Изобретение касается производных имидазола.
Гормон ангиотензин II считается одним из наиболее сильных вазопрессорных веществ, вызывающих гипертензию у млекопитающих. Действие фермента на белковый субстрат плазмы ангиотензиноген приводит к образованию неактивного декапептида, ангиотензина I, который под действием неизбирательного избирательного ангиотензин-конвертирующего фермента (АСЕ) дает ангиотензин II, активный гормон (Regolli et al., Pharm. Rev., 26, 69, 1974).
Ангиотензин II вызывает вазоконстрикцию и стимулирует секрецию альдостерона (надпочечниками), что приводит к увеличению объема циркулирующей крови и повышению артериального давления. Следовательно, ингибиторы ангиотензина II полезны для лечения гипертензии, застойной сердечной недостаточности, почечной недостаточности, связанной с диабетической или гипертонической нефропатией, и глаукомы (Garrison et al., in the Pharmacological Basis of Therapeufics, 8th Edinion, Eds. A.G.dilman, E.S.Goodman, T.W.Rall, A. S.Nies, P.Taylor, Pergamon Press, New York, 1990: p. 761-762; and Dzau V.J., The New York Eng J. Med. 324: 1124 - 1130, 1991).
Ангиотензин II также может действовать на другие органы, такие как головной мозг (Fitzsimmons, Rev. Physiol. Biochem. Pharmocol. 87, 117, 1980). Антагонисты ангиотензина II, следовательно, полезны для улучшения умственной работы у больных, страдающих такими заболеваниями, как возрастное слабоумие или болезнь Альцгеймера, и для лечения таких нарушений психики, как тревожные состояния (Dennis et al., Brit. J. Pharmacol., 105: 88 p., april 1992; Barnes J.M., et al., FASEB J., 5:678, march, 1991).
К тому же ангиотензин II действует на различные железы, включая почки, печень и яичники. Антагонисты ангиотензина II применяются для лечения состояний, нарушений или заболеваний этих тканей, связанных с избытком или неконтролируемой активностью ангиотензина II. Антагонисты ангиотензина II применяются также при лечении повреждения почек нестероидными противовоспалительными средствами.
Ангиотензин II играет роль в регуляции скорости роста и дифференцировки клеток. Ингибиторы ангиотензина II, следовательно, полезны для лечения нарушений, отмеченных чрезмерной полиферацией клеток, таких как рестеноз (Naftilan et al. , J. Clin. Invest., 83:1419, 1989; Kauffman et al., Lafe sciences, 49:223-228, 1191; Jackson et al., Nature, 335, 437, 1988).
Некоторые антигипертензивные свойства действуют как ингибиторы АСЕ, блокируя образование ангиотензина II и препятствуя связанному с ним повышению артериального давления. Недавно были обнаружены рецепторы пептидных и непептидных антагонистов ангиотензина II (см., например, публикацию патентной заявки EPO 253310 и включенные в нее ссылки и Clin et al., J. Pharmacol. Exp. Ther, 250, 867, 1989). Хотя пептидные ангатонисты играют важную роль в раскрытии физиологического действия ангиотензина II, их терапевтическое применение было ограничено либо частичным активированием агонистов, либо метаболической нестабильностью, либо ими вместе (Ashurth R.W. Birkhauser Verlag 26, 1982).
Изобретение представляет новые сильные и эффективные соединения, которые являются антагонистами ангиотензина II на уровне рецепторов в организме и поэтому полезны для лечения состояний, связанных с избыточной или нерегулируемой активностью ангиотензина II, таких как гипертензия, застойная сердечная недостаточность, нарушения умственной деятельности, почечная недостаточность, связанная с диабетической или гипертонической нефропатией, глаукома, повреждение почек нестероидными противовоспалительными препаратами и рестеноз.
Это изобретение представляет соединения формулы 1: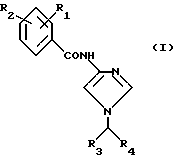
где
R1 - CO2H, SO3H или 5-тетразолил;
R2 - H или -OH;
R3 - неразветвленная цепь алкила C4-C9, неразветвленная цепь трифторалкила C4-C9;
R4 - -CONH(C1-C4)алкил, -CONH(C1-C4)трифторалкил, -CONH-гидрокси-(C1-C4)-алкил; CONH-пиридил-2, N-оксо-пролин,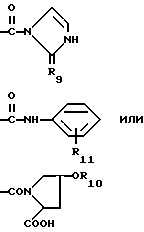
где
R9 - кислород или сера;
R10 - представляет водород, C1-C4-алкил, 3-пиридил, изоксазолил, хинолил, бензофуранил, возможно замещенный C1-C4-алкилом или карбоксигруппой, дибензофуранил, бензотиофенил, нафтил, возможно замещенный C1-C4-алкоксигруппой, тетрагидронафтил, инданил, бензодиоксоланил, ди-C1-C4-алкоксифенил, фенил, возможно замещенный в положении 4 C1-C4-алкилом, или C3-C6-циклоалкилом, или галогеном, или трифтор-(C1-C4)алкилом, или фенилом, или гидроксилом, или C1-C4-алкоксигруппой, гидрокси-(C1-C4)алкилом, или карбокси-(C1-C4)-алкилом, или тио(C1-C4)-алкилом, или S(O)n(C1-C4)-алкилом, где n - 1, 2, или карбоксигруппой, или аминокарбонилом, или нитрилом, или N-содержащим пятичленным гетероциклом, или группой SO2NCH3,
R11 - представляет гидрокси или карбоксигруппу.
Это изобретение также представляет способ лечения гипертензии, который заключается во введении млекопитающему, которое нуждается в таком лечении, антигипертензивного количества соединения формулы 1.
Это изобретение далее предоставляет способы лечения застойной сердечной недостаточности, почечной недостаточности, связанной с гипертонической или диабетической нефропатией, рестеноза, почки, поврежденной нестероидными противовоспалительными препаратами, тревожных состояний и глаукомы, которые заключаются во введение млекопитающему, нуждающемуся в лечении, фармацевтически эффективного количества соединения формулы 1.
Дальнейшим аспектом изобретения является способ повышения умственной работоспособности, который заключается во введении млекопитающему, нуждающемуся в этом, фармацевтически эффективного количества соединения формулы 1.
Также предоставляются фармацевтические прописи, включающие соединение формулы 1 вместе с одним или несколькими фармацевтически приемлемыми наполнителями, носителями или разбавителями.
Дополнительным аспектом этого изобретения является процесс получения предпочтительного стереизомера формулы 1, включающий
сочетание соединения формулы (XXI)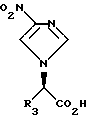
где
R3 - алкил с неразветвленной цепью C4-C9, трифторалкил с неразветвленной цепью C4-C9;
с соединением формулы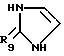
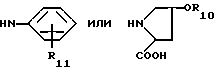
где значения R9, R10 и R11 такие же, как указано ранее;
восстановление нитро-группы соединения формулы (XXI) для того, чтобы получить аминоимидазол;
сочетание аминоимидазола с соединением формулы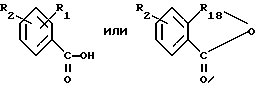
где значения R2 и R1 такие же, как указано ранее;
R18 - SO2 или C=O.
Как отмечено выше, это изобретение представляет соединения формулы I, которые являются антагонистами ангиотензина II на уровне рецепторов в организме. Предпочтительными соединениями этого изобретения являются те, у которых в формуле I R4 представлен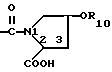
Особенно предпочтительными соединениями этого изобретения являются соединения формулы Ia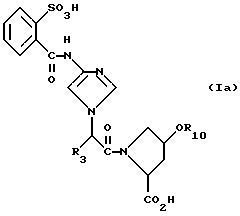
где R3 - алкил с неразветвленной цепью C4-C9;
значения R10, как определено выше.
Примеры предпочтительных соединений включают следующие:
1-[1-оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил] - 4-цис-(4-карбоксифенокси)-L-пролин;
1-[1-оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил] - 4-цис-(4-карбоксиметилфенокси)-L-пролин;
1-[1-оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил] - 4-цис-(4-трет-бутилоксифенокси)-L-пролин;
1-[1-оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил] - 4-цис-(4-метилсульфонилокси)-L-пролин;
1-[1-оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил] - 4-цис-(5-бензофуранокси)-L-пролин;
1-[1-оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил] - 4-цис-(2-нафтокси)-L-пролин;
1-[1-оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил] - 4-цис-(4-карбоксиметоксифенокси)-L-пролин;
1-[1-оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил] - 4-цис-(2-карбоксибензофуран-5-ил-окси)-L-пролин;
1-[1-оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил] - 4-цис-[(4-метиленфосфоновая кислота)-фенокси]-L-пролин.
В силу наличия кислотных группировок соединения формулы 1 включают основные сложные соли. Такие соли включают соли, образованные с неорганическими основаниями, такими как аммоний и гидроксиды щелочных и щелочно-земельных металлов, карбонаты, бикарбонаты и т.п., а также соли, образованные с основными органическими аминами, такими как алифатические и ароматические амины, алифатические диамины, гидроксиалкамины и т. п. Такие основания, пригодные для приготовления солей этого изобретения, таким образом, включают гидроксид аммония, карбонат калия, бикарбонат натрия, гидроксид кальция, метиламин, диэтиламин, этилендиамин, циклогексамин, этаноламин и т.п. Соли калия и натрия являются особенно предпочтительными.
Из-за наличия гетероциклической части соединения формулы 1 могут также существовать в виде фармацевтически приемлемых кислых сложных солей. Кислоты, обычно применяемые для образования таких солей, включают неорганические кислоты, такие как соляная, бромистоводородная, йодистоводородная, серная и фосфорная кислота, а также органические кислоты, такие как пара-толуолсульфоновая, метансульфоновая, щавелевая, пара-бромфенилсульфоновая, угольная, янтарная, лимонная, бензойная и уксусная кислота и родственные неорганические и органические кислоты. Такие фармацевтические приемлемые соли, таким образом, включают сульфат, пиросульфат, бисульфат, сульфит, бисульфит, фосфат, моногидрогенфосфат, дигидрогенфосфат, метафосфат, пирофосфат, хлорид, бромид, йодид, ацетат, пропионат, деканоат, каприлат, акрилат, формиат, изобутират, капроат, гептаноат, припиолат, оксалат, малонат, сукцинат, суберат, себакат, фумарат, малеат, 2-бутен-1,4-диоат, 3-гексин-2,5-диоат, бензоат, хлорбензоат, гидроксибензоат, метоксибензоат, фталат, ксилолсульфонат, фенилацетат, фенилпропионат, фенилбутират, цитрат, лактат, гиппурат, β - гидроксибутират, гликолат, малеат, тартрат, метансульфонат, пропансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат, манделат и тому подобные соли.
Фармацевтически приемлемые соли соединений формулы 1 могут также существовать в виде различных сольватов, таких как образующиеся с водой, метанолом, этанолом, диметилформамидом, этилацетатом и т.п. Можно также готовить смеси таких сольватов. Источником такого сольвата может быть растворитель кристаллизации, растворитель, использующийся для приготовления или кристаллизации или сопутствующий такому растворителю.
Выяснено, что существуют разные стереизомерические формы соединений формулы 1, например хиральный атом углерода, который присоединяется к имидазолу, R4 и R5. Это изобретение не ограничивается каким-либо отдельным стереоизомером, но включают все возможные отдельные изомеры и их смеси.
Синтез и применение 1,3-имидазолов в качестве антагонистов ангиотензина 11 описаны в патенте США N 5073566.
Тетразолильные части R1 в формуле 1 (предпочтительно, чтобы R1 был защищен в виде нитрила в ходе реакций сочетания) можно получить обработкой циано-промежуточных соединений азидом щелочного металла, таким как азид натрия, хлоридом аммония или гидрохлоридом триэтиламина и (факультативно) хлоридом лития в инертном растворителе с высокой температурой кипения, таком как N,N-диметилформамид (ДМФ), предпочтительно при температуре порядка 60 - 125oC.
Предпочтительно, вместо азида щелочного металла, хлорида аммония, хлорида лития и ДМФ пользоваться азидом три-(н-бутил) олова или азидом тетраметилгуанадиния, в чистом виде или в растворителе, таком как тетрагидрофуран, диметоксиэтан, диэтоксиэтан и т.п.
Карбоновые кислоты формулы 1 можно получить гидролизом цианопромежуточных соединений (R1 защищен в виде нитрила во время реакций сочетания). Гидролиз происходит при нагревании циано-производного в водном алкоголе в присутствии основания, такого как гидроксид натрия или калия. Соли карбоновой кислоты и конечный тетразоловый продукт производятся путем реакции свободной кислоты или тетразола с подходящим основанием по стандартным методикам.
Заданные продукты открытых реакций можно выделить общепринятыми средствами, предпочтительно хроматографией. Колоночная хроматография является предпочтительным способом. Колоночная хроматография высокого давления с силикагелем и обращенно-фазовая хроматография высокого давления дают самый эффективный способ очистки конечных продуктов. Альтернативно можно использовать кристаллизацию кислоты, тетразола или солей для очистки заданного конечного продукта.
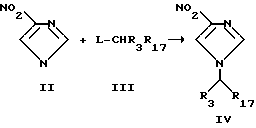
Одним из способов получения соединений формулы 1 является алкилирование имидазола алкилирующим реагентом III, представленное на схеме 1.
 .
.
Значение R3 такое же, как установлено ранее. R17 является защищенной карбокси группой, такой как эфир (Grune T.W., Protective Groups in Organic Synthesis, Yohn Wiley and Sons, New York., 1981).
Чтобы получить амиды формулы 1, R17 должен быть защищенной карбокси.
L является легко удаляемой группой, такой как хлор-, бром-, йод-, мезил, тозил и т.п. L также может быть гидрокси или предшественником, который легко превращается в покидающую группу приемами, известными специалисту.
В этой реакции обычно принимают участие эквимолярные количества двух реагентов, хотя действенными являются и другие соотношения, особенно те, в которых алкилирующий реагент присутствует в избытке. Реакцию лучше всего проводить в полярном апротическом растворителе, когда соединение является солью щелочного металла или при других общепринятых условиях алкилирования. Если покидающая группа является бром- или хлор-, для ускорения реакции можно добавить каталитическое количество йодида, такого как йодид калия. Предпочтительные условия реакции включают следующее: бромид лития и диметилформамид, фторид калия на оксиде алюминия в ТГФ, бикарбонат в диметилформамиде, гидрид натрия в диметилформамиде, карбонат калия, йодид калия и либо метилэтилкетон, либо ацетон. Температура реакции предпочтительна от почти комнатной температуры до почти температуры кипения с обратным холодильником. Когда используются повышенные температуры, реакция обычно завершается за 1 - 4 ч.
При получении амидов формулы 1 защищенная карбокси группа, R17 легко превращается в карбоновую кислоту, а затем в галоидангидрид общеизвестными методами (Grune T.W., Protective groups in Organic Synthesis, p. 152). Превращение кислоты в соответствующий хлорангидрид, например, может происходить в результате обработки реагентом, таким как тионил хлорид или оксалил хлорид, факультативно, в присутствии апротонного инертного растворителя. Предпочтительные сочетания включают обработку тионил хлоридом с последующей реакцией амина с карбонатом калия в тетрагидрофуране или реакцию оксалил хлорида с карбоновой кислотой.
Галоидангидрид соединения IV может затем вступить в реакцию с нужным амином, чтобы обработать амиды (R4 является амидом) настоящего изобретения. Эта реакция представлена на схеме 2.
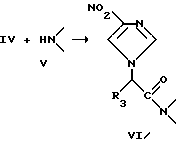
R3 имеет те же значения, что установлены ранее. Когда амин (соединение V) содержит карбонильную группу, предпочтительно чтобы карбонил был защищен во время реакции.
Какой амин использовать в этой схеме зависит от того, какой нужен амид формулы I. Например, для того чтобы получить производное, замещенное пролином, хлорангидрид соединения IV можно ввести в реакцию с метиловым эфиром, замещенным пролином (V). Подобным образом, для того, чтобы получить трифторпропиламид, галоидангидрид вводят в реакцию с трифторпропиламином.
Реакция сочетания между галоидангидридом соединения IV и амином может осуществляться любым из известных способов. Предпочтительным способом по этой схеме является реакция галоидангидрида, предпочтительно хлорангидрида, с амином непосредственно в ТГФ или метиленхлориде, в присутствии триэтиламина.
Образовавшийся амид можно превратить в соединения формулы 1 по известным методикам (Duncia et al., J. Org. Chem. 56 : 2395 - 2400, 1991).
Альтернативно, соединение III можно превратить в хлорангидрид и ввести в реакцию по схеме 2, для того чтобы образовать амид. Это промежуточное соединение можно затем алкилировать в соответствии с условиями, описанными в схеме I, для того чтобы образовать нитроимидазол.
Соединения этого изобретения, содержащие связь карбоксамидного типа, можно получить согласно схеме 3.
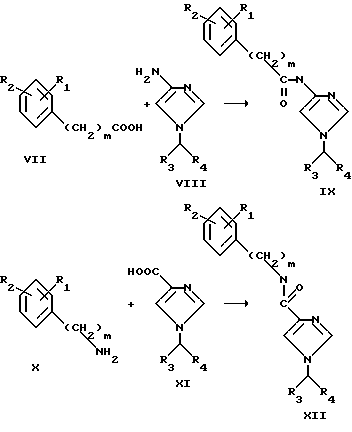
где значения R1, R2, R3, R4 и m такие же, как определено ранее.
Трансформация, изображенная на вышеприведенной схеме 3, может осуществляться любым из известных способов сочетания карбоновых кислот с аминами. Например, карбоновая кислота VII или XI может быть превращена в соответствующий галоидангидрид, особенно в хлорангидрид, и затем вступить в реакцию с соответствующим амином, для того чтобы получить амиды IX или XII, как обсуждалось ранее.
Альтернативно, можно использовать также другие реагенты, конденсирующие амид, такие как 1,1'-карбонилдиимидазол или 1,3-дициклогексилкарбодиимид. Эти реагенты обычно применяются в инертном растворителе с высокой точкой кипения, таком как диметилформамид и, факультативно, в присутствии таких реагентов, как диизопропилэтиламин, гидроксибензотриазол и другие, для того, чтобы облегчить реакцию.
Если R4 содержит карбокси, реакцию лучше всего проводить при защите карбокси группы в виде эфира. После завершения присоединения этот эфир легко превращается в кислоту известными специалисту способами. Например, эфир радикала можно подвергнуть гидролизу водным основанием, таким как 2 н. NaOH в метаноле. Добавлением 5 н. HCl pH снижается до 3,0. Кислый продукт можно затем извлечь общепринятыми средствами.
Кетон-содержащие соединения формулы I можно получить при реакции либо ангидрида (соединение XIII), либо хлорангидрида (соединение VII с соединением XIV) при условии, что соответствующие кетоны XV и XVI, соответственно, такие, как описано в схеме 4.
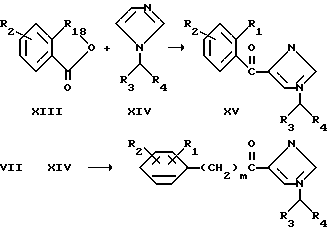
В схеме 4, приведенной выше, значения R2, R3, R4 и m такие же, как определено ранее. R18 является SO2 или CO, R1 - SO3H или CO2H.
Реакции, отображенные в схеме 4 общеизвестны как реакции Фриделя-Крафтса. В этих реакциях принимают участие приблизительно эквимолярные количества хлорангидрида соединения VII или ангидрида (XIII) и реагента XIV в присутствии кислоты Льюиса, такой же как хлорид алюминия, в инертном полярном растворителе, таком как диметилформамид или метиленхлорид.
Предпочтительные амид-содержащие соединения формулы I можно получить по схеме 5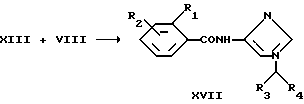
По схеме 5 амин затем вводится в реакцию с подходящим ангидридом (XIII) путем смешивания двух реагентов в одном или нескольких инертных растворителях, таких как диметилформамид. Эта реакцию дает продукты, подобные тем, которые находятся в вышеприведенной схеме 3, частично являются предпочтительными соединениями формулы I. Альтернативно, ангидрид (XIII) может вступать в реакцию с одним эквивалентом алкоголя, давая моноэфир с одним кислым радикалом (соединение VII), который можно ввести в реакцию по схеме 3.
Соединения этого изобретения, содержащие аминосвязь (X- - NH-) можно получить по известным специалисту методикам. Например, можно проводить реакцию Ульмана с соединением формулы
где R19 является защитной группой имидазола, такой как бензиловая функциональность. Реакция проводится в присутствии медной бронзы или хлорида меди в пиридине или диметилформамиде. Образовавшийся продукт может подвергаться снятию защиты и алкилированию способом, аналогичным схеме I.
Защитные фенокси производные пролина можно легко получить по схеме 6.
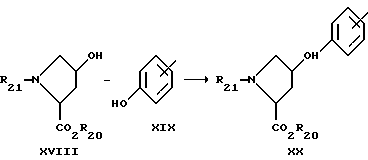
В вышеприведенной схеме 6 R21 является амино-защитной группой, предпочтительно карбобензилокси; R20 - карбокси-защитная группа, предпочтительно C1 - C4 алкил, образующий эфир. Фенольное соединение XIX вступает в реакцию по этой схеме, чтобы получить соединения формулы I, где R10 является замещенным фенилом, как определено ранее.
Изображенная реакция известна специалистам как реакция Мицунобу (Mitsunobu O., Synthesis. 1, 1981). Предпочтительно эта реакция проводится в присутствии трифенилфосфина и диэтилазодикарбоксилата в апротическом растворителе, таком как ТГФ. При завершении реакций по этой схеме с соединения XX можно снять защиту, чтобы образовать амин и ввести его в дальнейшем в реакции по схеме 2.
Как отмечалось выше, соединения этого изобретения содержат по крайней мере один хиральный центр, которым является атом углерода, присоединенный к имидазолу, заместители R3 и R4. Хотя все вышеприведенные схемы относятся к реакциям рацемических реагентов и продуктов, каждую из реакций можно производить, пользуясь хиральным исходным материалом, для того чтобы получить отдельный энантиомер, представляющий интерес. Альтернативно, отдельные изомеры можно выделять из рацематов стандартными способами, такими как фракционная кристаллизация, жидкостная хроматография высокого давления, оращенно-фазовая хроматография и т.п. Такое разделение можно осуществить либо в конечном продукте формулы I, либо в промежуточных соединениях на любых этапах синтеза, либо в производных конечного и промежуточного продуктов. Предпочтительно разделять соединение IV на энантиомеры до реакции соединения по схеме 2. Соединение IV разделяется оптическим разрешением при помощи (-)-синконидина в качестве разрешающего вещества.
Ясно, что соединение замещенной бензойной кислоты или замещенного ангидрида с имидазолилом по схеме 3 или 4 может происходить в любое время в течение синтеза. Предпочтительно, присоединение по схеме 2 происходит до реакций по схеме 3 или 4. Однако специалист должен признать, что порядок проведения реакций не является решающим, если только используются подходящие амино- и карбокси-защитные группы.
Во всех вышеприведенных схемах предпочтительно, чтобы реакции проводились, когда все R1 группы защищены во время реакции сочетания с последующим снятием защиты. Например, если R1 будет тетразолом, реакцию лучше всего проводить с циано-промежуточным соединением. Однако специалист согласится, что многие из этих реакций можно проводить со свободной кислотой или тетразолом, если пользоваться подходящими условиями реакций, блокирующими реагентами и т.п. Так как радикалы R1 значительно отличаются чувствительностью к гидролизу, последовательность превращения промежуточных соединений формулы II до конечных продуктов, обладающих как кислотной, так и тетразольной группой, не является решающей.
Соединения II, III, V, VII, X, XI, XIII, XIV, XVIII, XVIV и любые другие реагенты, требующиеся для их превращения, либо имеются в продаже и известны специалистам, либо их можно получить известными специалистам способами.
Следующие примеры и препараты представлены только для дальнейшей иллюстрации изобретения. Рамки изобретения не предназначены для включения только следующих примеров.
В следующих примерах и препаратах точка плавления, спектры ядерного магнитного резонанса, масс-спектрометрия, жидкостная хроматография высокого давления с силикагелем, N,N-диметилформамид, палладий на угле, гидрид диизобутилалюминия и тетрагидрофуран обозначены аббревиатурами: Т. пл., ЯМР, МС, ЖХВД, ДМФ, Пд/У, ДИБАЛ и ТГФ соответственно. Термины ЯМР и МС указывают, что спектр соответствует заданной структуре.
Препарат 1
2-Карбокси-6-гидроксибензолсульфокислота
Метил 2-гидрокси-3-метоксибензоат (0,027 моль, 5,0 г) добавляли к суспензии гидрида натрия (0,03 моль, 1,45 г, 50% в минеральном масле) в 50 мл ДМФ и перемешивали при комнатной температуре в течение 1 ч. Диметилтиокарбамоил хлорид (0,03 моль, 3,73 г) в 40 мл ДМФ добавляли по каплям в течение 1 ч. Реакционную смесь перемешивали в течение 18 ч. Добавляли этилацетат. Раствор тщательно промывали солевым раствором, осушали и выпаривали. Остаток очищали ЖХВД с силикагелем, элюировали 50% этилацетатом в гексане, чтобы получить 0,9 г O-(2-карбометокси-6-метоксифенил)-N,N-диметилтиокарбамата.
МС для C12H15NO4S:
Рассчитано, %: C 53,52; H 5,61 ; N 5,27.
Найдено, %: C 53,35; H 5,54; N 5,07.
O-(2-Карбометокси-6-метоксифенил)-N, N-диметилтиокарбамат карбамат (720 мг) нагревали при 220oC в течение 100 мин и охлаждали, чтобы получить 700 мг S-(2-карбометокси-6-метоксифенил)- N,N-диметилтиокарбамата.
МС для C12H15NO4S:
Рассчитано, %: C 53,52; H 5,61; N 5,20.
Найдено, %: C 53,74; H 5,60; N 4,92.
S-(2-Карбометокси-6-метоксифенил)-N, N-диметилтиокарбамат (14,4 ммоль, 3,9 г) разводили в 66 мл муравьиной кислоты. По каплям добавляли перекись водорода (24 мл, 30% раствора), охлаждая по мере необходимости. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч и выпаривали. К остатку добавляли толуол (100 мл). Раствор в толуоле сгущали. Сухой остаток размачивали эфиром и отфильтровывали, получая 3,0 г диметиламиновой соли 2-карбометокси-6-метоксибензолсульфокислоты.
Диметиламиновую соль 2-карбометокси-6-метоксибензолсульфокислоты (930 ммоль, 2,6 г) добавляли каплями при -20oC к раствору трибромида бора (27 ммоль, 3,8 мл) в 50 мл метиленхлорида и перемешивали при -20oC в течение 10 мин при комнатной температуре в течение ночи. Реакцию гасили водой. Доводили pH до 8,0 2 н. раствором NaOH. Водный раствор промывали метиленхлоридом. Доводили pH водного раствора до 1,0 2 н. раствором HCl. Промежуточное соединение экстрагировали этилацетатом и выпаривали. Сухой остаток растирали с этилацетатом и фильтровали получая 1,6 г 2-карбокси-6-гидроксибензолсульфокислоты.
Препарат 2
Метиловый эфир N-карбобензилокси-4-транс-гидрокси-L-пролина
Раствор оксида серебра (I) (1,08 ммоль, 250 г) в 1500 мл ацетона охлаждали до -5 - 0oC. Добавляли N-карбобензилокси-4-транс-гидрокси-L-пролин (0,5 ммоль, 132,6 г). Раствор перемешивали 25 мин. Добавляли метилйодид (1,2 ммоль, 170,4 г) при -6oC в течение 25 мин. Реакционную смесь перемешивали при комнатной температуре в течение 5 ч, отфильтровывали и выпаривали. Промежуточное соединение растворяли в этилацетате, фильтровали через силикагель и выпаривали.
МС для C14H17NO5:
Рассчитано, %: C 60,21; H 6,13; N 5,01.
Найдено, %: C 60,40; H 6,26; N 5,06.
Препарат 3
Метиловый эфир N-карбобензилокси-4-цис-фенокси-L-пролина
Метиловый эфир N-карбобензилокси-4-транс-гидрокси-L-пролина (0,267 ммоль, 74,5 г), фенол (0,282 ммоль, 26,5 г) и трифенилфосфин (0,279 ммоль, 73,3 г) растворяли в 750 мл ТГФ и охлаждали до -3oC. Диэтилазидодикарбоксилат (0,284 ммоль, 45 мл) добавляли каплями в течение 2 ч. Реакционную смесь перемешивали при комнатной температуре в течение ночи и затем выпаривали. Остаток растворяли в эфире, фильтровали и выпаривали. Промежуточное соединение хроматографировали с силикагелем, элюируя градиентом 0 - 40% этилацетата в гексане, получая 41,0 г (ЯМР).
Препарат 4
4-Бром-трет-бутоксибензол
4-Бромфенол (57,8 ммоль, 10,0 г) добавляли к раствору изобутилена (40 мл) с метиленхлоридом (50 мл) с температурой -30oC и затем охлаждали до -78oC. Добавляли трифторметансульфокислоту (4 ммоль, 0,35 мл). Смесь выдерживали при -78oC 4ч и затем давали ей согреться до комнатной температуры. Добавляли триэтиламин (0,5 мл) и растворитель удаляли. Остаток хроматографировали с силикагелем, элюируя 1% этилацетатом в гексане, получая 12,4 г.
МС для C10H13BrO
Рассчитано, %: C 52,42; H 5,72.
Найдено, %: C 52,69; H 5,67.
Препарат 5
4-трет-Бутоксифенол
Втор-бутиллитий (53,2 ммоль, 41 г, 1,3 М в гексане) добавляли каплями при -78oC к 4-бром-трет-бутоксибензолу (53,2 ммоль, 12,2 г) в 200 мл ТГФ, перемешивали при -78oC в течение 1 ч и добавляли медленно к раствору триизопропилбората (58,5 ммоль, 11,0 г) в 50 мл ТГФ при сохранении температуры ниже -60oC. Смеси давали постепенно нагреться до -20oC. Добавляли ледяную уксусную кислоту (80 ммоль, 9,6 мл). Поддерживая температуру ниже 0oC, добавляли каплями в течение 15 мин перекись водорода (58,5 ммоль, 5,9 мл 30% раствора, разведенного 5 мл воды) После 10-минутного перемешивания раствор промывали раствором сульфата аммония, осушали и выпаривали. Осадок растирали с гексаном и фильтровали, получая 4,2 г 4-трет-бутоксифенола.
МС для C10H14O2
Рассчитано, %: C 72,26; H 8,49.
Найдено, %: C 72,54; H 8,27.
Препарат 6
Диэтил-(4-гидрокси)-фенэтилфосфонат
Раствор тетраэтилметилендифосфоната (6,22 г, 21,6 ммоль) в 30 мл безводного ТГФ при -30oC в атмосфере азота обрабатывали н-бутиллитием (15,0 мл, 1,6 М раствора в гексанах) каплями через шприц. Образовавшийся раствор нагревали до 0oC за 30 мин и затем снова охлаждали до -30oC. Затем вводили через канюлю бензилоксибензальдегид в виде раствора в 15 мл безводного ТГФ. После нагревания до комнатной температуры и перемешивания в течение 2 ч реакцию гасили вливанием в воду (200 мл). Воду экстрагировали этилацетатом (3 • 100 мл). Органическую часть осушали (сульфат натрия) и выпаривали в вакууме до образования масла. Неочищенный продукт хроматографировали (силикагель, 25% гексан/этилацетат), получая 6,1 г (82%) ненасыщенного фосфоната в виде светло-желтого масла, затвердевающего при стоянии.
МС для C19H23O4P:
Рассчитано, %: C 65,89; H 6,69.
Найдено, %: C 66,15; H 6,59.
Фосфонат, полученный в предшествующей реакции (6,1 г, 17,5 ммоль), растворяли в 100 мл абсолютного этанола и обрабатывали 1,15 г 5% Пд/У. Смесь гидрогенизировали при 40 psi в течение 1 ч и затем пропускали прокладку из целита. Фильтрат сгущали в вакууме до получения 4,5 г (100%) диэтил-(4-гидрокси)-фенэтилфосфоната в виде светло-желтого масла.
Препарат 7
Диэтил-(4-гидрокси)-фенилфосфат
4-Бензилоксифенол (15,0 г, 75 ммоль) растворяли в 100 мл безводного ТГФ и охлаждали до 0oC. Затем маленькими порциями вводили через NaH (3,0 г, 75 ммоль, 60% дисперсия в минеральном масле). Когда прекращалось образование газа, каплями через шприц вводили диэтилхлорфосфат. После перемешивания в течение 1 ч реакционную смесь вливали в H2O/этилацетат (150 мл еа). Соли разделяли и органический слой промывали 0,1 н. NaOH (2 • 100 мл). Органическую часть осушали (сульфат натрия) и сгущали в вакууме до получения светло-желтой жидкости. Хроматография (силикагель, сначала 20% этилацетат/гексаны, затем 40% гексаны/этилацетат) давала 23,4 г (93%) диэтил-(4-бензилокси)-фенилфосфата в виде бесцветной жидкости.
Диэтил-(4-бензилокси)-фенилфосфат (15,0 г, 44,7 ммоль) растворяли в 150 мл 30% этилацетата в этаноле с 0,5 мл концентрированной соляной кислоты. К этому раствору добавляли 3,0 г 10% Пд/У. Смесь гидрогенизировали при 1 атм в течение 18 ч и затем пропускали через прокладку из целита для удаления катализатора. Фильтрат сгущали в вакууме и остаток хроматографировали (силикагель, этилацетат), получая 10,4 г (94%) диэтил-4-гидроксифенилфосфата в виде янтарной жидкости.
Препарат 8
Диэтил-(4-гидрокси)-бензолфосфонат
К раствору 4-бензилоксибромбензола (10,0 г, 38 ммоль) в 150 мл безводного ТГФ при -78oC в атмосфере азота добавляли н-бутиллитий (26,1 мл, 41,8 ммоль, 1,6 М в гексанах) каплями в течение 30 мин. После перемешивания в течение 15 мин каплями через шприц добавляли диэтилхлорфосфат (6,0 мл, 41,8 ммоль). Образовавшейся смеси давали постепенно нагреться до комнатной температуры, после чего реакцию гасили вливанием в H2O/этилацетат (200 мл еа). Слои разделяли и водный слой экстрагировали этилацетатом (2 • 100 мл). Органическую часть осушали (сульфат натрия) и сгущали в вакууме до желтой жидкости. Хроматография (силикагель, 50 - 100% этилацетат/гексаны) давали 11,1 г (91%) диэтил-(4-бензилокси)-бензолфосфоната в виде бесцветной жидкости, МС.
Диэтил-(4-бензилокси)-бензолфосфонат (11,0 г, 34 ммоль) гидрогенизировали, как описано в предыдущем примере. Хроматография неочищенного восстановленного продукта давала 4,3 г (52%) диэтил-(4-гидрокси)-бензолфосфоната в виде светло-желтой жидкости. МС.
Препарат 9
4-(Пирролидинсульфонил)-фенол
К раствору пирролидина (17 мл, 237 ммоль) в 20 мл воды при комнатной температуре добавляли пара-фторбензолсульфонилхлорид (15 г, 79 ммоль) по частям за 5-минутный период. Через 1 ч раствор разбавляли 100 мл воды и экстрагировали этилацетатом (3 х 50 мл). Органическую часть осушали (сульфат натрия) и выпаривали в вакууме до получения 12,3 г (72%) (4-(пирролидинсульфонил)-фторбензола в виде бесцветного масла, затвердевающего при стоянии. Это вещество без дальнейшей очистки использовалось в последующей реакции. МС.
К раствору бензилового спирта (6,63 мл, 62,0 ммоль) в 200 мл безводного ДМФ при комнатной температуре маленькими порциями добавляли NaH (2,40 г, 60,0 ммоль, 60% дисперсия в минеральном масле). После перемешивания в течение 30 мин добавляли 4-(пирролидинсульфонил)-фторбензол (11,0 г, 51,2 ммоль) за период в 10 мин. Через 30 мин образовался белый осадок. Затем реакционную смесь разводили 100 мл воды и продукт выделяли вакуум-фильтрацией. Осадок высушивали в вакууме, получая 14,85 г (95%) 4-(пирролидинсульфонил)-фенилбензилэфира в виде сухого вещества белого цвета. МС.
Раствор 4-(пирролидинсульфонил)-фенилбензилэфира (10,0 г, 33,1 ммоль) растворяли в 100 мл абсолютного этанола. Этот раствор обрабатывали 2,5 г 10% Пд/У. Смесь гидрогенизировали при 40 psi в течение 2 ч. Затем катализатор удаляли пропусканием реакционной смеси через прокладку целита. Фильтрат выпаривали в вакууме до получения 6,8 г (90%) 4-(пирролидинсульфонил)-фенола в виде белого сухого остатка. МС.
4-(Метиламиносульфонил)-фенол получали таким же образом.
Препарат 10
Метиловый эфир N-(4-гидроксибензамидо)-L-пролина
Гидрохлорид метилового эфира L-пролина (7,2 г, 43,8 ммоль) растворяли в 100 мл безводного ДМФ при 0oC. К этому раствору добавляли триэтиламин (4,2 г, 43,8 ммоль). После энергичного перемешивания в течение 1 ч, осадок гидрохлорида триэтиламина удаляли фильтрацией. К фильтрату добавляли 4-бензилоксибензойную кислоту (10,0 г, 43,8 ммоль) и следом ДЦК (9,9 г, 48,2 ммоль). Реакционную смесь оставляли на ночь перемешиваться при комнатной температуре. Осадок ДЦУ затем удаляли фильтрацией и отфильтрованное распределяли между водой и этилацетатом (300 мл еа). Органическую часть промывали несколько раз водой, порциями по 200 мл для удаления ДМФ. Органическую часть осушали (сульфат натрия) и выпаривали в вакууме до сухого остатка, который хроматографировали (силикагель, 15-100% этилацетат/гексаны). После выделения получали 5,3 г (35%) метилового эфира N-(4-бензилоксибензамидо)-L-пролина в виде белого сухого остатка. МС.
Вышеуказанный амид (10,0 г, 29,4 ммоль) растворяли в 75 мл абсолютного этанола. К этому раствору добавляли 3 г 10% Пд/У. Смесь гидрогенизировали при 1 атм в течение 5 ч. Затем катализатор удаляли, пропуская реакционную смесь через прокладку из целита. Выпаривание фильтрата давало неочищенный метиловый эфир N-(4-гидроксибензамидо)-L-пролина, который очищали хроматографией (силикагель, 30% этилацетат/гексан), получая 6,3 г (86%) сухого вещества белого цвета. МС.
Препарат 11
(-)-Синконидиновая соль (R) -α -гексил-4-нитро-1H-имидазол-1- уксусной кислоты
К суспензии 5,89 г (0,02 ммоль) (-)-синконидина в 80 мл воды добавляли 2,78 мл (2,02 г, 0,02 ммоль) триэтиламина. Смесь нагревали приблизительно до 40-45oC. К подогретой суспензии, помешивая, добавляли раствор 10,21 г (0,04 ммоль) рацемической смеси α -гексил-4-нитро-1H-имидазол-1-уксусной кислоты в 40 мл неразведенного технического этанола. (pH этой смеси доводили до 6,9 - 7,4 добавлением триэтиламина или водной соляной кислоты по необходимости). Образовавшуюся суспензию затем нагревали примерно до 85oC. Образовавшемуся раствору давали постепенно остыть до комнатной температуры, медленно помешивая. Осажденную соль отфильтровывали, промывали примерно 30 мл смеси этанола водой (1 : 2) и сушили в вакууме при 50oC до постоянного веса. Эта реакция давала 9 г (-)-синконидиновой соли (R)- α -гексил-4-нитро-1H-имидазол-1-уксусной кислоты. Часть продукта превращали в свободную соль и затем в производное, такое как метиловый эфир (диазометан) и анализировали ЖХВД на хиральной колонке. Анализ показывает, что кислота, образованная из продукта, имеет ее 94%. Перекристаллизация соли продукта из смеси этанол-вода (1:1) дает 7,4 г чистой соли, ее 99% (ЖХВД), Т.пл. 205oC (разрушается). (ЯМР).
Анализ для C30H39N5O5:
Рассчитано,%: C 65,55; H 7,15; N 12,74.
Найдено,%: C 65,32; H 7,25; N 12,74.
Препарат 12
(R)- α -Гексил-4-нитро-1H-имидазол-1-уксусная кислота
Часть чистой синконидиновой соли, полученной способом, описанным в примере 1, 2,80 г смешивали с 20 мл 1 М HCl. Образовавшуюся суспензию экстрагировали 30 мл этилацетата. Фазу этилацетата осушали (MgSO4) и выпаривали до сухости, получая 0,82 г (63%) (R)- α -гексил-4-нитро-1H-имидазол-1-уксусной кислоты. Т.пл. 112-114oC.
Пример 1. N-Этил-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил]- октанамид
4-Нитроимидазол (0,29 ммоль, 32,9 г в 30 мл ДМФ) обрабатывали частями гидридом натрия (0,29 ммоль, 11,6 г, 60% в минеральном масле) и перемешивали 45 мин. В течение 1 ч каплями добавляли этил-2-бромоктаноат, (0,29 ммоль, 73,1 г). Реакционную смесь перемешивали в течение ночи при комнатной температуре, вливали в ледяную воду и экстрагировали этилацетатом. Органическую фазу промывали солевым раствором, осушали и выпаривали до получения 91 г этил-2-(4-нитро-1H-имидазол-1-ил)-октаноата. МС.
Этил 2-(4-нитро-1H-имидазол-1-ил)октаноат (17 ммоль, 5 г) и этиламино (20 мл) перемешивали в 150 мл этанола при комнатной температуре в течение 16 ч. Реакцию добавляли к ледяной воде, экстрагировали этилацетатом, промывали водой, осушали над сульфатом натрия и выпаривали. Масло при стоянии давало кристаллы с выходом 3,9 г N-этил-2-(4-нитро-1H-имидазол-1-ил)-октанамида.
МС для C13H22N4O3•1/4 H2O:
Рассчитано,%: C 54,40; H 7,83; N 19,52.
Найдено,%: C 54,45; H 7,73; N 19,12.
N-Этил-2-(4-нитро-1H-имидазол-1-ил)октанамид (5,3 ммоль, 1,5 г) восстанавливали гидрогенизацией при 40 psi над Пд/У. Реакционную смесь фильтровали и выпаривали. Остаток растворяли в 10 мл ТГФ и добавляли к раствору циклического ангидрида 2-сульфобензойной кислоты (5,3 ммоль, 0,98 г) в 10 мл ТГФ. После перемешивания в течение 10 мин при комнатной температуре продукт выпадал в осадок. Осадок собирали фильтрацией, промывали эфиром и высушивали до получения 1,1 г продукта.
Т.пл.: разрушается при 235oC.
МС для C20H28N4O5S:
Рассчитано,%: C 55,03; H 6,46; N 12,83.
Найдено,%: C 54,75; H 6,49; N 13,08.
Пример 2. N-Пропил-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октанамид
Этил 2-(4-нитро-1H-имидазол-1-ил)октаноат (17 ммоль, 5,0 г) вводили в реакцию с пропиламином, как в примере 1, получая 2,5 г N-пропил-2-(4-нитро-1H-имидазол-1-ил)октанамида. МС.
N-Пропил-2-(4-нитро-1H-имидазол-1-ил)октанамид (2,3 ммоль, 0,7 г) восстанавливали в амин в вводили в реакцию с циклическим ангидридом 2-сульфобензойной кислоты (2,3 ммоль, 0,423 г), как в примере 1, получая 0,70 г продукта.
Т.пл.: разрушается при 235oC.
МС для C21H30N4O5S • 1,5 H2O
Рассчитано,%: C 52,78; H 6,91; N 11,71.
Найдено,%: C 52,86; H 6,37; N 11,16.
Пример 3. N-(2,2,2-Трифторэтил)-2-[4-(2-сульфобензоил)амино-1H- имидазол-1-ил]октанамид
2-(4-Нитро-1H-имидазол-1-ил)октановую кислоту (7,8 ммоль, 2,0 г) обрабатывали 25 мл оксалилхлорида, выпаривали и добавляли к раствору трифторэтиламиногидрохлорида (7,8 ммоль, 1,06 г) и триэтиламина (2 мл) в 50 мл ТГФ. После перемешивания при комнатной температуре в течение 16 ч смесь добавляли ко льду, экстрагировали этилацетатом, осушали над сульфатом натрия и выпаривали. Это промежуточное соединение хроматографировали с силикагелем, получая 0,6 г N-(2,2,2-трифтор)-этил-2-(4-нитро-1H-имидазол-1-ил)октанамида.
МС для C11H19F3N4O3:
Рассчитано,%: C 46,43; H 5,69; N 16,66.
Найдено,%: C 46,56; H 5,88; N 16,37.
N-(21,2,2-Трифтор)этил-2-(4-нитро-1H-имидазол-1-ил)-октанамид (1,5 ммоль, 0,5 г) восстанавливали и вводили в реакцию с циклическим ангидридом 2-сульфобензойной кислоты (1,4 ммоль, 0,27 г), как в примере 1, получая 500 мг продукта.
Т.пл.: разрушается при 257 - 259oC.
МС для C20H25F3N4O5S • H2O:
Рассчитано,%: C 47,26; H 5,35; N 11,01.
Найдено,%: C 47,39; H 5,01; N 10,82.
Пример 4. N-[2-(1-Гидрокси-2-метил)пропил] 2-[4-(2-сульфобензоил) амино-1H-имидазол-1-ил]октанамид
2-(4-Нитро-1H-имидазол-1-ил)октановую кислоту (3,9 ммоль, 1,0 г) превращали в кислый хлорид и вводили в реакцию с 2-амино-2-метил-1-пропанолом (5,85 ммоль, 0,52 г), как в примере 3, получая 0,465 г N-[2-1-гидрокси-2-метил)пропил]-2-(4-нитро- 1H-имидазол-1-ил)октанамида.
N-[2-(1-Гидрокси-2-метил)пропил] -2-(4-нитро-1H-имидазол-1-ил) октанамид (0,52 ммоль, 170 мг) восстанавливали и вводили в реакцию с 2-сульфобензоной кислотой (0,52 ммоль, 96 мг), как в примере 1, получая 53 мг продукта.
Т.пл.: разрушается при 148 - 158oC.
МС для C22H32N4O6S:
Рассчитано,%: C 54,98; H 6,71; N 11,66.
Найдено,%: C 51,49; H 5,53; N 8,32.
Пример 5. 2-[[[1-[1-2-Оксо-1-имидазолидинил)карбонил]-гептил]-1H-имидазол-4- ил]амино]карбонил]бензолсульфокислота
Готовилась, как в примере 3. Выход продукта 17%.
Т.пл. 219 - 228oC.
МС для C21H27N5O6S:
Рассчитано,%: C 52,82; H 5,70; N 14,66.
Найдено,%: C 51,19; H 5,54; N 12,65.
Пример 6. 2-[[[1-[1-[(2-Тиоксо-1-имидазолидинил)карбонил]-гептил]- 1H-имидазол-4-ил]амино]карбонил]бензолсульфокислота
Готовилась, как в примере 3. Выход продукта 11%.
Т.пл. 203 - 211oC.
МС для C21H27N5O5S:
Рассчитано,%: C 51,10; H 5,51; N 14,19.
Найдено,%: C 49,30; H 5,71; N 13,69.
Пример 7. N-(2-Пиридил)-2-[4-(2-сульфобензоил)амино- 1H-имидазол-1-ил] октанамид
Готовился, как в примере 3. Выход продукта 23%.
Т.пл. 165 - 173oC.
МС для C23H27N5O5S:
Рассчитано,%: C 56,89; H 5,61; N 14,42.
Найдено,%: C 52,50; H 4,75; N 9,72.
Пример 8. N-(2-Гидроксифенил)-2-[4-(2-сульфобензоил)амино- 1H-имидазол-1-ил]октанамид
Готовился, как в примере 3. Выход продукта 33%
Т.пл.: 138 - 147oC.
МС для C24H28N4O6S • 1,25 HCl.
Рассчитано,%: C 52,78; H 5,40; N 10,26.
Найдено,%: C 52,75; H 5,23; N 9,93.
Пример 9. N-(2-Карбоксифенил)-2-[4-(2-сульфобензоил)амино- 1H-имидазол-1-ил]октанамид
N-(2-Карбоэтоксифенил)-2-[4-(2-сульфобензоил)амино-1H-имидазол- 1-ил] октанамид готовился, как в примере 3. Этот эфир (100 мг) подвергали гидролизу в 1 н. NaOH и 0,2 мл метанола в течение 1 ч при комнатной температуре и подкисляли 1 н. HCl. Осадок отфильтровывали и высушивали с 88% выхода.
Т.пл.: 163 - 168oC.
МС для C25H28N4O7S • 1,25 H2O:
Рассчитано,%: C 54,49; H 5,57; N 10,17.
Найдено,%: C 54,58; H 5,18; N 9,75.
Пример 10. 2-[4-(3-Гидрокси-2-сульфобензоил)амино-1H-имидазол-1- ил]октановая кислота
2-Карбокси-6-гидроксибензолсульфокислоту (1,8 ммоль, 400 мг) растворяли в 15 мл оксалихлорида. Добавляли одну каплю ДМФ. Реакционную смесь перемешивали 30 мин при комнатной температуре. Растворитель удаляли в вакууме и добавляли 20 мл ТГФ. По каплям добавляли раствор этил-2-(4-амино-1H-имидазол-1-ил)октаноата (полученного восстановлением 1,8 ммоль этил 2-(4-нитро-1H-имидазол-1-ил)октаноата в этаноле с 5% Пд/У) в 40 мл ТГФ и 2,0 ммоль триэтиламина. Реакционную смесь перемешивали при комнатной температуре в течение 3 ч; добавляли этилацетат. Раствор промывали водой, осушали над сульфатом натрия и выпаривали. Промежуточное соединение размачивали в эфире, фильтровали, хроматографировали с силикагелем, элюируя 15% метанолом в метиленхлориде. Эфир подвергали гидролизу в 20 мл метанола и 45 мл 2 н. NaOH, при комнатной температуре в течение 2 ч. Растворитель удаляли; добавляли воду. При помощи 5 н. HCl доводили pH до 2,0. Продукт экстрагировали этилацетатом, осушали над сульфатом натрия и выпаривали, получая 2-[4-(3-гидрокси-2-сульфобензоил)амино-1H-имидазол-1-ил]октановую кислоту.
Т.пл.: 238 - 240oC.
МС для C18H23N3O7S • 1/2 H2O:
Рассчитано,%: C 49,71; H 5,50; N 9,60.
Найдено,%: C 49,73; H 5,38; N 9,30.
Пример 11. 1-[1-Оксо-1-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил]октил] - D-пролин
Смесь стереоизомеров готовили, как в примере 21.
Изомер A: выход 9%.
Т.пл.: 145 - 150oC.
МС для C23H30N4O7S:
Рассчитано,%: C 54,53; H 5,97; N 11,06.
Найдено,%: C 54,34; H 6,06; N 11,03.
Изомер B: выход 5%.
Т.пл.: 148 - 155oC.
МС для C23H30N4O7:
Рассчитано,%: C 54,53; H 5,97; N 11,06.
Найдено,%: C 54,52; H 6,08; N 10,93.
Пример 12. 1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H- имидазол-1-ил]октил]-4-цис-фенокси-L-пролин
Метиловый эфир N-карбобензилокси-4-цис-фенокси-L-пролина (0,115 ммоль, 41 г) гидрогенизировали в этаноле над 5% Пд/У и выпаривали.
2-Бромоктановую кислоту (0,116 ммоль, 26 г) добавляли каплями к раствору оксалилбромида (75 г) в 50 мл метиленхлорида при температуре ледяной бани; добавляли 1 каплю ДМФ. Раствор перемешивали при комнатной температуре в течение 1 ч и выпаривали. Остаток растворяли в ТГФ и по каплям прибавляли в раствор пролина и триэтиламина (45 мл) в ТГФ при температуре ледяной бани. Реакционную смесь перемешивали в течение ночи при комнатной температуре, фильтровали и выпаривали. Масло растворяли в этилацетате, промывали солевым раствором, осушали над сульфатом натрия и выпаривали. Продукт хроматографировали с силикагелем, элюируя 0-30% этилацетатом в гексане, получая 26 г метилового эфира 1-(2-бром-1-оксо)октил-4-цис-фенокси-L-пролина. (ЯМР).
4-Нитроимидазол (66,3 ммоль, 7,5 г) растворяли в 200 мл ДМФ. Порциями добавляли гидрид натрия (75 ммоль, 3,0 г 60% в минеральном масле). Раствор перемешивали в течение 1 ч. Добавляли метиловый эфир 1-(2-бром-1-оксо)октил-4-цис-фенокси-L-пролина (60 ммоль, 25,6 г). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь выпаривали, остаток растворяли в этилацетате, дважды промывали солевым раствором, осушали над сульфатом натрия и выпаривали. Промежуточное соединение хроматографировали с силикагелем, элюируя 25-75% этилацетата в гексане.
Изомер A: выход 40%.
МС для C23H30N4O6:
Рассчитано,%: C 60,16; H 6,67; N 11,95.
Найдено,%: C 60,25; H 6,59; N 12,21.
Изомер B: выход 16%.
МС для C23H30N4O6:
Рассчитано,%: C 60,25; H 6,59; N 12,22.
Найдено,%: C 60,43; H 6,63; N 12,26.
Метиловый эфир 1-[1-оксо-2-(4-нитро-1H-имидазол-1-ил)октил] -4-цис-фенокси-L-пролина (изомер A, 22,7 ммоль, 10,4 г) восстанавливали в этаноле с Пд/У и вводили в реакцию с циклическим ангидридом 2-сульфобензойной кислоты (34,2 ммоль, 6,5 г), как в примере 1, получая 9,2 г эфира. МС. Эфир подвергали гидролизу в 25 мл этанола и 100 мл 1 н. гидроксида натрия при комнатной температуре в течение 1 ч и выпаривали. Остаток растворяли в минимальном объеме воды. При помощи 2 н. HCl pH доводят до 2,4. Осадок отфильтровывали и высушивали, получая 6,0 г продукта.
Т.пл.: 180 - 190oC.
МС для C29H34N4O8S:
Рассчитано,%: C 58,18; H 5,72; N 9,36.
Найдено,%: C 58,18; H 5,78; N 9,50.
Изомер B обрабатывали таким же образом, получая 90% выхода кислоты.
Т.пл.: > 200oC.
МС для C29H34N4O8S • 1/2 H2O • 1/2 NaCl
Рассчитано,%: C 54,69; H 5,54; N 8,80.
Найдено,%: C 54,21; H 5,46; N 8,77.
Пример 13. 1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол- 1-ил]гептил]-4-цис-фенокси-L-пролин
2-(4-Нитро-1H-имидазол-1-ил)гептановую кислоту (4 ммоль, 0,98 г, приготовленную, как в примере 1, перемешивали в 25 мл оксалилхлорида в течение 1 ч и выпаривали. Остаток растворяли в метиленхлориде (100 мл) и добавляли каплями к раствору метилового эфира 4-цис-фенокси-1-пролина (4 ммоль, 0,9 г) и триметиламина (0,56 мл) в 100 мл метиленхлорида. Реакционную смесь перемешивали 2 ч при комнатной температуре и затем добавляли к ледяной воде. Органический слой промывали водой, осушали над сульфатом натрия, выпаривали. Остаток хроматографировали с силикагелем, элюируя градиентом 50-75% этилацетата в гексане.
Изомер A промежуточного соединения: выход 41%, МС.
Изомер B промежуточного соединения: выход 28%, МС.
С изомером A промежуточного соединения далее проводили такие реакции, как в примере 12, чтобы получить кислый продукт,
МС для C28H32N4O8S:
Рассчитано,%: C 57,52; H 5,52; N 9,58.
Найдено,%: C 56,87; H 6,13; N 10,33.
Пример 14. 1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1- ил]гексил]-4-цис-фенокси-L-пролин
Готовился, как в примере 12.
Изомер A: выход 48%.
Т.пл.: разрушается при 215 - 220oC.
МС для C27H30N4O8S • H2O • NaCl:
Рассчитано,%: C 50,12; H 4,98, N 8,66.
Найдено,%: C 49,79; H 4,81; N 8,71.
Изомер B: выход 29%.
Т.пл.: разрушается при 210oC.
МС для C27H30N4O8S • H2O:
Рассчитано,%: C 55,09; H 5,48; N 9,52.
Найдено,%: C 55,39; H 5,36; N 9,15.
Пример 15. 1-[1-Оксо-2-[2-сульфобензоил)амино-1H-имидазол-1- ил] -8,8,8-трифтороктил]-4-цис-фенокси-L-пролин
6-Бромгексановую кислоту (0,51 ммоль, 100 г) нагревали под SF4 при 130oC 8 ч. Добавляли метиленхлорид. Раствор фильтровали и выпаривали. Образовавшееся черное масло 6-бром-1,1,1-трифторгексана дистиллировали.
Т.по.: 158 - 164oC / 760 мм.
6-Бром-1,1,1-трифторгексан (0,228 ммоль, 50 г) добавляли к раствору йодида натрия (51 г) в 250 мл ацетона и перемешивали в течение 1 ч при комнатной температуре. Раствор фильтровали и выпаривали. Осадок размачивали в эфире, фильтровали и выпаривали до получения 56 г, 92% йодида. Этилацетат (0,125 ммоль, 16,45 г) медленно добавляли к гидриду натрия (0,126 ммоль, 5,06 г 60% в минеральном масле). Добавляли йодид (0,115 ммоль, 30,6 г). Реакционную смесь нагревали при 50oC в течение 16 ч и затем вливали в ледяную воду. Промежуточное соединение экстрагировали этилацетатом, осушали над сульфатом натрия и выпаривали. Остаток хроматографировали с силикагелем, элюировали этилацетатом в гексане, получая 13,3 г этил 6,6,6-трифторгексилацетоацетата. МС.
Этил 6,6,6-трифторгексилацетоацетат добавляли при -35oC к раствору натрия (50 ммоль, 1,15 г) в 150 мл этанола и перемешивали 15 мин. Добавляли N-бромсукцинимид (50 ммоль, 8,9 г). Раствору давали нагреться до комнатной температуры и перемешивали его в течение 2,5 ч. Смесь вливали в воду. Промежуточное соединение экстрагировали гексаном. Растворитель удаляли. Масло хроматографировали с силикагелем, элюируя гексаном, чтобы получить 13,6 г этил 2-бром-8,8,8-трифтороктаноат. МС.
4-Нитроимидазол (43 ммоль, 4,86 г) вводили в реакцию с гидридом натрия (43 ммоль, 1,72 г) и затем с этил 2-бром-8,8,8- трифтороктаноатом (43 ммоль, 13,2 г), как в примере 1. Эфир подвергали гидролизу в 10 мл метанола и 30 мл 2 н. NaOH, получая значительный выход 2-(4-нитро-1H-имидазол-1-ил)-8,8,8- трифторактановой кислоты (ЯМР).
Далее с кислотой проводили реакции, как в примере 13, чтобы получить стереоизомеры метилового эфира 1-[1-оксо-2-(4-нитро- 1H-имидазол-1-ил)-8,8,8-трифтороктил]-4-цис-фенокси-L-пролина. (ЯМР). Изомеры разделяли, как в примере 1; изомер А далее вводили в реакции, как описывалось ранее. Продукт хроматографировали с силикагелем, элюировали 5% метанолом в хлороформе, получая 1-[1-оксо-2-[4-(2- сульфобензоил)амино-1H-имидазол-1-ил]-8,8,8-трифтороктил]-4-цис- фенокси-L-пролин.
Изомер A: выход 13%.
МС для C29H30F3N4O8S • 0,6 HCl:
Рассчитано,%: C 51,64; H 4,72; N 8,31.
Найдено,%: C 51,58; H 4,80; N 8,18.
Пример 16. 1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1- ил]-7,7,7-трифторгептил]-4-цис-фенокси-L-пролин
2-(4-Нитро-1H-имидазол-1-ил)-7,7,7-трифторгептановая кислота готовилась, как в примере 15. (ЯМР). Далее кислоту вводили в реакции, как в примере 13, чтобы получить метиловый эфир 1-[1-оксо-2-(4-нитро- 1H-имидазол-1-ил)-7,7,7-трифторгептил]-4-цис-фенокси-1-пролина. (ЯМР).
Эфир вводили в реакции, как в примере 15, чтобы получить 1-[1-оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] - 7,7,7-трифторгептил] -4-цис-фенокси-L-пролин.
Изомер A: выход 21%.
МС для C28H29F3N4O8S • 0,9 HCl:
Рассчитано,%: C 50,09; H 4,49; N 8,44.
Найдено,%: C 50,13; H 4,66; N 8,44.
Изомер B: выход 26%.
МС для C28H29F3N4O8:
Рассчитано,%: C 52,66; H 4,58; N 8,77.
Найдено,%: C 52,80; H 4,85; N 8,63.
Пример 17. 1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1- ил]-октил]-4-цис-(3-пирридилокси)-L-пролин
Метиловый эфир 4-цис-(3-пиридилокси)-L-пролина (5,32 ммоль, 1,18 г) (приготовленный, как в Препарате 3 с последующим снятием защиты с метилового эфира N-карбобензилокси-4-цис(3-пиридилокси)-L-пролина в этаноле с 5% Пд/у), 2-(4-нитро-1H-имидазол-1-ил)октановую кислоту (5,32 ммоль, 1,36 г) и гидроксибензотриазол (5,85 ммоль, 0,8 г) растворяли в 5 мл ДМФ. Через 5 мин добавляли дициклогексилкарбодиимид (5,85 ммоль, 1,21 г). Реакционную смесь перемешивали в течение 60 ч при комнатной температуре. Добавляли этилацетат (15 мл). Раствор фильтровали, промывали водой, осушали над сульфатом натрия и выпаривали. Остаток хроматографировали с силикагелем, элюируя 1% метанолом в хлороформе, получая метиловый эфир 1-[1-оксо-2-(4-нитро- 1H-имидазол-1-ил)октил]-4-цис-(3-пиридилокси)-1-пролина.
Изомер A: 0,63 г; МС.
Изомер B: 0,36 г; МС.
Каждый изомер вводили в реакцию, как в примере 1, чтобы получить 1-[1-оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил] -4- цис-(3-пиридилокси)-L-пролин.
Изомер A: выход 3 %, МС.
Изомер B: выход 3 %, МС.
Пример 18. 1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1- ил]октил]-4-цис(4-метоксифенилокси)-L-пролин
Метиловый эфир N-карбобензилокси-4-цис-(4-метоксифенокси)-L- пролина (приготовленный, как в Препарате 3) вводили в такие реакции, как в примере 12, чтобы получить 1-[1-оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил-4-цис-(4- метоксифенокси)-L-пролин.
Изомер A: выход 42 %.
МС для C30H36N4O9S • 0,5 H2O:
Рассчитано,%: C 56,50; H 5,80; N 8,78.
Найдено,%: C 56,36; H 6,12; N 8,67.
Пример 19. 1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1- ил]октил]-4-цис-(4-гидроксифенокси)-L-пролин
Метиловый эфир N-карбобензилокси-4-цис-(4-трет-бутоксифенокси)-L-пролина (приготовленный, как в Препарате 3) вводили в такие реакции, как в примере 13, чтобы получить 1-[1-оксо-2-(4-нитро-1H-имидазол-1-ил)октил]-4-цис-(4-трет- бутоксифенокси)-L-пролин.
Изомер A: выход 34 %.
МС для C27H38N4O7 • H2O:
Рассчитано,%: C 59,50; H 7,32; N 10,27.
Найдено,%: C 59,60; H 7,04; N 9,98.
Изомер B': выход 33 %.
МС для C27H38N4O7:
Рассчитано,%: C 61,12; H 7,22; N 10,56.
Найдено,%: C 61,37; H 7,32; N 10,59.
Изомер A'вводился в дальнейшие реакции, как в примере 12, чтобы получить этиловый эфир 1-[1-оксо-2-[4-(2-сульфобензоил)амино-1H- имидазол-1-ил]октил] -4-цис(4-трет-бутоксифенокси)-L-пролина. Этиловый эфир трет-бутоксифеноксипролина перемешивали в трифторуксусной кислоте (ТФК, 3 мл) в течение трех часов при комнатной температуре. Избыток ТФК удаляли и осадок подвергали гидролизу, как в примере 12, чтобы получить продукт.
Изомер A: Т.пл.: 180 - 194oC.
МС для C29H34N4O9S:
Рассчитано,%: C 56,67; H 5,58; N 9,12.
Найдено,%: C 56,95; H 5,69; N 8,93.
Пример 20. 1-[1-Оксо-2-[4-(2-карбокси-3-гидробензоил)амино-1H- имидазол-1-ил]октил]-4-цис-фенокси-L-пролин
Изомер A 1-[1-оксо-2-(4-нитро-1H-имидазол-1-ил)-октил]-4- цис-фенокси-L-пролина (1,09 ммоль, 0,5 г, приготовленного, как в примере 12) гидрогенизировали в этаноле над 5 % Пд/У, фильтровали и выпаривали. Остаток растворяли в 25 мл ацетонитрила и добавляли к раствору 3-гидроксифталевого ангидрида в 25 мл ацетонитрила. После перемешивания при комнатной температуре в течение 2 ч осадок собирали и высушивали с выходом эфира 21%. Эфир (0,24 ммоль, 0,14 г) подогревали в течение 15 мин в 5 мл этанола и 5 мл 1 н. NaOH, перемешивали при комнатной температуре в течение 1 ч и сгущали. К остатку добавляли воду (20 мл). Доводили pH до 3,0 при помощи 5 н. HCl. Осадок отфильтровывали и высушивали до 86% выхода 1-[1-оксо-2-[4-(2-карбокси-3-гидроксибензол)амино-1H-имидазол-1-ил]октил]- 4-цисфенокси-L-пролина.
Т.пл.: 155 - 170oC.
МС для C30H34N4O8:
Рассчитано, %: C 62,27; H 5,92; N 9,68.
Найдено, %: C 62,01; H 5,66; N 9,62.
Пример 21. 1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил]-октил]- L-пролин
Диизопропилэтиламин (38,6 ммоль, 5,1 г) добавляли к раствору гидрохлорида бензилового эфира L-пролина (39,6 ммоль, 10,1 г) в 20 мл ДМФ при 0oC и перемешивали в течение 1 ч. Этот раствор добавляли к 2-(4-нитро-1H-имидазол-1-ил)октановой кислоте (39,6 ммоль, 10,1 г) и гидроксибензотриазолу (43 ммоль, 5,8 г) в 10 мл ДМФ и перемешивали 30 мин. Дициклогексилкарбодиимид (43 ммоль, 8,97 г) добавляли порциями на протяжении 2 ч. Добавляли этилацетат (50 мл). Раствор фильтровали, осушали над сульфатом натрия и выпаривали. Масло хроматографировали с силикагелем, элюируя 40% этилацетатом в гексане, получая 5,32 г бензилового эфира 1-[1-оксо-2-(4-нитро-1H-имидазол-1- ил)октил]-L-пролина.
Бензиловый эфир 1-[1-оксо-1-(4-нитро-1H-имидазол-1-ил)октил]-L-пролина (2,46 ммоль, 0,89 г, изомер А) восстанавливали в этаноле с 0,5 г 10% Пд/У. Катализатор отфильтровывали и раствор выпаривали. Амин растворяли в 5 мл ТГФ. Сульфобензойный ангидрид (2,46 ммоль, 0,46 г) добавляли и перемешивали в течение 30 мин. Растворитель удаляли и осадок растирали с эфиром. Осадок растворяли в 3 мл 1 н. NaOH и перемешивали в течение 2 ч, подкислив 1 н. HCl до pH 3,5. Продукт фильтровали и хроматографировали с обращенными фазами на силикагеле, получая 96 мг 1-[1-оксо-2-[4-(2-сульфобензоил)-амино-1H-имидазол-1- ил]октил]-L-пролина.
Т.пл.: 190 - 195oC.
МС для C23H30N4O7S:
Рассчитано, %: C 54,53; H 5,97; N 11,06.
Найдено, %: C 54,46; H 6,03; N 11,08.
Изомер B обрабатывали таким же образом, получая 115 мг. Т.пл.: 163 - 170oC.
МС, найденные значения: C 54,37; H 6,00; N 11,96.
Следующие примеры 22-90 (см. табл. 1) готовили таким же образом и иллюстрируют далее синтез соединений изобретения, определяемых формулой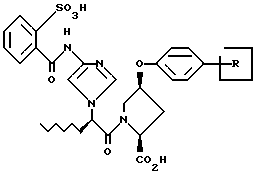
Пример 91. I-[I-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1- ил]октил]-4-цис-(4-метиленфосфоновая кислота)-фенокси)-L-пролин.
К раствору диметилфосфита (22,4 мл, 244 ммоль) в 400 мл безводного ТГФ при 0oC добавляли гидрид натрия (9,3 г, 232 ммоль, 60% дисперсия в минеральном масле) маленькими порциями. Затем через канюлю вводили бензилоксибензилхлорид в виде раствора 53,7 г 232 ммоль в 100 мл безводного ТГФ. Образовавшуюся смесь подогревали до комнатной температуры и перемешивали в течение ночи. Затем растворитель выпаривали в вакууме и образовавшееся масло распределяли между водой и эфиром (по 300 мл каждого). Слой разделяли, водный слой экстрагировали эфиром (2 x 200 мл). Органические части объединяли, осушали над сульфатом натрия и сгущали до получения 78 г густого масла. Хроматография (силикагель, 75% этилацетата/25% гексана) давала 36,6 г (52%) диметил-(4-бензилокси)-бензилфосфоната в виде сухого остатка.
МС для C16H19O4P:
Рассчитано, %: C 62,74; H 6,25.
Найдено, %: C 62,96; H 6,23.
Раствор диметил-(4-бензилокси)-бензилфосфоната 19,4 г, 63 ммоль) в 100 мл 1% концентрированной соляной кислоты в этаноле обрабатывали 840 мг 5% Пд/У. Смесь гидрогенизировали при 40 psi в течение 30 мин. Затем реакционную смесь фильтровали через прокладку из целита и фильтрат выпаривали в вакууме, получали 13,6 г (100%) диметил-(4-гидрокси)-бензилфосфоната в виде белого сухого остатка.
Т.пл.: 126 - 129oC.
МС для C9H13O4P:
Рассчитано, %: C 50,01; H 6,06.
Найдено, %: C 50,21; H 6,09.
К раствору метилового эфира N-карбобензилокситранс-4-гидроксипролина (10,0 г, 35,8 ммоль) в 400 мл безводного ТГФ в атмосфере азота при 0oC добавляли трифенилфосфин (10,6 г, 39,4 ммоль) и диметил-(4-гидрокси)-бензилофосфонат (7,9 г, 37,8 ммоль). К этой смеси по каплям в течение 30 минутного периода добавляли диэтилазодикарбоксилат (6,3 мл, 39,4 ммоль). Реакционной смеси затем давали согреться до комнатной температуры и перемешивали ее 18 ч. Затем растворитель удаляли в вакууме и остаток хроматографировали (силикагель, 50-100% этилацетат/гексан), получая 13,3 г (75%) метилового эфира N-карбобензилокси-4-(цис)-(диметил-4-оксобензилфосфонат)- L-пролина в виде густого масла.
МС для C23H28NO8P:
Рассчитано, %: C 57,86; H 5,91; N 2,93.
Найдено, %: C 57,66; H 6,04; N 3,02.
Раствор метилового эфира N-карбобензилокси-4-(цис)-(диметил-4- оксобензилфосфонат)-L-пролина (6,6 г, 13,8 ммоль) в 100 мл 1% концентрированной соляной кислоты в этаноле обрабатывали 1 г 10% Пд/У. Смесь гидрогенизировали при 40 psi в течение 2 ч и затем пропускали через прокладку из цеолита, чтобы удалить катализатор. Фильтрат выпаривали до масла и затем распределяли между CHCl3 и насыщенным NaHCO3 (по 100 мл каждого). Слои разделяли и органический слой осушали (Na2SO4) и сгущали в вакууме, получая неочищенный эфир пролина со снятой защитой в виде бледно-желтого масла.
В отдельной колбе растворяли 2-(4-нитроимидазол)-октановую кислоту (3,7 г, 14,5 ммоль) в 25 мл безводного CH2Cl2. К этому раствору добавляли оксалилхлорид (1,7 мл, 18,8 ммоль) следом - 3 капли ДМФ. Когда прекращалось образование газа, растворитель удаляли в вакууме, получая кислый хлорид в виде янтарного масла, которое выпаривали из дополнительных 20 мл CH2Cl2. Этот кислый хлорид немедленно использовали в следующей реакции.
К раствору вышеуказанного эфира пролина в 20 мл безводного CH2Cl2 при 10oC добавляли N,N-диизопропилэтиламин (2,7 мл, 15,1 ммоль). Затем через отдельную воронку каплями вводили кислый хлорид в виде раствора в 10 мл CH2Cl2. Образовавшуюся смесь подогревали до комнатной температуры и перемешивали в течение 18 ч. После этого реакционную смесь распределяли между этилацетатом и водой (по 200 мл). Разделяли слои и водный экстрагировали этилацетатом (3 x 100 мл). Органические растворы объединяли и промывали соляным раствором, затем водой. После этого органическую часть осушали (сульфат натрия) и выпаривали в вакууме до получения метилового эфира 1-[1-оксо-2-(4-нитро-1H-имидазол-1-ил)-октил] -4-цис-[(4- диметилметиленфосфонат)-фенокси]-L-пролина в виде масла. Диастереоизомеры октаноамида разделяли хроматографией (силикагель, 1% метанол/этилацетат), получая 1,57 г (19%) изомера R, S, S и 1,225 г изомера S, S, S, и 1,12 г смеси изомеров.
Данные изомера R, S, S:
МС для C26H37N4O9P:
Рассчитано, %: C 53,79; H 6,42; N 9,65.
Найдено, %: C 53,26; H 6,58; N 9,18.
Данные изомера S, S, S:
МС для C26H37N4O9P:
Рассчитано, %: C 53,79; H 6,42; N 9,65.
Найдено, %: C 53,55; H 6,47; N 9,38.
К раствору октаноамида 4-нитроимидазола (4,0 г, 6,9 ммоль) в 50 мл абсолютного этанола добавляли 1,0 г 5% Пд/У. Смесь гидрогенизировали при 40 psi в течение 30 мин. Затем катализатор удаляли, пропуская смесь через прокладку из целита. Фильтрат выпаривали до янтарного масла, которое дважды азеотропировали из безводного ТГФ.
В отдельной колбе, в 5 мл безводного ТГФ растворяли сульфобензойный ангидрид (1,4 г, 7,6 ммоль) в атмосфере азота. К этому раствору прибавляли вышеуказанный аминоимидазол в виде раствора в 5 мл безводного ТГФ. После перемешивания в течение 30 мин раствор растирали с эфиром/гексанами до получения 4,70 г (93%) сульфокислоты в виде светло-желтого осадка, который собирали фильтрацией. Этот продукт, метиловый эфир 1-[1-оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил]октил]-4-цис-[(4- диметилметиленфосфонат)-фенокси] -L-пролина без дальнейшей очистки использовался в следующей реакции.
Т.пл.: 110oC (разрушается).
МС для C33H43N4O11PS:
Рассчитано, %: C 53,94; H 5,90; N 7,62.
Найдено, %: C 53,66; H 5,94; N 6,85.
К раствору вышеуказанного метилового эфира (4,70 г, 6,5 ммоль) в 25 мл безводного CH2Cl2 при 0oC добавляли каплями в течение 15 мин триметилсилилбромид (5,0 г, 32,4 ммоль). Образовавшуюся смесь подогревали до комнатной температуры и перемешивали в течение 1 ч. Затем растворитель удаляли в вакууме и осадок растворяли в 16 мл 2 н. NaOH. После перемешивания в течение 1 ч реакционную смесь подкисляли 5 н. HCl до pH 1,0. Водную часть экстрагировали 10% этанол-этилацетатом (3 x 50 мл). Органическую часть осушали (сульфатом натрия) и выпаривали до сухого остатка, который растворяли в минимальном количестве абсолютного этанола и растирали с эфиром/гексанами. Фильтрацией выделяли 2,79 г (61%) названной в заголовке сульфокислоты в виде светло-желтого осадка.
Т.пл.: 190oC (разрушается).
МС для C30H37N4O11PS:
Рассчитано, %: C 52,02; H 5,38; N 8,09.
Найдено, %: C 51,80; H 5,42; N 7,91.
Пример 92. 1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1- ил]октил]-4-цис[(4-N-метансульфоамидо)-фенокси]-L-пролин
К раствору метилового эфира N-Вос-транс-4-гидроксипролина (10,0 г, 41 ммоль) в 150 мл безводного ТГФ при 0oC в атмосфере азота добавляли трифенилфосфин (12,7 г, 48 ммоль) и 4-нитрофенол (6,7 г, 48 ммоль). К этой смеси каплями в течение 30 мин добавляли диэтилазодикарбоксилат (7,7 мл, 48 ммоль). Смесь подогревали до комнатной температуры. После перемешивания в течение 2 дней растворитель удаляли в вакууме и неочищенное масло обрабатывали последовательно толуолом и эфиром, порциями по 200 мл, чтобы удалить кристаллизацией оксид трифенилфосфина и диэтилазодигидразид. Затем получившееся масло хроматографировали (силикагель, 15-50% этилацетат/гексаны), получая метиловый эфир N-Вос-4-(цис)-(4-нитрофенокси)-L-пролина в виде светло-желтого масла.
МС для C17H22N2O7:
Рассчитано, %: C 55,73; H 6,05; N 7,65.
Найдено, %: C 55,94; H 6,09; N 7,59.
К раствору метилового эфира N-Вос-4-(цис)-(4-нитрофенокси)-L-пролина (9,0 г, 24,7 ммоль) в 100 мл этанола добавляли 1,5 г 10% Пд/У. Эту смесь гидрогенизировали в течение 3 ч при 40 psi. Затем катализатор удаляли, пропуская реакционную смесь через прокладку из целита. Выпаривание в вакууме давало масло, которое немедленно использовалось в следующей реакции.
Вышеуказанное масло растворяли в 50 мл безводного CH2Cl2 вместе с 11,5 мл (65,5 ммоль) N,N-диизопропилэтиламина. К этой смеси через отдельную воронку каплями прибавляли метансульфонилхлорид (6,4 г, 55 ммоль) в виде раствора в 10 мл CH2Cl2. После перемешивания в течение 2 ч реакционную смесь выливали в воду (200 мл). Водную часть экстрагировали этилацетатом (3 x 100 мл). Органическую часть осушали (сульфат натрия) и выпаривали в вакууме до масла. Хроматография (силикагель, 25% этилацетат/гексаны) давала 3,32 г (27%) метилового эфира N-Вос-4-(цис)-((4-N,N-бисметансульфонамидо)- фенокси)-L-пролина в виде бесцветного масла.
МС для C19H28N2O9S2:
Рассчитано, %: C 46,33; H 5,73; N 5,69.
Найдено, %: C 46,16; H 5,48; N 5,45.
К раствору метилового эфира N-Вос-4-(цис)-[(4-N,N-бисметансульфонамидо)- фенокси] -L-пролина (3,0 г, 6,1 ммоль) в 40 мл безводного CH2Cl2 при комнатной температуре добавляли трифторуксусную кислоту (1,5 мл, 18 ммоль). После размешивания в течение 3 ч растворитель удаляли в вакууме, получая масло, которое распределялось между насыщенным раствором NaHCO3 и этилацетатом (по 100 мл каждого). Органическую часть осушали (сульфатом натрия) и выпаривали, получая 2,27 г метилового эфира 4-(цис)-[(4-N,N-бисметансульфонамидо)-фенокси] -L- пролина в виде осадка, который немедленно использовался в следующей реакции.
2-(4-Нитроимидазол)-октановая кислота (1,7 г, 6,7 ммоль) превращалась в кислый хлорид и вводилась в реакцию с вышеуказанным эфиром пролина согласно ранее описанному способу. Хроматография (силикагель, 70 - 100% этилацетат/гексаны) неочищенной реакционной смеси давала 1,66 г метилового эфира (R)-1-[1-оксо-2-(4-нитро-1H-имидазол- 1-ил)октил] 4-(цис)-[(4-N,N-бисметансульфонамидо)фенокси]-L-пролина в виде белого сухого остатка.
Т.пл.: 106 - 109oC.
МС для C25H35N5O10S2:
Рассчитано, %: C 47,69; H 5,60; N 11,12.
Найдено, %: C 47,88; H 5,51; N 11,22.
К раствору метилового эфира (R)-1-[1-оксо-2-(4-нитро-1H-имидазол-1- ил)октил] -4-(цис)-[(4-N,N-бисметансульфонамидо)-фенокси]-L-пролина (1,26 г, 2,0 ммоль) в 50 мл абсолютного этанола добавляли 0,5 г 5% Пд/У. Смесь гидрогенизировали при 40 psi в течение 1,5 ч. Затем катализатор удаляли, пропуская смесь через прокладку из целита. Фильтрат выпаривали до янтарного масла, которое азеотропировали дважды из безводного ТГФ.
В отдельной колбе в атмосфере азота растворяли сульфобензойный ангидрид (0,40 г, 2,2 ммоль) в 5 мл безводного ТГФ. После перемешивания в течение 30 мин раствор растирали с эфиром/гексанами, получая 1,50 г (95%) метилового эфира (R)-1-[1-оксо-2-[4-(2- сульфобензоил)амино-1H-имидазол-1-ил]октил]-4-(цис)-[(4-N,N- бисметансульфонамидо)-фенокси]-L-пролина в виде светло-желтого осадка, который собирали фильтрацией. Это промежуточное соединение без дальнейшей очистки использовалось в следующей реакции.
Т.пл.: 165 - 170oC.
МС для C32H41N5O12S3•1,0 H2O:
Рассчитано, %: C 47,93; H 5,40; N 8,73.
Найдено, %: C 48,31; H 5,40; N 8,44.
Метиловый эфир (R)-1-[1-оксо-2-[4-(2-сульфобензоил)-амино-1H-имидазол- 1-ил] октил] -4-(цис)-[(4-N,N-бисметансульфонамидо)-фенокси]-L-пролина (0,40 г, 0,52 ммоль) растворяли в 1 н. растворе NaOH (3 мл) и ТГФ (2 мл) при комнатной температуре. Раствор перемешивали в течение ночи. Затем 1 н. HCl pH раствора доводили до 1,0. Образовавшийся осадок экстрагировали из водной среды при помощи 10% этанол/этилацетатной смеси. Осушение (сульфат натрия) и выпаривание в вакууме давало осадок, который растирали в метаноле/эфире, извлекая 0,27 г (75%) (R)-1-[1-оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил]-4-(цис)- ((4-N-метансульфонамидо)-фенокси)-L-пролина в виде осадка белого цвета.
Т.пл.: 195 - 198oC.
МС для C30H37N5O10S2:
Рассчитано, %: C 52,09; H 5,39; N 10,12.
Найдено, %: C 51,82; H 5,47; N 10,28.
Пример 93. (R)-1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1- ил] октил]-4-(цис)-((4-трифторметансульфонамидо)-фенокси)-L-пролин
(R)-1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1- ил] октил]-4-(цис)-((4-N-трифторметансульфонамидо)-фенокси)-L-пролин готовился таким же способом, как в примере 92.
Т.пл.: 145oC (разрушается).
МС для C30H33N5O10S2F3•1,0 NaCl:
Рассчитано, %: C 46,93; H 4,84; N 9,99.
Найдено, %: C 46,97; H 4,58; N 9,75.
Пример 94. (R)-1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1- ил] -октил]-4-(цис)-[(4-N,N-метил-метансульфонамидо)-фенокси)-L-пролин
К раствору 4-бензилоксианилина (21,7 г, 108 ммоль) в 100 мл безводного CH2Cl2 добавляли N,N-диизопропилэтиламин (31,0 мл, 42,5 ммоль). После этого смесь охлаждали до 10oC и обрабатывали метансульфонилхлоридом (18,2 мл, 234 ммоль). После перемешивания в течение 1 ч реакционную смесь распределяли между водой/этилацетатом. Образующийся при этом осадок собирали фильтрацией. Высушиванием в вакууме давало 25,7 г (68%) 4-бензилокси-N,N-бисметансульфонамидо-бензола в виде коричневого осадка.
Т.пл.: 212 - 215oC.
МС для C15H17NO5S2:
Рассчитано, %: C 50,69; H 4,82; N 3,94.
Найдено, %: C 50,87; H 4,85; N 3,91.
К раствору 4-бензилокси-N,N-бисметансульфонамидо бензола (25,0 г, 71,0 ммоль) в 300 мл ТГФ добавляли 1 н. NaOH (250 мл). Образовавшийся раствор перемешивали в течение 2 ч при 70oC. Охладив, смесь подкисляли 5 н. HCl до pH 1,0. Экстракция CHCl3 (5 x 250 мл) с последующим осушением (сульфатом натрия) и выпариванием в вакууме давали 15,05 г (51%) 4-бензилокси-N-метансульфонамидобензола в виде белого осадка.
Т.пл.: 155 - 158oC.
МС для C14H15NO3S:
Рассчитано, %: C 60,63; H 5,45; N 5,05.
Найдено, %: C 60,59; H 5,46; N 5,01.
К раствору вышеуказанного соединения (15,0 г, 55,0 ммоль) в 45 мл безводного ДМФ добавляли углекислый калий (15,3 г, 110 ммоль). После перемешивания при комнатной температуре в течение 18 ч образовывался осадок, который собирали сначала разведением реакционной смеси водой, потом вакуумной фильтрацией. Высушивание давало 15,7 г (97%) 4-бензилокси-N,N-метилметансульфонамидобензола в виде белого осадка.
Т.пл.: 175 - 178oC.
МС для C15H17NO3S:
Рассчитано, %: C 61,83; H 5,88; N 4,81.
Найдено, %: C 62,04; H 5,96; N 4,97.
К раствору 4-бензилокси-N, N-метилметансульфонамидобензола (14,0 г, 48 ммоль) в 100 мл этанола добавляли 4,0 г 5% Пд/У. Смесь гидрогенизировали при 40 psi в течение 2 ч. Затем ее пропускали через прокладку из целита и выпаривали фильтрат в вакууме, получая 9,93 г (100%) 4-N,N-метилметансульфонамидофенола в виде белого осадка.
Т.пл.: 117 - 120oC.
МС для C8H11NO3S:
Рассчитано, %: C 47,75; H 5,51; N 6,96.
Найдено, %: C 47,65; H 5,28; N 6,88.
Это вещество использовалось так же, как в примере 92, для того, чтобы приготовить соединение, названное в заголовке.
Пример 95. (R)-1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил]- октил]-4-(цис)-[(4-метилен-N-метансульфонамидо)-фенокси]-L-пролин
К раствору метилового эфира N-карбобензилокси-транс-4-гидрокси-L-пролина (10,0 г, 35,8 ммоль) в 200 мл безводного ТГФ в атмосфере азота добавляли трифенилфосфин (10,6 г, 39,4 ммоль) и 4-цианофенол (4,7 г, 39,4 ммоль). Этот раствор охлаждали до 0oC и затем обрабатывали диэтилазодикарбоксилатом (6,3 мл, 39,4 ммоль), добавляя его каплями в течение 30 мин. Реакционную смесь согревали до комнатной температуры и размешивали в течение 2 дн. Растворитель удаляли в вакууме и остаток хроматографировали (силикагель, 30% этилацетат/гексаны), чтобы получить 12,1 г (89%) метилового эфира N,N-карбобензилокси-4-(цис)-(4-цианофенокси)-L-пролина в виде бесцветного масла.
МС для C21H20N2O5:
Рассчитано, %: C 66,31; H 5,30; N 7,36.
Найдено, %: C 66,10; H 5,34; N 7,50.
К раствору метилового эфира N-карбобензилокси-4-(цис)-(4-цианофенокси)-L-пролина (3,8 г, 10 ммоль) в 75 мл метанола добавляли COCl2 (2,6 г, 20 ммоль). Этот раствор охлаждали до 0oC и затем обрабатывали NaBH4 (3,8 г, 100 ммоль), добавляя его маленькими порциями. После перемешивания в течение 2 ч добавляли 50 мл 3 н. HCl. После перемешивания этого раствора в течение 15 мин реакцию распределяли между водой и эфиром (200 мл каждого). Разделяли слои и водную фазу экстрагировали эфиром (2 x 100 мл). Водную фазу затем ощелачивали концентрированным раствором NH4OH. Экстракция этилацетатом (3 x 100 мл) с последующим осушением (сульфатом натрия) и выпариванием в вакууме дает 3,50 г (90%) метилового эфира N-карбобензилокси-4-(цис)-(4-аминометил)-фенокси-L-пролина в виде масла. Это вещество без дальнейшей очистки используется в следующей реакции.
МС для C21H24N2O5:
Рассчитано, %: C 65,61; H 6,29; N 7,29.
Найдено, %: C 65,87; H 6,04; N 7,03.
К раствору метилового эфира N-карбобензилокси-4-(цис)-(4-аминометил)- фенокси-L-пролина (0,90 г, 2,34 ммоль) в 15 мл безводного CH2Cl2 добавляли N, N-диизопропиламин (0,06 мл, 3,4 ммоль). Этот раствор охлаждали до 0oC и затем обрабатывали метансульфонилхлоридом (0,22 мл, 2,8 ммоль) в виде раствора в 5 мл CH2Cl2. После перемешивания в течение 1,5 ч реакцию распределяли между этилацетатом/водой (50 мл каждого). Слои разделяли и водный экстрагировали этилацетатом (2 x 50 мл). Органическую часть осушали (сульфатом натрия) и выпаривали в вакууме, чтобы получить неочищенное масло. Хроматография (силикагель, 50/50 этилацетат/гексаны) давала 0,74 г (70%) метилового эфира N-карбобензилокси-4-(цис)-[(4-(метилен-N-метансульфонамидо))-фенокси]-L- пролина в виде бесцветного масла. МС.
К раствору (-)-синконидина (48,0 г, 163 ммоль) в 880 мл дистиллированной воды при комнатной температуре добавляли 2-(4-нитро-1H-имидазол-1-ил)-октановую кислоту (83,0 г, 326 ммоль) в виде раствора в 440 мл этанола. К этой смеси добавляли триэтиламин (11,7 мл). Затем смесь нагревали до 80oC, и поддерживали pH между 6,9 и 7,1 добавлением триэтиламина каплями (5-10 мл). После стабилизации pH на уровне 7,01 раствору давали остыть до комнатной температуры и оставляли стоять на ночь, после чего происходила кристаллизация синконидиновой соли (R)-2-(4-нитро-1H-имидазол-1-ил)-октановой кислоты. Кристаллическую соль собирали фильтрацией. Затем соль суспендировали в смеси этилацетат/вода (по 200 мл каждого). К этой суспензии добавляли 1 н. соляную кислоту (750 мл). Разделяли слои и водный экстрагировали этилацетатом (2 x 500 мл). Органические части объединяли, осушали (сульфатом натрия) и выпаривали в вакууме, чтобы получить 29,9 г (72%) (R)-2-(4-нитро-1H-имидазол-1-ил)-октановой кислоты в виде беловатого осадка.
Т.пл.: 116 - 118oC.
МС для C11H17N3O4:
Рассчитано, %: C 51,76; H 6,71; N 16,46.
Найдено, %: C 51,89; H 6,76; N 16,20.
[a]D -29,9 (c = 1,00, этанол).
Избыток энантиомера устанавливается в 96% превращением кислоты в ее метиловый эфир (диазометан) с последующей ЖХВР с хиральной колонкой.
(R)-2-(4-Нитро-1H-имидазол-1-ил)-октановую кислоту (16,0 г, 63,0 ммоль) растворяли в 1 л безводного метанола. К этому раствору добавляли pTs OH (300 мг). Затем реакционную смесь нагревали до кипения в течение 16 ч. Охладив, растворитель удаляли в вакууме, получая масло, которое растворяли в 300 мл этилацетата. Раствор промывали насыщенным раствором NaHCO3 (2 x 250 мл). Затем органическую часть осушали (сульфатом натрия) и выпаривали в вакууме, получая 13,2 г (78%) (R)-метил-2-(4-нитро-1H-имидазол-1-ил)-октаноата в виде янтарного масла.
МС для C12H19N3O4:
Рассчитано, %: C 53,32; H 7,11; N 15,60.
Найдено, %: C 53,23; H 7,05; N 15,39.
(R)-Метил-2-(4-нитро-1H-имидазол-1-ил)-октаноат (13,0 г, 45,7 ммоль) растворяли в 150 мл абсолютного этанола. К этому раствору добавляли 2,0 г 10% Пд/У. Смесь гидрогенизировали при 40 psi в течение 2 ч. Затем удаляли катализатор, пропуская реакцию через целитовую прокладку. Потом фильтрат сгущали до масла, которое дважды выпаривалось из безводного ТГФ (100 мл). Неочищенный продукт растворяли затем в 100 мл безводного ТГФ и обрабатывали KOAc (4,44 г) и K2CO3 (3,12 г). К этой смеси добавляли сульфобензойный ангидрид (8,83 г, 47,7 ммоль). Реакцию перемешивали в течение 4 ч, после чего обрабатывался осадок. Смесь разбавляли ТГФ (100 мл) и осадок собирали фильтрацией. Высушивание в вакууме давало 22,5 г неочищенной калиевой соли (R)-метил[(2-сульфобензоил)амино-1H-имидазол-1-ил] октаноата. Это вещество переносили в следующую реакцию без дальнейшей очистки.
Калиевую соль (22,5 г) растворяли в смеси из 200 мл воды и 100 мл этанола. К этому раствору добавили 1 н. NaOH (53 мл). Реакцию смешивали в течение 3 ч. Затем этанол удаляли в вакууме и водный раствор подкисляли до pH 1,5 5 н. HCl. Этот раствор экстрагировали 10 % этанолом/этилацетатом (3 х 200 мл). Органическую часть осушали (сульфатом натрия) и выпаривали в вакууме, получали 8,65 г (46 % за две стадии) (R)-[(2-сульфобензоил)амино-1H-имидазол-1-ил]-октановой кислоты в виде белого остатка.
МС для C18H23N3O6S:
Рассчитано,%: C 52,80; H 5,66; N 10,26.
Найдено,%: C 52,53; H 5,59; N 10,27.
К раствору метилового эфира N-карбобензилокси-4-(цис)-[(4-метилен-N-метансульфонамидо))-фенокси]-L- пролина (1,5 г, 3,25 ммоль) в 50 мл абсолютного этанола добавляли 0,5 г 5 % Пд/У. Смесь гидрогенизировали при 40 psi в течение 1,5 ч. Затем реакционную смесь пропускали через прокладку из целита и фильтрат выпаривали в вакууме, получая 1,7 г метилового эфира 4-(цис)-(метилен-N-метансульфонамидо)-фенокси-L-пролина в виде масла. МС. Это вещество немедленно использовалось в следующей реакции.
К раствору вышеуказанного амино в 10 мл безводного ДМФ добавляли (R)-[2-сульфобензоил)амино-1H- имидазол-1-ил] -октановую кислоту (1,00 г, 2,45 ммоль) и гидроксибензотриазол (0,37 г, 2,77 ммоль). Эту смесь охлаждали до 0oC и затем обрабатывали дициклогексилкарбодиимидом (0,56 г, 2,70 ммоль). Образовавшийся раствор согревали до комнатной температуры и перемешивали в течение 48 ч. После удаления фильтрацией дициклогексилмочевины фильтрат разбавляли 100 мл этилацетата и промывали несколько раз водой. Органическую часть осушали (сульфатом натрия) и сгущали в вакууме до масла. Хроматография (силикагель, 5 % метанол/CHCl3) давала 0,84 г (34 %) метилового эфира (R)-1-[1-оксо-2-[4-(2-сульфобензоил)-амино-1H-имидазол-1-ил] октил] -4- (цис)-[(4-метилен-N-метансульфонамидо)-фенокси]-L-пролина в виде белого осадка.
Т.пл.: 150oC (разрушается).
Рассчитано,%: C 53,40; H 5,74; N 9,73.
Найдено %: C 53,66: H 5,97: N 9,50.
Метиловый эфир (R)-1-[1-оксо-2-[4-(2-сульфобензоил)-амино-1H-имидазол-1-ил] октил] -4- (цис)-[(4-метилен-N-метансульфонамидо)-фенокси] -L-пролина (0,37 г, 0,52 ммоль) растворяли в смеси из 1 н. NaOH (3,0 мл) и ТГФ (7 мл). Этот раствор перемешивали в течение 1 ч. Затем ТГФ удаляли в вакууме и водную часть подкисляли pH 1,0, пользуясь 1 н. HCl. Экстракция 5 % этанол/этилацетат (2 х) с последующим осушением органической части и выпариванием давали сухой остаток. Растиранием в этаноле/этилацетате/эфире получали 0,26 г (74 %) (R)-1-[1-оксо-2-[4-(2-сульфобензоил)-амино-1H-имидазол-1-ил] -октил] -4- (цис)-[(4-метилен-N-метансульфонамидо)-фенокси]-L-пролина в виде белого осадка.
Т.пл.: 172 - 176oC.
МС для C31H39N5O10S2:
Рассчитано,%: C 52,75; H 5,57; N 9,92.
Найдено,%: C 52,54; H 5,53; N 10,15.
Как обсуждалось ранее, соединения формулы I являются сильными эффективными антагонистами ангиотензина П. Способность репрезентативных соединений формулы I блокировать связывание рецепторов ангиотензина П устанавливалась по состоянию клубочковой зоны надпочечников. Способность противодействовать вазоконстрикции, вызванной ангиотензином, оценивалась испытанием на аорте кролика.
Тест-система клубочковой зоны надпочечника
Фиксация ангиотензина П, меченного I125, к мембранам надпочечника обычно проводилась в 96-гнездовых фильтрационных планшетах. Мембраны надпочечника готовили из частей, прилежащих к капсуле (примыкающих к клубочковой зоне) надпочечников крыс дифференциальным центрифугированием. Капсулы гомогенизировали в растворе, содержащем сахарозу (250 мМ), хлористый магний (1 мМ) и трис (5 мМ), pH 7,5 при 4oC, пользуясь политроном на осаждение 5 за 20 с. Гомогенат осторожно перемешивали в течение 15 мин при 40oC и затем центрифугировали 10 мин при 1000 х g, 4oC. Супернатант центрифугировали 30 мин при 30 000 х g, 4oC и образовавшийся осадок ресуспендировали в 50 мМ трисбуфера. Препараты мембраны хранили разделением на части при -70oC до употребления. Фиксацию меченного I125 ангиотензина П к мембранам надпочечника производили при комнатной температуре в течение 90 мин в 96-гнездных планшетах, содержащих гидрофильную оболочку из поливинилиденфторида (0,45 мкм, millipore- gvmultiscreen). В каждом гнезде содержалось по 250 мкл инкубационной среды, включающей (итоговая концентрация): трис (50 мМ), хлористый натрий (120 мМ), хлористый магний (5 мМ), дитиотриетол (1 мМ), бычий сывороточный альбумин (0,05 %), I125-ангиотензина П (0,1 нМ) и белок мембран надпочечника (8 - 15 мкг). Антагонисты вводились в концентрациях от 10 до 100 мкМ. Неспецифическое связывание измеряли в присутствии 0,1 мкМ Sar1, 11exангиотензина П.
Связывание заканчивалось вакуум-фильтрацией. Комплекс рецептор-лиганд, захваченный фильтратами, трижды промывался 300 мкл охлажденного на льду промывающего раствора (трис, 50 мМ; хлористый натрий, 120 мМ; хлористый магний, 5 мМ; дитиотриетол 1 мМ). Диски фильтров сушили, выбивали пробойником и подсчитывали в гамма-камере с эффективностью счета 52 %. Специфическое связывание представляло 96 % общей фиксации (приблизительно 150 фемтомоль ангиотензина П/мг белка). Молярная концентрация (IC50) ингибитора, которая вытесняет 50 % связанного I125-ангиотензина П, для каждого соединения подсчитывалась по логической модели с 4 параметрами (NonLin SAS Institute). Данные выражались в виде K1, подсчитанного по уравнению Ченга-Прусоффа (см. Cheng et al., Biochem. Pharmacol., 22 : 3099, 1973).
Тест-система аорты кролика
Новозеландских белых кроликов (Hazelton, 2-3 кг) забивали смещением шейных позвонков. Удаляли грудной отдел аорты и освобождали его от лишнего жира и соединительной ткани. Кольца ткани (шириной 3 мм) закрепляли в ванночках для культуры ткани емкостью 10 мл между 2 крюками I-образной формы из нержавеющей стали. Нижний крючок прикреплялся к неподвижному стержню. Верхний крючок прикреплялся к датчику силы смещения (модель Grass FT. 03). Ванночки держали при 37oC, аэрирования 95 % O2/5 % CO2 и наполняли физиологическим раствором следующего состава (мМ): NaCl 117; глюкоза 5,5; NaH2PO4 1,0; MgSO4 0,7; KCl 5,2; CaCl2 1,8; NaHCO3 26 и фентоламин HCl 0,003.
Кольца уравновешивали в течение 1 ч с напряжением в 2 г. Во время уравновешивания ткани омывались сверху каждые 15 мин. Затем на кольца воздействовали 10-8 М ангиотензина П (АП) и ожидали, пока они придут в состояние постоянного сокращения. Затем вычерчивали кривую ответа на кумулятивную концентрацию АП (от 10-10 до 10-7 М). После окончания построения кривой ответа на концентрацию АП ткани промывали каждые 2 мин до тех пор, пока натяжение не возвращалось к исходному уровню, затем каждые 15 мин в течение 30 мин. Соединения добавляли в объем 10 мкл диметилсульфоксида и выжидали 30 мин, прежде чем повторно получали кривую ответа на концентрацию АП. Сокращения в ответ на АП выражались в процентах к максимальному сокращению, полученному на контрольной кривой (первая кривая ответа на концентрацию АП). EC50 (концентрация, вызывающая сокращение тканей до 1/2 максимальной в контроле) для каждой кривой подсчитывались по логической модели с 4 параметрами (NonLin, SAS Instititute). Данные активности для каждого испытанного соединения выражались в виде pA2 (определяемого как log Kb, где Kb = [молярная концентрация антагониста]/[(EC50 АП с антагонистом/EC50 без антагониста)-1] ).
При помощи описанной методики репрезентативные соединения настоящего изобретения были оценены и было установлено, что они проявляют активность, измеряемую по pA2 не менее 4,1 в тест-системе аорты кролика, тем самым демонстрируя и подтверждая пригодность соединений изобретения в качестве эффективных антагонистов ангиотензина П (см. табл. 2).
Термин "фармацевтически эффективное количество" здесь используется для обозначения количества соединения изобретения, которое способно блокировать рецепторы ангиотензина П у млекопитающих. Конкретная доза соединения, введенного в соответствии с этим изобретением, будет, конечно, определяться конкретными обстоятельствами случая, включая то, какое соединение введено, способ введения, состояние, по поводу которого проводится лечение, и тому подобные соображения. Соединения могут вводиться различными путями, включая пероральный, ректальный, чрескожный, подкожный, внутривенный, внутримышечный или интранзальный. Обычная суточная доза должна содержать от 0,01 до 20 мг/кг активного соединения этого изобретения. Предпочтительные суточные дозы должны быть от 0,05 до 10 мг/кг, идеально от 0,1 до 5 мг/кг. Термин "лечение" используется здесь для описания ведения и ухода с целью борьбы с заболеванием, состоянием или нарушениями у больного.
Термин "лечение" включает введение соединения настоящего изобретения, чтобы предупредить появление симптомов, облегчить симптоматику или устранить заболевание, болезненное состояние или нарушение.
Термин "улучшение умственной деятельности" употребляется здесь для обозначения облегчения запоминания и обучения у больных, которые нуждаются в таком лечении. Примеры включают больных, страдающих нарушениями умственной деятельности, подобными возрастным нарушениям психики и болезни Альцгеймера.
Соединения формулы I предпочтительно приводить в препаративную форму до введения. Поэтому еще одним воплощением настоящего изобретения является создание фармацевтической препаративной формы, содержащей соединение формулы I и один или несколько фармацевтически приемлемых носителей, разбавителей или наполнителей.
Настоящие фармацевтические препаративные формы готовятся по известным технологиям с использованием широко известных и доступных ингредиентов. При изготовлении композиции настоящего изобретения активный ингредиент обычно должен смешиваться с носителем, или должен быть разбавлен носителем, или включен в носитель, который может быть в виде капсулы, пакета, облатки или другого вместилища. Когда носитель служит разбавителем, он может быть твердым, полужидким или жидким веществом, которое действует как средство доставки, наполнитель или среда для активного ингредиента. Так, композиции могут быть в форме таблеток, пилюль, порошков, лепешек, пакетов, крахмальных облаток, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (в виде твердой или жидкой среды), мазей, содержащих, например, до 10% по массе активного соединения, мягких и твердых желатиновых капсул, суппозиториев, стерильных растворов для инъекций и стерильно расфасованных порошков.
Некоторые примеры подходящих носителей, наполнителей и разбавителей включают лактозу, декстрозу, сахарозу, сорбитол, маннитол, крахмалы, гумми акации, фосфат кальция, альгинаты, трагакант, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, водный сироп, метилцеллюлозу, метил- и пропилгидроксибензоаты, тальк, стеарат магния и минеральное масло. Препаративные формы дополнительно могут включать смазывающие вещества, увлажняющие вещества, вещества эмульгирующие и суспендирующие, вещества консервирующие, подслащающие или ароматизирующие вещества. Композиции изобретения предпочтительно создавать в препаративной форме с унифицированной дозировкой, содержащей каждая от 5 до 500 мг, чаще от 25 до 300 мг активного ингредиента. Термин "формула унифицированной дозировки" относится к физически отдельным единицам, пригодным в качестве единичных доз для людей и других млекопитающих, каждой единице, содержащей определенное количество активного вещества, рассчитанное для достижения желаемого терапевтического эффекта, в сочетании с подходящим фармацевтическим носителем.
Следующие примеры препаративных форм только иллюстрируют изобретение, а не предназначены каким-либо образом ограничить рамки изобретения.
Препаративная форма 1
Твердые желатиновые капсулы готовятся с использованием следующих ингредиентов, (мг/капсула):
1-[1-Оксо-2-[4-(2-сульфобензоил)-амино-1H-имидазол-1-ил] октил] -4-цис- (2-нафтокси)-L-пролин - 250
Высушенный крахмал - 200
Стеарат магния - 10
Всего - 460
Высушенные ингредиенты смешиваются и помещаются в твердые капсулы в количестве 460 мг.
Препаративная форма 2
Таблетка, приготовленная из нижеперечисленных ингредиентов (мг/капсула):
1-[1-Оксо-2-[4-(2-сульфобензоил)-амино-1H-имидазол-1-ил] октил]-4-цис-[(4-метиленфосфоновая кислота)-фенокси]-L-пролин - 250
Микрокристаллическая целлюлоза - 400
Диоксид кремния, плавленный - 10
Стеариновая кислота - 5
Всего - 665
Компоненты смешиваются и прессуются в виде таблеток весом по 665 мг каждая.
Препаративная форма 3
Аэрозольный раствор готовится с содержанием следующих компонентов (мг/капсула):
1-[1-Оксо-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил] -4-цис-(4-третбутилоксифенокси)-L-пролин - 0,25
Этанол - 29,75
Газ-вытеснитель 22 (хлордифторметан) - 70,00
Всего - 100,00
Активное соединение смешивается с этанолом. Смесь добавляется к порции газа-вытеснителя 22, охлажденного до -30oC, и переносится в наполняющее устройство. Необходимое количество затем заправляется в контейнер из нержавеющей стали и разбавляется остатком газа-вытеснителя. Потом к контейнеру прикрепляется клапан.
Препаративная форма 4
Таблетки, содержащие каждая по 60 мг активного ингредиента, делаются следующим образом (мг/капсула):
1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил] -4-цис-(4-метил-сульфонилфенокси)-L-пролин - 60
Крахмал - 45
Микрокристаллическая целлюлоза - 35
Поливинилпирролидон (в виде 10% раствора в воде) - 4
Карбоксиметил натрия крахмал - 4,5
Стеарат магния - 0,5
Тальк - 1,0
Всего - 150
Активный ингредиент, крахмал и целлюлозу пропускают через сито с ячейками N 45 США и тщательно смешивают. С образовавшимися порошками смешивают раствор поливинилпирролидона и пропускают через сито с ячейками N 14 США. Образованные таким образом гранулы сушат при 50oC и пропускают через сито с ячейками N 18 США. Натриевый карбоксиметиловый крахмал, стеарат магния и тальк, предварительно пропущенные через сито с ячейками N 60 США, добавляются затем к гранулам, которые после смешивания прессуются таблетировочной машиной, производящей таблетки, весящие по 150 мг каждая.
Препаративная форма 5
Капсулы, содержащие по 80 мг медикамента каждая, делаются следующим образом (мг/капсула):
1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил] -4-цис(5-бензофуранокси)-L-пролин - 80
Крахмал - 59
Микрокристаллическая целлюлоза - 59
Стеарат магния - 2
Всего - 200
Активный ингредиент, целлюлозу, крахмал и стеарат магния смешивают, пропускают через сито с ячейками N 45 США и наполняют твердые желатиновые капсулы массой по 200 мг.
Препаративная форма 6
Суппозитории, содержащие по 225 мг активного ингредиента каждый, можно изготовить следующим образом (мг/капсула):
1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил]-4-цис-(5-бензотиофенокси)-L-пролин - 225
Глицериды насыщенной жирной кислоты - 2,000
Всего - 2,225
Активный ингредиент пропускают через сито с ячейками N 60 США и суспендируют в глицеридах насыщенной жирной кислоты, предварительно расплавленных при минимуме необходимого нагревания. Затем смесь выливают в форму для суппозиториев с номинальной емкостью в 2 г и дают застыть.
Препаративная форма 7
Суспензии, содержащие по 50 мг медикамента на 5 мл каждая, готовятся следующим образом (мг/капсула):
1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил] - 4-цис-(4-карбоксиметилфенокси)-L-пролин - 50
Натриевая карбоксиметиловая целлюлоза - 50
Сироп - 1,25
Раствор бензойной кислоты - 0,10 мл
Ароматизатор
Краситель
Очищенная вода до общего объема - 5
Медикамент пропускают через сито с ячейками N 45 США и смешивают с натриевой карбоксиметилцеллюлозой и сиропом до образования однородной пасты. Раствор бензойной кислоты, ароматизатор и краситель разводят водой и добавляют, помешивая, к пасте. Затем водой доводят до требуемого объема.
Препаративная форма 8
Препаративная форма для внутреннего введения готовится следующим образом (мг/капсула):
1-[1-Оксо-2-[4-(2-сульфобензоил)амино-1H-имидазол-1-ил] октил] 4-цис- (4-гидроксифенокси)-L-пролин - 250
Изотонический солевой раствор - 1000
Раствор вышеописанных ингредиентов вводится внутривенно со скоростью 1 мл в минуту лицу, нуждающемуся в лечении.
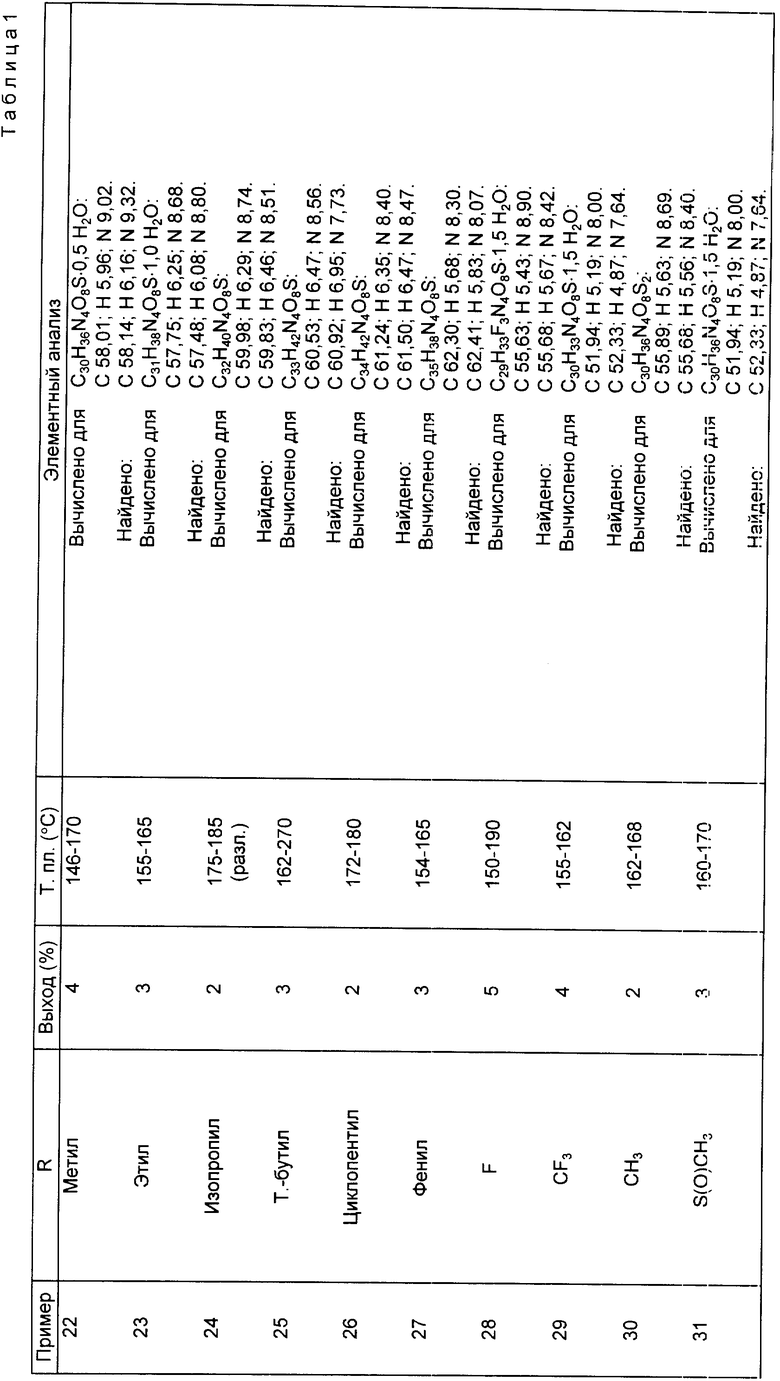
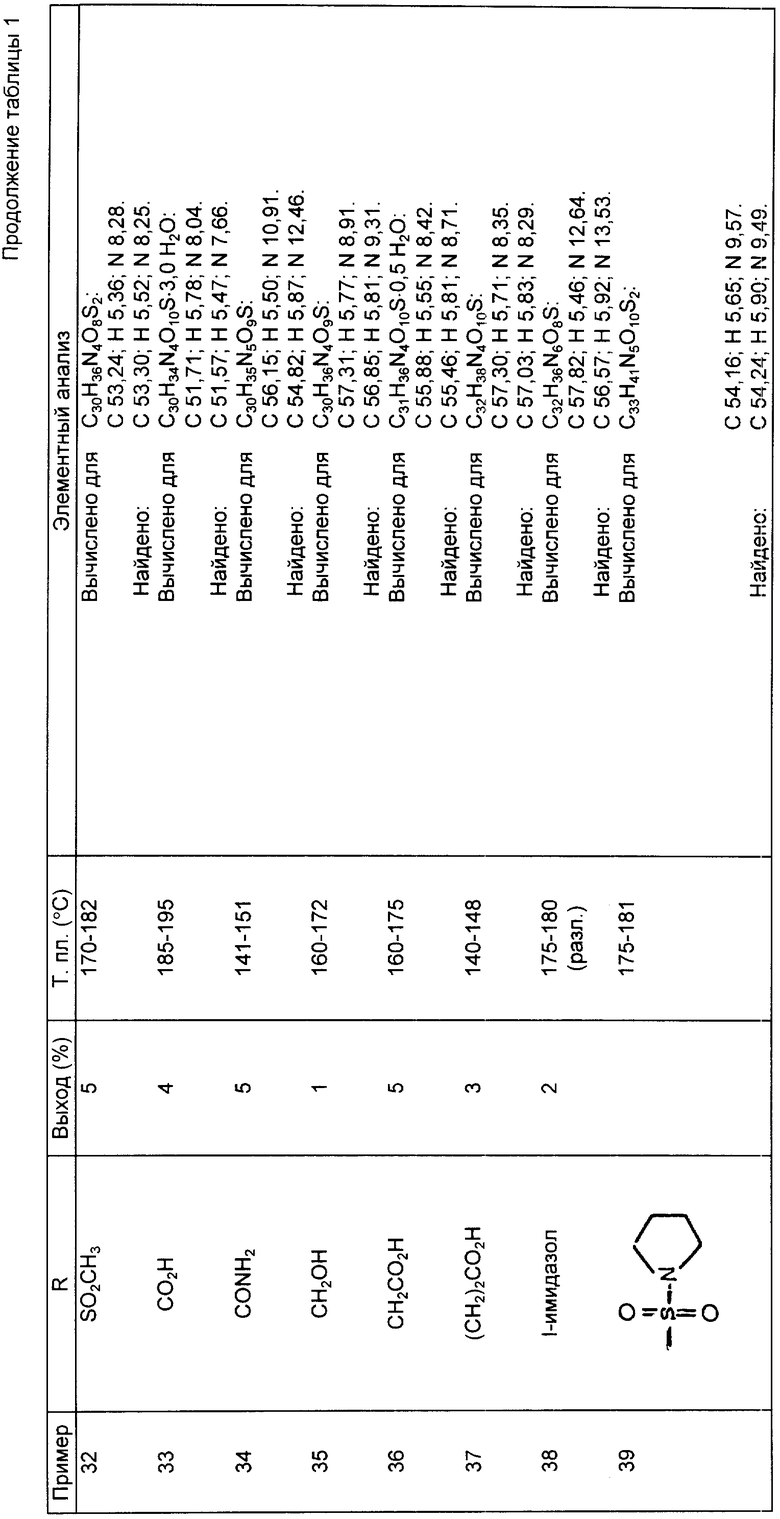

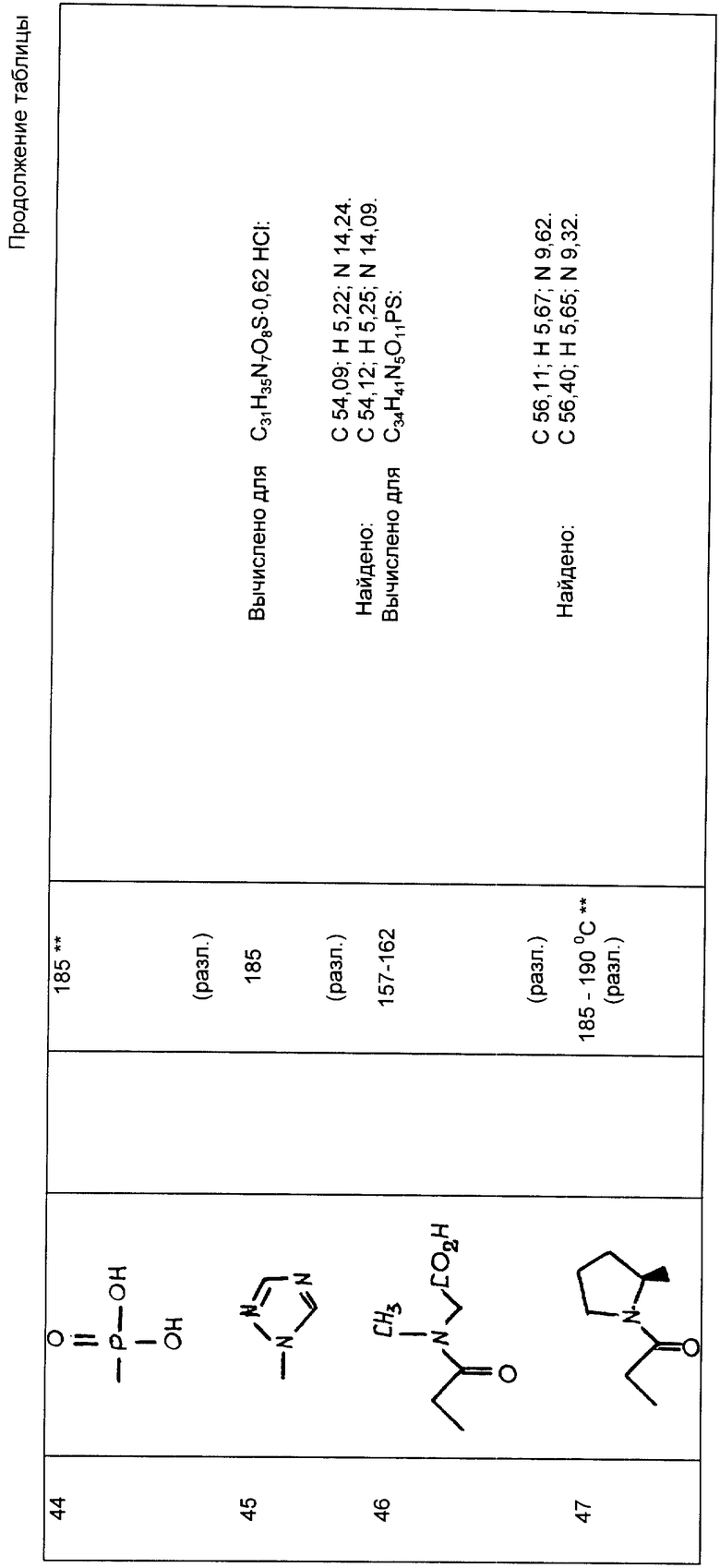
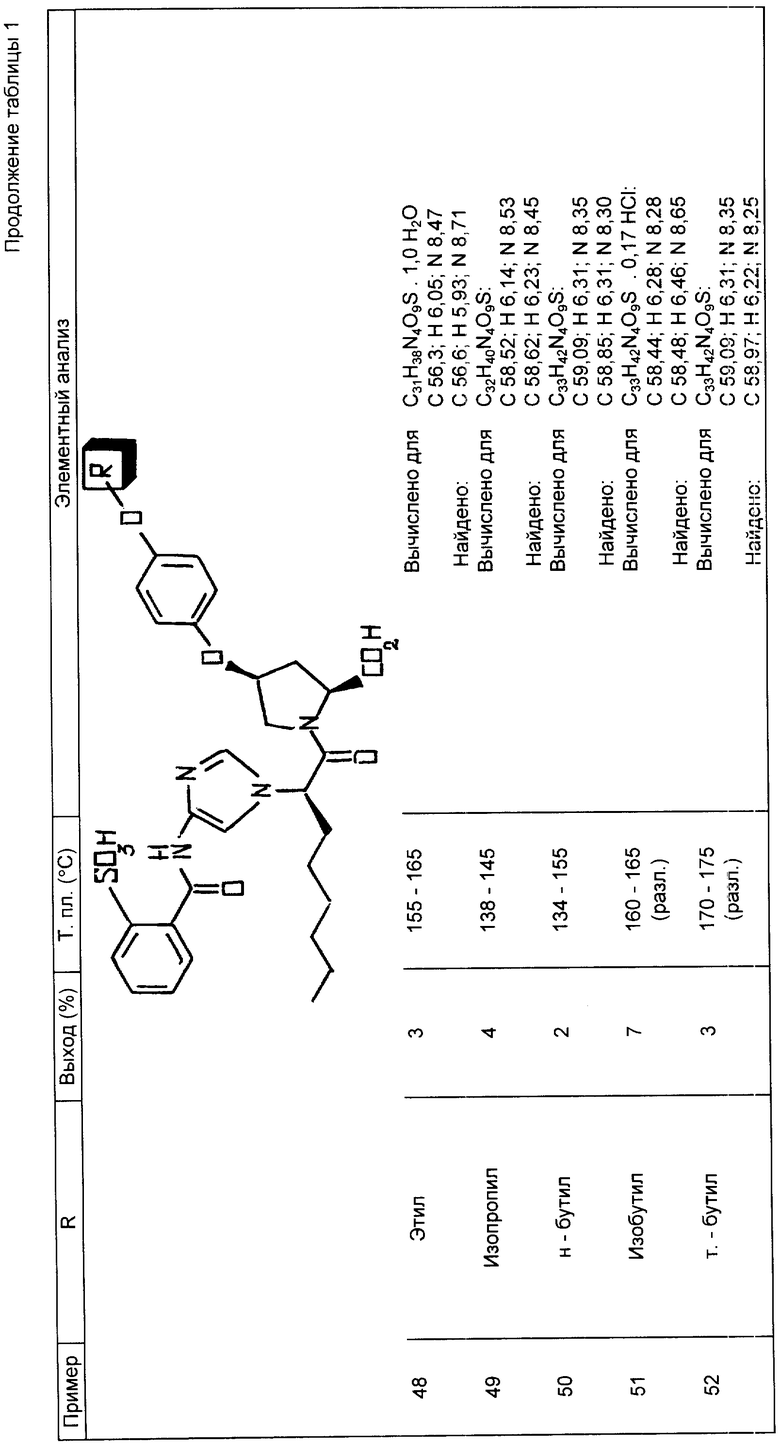
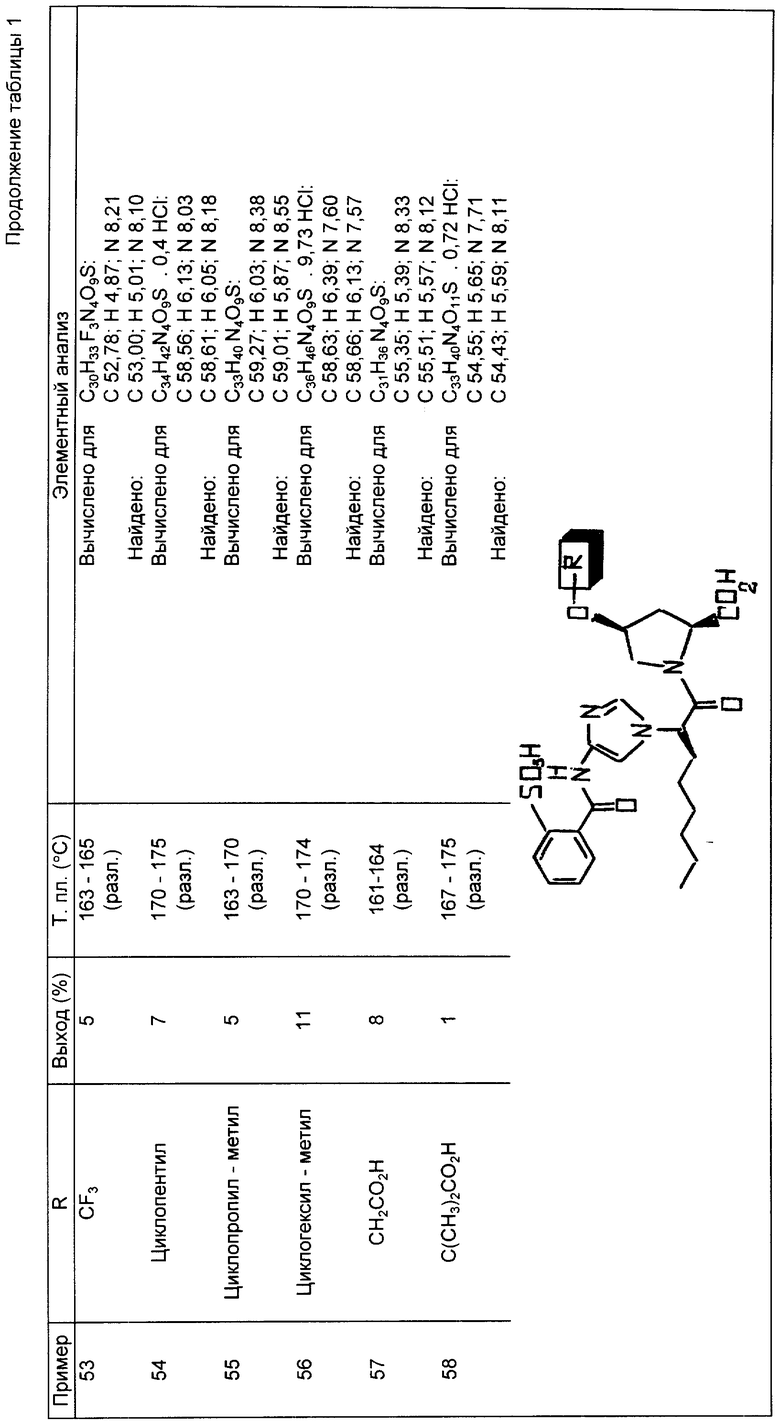
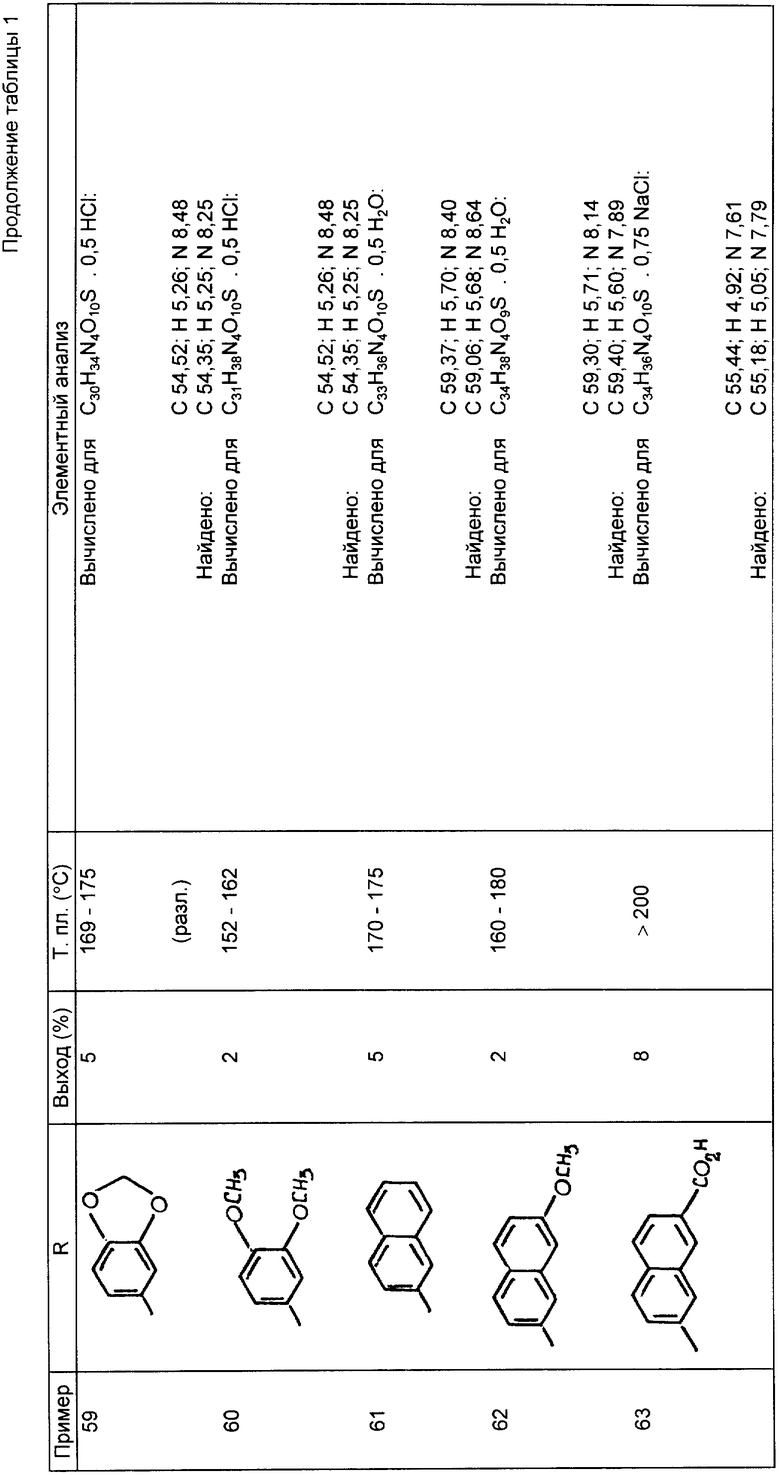
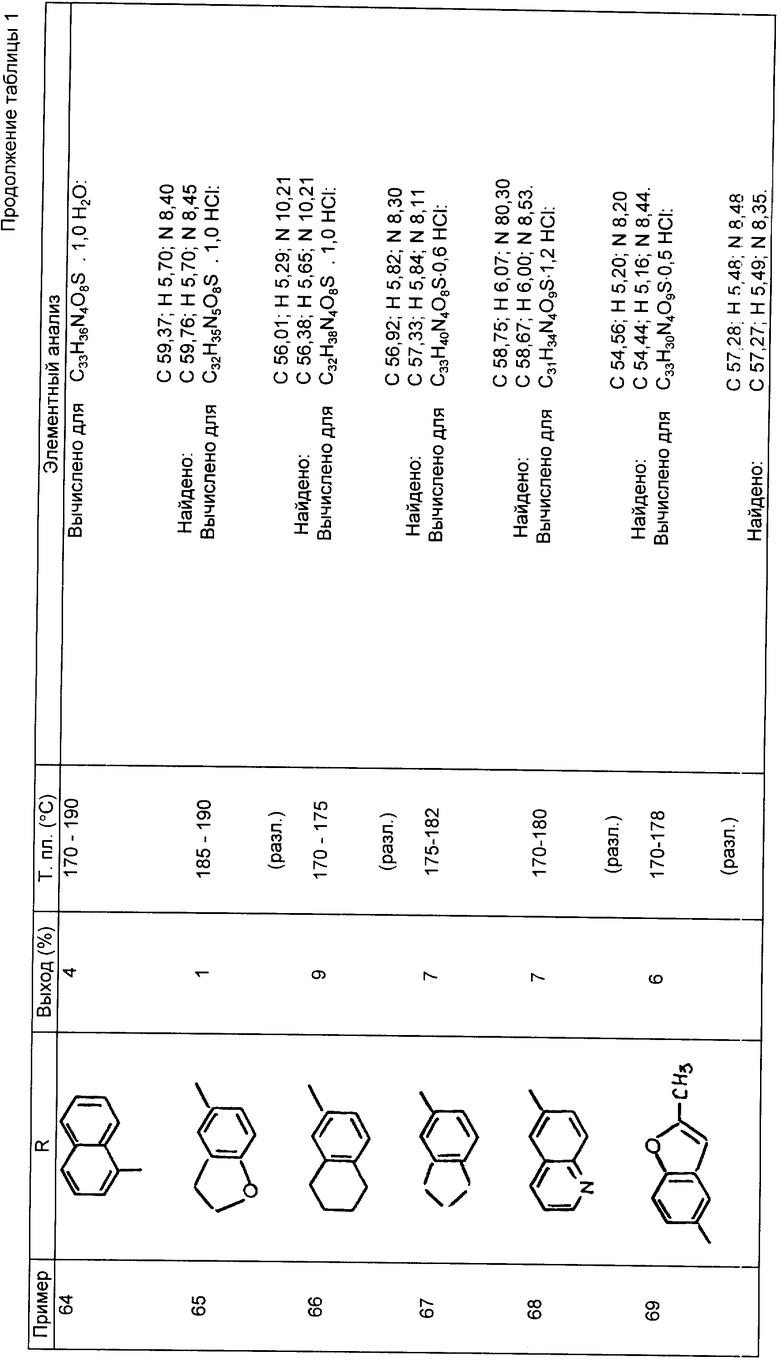
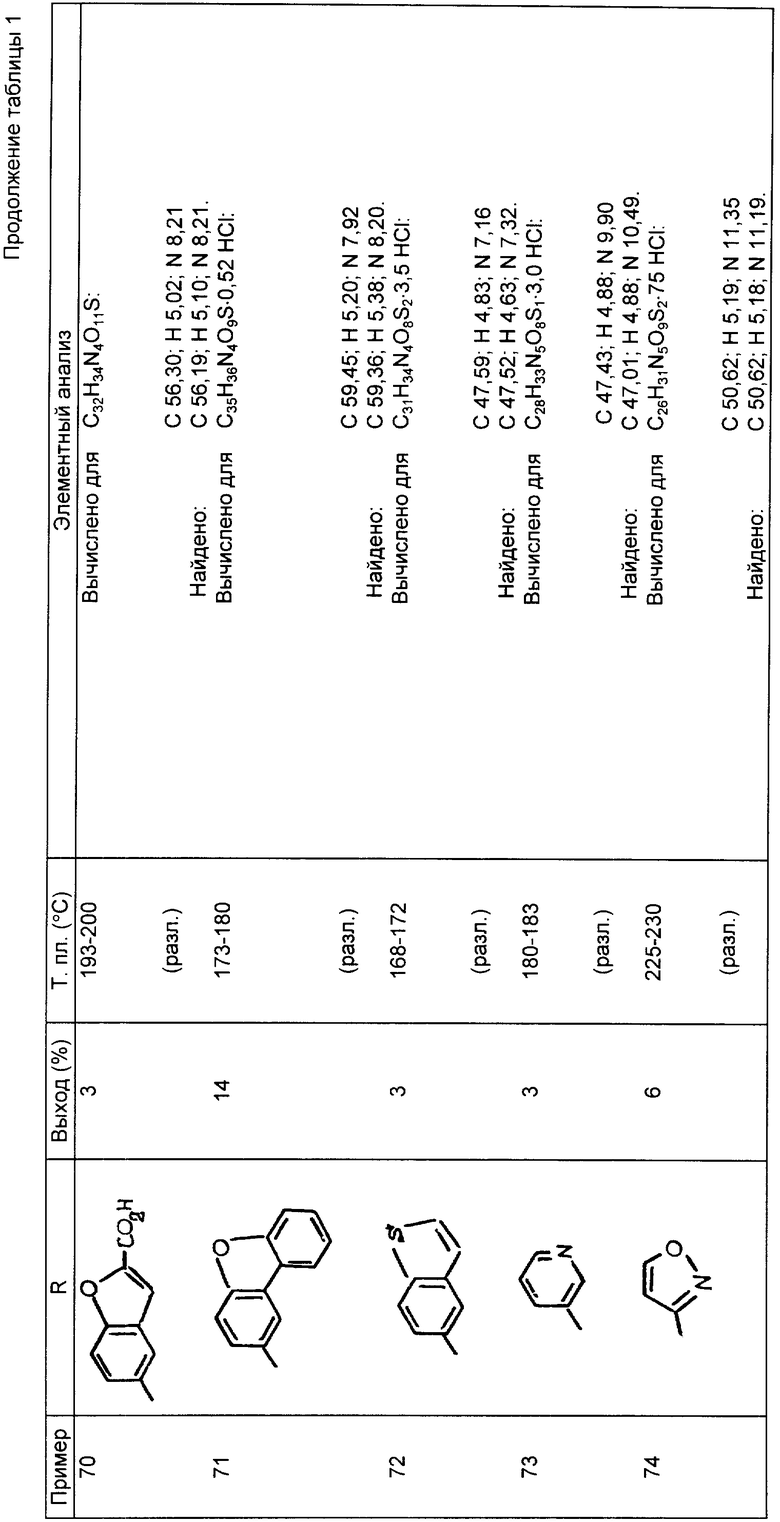

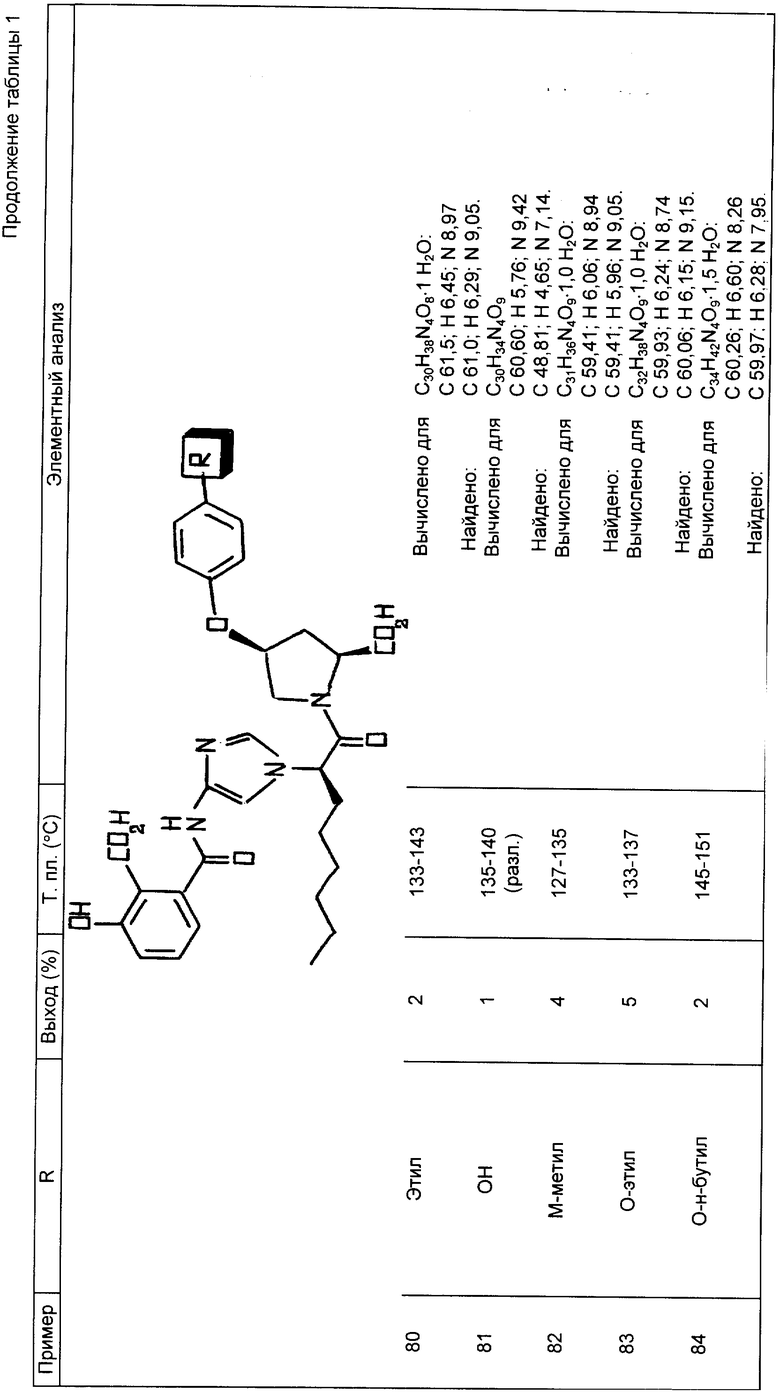
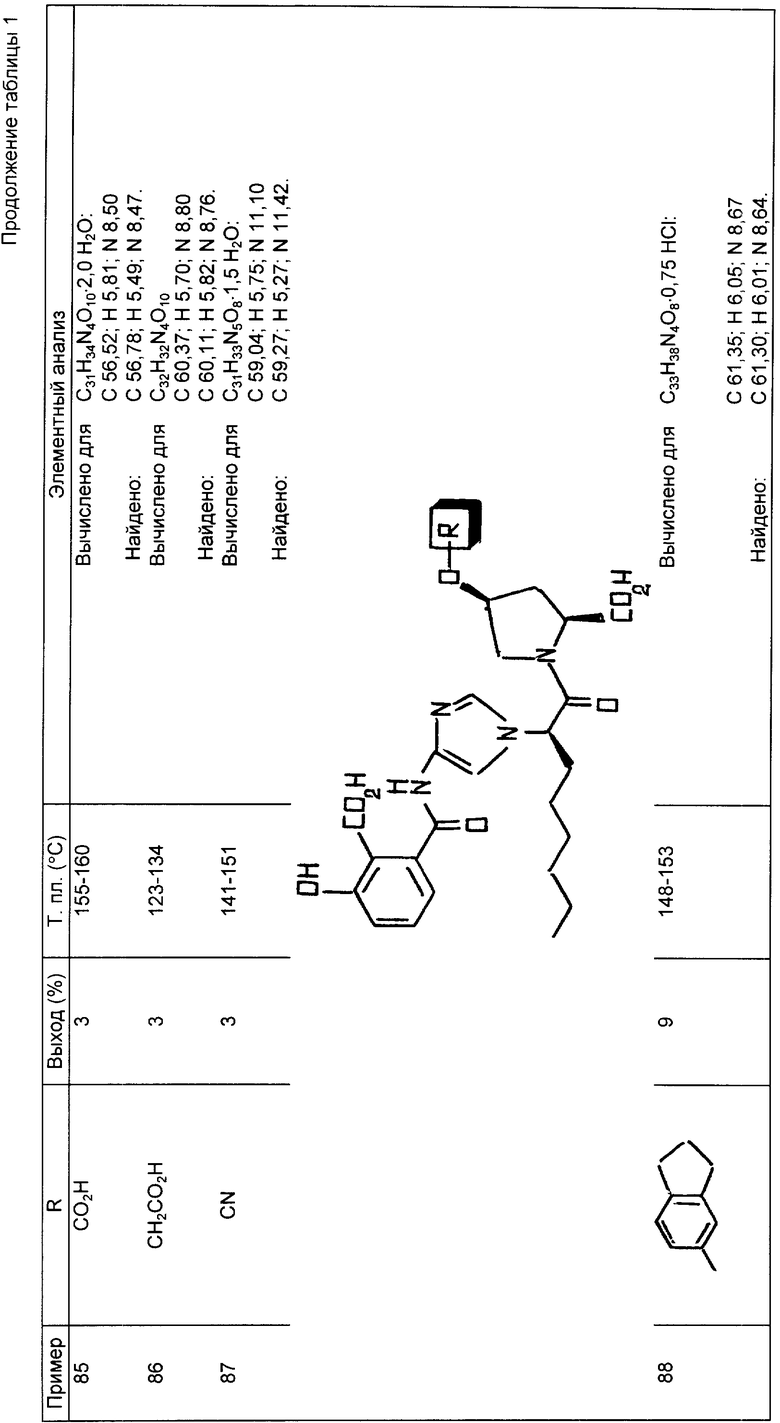
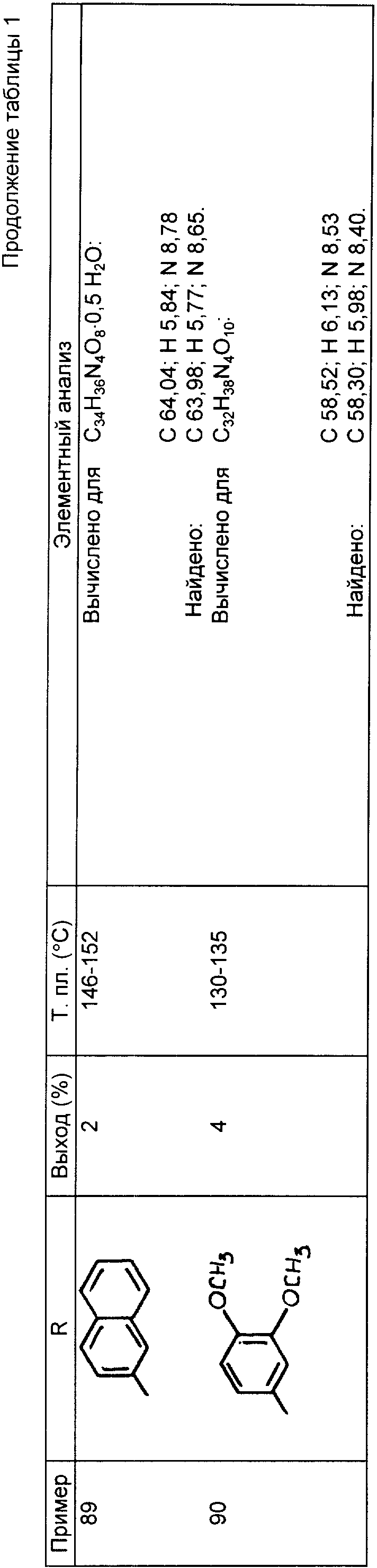
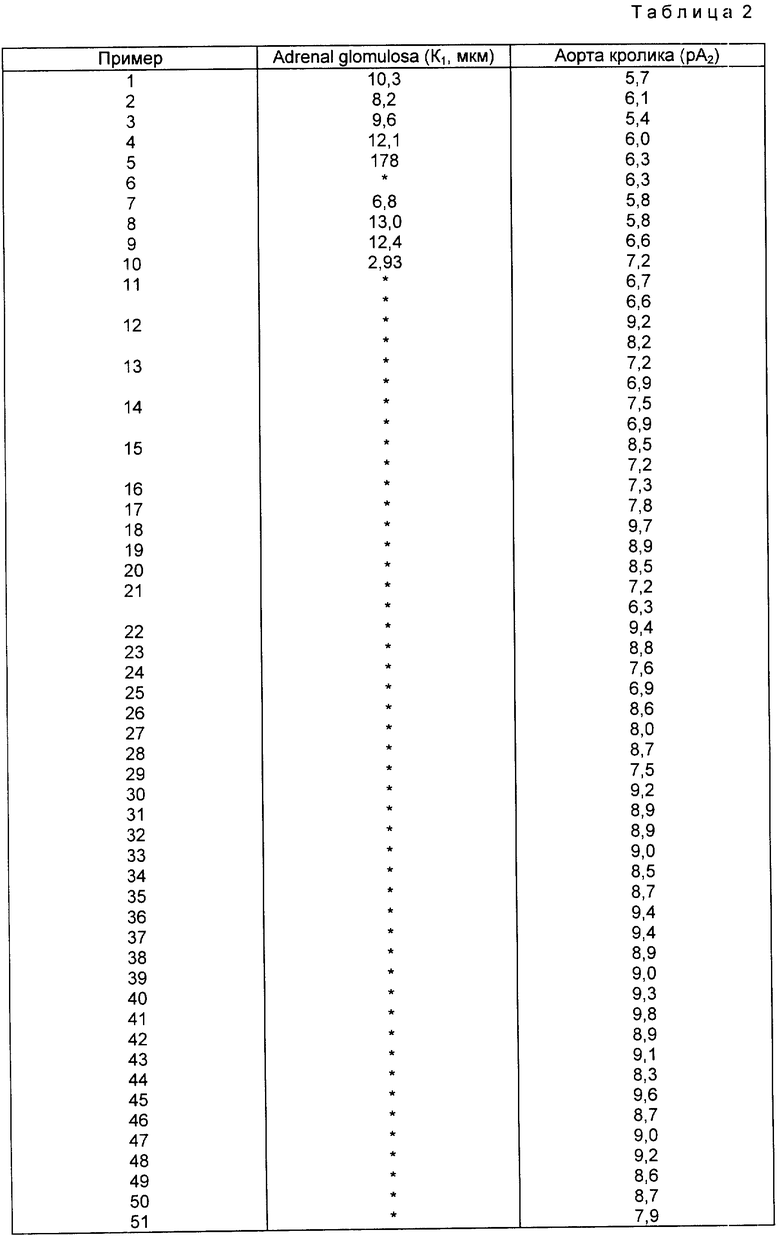

Новые производные имидазола формулы 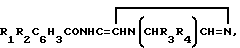 обладающие активностью антагониста ангиотензина П, и фармацевтическая композиция, включающая помимо обычных целевых добавок эффективное количество активного соединения вышеуказанной формулы. Наиболее предпочтительны (R)-1-[1-оксо-2-[4-(2-сульфобензоил)амино-IH-имидазол -1-ил] октил] -4-цис(4-карбоксиметилфенокси)-L-пролин и (R)-1-(1-оксо-2-[4-2-сульфобензоил)амино-IH-имидазол-1-ил] октил) -4-цис-[(4-метиленфосфоновая кислота)-фенокси] -L- пролин. 2 с. и 5 з. п. ф-лы. 2 табл.
обладающие активностью антагониста ангиотензина П, и фармацевтическая композиция, включающая помимо обычных целевых добавок эффективное количество активного соединения вышеуказанной формулы. Наиболее предпочтительны (R)-1-[1-оксо-2-[4-(2-сульфобензоил)амино-IH-имидазол -1-ил] октил] -4-цис(4-карбоксиметилфенокси)-L-пролин и (R)-1-(1-оксо-2-[4-2-сульфобензоил)амино-IH-имидазол-1-ил] октил) -4-цис-[(4-метиленфосфоновая кислота)-фенокси] -L- пролин. 2 с. и 5 з. п. ф-лы. 2 табл.
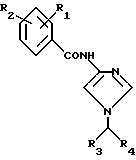
где R1 - CO2H, SO3H или 5-тетразолил;
R2 - Н или -ОН;
R3 - неразветвленная цепь алкила С4 - С9, неразветвленная цепь трифторалкила С4 - С9;
R4 - CONH (С1 - С4)алкил, -CONH(С1 - С4)трифторалкил, -CONH-гидрокси-(С1 - С4)алкил, CONH-пиридин-2, N-оксопролин,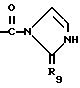
или
или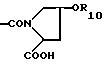
где R9 - -О- или -S-;
R1 0 - водород, С1 - С4-алкил, 3-пиридил, изоксазолил, хинолил, бензофуранил, возможно замещенный С1 - С4-алкилом или карбоксигруппой, дибензофуранил, бензотиофенил, нафтил, возможно замещенный С1 - С4-алкоксигруппой, тетрагидронафтил, инданил, бензодиоксоланин, ди-С1 - С4-алкоксифенил, фенил, возможно замещенный в положении 4 С1 - С4-алкилом, или С3 - С6-циклоалкилом, или галогеном, или трифтор-(С1 - С4)алкилом, или фенилом, или гидроксилом, или С1 - С4-алкоксигруппой, гидрокси-(С1 - С4)алкилом, или карбокси-(С1 - С4)-алкилом, или тио-(С1 - С4)-алкилом, или S(O)n(С1 - С4)-алкилом, где n = 1, 2, или карбоксигруппой, или аминокарбонилом, или нитрилом, или N-содержащим пятичленным гетероциклом, или группой SO2NHCH3,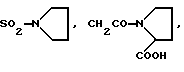
CH2CON(CH3)CH2COOH, P(O)(OH)2 или OP(O)(OH)2;
R1 1 - гидрокси- или карбоксигруппа.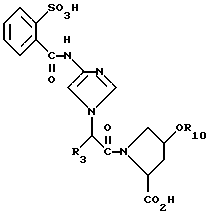
где R3 - С4 - С9-алкил с неразветвленной цепью;
R1 0 - незамещенный или паразамещенный фенил, хинолил, бензофуранил, возможно замещенный С1 - С4-алкилом или карбоксигруппой, дибензофуранил, бензотиофенил, инданил или бензодиоксоланил.
Приоритеты по пунктам и признакам:
03.06.92 - остальные признаки кроме указанного ниже, по п.1 и по пп.2 - 7.
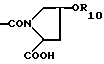
| EP, 438869, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, 324377, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| US, 4880804, кл | |||
| Устройство для сортировки каменного угля | 1921 |
|
SU61A1 |
| SU, 646908 А2, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
