Изобретение относится к хелатам марганца(II), к указанным хелатам марганца(II), присоединенным к другим молекулам, и к их применению в качестве контрастных агентов в магнитно-резонансной визуализации (МРВ).
МРВ представляет собой метод медицинской визуализации, при котором участки организма визуализируют посредством ядер выбранных атомов, в частности ядер водорода. МРВ сигнал зависит от среды, окружающей визуализируемые ядра, и времен их продольной и поперечной релаксации, Т1 и Т2. Так, в том случае, когда визуализируемым ядром является протон, интенсивность МРВ сигналов будет зависеть от таких факторов, как плотность протонов и химическое окружение протонов. Контрастные агенты часто используют в МРВ с целью улучшения контраста изображения. Они работают, воздействуя на время релаксации Т1, Т2 и/или Т2*, и, следовательно, влияют на контраст изображений.
Времена релаксаций Т1, Т2 и/или Т2* хелатированного парамагнитного контрастного агента могут быть оптимизированы путем структурной модификации. Особенно важным является присутствие и время пребывания молекулы воды, связанной с парамагнитным ионом, и время корреляции вращательного движения контрастного агента. Присутствие и время пребывания молекулы воды, связанной с парамагнитным ионом, может быть модулировано путем выбора парамагнитного иона и хелатирующей группировки. Время корреляции вращательного движения может быть модулировано путем варьирования размера контрастного агента.
Парамагнитный ион может вмешиваться в биологические пути и индуцировать токсичность, поэтому необходимо, чтобы парамагнитный ион оставался в хелате. Стабильность парамагнитного комплекса может быть модулирована путем разработки структуры хелатирующей группировки. Особый интерес представляет кинетическая стабильность, измеряемая как период полудиссоциации, который показывает степень инертности по отношению к измененному химическому окружению (т.е. к эндогенным ионам).
Несколько типов контрастных агентов были использованы в МРВ. Ангиографические МР контрастные агенты, например частицы суперпарамагнитного оксида железа, удерживаются в сосудистой сети в течение длительного времени. Было доказано, что они чрезвычайно полезны для усиления контраста в печени, а для обнаружения аномалий проницаемости капилляров, например "утечку" из стенок капилляров в опухолях, которая приводит к ангиогенезу опухоли.
Водорастворимые парамагнитные хелаты, т.е. комплексы хелатирующего агента и иона парамагнитного металла, например хелаты гадолиния, такие как OmniscanTM (GE Healthcare), являются широко применяемыми МР контрастными агентами. Благодаря их низкой молекулярной массе они быстро распределяются во внеклеточное пространство (т.е. в кровь и интерстиций) при введении в сосудистую сеть. Они также быстро выводятся из организма.
Растворимость парамагнитного хелата в воде также является важным фактором, когда их применяют в качестве контрастных агентов для МРВ, поскольку их вводят пациентам в относительно больших дозах. В случае парамагнитных хелатов с высокой растворимостью в воде требуется меньший объем для инъекции, их, следовательно, легче вводить пациенту, и они вызывают меньший дискомфорт.
В документах предшествующего уровня техники по парамагнитным хелатам часто упоминаются парамагнитные ионы в общем, но обычно в них идет речь о гадолинии и разработках с использованием гадолиния. Так как структурный дизайн хелата специфичен для каждого парамагнитного иона, хелат, разработанный для гадолиния, не будет оптимальным, в отношении релаксивности или стабильности, для других парамагнитных ионов, таких как марганец(II) или железо(III). Гадолиний(III) является самым широко используемым ионом парамагнитного металла для хелатов для МРВ.
В WO 2006/080022 (Degani et al.) раскрыты бифункциональные конъюгаты, содержащие группировку лиганда рецептора, ассоциированного со злокачественными опухолями, и металл-связывающую группировку, и их комплексы с парамагнитными лантаноидами или переходными металлами.
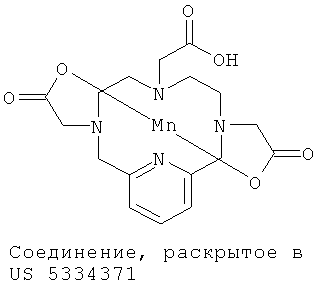
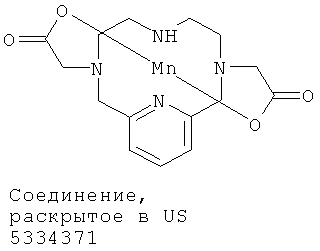
В US 5334371 (Cries et al.) раскрыты макроциклические полиазабицикликлические соединения, содержащие ионы марганца(II). Эти раскрытые соединения марганца(II) уступают соединениям по настоящему изобретению с точки зрения стабильности и способности генерировать сигналы и поэтому меньше подходят для применения в качестве визуализирующих агентов.
Ион марганца(II) представляет собой парамагнитный ион с высоким спиновым числом и длительным временем электронной релаксации, и в литературе были сообщения о потенциале контрастного агента с высокой релаксивностью на основе марганца(II). Однако низкая стабильность хелатов марганца, как было доказано, является проблемой и, следовательно, ограничивает осуществимость применения таких хелатов в контрастной среде.
Задача настоящего изобретения заключается в создании хелатов на основе марганца(II), которые являются кинетически стабильными и демонстрируют оптимальную кинетику водного обмена и которые могут быть использованы в качестве МР контрастных агентов.
Таким образом, в первом аспекте настоящего изобретения предложено соединение формулы (I):
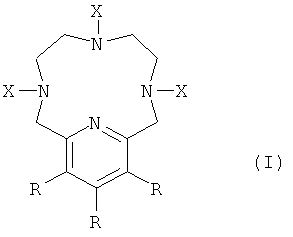
где
Х представляет собой алкил, C(Y)2COOZ или линкер;
Y представляет собой алкил, -PEG, -COOH, -РО(ОН)2, -Н или линкер;
R представляет собой алкил, -PEG, -N(алкил)2, -N(PEG)2, -О(алкил), -O(PEG), -NMe2, -NH2, -OMe, -OH, -H или линкер;
Z представляет собой Mn(II) или -H,
при условии, что две из присутствующих трех групп X представляют собой C(Y)2COOZ,
с дополнительным условием, что если Z представляет собой Mn(II), то две Z-содержащие группы X имеют один общий Mn(II).
Группа X, не являющаяся группой C(Y)2COOZ, представляет собой алкильную группу или линкер.
Термин "алкил" сам по себе или как часть другого заместителя относится к углеводороду, предпочтительно низшему алкилу, например C1-C6алкилу и более предпочтительно -CH3.
Термин PEG означает полиэтиленгликоль любой молекулярной массы, предпочтительно PEG-единицы от 1 до 5 кДа.
Y может представлять собой предпочтительно метил, -COOH, -РО(OH)2, -H или линкер. Наиболее предпочтительно, Y представляет собой -H или линкер.
R может представлять собой предпочтительно метил, -NMe2, -NH2, -OMe, -OH или линкер. Наиболее предпочтительно, R представляет собой линкер.
Линкер, если он присутствует, присоединяет соединение формулы (I) к другой молекуле. На выбор указанной другой молекулы будут влиять биораспределение и уровень сигнала контрастного агента.
Предпочтительно, линкерная группировка может быть выбрана из группы, включающей:
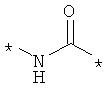
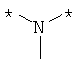
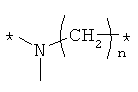
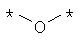
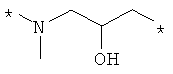
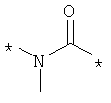
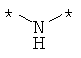
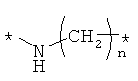
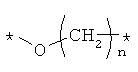
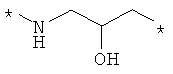
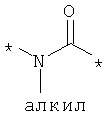
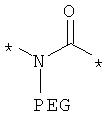
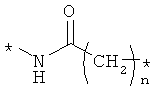
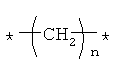 ,
,
где n означает целое число от 1 до 7.
* означает положение, в котором линкер присоединен к соединению формулы (I) и другой молекуле, определенной выше.
Предпочтительно, линкер выбран из группы, включающей:
.
Наиболее предпочтительно, линкер представляет собой
.
Таким образом, предпочтительным воплощением соединения формулы (I) является соединение формулы (III)
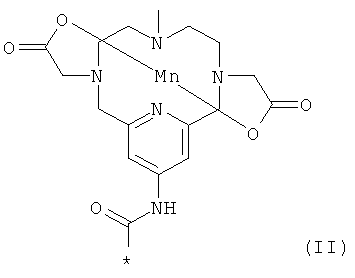 ,
,
где * показывает точку присоединения указанной другой молекулы.
В этом воплощении один X представляет собой CH3, один X представляет собой C(Y)2COOZ, и один X представляет собой C(Y)2COO-; Z представляет собой Mn(II); Y представляет собой -H; и линкер представляет собой:
Соединения по настоящему изобретению могут быть использованы в качестве МР контрастных агентов и, как объяснено выше, могут быть присоединены или могут не быть присоединены к другим молекулам, таким как природные или синтетические пептиды, аминокислоты или их производные, полиамины или их производные, пептидомиметики, полипептиды, белки или антитела. В результате связывания соединений по настоящему изобретению с этими молекулами могут быть получены МР контрастные агенты для направленной доставки к мишени, если, например, пептид или белок является вектором, который связывается с мишенью, такой как рецептор или клеточный поверхностный маркер. Соединения по настоящему изобретению также могут быть присоединены к полимерным группировкам, таким как природные или синтетические полимеры или дендримеры. Такое связывание придает соединениям по настоящему изобретению дополнительно пониженную молекулярную подвижность и, следовательно, увеличивает их релаксивность при высокой напряженности поля, используемой в современных МРВ-сканерах. В другом воплощении соединения по настоящему изобретению могут быть присоединены к липофильным соединениям, и полученные в результате амфифильные соединения могут быть дисперсными. Такие дисперсии могут быть использованы в качестве МР контрастного агента для визуализации опухолей. В еще одном воплощении соединения по настоящему изобретению могут быть присоединены к наночастицам. И снова такое связывание придает соединениям по настоящему изобретению дополнительно пониженную молекулярную подвижность и, следовательно, увеличивает их релаксивность.
Таким образом, во втором аспекте данного изобретения предложено соединение формулы (I), как оно определено выше, присоединенное к другой молекуле через группу X, Y или R. В данном контексте термин "другая молекула" включает атомы. В предпочтительном воплощении указанная другая молекула представляет собой О, S, Р или N, наиболее предпочтительно N. В другом предпочтительном воплощении указанная другая молекула представляет собой ароматическое кольцо, инозит или углевод, или любое их производное. В другом предпочтительном воплощении указанная другая молекула представляет собой природный или синтетический пептид, аминокислоты или их производные, полиамины или их производные, пептидомиметик, полипептид, белок, антитело, природный или синтетический полимер, дендример, наночастицу или липофильное соединение. За счет присоединения соединений формулы (I) к другим молекулам биораспеределение будет изменено, и контрастный агент может интернализоваться и связываться с клетками с аффинностью к молекуле, присоединенной к соединению формулы (I).
Для специалиста в данной области техники будет очевидно, что линкерная группировка X, Y или R может быть присоединена к другим молекулами любым способом, известным в данной области техники.
В одном предпочтительном воплощении соединения по настоящему изобретению присоединены к дендримеру. Дендример представляет собой многократно разветвленную молекулу и будет служить в качестве каркасной структуры для многократного присоединения соединений формулы (I). В его простейшей форме дендример может состоять только из каркаса-ядра, но допускает многократное присоединение соединений формулы (I). Дендримерные конструкции будут иметь измененное биораспределение и усиленный сигнал по сравнению с мономерными хелатами.
Подходящими дендримерными соединениями с присоединенными соединениями формулы (I) являются:
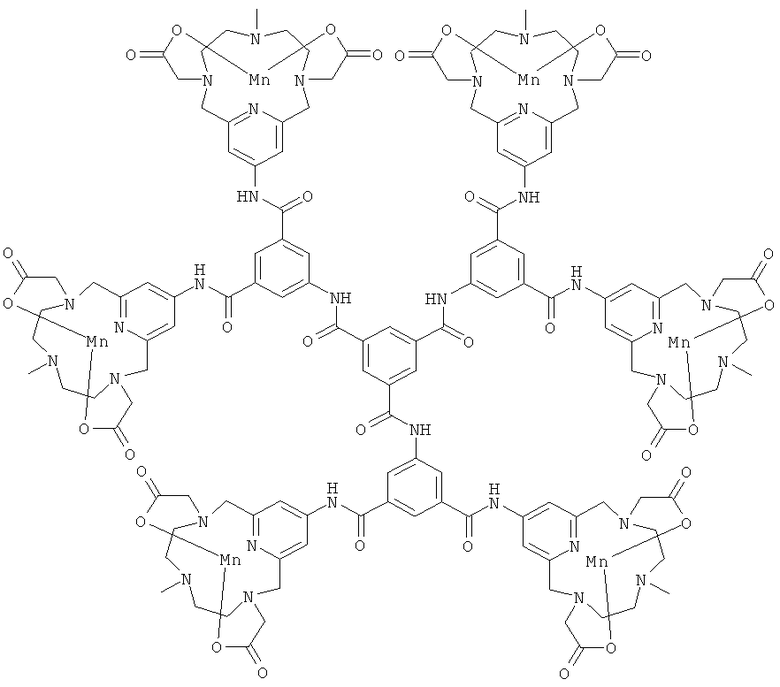
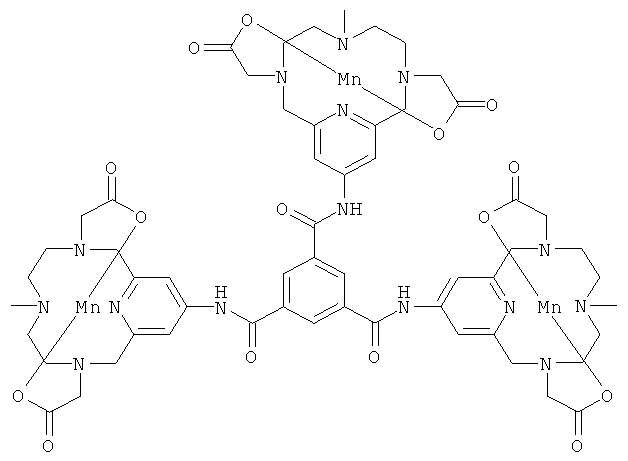
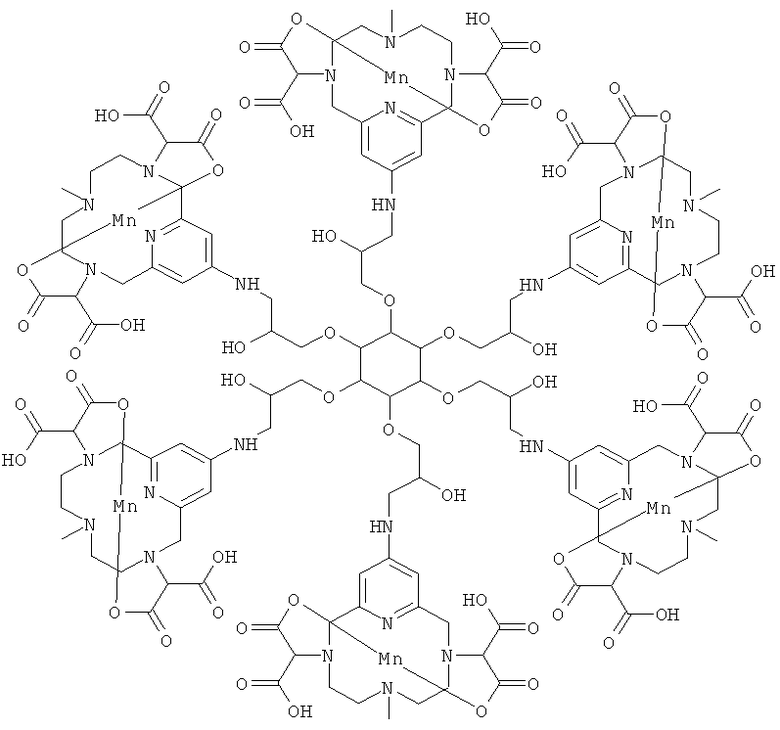
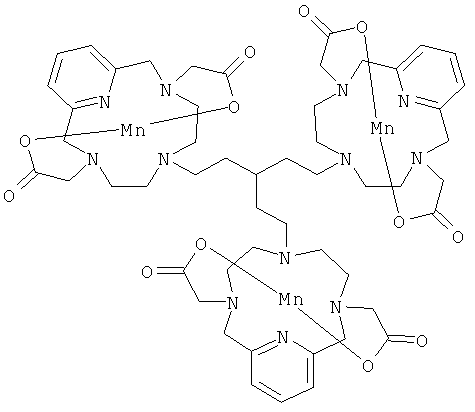
Подходящими производными природных или синтетических пептидов, аминокислот и их производных, полиаминов и их производных, пептидомиметиков или полипептидов с присоединенными соединениями формулы (I) являются:
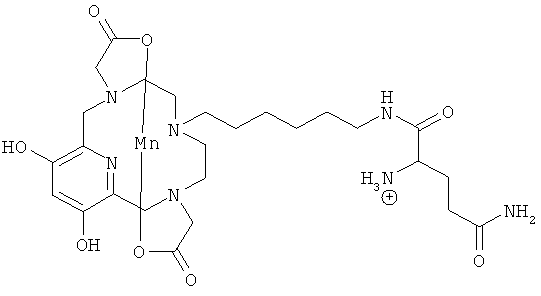
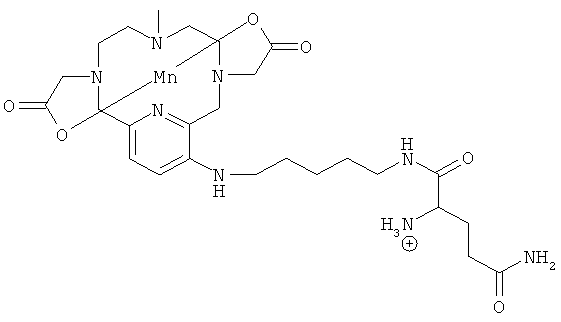
В случае если указанная другая молекула представляет собой группировку-лиганд рецептора, ассоциированного со злокачественными опухолями, то R не может быть линкером, содержащим C2-C10 гидроксикарбиленовую цепь.
Соединения формулы (I) могут быть синтезированы несколькими путями синтеза, известными специалисту в данной области техники, из коммерчески доступных исходных вещества по следующей общей методике:
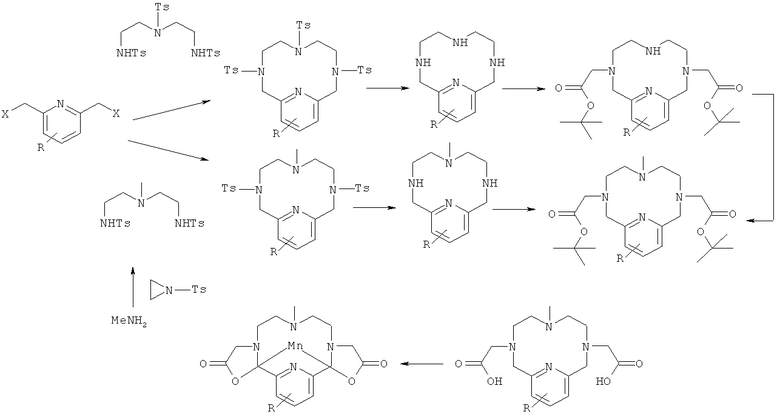
2,6-бис-метил-пиридиновое соединение с уходящими группами (X) в положениях метилов могут быть получены из коммерчески доступных веществ способами, известными специалистам в данной области техники. Примерами подходящих уходящих групп являются хлоро-, бромо-, тозил-нозил- и мезил-группы. Пиридиновое кольцо может быть замещено группой (R), которая может быть использована в качестве точки присоединения, если хелат предполагается связывать с другой молекулой. Примером подходящей группы R является -NH2. Группа R может представлять собой также инертный предшественник для группы, используемой в последующей реакции сочетания, т.е. группа -NO2 является предшественником группы -NH2. Могут быть также предусмотрены группы R с единственной функцией увеличения стабильности последующего марганцевого комплекса, за счет электронной делокализации к азоту пиридина. Примерами таких групп R являются -CH3 и -OCH3. 2,6-бис-метил-пиридиновое соединение с подходящими уходящими группами, как определено выше, затем подвергают циклизации путем взаимодействия с подходящим бис-амино-нуклеофилом с образованием тетраазамакроцикла способами, известными специалистам в данной области техники. К нуклеофильным атомам азота могут быть присоединены тозилатные группы, которые способствуют протеканию реакции циклизации, как известно специалистам в данной области. Последующее удаление тозилатных групп может быть осуществлено способами, известными в данной области техники. Введение двух карбоксиметильных групп на два из трех алифатических азотов может быть осуществлено путем аккуратного региоселективного алкилирования, предпочтительно осуществляемого под строгим контролем рН, способами, общеизвестными специалистам в данной области техники. Оставшийся вторичный азот впоследствии может быть алкилирован углеродным электрофилом. Примерами таких углеродных электрофилов являются CH3I и CF3SO2OCH3. Алкильная группа может быть также введена до реакции циклизации путем осуществления взаимодействия алкилированного бис-тозиламидного производного с электрофильным пиридиновым соединением. Раннее введение алкильной группы исключает необходимость в региоселективном введении карбоксиметильных групп на следующей реакционной стадии. Карбоксиметильные группы могут быть защищены в форме сложного эфира. Это особенно полезно, когда хелатируемое соединение предназначено для реакции сочетания с другой молекулой через группу R с использованием способов, общеизвестных специалистам в данной области техники, так как растворимость в растворителях, используемых в реакции сочетания, значительно более низкая у производного, представляющего собой свободную карбоновую кислоту. Примерами таких сложноэфирных групп являются трет-бутиловые, этиловые и метиловые сложные эфиры. Удаление сложноэфирных групп известно специалистам в данной области техники. Образование комплекса может быть осуществлено в водном растворе в результате реакции с использованием подходящего источника иона марганца(II) способами, известными специалистам в данной области техники.
Соединения по настоящему изобретению, присоединенные к другим молекулам через группу X, Y или R, могут быть получены способами, известными в данной области техники. Если, например, указанная другая молекула представляет собой пептид, полипептид или белок, то соединения формулы (I) без труда могут быть присоединены к подходящим функциональным группам в указанных других молекулах, например к карбоксильным группам. Возможно, что функциональные группы в указанных других молекулах необходимо активировать, например путем образования ацилхлорида из карбоксильной группы. Способы активации функциональных групп с целью усиления их реакционной способности известны специалисту в данной области техники (смотри, например, Sandier and Karo, eds. Organic Functional Group preparation, Academic Press, San Diego 1998).
Соединения формулы (I) и соединения формулы (I), присоединенные к другим молекулам, предпочтительно к природным или синтетическим пептидам, аминокислотам и их производным, полиаминам и их производным, пептидомиметикам, полипептидам, белкам, антителам, природным или синтетическим полимерам, дендримерам, липофильным соединениям или наночастицам, могут быть использованы в качестве МР контрастных агентов. Таким образом, в третьем аспекте настоящего изобретения предложены соединения для применения в качестве МР контрастных агентов.
Для этой цели соединения формулы (I) и соединения формулы (I), присоединенные к другим молекулам, готовят в виде композиции с традиционными физиологически переносимыми носителями, такими как водные носители, например вода и буферные растворы, и возможно с эксципиентами. Полученную композицию называют "МР контрастная среда".
В четвертом аспекте данного изобретения предложена композиция, содержащая соединение формулы (I) или соединение формулы (I), присоединенное к другим молекулам, и по меньшей мере один физиологически переносимый носитель. Указанная композиция может быть использована в качестве MR контрастной среды для МРВ.
Для применения в качестве MR контрастной среды для МРВ организма человека или животного, не являющегося человеком, необходимо, чтобы указанная MR контрастная среда была подходящей для введения в указанный организм. Соответственно, соединения формулы (I) или соединения формулы (I), присоединенные к другим молекулам, и возможно фармацевтически приемлемые эксципиенты и добавки могут быть суспендированы или растворены в по меньшей мере одном физиологически переносимом носителе, например в воде или в буферном(ых) растворе(ах). Подходящие добавки включают, например, физиологически совместимые буферные агенты, такие как гидрохлорид трометамина, хелатирующие агенты, такие как DTPA (диэтиленпентауксусная кислота), DTPA-BMA (диэтиленпентауксусная кислота-N,N′-бис(метиламид)), или соединения формулы (I), слабые комплексы физиологически переносимых ионов, такие как кальциевые хелаты, например кальций-DTPA, CaNaDTPA-BMA, соединения формулы (I), где Х образует комплекс с Са2+, или Ca/Na соли соединений формулы (I), кальциевые или натриевые соли, такие как хлорид кальция, аскорбат кальция, глюконат кальция или лактат кальция. Эксципиенты и добавки дополнительно описаны, например, в WO-A-90/03804, ЕР-А-463644, ЕР-А-258616 и US 5876695, содержание которых включено в данное описание посредством ссылки.
Пятым аспектом данного изобретения является способ МР визуализации, при котором субъекту вводят композицию, содержащую соединение формулы (I) или соединение формулы (I), присоединенное к другой молекуле, и по меньшей мере один физиологически переносимый носитель, и этого субъекта подвергают МР обследованию, при котором детектируют МР сигналы от субъекта или частей тела субъекта, в которых распределена композиция, и возможно формируют МР изображения и/или МР спектры из детектированных сигналов. В предпочтительном воплощении субъектом является организм живого человека или животного, не являющегося человеком.
В предпочтительном воплощении композицию вводят в количестве, эффективном в усилении контраста, т.е. количество, которое является подходящим для усиления контраста в способе МР визуализации.
В другом предпочтительном воплощении субъектом является живой человек или животное, не являющееся человеком, и способ МР визуализации представляет собой способ МР обнаружения опухоли или способ визуализации для выявления опухоли.
В шестом аспекте данного изобретения предложен способ МР визуализации, при котором субъекта, которому предварительно была введена композиция, содержащая соединение формулы (I) или соединение формулы (I), присоединенное к другой молекуле, и по меньшей мере один физиологически переносимый носитель, подвергают МР обследованию, при котором детектируют МР сигналы от субъекта или частей тела субъекта, в которых распределена композиция, и возможно формируют МР изображения и/или МР спектры из детектированных сигналов.
Термин "предварительно введена" означает, что любая стадия, требующая введения композиции пациенту лицом, проводящим медицинское обследование, уже была выполнена до начала осуществления способа МР визуализации и/или MR спектроскопии согласно изобретению.
В седьмом аспекте данное изобретение относится к применению соединения по изобретению в изготовлении диагностического агента для использования в качестве МР контрастной среды.
Далее изобретение будет описано более подробно посредством приведенных не ограничивающих примеров.
ПРИМЕРЫ
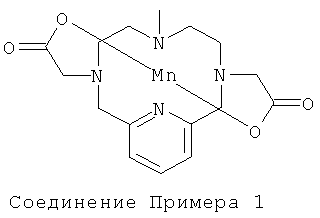
Пример 1
а) Синтез 3,6,9-трис-(толуол-4-сульфонил)-3,6,9,15-тетрааза-бицикло[9.3.1]пентадека-1(14),11(15),12-триена
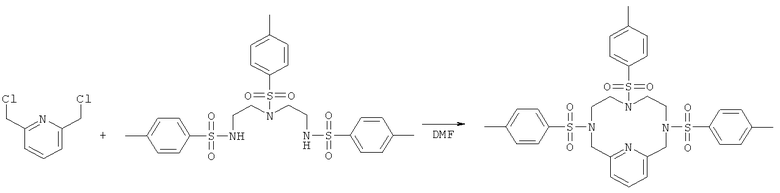
Динатриевую соль N,N′,N″-три-пара-тозилдиэтилен-триамина (6,90 г, 11,4 ммоль) в диметилформамиде (DMF) (92 мл) нагревали до 100°C в атмосфере N2. По каплям в течение 45 мин добавляли 2,6-бис(хлорметил)пиридин (2,01 г, 11,4 ммоль) в DMF (37 мл). После окончания добавления реакционную смесь перемешивали при 40°C в атмосфере N2 в течение 12 часов. Затем в реакционную смесь добавляли 75 мл воды. Полученную суспензию затем фильтровали, и твердое вещество промывали водой и сушили в вакууме. Неочищенный продукт пытались растворить в смеси вода : ацетонитрил 1:1. Белый осадок отфильтровывали и сушили с получением 5,52 г (72%) продукта.
Продукт анализировали методом ЖХ/МС (жидкофазная хроматография/масс-спектрометрия) (обнаружено m/z: 669,3 МН+, вычислено m/z: 669,2).
б) Синтез HBr-соли 3,6,9,15-тетрааза-бицикло[9.3.1]пентадека-1(14),11(15),12-триена
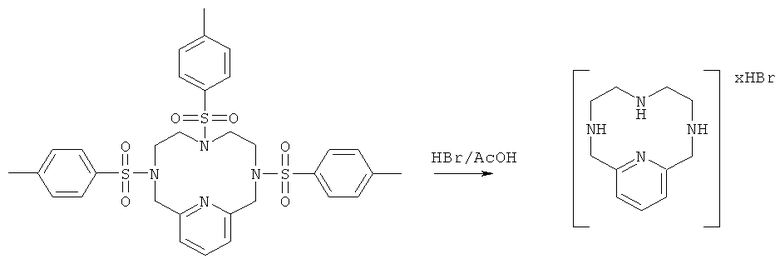
Раствор HBr и уксусной кислоты готовили путем смешивания 48% HBr и ледяной уксусной кислоты в соотношении 65:35. К 111 мл смеси HBr/AcOH добавляли продукт, полученный на стадии 1а (5,0 г, 7,5 ммоль), и эту реакционную смесь нагревали с обратным холодильником при постоянном перемешиванием в течение 80 часов. Реакционную смесь затем охлаждали до комнатной температуры и концентрировали до приблизительно 1/10 первоначального объема. Остаточную смесь энергично перемешивали, и в нее добавляли 50 мл диэтилового эфира. Образовавшееся не совсем белое твердое вещество отфильтровывали, промывали диэтиловым эфиром и сушили в вакууме с получением 3,8 г неочищенного продукта. Этот неочищенный продукт использовали без очистки. Неочищенный продукт анализировали методом ЖХ/МС (обнаружено m/z: 207,3 МН+, вычислено m/z: 207,2).
в) Синтез 3,6,9,15-тетрааза-3,9-бис-(трет-бутилкарбонилметил)-бицикло[9.3.1]пентадека-1(14),11(15),12-триена
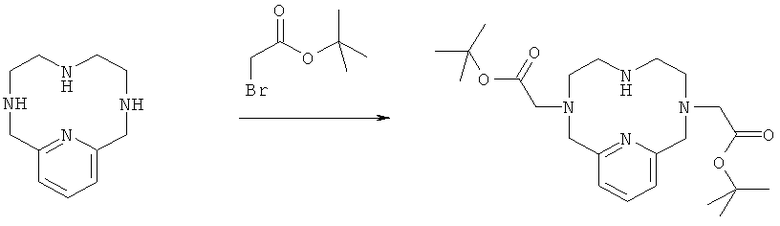
Соединение, полученное на стадии 16 (1,5 г, 3,3 ммоль), растворяли в смеси диоксан : вода 1:1 (30 мл), и рН доводили до значения 3 добавлением NaOH (2M), затем добавляли трет-бутилбромацетат (0,66 мл, 4,5 ммоль) в смеси диоксан : вода (35 мл). После добавления рН доводили до значения 9 добавлением NaOH (2M). Через 3,5 часа снова добавляли трет-бутилбромацетат (0,10 мл, 0,68 ммоль), и рН доводили до значения 9 добавлением NaOH (2M). Добавление трет-бутилбромацетата повторяли дважды (2×0,116 мл, 0,79 ммоль) через 14 и через 17 часов. рН также доводили до значения 9. Реакционную смесь наносили на препаративную колонку С18, и продукт очищали препаративной ВЭЖХ. Выделили 0,9 г (63%) чистого соединения.
Продукт анализировали методом ЖХ/МС (обнаружено m/z: 435,1 МН+, вычислено m/z: 435,3).
г) Синтез 3,6,9,15-тетрааза-3,9-бис-(трет-бутилкарбонилметил)-6-метил-бицикло[9.3.1]пентадека-1(14),11(15),12-триена
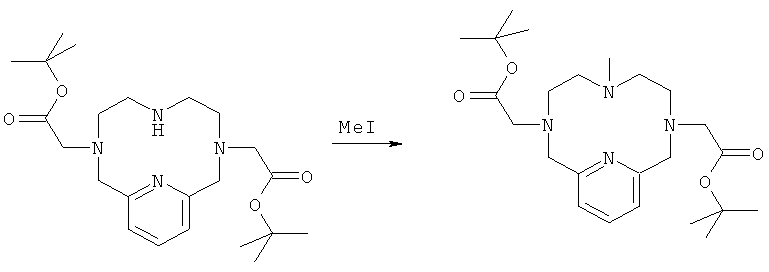
К соединению, полученному на стадии 1в (200 мг, 0,460 ммоль), в диметилформамиде (10 мл) добавляли триэтиламин (65,3 мг, 64,1 мкл, 0,460 ммоль) и йодметан (65,3 мг, 28,7 мкл, 0,460 ммоль). Добавляли аликвоты 1,1,3,3-тетраметилгуанидина (58 мкл, 0,46 ммоль) и йодметана (29 мкл, 0,46 ммоль). Реакционную смесь анализировали методом ЖХ/МС и затем добавляли воду. Реакционную смесь очищали препаративной ВЭЖХ. Выделили 106 мг, 0,24 ммоль (51%) чистого соединения. Продукт анализировали методом ЖХ/МС (обнаружено m/z: 449,1 МН+, вычислено m/z: 449,3).
д) Синтез 3,6,9,15-тетрааза-3,9-бис-(карбоксиметил)-6-метил-бицикло[9.3.1]пентадека-1(14),11(15),12-триена
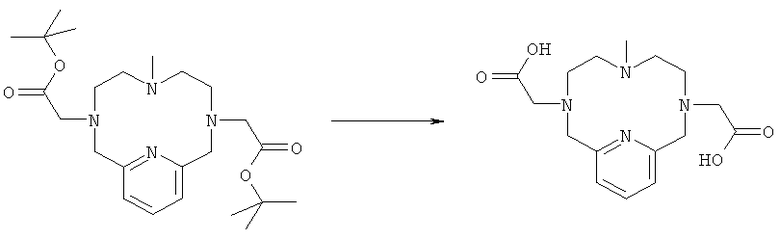
Соединение, полученное на стадии 1г (0,11 г, 0,24 ммоль), в муравьиной кислоте (30 мл) нагревали до образования флегмы и охлаждали до комнатной температуры. Муравьиную кислоту выпаривали при пониженном давлении. Добавляли толуол (2×20 мл) и выпаривали при пониженном давлении. Неочищенный продукт использовали на следующей стадии без очистки. Продукт анализировали методом ЖХ/МС (обнаружено m/z: 337,1 МН+, вычислено m/z: 336,2).
е) Синтез комплекса 3,6,9,15-тетрааза-3,9-бис-(карбоксиметил)-6-метил-бицикло[9.3.1]пентадека-1(14),11(15),12-триена с марганцем(II)
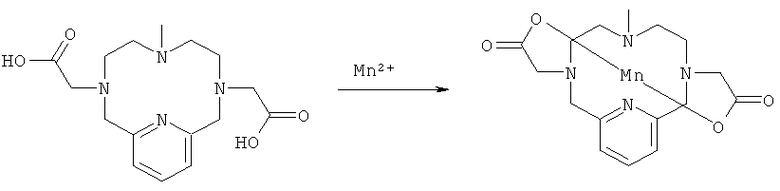
Соединение, полученное на стадии 1д (81 мг, 0,24 ммоль), растворяли в дегазированной воде (15 мл), после чего рН доводили до значения от 2,8 до 6,7, используя 200 мкл NaOH в дегазированной воде (2,35 М). Добавляли 1,1 мл хлорида марганца(II) в дегазированной воде (465 мг в 10 мл, 0,23 М). рН снижался до значения 3,5. рН доводили до значения 5,9 аликвотами NaOH (водн.). Через 15 минут рН доводили до 9,3 аликвотами NaOH (водн.), и смесь оставляли стоять в течение 15 минут. pH доводили до значения 7,0, используя аликвоты HCl (150 мкл в 1 мл дегазированной воды), после чего раствор фильтровали, наносили на препаративную колонку С18, и продукт очищали препаративной ВЭЖХ. Выделили 0,11 ммоль, 42 мг (45%) чистого соединения. Продукт анализировали методом ЖХ/МС (обнаружено m/z: 390,0 МН+, вычислено m/z: 390,1).
Пример 2
а) Синтез диметил-4-аминопиридин-2,6-дикарбоксилата
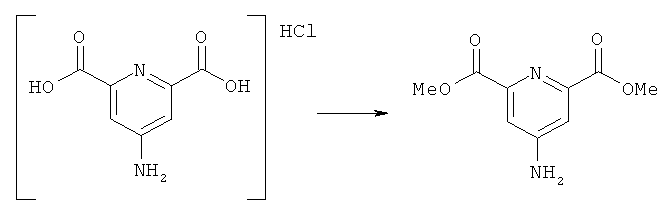
Тионилхлорид (2,5 мл) медленно добавляли к охлажденному льдом метанолу (20 мл) в атмосфере азота. Затем в охлаждаемый льдом раствор порциями добавляли гидрохлоридную соль 4-аминопиридин-2,6-дикарбоновой кислоты (2,5 г, 11,5 ммоль). Затем реакционную смесь кипятили с обратным холодильником в течение 4 ч и затем концентрировали с получением желтого аморфного твердого вещества. Это твердое вещество растворяли в водном растворе HCl (0,8М, 50 мл), и раствор фильтровали и подщелачивали до рН 9. Образовавшийся осадок отфильтровывали с получением целевого продукта (1,6 г, 66%). Продукт анализировали методом ЯМР ((Cd3)2SO), 7.36 (s, 2H), 6.70 (bs, 2H), 3.84 (s, 6H)).
б) Синтез (4-аминопиридин-2,6-диил)диметанола
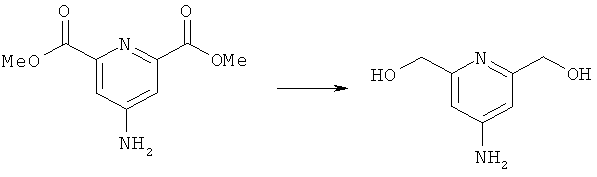
К соединению, полученному на стадии 2а (2,8 г, 13,3 ммоль), добавляли метанол (100 мл). Периодически добавляя по каплям боргидрид натрия (всего 4 г, 11 ммоль) в течение 1 ч, получили прозрачный раствор. Реакционную смесь затем кипятили с обратным холодильником и периодически по каплям добавляли боргидрид натрия (дополнительно 4 г, 11 ммоль) в течение 1 ч. Затем добавляли воду (25 мл), и реакционную смесь концентрировали с получением белого аморфного порошка. Это твердое вещество подвергали экстракции в аппарате Сокслета в этилацетате в течение 72 ч. Органическую фазу затем концентрировали и добавляли воду (100 мл). рН доводили до значения 11, и полученную суспензию нагревали при 75°C в течение 1 ч с получением прозрачного раствора. рН доводили до значения 12, и раствор охлаждали до 0°C. Образовавшийся осадок отфильтровывали с получением целевого продукта в виде аморфного твердого вещества (1,3 г, 66%). Продукт анализировали методом ЖХ/МС (обнаружено m/z: 155,1 МН+, вычислено m/z: 155,1).
в) Синтез 2,6-бис(хлорметил)пиридин-4-амина
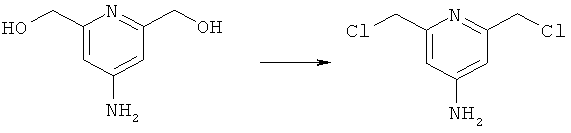
К соединению, полученному на стадии 2б (2,3 г, 14,9 ммоль), добавляли тионилхлорид (15 мл), и затем полученный пенящийся гель кипятили с обратным холодильником в течение 2 ч. Затем реакционную смесь концентрировали с получением аморфного твердого вещества, к которому добавляли воду (30 мл). Полученную суспензию подщелачивали до рН 8, и осадок отфильтровывали с получением целевого продукта (2,6 г, 88%). Продукт анализировали методом ЖХ/МС (обнаружено m/z: 191,0 МН+, вычислено m/z: 191,0).
г) Синтез 13-амино-3,6,9,15-тетрааза-3,9-бис-(толуол-4-сульфонил)-6-метил-бицикло[9.3.1]пентадека-1(14),11(15),12-триена
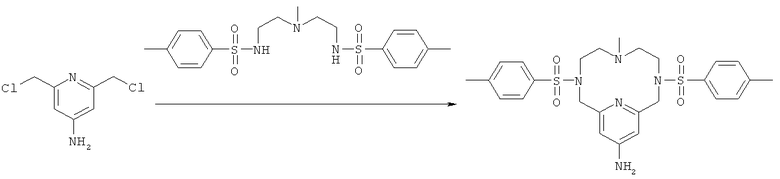
К соединению, полученному на стадии 2в (2,5 г, 13,1 ммоль), добавляли N,N′-(2,2′-(метилазандиил)бис(этан-2,1-диил))бис(4-метилбензолсульфонамид) (2,8 г, 6,6 ммоль) и K2CO3 (11 г, 79,7 ммоль), а затем диметилформамид (250 мл). Полученную суспензию перемешивали при 100°C в атмосфере азота в течение 2 ч. Затем дополнительно добавляли N,N′-(2,2′-(метилазандиил)бис(этан-2,1-диил))бис(4-метилбензолсульфонамид) (2,8 г, 6,6 ммоль), и нагревание продолжали в течение 4 ч. Затем 125 мл диметилформамида выпаривали, и оставшуюся суспензию по каплям добавляли в сосуд, содержащий воду (1,6 л). Полученную суспензию подкисляли до рН 1 и затем нагревали до 75°C. Раствору давали возможность охладиться до комнатной температуры в течение ночи, и полученный осадок отфильтровывали с получением целевого продукта (5,1 г, 72%). Продукт анализировали методом ЖХ/МС (обнаружено m/z: 544,1 МН+, вычислено m/z: 544,2).
д) Синтез 13-амино-3,6,9,15-тетрааза-3,9-бис-(трет-бутилкарбонилметил)-6-метил-бицикло[9.3.1]пентадека-1(14),11(15),12-триена
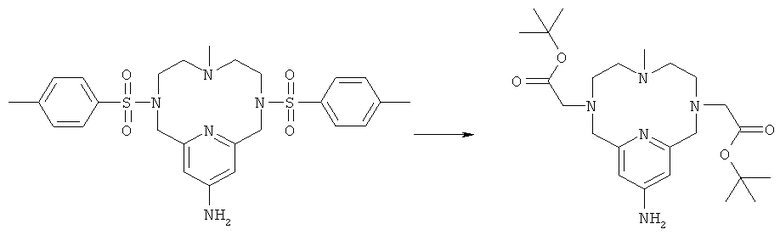
К соединению, полученному на стадии 2г (2,5 г, 4,6 ммоль), добавляли концентрированную серную кислоту (15 мл). Полученную суспензию перемешивали при 100°C в течение 8 ч и затем вливали в воду, охлажденную раскрошенным льдом (40 мл). Затем добавляли водный раствор NaOH (25%, 90 мл) с получением белой суспензии. В эту суспензию добавляли ацетонитрил (50 мл), K2CO3 (1,2 г, 8,7 ммоль) и трет-бутилбромацетат (1,36 мл, 9,2 ммоль). Двухфазную суспензию энергично перемешивали в течение 5 ч и затем снова добавляли трет-бутилбромацетат (1,36 мл, 9,2 ммоль). Через 12 ч органическую фазу отделяли и добавляли к буферному раствору (0,1М NaHCO3/Na2CO3 при рН 10), насыщенному NaCl. В этот двухфазный раствор добавляли трет-бутилбромацетат (1,36 мл, 9,2 ммоль), и затем смесь энергично перемешивали в течение 24 ч. Затем снова добавляли трет-бутилбромацетат (1,36 мл, 9,2 ммоль), и смесь энергично перемешивали в течение 24 ч. Органическую фазу отделяли и добавляли к фосфатному буферу (300 мл, 0,1М, рН 7). Водный раствор затем повторно экстрагировали дихлорметаном. Объединенные органические фазы сушили с использованием сульфата магния, фильтровали и концентрировали с получением целевого продукта (2,7 г). Продукт анализировали методом ЖХ/МС (обнаружено m/z: 464,2 МН+, вычислено m/z: 464,3).
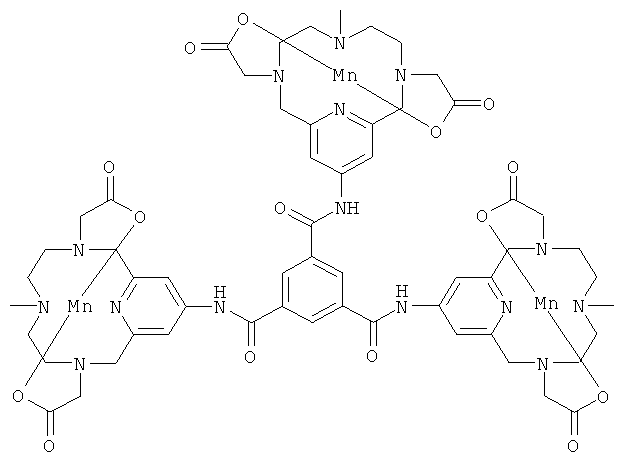
е) Синтез комплекса N1,N3,N5-трис[3,6,9,15-тетрааза-3,9-бис-(карбонилметил)-6-метил-бицикло[9.3.1]пентадека-13-триенил]бензол-1,3,5-трикарбоксамида с марганцем(II)
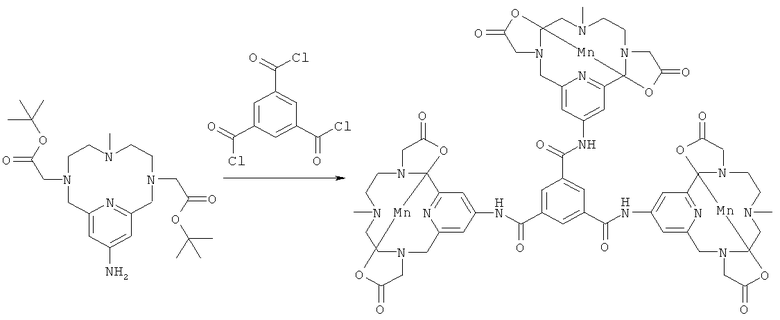
Соединение, полученное на стадии 2д (1,0 г, 1,1 ммоль), растворяли в ацетонитриле (10 мл), затем добавляли N-этилдиизопропиламин (185 мкл, 1,1 ммоль) и хлорангидрид бензолтрикарбоновой кислоты (64 мкл, 0,36 ммоль), и эту реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь затем концентрировали с получением белого аморфного твердого вещества. Это твердое вещество затем растворяли в муравьиной кислоте (10 мл) и нагревали до образования флегмы в течение 1 ч. Реакционную смесь затем концентрировали, и полученное вещество растворяли в насыщенном буферном растворе NaHCO3 и нагревали при 80°C в течение 24 ч. Реакционную смесь нейтрализовали и затем добавляли MnCl2 (435 мг, 2,2 ммоль). рН затем доводили до значения 7,6, и смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь затем фильтровали и концентрировали. После препаративной ВЭЖХ получили трехмерный комплекс с марганцем(II) (230 мг, 47% за три стадии). Продукт анализировали методом ЖХ/МС (обнаружено m/z: 457,0 M3H+, вычислено m/z: 457,1).
Пример 3
Анализ релаксивности
Измерения релаксивности выполняли с использованием растворов комплекса в концентрации в пределах от 0,05 до 1 мМ на Bruker Minispec PC 120b - NA 770 при 20 МГц или на Bruker Minispec Mq 60m при 60 МГц и 37°C. Растворы готовили путем растворения соответствующего количества хелата в дегазированной воде Millipore или в сыворотке крови человека.
Измерения водного обмена
Измерения водного обмена выполняли с использованием 6-15 мМ растворов комплексов. Растворы готовили путем растворения соответствующего количества комплекса в 0,6 мл буферного раствора (TRIS, 0,05 М, рН 7,4, в дегазированной воде Millipore при приблизительно 3 атомн. % 17O-обогащении). 17O ЯМР спектры с Фурье-преобразованием при варьируемой температуре регистрировали при частоте 40,7 МГц на спектрометре Varian Unity 300 MHz. Температуры измеряли с использованием метанола и этиленгликоля в качестве стандартов. Температурную зависимость уширения спектральной линии 17O для каждой системы измеряли в температурном диапазоне от 273,7 до 356,1 К. Каждую температуру калибровали с использованием метанола или этиленгликоля в качестве стандарта.
Пример 4 Анализ стабильности
Кинетическую стабильность марганцевых хелатов определяли по скоростям реакции обмена с Zn2+ в ацетатном буфере (10 мМ) с различными значениями рН. Увеличение свободных ионов марганца отслеживали на Bruker Minispec PC 120b - NA 770 при 20 МГц и 25°C. Общую ионную силу доводили до 0,5М с использованием KCl. Концентрация хелата составляла приблизительно 0,5 мМ для всех измерений.
Изменение концентрации хелатированного марганца при введении ионов цинка отслеживали спектроскопическим анализом. При замещении иона марганца ионом цинка наблюдается общее увеличение релаксивности, так как свободный ион марганца имеет релаксивность примерно 10 мМ-1с-1, тогда как исследованные хелаты марганца имеют более низкую релаксивность.
Константы скорости реакции псевдо-первого порядка (kobs) вычисляли из соответствующих данных по релаксивности по следующему уравнению:
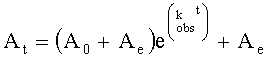
где At, A0 и Ае представляют собой значения релаксивности в момент времени t, в начале реакции и при равновесии соответственно. Период полудиссоциации получали из kobs.
Пример 5
а) Синтез N,N′,N″,N′″,N″″,N′″″-(((нитрилотрис(этан-2,1-диил))трис(азантриил))гексакис(этан-2,1-диил))гексакис(4-метилбензолсульфонамида)
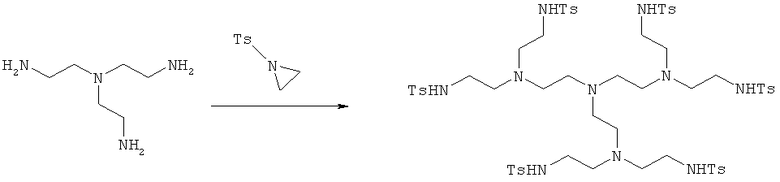
Трис-(2-аминоэтил)-амин (10 г, 68,5 ммоль) растворяли в ацетонитриле (500 мл) и затем медленно добавляли тозилазиридин (97,2 г, 0,49 моль).
Раствор перемешивали в течение 24 ч при комнатной температуре, затем добавляли NaOH (2,7 г, 68,5 ммоль), и перемешивание продолжали в течение еще 24 ч. Реакционную смесь затем вливали в воду (2 л), и полученный осадок отфильтровывали с получением чистого продукта.
б) Синтез 6,6′,6″-трис-этил-[3,6,9,15-тетрааза-3,9-бис-(4-метилбензолсульфонамид)-бицикло[9.3.1]пентадекатриен]-амина
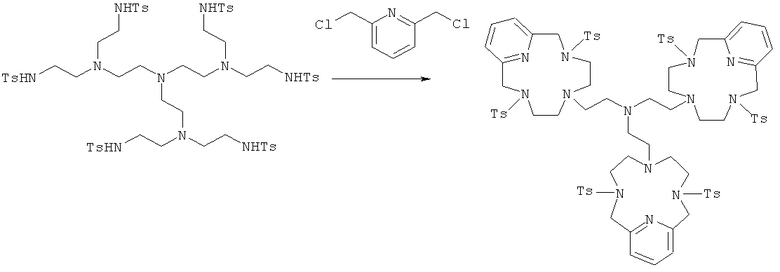
Соединение, полученное в Примере 5а (10 г, 7,5 ммоль), растворяли в DMF (200 мл) и затем добавляли 2,6-бис(хлорметил)пиридин (8,1 г, 46 ммоль) и карбонат калия (12,7 г, 92 ммоль). Реакционную смесь нагревали до 80°C в течение 24 ч и затем вливали в воду (1 л). Осадок отфильтровывали с получением чистого продукта.
в) Синтез марганцевого комплекса 6,6′,6″-трис-этил-[3,6,9,15-тетрааза-3,9-бис-(4-метилбензолсульфонамид)-бицикло[9.3.1]пентадекатриен]-амина
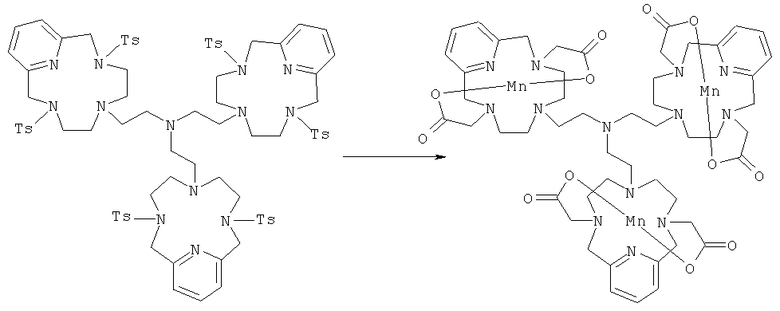
К соединению, полученному в Примере 66 (10 г, 6,1 ммоль), добавляли концентрированную серную кислоту (50 мл), и полученную суспензию нагревали при 100°C в течение 12 ч и затем вливали в охлажденную ледяную воду (1 л). Затем добавляли водный раствор NaOH (25%, 300 мл) с получением белой суспензии. В эту суспензию добавляли ацетонитрил (400 мл), K2CO3 (5,1 г, 37 ммоль) и трет-бутилбромацетат (22,2 мл, 0,15 моль). Двухфазную суспензию энергично перемешивали в течение 15 ч, и затем водную фазу отделяли и добавляли к фосфатному буферу (1 л, 0,1М, рН 7). Водный раствор затем неоднократно экстрагировали дихлорметаном. Объединенные органические фазы концентрировали с получением аморфного твердого вещества. Это твердое вещество растворяли в муравьиной кислоте (50 мл) и кипятили с обратным холодильником в течение 5 ч. Реакционную смесь концентрировали, полученное аморфное твердое вещество растворяли в воде (100 мл), нейтрализовали и затем добавляли MnCl2 (3,5 г, 27,5 ммоль). рН затем доводили до значения 7,6, и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь затем фильтровали и концентрировали. Аморфное твердое вещество кристаллизовали из кипящего этанола с получением марганцевого комплекса 6,6′,6″-трис-этил-[3,6,9,15-тетрааза-3,9-бис-(карбонилметил)-бицикло[9.3.1]пентадекатриен]-амина.
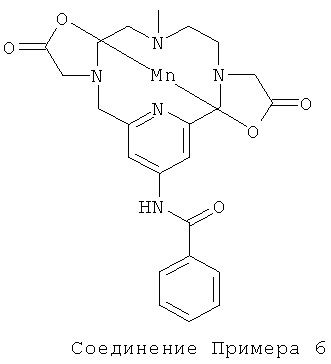
Пример 6
а) Синтез N,13-бензамид-3,6,9,15-тетрааза-3,9-бис-(трет-бутилкарбонилметил)-6-метил-бицикло[9.3.1]пентадека-1(14),11(15),12-триена
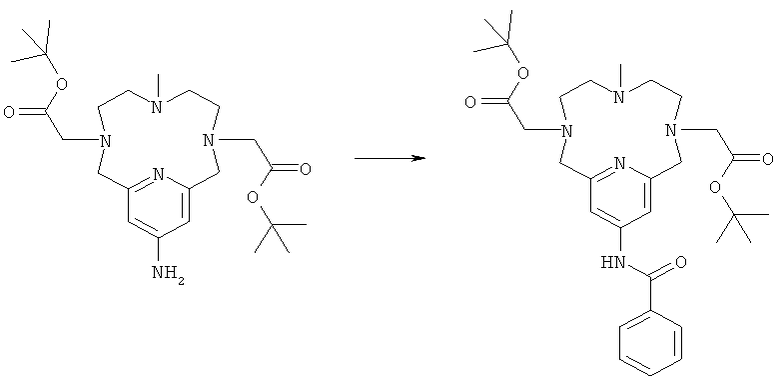
Соединение, полученное в Примере 2д (160 мг, 0,35 ммоль), растворяли в ацетонитриле (10 мл) и охлаждали до 0°C. Затем добавляли бензоилхлорид (93 мкл, 0,80 ммоль) и карбонат калия (48 мг, 0,35 ммоль), и реакционной смеси давали возможность достичь комнатной температуры. Через 24 ч реакционную смесь концентрировали с получением неочищенного продукта. Его использовали на следующей стадии без очистки.
б) Синтез марганцевого комплекса N,13-бензамид-3,6,9,15-тетрааза-3,9-бис-(карбоксиметил)-6-метил-бицикло[9.3.1]пентадека-1(14),11(15),12-триена
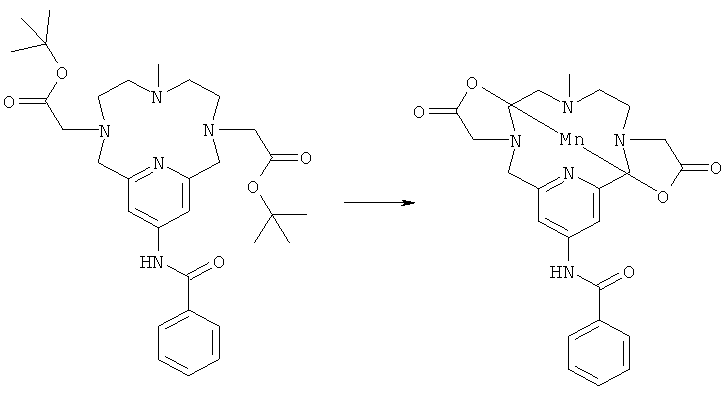
Неочищенное соединение, полученное в Примере 6а, растворяли в муравьиной кислоте, и реакционную смесь кипятили с обратным холодильником в течение 1,5 ч, и затем растворитель выпаривали. Неочищенное твердое вещество растворяли в воде (10 мл), и рН доводили до значения 7,1, используя NaOH (1M). Затем добавляли MnCl2 (69 мг, 0,35 ммоль), и рН доводили до значения 7, используя NaOH (1M). Через 2 ч реакционную смесь концентрировали и очищали препаративной ВЭЖХ с получением марганцевого комплекса N,13-бензамид-3,6,9,15-тетрааза-3,9-бис-(карбоксиметил)-6-метил-бицикло[9.3.1]пентадека-1(14),11(15),12-триена (60 мг, 33%).
Изобретение относится к соединениям формулы (I)
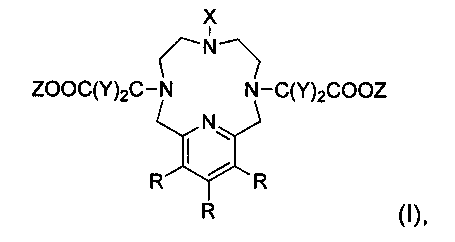
в которой X представляет собой C1-С6алкил; Y представляет собой C1-С6алкил или -Н; R представляет собой С1-С6алкил, -N(С1-С6алкил)2, -O(С1-С6алкил), -NMe2, -NH2, -ОМе, -ОН или -Н; Z представляет собой Mn(II) или -Н, при условии, что если Z представляет собой Mn(II), то две Z-содержащие группы X имеют один общий Mn(II). Изобретение также относится к композиции для применения в качестве MP контрастной среды для МРВ, содержащей соединение изобретения, к способу MP визуализации и к применению соединения изобретения в изготовлении диагностического агента для использования в качестве MP контрастной среды. Технический результат: получение хелатов на основе марганца (II), которые могут применяться в качестве магнитно-резонансных контрастных агентов. 5 н. и 5 з.п. ф-лы, 6 пр.
1. Соединение формулы (I)
где
X представляет собой C1-С6алкил;
Y представляет собой C1-С6алкил или -Н;
R представляет собой С1-С6алкил, -N(С1-С6алкил)2, -O(С1-С6алкил), -NMe2, -NH2, -ОМе, -ОН или -Н;
Z представляет собой Mn(II) или -Н,
при условии, что если Z представляет собой Mn(II), то две Z-содержащие группы X имеют один общий Mn(II).
2. Соединение по п. 1, где Y представляет собой метил или -Н.
3. Соединение по п. 2, где Y представляет собой -Н.
4. Соединение по п. 1, где Z представляет собой Mn(II).
5. Соединение по п. 1, где R представляет собой метил, -NMe2, -NH2, -ОМе или -ОН.
6. Соединение, представляющее собой
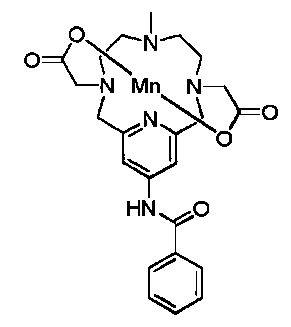 или
или 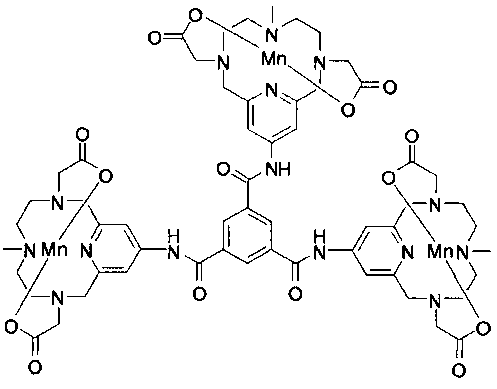 .
.
7. Соединение по любому из пп. 1-6 для применения в качестве MP (магнитно-резонансных) контрастных агентов.
8. Композиция для применения в качестве MP контрастной среды для МРВ (магнитно-резонансная визуализация), содержащая соединение по любому из пп. 1-6 и по меньшей мере один физиологически переносимый носитель.
9. Способ MP визуализации, при котором субъекту вводят композицию по п. 8, и этого субъекта подвергают MP исследованию, при котором детектируют MP сигналы от субъекта или частей тела субъекта, в которых распределена композиция, и возможно формируют MP изображения и/или MP спектры из детектированных сигналов.
10. Применение соединения по любому из пп. 1-6 в изготовлении диагностического агента для использования в качестве MP контрастной среды.
| Silvio Aime et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Зубчатое колесо со сменным зубчатым ободом | 1922 |
|
SU43A1 |
| ПРОИЗВОДНЫЕ БИЦИКЛОПОЛИАЗАМАКРОЦИКЛОФОСФОНОВЫХ КИСЛОТ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2114115C1 |
| RU 95122737 A, 10.01.1998 | |||
| WO 9426313 A1, 24.11.1994 | |||
| WO 9311800 A1, 24.06.1993 | |||
| US 5334371 A, 02.08.1994 | |||

