кислоты, который предварительно приготовляют in situ. Охлаждающую баню удаляют, и осуществляют перемешивание в течение 1 ч. Реакционную смесь добавляют в раствор лед/NaHSO4, экстрагируют EtOAc, органические фазы промывают насыщенным водным раствором NaHCO3 и насыщенным водным раствором NaCl, и затем сушат над Na2SO4 и концентрируют в вакууме. Полученный сырой материал хроматографируют на силикагеле (гексан/EtOAc, 3:1). Получают названное в заголовке соединение в виде белого твердого вещества; масс-спектр: 433 (M + H)+.
E) [3(2S,4S),4R]-3-[5-азидо-4-{[(1,1-диметилэтил)- диметилсилил]окси}-2-трет-бутилоксикарбонилметил-1-оксопентил]-4- фенилметил-2-оксазолидинон
Оптически чистый имид со стадии (D) примера 7 (19,05 г, 44 ммоль) растворяют в 44 мл THF, и добавляют к охлажденному до -78oC раствору LiHMDS (48,4 мл, 1,0 М раствора) в 66 мл THF. После 1/2-часовой выдержки при -78oC, трет-бутиловый эфир бромуксусной кислоты (9,7 мл, 66 ммоль) добавляют по каплям в течение 20 мин в енолят, который охлаждают до -78oC. После выстаивания в течение 1 ч при -78oC, смесь нагревают до 0oC, и затем в реакционную смесь добавляют насыщенный водный раствор NH4Cl. После экстракции эфиром объединенные органические фазы промывают насыщенным водным раствором NaHCO3 и насыщенным водным раствором NaCl, сушат над Na2SO4 и концентрируют в вакууме. В соответствии с 1Н-ЯМР, продукт существует в виде диастереоизомерической смеси (90: 10) [3(2S,4S),4R] : [3(2R,4S)4R]. После хроматографии на силикагеле (гексан/EtOAc, 6:1), получают названное в заголовке соединение в чистой диастереоизомерической форме в виде бесцветного масла; масс-спектр: 521 (M - N2 + 3H)+.
F) трет-Бутиловый эфир (3S,5S)-6-азидо-5-{[(1,1-диметилэтил- диметилсилил]окси}-3-оксикарбонилгексановой кислоты
Соединение, названное в заголовке стадии (E) примера 7 (21,5 г, 39,4 ммоль), растворяют в 500 мл THF, смешивают последовательно со 180 мл H2O, 16,7 мл H2O2 (30 % раствор) и LiOH (3,3 г, 78,8 ммоль), и перемешивают в течение 1,5 ч при 0oC. Затем добавляют Na2SO3 (17,1 г, 136 ммоль) в 120 мл H2O, и осуществляют перемешивание в течение 5 мин при 0oC. Реакционную смесь подкисляют 1N водным раствором NaHSO4, и несколько раз экстрагируют эфиром. Объединенные органические фазы промывают насыщенным водным раствором NaCl, сушат над Na2SO4 и концентрируют в вакууме. Полученный сырой продукт растворяют в гексане. Образующийся (4R)-4-фенилметил-2-оксазолидинон отфильтровывают, и путем концентрирования фильтрата и высушивания в высоком вакууме, получают производное трет-бутилового эфира гексановой кислоты в чистом виде. Масс-спектр: 410 (M + H)+ из Na-соли.
G) трет-Бутиловый эфир (3S,5S)-6-азидо-5-{[(1,1-диметилэтил)- диметилсилил]окси}-3-бензоилоксикарбонил-аминогексановой кислоты
Соединение, названное в заголовке стадии (F) примера 7 (1,55 г, 4,0 ммоль), растворяют в 20 мл безводного толуола, и смешивают последовательно с DPPA (956 мкл (95 %), 4,2 ммоль) и триэтиламином (613 мкл, 4,4 ммоль), и перемешивают при температуре кипения с обратным холодильником в течение 1/2 ч. После охлаждения примерно до 40oC, добавляют бензиловый спирт (4,14 мл, 40 ммоль), и смесь перемешивают при температуре кипения с обратным холодильником еще в течение 60 мин. Реакционный раствор охлаждают до комнатной температуры, смешивают с дополнительным количеством толуола, промывают водным насыщенным раствором NaHCO3, 10%-ным раствором винной кислоты и водным насыщенным раствором NaCl, сушат над Na2SO4 и концентрируют в вакууме. Сырой продукт хроматографируют на силикагеле (гексан/EtOc, 6:1), и получают в чистом виде соединение, названное в заголовке; масс-спектр: 493 (M + H)+.
H) трет-Бутиловый эфир (3S,5S)-6-[4-(трет-бутилоксикарбонил)- амидинофенилацетиламино] -5-[{(1,1-диметилэтил)диметилсилил}-окси]-3- бензилоксикарбонил-аминогексановой кислоты
Соединение, названное в заголовке стадии (G) примера 7 (1,18 г, 2,4 ммоль), растворяют в THF (12,0 мл) и вводят во взаимодействие с трифенилфосфином (0,66 г, 2,52 ммоль) аналогично методике, описанной в литературе (Tetrahedron Lett. 24, 763, 1983), сначала при комнатной температуре (17 ч) и затем при температуре кипения с обратным холодильником. После кипячения с обратным холодильником в течение 1 ч, добавляют 3,6 моль эквивалента H2O, и смесь перемешивают еще в течение 4 ч при температуре кипения с обратным холодильником. Реакционную смесь охлаждают, концентрируют в вакууме, остаток разводят в гексане, и отфильтровывают нерастворимый трифенилфосфиноксид. Фильтрат концентрируют в вакууме, и образовавшийся в результате амин выделяют в виде масла, которое затем сразу же вводят в реакцию. Амин растворяют в 8 мл DMF вместе с 4-(трет-бутилоксикарбонил)-амидинофенилуксусной кислотой (733 мг, 2,64 ммоль) и HOBT (4,89 мг, 3,19 ммоль), и вводят во взаимодействие при комнатной температуре с DCC (494 мг, 2,4 ммоль). После 16-часовой выдержки при комнатной температуре полученную суспензию охлаждают до 0oC, и выпавшую в осадок дициклогексилмочевину отфильтровывают. Фильтрат разбавляют EtOAc и промывают насыщенным водным раствором NaHCO3 и насыщенным водным раствором NaCl. После высушивания над Na2SO4, продукт концентрируют в вакууме. Полученный сырой материал хроматографируют на силикагеле (гексан/EtOAc, 30: 70). Получают соединение, названное в заголовке, в виде белого твердого вещества; масс-спектр: 727 (M + H)+.
I) трет-Бутиловый эфир (3S,5S)-6-[4-(трет-бутилоксикарбонил)- амидинофенилацетиламино] -5-[{ (1,1-диметилэтил)-диметилсилил} окси]-3-(4-метоксифенилпропиониламино)гексановой кислоты
Соединение со стадии (H) примера 7 (1,23 г, 1,69 ммоль) растворяют в 8,5 мл этанола, и гидрируют над 10-%-ным Pd/C. После 2-часовой выдержки при комнатной температуре катализатор удаляют фильтрованием, и фильтрат концентрируют в вакууме. Полученный свободный амин растворяют в 5,6 мл DMF вместе с 4-метоксифенилпропионовой кислотой (335 мг, 1,86 ммоль) и HOBT (345 мг, 2,25 ммоль), и затем смешивают с DCC (349 мг, 1,69 ммоль). После выстаивания при комнатной температуре в течение 63 ч суспензию охлаждают до 0oC, и действуют, аналогично процессу, описанному в стадии (H) примера 7.
Полученный сырой продукт хроматографируют на силикагеле (гексан/EtOAc). Соединение, названное в заголовке, выделяют в виде белой пены; масс-спектр: 755 (M + H)+.
J) δ -Лактон (3S, 5S)-6-(4-амидинофенилацетиламино)-3- (4-метоксифенилпропиониламино)гексановой кислоты
Соединение со стадии (I) примера 7 (639 мг, 0,846 ммоль) смешивают с 0,21 мл этандитиола, 0,21 мл анизола и 4,23 мл смеси TFA/H2O (95:5) при 0oC. После 3-часовой выдержки при комнатной температуре смесь снова охлаждают до 0oC, добавляют эфир, и выпавшее в осадок твердое вещество отфильтровывают. Полученное твердое вещество промывают эфиром и затем перекристаллизовывают. Получают трифторацетат соединения, названного в заголовке, в виде белого кристаллического вещества, Т. пл. 216 - 218oC; [α]D = +13,5o (C = 0,48, MeOH), масс-спектр: 467 (M + H)+. Свободное соединение получают из трифторацетат известным способом.
Пример 8 δ -Лактон (3S,5S)-6-(4-амидинофенилацетиламино)-3- (3-метилбутириламино)гексановой кислоты
A) трет-Бутиловый эфир (3S,5S)-6-[4-(трет-бутилоксикарбонил) амидинофенилацетиламино] -5-[{ (1,1-диметилэтил)диметилсилил}-окси]-3- (3-метилбутириламино)-гексановой кислоты
Соединение со стадии (H) примера 7 (0,6 г, 0,82 ммоль) растворяют в 5 мл этанола и гидрируют над 10-%-ным Pd/C. После 2-часовой выдержки при комнатной температуре катализатор удаляют фильтрованием, и фильтрат концентрируют в вакууме. Полученный свободный амин растворяют в 3 мл DMF вместе с изовалериановой кислотой (110 мкл, 0,9 ммоль) и HOBT (172,5 мг, 1,13 ммоль), и смешивают с DCC (175 мг, 0,82 ммоль). После 16-часовой выдержки при комнатной температуре с реакционной смесью работают так, как описано в процессе стадии (I) примера 7. Полученный сырой продукт хроматографируют на силикагеле (гексан/EtOAc). Соединение, названное в заголовке, получают в виде белой пены. Масс-спектр: 677 (M + H)+.
B) δ -Лактон (3S,5S)-6-(4-амидинофенилацетиламино)-3- (3-метилбутириламино)гексановой кислоты
Продукт со стадии (A) примера 8 (320 мг, 0,473 ммоль) обрабатывают TFA аналогично процессу стадии (J) примера 7, и таким образом удаляют защитную группу. Дальнейшим осаждением с эфиром и последующей перекристаллизацией получают трифторацетат соединения, названного в заголовке, в виде белых кристаллов.
Т. пл.: 236,7 - 237,7oC; [α]d = +15,0o (c = 0,5 MeOH); масс-спектр: 389 (M + H)+.
Свободное соединение получают из трифторацетата известным способом.
Пример 9 (S)-3-N-[3-(п-метоксифенил)пропионил]амино-7-[N- (п-амидинобензоил)амино]гептановая кислота
A) Метиловый эфир (S)-3-N-[3-(п-метоксифенил)пропионил]амино- 7-N-Z-аминогептановой кислоты
Трифторацетат (1,18 г), полученный в соответствии со стадией (A) примера 6, вводят в взаимодействие с 0,43 г 3-п-метоксифенилпропионовой кислоты, 0,4 г HOBT, 0,33 мл триэтиламина и 0,46 г DCC, и действуют по способу, описанному в стадии (B) примера 6. Полученное сырое вещество перекристаллизовывают из эфира, и получают названное в заголовке соединение в виде белых кристаллов; масс-спектр: 471 (M + H)+.
B) трет-Бутиловый эфир (S)-3-N-[3-(п-метоксифенил)- пропионил]-амино-7-N-Z-аминогептановой кислоты
Используя способ, соответствующий стадии (B) примера 4, метиловый эфир (0,7 г), полученный на стадии (A) примера 9, гидролизуют с 0,21 г LiOH/H2O, и продукт гидролиза вводят во взаимодействие с 0,82 г трет-бутил-2,2,2-трихлорацетимидата в присутствии 0,1 мл трифторэфирата бора. Последующей хроматографией на силикагеле (этилацетат/гексан = 1:1) получают соединение, названное в заголовке, в виде бесцветного масла; масс-спектр: 513 (M + H)+.
C) (S)-3-N-[3-(п-метоксифенил)пропионил] амино-7-[N-(п- амидинобензоил)амино]гептановая кислота
Соединение (0,55 г), полученное в соответствии со стадией (B) примера 9, гидрируют по способу, описанному в стадии (C) примера 4, и полученный таким образом амин вводят во взаимодействие с 0,28 г N-Boc-п-амидинобензойной кислоты, 0,18 г HOBT, 0,15 мл триэтиламина и 0,22 г DCC, используя способ, описанный в стадии (C) примера 4. Полученный сырой продукт очищают хроматографически (силикагель, этилацетат), и удаляют защитную группу с 5 мл TFA /анизол (95: 5). После осаждения эфиром, получают трифторацетат соединения, названного в заголовке, в виде белого порошка. Масс-спектр: 469 (M + H)+.
Свободное соединение получают из трифторацетата известным способом.
Пример 10 δ -Лактон (3S,5S)-6-(4-амидинофенилацетиламино)-3-N- [2-(п-метоксифенил)этаносульфониламино]гексановой кислоты
A) трет-Бутиловый эфир (3S,5S)-6-[4-(трет-бутилоксикарбонил) амидинофенилацетиламино] -5-[{ (1,1-диметилэтил)диметилсилил} окси]-3- аминогексановой кислоты
Продукт (1,09 г, 1,50 ммоль), названный в заголовке стадии (H) примера 7, смешивают с 75,0 мг PtO2 в 7,50 мл EtOH, и проводят гидрирование, сначала нагревая смесь при температуре кипения с обратным холодильником в течение 3/4 ч, после чего добавляют 75 мг Pd/C (10 %),и смесь нагревают при температуре кипения с обратным холодильником еще в течение 3/4 ч. Катализатор удаляют фильтрованием, добавляют к фильтрату 150 мг Pd/C (10 %), и греют еще при температуре кипения с обратным холодильником в течение 1/2 ч, после чего добавляют еще 75 мг Pd/C и продолжают нагревание при температуре кипения с обратным холодильником еще в течение 1 ч. Катализатор снова удаляют фильтрованием, добавляют еще 150 мг Pd/C (10 %), и продолжают нагревание при температуре кипения с обратным холодильником в течение 1,5 ч. Затем извлекают названный в заголовке продукт, удаляя катализатор фильтрованием и упаривая фильтрат досуха. Сырой продукт (890 мг) имеет почти 100%-ную чистоту, как оценивает TCX, и его используют в последующих синтезах без дополнительной очистки.
B) трет-Бутиловый эфир (3S,5S)-6-[4-(трет-бутилоксикарбонил) амидинофенилацетиламино]-3-N-[2-(п-метоксифенил)этансульфониламино] гексановой кислоты
Соединение (890 мг, 1,50 ммоль), названное в заголовке части (A) примера 10, и 99 мкл NMM (0,6 мол.•экв.) в 7,5 мл THF энергично перемешивают со 193,5 мг (0,55 мол.•эквив.) 2-(п-метоксифенил)этансульфонилхлорида в атмосфере азота при комнатной температуре в течение 2,5 ч. Далее, добавляют 193,5 мг сульфонилхлорида и 99 мкл NMM, и смесь, как и прежде, перемешивают при комнатной температуре в течение 1 ч, после чего смесь оставляют стоять при комнатной температуре в течение 18 ч. Образовавшуюся реакционную смесь суспендируют в EtOAc и экстрагируют один раз 10%-ным водным раствором уксусной кислоты, один раз водным раствором NaHCO3, и один раз водным раствором NaCl. Объединенные водные фазы еще один раз экстрагируют EtOAc. Объединенные органические фазы затем сушат и выпаривают досуха в роторном испарителе. Операция дает выход 1,10 г сырого продукта, что соответствует выходу 92,70%. Этот сырой продукт очищают на хроматографической колонке 102,5 мл (силикагель 25 - 40 мкм, элюент гексан/EtOAc 3:7; скорость перемещения 10 мл/мин; фракции 10 мл, поглощательная способность (absorbance) 0,02/200 mv). Фракции с 11 до 20 объединяют, и после выпаривания растворителя извлекают продукт, названный в заголовке (257 мг). MA+ 791; [MA-BOC]+ 691; [MA-(BOC + C4H8)]+ 635.
C) Соединение {трифторуксусная кислота}• { δ -лактон (3S,5S)-6-(4-амидинофенилацетиламоно)-3-N-{ 2-(п-метоксифенил) этансульфониламино]гексановой кислоты}
Продукт (257 мл) (B) примера 10, смешивают с анизолом (100 мкл) и этандитиолом (100 мкл) в 2 мл 95%-ной водной трифторуксусной кислоты, и оставляют стоять при комнатной температуре на 1 1/3 ч. Образовавшуюся суспензию, при охлаждении на ледяной бане, энергично перемешивают с 20 мл диэтилового эфира. Выкристаллизовавшийся продукт отфильтровывают, промывают диэтиловым эфиром и сушат в вакууме, создаваемом водоструйным насосом, при 40oC. Полученный в результате сырой продукт растворяют в теплом EtOH, раствор охлаждают, продукт перекристаллизовывают и получают в чистом виде продукт, названный в заголовке (158 мг). Конечный продукт анализируют и получают следующие характеристики:
F : Ab 160oC;
[α]
c = 0,25 в MeOH; 221 - 896 Hf MA+ 503.
Свободное соединение получают из трифторацетата известным способом.
Пример 11 (3S, 5S)-6-(4-Амидинофенилацетиламино)-3-(4-метоксифенилпропиониламино) гексаноат лития
Соединение (153 мг, 264 мкмоль), названное в заголовке примера 7, растворяют в 528 мкл воды и переводят в форму ацетата методом анионообменной хроматографии, используя ацетатную форму анионообменной смолы Bio-Rad ACr I-X2, 100 - 200 меш. Раствор уксусной кислоты (2 М) в смеси вода/ацетонитрил (1: 1) применяют в качестве элюента, фракции, содержащие продукт, упаривают, остаток растворяют в минимальном количестве этилацетата, и продукт осаждают, добавляя диэтиловый эфир. Оставшийся аморфный продукт растворяют в воде, и смешивают с 264 мкл 2 N LiOH (2,00 мол.•экв.), и оставляют стоять при комнатной температуре на 15 ч. Из образовавшегося в результате раствора выпариванием и кристаллизацией из EtOH получают названный в заголовке продукт (87 кг, приблизительно 67%-ный выход). В результате анализа находят, что продукт имеет следующие характеристики:
Т. пл. = 227 - 230oC;
[α]
(c = 0,31 в H2O); /M + Li/ + соли лития 497 - COOLi 491 MH+ - COOH 485.
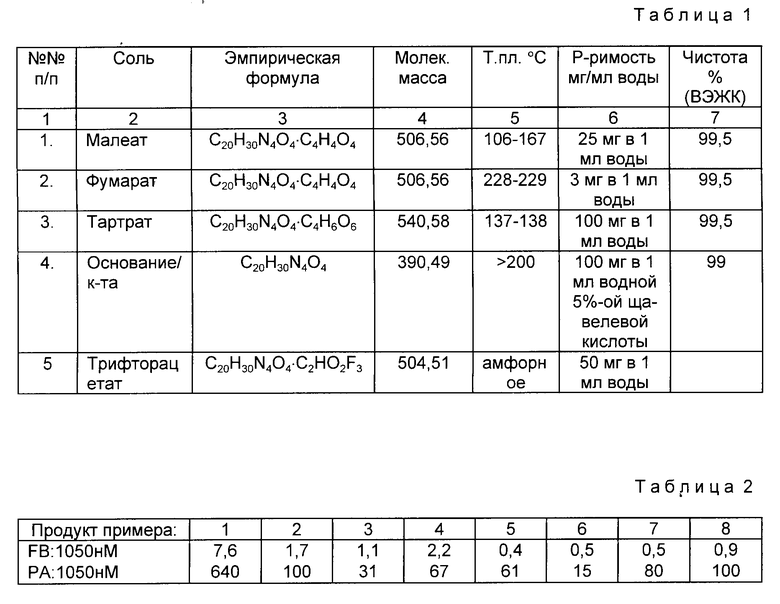
Солевые формы (S-3-/N-/3-метилбутирил/-амино/-7-/N-амидинобензоил)амино / гептановой кислоты /пример 6/ указаны в таблице I.
Соединение формулы I являются замечательными соединениями в силу их ценных терапевтических свойств. В частности, соединения формулы I обладают способностью ингибировать связывание фибриногена с GPIIb/IIIa, и таким образом, предотвращать агрегацию тромбоцитов.
Полезные свойства соединений формулы I, заключающиеся в ингибировании связывания фибриногена с изолированным и неподвижным GPIIb/IIIa, и в ингибировании агрегации тромбоцитов человека, индуцированной A P в присутствии фибриногена, демонстрируются следующими далее опытами.
a) Ингибирование связывания фибриногена с изолированным и неподвижным GPIIb/IIIa.
GPIIb/IIIa выделяют из оболочек тромбоцитов человека экстракцией тринитротолуолом X-100, и очищают хроматографически на ионитах и гель-фильтрацией. Полученный таким образом рецептор протеина закрепляют на пластинах микротитратора. Ингибирование связывания с рецептором помеченного биотином фибриногена, в присутствии ингибитора, определяют количественно.
b) Ингибирование агрегации тромбоцитов человека, индуцированной ADP в присутствии фибриногена.
Тромбоциты выделяют методом центрифугирования из свежезаготовленной цельной крови и промывают. Промытые тромбоциты снова переводят во взвешенное состояние в присутствии PC12 и аспиразы, и стимулируют ADP (10 мМ) в присутствии фибриногена. Способность тромбоцитов к агрегации в присутствии или отсутствии ингибиторов устанавливают количественно с помощью агрегометра.
Соединения формулы I осуществляют связывание фибриногена GP IIb/IIIa в области IC50 (концентрация соединений формулы I, которая уменьшает связывание фибриногена с рецептором на 50%) в промежутке от 0,5 до 20 нМ (nM).
Ингибирование агрегации тромбоцитов, индуцированной ADP, вызывается соединениями формулы I в области IC50 концентрация соединений формулы I, при которой агрегация тромбоцитов ингибируется на 50% в промежутке от 20 до 100 нМ.
Проводят испытания соединений примеров 1 - 8, чтобы определить величины их IC50 при ингибировании связывания фибриногена GP IIb/IIIa /FB/ и ингибировании агрегации тромбоцитов, индуцированной ADP (PA), в соответствии с методиками, описанными выше. Полученные результаты приводятся в табл. 2.
Благодаря активности, продемонстрированной в этих испытаниях, соединения формулы I могут быть использованы для профилактики и оказания срочной помощи при тромбозах. Положительные результаты получают при дозах от 0,1 до 20 мг/кг, предпочтительно от 061 до 3 мг/кг в день для взрослых.
Соединения формулы I могут также использоваться в форме их солей, которые получают при их взаимодействии с фармакологически приемлемыми кислотами, такими как уксусная кислота, трифторуксусная кислота, соляная кислота и т.д.
Соединения формулы I, их сольваты или соли могут вводиться энтерально, например, орально (в таком виде, как таблетки, капсулы и т.п.) или ректально, или в виде аэрозолей. Возможно также парэнтеральное введение в виде инъекций растворов или вливаний растворов.
Примеры фармацевтических композиций.
А. Таблетка
Соединение по пример 6 - 25 мг
Инертный твердый носитель - до общего веса 1 г.
Б. Питьевой раствор
Соединение по примеру 6 - 50 мг
Вода - до общего веса 1 г.
Использование: в медицине для профилактики и лечения тромбоза. Сущность изобретения: псевдопептиды или их соли формулы I: A - NH - (CH2)m - x - y - CH (CH2 - R1) NHB, где R1 : COON, COOL1, X = CH2- или R1 и X вместе - >CH - O - CO; y = -/CH2/m-, m = 1 или 2, A = -CO-/CH2/p - C6H4 - C 1 = NH1 NH2, где p = 0 или 1; B = -CO - /CH2/q - R3, где q = 1 или 2; R3 = /C1 - C4/ алкил, 1-адамантил, /C1 - C4/ алкоксифенил или B = -SO2 - (CH2)r - R4, где r = 0,1 или 2, R4 = n - /C1 - C4/ алкоксифенил; 2 способа получения соединений I; фармацевтическая композиция, содержащая в качестве активного ингредиента псевдопептид формулы I или его соль в эффективном количестве. 4 н.пункта, 4 з, пункта формулы. 2 с. и 6 з.п. ф-лы, 2 табл.
в которой R1 означает группу, имеющую одну из формул -COOH, -COOLi;
X означает -CH2 или X и R1 вместе означают группу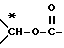
Y означает - (CH2)m-; и m равен 1 или 2,
A означает группу формулы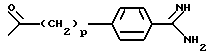
где p равен 0 или 1,
B означает группу формулы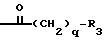
в которой q равен 1 или 2, и
R3 означает (C1 - C4)-алкил, 1-1а-дамантил, или (C1 - C4)-алкоксифенил, или
B означает группу формулы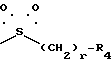
в которой r = 0, 1 или 2, и
R4 означает п-(C1 - C4)-алкоксифенил или толил.
(S)-3-(N-тозиламино)-6-/N-(n-амидинофенилацетил)-амино/-гексановая кислота;
(S)-3-/N-(3-метилбутирил)амино/6-/N-(п-аминодифенилацетил) амино/гексановая кислота;
(S)-3-N/3-(п-метоксифенил) пропионил/-амино-6-/N-(п-амидинофенилацетил)амино/гексановая кислота;
(S)-3-/N-(адамант-1-илацетил)-амино/-6-/N-(п-амидинофенилацетил)амино/гексановая кислота;
(S)-3-/N-(3-адамант-1-илпропионил)амино/-6-/N-(п-амидинофенилацетил)амино/гексановая кислота;
(S)-3-/N-(3-метилбутирил)амино/-7-/N-(п-амидинобензоил)амино/гептановая кислота;
δ-лактон (3S, 5S)-6-(4-амидинофенилацетиламино (-5-гидрокси-3-(4-метоксифенилпропиониламино)гексановой кислоты;
δ-лактон (3S, 5S)-6-(4-амидинофенилацетиламино)-5-гидрокси-3-(3-метилбутириламино) гексановой кислоты;
(S)-3-N-/3-(п-метоксифенилпропионил) /амино-7-/N-/п амидинобензоил)амино/ гептановая кислота;
δ-лактон (3S, 5S)-6-(4-амидофенилацетиламино)-5-гидрокси-3-N-/2-(п-метоксифенил)этансульфониламино/гексановой кислоты;
(3S, 5S)-6-(4-амидинофенилацетиламино)-5-гидрокси-3-(4-метоксифенилпропиониламино)гексаноат лития.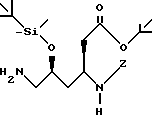
в которой Z обозначает защитную группу, соединяют с подходящим радикалом A, при этом A имеет значение, как определено выше, защитную группу Z отщепляют методом гидрогенолиза, продукт реакции соединяют с подходящим радикалом B, при этом B имеет значения, как определено выше, и впоследствии защитные группы отщепляют с помощью трифторуксусной кислоты, и соединение формулы I получают, необязательно, из полученного таким образом трифторацетата.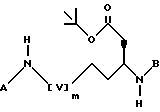
в которой [V] m означает группу [CH2]m, A и B имеют значения, установленные в п.1, и m равен 1 или 2, вводят во взаимодействие с трифторуксусной кислотой при комнатной температуре, и соединение формулы I в последствии получают, необязательно, из полученного таким образом трифторацетата.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Шредер Э., Любке К | |||
| Пептиды - М.: Мир, 1967, ч | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ получения бензидиновых оснований | 1921 |
|
SU116A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| GB, патент, 2219501, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| CH, патент, 678059, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| EP, заявка, 0454651, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
