Изобретение относится к медицинской технике и фармацевтической промышленности, а более конкретно к способу определения в анализируемой жидкости биологически активного вещества и устройству для его осуществления.
Предлагаемое изобретение может быть использовано в медицинской и клинической биохимии, а также в молекулярной фармакологии при исследовании фармакокинетики биологически активных соединений фармацевтической промышленности и экологии. Наиболее эффективно использование настоящего изобретения в клинической биохимии.
Основная проблема при рациональной фармакокинетике биологически активных веществ (БАВ), в частности синтетических и полусинтетических противоопухолевых соединений, влияющая на возможность эффективной терапии, сводится к быстроте и точности определения концентрации этих соединений в биологических жидкостях (кровь, плазма крови, моча и так далее) после введения пациентам определенного количества противоопухолевого соединения.
Известны две основные группы методов определения наличия и эффективности действия БАВ, основной мишенью которых являются молекулы двухцепочечных нуклеиновых кислот.
Первую группу составляют биологические методы [Современные методы в биохимии. /Под ред. В.Н.Ореховича. М.: Медицина. 1968, с. 372; Методы практической биохимии. /Под ред. Б. Уильямса, К. Уильсона. М.: Мир, 1978, с. 256], заключающиеся в том, что на изученных генетических системах (бактерии, бактериофаги, культуры клеток) проверяют действие веществ, вызывающих хорошо регистрируемые изменения в названных системах. Эти методы осуществляются посредством стандартных микроскопов. Однако на пути проникновения БАВ может происходить модификация их структур, влияющая на точность установления корреляции между структурой вещества, его концентрацией и биологической активностью. Кроме того, указанные методы отличаются длительностью проведения самого эксперимента (от суток до недель).
Вторую группу методов составляют физико-химические методы или различные комбинации таких методов. Начиная с 1980 г., предложено несколько методов определения биологически активных веществ, в частности производных антрахиноновой группы. Среди них различные варианты радиоиммунного анализа [Nicolau G., Szucs-Myers V., McWilliams W., Morrison J., Lanzilotti A. (1985). Investigational New Drugs, 3: pp. 51-55], тонкослойной хроматографии высокого разрешения [Avramis V. (1982), Abstract. Pharmacologist, 24, pp. 241,; Ehninger G., Proksch B., Hartmann F., Gartner H.V., Wilms K., (1984). Cancer Chemotherapy and Pharmacology, 12, pp. 50-52], метод вытеснения этидиумбромида, связанного с молекулой ДНК [Horvath J., Gueguetchkeri M., Gupta A., Penumatchu D., Weetall H. H., (1995), Biosensor and Chemical Sensor Technology, ed. Rogers K.R. et al., Washington, ASC Symp. Ser. 613. pp. 45-60]. Наибольшее распространение получила колоночная жидкостная хроматография высокого разрешения (HPLC) [Chiccarelli F.S., Morrison J.A., Cosulich D.B., Perkinson N.A., Ridge D.N. (1986), Cancer Research, 46, pp. 4858-4861; Lin K.T. Rivard G.E., Leclerc J.M. (1989). J. Chromatographe, 465, pp. 75-86], осуществляемая при помощи стандартных хроматографов.
Известна модификация метода HPLC для определения одного из важных антрахинонов - митоксантрона (МХ), которая описана сравнительно недавно [Nicoletto M.O., Padrini R., Ferrazi E., Nascimben O., Visona E., Tumolo S., Palumbo M. , Cossta L., Vinante O., Monfardini S., Fiorentino M.V. (1993). Eur. J. Cancer. 29A. pp. 1242-1248]. Согласно этому методу МХ и продукты его метаболизма экстрагируют из крови пациентов, затем их концентрируют и проводят хроматографическое выделение МХ с последующим спектрофотометрическим определением концентрации МХ. Несмотря на точность определения МХ, достигающую несколько десятков нг/мл, указанный метод характеризуется:
- длительностью всего процесса определения, достигающей двух суток, что обусловлено необходимостью специальной подготовки образцов (обработка крови пациентов, экстракция и концентрирование МХ и так далее).
- применением дорогостоящего оборудования, а именно хроматографов высокого давления и тому подобных устройств.
- необходимостью использовать высококвалифицированный персонал для проведения всего цикла анализа.
Для лабораторных растворов известен способ определения окрашенных биологически активных веществ, мишенью которых являются молекулы двухцепочечных ДНК, учитывающий взаимодействие БАВ с молекулами ДНК, образующими жидкие кристаллы, иммобилизованными в составе пленочных (гелевых) биодатчиков [1].
Этот способ включает формирование лиотропной жидкокристаллической дисперсии ДНК в водносолевом растворе, содержащем нейтральный полимер, добавление к нему специальных мономеров, способных к полимеризации, и полимеризацию полученной смеси, получение пленки (геля), имеющей форму и размер, удобные для экспериментатора, погружение пленки (геля) в анализируемый лабораторный раствор, содержащий БАВ, и выдерживание пленки в этом растворе в течение промежутка времени, достаточного для диффузии БАВ в пленку и взаимодействия с молекулами ДНК, регистрацию спектра кругового дихроизма (аномальный оптической активности) в области поглощения БАВ, определение наличия БАВ по форме спектра кругового дихроизма.
Однако точное определение концентрации исследуемого БАВ, а следовательно, и практические возможности указанного способа определения БАВ ограничены следующими факторами:
- трудностью создания пленок (гелей), отвечающих определенным физикохимическим требованиям (нейтральный по отношению к ДНК характер пленки, ее прозрачность, оптически изотропные свойства и т.д.),
- трудностью в поддержании неизменных свойств пленок, даже в течение промежутка времени диффузии БАВ,
- значительным промежутком времени, после которого становится возможным регистрация заметной величины оптического сигнала, возникающего в результате диффузии молекул БАВ в полимерную пленку и их последующего взаимодействия с молекулами нуклеиновых кислот, образующими жидокристаллическую дисперсию,
- невозможностью точного определения величины аномального оптического сигнала в УФ-области спектра (в полосе поглощения ДНК), что обусловлено недостаточной прозрачностью пленок (гелей) в этой области спектра. Поэтому, хотя наличие исследуемого соединения может быть зарегистрировано, точное определение его концентрации крайне затруднительно,
- применением для регистрации аномального оптического сигнала дорогостоящих стационарных дихрографов (Jobin-Yvon, Mark III или MarkV; Jasco - 710-720), имеющихся, как правило, только в специализированных научных лабораториях. Недостатком этих приборов является не только их высокая стоимость, но и невысокая скорость регистрации величины оптического сигнала [Mark III, Performances; Jasco J-710/720 Spectropolarimeter, Instruction Manual].
Известный дихрограф фирмы Jasco [2], который может быть применен как устройство для определения БАВ в анализируемой жидкости, содержит установленные последовательно источник светового излучения; селектор, формирующий световой поток определенной длины волны; поляризатор, формирующий из указанного потока линейно поляризованный световой поток, модулятор поляризации, преобразующий линейно поляризованный световой поток в циркулярно поляризованный световой поток с периодически изменяющимся направлением вращения вектора поляризации; ячейку для размещения исследуемой пробы; фотодетектор, преобразующий оптический сигнал, генерируемый компонентами исследуемой пробы, в пропорциональный ему электрический сигнал; синхронный усилитель, усиливающий указанный электрический сигнал; средство для обработки полученного электрического сигнала и вычисления концентрации биологически активного вещества; блок управления.
При этом в качестве селектора указанный дихрограф содержит имеющий низкий коэффициент пропускания светового потока двойной призменный монохроматор, основанный на синхронном вращении двух полупризм посредством электромеханического привода, содержащего рычажный механизм, приводящийся в действие посредством электродвигателя. Световой поток от источника светового излучения попадает в двойной монохроматор, две призмы которого настроены на определенную длину волны. Наличие рычажного механизма дает возможность перестраивать указанный монохроматор на разные длины волн выходящего из него светового потока.
Однако из-за высокой инерционности указанного привода, содержащего рычажный механизм, указанный селектор обладает малой скоростью перестройки на разные длины волн. В результате этого использование указанного дихрографа для определения БАВ значительно увеличивает время проведения анализа исследуемой пробы, что при исследовании биологических жидкостей, таких как кровь, моча и так далее, может нанести вред здоровью, а в некоторых случаях и жизни пациентов.
Кроме того, из-за сложной конструкции монохроматора он имеет большие потери на оптических элементах, в результате чего обладает низким коэффициентом пропускания светового потока, что приводит к ухудшению чувствительности устройства в целом и не дает ему определить низкие концентрации БАВ в анализируемых жидкостях. При этом указанное устройство многофункционально и обладает большими габаритами и весом, что требует специально оборудованного помещения для его установки и эксплуатации. В результате указанное устройство не обладает мобильностью и не может быть использовано для оперативного проведения срочных анализов в лабораториях клиник или непосредственно в больничной палате.
Названные выше факторы существенно затрудняют быстрое получение информации о концентрации БАВ в анализируемых растворах и ограничивают широкое применение оптических систем для такого рода анализов в условиях клиник и лабораторий.
В основу предлагаемого изобретения положена задача создать способ определенной БАВ в анализируемой жидкости с такими условиями его проведения и такое устройство для осуществления этого способа, которые позволили бы быстро и точно определять концентрацию, в том числе низкую, биологически активного вещества, способного взаимодействовать с линейными двухцепочечными молекулами ДНК, в любых жидкостях, в том числе биологических жидкостях, таких как плазма крови, цельная кровь и тому подобное.
Эта задача решена созданием способа определения в анализируемой жидкости биологически активного вещества, взаимодействующего с лиотропной жидкокристаллической холестерической дисперсией ДНК, сформированной в нейтральном по отношению к ДНК полимере, в котором согласно изобретению лиотропную жидкокристаллическую холестерическую дисперсию формируют в водно-солевом растворе указанного полимера из линейных двухцепочечных молекул ДНК низкой молекулярной массы непосредственно перед смешением с анализируемой жидкостью, содержащей определяемое БАВ, при этом анализируемую жидкость предварительно смешивают с указанным полимером в условиях, при которых оптические свойства лиотропной жидкокристаллической дисперсии ДНК не нарушается, затем через пробу, полученную в результате указанного смешения подготовленной анализируемой жидкости с указанной жидкокристаллической дисперсией ДНК, пропускают поток светового циркулярно поляризованного излучения и регистрируют оптический сигнал, генерируемый жидкокристаллической дисперсией на двух длинах волн, одна из которых находится в области поглощения азотистых оснований ДНК, а другая - в области поглощения определяемого биологически активного вещества, после чего вычисляют соотношение между этими сигналами на указанных длинах волн и по величине этого соотношения определяют концентрацию БАВ по калибровочной кривой. Теория формирования жидкокристаллических дисперсий ДНК в нейтральных по отношению к этой макромолекуле растворах полимеров описана в работе [Yevdokimov Yu. M. , Skuridin S.G., Salyanov V.l. (1988). Liquid Crystals, 3, p.p. 1443-1459], а экспериментальные граничные условия, при которых аномальная оптическая активность жидкокристаллических дисперсий ДНК сохраняется, приведены в работе [Евдокимов Ю.М., Скуридин С.Г., Акименко Н. М. (1984). Высокомолекулярные соединения, А24, стр. 2403-2410].
Использование предлагаемого способа позволяет быстро, просто, с высокой точностью и чувствительностью определять любые биологически активные вещества, способные взаимодействовать с линейными двухцепочечными молекулами ДНК в любых жидкостях, в которых можно создать условия для сохранения жидкокристаллических дисперсий ДНК, при этом анализ можно осуществлять в любых лабораториях, где не требуется специально организованного места и специальной квалификации обслуживания персонала.
Предлагаемый способ особенно ценен для быстрого, высокоточного и высокочувствительного определения наличия и концентрации (в том числе низкой) БАВ (противоопухолевые препараты, антибиотики, белки и так далее) в крови пациентов в практике онкологии, хирургии, гинекологии, при медико-экологическом скрининге и может спасти здоровье и жизнь пациентов в тех случаях, когда другие способны не применимы или не дают надлежащего результата.
Целесообразно в качестве нейтрального полимера использовать полиэтиленгликоль, поскольку этот полимер является безвредным для экспериментатора, химически нейтральным по отношению к ДНК, обладает высокой растворимостью, необходимой для создания условий фазового исключения ДНК, оптической изотропией и высокой прозрачностью, необходимой для измерения спектров кругового дихроизма, причем препараты этого полимера разной молекулярной массы доступны по разумной цене.
Желательно в качестве анализируемой жидкости использовать биологическую жидкость, поскольку фармакокинетика биологически активных соединений нацелена на применение в таких жидкостях, как кровь, моча и т.д.
Благоприятно в качестве биологической жидкости использовать плазму крови, поскольку в этом случае уменьшается число факторов, влияющих на точность определения биологически активного вещества.
Целесообразно, чтобы биологически активное вещество представляло собой противоопухолевое соединение антрахиноновой группы, поскольку соединения этой группы широко используются как сами, так и в различных комбинациях в химиотерапии онкологических заболеваний.
Желательно, чтобы противоопухолевое соединение антрахиноновой группы представляло собой митоксантрон, поскольку митоксантрон является одним из самых мощных противоопухолевых агентов широкого спектра действия.
Благоприятно, чтобы при определении биологически активного вещества в биологической жидкости с гетерогенным в ней его распределением концентрация биологически активного вещества, полученная по калибровочной кривой, корректировалась с учетом коэффициента его распределения между компонентами в биологической жидкости.
Таким образом, предлагаемый способ может быть использован для быстрого, высокоточного и высокочувствительного определения, наличия и концентрации различных биологически активных веществ (противоопухолевые препараты, антибиотики, белки и так далее) в различных жидкостях, в том числе в крови пациентов в практике онкологии, хирургии, гинекологии, при медико-экологическом скрининге и поможет спасти здоровье и жизнь пациентов в тех случаях, когда другие способны не применимы или не дают надлежащего эффекта.
Поставленная задача решена также созданием устройства для определения в анализируемой жидкости биологически активного вещества, содержащего установленные последовательно источник светового излучения, селектор, формирующий световой поток определенной длины волны, поляризатор, формирующий из указанного потока линейно поляризованный световой поток, модулятор поляризации, преобразующий линейно поляризованный световой поток в цикрулярно поляризованный световой поток с периодическим изменением направления вращения вектора поляризации, ячейку для размещения исследуемой пробы, фотодетектор, преобразующий оптический сигнал, генерируемый компонентами исследуемой пробы, в пропорциональный ему электрический сигнал, синхронный усилитель, усиливающий указанный электрический сигнал, средство для обработки полученного электрического сигнала и вычисления концентрации биологически активного вещества, блок управления, в котором согласно изобретению селектор содержит минимально необходимое число оптических элементов и электродинамический (гальванометрический) привод позиционного типа, обеспечивающий измерение сигнала на двух выбранных для определяемого БАВ длинах волн.
Указанное конструктивное выполнение позволяет по меньшей мере в десять раз сократить время селекции требуемой длины волны светового потока при уменьшении энергопотребления и позволяет быстро, точно и с высокой чувствительностью определять концентрацию (в том числе низкую) биологически активных веществ, способных взаимодействовать с молекулами двухцепочечных ДНК в анализируемых жидкостях, в том числе в биологических жидкостях, таких как цельная кровь, плазма крови и так далее.
Желательно, чтобы селектор представлял собой одинарный монохроматор с минимальным числом оптических элементов, включающих и дисперигрующий элемент, при этом один из указанных элементов закреплен на валу двигателя электродинамического (гальванометрического) привода [И.Н. Нестерук, О.Н. Компанец, В.И. Мишин (1988). Квантовая электроника, 15, N 3, стр. 455-459] с возможностью его углового позиционирования для быстрой и точной установки двух выбранных длин волн.
Наличие указанного привода и использование такого монохроматора упрощают конструкцию устройства в целом, они позволяют значительно уменьшить его габариты. Это приводит к значительному удешевлению устройства и позволяет оснастить этим устройством любые лаборатории, так как предлагаемое устройство не требует наличия специально организованного места и специальной квалификации обслуживающего персонала.
Кроме того, использование в качестве селектора простого монохроматора с минимальным числом оптических элементов позволяет упростить оптическую систему устройства, уменьшить потери света на оптических элементах, то есть увеличить коэффициент пропускания светового потока, и благодаря этому усилить чувствительность устройства, уменьшить его размеры и, следовательно, обеспечить мобильность устройства для его использования в любых условиях.
Благоприятно, чтобы диспергирующий элемент представлял собой дифрационную решетку. Это дает возможность обеспечить требуемую (1-5 нм) разрешающую силу монохроматора, а значит не потерять в спектральном разрешении селектора и в точности измерения оптического сигнала.
Возможно, чтобы селектор содержал набор узкополосных интерференционных фильтров, каждый из которых имеет полосу пропускания в области выбранной для определяемого БАВ длины волны, закрепленных на валу двигателя электродинамического (гальванометрического) привода позиционного типа с возможностью их поочередного введения в световой поток.
Использование в селекторе набора интерференционных фильтров, укрепленных на валу двигателя электродинамического (гальванометрического) привода позиционного типа, позволяет при сохранении высокой скорости селекции требуемой длины волны существенно упростить оптическую схему селектора и, следовательно, увеличить коэффициент пропускания светового потока и уменьшить габариты и вес всего устройства.
Целесообразно, чтобы электродинамический (гальванометрический) привод содержал двигатель, имеющий статор и ротор, и датчик угла поворота ротора, представляющий собой индуктивный дифференциальный преобразователь угла поворота ротора в электрический сигнал, при этом указанный датчик содержит модулятор, выполненный в виде кольца, закрепленного на валу ротора, двигатель с эксцентриситетом относительно его оси вращения.
Благодаря наличию указанного эксцентриситета появляется возможность реализовать определенный характер зависимости амплитуды сигнала датчика угла поворота, а значит и длины волны селектора, от угла поворота ротора двигателя, причем характер этой зависимости определяется формой (конструктивным выполнением) модулятора.
Таким образом, использование предлагаемого способа и предлагаемого устройства позволяет быстро, точно и с высокой чувствительностью определять наличие и концентрацию различных биологически активных веществ (противоопухолевые препараты, антибиотики, белки и так далее) в различных жидкостях, в том числе в биологических жидкостях, например, в крови пациентов в практике онкологии, хирургии, гинекологии, при медико-экологическом скрининге, и поможет спасти здоровье и жизнь пациентов в тех случаях, когда другие способы не применимы или не дают надлежащего эффекта.
Кроме того, предлагаемое изобретение позволяет значительно уменьшить размеры и вес устройства, а следовательно, обеспечить его мобильность для использования в любых условиях, не требующих специально оборудованного места.
Предлагаемый способ определения концентрации БАВ осуществляют следующим образом:
- непосредственно перед определением БАВ в анализируемой жидкости формируют в водно-солевом растворе нейтрального полимера лиотропную холестерическую жидкокристаллическую дисперсию линейных двухцепочечных молекул ДНК низкой молекулярной массы;
- смешивают жидкость, содержащую БАВ, с раствором полимера таким образом, чтобы в полученной смеси создавались физико-химические условия, обеспечивающие сохранение аномальных оптических свойств, в частности кругового дихроизма, характерных для жидких кристаллов ДНК;
- центрифугируют полученную смесь и отбирают супернатант, содержащий молекулы БАВ, не взаимодействующие с компонентами анализируемой жидкости;
- смешивают супернатант с жидкокристаллической дисперсией ДНК, сформированной в водно-солевом растворе нейтрального полимера, что приводит к получению пробы для анализа, в которой молекулы БАВ быстро (за время 10-6 с) реагируют с ДНК;
- облучают пробу циркулярно поляризованным светом;
- измеряют аномальную оптическую активность пробы на двух длинах волн, одна из которых находится в области поглощения азотистых оснований ДНК, другая - в области поглощения БАВ (при этом полоса в области поглощения ДНК используется не только как внутренний стандарт качества сформированной жидкокристаллической дисперсии, но и отражает стабильность и правильность работы предлагаемого устройства, полоса в спектре кругового дихроизма в области поглощения БАВ выступает в качестве индикатора его наличия в исследуемой жидкости, а амплитуда этой полосы отражает концентрацию БАВ);
- вычисляют соотношение между величинами оптических сигналов при названных выше длинах волн;
- определяют точную концентрацию БАВ по величине этого соотношения с использованием калибровочной кривой зависимости отношения величин сигналов от концентрации БАВ, предварительно построенной по описанному выше способу.
Для лучшего понимания настоящего изобретения ниже приведены примеры, характеризующие различные стадии заявляемого способа со ссылками на прилагаемые чертежи.
Фиг. 1 характеризует спектр поглощения исходного водно-солевого раствора линейной двухцепочечной ДНК, использованной для приготовления частиц холестерической жидкокристаллической дисперсии в координатах - оптическая плотность, A - длина волны, λ; CДНК 80 мкг/мл; ДНК эритроцитов цыплят ("Reanal", Венгрия); Мол. масса ДНК (3-5)•105Da.
Фиг. 2 характеризует спектры кругового дихроизма исходной ДНК и ее холестерической жидкокристаллической дисперсии в координатах Δε = εL-εR - длина волны λ, где величина Δε = ΔA/CДНК - молярный круговой дихроизм; εL - молярный дихроизм для левоциркулярно поляризованного света εR - молярный дихроизм для правоциркулярно поляризованного света; ΔA - экспериментально измеренный круговой дихроизм; CДНК - концентрация ДНК; кривая 1 - спектр КД холестерической жидкокристаллической дисперсии ДНК; кривая 2 - спектр ДНК водно-солевого раствора исходной линейной ДНК.
Кривая 1 (правая ордината): CПЭГ= 170 мг/мл, мол. масса ПЭГ=4000 ("Ferak", Германия); 0,3 M NaCl + 10-2 M фосфатный буфер; pH 7,0.
Кривая 2 (левая ордината): 0,3 M NaCl + 10-2M фосфатный буфер; pH 7,0.
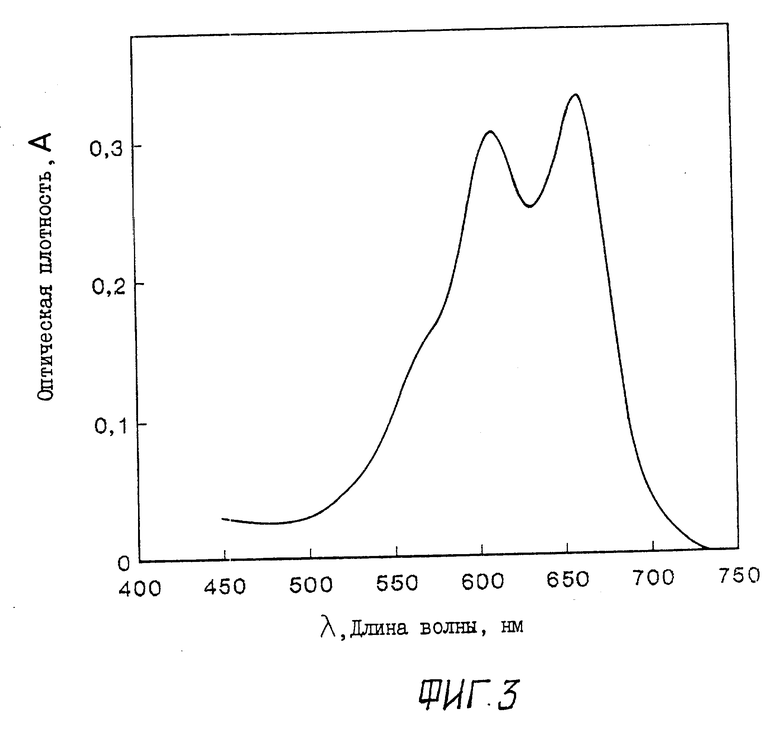
Фиг. 3 характеризует спектр поглощения митоксантрона, использованного в качестве примера биологически активного соединения в координатах - оптическая плотность, круговой дихроизм A - длина волны λ ; 0,3 M NaCl + 10-2 M фосфатный буфер; pH 7,0; CtMX=1,5•10-5 M.
Фиг. 4 характеризует спектры кругового дихроизма жидкокристаллической дисперсии ДНК, обработанной разными концентрациями митоксантрона, в координатах круговой дихроизм ΔA = AL-AR - длина волны λ , где AL - дихроизм для левоциркулярно поляризованного света, AR - дихроизм для правоциркулярно поляризованного света, ΔA - экспериментально измеренный круговой дихроизм;
1 - CtMX = 0; 2 - CtMX = 1,55 • 10-6 M;
3 - CtMX = 3,08 • 10-6 M; 4 - CtMX = 5,35 • 10-6 M.
CДНК = 20 мкг/мл; CПЭГ = 170 мг/мл;
0,3 M NaCl + 10-2 M фосфатный буфер; pH 7,0.
ΔA = AL-AR в мм ; 1 мм = 5 • 10-5 оптических единиц; L = 1 см.
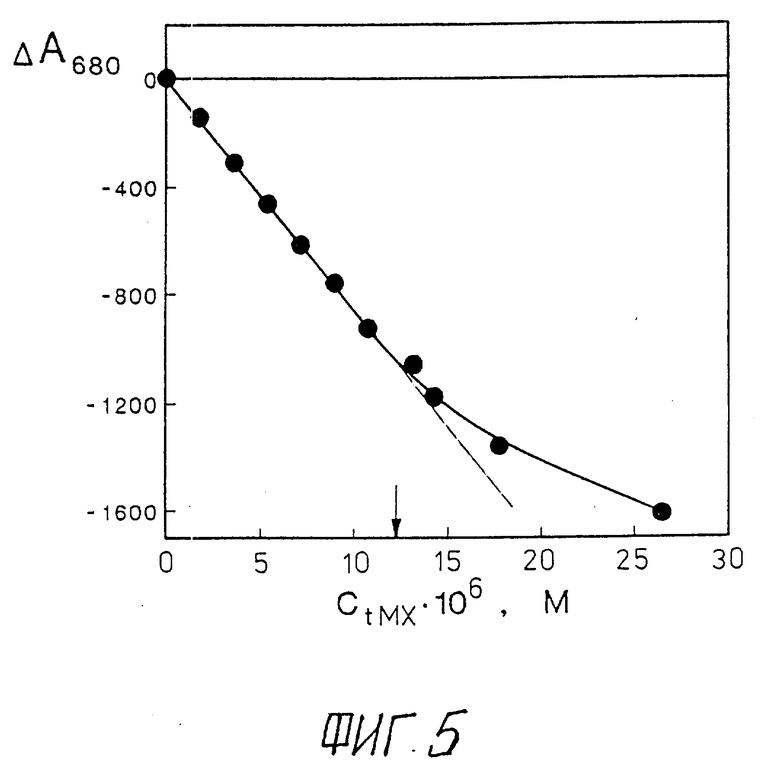
Фиг. 5 отражает зависимость амплитуды ΔA680 отрицательной полосы при λ 680 нм в спектре кругового дихроизма жидкокристаллической дисперсии ДНК от концентрации (CtMX) добавленного митоксантрона, причем стрелкой на рисунке обозначена концентрация митоксантрона, вплоть до которой между величинами ΔA680 и CtMX наблюдается прямо пропорциональная зависимость.
CДНК = 20 мкг/мл; CПЭГ = 170 мг/мл; 0,3 M NaCl + 10-2 M фосфатный буфер; pH 7,0; ΔA680 в мм ; 1 мм = 1 • 10-6 оптических единиц; L = 1 см.
Фиг. 6 показывает зависимость амплитуды полосы в спектрах кругового дихроизма жидкокристаллических дисперсий комплексов (ДНК-МХ) при λ 680 нм от величины CtMX в анализируемых пробах и в контрольных растворах (соответственно прямая 1 и прямая 2). Стрелками на фиг. 6 (A → Б → B = CtMX) показан способ определения величины CtMX в анализируемой пробе, учитывающий связывание митоксантрона с форменными элементами крови и высокомолекулярными компонентами плазмы разной химической природы;
CДНК = 20 мкг/мл; CПЭГ = 170 мг/мл.
0,225 M NaCl + 7,5 • 10-3 M фосфатный буфер; pH 7,0.
Δ A680 в мм; 1 мм = 2 • 10-6 оптических единиц; L = 1 см.
Фиг. 7 отражает зависимость величины C
CДНК = 20 мкг/мл; CПЭГ = 170 мг/мл; 0,225 M NaCl + 7,5 • 10-3 M фосфатный буфер; pH 7,0.
Фиг. 8 показывает зависимость отношения полос в спектрах КД жидкокристаллических дисперсий комплексов (ДНК-МХ) при 680 нм и λ1 680 нм и λ2 275 нм 275 нм от величины C
CДНК = 20 мкг/мл; CПЭГ = 170 мг/мл; 0,225 M NaCl + 7,5 • 10-3 M фосфатный буфер; pH 7,0; ΔA680 и ΔA275 в мм ; 1 мм = 2 • 10-6 оптических единиц; L = 1 см.
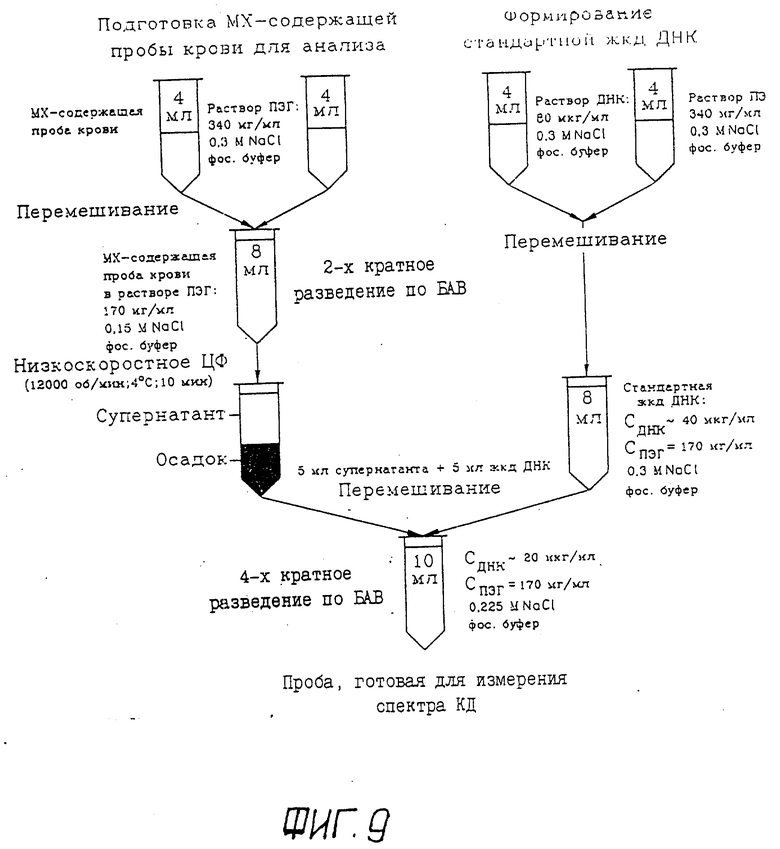
Фиг. 9 изображает принципиальную схему подготовки пробы крови для определения наличия БАВ, взаимодействующих с жидкокристаллической дисперсией ДНК. (Следует обратить внимание на то, что в результате предложенного способа подготовки крови исходная концентрация БАВ уменьшается в четыре раза).
Пример 1. Формирование жидкокристаллической дисперсии ДНК.
1.1. Навески NaCl (17,532 г), NaH2PO4•2H2O (0,78 г) и Na2HPO4•12H2O (1,79 г) помещают в мерную колбу (V = 1000 мл) и растворяют в дистиллированной воде; доводят объем раствора до метки.
Таким образом готовят 1 л 0,3 M раствора NaCl, содержащего 10-2 M фосфатный буфер.
1.2. 10 мг препарата двухцепочечной ДНК ("Reanal", Венгрия; (0,3-0,5)•105 Da) помещают в мерочную пробирку (V=10 мл) и растворяют в растворе, приготовленном по п. 1.1; доводят объем раствора до метки.
Таким способом готовят 10 мл водно-солевого раствора ДНК с фиксированной концентрацией. Концентрацию ДНК определяют спекрофотометрически, исходя из соотношения: 1 мг ДНК соответствует 20 оптическим единицам в мл 1 мл λмакс= 258,4 нм ; pH 7,0).
1.3. Навески NaH2PO4•2H2O (0,078 г) и Na2HPO4•12H2O (0,179 г), NaCl (1,7532 г) и полиэтиленгликоля (ПЭГ) ("Ferak", Германия; ПЭГ = 4000; 34 г) помещают в мерную колбу (V = 100мл) и растворяют в дистиллированной воде; доводят объем раствора до метки.
Таким способом готовят 100 мл водно-солевого раствора ПЭГ (CПЭГ = 340 мг/мл; 0,3 M NaCl+10-2 M фосфатный буфер).
1.4. В стеклянной пробирке (V = 10 мл) смешивают 4,6 раствора 1.1 и 0,4 мл раствора 1.2.
Таким образом готовят 5 мл водно-солевого раствора двухцепочечной линейной ДНК (CДНК=80 мкг/мл; 0,3 M NaCl+10-2 M фосфатный буфер).
После смешения растворов 1.1 и 1.2 регистрируют спектр поглощения водно-солевого раствора ДНК заданной концентрации, приготовленного по п. 1.4 (фиг. 1, где изображен спектр поглощения водно-солевого раствора ДНК),
CДНК - 80 мкг/мл; ДНК эритроцитов цыплят ("Reanal", Венгрия);
Мол. м. ДНК - (3-5)•105 Da;
1.5. В стеклянной пробирке (V=15 мл) смешивают 4 мл раствора (1.3) с 4 мл раствора (1.4); полученную смесь интенсивно перемешивают в течение 3 мин.
Таким способом готовят 8 мл стандартной жидкокристаллической дисперсии ДНК (CДНК=40 мкг/мл; CПЭГ=170 мг/мл; 0,3 M NaCl+10-2 M фосфатный буфер).
После смешения регистрируют спектр кругового дихроизма (КД) смеси, приготовленной по п. 1.5. Интенсивная отрицательная полоса в области поглощения азотистых оснований ДНК (фиг. 2, кривая 1) свидетельствует о том, что в результате смешениярастворов, приготовленных по п. 1.3 и 1.4, образуется жидкокристаллическая дисперсия ДНК холестерического типа.
Пример 2. Оптические свойства жидкокристаллической дисперсии ДНК, обработанной митоксантроном.
2.1. Навеску (0,4 мг) митоксантрона (МХ; противоопухолеволе соединение антрахиновой группы) помещают в пробирку (V=0,5 мл) и растворяют в 200 мкл раствора 1.1
Таким способом готовят 200 мкл водно-солевого раствора МХ (CМХ=2 мг/мл; 0,3 М NaCl + 10-2 M фосфатный буфер).
2.2. Концентрацию МХ в растворе 2.1. определяют спекрофотометрически при помощи спектрофотометра ("Specord" М 40, Германия) пользуясь, известным значением молярного коэффициента поглощения МХ ε = 21500 М-1см-1; λмакс= 660 нм. Молярная концентрация МХ в растворе 2.2 составляет 3,868 • 10-3 M.
На фиг. 3 приведен спектр поглощения водно-солевого раствора МХ.
2.2. В прямоугольную оптическую кварцевую кювету (V = 4 мл; длина оптического пути 1 см) помещают 2 мл стандартной жидкокристаллической дисперсии, приготовленной по п. 1.5, и записывают ее спектр КД в области длин волн 750-220 нм при помощи дихрографа "Jodin-Yvon, Mark III" (Франция).
2.3. В кювету, содержащую 2 мл дисперсии ДНК, приготовленной по п. 2.2, добавляют по 1 мкг раствора 2.1 (суммарный объем добавленного раствора 2.1 составляет 6 мкл); после каждой добавки раствора 2.1 полученную в оптической кювете смесь перемешивают и регистрируют спектр КД в области длин волн 750-220 нм.
На фиг. 4 спектр КД исходной жидкокристаллической дисперсии ДНК (кривая 1) сопоставлен со спектрами КД этой же дисперсии, в которую добавлены разные концентрации (CtМХ) МХ (CtМХ - общая концентрация МХ, добавленного в раствор жидкокристаллической дисперсии ДНК). Добавление МХ сопровождается появлением в спектре КД жидкокристаллической дисперсии ДНК дополнительной интенсивной отрицательной полосы в области поглощения МХ (λ 680 нм) . При этом амплитуда полосы при длине волны λ 680 нм связана с амплитудой полосы при λ 275 нм следующим образом: при одинаковой концентрации МХ амплитуда полосы при λ 680 нм тем выше, чем выше амплитуда полосы при λ 275 нм и наоборот.
Зависимость амплитуды полосы при λ 680 нм в спектре КД жидкокристаллической дисперсии комплексов (ДНК-МХ) от CtМХ (фиг.5) показывает, что между величиной ΔA680 и CtМХ наблюдается прямо пропорциональная зависимость в области концентраций МХ от 0 до 12•10-6 M. Поскольку амплитуда полосы в спектре КД в области поглощения МХ (ΔA680) зависит только от концентрации молекул МХ, связанных (CbМХ) в комплекс с молекулами ДНК, и поскольку между величинами CtМХ и CbМХ существует простое соотношение:
CtМХ = CbМХ + CfreeМХ,
где
CfreeМХ - концентрация в растворе свободных (не связанных в комплекс с молекулами ДНК) молекул МХ, то прямо пропорциональную зависимость между величинами ΔA680 и CtМХ можно рассматривать как указание на низкую концентрацию свободного МХ в растворе.
Величина CbМХ при фиксированной концентрации ДНК тем ближе к величине CtМХ, чем ниже концентрация последней. В области фармакологически значимых значений концентрации МХ (нг/мл) можно принять, что величина CbМХ приблизительно равна величине CtМХ.
Таким образом, интенсивная полоса в области поглощения МХ в спектре КД в сочетании с наличием прямо пропорциональной зависимости амплитуды этой полосы и концентрацией добавленного МХ может быть использована для определения наличия и концентрации МХ.
При определении концентрации молекул МХ, которые могут взаимодействовать с молекулами ДНК, а следовательно, концентрации МХ в биологических жидкостях при помощи спектров кругового дихроизма нужно учитывать, что на определяемую величину могут влиять два фактора:
1) распределение молекул МХ форменными элементами крови,
2) неодинаковость оптических свойств жидкокристаллических дисперсий ДНК в крови разных пациентов, влияющая на амплитуду полосы при λ 275 нм , а следовательно, и на амплитуду полосы при λ 680 нм .
Точное определение концентрации МХ требует учета влияния этих факторов.
Пример 3. Учет распределения митоксантрона в крови человека
При определении концентрации МХ в биологических жидкостях (кровь, плазма и др. ) необходимо учитывать возможное связывание поступающего в кровь МХ с форменными элементами крови и высокомолекулярными компонентами плазмы разной природы. Приведенный ниже пример позволяет оценивать степень связывания МХ.
3.1. По 2 мл крови человека разливают в 8 полипропиленовых центрифужных пробирок (V=12,5 мл).
3.2. В каждую пробирку (п. 3.1) добавляют соответственно 0; 1; 3; 5; 7; 9; 11 и 13 мкл раствора, приготовленного по п. 2.1.
3.3. После добавления раствора 2.1 в кровь каждая из полученных проб перемешивается.
Таким способом готовят серию МХ-содержащих проб крови, в которых CtМХ меняется от 0 до 24,98•10-6 M.
3.4. В каждую МХ-содержащую пробу крови, полученную в соответствии с п. 3.1.- 3.3, добавляют по 2 мл раствора, приготовленного по п. 1.3.
3.5. После добавления раствора 1.3 в пробы крови (п. 3.3) полученные смеси перемешиваются.
Таким способом получают МХ-содержащие пробы крови в водно-солевом растворе ПЭГ (CtМХ от 0 до 12,49•10-6 M; CПЭГ=170 мг/мл; 0,15 M NaCl+5•10-3 M фосфатный буфер).
3.6. Полученные в соответствии с п. 3.5 пробы крови центрифугируют (12000 об/мин; 4oC; центрифуга K 24D, Германия).
В результате центрифугирования образуется осадок, состоящий из форменных элементов крови и высокомолекулярных компонентов плазмы.
3.7. После низкоскоростного центрифугирования из каждой центрифужной пробирки (п. 3.6, 8 пробирок) отбирают по 2 мл супернатанта и переносят в 8 стеклянных пробирок (V=10 мл).
3.8. В каждую из 8 стеклянных пробирок, содержащих по 2 мл супернатанта (п. 3.7), добавляют по 2 мл стандартной жидкокристаллической дисперсии ДНК, сформированной по п. 1.5.
3.9. Полученные в соответствии с п. 3.8 пробы перемешиваются.
Таким способом получают серию проб, содержащих исходную жидкокристаллическую дисперсию ДНК и супернатант ПЭГ-содержащих проб крови с разным содержанием МХ (CДНК=20 мкг/мл; CПЭГ=170 мг/мл; 0,15 M NaCl+5•10-3 M фосфатного буфера).
3.10. Параллельно серии проб, указанной в п. 3.9, в соответствии с операциями по п. 3.1.-3.9 готовится контрольная серия, при приготовлении которой вместо крови используется раствор 1.1. (В контрольной серии CtМХ меняется от 0 до 6,245•10-6 M).
3.11. После перемешивания анализируемых проб (п. 3.9) и контрольных растворов (п. 3.10) при помощи дихрографа "Jobin-Yvon, Mark III" (Франция) регистрируют спектры КД анализируемой (п. 3.9) и контрольной (п. 3.10) серий проб.
Амплитуда полосы в спектре КД λ 680 нм ПЭГ-содержащих проб крови (п. 3.4) возрастает по мере увеличения CtМХ (фиг.6, прямая 1).
Различие между прямыми 1 и 2 (фиг. 6) показывает, что в анализируемых пробах содержится меньшая, по сравнению с контрольным раствором, концентрация МХ. Уменьшение концентрации МХ отражает тот факт, что часть молекул МХ связывается с форменными элементами крови и высокомолекулярными компонентами плазмы и становится недоступной (п. 3.6) для определения при помощи оптического метода.
Для учета расхождения между одними и теми же значениями концентраций МХ (CtМХ), добавленными в кровь и в контрольный раствор, пользуются приемом, показанным на фиг. 6.
В соответствии с этим приемом амплитуда полосы, характерная для спектра КД анализируемой пробы при λ 680 нм (прямая 1, точка А), переносится на прямую 2 (точка "Б"), которая представляет собой зависимость величины ΔA680 от CtМХ, для контрольной серии растворов:
(A _ _ → Б _ _ → B = C
Значению величины ΔA680/ в точке Б на оси абсцисс соответствует величина CtМХ (точка В), т.е. значение концентрации МХ в анализируемой пробе (C
На фиг. 7 приведена зависимость концентрации МХ в анализируемых пробах (C
Зависимость между этими величинами описывается прямой, тангенс угла наклона которой составляет 0,384. Это означает, что в состоянии, доступном для анализа при помощи оптических методов, остается только 38,4% от всего МХ, добавленного в кровь. На эту величину должна быть введена поправка при написании окончательного уравнения для определения концентрации МХ, добавленного в кровь.
Пример 4. Учет возможной неодинаковости оптических свойств жидкокристаллических дисперсий ДНК при вычислении концентрации митоксантрона в анализируемой биологической жидкости.
4.1. Чтобы исключить неоднородность оптических свойств жидкокристаллической дисперсии ДНК в крови разных пациентов, а следовательно, и величину оптического сигнала, генерируемого этими жидкокристаллическими дисперсиями при образовании комплекса (ДНК-МХ), значения величины ΔA680, полученные в экспериментах по определению МХ, были отнормированы на величину оптического сигнала, регистрируемого в спектре КД контрольной жидкокристаллической дисперсии ДНК при λ 275 нм (ΔA275) .
4.2. Зависимость отношения ΔA680/ΔA275 от величины C
Пример 5. Регламент определения концентрации биологически активного вещества в крови человека.
Подготовка взятой у пациента крови, содержащей биологически активное вещество, и оценка концентраций БАВ проводится по схеме, приведенной на фиг. 9. В соответствии с этой схемой, отработанной на примере определения концентрации МХ:
5.1. В полипропиленой центрифужной пробирке (V = 12,5 мл) смешивают 4 мл крови, взятой у пациента, с 4 мл раствора по п. 1.3.
5.2. Полученную смесь перемешивают.
Таким способом получают пробу крови, содержащую биологически активное вещество (БАВ), в водно-солевом растворе ПЭГ.
5.3. Приготовленную по п. 5.2 БАВ-содержащую пробу крови в водно-солевом растворе ПЭГ центрифугируют.
5.4. После низкоскоростного центрифугирования 5 мл супернатанта смешивают с 5 мл стандартной жидкокристаллической дисперсии ДНК, приготовленной по п. 1.1 - 1.5 (пример 1).
Таким способом получают пробу крови для анализа.
5.5. Параллельно с приготовлением пробы крови по п. 5.1 - 5.4 готовят контрольную жидкокристаллическую дисперсию ДНК, при получении которой вместо крови используется раствор по п. 1.1.
5.6. После перемешивания анализируемой пробы крови (п. 5.4) и контрольной жидкокристаллической дисперсии ДНК (п. 5.5) определяют значения амплитуд полос в спектрах КД соответственно при длине волны λ1 , соответствующей области поглощения БАВ (в данном случае при 680 нм (ΔA680)), и при длине волны, соответствующей области поглощения ДНК (λ2 275 нм, ΔA275)..
5.7. Определяют отношение ΔA680/ΔA275 .
5.8. Полученное отношение (п. 5.7) наносят на универсальную калибровочную кривую (фиг. 8, пример 4.2) и определяют соответствующее этому отношению значение величины C
5.9. Определив значение величины CtБАВ(MX) и учтя 4-х кратное разбавление пробы по концентрации МХ при помощи выражения:
C
определяют концентрацию БАВ (МХ) в крови пациента (CtБАВ(МХ)), не связанного с форменными элементами крови.
5.10. Для определения исходной концентрации БАВ (МХ) вводят поправку по п. 3.11.

Минимальная концентрация БАВ (МХ), определяемая в крови пациента при помощи предлагаемого регламента, основанного на измерении оптического сигнала, генерируемого жидкокристаллической дисперсией ДНК, при помощи дихрографа "Jobin-Yvon, Mark III" (Франция), составляет 5•10-7М.
Ниже приведен конкретный пример выполнения предлагаемого устройства для осуществления предлагаемого способа со ссылками на прилагаемые чертежи, на которых:
Фиг. 10 изображает блок-схему устройства, выполненного согласно изобретению.
Фиг. 11 схематично изображает селектор, представляющий собой одинарный монохроматор, выполненный согласно изобретению, вид сверху.
Фиг. 12 схематично изображает электродинамический (гальванометрический) привод позиционного типа, выполненный согласно изобретению, вид сверху со сложным сечением.
Фиг. 13 схематично изображает селектор, представляющий собой набор интерференционных фильтров, выполненный согласно изобретению, изометрия.
Фиг. 14 изображает вид спектра КД водно-солевого раствора н-пропиламмониевой соли d-10-камфорсульфоновой кислоты, зарегистрированного предлагаемым устройством:
концентрация C10H16O4S 0,15 мг/мл;
шаг развертки спектра по длинам волн 5 нм/дел.
Фиг. 15 изображает вид спектра КД водно-солевого раствора линейной B-формы ДНК, зарегистрированного предлагаемым устройством:
CДНК = 5 мкг/мл; 0,3 М NaCl + 10-2 фосфатный буфер; pH ≈ 7,0;
шаг развертки спектра по длинам волн 3,5 нм/дел.
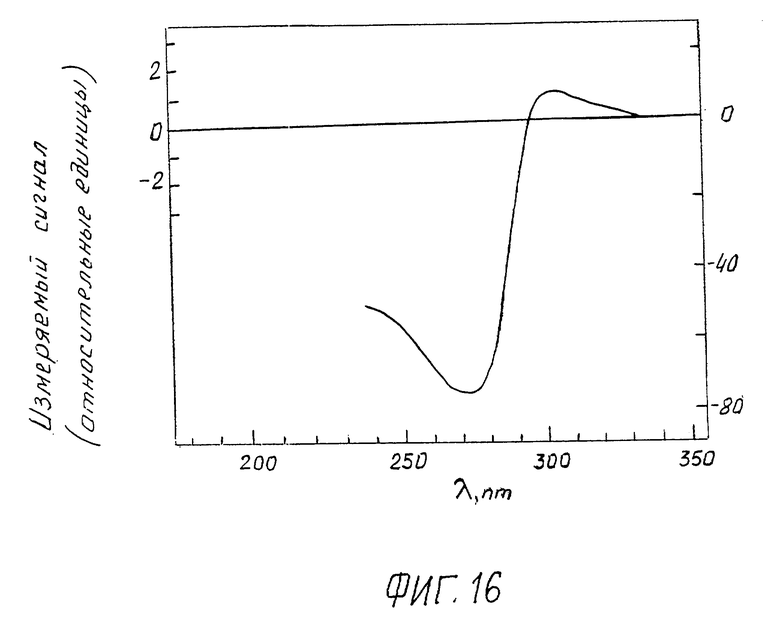
Фиг. 16 изображает вид спектра КД жидкокристаллической дисперсии ДНК, зарегистрированного предлагаемым устройством:
CДНК = 5 мкг/мл; CПЭГ = 170 мг/мл;
0,3 М NaCl + 10-2 M фосфатный буфер; pH ≈ 7,0;
шаг развертки спектра по длинам волн 10 нм/дел.
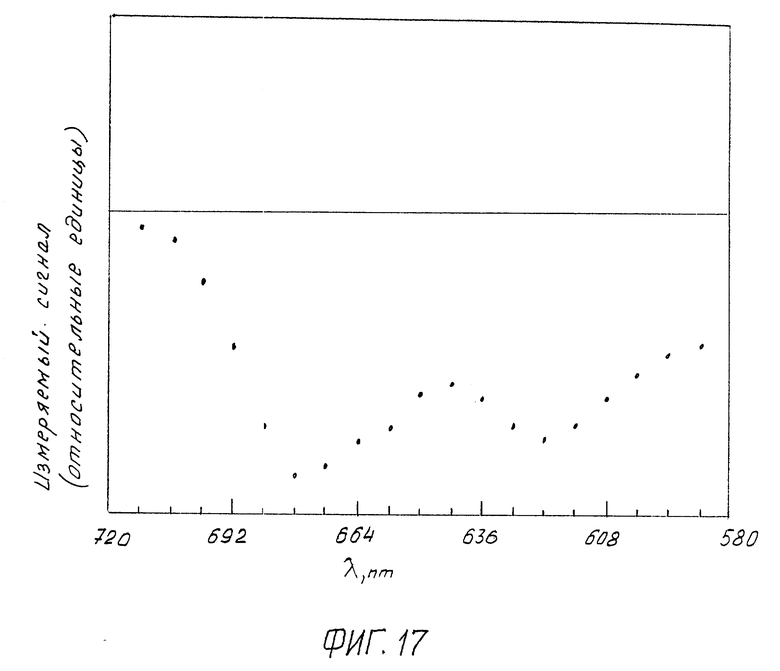
Фиг. 17 изображает вид спектра КД жидкокристаллической дисперсии комплекса (ДНК-МХ), зарегистрированного предлагаемым устройством:
CДНК = 5 мкг/мл; CПЭГ = 170 мг/мл;
0,3 М NaCl + 10-2 М фосфатный буфер; pH ≈ 7,0;
шаг развертки спектра по длинам волн 10 нм/дел.
Фиг. 17 изображает вид спектра КД жидкокристаллической дисперсии комплекса (ДНК-МХ), зарегистрированного предлагаемым устройством:
CДНК = 5 мкг/мл; CПЭГ = 170 мг/мл;
0,3 М NaCl + 10-2 М фосфатный буфер; pH ≈ 7,0; CtМХ = 8,623•10-7 М;
шаг развертки спектра по длинам волн 7 нм/дел.
Фиг. 18 изображает зависимость величины оптического сигнала, генерируемого жидкокристаллической дисперсией ДНК (λ 680 нм) от общей концентрации МХ. Разные черные точки обозначают данные, полученные предлагаемым устройством в разных сериях, отличающихся концентрацией МХ.
Устройство для определения биологически активного вещества в анализируемой жидкости, выполненное согласно изобретению, содержит источник 1 (фиг. 10) светового излучения, выполненный, например, на основе ксеноновой лампы с воздушным охлаждением; селектор 2 длин волн светового излучения, формирующий световой поток в определенном узком спектральном интервале длин волн; поляризатор 3, выполненный, например, в виде призмы из нелинейного кристаллического материала, закрепленный после селектора 2 и формирующий из указанного потока линейно поляризованный световой поток; модулятор 4 поляризации, например, фотоэластического типа, изготовленный из кварца, установленный за поляризатором 3 и преобразующий линейно поляризованный световой поток в циркулярно поляризованный световой поток с периодически изменяющимся направлением вращения вектора поляризации; ячейку 5 для размещения кюветы с исследуемой пробой, содержащей смесь подготовленных в соответствии с предлагаемым описанным выше способом анализируемой жидкости, содержащей биологически активное вещество, и лиотропной жидкокристаллической холестерической дисперсии ДНК; фотодетектор 6, фоточувствительная поверхность которого обращена к указанной ячейке 5 и который преобразует оптический сигнал, генерируемый указанной жидкокристаллической дисперсией, в пропорциональный ему электрический сигнал. Фотодетектор имеет два выхода 7 и 8, один из которых (7) соединен с входом управления источника 9 питания фотодетектора 6, а другой выход 8 подключен к первому входу 10 синхронного усилителя 11, выход 12 которого соединен с входом 13 средства 14 для обработки активного вещества, другой вход 15 указанного средства для обработки соединен с первым выходом 16 блока 17 управления, второй выход 18 блока 17 управления соединен с модулятором 4 поляризации, третий выход 19 блока 17 управления соединен с селектором 2, а четвертый его выход 20 подключен к второму входу 21 синхронного усилителя 11.
В качестве средства 14 для обработки полученного электрического сигнала и вычисления концентрации биологически активного вещества может быть использован, например, персональный компьютер или любое средство, предназначенное для аналогичных целей.
Селектор 2, формирующий световой поток определенной длины волны, может иметь различные конструктивные выполнения, общим преимуществом которых является минимальное число оптических элементов, необходимое для получения на выходе максимального светового потока при сохранении требуемых разрешения и точности измерения, и наличие в селекторе 2 электродинамического (гальванометрического) привода 22 позиционного типа, обеспечивающего измерение сигнала КД на двух выбранных для определяемого БАВ длинах волн.
Например, на фиг. 11 изображен селектор 2, представляющий собой одинарный перестраиваемый по длинам волн монохроматор 23, содержащий закрепленные на общем основании 24 входную щель 25, два зеркала 26 и 27 и диспергирующий элемент 28. Одно из зеркал 26 представляет собой коллимирующее зеркало, вогнутая поверхность 29 которого обращена в сторону входной щели 25 и рабочей поверхности 30 диспергирующего элемента 28, который представляет собой дифракционную решетку. Второе зеркало 27 представляет собой фокусирующее зеркало, вогнутая поверхность 31 которого обращена одновременно в сторону рабочей поверхности 30 дифракционной решетки и расположенной напротив него выходной щели 32, выполненной аналогично входной щели 25.
Электродинамический (гальванометрический) привод 22 позиционного типа имеет двигатель 33 (фиг. 12), содержащий статор (на чертеже не показан) и ротор 34, закрепленный на валу 35 двигателя 33. Кроме того, привод 22 имеет датчик 36 угла поворота ротора 34, расположенный на одной оси с двигателем 33 и представляющий собой индуктивный дифференциальный преобразователь угла поворота ротора 34 в электрический сигнал. Датчик 36 имеет модулятор 37, закрепленный на валу 35 двигателя 33 с эксцентриситетом "e" относительно оси 38 вращения ротора 34.
Ротор 34 двигателя 33 выполнен в виде кольца, состоящего из набора постоянных магнитов 39 и промежуточных магнитопроводов 40. Обмотки двигателя 33 образованы несколькими катушками 41 с проводом 42, расположенными неподвижно на статоре вдоль кольца ротора 34. Кольцо ротора 34 жестко связано с валом 35 двигателя 33, расположенным перпендикулярно плоскости фиг. 12.
Датчик 36 угла поворота содержит две катушки 43, размещенные на магнитопроводах 44 с полюсными наконечниками 45, расположенными диаметрально противоположно относительно модулятора 37, причем геометрический центр A модулятора 37 смещен относительно общей оси вращения ротора 34 и датчика 36 угла поворота на эксцентриситет "e". Указанный эксцентриситет "e" позволяет реализовать амплитудную зависимость выходного сигнала датчика 36 угла поворота от угла поворота ротора 34 двигателя 33.
По меньше мере один оптический элемент монохроматора 23 (зеркало 26, 27 или диспергирующий элемент 28) должен быть закреплен на валу 35 двигателя 33 электродинамического привода 22.
На фиг. 11 представлен вариант, когда на валу 35 двигателя 33 закреплен диспергирующий элемент 28, а именно дифракционная решетка. При этом в качестве диспергирующего элемента 28 может быть использован и любой другой элемент, предназначенный для аналогичных целей, например оптическая призма. Пунктиром на фиг. 11 изображен вариант, когда на валу 35 двигателя 33 закреплено одно из зеркал, а именно фокусирующее зеркало 27, при этом диспергирующий элемент 28 и второе зеркало, а именно коллимирующее зеркало 26, должны быть закреплены неподвижно.
В другом варианте выполнения настоящего изобретения селектор 46 (фиг. 13) содержит набор узкополосных интерференционных фильтров 47, каждый из которых имеет полосу пропускания в области выбранной для определяемого БАВ длины волны. Фильтры 47 закреплены на валу 35 электродинамического привода 22 посредством кассеты 48 с возможностью их поочередного введения в световой поток, направление которого изображено на фиг. 13 стрелкой B. Количество фильтров 47 зависит от числа разновидностей биологически активного вещества, которые требуется определить посредством предлагаемого устройства.
Устройство работает следующим образом.
В соответствии с описанным выше способом готовят серию проб с различными концентрациями известного вещества и проводят измерение сигналов кругового дихроизма последовательно на двух длинах волн для каждой пробы, одно измерение проводят на длине волны 270 нм, а другое - на длине волны, характерной для данного анализируемого вещества. Используя полученные значения, нормируют сигналы кругового дихроизма для характерных длин волн на сигналы, соответствующие длине волны 270 нм для каждой пары значений. Полученные нормировочные значения и соответствующие им концентрации анализируемого вещества заносят в память компьютера и строят график зависимости нормированного сигнала от концентрации вещества, то есть калибровочную кривую, которая закладывается в компьютер.
Далее, в соответствии с описанным выше способом, готовят исследуемую пробу, содержащую смесь подготовленной анализируемой жидкости, содержащей биологически активное вещество, и лиотропной жидкокристаллической дисперсии ДНК, сформированной в нейтральном по отношению к ДНК полимере. Размещают эту пробу в ячейке 5 предлагаемого устройства и включают его.
Источник 1 светового излучения излучает широкополосный световой поток, падающий на входную щель 25 селектора 2 длин волн, через выходную щель 32 которого излучается узкополосный световой поток, имеющий одну известную длину волны. Этот поток проходит через поляризатор 3, становится линейно поляризованным с заданным направлением вектора поляризации и попадает на оптический вход модулятора 4 поляризации, пройдя который он становится циркулярно поляризованным с периодически изменяющимся направлением вращения вектора поляризации, вращающегося в плоскости, перпендикулярной оптической оси устройства. Пройдя ячейку 5 с исследуемой пробой, обладающей свойством аномальной оптической активности или, другими словами, круговым дихроизмом, световой поток становится модулированным по интенсивности. Под действием света на выходах 7 и 8 фотодетектора 6 возникает электрический сигнал, причем на выходе 8 регистрируется переменная составляющая, пропорциональная ΔA (величине сигнала, порожденного аномальной оптической активностью), а на выходе 7 - постоянная составляющая, пропорциональная A (величине сигнала, характеризующей поглощение биологически активного вещества пробы), при этом частота переменной составляющей равна частоте модуляции поляризации излучения. В данном устройстве постоянная составляющая поддерживается на постоянном уровне путем регулирования напряжения питания фотодетектора 6, для чего сигнал постоянной составляющей с выхода 7 фотодетектора 6 заводится на вход управления источника 9 питания, то есть осуществляется режим стабилизации постоянной составляющей с помощью отрицательной обратной связи по постоянной составляющей с одновременным измерением переменной составляющей, что эквивалентно измерению их отношения, а значит, измерению сигнала кругового дихроизма исследуемой пробы. С выхода 8 фотодетектора 6 сигнал поступает на первый вход 10 синхронного усилителя 11, на второй вход 21 которого подается опорный сигнал с частотой модуляции поляризации. В синхронном усилителе 11 сигнал усиливается, преобразуется в постоянный ток и подается в средство 14 для обработки, где преобразуется в цифровую форму, обрабатывается, сравнивается с калибровочной кривой и выводится в виде значения концентрации исследуемого биологически активного вещества в пробе. Блок 17 управления осуществляет необходимое взаимодействие всех элементов устройства, реализует требуемый алгоритм обработки, вырабатывает напряжение с частотой модуляции для работы модулятора 4 поляризации, формирует опорный сигнал для функционирования синхронного усилителя 11. Способ обработки цифровой формы полученного сигнала зависит от используемого для этой цели приспособления.
Электродинамический (гальванометрический) привод 22 позиционного типа селектора 2 работает следующим образом.
Датчик 36 угла поворота ротора 34 вырабатывает электрический сигнал, амплитуда которого зависит от угла поворота вала 35 двигателя 33 благодаря наличию эксцентриситета "e" установки модулятора 37 датчика 36 относительно оси 38 вращения ротора 34. При повороте вала 35 двигателя 33 зазор между полюсными наконечниками 45 и модулятором 37 будет меняться и, следовательно, будет меняться амплитуда сигнала датчика 36 угла поворота ротора 34. Этот сигнал поступает в блок 17 управления, в котором его значение сравнивается со значением, соответствующим определенной длине волны селектора 2, и определяется их разность. В зависимости от этой разности блок 17 управления вырабатывает сигнал управления, поступающий на катушки 41 статора двигателя 33. Магнитное поле, вызванное током сигнала управления, протекающим по катушкам 41 статора двигателя 33, взаимодействует с магнитным полем постоянных магнитов 39 ротора 34 двигателя 33 таким образом, что ротор 34, а значит и вал 35, поворачиваются на угол, соответствующий заданной длине волны селектора 2, в зависимости от амплитуды сигнала управления.
В случае использования в качестве селектора 2 монохроматора 23 принцип действия последнего не отличается от принципа действия широко известных дифракционных монохроматоров, построенных по схеме Черни-Турнера [M.Czerny, A. F. Turner and M.V.R.K.Murty. Principles of Monochromators, Spectrometers and Spectrographs. Optical Engineering, vol. 13, N 1, 1974, pp. 23 - 38], отличие заключается в том, что в качестве приспособления для поворота одного из оптических элементов монохроматора 23 (зеркала 26 и 27 или диспергирующего элемента 28) для установки выбранных длин волн применяется электродинамический (гальванометрический) привод 22 позиционного типа, управляемый сигналом, поступающим из блока 17 управления предлагаемого устройства.
В случае использования в качестве селектора 2 набора интерференционных фильтров 47 работа осуществляется следующим образом. Световой поток источника 1 светового излучения проходит через один из вышеуказанных фильтров 47, установленный в данный момент пути потока, и преобразуется на выходе в поток света с заданной длиной волны, определяемой полосой пропускания данного фильтра 47. При необходимости изменения длины волны на двигатель 33 электродинамического привода 22 позиционного типа поступает сигнал управления из блока 17 управления, вызывающий поворот вала 35 ротора 34 двигателя 33 на другой угол, в результате чего в поток света вводится другой фильтр 47, изготовленный на другую длину волны, и выходной световой поток приобретает длину волны, определяемую полосой пропускания этого фильтра 47.
Ниже приведены примеры осуществления предлагаемого способа на предлагаемом устройстве.
Пример 6. Аналитические возможности устройства.
Для проверки правильности измерения КД и получения достоверной информации об оптических свойствах анализируемых жидкостей с помощью предлагаемого устройства проводили его калибровку с использованием водного раствора н-пропиламмониевой соли d-10-камфорсульфоновой кислоты и водно-солевого раствора линейной В-формы двухцепочечной ДНК.
6А. Калибровка устройства с использованием водного раствора н-пропиламмониевой соли d-10-камфоросульфоновой кислоты и водно-солевого раствора линейной В-формы двухцепочечной ДНК.
6А. 1 Водный раствор н-пропиламмониевой соли d-10-камфорсульфоновой кислоты (C10H16O4S) обычно применяется для калибровки стандартных фирменных дихрографов и спектрополяриметров. Спектр КД водного раствора этой кислоты (при определении концентрации и температуре) характеризуется наличием положительной полосы, расположенной в УФ-области (230 - 320 нм). Форма полосы, точное положение ее максимума, амплитуда этой полосы детально описаны [Gillon, M.F. Williams, R.E. (1975). Can. J. Chem., 53, pp. 2351 - 2353].
Для калибровки использовался водный раствор C10H16O4S с концентрацией 0,15 мг/мл. Для этого в прямоугольную оптическую кварцевую кювету устройства (длина оптического пути 1 см) помещали 2 мл водного раствора указанной концентрации и при помощи устройства регистрировали спектр КД раствора в области длин волн 250 - 350 нм.
На фиг. 14 приведен спектр КД раствора, наблюдаемый на экране монитора компьютера, связанного с предлагаемым устройством.
В спектре КД раствора в области длин волн 250 - 320 нм присутствует положительная полоса, амплитуда и положение максимума (λ 290 нм) которой полностью согласуется с литературными данными (см. выше).
6А. 2 Хорошо известный в литературе консервативный спектр КД линейной В-формы двухцепочечной ДНК характеризуется наличием двух полос разного знака, приблизительно равных по своей амплитуде. Положительная полоса имеет максимум при λ ~ 278 нм, отрицательная - при λ ~ 247 нм . Еще одной реперной длиной волны, характерной для спектра КД линейной В-формы двухцепочечной ДНК, является длина волны, при которой молекулярная оптическая активность водно-солевого раствора ДНК, меняя свой знак, обращается в нуль. Согласно литературным данным [сб. "Итоги науки и техники". /Под ред. Волькенштейна М. В. М. , Изд-во ВИНИТИ, (1975), т. 1, стр. 115] этот оптический эффект наблюдается при λ ~ 258 нм .
Для калибровки использовался водно-солевой раствор линейной В-формы двухцепочечной ДНК (CДНК - 5 мкг/мл; 0,3 M NaCl + 10-2 M фосфатный буфер), приготовленный согласно п. 1.1, 1.2 и 1.4.
В прямоугольную кварцевую кювету (длина оптического пути 1 см) помещали 2 мл водно-солевого раствора ДНК и при помощи устройства регистрировали его спектр КД в области длин волн 240 - 310 нм.
На фиг. 15 приведен спектр КД раствора, наблюдаемый на экране монитора компьютера, связанного с портативным дихрометром.
Регистрируемый с помощью устройства спектр КД полностью повторяет вышеописанные особенности консервативного спектра КД линейной В-формы двухцепочечной ДНК.
Таким образом, полученные в п. 6А результаты свидетельствуют о достоверности регистрации оптических свойств анализируемых растворов при помощи предлагаемого устройства.
6Б. Определение оптических свойств жидкокристаллической дисперсии ДНК и жидкокристаллической дисперсии комплексов (ДНК-МХ) при помощи устройства.
6Б.1 В прямоугольную кварцевую кювету (длина оптического пути 1 см) помещают 2 мл жидкокристаллической дисперсии ДНК (CДНК - 5 мкг/мл; CПЭГ = 170 мг/мл; 0,3 M NaCl + 10-2M фосфатный буфер), приготовленной согласно примеру 1, и при помощи устройства регистрируют ее спектр КД в области длин волн 240 - 310 нм.
На фиг. 16 приведен наблюдаемый спектр КД жидкокристаллической дисперсии ДНК, который характеризуется наличием интенсивной отрицательной полосы, форма, знак, амплитуда и положение максимума (λ ~ 270 нм) которой полностью соответствуют данным, приведенным на фиг. 2 (пример 1), полученным при помощи дихрографа фирмы "Jobin-Yvon".
6Б.2 В оптическую кювету, содержащую 2 мл жидкокристаллической дисперсии ДНК (п. 6Б. 1), добавляют 4 мкл раствора, приготовленного по п. 2.1; полученную в оптической кювете смесь перемешивают в течение 30 с и при помощи устройства регистрируют спектр КД смеси в области длин волн 580 - 720 нм.
На фиг. 17 приведен зарегистрированный в области длин волн от 580 до 720 нм спектр КД такой жидкокристаллической дисперсии, обработанной МХ. Добавление МХ сопровождается появлением в спектре КД интенсивной отрицательной полосы в области поглощения МХ, характеризующейся наличием двух максимумов при λ ~ 680 нм и λ ~ 620 нм, что соответствует литературным данным. Появление этой полосы свидетельствует о формировании жидкокристаллической дисперсии из комплекса (ДНК-МХ).
Таким образом, полученные результаты убедительно свидетельствуют о том, что устройство можно использовать для определения МХ в анализируемых пробах.
6В. Проверка аналитических возможностей устройства по жидкокристаллическим дисперсиям комплексов (ДНК-МХ).
6В. 1 В прямоугольную кювету (длина оптического пути 1 см) помещают 2 мл жидкокристаллической дисперсии ДНК (CДНК - 5 мкг/мл; CПЭГ = 170 мг/мл; 0,3 M NaCl + 10-2M фосфатный буфер) и при помощи устройства регистрируют оптическую активность при λ 270 нм .
6В.2 В кювету, содержащую 2 мл жидкокристаллической дисперсии ДН (п. 6В. 1), добавляют по 1 мкл водно-солевого раствора МХ (CМХ = 0,2 мг/мл (3,868 • 10-4M; 0,3 M NaCl + 10-2M фосфатный буфер) - всего 4 добавки; суммарный объем добавленного раствора составляет 4 мкл. После каждой добавки МХ смесь перемешивают (30 с) и при помощи устройства регистрируют аномальные оптические свойства системы при λ 270 нм .
6В. 3 Полученные в результате проведенного измерения при λ 270 нм данные (п. 6В.2) сравнивают с результатами измерения, проведенного в п. 6В.1. Если результаты этих измерений отличаются не более, чем на 5%, приступают к регистрации оптического сигнала при λ 680 нм , генерируемого жидкокристаллической дисперсией ДНК в результате образования комплекса с добавленными в раствор (п. 6В.2) молекулами МХ.
6В.4 В полном соответствии с пп. 6В.1 - 6В.3 проводится еще одно титрование жидкокристаллической дисперсии ДНК (п. 6В.1) водно-солевым раствором МХ с меньшей концентрацией CМХ = 0,02 мг/мл (3,868 • 10-5 M; 0,3 М NaCl + 10-2M фосфатный буфер).
Данные, полученные в ходе проверки аналитических возможностей устройства с использованием жидкокристаллических дисперсий комплексов (ДНК - МХ), показали (фиг. 18), что также, как и в случае данных, полученных при помощи дихрографа фирмы "Jobin Yvon" (фиг. 5, пример 2), между величиной оптического сигнала, генерируемого жидкокристаллической дисперсией ДНК при λ 680 нм при образовании комплекса (ДНК - МХ), и величиной CtМХ наблюдается прямо пропорциональная зависимость. Эта зависимость показывает, что при помощи устройства можно определять наличие и концентрацию МХ в анализируемых пробах.
Проведенное тестирование показало, что большим преимуществом устройства является тот факт. Что оно позволяет быстро и точно определять МХ в широкой области концентраций, в том числе в области концентраций от 5 • 10-8M до 5 • 10-7M, на порядок более низких, чем предельные концентрации, измеряемые известными приборами фирм "Jobin-Yvon" и "Jasco".
Таким образом, предлагаемые способ и устройство позволяют быстро, точно и с высокой чувствительностью определять наличие и концентрацию БАВ (МХ) в крови больных, терапия которых связана с применением противоопухолевых соединений.
Предлагаемый способ исключает использование сложного и дорогостоящего оборудования, а также наличие высококвалифицированного персонала, и вместе с предлагаемым устройством может быть использован для определения других биологически активных и фармакологических соединений, образующих интеркаляционные комплексы с парами оснований ДНК.
Промышленная применимость.
Предлагаемое изобретение может быть использовано в медицинской и клинической биохимии, а также в молекулярной фармакологии при исследовании фармако-кинетики биологически активных соединений, в фармацевтической промышленности и экологии. Наиболее эффективно его использование в клинической биохимии.
| название | год | авторы | номер документа |
|---|---|---|---|
| МОЛЕКУЛЯРНАЯ КОНСТРУКЦИЯ НА ОСНОВЕ ЖИДКОКРИСТАЛЛИЧЕСКОЙ ДИСПЕРСИИ НУКЛЕИНОВОЙ КИСЛОТЫ КАК ИНТЕГРАЛЬНЫЙ БИОДАТЧИК И СПОСОБ ЕЕ СОЗДАНИЯ | 1998 |
|
RU2139933C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ГЕПАРИНА | 1997 |
|
RU2123008C1 |
| БИОДАТЧИК ДЛЯ ОПРЕДЕЛЕНИЯ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ, ВЗАИМОДЕЙСТВУЮЩИХ С ДВУХЦЕПОЧЕЧНЫМИ МОЛЕКУЛАМИ НУКЛЕИНОВЫХ КИСЛОТ | 1991 |
|
RU2016888C1 |
| ЖИДКОКРИСТАЛЛИЧЕСКАЯ ДИСПЕРСИЯ НА ОСНОВЕ КОМПЛЕКСА (НУКЛЕИНОВАЯ КИСЛОТА - ХИТОЗАН) КАК ИНТЕГРАЛЬНЫЙ БИОДАТЧИК И СПОСОБ ЕЕ СОЗДАНИЯ | 2000 |
|
RU2169770C1 |
| ЖИДКОКРИСТАЛЛИЧЕСКИЙ БИОДАТЧИК ДЛЯ ОПРЕДЕЛЕНИЯ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ | 1989 |
|
RU2032895C1 |
| МНОГОФУНКЦИОНАЛЬНАЯ АНАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ ОПРЕДЕЛЕНИЯ ХАРАКТЕРИСТИК ОПТИЧЕСКОГО СИГНАЛА КРУГОВОГО ДИХРОИЗМА БИОЛОГИЧЕСКИ АКТИВНОГО МАТЕРИАЛА | 2013 |
|
RU2569752C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ФИЗИОЛОГИЧЕСКИХ КОНЦЕНТРАЦИЙ ГЕПАРИНА В АНАЛИЗИРУЕМЫХ ЖИДКИХ ПРОБАХ | 2010 |
|
RU2440575C1 |
| ОПТИЧЕСКИЙ ДИФФУЗОМЕТР ДЛЯ АНАЛИЗА ТРАНСПОРТА БИОЛОГИЧЕСКИ АКТИВНОГО ВЕЩЕСТВА, АНАЛИТИЧЕСКАЯ СИСТЕМА ДЛЯ ОПРЕДЕЛЕНИЯ БИОЛОГИЧЕСКИ АКТИВНОГО ВЕЩЕСТВА В ЖИДКОСТИ И СПОСОБ ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИИ БИОЛОГИЧЕСКИ АКТИВНОГО ВЕЩЕСТВА В ЖИДКОСТИ | 2010 |
|
RU2429465C1 |
| ОТРАЖАТЕЛЬНАЯ ДИФРАКЦИОННАЯ РЕШЕТКА | 1996 |
|
RU2105274C1 |
| СПЕКТРОФОТОМЕТР | 1995 |
|
RU2109255C1 |












Изобретение предназначено для определения биологически активных веществ (БАВ), способных взаимодействовать с линейными двухцепочечными молекулами ДНК, регистрацией кругового дихроизма. Способ включает смешение анализируемой жидкости, содержащей БАВ, с нейтральным по отношению к ДНК полимером в условиях, при которых оптические свойства лиотропной жидкокристаллической дисперсии ДНК не нарушаются. Затем непосредственно перед смешением с анализируемой жидкостью формируют лиотропную жидкокристаллическую холестеричную дисперсию ДНК из линейных двухцепочечных молекул ДНК низкой молекулярной массы в водно-солевом растворе упомянутого полимера. Смешивают раствор анализируемой пробы в полимере с раствором ДНК в полимере. Пропускают через полученный раствор циркулярно поляризованное излучение. Регистрируют оптический сигнал на двух длинах волн, одна из которых находится в полосе поглощения ДНК, другая - в области поглощения БАВ. О концентрации БАВ судят по величине отношения полученных сигналов. Устройство для осуществления способа представляет собой дихрограф, в котором селектор, формирующий излучение определенной длины волны, содержит минимальное число оптических элементов для обеспечения максимального светового потока. Селектор содержит электродинамический привод позиционного типа, обеспечивающий возможностью установлениях двух длин волн, на которых проводят измерения. 2 с. и 11 з.п.ф-лы, 18 фиг.
| RU, патент, 201688, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| ПРИБОР ДЛЯ ИЗМЕРЕНИЯ ГЛУБИНЫ ВЫЕМКИ | 1922 |
|
SU710A1 |

