Изобретение касается лигандов, которые представляют собой бициклополиазамакроциклофосфоновые кислоты, и их комплексов и конъюгатов, для использования в качестве контрастных агентов в магнитнорезонансной визуализации (MRI). Некоторые лиганды и комплексы также полезны в качестве средств ухода за полостью рта и в качестве агентов ингибирования образования накипи в системах водной обработки. Для лучшего понимания изобретения в следующем разделе дается краткий обзор или предпосылки, касающиеся MRI.
MRI является не-инвазивным приемом диагностики, который дает хорошо разрешенные изображения поперечного сечения мягких тканей животного организма, предпочтительно, человека. Данный прием основан на свойстве некоторых атомных ядер (например протонов воды), которые обладают магнитным моментом (как это определено с помощью математических уравнений: см. G.M. Barrow, physical Chemistry, 3-е издание, Mc Graw-Hill, HY (1973)), устанавливаться в одну линию (центрироваться) в магнитном поле. После центрирования данное состояние равновесия может отклоняться от нормы при применении внешнего радиочастотного (RF) импульса, который заставляет протоны уклоняться от выравнивания магнитным полем. Когда RF импульс прекращается (заканчивается), ядра возвращаются в свое состояние равновесия, и время, требуемое для того, чтобы это происходило, известно как время релаксации. Время релаксации состоит из двух параметров, известных как спинрешеточная (TI) и спинспиновая (T2), релаксация, и именно эти измерения релаксации дают информацию о степени молекулярной организации и взаимодействии протонов с окружающей средой.
Поскольку содержание воды в живой ткани является существенным, и существуют варьирования содержания и окружающей среды среди типов тканей, получаются диагностические изображения биологических организмов, которые отражают плотность протонов и периоды релаксации. Чем больше различия во времени релаксации (T1 и T2) протонов, присутствующих в проверяемой ткани, тем больше будет контраст получаемого изображения (J. Magnetic Resonance, 33, 83-106 (1979)).
Известно, что парамагнитные хелаты, обладающие симметричным электронным основным состоянием, могут оказывать резкое влияние на скорости релаксации T1 и T2 наложенных друг на друга водных протонов, и что эффективность хелата в данном отношении связана частично с числом непарных электронов, дающих магнитный момент (Magnetic Resonance Annual, 231-266, Raven Press, NY (1985). Было показано также, что, когда парамагнитный хелат данного типа вводится в организм животного, его влияние на T1 и T2 различных тканей можно непосредственно наблюдать по магнитнорезонансным (MR) изображениям, причем увеличенный контраст наблюдается в области локализации хелата. Поэтому, для увеличения диагностической информации, получаемой с помощью MRI, было предложено вводить животным стабильные нетоксичные парамагнитные хелаты (Frontiers of Biol. Energetics, 1, 752-759 (1978); J. Nucl. Med., 25, 506-513 (1984); Proc. of NMR Imaqinq Symp. (Oct. 26-27, 1980); F.A. Cotton et al. , Adv. Inorg. Chem., 634-639, 1966). Парамагнитные хелаты металлов, используемые данным образом, называют агентами усиления контраста или контрастными агентами.
Имеется ряд парамагнитных металлических ионов, которые могут учитываться, когда берутся за конструирование (разработку) контрастных агентов MRI. На практике, однако, наиболее полезными ионами парамагнитных металлов являются гадолиний (Gd+3), железо (Fe+3), марганец (Mn+2) и (Mn+3) и хром (Cr+3), потому что эти ионы оказывают наибольшее влияние на водные протоны благодаря их огромным магнитным моментам. В незакомплексованной форме (например, GdCl3) ионы этих металлов являются токсичными для животных, вследствие чего предотвращается их использование в форме простой соли. Поэтому основная роль органического хелатирующего агента (называемого также лигандом) заключается в том, чтобы сделать парамагнитный металл нетоксичным для животного при сохранении его желательного влияния на скорости релаксации T1 и T2 окружающих водных протонов.
Знания и опыт в области MRI являются весьма обширными, так что следующее ниже краткое описание, без намерения быть исчерпывающим, дается лишь в качестве обзора данной области и других соединений, которые являются возможно сходными по структуре. В патенте США 4899755 раскрывается метод чередования (смены) времени протонной ЯМР релаксации в печени или желчных протоках животных с использованием Fe+3-этилен-бис-(2-гидроксифенилглициновых) комплексов и их производных, и предлагается среди разнообразных других соединений возможное использование пиридин-макроциклометиленкарбоновой кислоты. В патенте США 4880008 (заявка с частичным продолжением патента США 4899755) раскрываются дополнительные данные визуализации ткани печени крыс, но не показаны какие-либо дополнительные комплексы. Патент США 4980148 раскрывает гадолиниевые комплексы для MRI, которые являются нециклическими соединениями (C.J. Broan et al., J. Chem. Soc., Chem. Commun., 1739-1741, 1990) описывают некоторые бифункциональные макроциклические фосфиновокислотные соединения (C. J. Broan и др., J. Chem. Soc., Chem. Commun., 1738-1739, 1990) описывают соединения, которые представляют собой триазабициклосоединения (I.K. Adzamli и др., J. Med. Chem., 32, 139-144, 1989) описывают ациклические фосфонатные производные гадолиниевых комплексов для ЯМР визуализации.
В настоящее время единственным промышленным контрастным агентом, доступным в США, является комплекс гадолиния с диэтилентриаминпентауксусной кислотой (ДТРА-Gd3+ - МАГНЕВИСТТМ фирмы Шеринг АГ). МАГНЕВИСТТМ считается неспецифичным перфузионным агентом, поскольку он свободно распространяется в межклеточной жидкости с последующим эффективным выводом через ренальную систему. МАГНЕВИСТТМ оказался весьма ценным в диагностике повреждений головного мозга, поскольку сопровождающий разрыв гематоэнцефалического барьера обеспечивает возможность перфузии контрастных агентов в пораженные области. В дополнение к МАГНЕВИСТТМ'у Guerbet выпускает промышленно для продажи макроциклический перфузионный агент (DOTAREMТМ), который в настоящее время доступен только в Европе. В различных стадиях разработки находится ряд других потенциальных контрастных агентов.
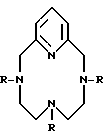
В настоящее время неожиданно было обнаружено, что контрастными агентами могут быть различные бициклополиазамакроциклофосфоновокислотные лиганды. Кроме того, эти лиганды могут видоизменять свой заряд, благодаря структуре лиганда и выбранному металлу, что может оказывать воздействие на их способность быть более специфичными к тому или иному участку (сайту). В частности, изобретение направлено на новые лиганды, которые представляют собой бициклополиазамакроциклофосфоновокислотные соединения формулы:
где
R представляет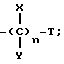
где
X и Y представляют независимо H, OH, (1 - 3)C алкил или COOH;
n - целое число 1, 2 или 3;
при условии, что: когда n представляет 2, тогда сумма X и Y должна быть равна двум или более H; и когда n составляет 3, тогда сумма X и Y должна быть равна трем или более H;
T представляет H, (1 - 18)C алкил, COOH, OH, SO3H, группу
где
R1 представляет OH, (1 - 5) C-алкил или -O- (1 - 5) C алкил;
R4 представляет H, NO2, NH2, изотиоцианато, семикарбазидо, тиосемикарбазидо, малеимидо, бромацетамидо или карбоксил;
R2 представляет H или OH; при условии, что, когда R2 представляет OH, тогда R, содержащий R2, должен иметь все X и Y равные H; при условии, что по крайней мере один T должен представлять P/O/R1OH, и при условии, что, когда один T представляет группу
тогда один радикал X или Y данного R радикала может представлять COOH, а все другие X и Y данного R радикала должны представлять H;
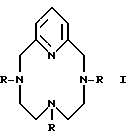
A представляет собой CH, N, C-Br, C-Cl, C-OR3, C-OR8, N+-R5X,
R3 представляет H, (1 - 5)C алкил, бензил, или бензил замещенный по крайней мере одним R4;
R4 имеет значения, определенные выше;
R5 представляет (1 - 16)C алкил, бензил, или бензил, замещенный по крайней мере одним R4;
R8 представляет (1 - 16)C алкиламино;
X- представляет Cl-, Br-, l- или H3CCO
Q и Z независимо представляют собой CH, N, N+-R5X, C-CH2-OR3 или C-C(O)-R6;
R5 имеет значения, определенные выше;
R6 представляет -O-(1 - 3)C алкил, OH или NHR7;
R7 представляет (1 - 5)C алкил или биологически активный материал;
X- имеет значения, определенные выше; или их фармацевтически приемлемые соли;
при условии, что:
a) когда Q, A или Z представляет N или N+-R5X, тогда другие две группы должны представлять CH;
b) когда A представляет C-Br, C-Cl, C-OR3 или C-OR8, тогда и Q и Z должны быть CH;
c) сумма радикалов R4, R7 и R8, когда они присутствуют, не может превышать одного; и
d) только один из Q или Z может быть C-C (O)-R6, и когда один из Q или Z представляет C-C (O)-R6, тогда A должен быть CH.
Когда указанные выше лиганды формулы 1 имеют по крайней мере два из R радикалов T равные PO3H2[P(O)R1OH, где R1 представляет OH, и третий T равен H, COOH или (1 - 18)C алкил; A, Q и Z представляют CH; n равно 1; и X и Y независимо представляют H или (1 - 3)C алкил; тогда лиганды полезны для ухода за полостью рта. Особенно предпочтительными являются те лиганды, в которых три R радикала T представляет P(O)R1OH, где R1 представляет OH, n равно 1; и X и Y представляют H. Использование этих лигандов обсуждается и заявляется в других находящихся совместно на рассмотрении заявках.
Когда указанные выше лиганды формулы 1 имеют:
в R радикале по крайней мере два T равные P(O)R1OH, где R1 представляет OH, а в другом R радикале, T представляет COOH или P/O/ R1OH, и n, R1, X, Y, A, Q и Z имеют указанные выше значения;
в по крайней мере одном R радикале T представляет P(O)R1OH, где R1 представляет OH, а в других двух R радикала T представляет COOH или P(O)R1OH, и n, R1, X, Y, A, Q и Z имеют указанные выше значения; или
в R радикале три T представляют P(O)R1OH, где R1 представляет (1 - 5)C алкил или -O-(1 - 5)C алкил, и n, R1, X, Y, A, Q и Z имеют значения, определенные выше; тогда лиганды являются полезными в качестве контрастных агентов.
Особенно предпочтительными являются те лиганды, формулы 1,
где X и Y представляют H; n представляет 1; или
A, Q и Z представляют CH.
Бифункциональные лиганды формулы 1 желательны для получения конъюгатов данного изобретения. Такие лиганды должны иметь:
один R радикал, в котором T фрагмент представляет
где
R2 и R4 имеют значения, определенные выше, особенно, когда в двух R радикалах, не содержащих R4 радикала, оба радикала T представляет P/O/R1OH, где R1 имеет значения, определенные выше, или где в двух R радикалах, не содержащих R4 радикала, один T представляет COOH, а другой T радикал представляет P(O)R1OH, где R1 имеет значения, определенные выше, предпочтительно фрагмент указанного выше T радикала, где один из X и Y данного радикала представляет COOH; и
предпочитаются также те лиганды, в которых n представляет 1 и/или остальные X и Y радикалы представляют H; или
A представляет C-OR3 или C-OR8, где и R3 и R8 имеют значения, определенные выше, или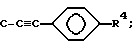
где
R4 имеет значения, определенные выше; или
A представляет CH, и один из Q или Z представляет CH, а другой представляет C-C/О/-R6, где R6 имеет значения, определенные выше;
особенно те лиганды, в которых R6 представляет NHR7, где R7 представляет биологически активный материал.
Лиганды формулы I могут быть закомплексованными с различными ионами металлов, такими как гадолиний /Gd+3/, железо /Fe+3/, и марганец /Mn+2/, причем, предпочтительным является Gd+3. Комплексы, образованные таким образом, могут использоваться сами по себе или могут присоединяться с помощью ковалентной связи к более крупным молекулам, таким как декстран, полипептид или биологически активные молекулы, включая антитела или их фрагменты, и использоваться для диагностических целей. Такие конъюгаты и комплексы являются полезными в качестве контрастных агентов.
Комплексы и конъюгаты изобретения могут конструироваться для обеспечения специфичного общего заряда, который успешно влияет на ин виво биолокализацию и контрастность изображения. Например, когда ионом металла является +3, могут получаться следующие:
(A) общий заряд - 2 или более - когда
в трех радикалах R T представляет P(O)R1OH, где R1 представляет OH, и n равно 1; или
в двух R радикалах T представляет P(O)R1OH, где R1 представляет OH, в третьем R радикале T представляет COOH, и n равно 1; или
в двух R радикалах T представляет P(O)R1OH, где R1 представляет OH, в третьем R радикале T представляет P(O)R1OH, где R1 представляет (1-5)C алкил, и n равно 1; или
в двух R радикал T представляет P(O) R1OH, где R1 представляет OH, в третьем R радикале T представляет P(O)R1OH, где R1 представляет -O-(1-5)C алкил, и n представляет 1; или
(B) Общий заряд -1 - когда
в одном R радикале T представляет P(O)R1OH, где R1 представляет OH, и в двух других R радикалах T представляет P(O)R1OH, где R1 представляет собой -O-(1-5)C алкил, и n равно 1; или
в одном R радикале T представляет P(O)R1OH, где R1 представляет OH, и в других двух R радикалах T представляет P(O)R1OH, где R1 представляет (1-5)C алкил, и n равно 1; или
в одном R радикале T представляет P(O) R1OH, где R1 представляет OH, и двух других R радикалах T представляет COOH, и n равно 1; или
(C) общий нейтральный заряд - когда
в трех R радикалах T представляет P(O)R1OH, где R1 представляет собой -O-((1-5)C алкил), и n равно 1; или
в трех R радикалах T представляет P(O)R1OH, где R1 представляет (1-5)C алкил, и n равно 1; или
(D) общий заряд +1 - когда
один из A, Q или Z представляет N+-R5X-, где R5 и X- имеют значения, определенные выше; и в одном R радикале T фрагмент представляет P(O)R1OH, где R1 представляет (1-5)C алкил или -O-/(1-5)C алкил/; и в других двух R фрагмент T представляет COOH или P(O) R1OH, где R1 представляет (1-5)C алкил, -O-((1-5)C алкил); и все X и Y радикалы представляют H.
Как комплексы, так и конъюгаты могут быть сформулированы в фармацевтически приемлемой форме для назначения животным.
Возможно использование лигандов формулы I с ионами других металлов для диагностики болезненных состояний, таких, как рак. Использование этих комплексов и конъюгатов обсуждается в еще одной заявке, находящейся совместно на рассмотрении.
Соединения формулы I пронумерованы для номенклатурных целей следующим образом: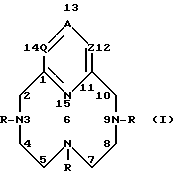
Один из аспектов изобретения касается разработки контрастных агентов, имеющих синтетические модификации парамагнитного хелата, обеспечивающих возможность сайт-специфичной доставки контрастного агента в желаемую ткань. Причем, преимущество заключается в увеличенной контрастности в областях, представляющих интерес, основанной на средстве к тканям, в противоположность контрасту, возникающему в результате неспецифичной перфузии, которая может быть или может не быть видимой с межклеточными агентами. Специфичность лиганда формулы I может регулироваться с помощью регулировки общего заряда и липофильного характера комплекса. Общий интервал заряда комплекса составляет от -3 до +1. Например, для комплекса, имеющего 2 или более групп PO3H2, общий заряд является высоко отрицательным и ожидается поглощение костью; тогда как, когда общий заряд комплекса составляет O (является таким образом нейтральным) комплекс может обладать способностью пересекать гематоэнцефалический барьер и может быть возможно поглощение головным мозгом.
Тканевая специфичность может также реализоваться с помощью ионного или ковалентного присоединения хелата к встречающейся в природе или к синтетической молекуле, имеющей специфичность в отношении желаемой целевой ткани. Одно из возможных применений данного подхода заключается в использовании сопряженных с хелатом моноклональных антител, которые транспортируют парамагнитный хелат к пораженной болезнью ткани, обеспечивая возможность визуализации с помощью MRI. В дополнение к этому, присоединение парамагнитного хелата к макромолекуле может дополнительно увеличивать эффективность контрастного агента, приводя в результате к улучшенному контрасту относительно несвязанного хелата. Недавняя работа автора Lauffer (патенты США 4880008 и 4899755) продемонстрировала, что вариации липофильности могут давать в результате специфичные к ткани агенты, и что увеличенный липофильный характер благоприятствует не-ковалентному взаимодействию с белками крови, приводя в результате к усилению релаксивности.
В дополнение к сказанному настоящие контрастные агенты формулы I, которые являются нейтральными по заряду, особенно предпочтительны для образования конъюгатов данного изобретения, поскольку сводятся до минимума нежелательные ионные взаимодействия между хелатом и белком, что сохраняет иммунореакционноспособность антитела. Кроме того, настоящие нейтральные комплексы уменьшают осмотическое давление относительно ДТРА-Gd+3, что может смягчать или устранять дискомфорт от инъекций.
Не желая быть связанными какой-либо теорией, считают, что когда получается заряженный комплекс изобретения (например, возможно -2 или -3 для кости, -1 для печени, или +1 для сердца), варьирования ионного заряда этого хелата могут влиять на биолокализацию. Таким образом, если антитело или другой направляющий фрагмент является также специфичным к одному и тому же участку или сайту, тогда конъюгат обнаруживает две доли для оказания помощи сайт-специфичной доставке.
Термины, использованные при определении формулы I, дополнительно определяются следующим образом, (1-3)C алкил, (1-5)C алкил, (1-18)C алкил включают алкильные группы, как с прямой, так и разветвленной цепью. Животное включает теплокровное млекопитающее, предпочтительно человека.
Биологически активный материал относится к декстранам, пептидам или молекулам, которые имеют специфическое сродство в отношении рецепторов, или предпочтительно антителам или фрагментам антител.
Антитело относится к любому поликлональному, моноклональному, химерному антителу или гетероантителу, предпочтительно к моноклональному антителу; "фрагменты антител" включают Fab фрагменты и F/ab'/2 фрагменты, и любую часть антитела, имеющую специфичность по отношению к желаемому эпитопу или эпитопам. Когда используется термин конъюгат хелата радиоактивного металла/антитела или конъюгат, имеется в виду, что антитело включает целые антитела и/или фрагменты антител, включая полусинтетические их варианты или варианты, полученные методами генетической инженерии. Возможными антителами являются 1116-NS-19-9 (анти-колоректальная карцинома), 1116-NS-3d (анти-CEA), 703D4 (анти-человеческий рак легких), 704AI (анти-человеческий рак легких), CC49 (анти-TAG-72), CC83 (анти-TAG-72) и B72.3. Гибридомные клеточные линии 1116-NS-19-9, 1116-NS-3d, 703D4, 704A1, CC49, CC83 и B72.3 депонированы в Американской коллекции культур под депозитарными номерами ATCC HB 8059, ATCC CRL 8019, ATCC HB 8301, ATCC HB 8302, ATCC HB 9459, ATCC HB 9453 и ATCC HB 8108, соответственно.
В том смысле, как он используется здесь, термин комплекс относится к комплексу соединения формулы I, связанного в комплекс или закомплексованного с ионом металла, где по крайней мере один атом металла хелатирован или связан в хелатный комплекс; конъюгат относится к хелату иона метила, который ковалентно присоединен к антителу или фрагменту антитела. Термины бифункциональный координатор, бифункциональный хелатирующий или хелатообразующий агент и функционализированный хелатирующий агент используются взаимозаменяемо и относятся к соединениям, которые имеют хелатирующий фрагмент, способный образовывать хелатный комплекс иона металла и фрагмента, ковалентно связанного с хелатирующим фрагментом, то есть способен служить в качестве средства ковалентного присоединения к антителу или фрагменту антитела.
Бифункциональные хелатирующие агенты, описанные здесь (представленные формулой 1), могут использоваться для образования хелата или связывания в хелатный комплекс ионов металлов, образуя хелаты металлических ионов (называемые здесь также комплексами). Комплексы, вследствие присутствия функционализирующего фрагмента (представленного заместителями R4 или R3 в формуле 1) могут ковалентно присоединяться к биологически активным материалам, таким как декстран, молекулы, которые обладают специфическим сродством к рецепторам или предпочтительно ковалентно присоединяться к антителам или фрагментам антител. Таким образом, комплексы, описанные здесь, могут ковалентно присоединяться к антителам или фрагментам антител или обладать специфическим сродством к рецепторам и называются здесь конъюгатами.
В том смысле, как он используется в данном описании, термин "фармацевтически приемлемые соли" означает любую соль или смеси солей соединения формулы 1, которые являются в достаточной степени нетоксичными, чтобы быть полезными в терапии или диагностике животных, предпочтительно млекопитающих. Таким образом, согласно данному изобретению полезными являются соли. Характерные представители этих солей, получаемых или образуемых с помощью стандартных реакций как с органическими, так и с неорганическими источниками, включают, например, соли серной, соляной, фосфорной, уксусной, янтарной, лимонной, молочной, малеионовй, фумаровой, пальмитиновой, холевой, памовой, слизевой, глютаминовой, глюконовой кислоты, o-камфорной, глутаровой, гликолевой, фталевой, винной, муравьиной, лауриновой, стеариновой, салициловой, метансульфоновой, бензолсульфоновой, сорбиновой, пикриновой, бензойной, коричной кислот и других подходящих кислот. Включаются также соли, образуемые с помощью стандартных реакций из органических или неорганических источников, таких как аммоний или 1-дезокси-1-/метиламино/-Д-глюцитол, ионы щелочных металлов, ионы щелочноземельных металлов и других аналогичных ионов. Особенно предпочтительными являются соли соединений формулы 1, когда солью является соль калия, натрия, аммония. Включены также смеси указанных выше солей.
Соединения формулы 1 получаются с помощью различных процессов. Типичный общий синтетический путь или подход к таким процессам показан с помощью схем реакций, приведенных ниже.
Согласно схеме 1 (см. в конце текста) получаются соединения формулы 1, где X и y = H, n = 1 (но также применим случай, если n = 2 или 3, с соответствующей заменой реагента), T = PO3H2, и Q, A и Z = CH.
Согласно схеме 2 (см. в конце текста) получаются соединения формулы 1, в которой X и Y = H, n = 1 (но метод применим также, если n = 2 или 3 при соответствующей замене реагента), T =
где
R1 = -О-(1-5)C алкил; и Q, A и Z = CH.
По схеме 3 (см. в конце текста) получаются соединения формулы 1, в которой X и Y = H, n = 1 (но метод применим также, когда n = 2 или 3, с соответствующим изменением реагента), T =
где
R1 = (1-5)C алкил; и Q, A и Z = CH.
В соответствии со схемой 4 (см. в конце текста) получаются соединения формулы 1, в которой X и Y = H, n = 1 (но метод применим также, если n = 2 или 3, с соответствующей заменой реагента), T =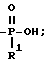
где
R1 = -О-(1-5)C алкил или (1-5)C алкил; A = C-Br, и Q и Z = CH.
По схеме 5 (см. в конце текста) получаются соединения формулы 1, в которой X и Y = H, n = 1 (но это применимо также, если n = 2 или 3, с соответствующей заменой реагента), T =
где
R1 = -О-(1-5)C алкил или (1-5)C алкил; A =
R4 = H, NO2, NH2 или Q и Z = CH.
Согласно схеме 6 (см. в конце текста) получают соединения формулы 1, где X и Y = H, n = 1 (но это применимо также, если n = 2 или 3, с соответствующей заменой реагента) T =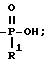
где
R1 = -О-(1-5)C алкил/ или (1-5)C алкил;
A = C-OR8, где R8 = (1-5)C алкиламино; и Q и Z = CH.
Согласно схеме 7 (см. в конце текста) получают соединения формулы 1, в которой X и Y = H, n = 1, но это применимо также, если n = 2 или 3, с соответствующей заменой реагента), T =
где
R1 = -OH, -O-/(1-5)C алкил/, или (1-5)C алкил;
Z = C-C(O)-R6, где R6 = OH; и Q и A = CH.
Согласно схеме 8 (см. в конце текста) получают соединения формулы 1, в которой X и Y = H, n = 1 (но метод применим также, если n = 2 или 3, с соответствующей заменой реагента), T =
где
R1 = -OH, -O-((1-5)C алкил) или (1-5)C алкил;
Z = C-CH2-OR3, где R3 = бензил; и Q и A = CH.
По схеме 9 (см. в конце текста) получают соединения формулы 1, в которой X и Y = H, n = 1 (но метод также применим, если n = 2 или 3, с соответствующей заменой реагента), T =
где
R1 = -OH, -O-(1-5)C алкил, (1-5)C алкил;
A = N или N - R5; R5 = (1-16)C алкилгалогенид; и Q и Z = CH.
Согласно схеме 10 (см. в конце текста) получают соединения формулы 1, в которой X и Y = H, n = 1 (но метод применим также, если n = 2 или 3, с соответствующей заменой реагента), T =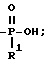
где
R1 = -OH, -O-(1-5)C алкил или (1-5)C алкил;
Q = N - R5; R5 = (1-16)C алкилгалогенид; и A и Z = CH.
Согласно схеме 11 (см. в конце текста) получают соединения формулы 1, в которой X и Y = H, n = 1 (но метод применим также, если n = 2 или 3, с соответствующей заменой реагента), T =
где
R1 = -OH, -O-(1-5)C алкил или (1-5)C алкил;
Q = N - R5; R5 = (1-16)C алкилгалогенид; и A и Z = CH.
По схеме 12 (см. в конце текста) получаются соединения формулы 1, в которой X и Y = H, n = 1 (но метод применим также, если n = 2 или 3, с соответствующей заменой реагента), R в положении 3 имеет T =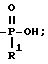
где
R1 = -OH или -O-(1-5)C алкил; а другие два R радикала имеют T = COOH; и A, Q и Z = CH.
По схеме 13 (см. в конце текста) получают соединения формулы 1, в которой X и Y = H, n = 1 (но метод применим также, если n = 2 или 3, с соответствующей заменой реагента), R в 3-положении и 6 положении имеет T =
где
R1 = OH или -O-(1-5)C алкил, а другой R радикал в 9 положении имеет T = COOH; и A, Q и Z = CH.
По схеме 14 (см. в конце текста) получают соединения формулы 1, где X и Y = H, n = 1 (но метод применим также, если n = 2 или 3, с соответствующей заменой реагента), R радикалы в 3 и 9 положении имеют T =
где
R1 = -OH или -O-(1-5)C алкил; и другой R радикал в 6 положении имеет T = COOH; и A, Q и Z = CH.
По схеме 15 (см. в конце текста) получают соединения формулы 1, где n = 1 (но метод применим также, если n = 2 или 3, с соответствующей заменой реагента), R радикалы в 3 и 9 положении имеют T =
где
R1 = -OH или -O-(1-5)C алкил; и X и Y = H; R радикал в 6 позиции имеет T =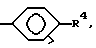
где
R4 = NO2 или NH2 и один из X или Y = H и другой = COOH; и A, Q и Z = CH.
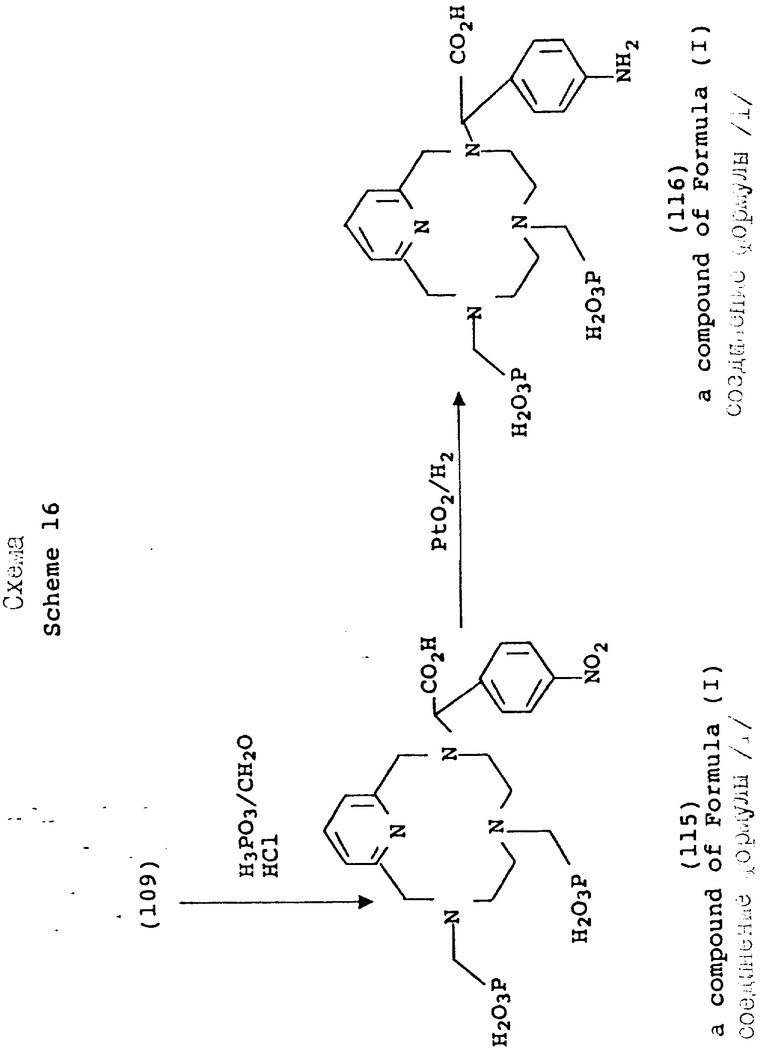
По схеме 16 (см. в конце текста) получают соединения формулы 1, где n = 1 (но метод применим также, если n = 2 или 3, с соответствующей заменой реагента), R радикалы в 3 и 6 положении имеют T =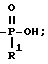
где
R1 = -OH или -O-(1-5)C алкил; и X и Y = H;
R радикал в 9 положении имеет T =
где
R4 = NO2 или NH2; и один из X или Y = H и другой = COOH;
A, Q и Z = CH.
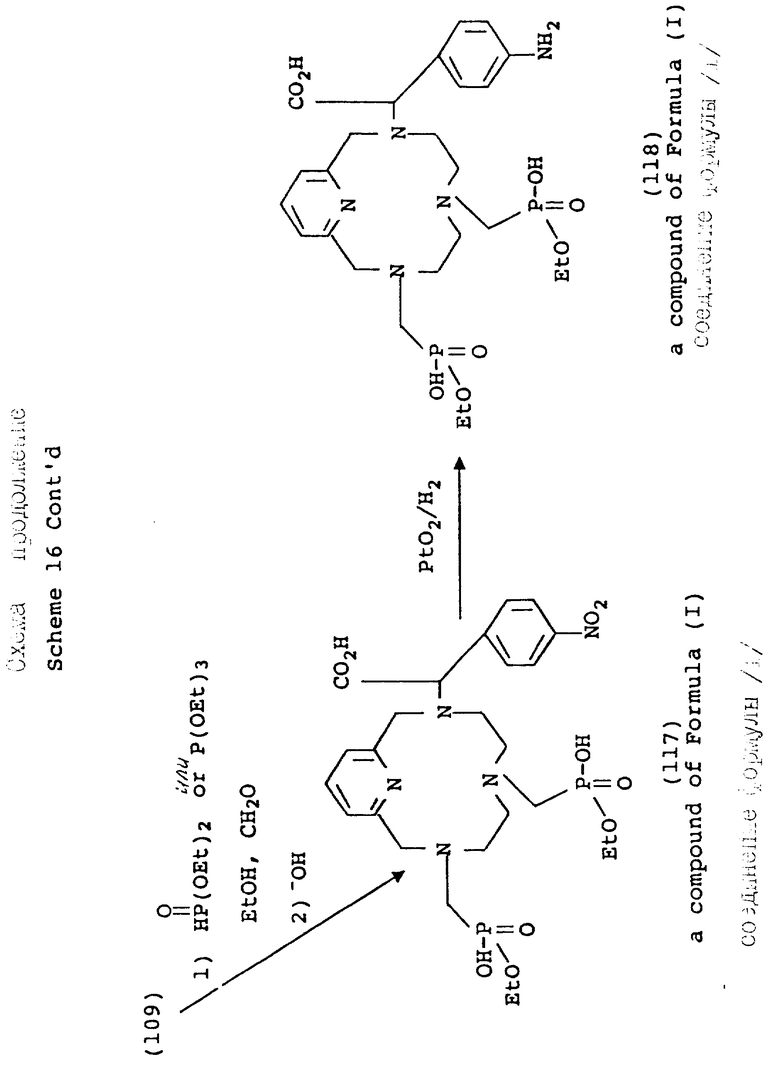
По схеме 17 (см. в конце текста) получают соединения формулы 1, где n=1 (но метод применим также, если n=2 или 3, с соответствующей заменой реагента).
R радикал в 6 положении имеет T=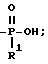
где
R1=-OH; И X и Y = H;
R радикал в 3 и 9 позициях имеет T = COOH; и A, Q и Z = CH.
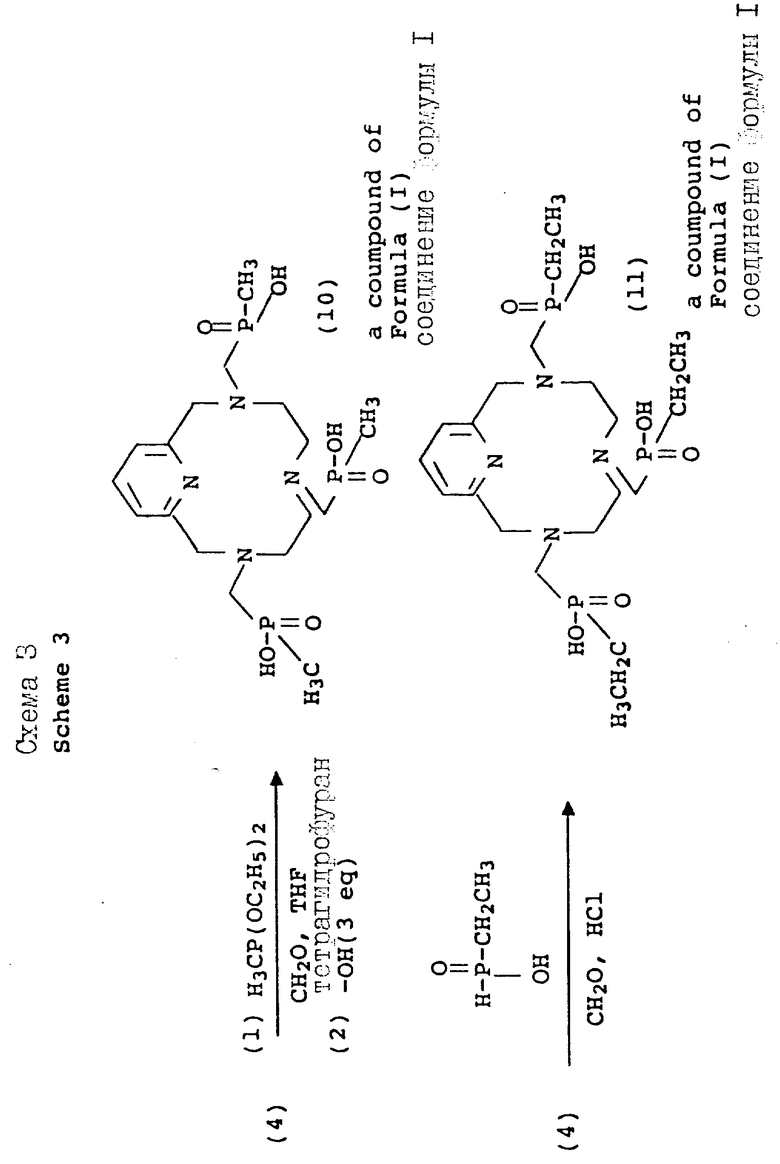
На приведенных выше схемах описание общего процесса иллюстрирует конкретные стадии, которые могут использоваться для выполнения требуемой стадии реакции.
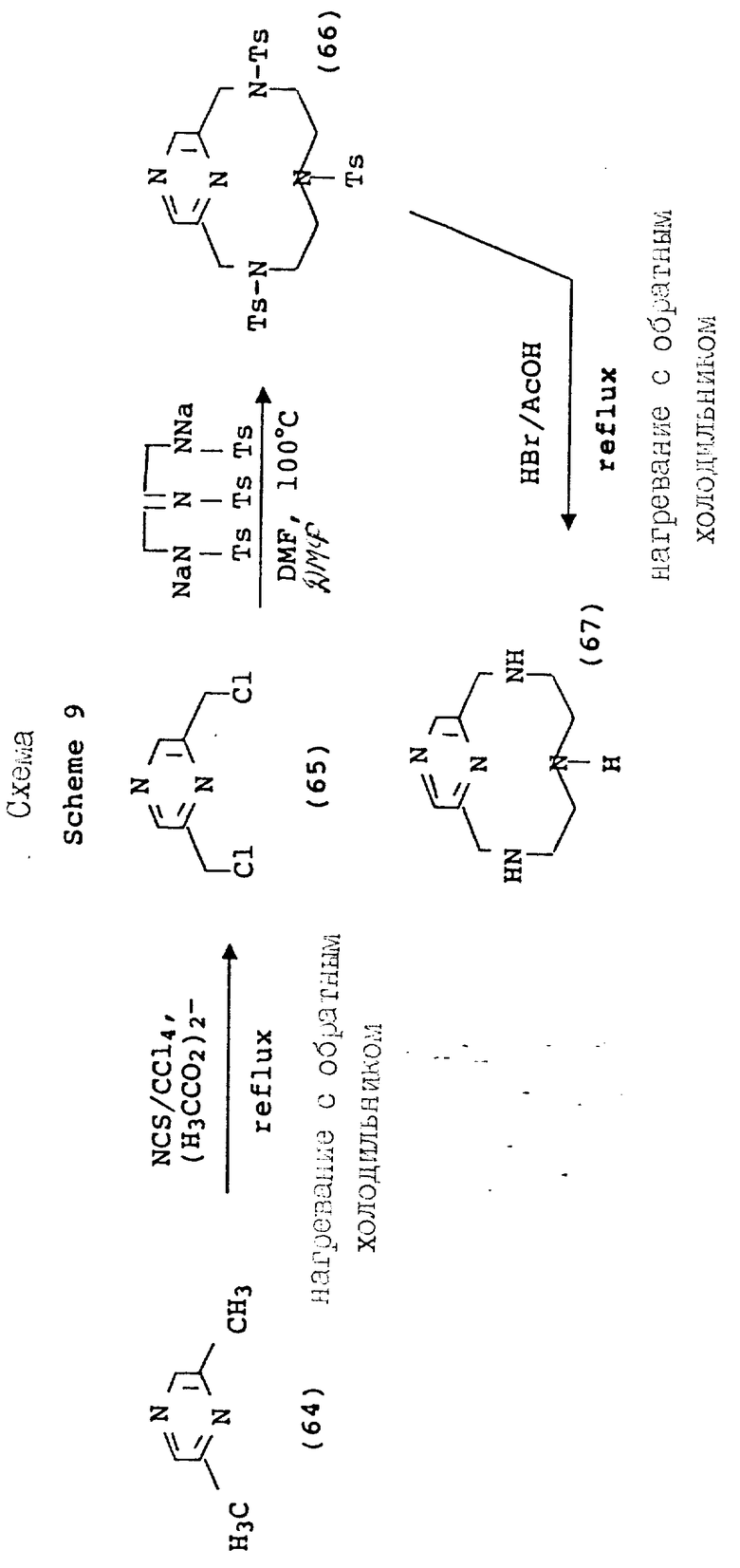
Схема синтеза 1 начинается с галоидирования промышленно доступного бис-пиридилового спирта 1 с использованием тионилхлорида. Сходные процедуры превращения спирта в электрофильный субстрат, такие как обработка толуолсульфонилхлоридом, HBr или HCi, также должна давать в результате аналогичным образом реакционноспособный продукт, который хорошо действует в последующих реакциях замыкания кольца. В литературе описаны многочисленные процедуры макроциклизации, и желаемый тетраазамакроцикл приготавливался в соответствии с методом Stetter и др., Tetrahedron, 37, 767 - 772 1981. С тех пор были опубликованы более общие приемы, которые дают хороший выход аналогичных макроциклов с использованием более мягких условий (A.D. Sherry и др., J. Ong. Chem. , 54, 2990 - 2992, 1989). Детозилирование промежуточного макроцикла [(3) для получения (4)] выполнялось в кислотных условиях с хорошим выходом. Приемы восстановительного детозилирования также хорошо известны в литературе и могут быть приспособлены для настоящей реакционной последовательности. Фосфонометилирование для получения производного трис-аминофосфоновой кислоты (5, РСТМР) проводилось в типичных условиях использования основания Манниха с использованием фосфористой кислоты и формальдегида.
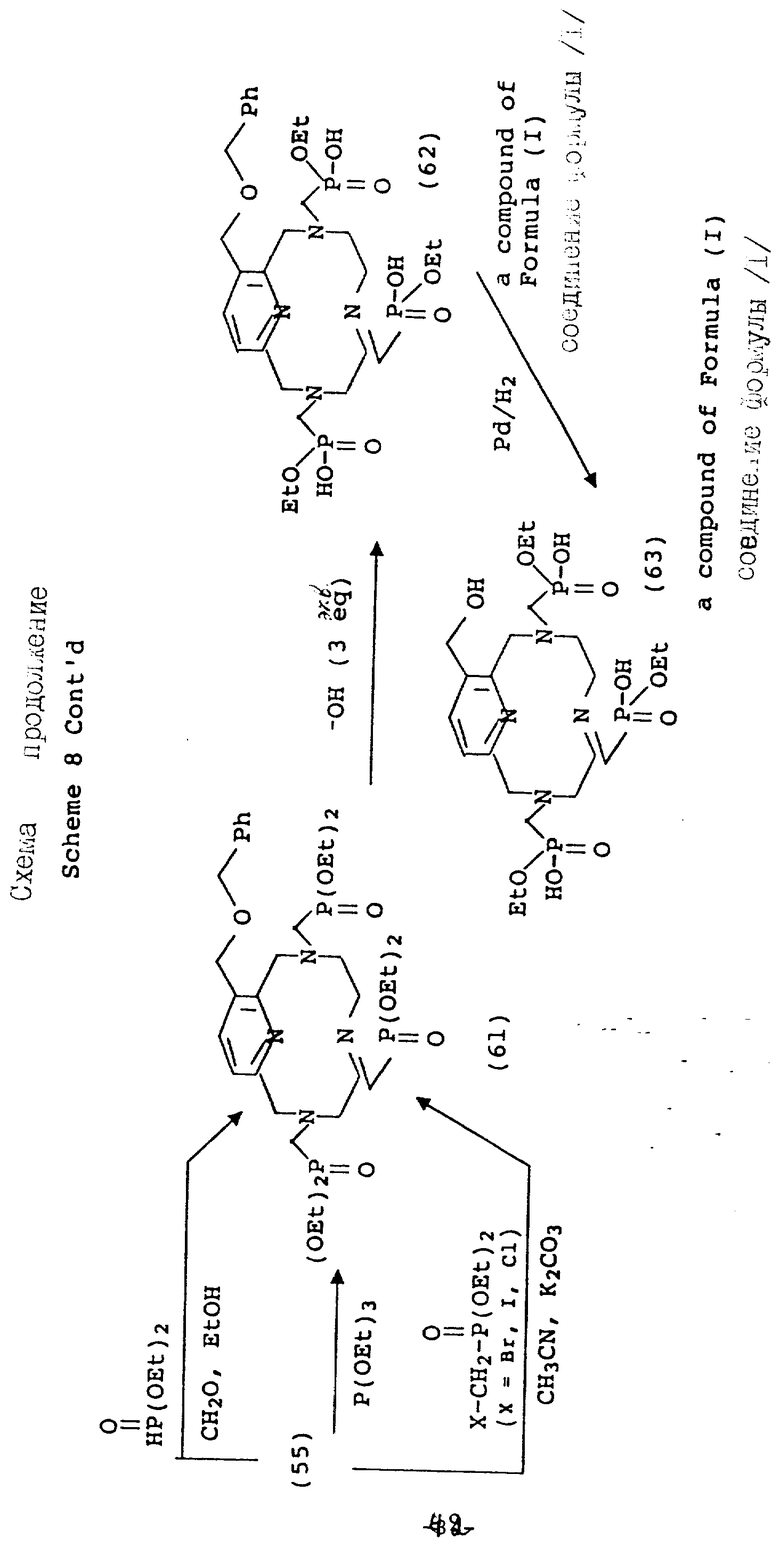
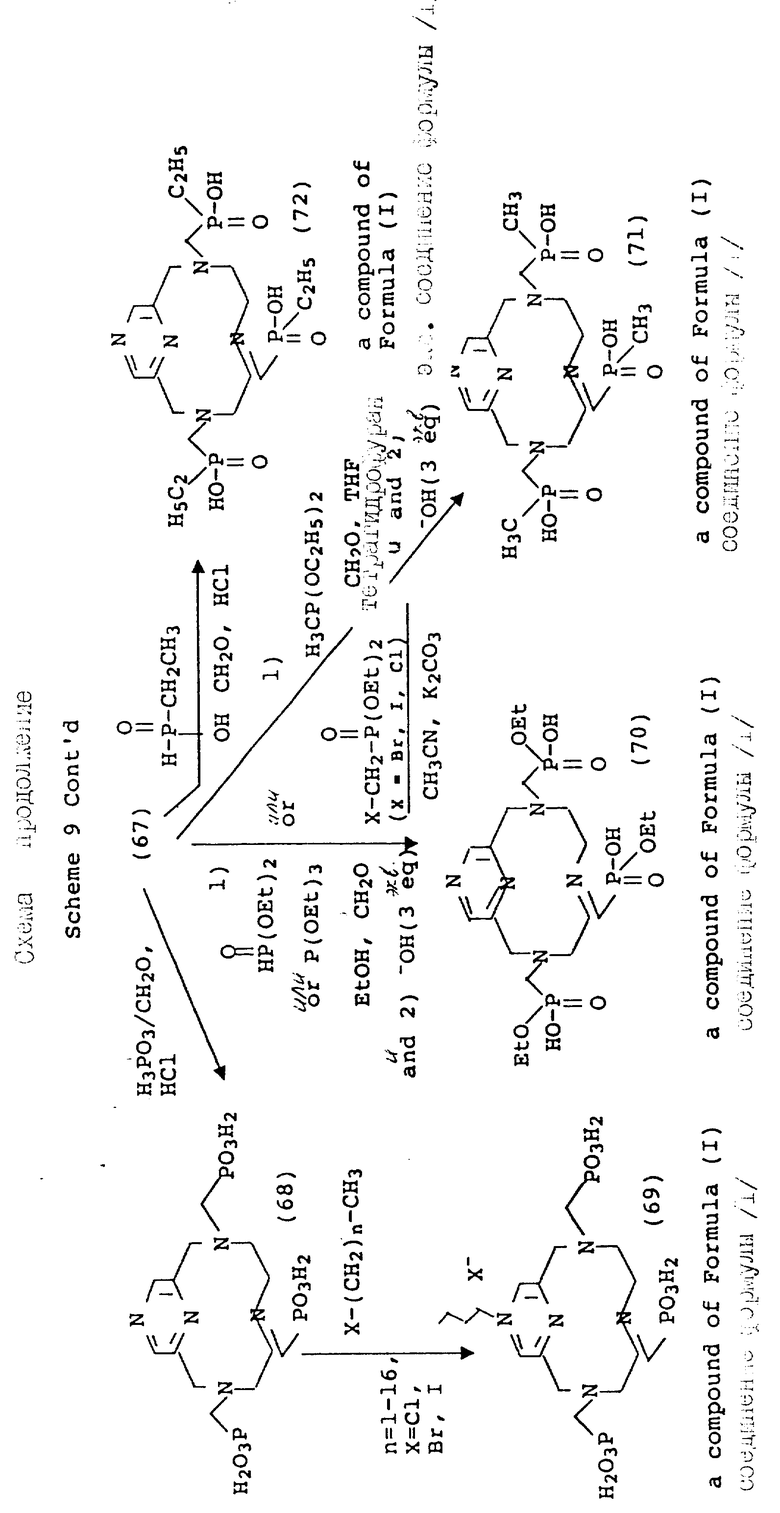
В дополнение к производным фосфоновой кислоты, фосфонатные сложные эфиры (например, формулы 6 могут быть получены в органических условиях в спиртах или апротонных растворителях (например, ацетонитриле, бензоле, толуоле, тетрагидрофуране) и с использованием желаемого диалкилфосфита в качестве нуклеофильного агента (см. Схему 2). В зависимости от реакционноспособности амина, эти реакции могут проводиться при температуре между -10 и примерно 100oC. В дополнение к сказанному триалкилфосфиты могут применяться в аналогичных условиях Манниха, давая фосфонатный сложный эфир путем окисления фосфора (III) в фосфор (V) с одновременным вытеснением одного моля спирта (реакция Арбузова). Эти реакции могут проводиться в присутствии или в отсутствии растворителя. Когда в качестве растворителя применяются спирты для реакций или с диалкил- или триалкил-фосфитом, благоприятно использовать спирт, из которого происходит соответствующий сложный фосфонатный эфир, для того, чтобы избежать альтернативных продуктов, возникающих в результате переэтерификации. Сложные эфиры данного типа получаются также с помощью N-алкилирования галоиддиалкилпросфонатов в таких растворителях, как ацетонитрил, хлороформ, диметилформамид, тетрагидрофуран или 1,4-диоксан, с добавлением или без добавления ненуклеофильного основания, такого как карбонат калия, при комнатной температуре или выше. Получающееся в результате сложноэфирное промежуточное соединение затем легко гидролизуется в основных условиях (водная гидроокись, pH 8 - 14, 30 - 110oC), давая соответствующее полу-кислотное производное.
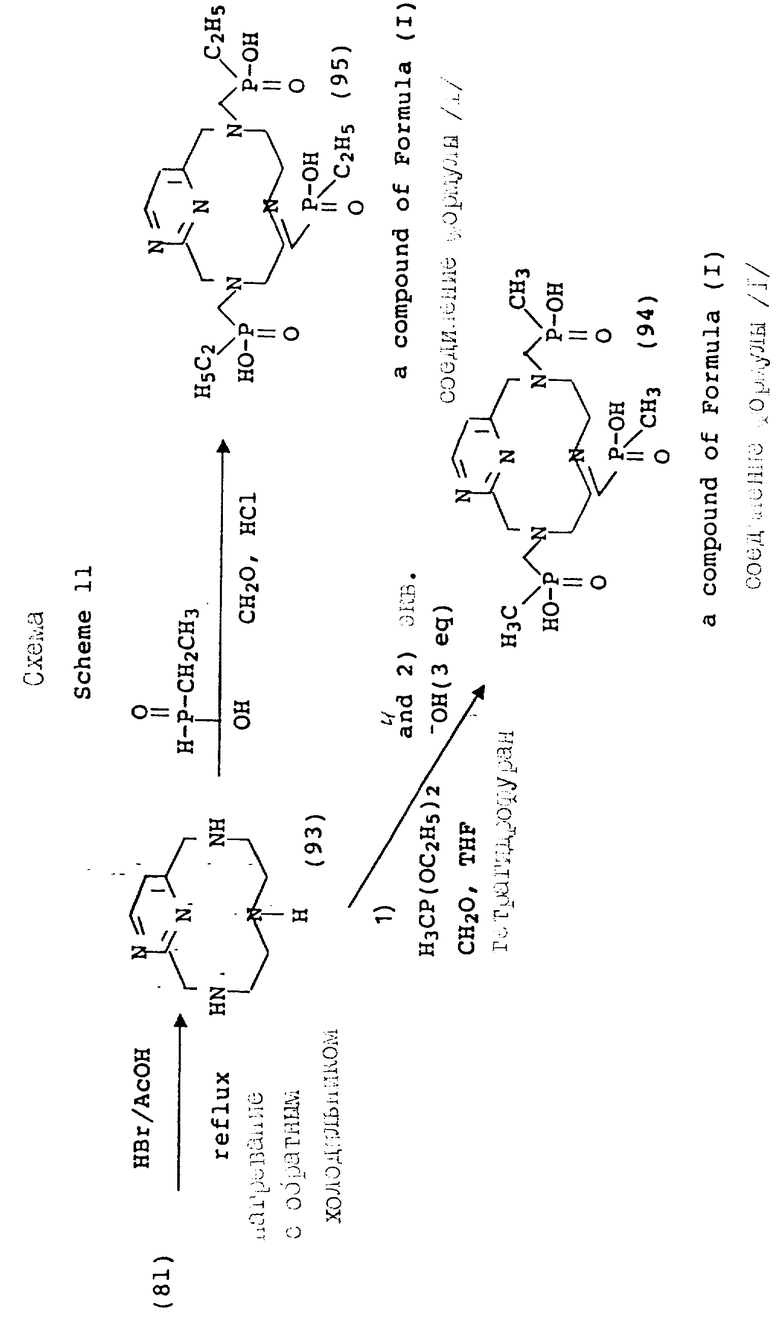
Согласно схеме 3 макроциклические метилфосфиновые кислоты (10 и 11) получаются в условиях, аналогичных условиям, описанным на схеме 2. Конденсация с использованием диэтоксиметилфосфина в качестве нуклеофильного агента и параформальдегида может проводиться в растворителях, таких как тетрагидрофуран, диметилформамид, диоксан, ацетонитрил или в спиртовых средах. Получающийся в результате фосфинатный эфир затем гидролизуется в кислотных (6 норм. HCl, 80 - 100oC) или основных (стехиометрические количества основания, 40 - 100oC) условиях, давая соответствующую метилфосфоновую кислоту. Альтернативно, для получения фосфинатных производных, имеющих повышенный липофильный характер, может использоваться метод, разработанный авторами A.D. Sherry и др. (Inorg. Chem., 1991) с использованием этилфосфоновой кислоты, получаемой ин ситу.
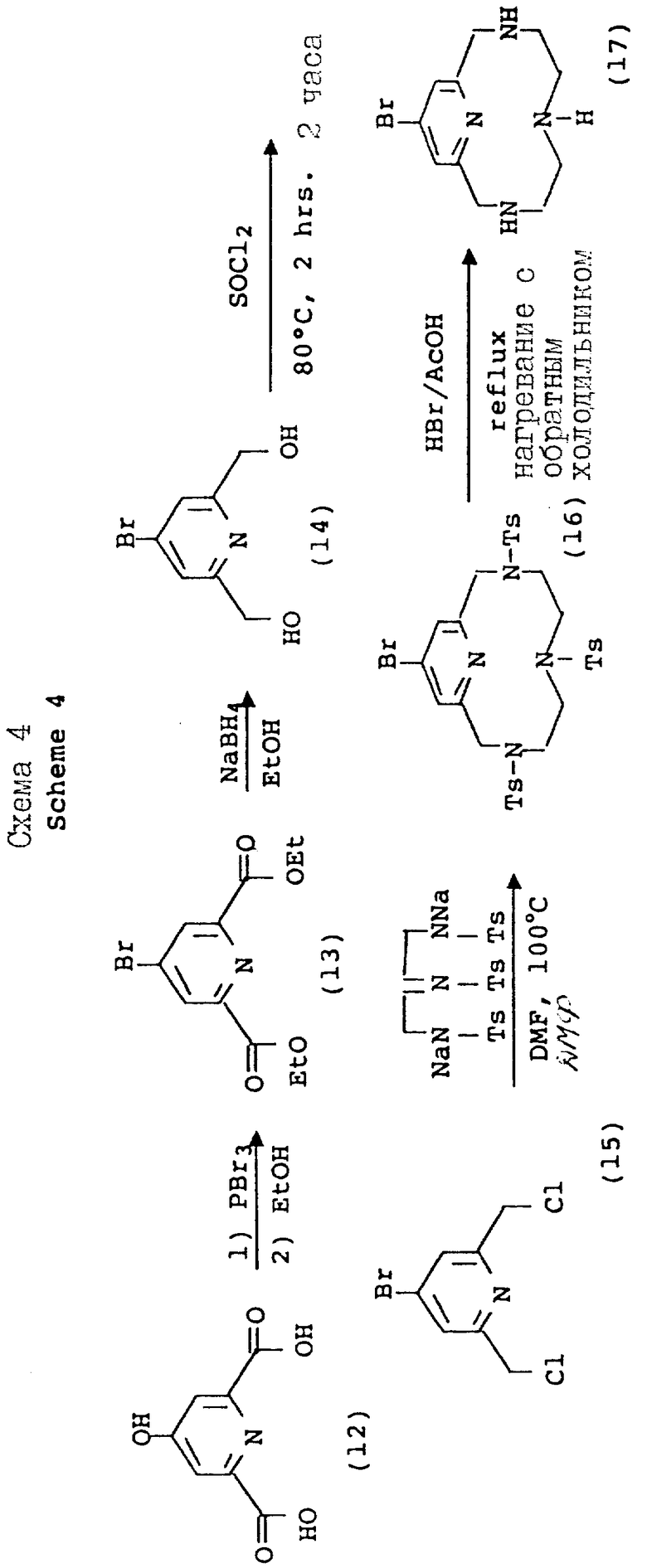
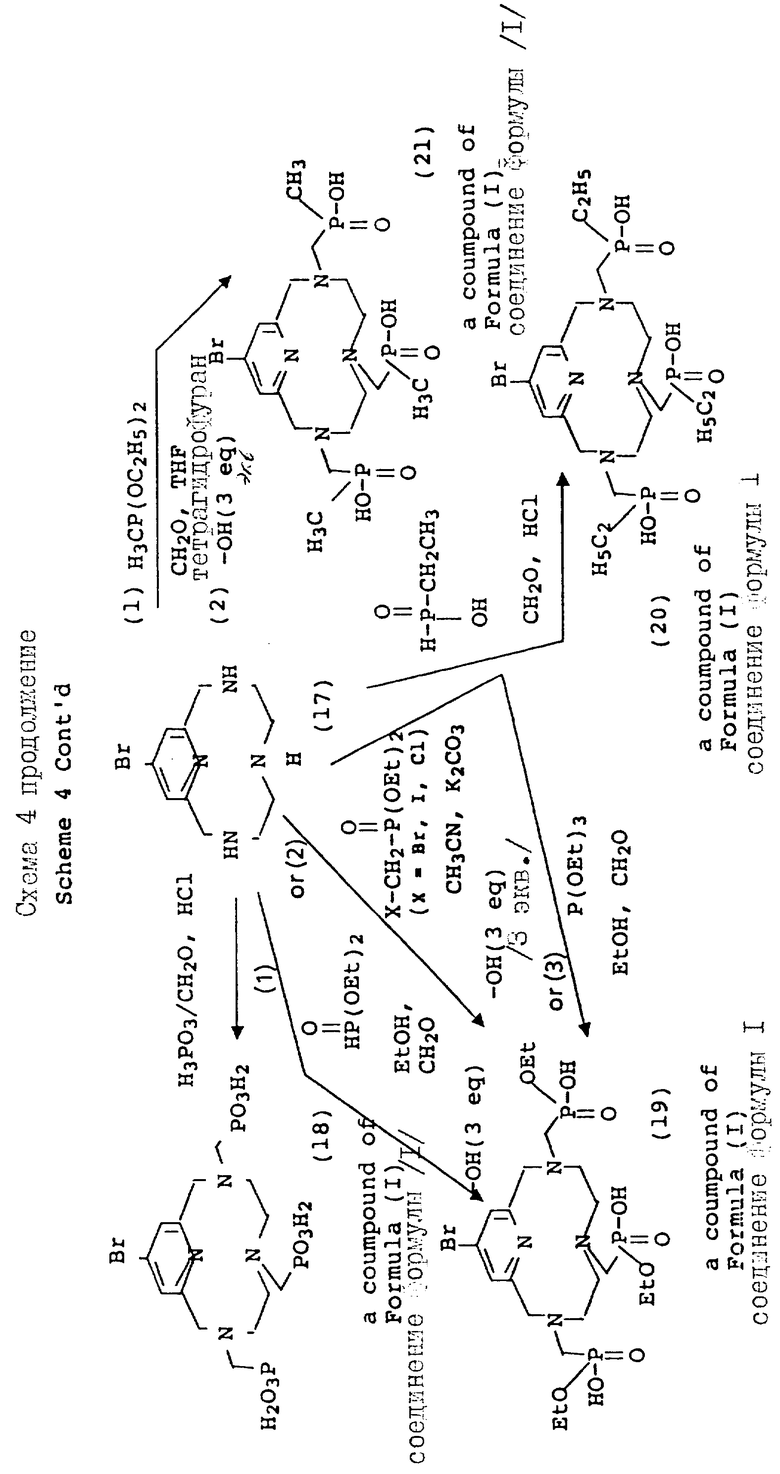
Схема 4 иллюстрирует введение дополнительной функциональности в пиридиновое звено 12-членного тетраазамакроцикла. Так, хелидамовая кислота (Сигма Кемикал Компани, 12) может превращаться в бис-галоидметильное производное (13), имеющее соответствующее замещение в 4-положении пиридила. Преобразования, ведущие к данному промежуточному соединению, являются общими по своей природе, и его получение описывается авторами Takalo и др. (Acta Chemica Scandinavica B 42, 373 - 377, 1988). Последующая макроциклизация с использованием данного промежуточного продукта (15) может проводиться с помощью стандартной реакции в ДМФ при 100oC с натрий-тритозилированным триамином, или при комнатной температуре с тритозилированным свободным основанием и карбонатом калия, карбонатом натрия или карбонатом цезия в качестве основания, давая продукты аналогичным продуктам, описанным ранее. Последующие реакции, ведущие к фосфонатным полу-кислотам и фосфинатной функциональности, являются идентичными преобразованиями и условиях, описанным на предыдущих схемах.
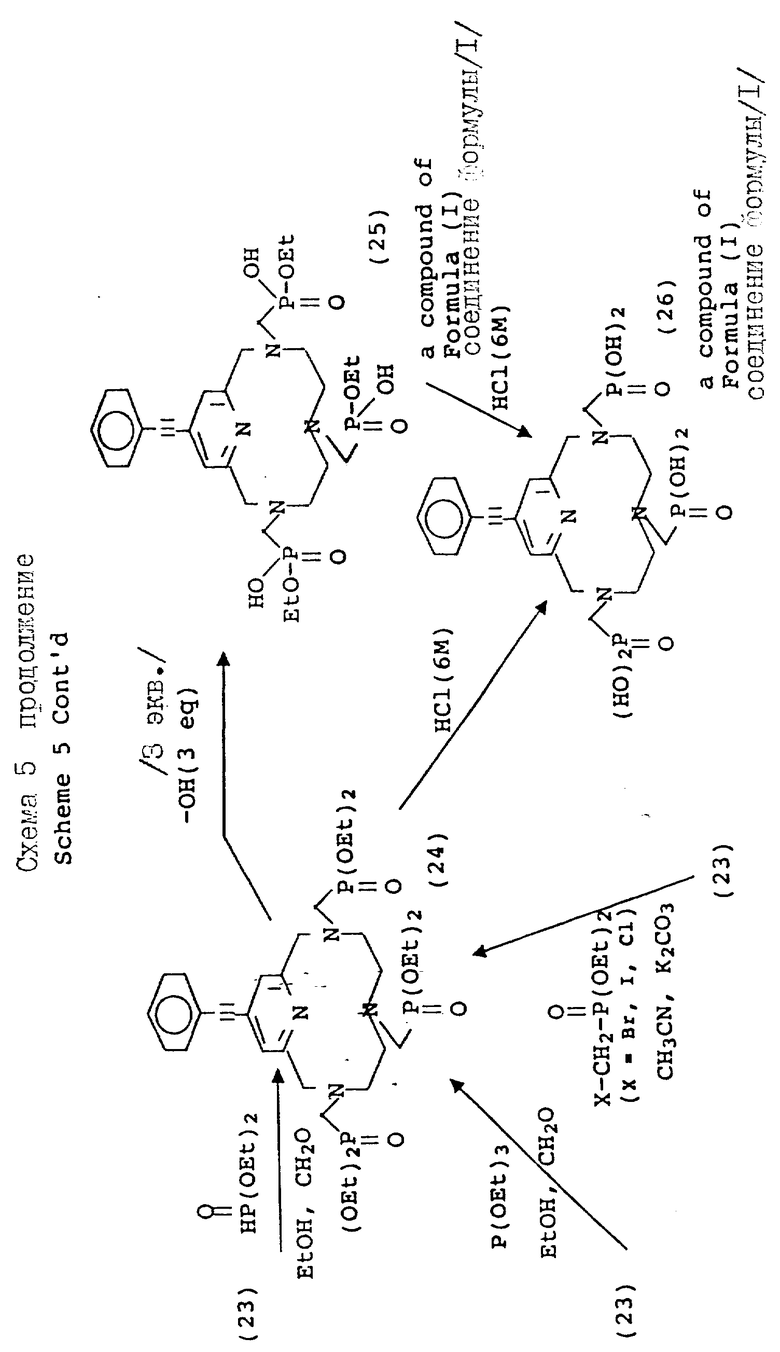
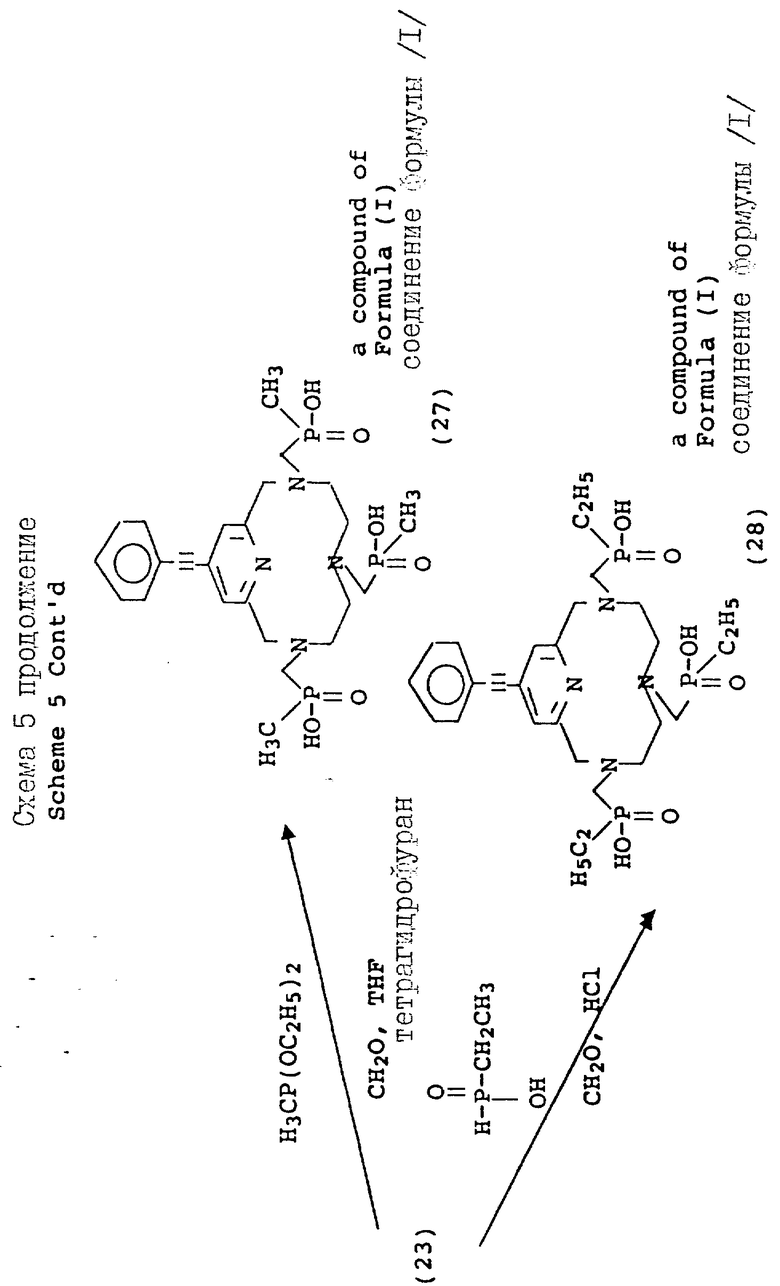
На схеме 4 описываются 4-галоидпиридил замещенные макроциклы (16), которые могут подвергаться замещению в 4 положении пиридильного фрагмента, как описано на схеме 5. Так, органометаллические Pd (II) комплексы могут применяться для ускорения или облегчения реакции сочетания между фенилацетиленом и производными фенилацетилена и пиридильным макроциклом. Согласно типичным условиям реакции для данного преобразования используются безводные условия с триэтиламином в качестве растворителя и температурой реакции примерно между 10 и 30oC для достижения оптимальных выходов. Идентичный продукт может также получаться при использовании фенилацетилида Cu /I/ в безводном пиридине при температуре между примерно 80 и 110oC. Помимо этого, для проведения замещения в пиридиновом ядре могут применяться стандартные приемы анионного алкилирования, с применением, например, алкоголятов натрия в ДМФ или диоксане при температуре примерно от 80 до 100oC, с использованием оснований, таких как карбонат калия или гидроокись натрия. Макроциклические тетраазамакроциклические соединения (24, 25, 26, 27, 28), дериватизированные данным образом, совместимы с преобразованиями, описанными на предыдущих схемах, дающими в результате аналогичные фосфонатные хелатирующие агенты.
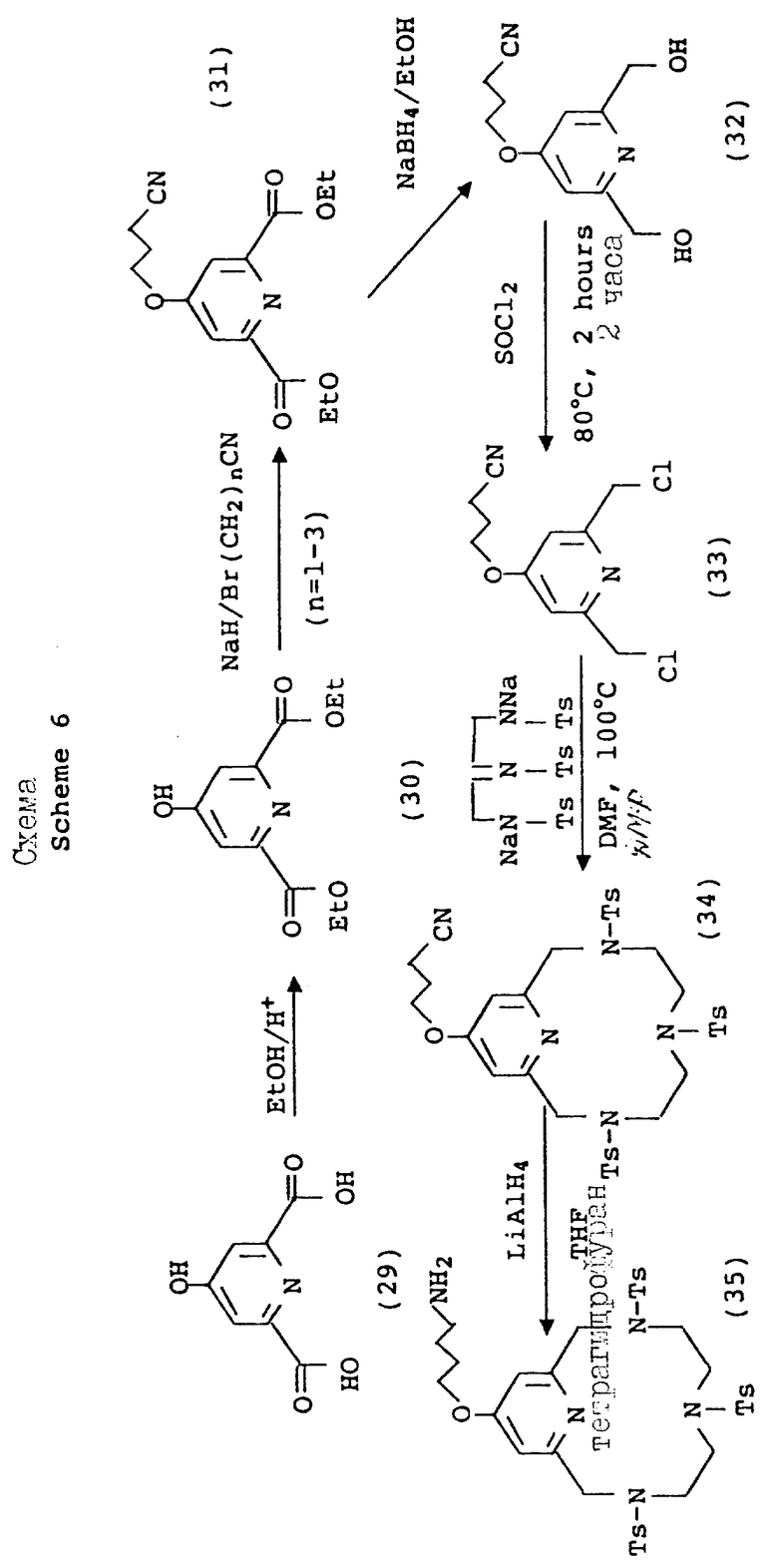
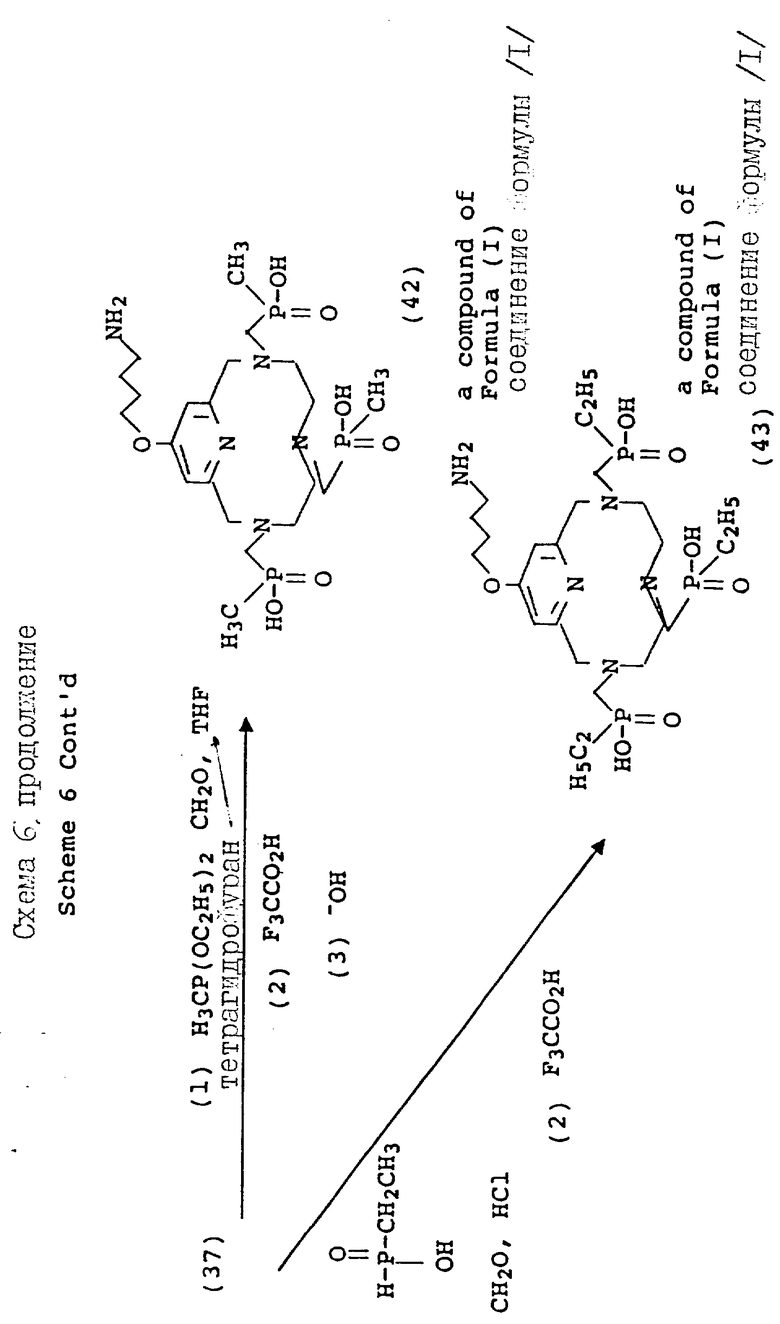
Варьирование 4-пиридильного замещения описывается на схеме 6, по которой 4-гидроксипиридильный фрагмент (29) алкилируется бромалкилнитрилом, давая связанный с промежуточным эфиром натрия (31), который впоследствии превращается в макроциклическую структуру. Данный тип приема алкилирования наилучшим образом проводится в безводных условиях в апротонном растворителе, таком как тетрагидрофуран (ТГФ) и с использованием ненуклеофильного основания, такого как гидрид натрия или бутиллитий, при температурах между примерно -30 и 80oC. Применимость данного подхода описана вторыми Chaubet и др. для ациклических аналогов (Tetrahedron Letters, 31 (40), 5729 - 5732, 1990). Макроциклический нитрил, полученный данным способом, может восстанавливаться в первичный амин (36) с помощью стандартных процедур с последующей защитой первичного амина 2-/трет-бутоксикарбонилоксиимино/-2-фенилацетонитрилом (BOC-ON; 37). Последующая функцтионализация макроциклических вторичных аминов (38, 39, 40, 41, 42, 43) может затем проводиться с помощью обсуждаемых приемов с дополнительным требованием удаления BOC защитной группы с использованием трифторуксусной кислоты, как описано на схеме 6.
Как проиллюстрировано на схеме 7, функционализация может также осуществляться в 3-положении пиридинового кольца в макроциклической структуре. Newkome и др. (Tetrahedron, 39(12), 2001 - 2008, 1983) ранее описали синтез этил 2,6-галоидметилникотината (45), который служит в качестве начального исходного материала в данном синтетическом способе. Так, тристозилированное макроциклическое промежуточное соединение (46) может детозилироваться в кислотных условиях (HBr) AcOH, 25 - 115oC) с одновременным гидролизом, давая производное никотиновой кислоты (48), или восстановлением сложного эфира в кипящем этаноле перед детозилированием приводит в результате к 3-гидроксиметильному промежуточному соединению (47). Никотиновокислотный макроцикл может затем замещаться согласно общей схеме функционализации вторичного амина, давая разнообразные типы фосфонатных хелатирующих агентов формулы 1 (49, 50, 51, 52, 53).
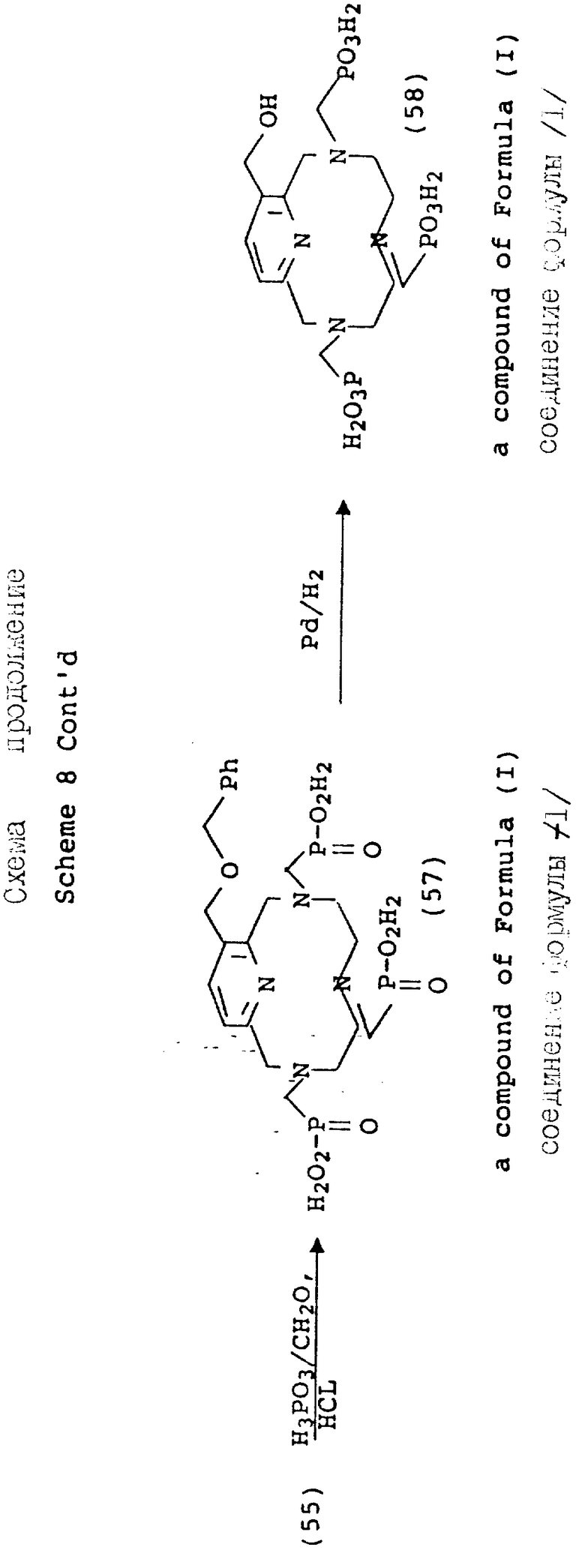
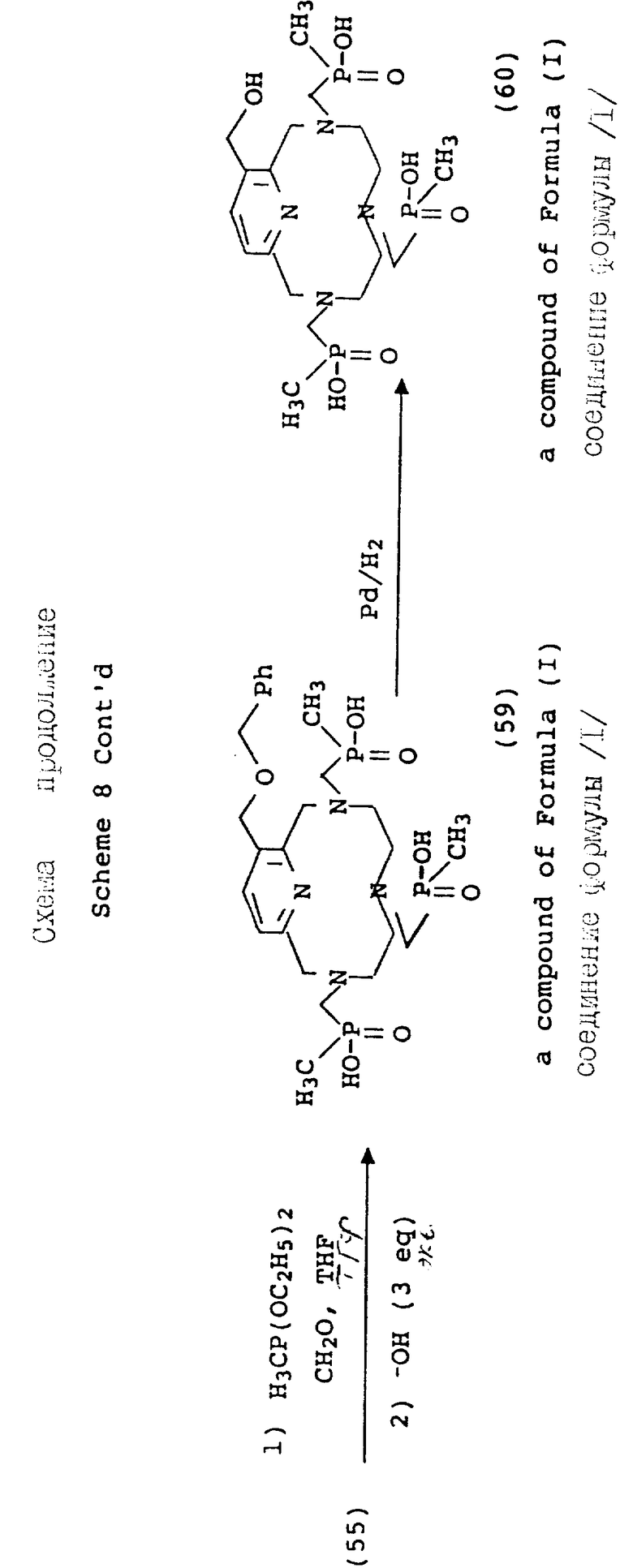
В противоположность сказанному, 3-гидроксиметильный аналог благоприятного защищается перед функционализацией макроциклических аминов. На схеме 8 показана бензильная (Bz) защитная группа, поскольку она должна быть стойкой к жестким кислотным условиям, имеющим место на стадии детозилирования. После того, как будет проведена функционализация вторичных аминов, как описано на предыдущих схемах, бензильная группа удаляется в мягких условиях каталитического гидрирования (58).
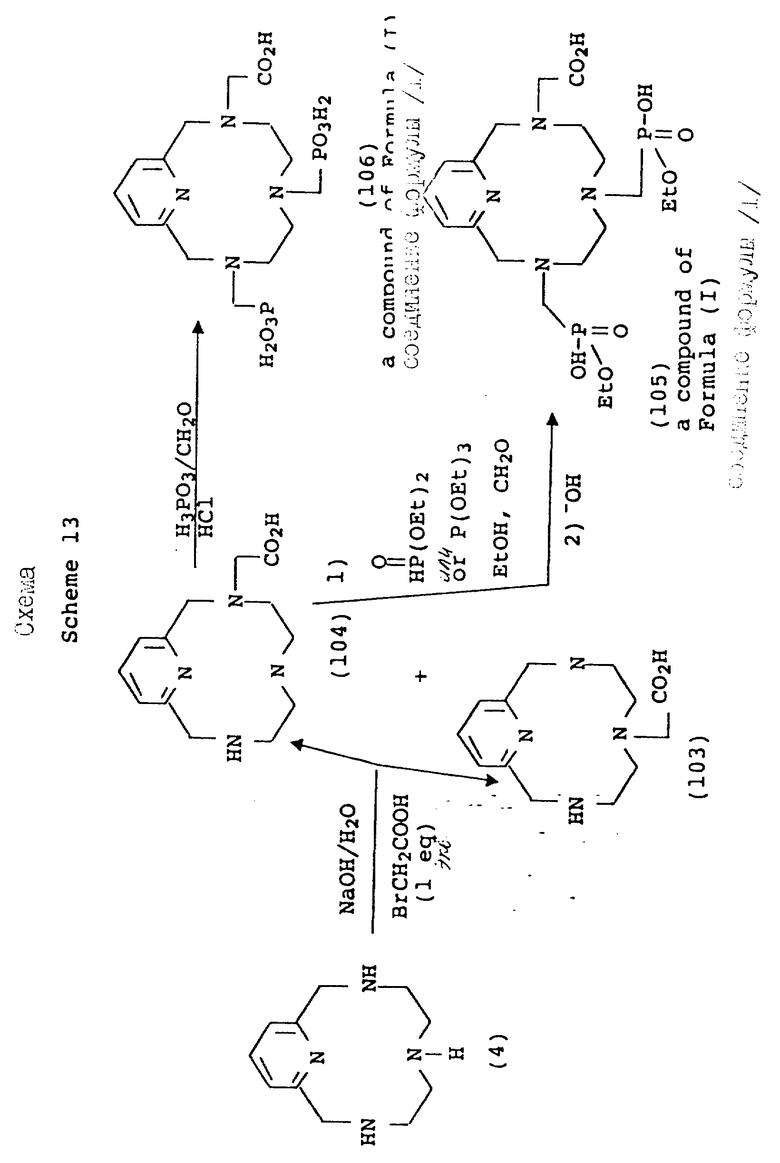
Микроциклические производные могут также получаться, как показано на схемах 12-14, когда в одной и той же молекуле присутствует, как карбоксилатная, так и фосфонатная хелатирующая функциональность. Так, различные степени карбоксилатной функциональности могут вводиться с помощью типичных процедур водного алкилирования с использованием бромуксусной кислоты. После данной стадии остальные амины могут фосфонометилироваться с помощью процедур, обсуждаемых на предыдущих схемах, с использованием формальдегида и фосфористой кислоты, диалкилфосфонатов или триалкилфосфитов.
На схемах 15 и 16 показан синтетический метод, по которому в одно из положений макроциклического азота вводится ароматический нитробензильный заместитель. В типичном случае макроциклический амин моно-N-функционализируется в органическом растворителе, таком как ацетонитрил или ДМФ, при комнатной температуре с использованием ненуклеофильного основания, такого как карбонат калия. Дополнительная функционализация остальных положений азота затем проводится с помощью методов и условий, описанных на предыдущих схемах. После введения желаемых хелатирующих фрагментов, нитро группа восстанавливается с использованием окиси платины и водорода в воде. В данной форме хелатирующий агент совместим с технологией конъюгирования, которая делает возможным присоединение к более крупным синтетическим или природным молекулам.
Схема 17 иллюстрирует синтез макроциклических соединений 4, при котором амины в положениях 3 и 9 вводятся в реакцию с по крайней мере двумя молями натриевой соли гидроксиметансульфоновой кислоты в воде при pH примерно 9, давая соответствующее макроциклическое соединение, в котором положения 3 и 9 представляют натриевую соль метансульфоновой кислоты (119). Группа сульфоновой кислоты затем замещается с использованием цианида натрия, образуя соответствующее цианометановое производное (120). Циано группа гидролизуется в карбоновую кислоту; или одновременно с добавлением фосфористой кислоты и формальдегида, или с помощью последовательной реакции с производным фосфористой кислоты и формальдегида с образованием фосфоновой кислоты в 6-положении (121), с последующим кислотным гидролизом, при повышенной температуре, цианатных групп и любого присутствующего производного фрагмента фосфористой кислоты. Получающееся в результате соединение представляет макроцикл с двумя группами карбоновой кислоты в положениях 3 и 9 и фосфоновокислотной группы в положении 6. Фосфонометилирование может также проводиться с помощью методов, обсуждаемых выше.
Ионы металлов, используемые для образования комплексов данного изобретения, представляют собой Gd+3, Mn+2, Fe+3 и являются промышленно или коммерчески доступными, например, поставляются фирмой Олдрич Кемикал Компани. Присутствующим анионом является галогенидный ион, предпочтительно хлоридный, или свободный от соли (окись металла).
Парамагнитный нуклид изобретения означает ион металла, который обнаруживает спин-ангулярный момент и/или орбитально-ангулярный момент. Два типа моментов объединяются, давая наблюдаемый парамагнитный момент способом, который зависит в огромной степени от атомов, несущих непарный электрон, и в меньшей степени,от окружения таких атомов. Парамагнитными нуклидами, которые найдены полезными в практике данного изобретения, являются гадолиний (Gd+3, железо (Fe+3) и марганец (Mn+2), причем, Gd+3 является предпочтительным.
Комплексы получаются по способам, хорошо известным в данной области техники. Так, например, смотрите работу Chelating Agents and Metal Chelates, Dwyer и Mellor, Academic Press (1964), глава 7. Смотрите также способы получения амино-кислот в работе Synthetic Production and Utilization of Amino Acids (изданий Камеко и др.), Джон Вили анд Санз (1974). Один из примеров получения комплекса включает взаимодействие бициклополиазамакроциклофосфоновой кислоты с металлическим ионом в водных условиях при pH от 5 до 7. Комплекс образуется с помощью химической связи и дает в результате стабильный парамагнитный нуклидный состав (композицию), например стабильный к дисассоциированию парамагнитного нуклида от лиганда.
Комплексы настоящего изобретения назначаются при молярном отношении лиганда к металлу по крайней мере около 1:1, предпочтительно от 1:1 до 3:1, более предпочтительно от 1:1 до 1,5:1. Большой избыток лиганда является нежелательным, поскольку незакомплексованный лиганд может быть токсичным для животного или может приводить в результате к остановке сердца или гипокальцемическим конвульсиям.
Антитела или фрагменты антител, которые могут использоваться в конъюгатах, описанных в данном описании, могут получаться с помощью приемов, хорошо известных в технике. Высоко специфичные моноклональные антитела могут получаться с помощью приемов гибридизации, хорошо известных в технике (Kohler and Milsten Nature, 256, 495-497 (1975); и Eur. J. Immunol., 6, 511-519, 1976). Такие антитела обычно имеют высоко специфичную реакционноспособность. В нацеленных на данное антитело конъюгатах могут использоваться антитела, направленные против любого желаемого антигена или гаптена. Предпочтительно антителами, которые используются в конъюгатах, являются моноклональные антитела, или их фрагменты, имеющие высокую специфичность к желаемому эпитопу (эпитопам). Антитела, используемые в настоящем изобретении, могут быть направлены, например, против опухолей, бактерий, грибков, вирусов, паразитов, микоплазмы, антигенов дифференциации и других антигенов клеточных мембран, патогенных поверхностных антигенов, токсинов, ферментов, аллергенов, лекарств и любых биологически активных молекул. Некоторыми примерами антител или фрагментов антител являются 1116-NS-19-9, 1116-NS-3d, 703D4, 704A1, CC49, СС83 и B72.3. Все эти антитела депонированы в АТСС. Более полный перечень можно найти в патенте США 4193983. Конъюгаты настоящего изобретения особенно предпочтительны для диагностики разнообразных раковых заболеваний.
Изобретение используется с физиологически приемлемым носителем, эксципиентом или заполнителем для него. Способы получения таких готовых препаративных форм хорошо известны. Препаративные формы могут быть в виде суспензии, инъецируемого раствора или других подходящих препаративных форм. Могут использоваться физиологически приемлемые суспендирующие среды с адъювантами или без них.
Для диагностики используется "эффективное количество" препаративной формы. Рассматриваются также методы диагностики ин виво, с использованием препаративных форм изобретения.
Другие области использования некоторых хелатирующих агентов настоящего изобретения включают удаление нежелательных металлов (например, железа) из организма, присоединение к полимерным носителям для различных целей, например, в качестве диагностических агентов, и удаление ионов металлов с помощью селективной экстракции. Лиганды формулы 1, имеющие по крайней мере в двух R радикалах T, равный T/O/R1OH, могут использоваться в меньших, чем стехиометрические количества. Сходные области использования известны для соединений (патенты США NN 2609390, 3331773, 3336221 и 3434969).
Изобретение дополнительно будет пояснено с учетом следующих ниже примеров, целью которых является чисто иллюстрация изобретения.
Некоторые термины, используемые в следующих ниже примерах, определяются следующим образом:
ЖХ = жидкостная хроматография, очистка осуществлялась при низком давлении с использованием системы Дионекс 2010, смонтированной с наполненной вручную Q-Сефарозойтм анионообменной колонкой (23х2 см).
ДМФ = диметилформамид,
AcOH = уксусная кислота,
1CP = индуктивно спаренная плазма,
г = грамм/ы/,
мг = миллиграммы,
кг = килограммы,
мл = миллилитр/ы/,
мкл = микролитр/ы/.
Общая процедура pH стабильности
Приготавливался исходный раствор 159Gd Cl3 (или 153 SmCl3) с помощью добавления 2 мкл 3•10-1 М 159 Gd Cl3 в 0,1 норм. HCl к 2 мл 3•10-4 М GdCl3 раствора носителя. Затем приготавливались соответствующие растворы лигандов в деионизированной воде. Затем приготавливались комплексы 1:1 лиганд/металл путем объединения лигандов, растворенных в 100 - 500 мкл деионизированной воды, с 2 мл исходного 159 GdCl3 раствора с последующим тщательным смешением с получением кислого раствора (рH 2). Величина pH раствора затем поднималась до 7,0 с использованием 0,1 норм. NaOH. Затем определялся процент металла в виде комплекса путем пропускания образца раствора комплекса через Сефадекстм G - 50 колонку, элюирования 4:1 молевым раствором (85% NaCl/NH4OH) и сбора 2х3 мл фракций. Количественный показатель радиоактивности в объединенных элюированных растворах затем сравнивался с количеством, оставшимся на смоле (на смоле оставался незакомплексованный металл). Профиль pH стабильности получался путем доведения величины pH аликвоты раствора комплекса с использованием IM NaOH или IM HCl и определения процента металла, существующего в виде комплекса, с использованием метода ионного обмена, описанного выше. По данным экспериментального сравнения известно, что результаты Sm идентичны в случае комплексообразования и биораспределения дигандов данного изобретения, исходные материалы.
Пример A. Получение 2,6-бис(хлорметил) пиридина.
К 100 мл тионилхлорида, который охлаждался (на ледяной бане), добавлялось 24 г (0,17 моль) 2,6-бис (гидроксиметил) пиридина. Спустя 30 мин реакционная смесь подогревалась до комнатной температуры, затем нагревалась с обратным холодильником в течение 1,5 ч. После охлаждения реакционной смеси до комнатной температуры твердое вещество, которое образовалось, отфильтровывалось, промывалось бензолом и сушилось в вакууме. Твердое вещество затем нейтрализовалось насыщенным бикарбонатом натрия, отфильтровывалось и сушилось, давая 23,1 г (71,5%) целевого продукта в виде не совсем белого кристаллического твердого вещества, т.пл. 74,5 - 75,5oC, и далее характеризовалось следующими данными:
1H ЯМР (CDCl3) δ 4,88 (с., 4H), 7,25 - 7,95 (м., 3H).
Пример B. Получение 3,6,9-трис/п-толилсульфонил/-3,6,9,15-тетраазабицикло [9.3.1] пентадека-1 (15), 11, 13-триена.
Раствор в диметилформамиде (92 мл) 6,9 г (11,4 ммоль) динатриевой соли 1,4,7-трис/п-толилсульфонил/диэтилентриамина перемешивался и нагревался до 100oC в атмосфере азота. К раствору добавлялось по каплям на протяжении 45 мин 2 г (11,4 ммоль) 2,6-бис(хлорметил/пиридина (полученного с помощью процедуры примера A) в 37 мл ДМФ. Когда добавление завершалось, реакционная смесь перемешивалась при 40oC в течение 12 ч. К реакционной смеси добавлялось затем 50 - 75 мл воды, что приводило в результате к немедленному растворению NaCl с последующим осаждением целевого продукта. Получающаяся в результате суспензия затем фильтровалась, и твердое вещество промывалось водой и сушилось в вакууме. Целевой продукт получался в виде светло-рыжеватокоричневого порошка, 6,5 г (86%), т. пл. 168 - 170oC разлож. и далее характеризовался следующим образом:
1H ЯМР (CDCl3) δ 2,40 (с., 3H), 2,44 (с., 6H), 2,75 (м., 4H), 3,30 (м., 4H), 4,28 (с., 4H), 7,27 (д., 2H), 7,34 (д., 4H), 7,43 (д., 2H), 7,65 (д., 4H), 7,75 (т., 1H); и
13C ЯМР δ 21,48, 47,29 50,37, 54,86, 124,19, 127,00, 127,11 129,73, 135,04, 135,74, 138,95, 143,42, 143,73, 155,15.
Пример C. Получение 3, 6, 9, 15-тетраазабицикло [9.3.1.] пентадека-1(15), 11, 15-триена.
Раствор HBr и AcOH приготавливался с помощью смешения 48% HBr и ледяной AcOH в соотношении 64: 35. К 112 мл HBr/AcOH смеси добавлялось 5,5 г (8,2 ммоль) 5,6,9-трис/п-толилсульфонил/-3,6,9,15-тетраазабицикло [9.3.1.] пентадека-1(15), 11,13-триена (полученного с помощью процедуры примера B), и реакционная смесь нагревалась в условиях мягкой дефлегмации при постоянном перемешивании в течение 72 ч. Реакционная смесь затем охлаждалась до комнатной температуры и концентрировалась до приблизительно 1/10 первоначального объема. Оставшийся раствор перемешивался энергично, и добавлялось 15 - 20 мл диэтилового эфира. Образовалось не совсем белое твердое вещество, которое отфильтровывалось диэтиловым эфиром и сушилось в вакууме. Сухая тетрагидробромидная соль затем растворялась в 10 мл воды, доводилась до pH 9,5 с помощью NaOH (50% вес/вес) и непрерывно экстрагировалась хлороформом в течение 4 часов. После сушки над безводным сульфатом натрия хлороформ выпаривался, давая светло-рыжеватокоричневое масло, которое постепенно кристаллизовалось при стоянии при комнатной температуре, давая 1,2 г (71%) целевого продукта, т. пл. 86 - 88oC, который характеризовался следующими данными:
1H ЯМР (CDCl3) δ 2,21 (м., 4H), 2,59 (м., 4H), 3,06 (с., 3H), 3,85 (с., 4H), 6,89 (д., 2H), 7,44 (т., 1H), и
13C ЯМР δ 48,73, 49,01, 53,63, 119,67, 136,29, 159,54.
Пример D. Получение 3,6,9,15-тетразабицикло [9,3,1]-пентадека-1 (15), 11,13-триен-3,9-диметиленсуофльновой кислоты.
Суспензия 500 мг (2,4 ммоль) 3,6,9,15-тетраазабицикло [9.3.1]-пентадека-1(15), 11,13-триена (полученного с помощью процедуры примера C) перемешивалась в 6 мл воды, и величина pH доводилась до 3 с использованием 6 М HCl. К смеси добавлялось 688 мг (5,1 ммоль) натриевой соли гидроксиметансульфоновой кислоты, и величина pH доводилась до 9 с помощью 50% водной гидроокиси натрия. После перемешивания в течение 3 ч при комнатной температуре данные 13C ЯМР указывали на полное превращение в целевой бис-метиленсульфоновокислотный продукт.
Пример E. Получение 3,6,9,15-тетраазабицикло [9.3.1]-пентадека-1 (15), 11,13-триен-3,9-диметиленнитрила.
К реакционной смеси, содержащей 3,6,9,15-тетраазабицикло [9.3.1]-пентадека-1(15), 11,13-триен-3,9-диметиленсульфоновую кислоту из примера D, добавлялось 47 мг (9,6 ммоль) цианида натрия. Реакционная смесь перемешивалась при комнатной температуре в течение 24 ч.
13C ЯМР данные показали, что преобразование в бис-нитрил завершалось. Реакционная смесь затем фильтровалась, экстрегировалась три раза 25 мл порциями хлороформа, сушилась над безводным сульфатом магния и концентрировалась, давая вязкое масло. Масло затем растворялось в хлороформе, растиралось с циклогексаном и концентрировалось, давая в виде белого порошка, 530 мг (78%) целевого диметиленнитрильного продукта.
Пример F. Получение 3 ,9-бис/натрий метиленсульфонат/-3,6,9,15-тетраазабицикло-[9.3.1] пентадека-1/15/, 11,13-триена (PC25).
Водный раствор (10,0 мл) 3,6,9,15-тетраазабицикло [9.3.1] пентадека-1(15), 11,13-триена (полученного с помощью процедуры примера C), 1,03 г (5,0 ммоль), добавлялось 0,5 мл концентрированной HCl, и смесь перемешивалась в течение 10 мин для обеспечения полного растворения. Получающийся в результате раствор имел величину pH 8,6. К раствору затем добавлялось 1,37 г (10,2 ммоль) HOCH2SO3Na с 5 мл деионизированной воды. Раствор нагревался при 60oC в течение 10 мин, и величина pH падала до 5,6. После охлаждения величина pH доводилась до 9,0 с помощью 1М водной гидроокиси натрия, затем осуществлялась лиофилизация, давая целевой продукт в виде белого твердого вещества с количественным выходом, которое характеризовалось следующими данными:
1H ЯМР (D2O) δ 2,87 (т, 4H), 3,18 (т., 4H), 8,85 (с., 4H), 4,11 (с., 4H), 7,03 (д., 2H), 7,55 (т., 1H); и
13C ЯМР (D2O) δ 48,52, 54,04, 58,92, 79,09, 123,90, 141,37, 161,89.
Пример G. Получение 3,9-бис(метиленнитрил)-3,6,9,15-тетраазабицикло [9.3.1]-пентадека-1(15), 11,13-триена.
К водному раствору, 10,0 мл, 3,9-бис/натрий метиленсульфонат/-3,6,9,15-тетраазабицикло [9.3.1] пентадека-1 (15), 11,13-триена (полученного с помощью процедуры примера F), 2,26 г (5 ммоль) добавлялось 0,6 г (12,24 ммоль) цианида натрия. Смесь перемешивалась в течение 3 ч при комнатной температуре. Величина pH реакционной смеси составляла примерно 10. Величина pH доводилась до выше 13 с помощью концентрированной водной гидроокиси натрия. Продукт выпадал в осадок и экстрагировался хлороформом (3х20 мл), сушился над безводным сульфатом магния и фильтровался. После удаления растворителя и концентрирования в вакууме желаемый продукт выделялся в виде воскообразного белого порошка, 1,0 г (71%) и характеризовался следующими данными:
1H ЯМР (CDCl3) δ 2,03 (шир., с., 4H), 2,64 (м., 4H), 3,82 (с., 4H), 3,90 (с., 4H), 7,14 (д., 2H), 7,62 (т., 1H); и
13C ЯМР (CDCl3) δ 46,08, 46,64, 52,89, 60,78, 115,31, 122,02, 137,57, 157,33.
Пример H. Получение 3,9-бис/метиленнитрил/-6- /метилендиметилфосфонат/-3,6,9,15-тетраазабицикло [9.3.1] -пентадека-1(15), 11,13-триен-3,9-диметиленнитрила.
3,9-бис/метиленнитрил/-3,6,9,15-тетраазабицикло [9.3.1] пентадека-1(15), 11, 13-триен (полученный с помощью процедуры примера G), 285 мг (1,0 ммоль), объединялся с 60 мл (2,0 ммоль, избыток) параформальдегида и 0,354 мл (372 мг, 3,0 ммоль, избыток) триметилфосфита. Смесь осторожно перемешивалась в течение 10 мин с получением суспензии, затем нагревалась до 90oC в течение 1 ч. После того, как избыток реагентов и побочные продукты удалялись в вакууме (1 ч при 125oC/0,01 мм рт. ст.) получающийся в результате темно-коричневый остаток растворялся в 20 мл хлороформа и промывался деионизированной водой (5х15 мл). Органический слой сушился над безводным сульфатом магния, фильтровался, и избыток растворителей выпаривался в вакууме, давая требуемый продукт в виде желтого воскообразного твердого вещества, 168 мг (41%), которое характеризовалось следующими данными:
1H ЯМР (CDCl3) δ 2,61 (шир.с., 8H), 2,73 (д., 2H), 3,62 и 3,68 (с., 6H), 3,73 (с., 4H), 3,84 (с., 4H), 7,06 (д., 2H), 7,57 (т., 1H); и
13C ЯМР (CDCl3) δ 44,44, 50,74, 51,03, 51,85, 52,51, 60,28, 115,61, 122,27, 137,24, 156,61.
Пример 1. Получение 3,6,9,15-тетраазабицикло[9.3.1] пентадека-1(15), 11,13-триен-3,6,9-метилендиэтилфосфоната.
Смесь 1 г (4,8 ммоль) 3,6,9,15-тетраазабицикло[9.3.1] пентадека-1(15), 11,13-триена (полученного с помощью процедуры примера C), 4,8 г (28,8 ммоль) триэтилфосфита и 864 мг (28,8 ммоль) параформальдегида нагревалась при 90oC при постоянном перемешивании в течение 45 мин. Реакционная смесь концентрировалась в вакууме, и вязкое масло хроматографировалось на колонке с основной окисью алюминия, при элюировании хлороформом. После концентрирования органического элюента желаемый продукт выделялся в виде бесцветного масла, 2,0 г (64%) и характеризовался следующими данными:
1H ЯМР (CDCl3) δ 1,23 (м., 18H), 2,77 (м., 12H), 3,04 (д., 6H), 4,13 (м. , 12H), 7,17 (д., 2H), 7,60 (т., 1H); и
13C ЯМР (CDCl3) δ 16,43, 50,03, 50,31, 50,43, 50,77, 51,23, 51,38, 52,63, 53,30, 60,86, 60,92, 61,63, 61,74, 61,83, 61,93, 62,32, 76,46, 76,97, 77,18, 77,48, 122,50, 137,10, 157,18; и
31P ЯМР δ 24,92 (с., 2P), 24,97 (с., 1P).
Пример J. Получение 3,6,9,15-тетраазабицикло [9.3.1] пентадека-1(15), 11,13-триен-3,6,9-метиленди/н-пропил/фосфоната.
К 3 мл хлороформ (диоксанового раствора (1:1) добавлялось 100 мг (0,48 ммоль) 3,6,9,15-тетраазабицикло[9.3.1] пентадека-1(15), 11,13-триена (полученного с помощью процедуры примера C), 318 мл (1,53 ммоль) трипропилфосфита и 46 мг (1,53 ммоль) параформальдегида. Реакционная смесь нагревалась при 90oC при перемешивании в течение 1 ч. Получающийся в результате гомогенный раствор концентрировался в вакууме, давая вязкое масло, которое хроматографировалось на колонке с нейтральной окисью алюминия, при элюировании хлороформом. После концентрирования органического элюента желаемый продукт выделялся в виде бесцветного масла, 320 мг (90%), и характеризовался следующими данными:
1H ЯМР (CDCl3) δ 0,88 (м., 18H), 1,61 (м., 12H), 2,72 (м., 12H), 3,03 (д., 6H), 3,97 (м., 12H), 7,13 (д.. 2H), 7,55 (т., 1H); и
13C ЯМР (CDCl3) δ 9,96, 23,73, 49,84, 50,14, 50,26, 50,57, 51,11, 51,23, 52,43, 53,01, 60,78, 60,84, 67,27, 67,40, 122,48, 137,04, 157,16; и
31P ЯМР δ 24,98 (3P).
Пример K. Получение 3,6,9,15-тетраазабицикло[9.3.1] пентадека-1(15), 11,13-триен-3,6,9-метиленди/н-бутил/фосфоната.
Смесь 500 мг (2,4 ммоль) 3,6,9,15-тетраазабицикло[9.3.1] пентадека-1(15), 11,13-триена (полученного с помощью процедуры примера C), 2,0 г (8 ммоль) трибутилфосфита и 240 мг (8 моль) параформальдегида нагревалась при 100oC при перемешивании в течение 1 ч. Получающийся в результате вязкий раствор концентрировался в вакууме, давая масло, которое хроматографировалось на колонке с основной окисью алюминия при элюировании хлороформом. После концентрирования органического элюента желаемый продукт выделялся в виде бесцветного масла, 1,25 г (65%), которого характеризовалось следующими данными:
1H ЯМР (CDCl3) δ 0,84 (м., 18H), 1,27 (м., 12H), 1,58 (м., 12H), 2,57 (м., 12H), 3,01 (д.. 6H), 3,99 (м., 12H), 7,12 (д., 2H), 7,54 (т., 1H); и
13H ЯМР (CDCl3) δ 13,42, 13,46, 18,50, 18,59, 32,16, 32,43, 49,88, 50,03, 50,16, 50,63, 51,11, 51,27, 52,48, 53,16, 60,71, 60,78, 65,38, 65,48, 65,58, 122,46, 136,96, 157,14; и
31P ЯМР δ 24,88 (2P), 24,93 (1P).
Пример L. Получение 3,6,9,15-тетраазабицикло [9.3.1] пентадека-1(15), 11,13-триен-3[(4-нитрофенил)метилацетата].
К раствору 2,5 мл хлороформа, который быстро перемешивался, и 200 мг (0,97 ммоль) 3,6,9,15-тетраазабицикло [9.3.1] пентадека-1(15), 11,13-триена (полученного с помощью процедуры примера C) добавлялось в виде одной порции 266 мг (0,97 ммоль) бром-/4-нитрофенил/метилацетата и 2,5 мл хлороформа. Реакционная смесь перемешивалась в течение 24 ч при комнатной температуре. Раствор концентрировался в вакууме, давая полутвердое вещество, которое хроматографировалось на силикагельной колонке при элюировании смесью хлороформ/метанол/гидроокись аммония (16: 4: 1). После концентрирования органического элюента выделялся желаемый продукт в виде светло-желтого твердого вещества, 250 мг (64%), которое характеризовалось следующими данными:
13C ЯМР (CDCl3) δ 45,67, 45,90, 45,97, 51,65, 52,08, 52,28, 53,78, 69,54, 119,03, 119,23, 122,85, 130,30, 137,06, 143,27, 147,05, 159,59, 160,41, 171,70.
Конечные продукты
Пример 1. Получение 3, 6, 9, 15-тетраазабицикло [9.3.1] пентадека-1 (15), 11, 13-триен-3,6,9-триметиленфосфоновой кислоты (РСТМР).
Смесь 2,06 г (10 ммоль) 3, 6, 9, 15-тетраазабицикло [9.3.1]-пентадека-1(15), 11, 13-триена (полученного с помощью процедуры примера C), 11,3 г (138 ммоль) фосфорной кислоты и 15 г (152 ммоль) концентрированной HCl нагревалась до осторожного кипения (103oC) при постоянном перемешивании с последующим добавлением по каплям (2 мл/мин) 12,2 г (150 ммоль, 15 мл) водного формальдегида (37%). После полного добавления реакционная смесь перемешивались при температуре дефлегмации в течение 16 ч, охлаждалась до комнатной температуры и концентрировалась до густого вязкого масла. Продукт затем очищался с помощью ЖХ анионообменной хроматографии (0-30%-ная муравьиная кислота, 3 мл/мин, время удержания = 32 мин). Объединенные фракции сушились вымораживанием, давая 4,8 г (99%) целевого продукта в виде белого твердого вещества, т.пл. 275-280oC, которое характеризовалось следующими данными:
1H ЯМР (D2O) δ 2,83 (м., 6H), 3,46 (м., 10H), 7,28 (д., 2H) 7,78 (т., 1H); и
13C ЯМР δ 53,61, 53,81, 55,27, 57,93, 62,20, 125,48, 143,08, 152,31; и
31P δ 8,12 (2P), 19,81 (1P).
Пример 2. Получение комплекса 153 Sm-3,6,9,15-тетраазабицикло [9.3.1] пентадека-1(15), 11, 13-триен-3,6,9-триметиленфосфоновой кислоты (153Sm-РСТМР).
Раствор лиганда примера 1 приготавливался с помощью растворения 3,8 мг лиганда на 0,517 мл деионизированной воды (pH 2). Затем приготавливался 1:1 лиганд/металл комплекс с помощью объединения 40 мкл раствора лиганда с 2 мл водного SmCl3•H2O (3•10-4М в 0,01 норм. HCl), содержащего изотопный индикатор 153 SmCl3. После тщательного смешения определялся процент металла в виде комплекса путем пропускания образца раствора комплекса через колонку с Сефадекстм при элюировании смесью 4:1 солевого раствора (0,85% NaCl (NH4OH) и сбора 2х3 мл фракций. Количество радиоактивности в объединенных растворах после элюирования сравнивалось с величиной вещества, оставшегося на смоле. В этих условиях комплекс удалялся элюентом, а незакомплексованный металл остается на смоле. По данному методу определялось, что комплексообразование составило 98%. Образец раствора, который пропускался через смолу, использовался для исследования pH. Затем определялась pH стабильность с использованием общей процедуры, описанной выше.
Пример 3. Получение 3,9-диуксусная кислота-6-/метиленфосфоновая кислота/-3,6,9,15-тетраазабицикло [9.3.1] пентадека-1 (15), 11, 13-триена (РС2А1Р).
Концентрированный солянокислотный раствор (37%, 5 мл) 3,9-бис/метиленнитрил/-6-/метилендиметилфосфонат/-3,6,9,15-тетраазабицикло [9.3.1]пентадека-1(15), 11, 13-триена (полученного в примере H), 168 мг (1,0 ммоль), нагревался в условиях дефлегмации в течение 16 ч. После охлаждения раствор выпаривался досуха, с последующим совместным выпариванием и деионизированной водой (2х10 мл) для удаления избытка соляной кислоты. Конечный продукт выделялся в виде темно-коричневого твердого вещества после лиофилизации концентрированного водного раствора, которое характеризовалось следующими данными:
1H ЯМР (D2О) δ 2,68 (шир.с., 4H), 3,31 (шир.с., 4H), 4,08 (с., 4H), 4,55 (с., 4H), 7,16 (д., 2H), 7,68 (т., 1H); и
13C ЯМР (D2O) δ 52,35, 54,04, 57,02, 59,24, 62,26, 125,52, 143,64, 152,36, 171,54; и
31P ЯМР (D2O) δ 20,03.
Пример 4. Получение 3,6,9,15-тетраазабицикло [9.3.1.] пентадека-1(15), 11,13-триен-3,6,9-метиленэтилфосфонат трис (калиевой соли (РМЕНЕ).
К водному 0,1 норм. раствору гидроокиси калия (2 мл) добавлялось 250 мг (0,38 ммоль) 3,6,9,15-тетраазабицикло [9.3.1] пентадека-1(15), 11,13-триен-3,6,9-метилендиэтилфосфоната (полученного с помощью процедуры примера 1). Раствор нагревался при 90oC в течение 5 ч. Реакционная смесь охлаждалась до комнатной температуры, фильтровалась, сушилась вымораживанием, давая целевой продукт в виде не совсем белого твердого вещества, 252 мг (97%), который характеризовался следующими данными:
13C ЯМР (D2O) δ 18,98, 19,82, 51,78, 52,06, 53,08, 54,46, 54,68, 57,01, 58,22, 60,24, 63,19, 63,25, 63,36, 63,49, 63,59, 63,95, 64,18, 64,25, 66,80, 126,62, 141,63, 159,40; и
31P ЯМР δ 20,58 (с., 2P), 20,78 (с., 1P).
Пример 5. Получение 3,6,9,15-тетраазабицикло [9.3.1] пентадека-1(15), 11,13-триен-3,6,9-метилен/н-пропил/фосфонат трис/калиевой соли (РМРНЕ).
К водному раствору гидроокиси калия (0,5 мл 1 норм./диоксан /0,5 мл/) добавлялось 81 мг (0,108 ммоль) 3,6,9,15-тетраазабицикло [9.3.1]-пентадека-1(15), 11,13-триен-3,6,9-метилен/н-пропил/фосфоната /полученного с помощью процедуры примера J). Раствор нагревался в условиях дефлегмации в течение 24 ч. Реакционная смесь охлаждалась до комнатной температуры и экстрагировалась диэтиловым эфиром. Эфирный экстракт затем концентрировался в вакууме, давая желаемый продукт в виде не совсем белого твердого вещества, 48,6 мг (60%), который характеризовался следующими данными:
31P ЯМР δ 20,49 (с., 3P).
Пример 6. Получение 3,6,9,15-тетраазабицикло [9.3.1] пентадека-1(15), 11.13-триен-3,6,9-метилен/н-бутил/фосфоната трис/калиевой соли/ (PMBHE).
К водному раствору 35 мл 1 норм. гидроокиси калия добавлялось 3,21 г (3,88 ммоль) 3,6,9,15-тетраазабицикло /9.3.1/ пентадека-1 (15),11.13-триен-3,6,9-метиленди/н-бутил/фосфаната (полученного с помощью процедуры примера К). Раствор нагревался в условиях дефлегмации в течение 5 дней. Реакционная смесь охлаждалась до комнатной температуры, фильтровалась, и фильтрат подвергался сушке вымораживанием, давая окрашенное в кремовый цвет твердое вещество. Данное вещество затем суспендировалось в 160 мл метанола и перемешивалось в течение 12 ч при комнатной температуре. Суспензия затем фильтровалась и фильтрат концентрировался, давая полу-твердое вещество. Данное вещество бралось в 150 мл хлороформа м сушилось над безводным сульфатом натрия и фильтровалось. После концентрирования в вакууме продукт выделялся в виде не совсем белого твердого вещества, 1,86 г (62%), и характеризовался следующими данными:
IH ЯМР (D2O) δ, 0,68 (м., 9H), 1,14 (м., 6H), 1,37 (м., 6H), 2,76 (д., 6H), 3,41 (м., 12H), 3,73 (м., 6H), 7,24 (д., 2H), 7,76 (т., 1H); и
13C ЯРМ (D2O) δ 15,76, 15,80, 21,12, 21,20, 34,96, 35,06, 35,14, 52,08, 52,53, 53,38, 53,48, 54,49, 54,75, 57,70, 57,76, 61,86, 67,65, 67,75, 67,98, 68,08, 125,15, 142,93, 152,25; и
31P ЯРМ δ 9,73 (с., 2P), 21,00 (с., 1P).
Пример 7. Получение 3,6,9,15-тетраазабицикло[9.3.1] пентадека-1(15), 11,13-триен- 3[(4-нитрофенил)метилацетат]-6,9-метилендиэтилфосфоната.
Раствор 250 мг (0,62 ммоль, 3,6,9,15-тетраазабицикло [9.3.1] пентадека-1 (15), 11,13-триен-3-[(4-нитрофенил) метилацетата] (полученного с помощью процедуры примера L), 624 мг (3,7 ммоль) триэтилфосфита и 111 мг (3,7 ммоль) параформальдегида перемешивался при 100oC в течение 1 ч. Получающийся в результате гомогенный раствор концентрировался в вакууме, давая вязкое масло. Масло растворялось в 10 мл хлороформа и промывалось водой (3х5 мл). Органический слой сушился над безводным сульфатом магния, фильтровался, и фильтрат концентрировался в вакууме, давая продукт в виде вязкого масла, 326 мг (96%), который характеризовался следующими данными:
31P ЯРМ (CDCl3) δ 24,67, (с., 2P), 24,88 (с., 1P).
Биораспределение. Общая процедура.
Крысы Sprague Dawley оставлялись для акклиматизации в течение пяти дней, затем инъецировались 100 мкл раствора комплекса через хвостовую вену. Во время инъекции крысы весили между 150 и 200 г. Спустя 30 мин крыс убивали путем цервикального смещения и разрезали. Количество радиоактивности каждой ткани определялось путем подсчета в NaI сцинтилляционном счетчике, подсоединенном к многоканальному анализатору. Число подсчетов сравнивалось с подсчетами в 100 мкл стандартных пробах для того, чтобы определить процент дозы в каждой ткани или органе.
Процентная доза в крови оценивалась при принятии крови за 7% от веса тела. Процентная доза в кости оценивалась путем умножения процентной дозы в бедрах на 25. Процентная доза в мышцах оценивалась при принятии мышц за 43% от веса тела.
В дополнение к биораспределению в органах хелаты соединения формулы I оценивались на эффективность локализации в костях, поскольку фосфонаты известны за их способность связываться в гидроксиапатит.
Пример 1. Процент инъецированной дозы комплекса примера 2 (153 Sm-PCTMP) в нескольких тканях дан ниже). Числовые величины представляют среднюю величину от минимум 3 крыс через 2 ч после инъекции:
Ткань - Средняя величина
Кости - 34,87
Печень - 0,99
Почки - 1,42
Селезенка - 0,07
Мышцы - 4,77
Кровь - 6,27
Пример 2. Процент инъецированной дозы комплекса примера 5 (153 Sm-PMPHE) в нескольких тканях дан ниже. Числовые величины представляют среднее от минимум 3 крыс через 2 ч после инъекции:
Ткань - Среднее
Кости - 10,86
Печень - 4,14
Почки - 1,55
Селезенка - 0,05
Мышцы - 1,19
Кровь - 0,25
Сердце - 0,12
Легкие - 0,12
Головной мозг - 0,00
Желудок - 0,44
Тонкий кишечник - 10,71
Толстая кишка - 2,17
Пример 3. Процент инъецированной дозы комплекса примера 6 (153 Sm-PMBHE) в нескольких тканях дан ниже. Числовые величины представляют среднее от минимум 3 крыс через 2 ч после инъекции:
Ткань - Среднее
Кости - 3,73
Печень - 2,70
Почки - 0,43
Селезенка - 0,05
Мышцы - 1,09
Кровь - 0,14
Сердце - 0,02
Легкие - 0,04
Головной мозг - 0,00
Желудок - 0,08
Тонкая кишка - 57,89
Толстая кишка - 0,77
Пример 4. Процент инъецированной дозы комплекса примера 3 (153 Sm-PC2A1) в некоторых тканях (дан ниже). Числовые значения представляют среднее от минимум 3 крыс через 2 ч после инъекции:
Ткань - Среднее
Кости - 47,98
Печень - 1,46
Почки - 0,93
Селезенка - 0,02
Мышцы - 1,00
Кровь - 0,36
Сердце - 0,04
Легкие - 0,06
Головной мозг - 0,01
Желудок - 0,25
Тонкая кишка - 13,10
Толстая кишка - 0,12
Эксперименты по визуализации. Общая процедура.
Сначала приготавливались инъецируемые растворы (0,5М) путем растворения соответствующего количества каждого комплекса в 2 мл деионизированной воды. Величины pH растворов затем доводились до 7,4 с использованием 1М HCl или NaOH в зависимости от необходимости. Затем определялось общее содержание Gd каждого раствора с помощью анализа ICP.
Анестезированным крысам Sprague Dawley инъецировали внутримышечно один из растворов металла, описанных выше, в дозе 0,05 - 0,1 ммоль Gd/кг веса тела. Затем через различные интервалы времени снимались изображения и сравнивались с неинъецированным контролем в 0 момент времени.
Пример 2. Комплекс Gd-РСТМР (полученный в примере 2) показал почечную активацию и костную локализацию в плечах, позвоночнике и грудине.
Другие воплощения изобретения станут очевидными (понятными) специалистам в данной области при рассмотрении данного описания или практической реализации описанного в нем изобретении. Следует иметь в виду, что описания и примеры служат лишь в качестве иллюстрации, истинное же существо и объем изобретения показан в формуле изобретения.


















Использование: в качестве контрастных агентов в магниторезонансной визуализации. Продукты: производные бициклополиазамакроциклофосфоновых кислот формулы I, где R - группа - (C)n (x) (y) - T, x и y - H или COOH, п - целое число 1,2 или 3, T - COOH, - C6H4R4 или P (O) (R1) OH, R1 - OH или -O-(C1 - C5)алкил, R4 - NO2 или NH2, при условии, что один T должен быть группой P(O) (R1)OH, или их фармацевтически приемлемые соли. Реагент 1: соединение формулы 1, в которой по крайней мере одна группа R представляет собой водород. Реагент 2: фосфористая кислота или ее эфир. Условия реакции: в среде растворителя в присутствии формальдегида. 2 с. и 18 з.п.ф-лы, 4 табл.
соединение формулы I.
где R - группа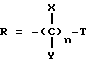
где X и Y - независимо H или COOH;
n - целое число 1, 2 или 3, при условии, что когда n = 2, тогда сумма X и Y должна быть равна двум или более H, когда n = 3, тогда сумма X и Y должна быть равна трем или более H;
T - COOH, группа
где R1 - -OH или -O-C1 - C5 -алкил;
R4 - NO2 или NH2, при условии, что по крайней мере один T должен быть группой P(O)R1OH, и при условии, что когда один T представляет
тогда один X или Y указанного R радикала может представлять COOH, а все другие X и Y радикала R должны представлять H, и при условии, что сумма R4 радикалов, когда они присутствуют, не может превышать 1,
или их фармацевтически приемлемые соли.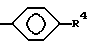
где R4 имеет значения, определенные в п. 1,
и два других T имеют значения, определенные в п. 1.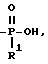
где R1 представляет OH или -O-C1-C5 - алкил.
| EP, заявка, 0391766, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, заявка, 0438206, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, заявка, 0352218, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| WO, заявк а, 91/10669, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| WO, заявка, 91/10645, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
