Изобретение касается амидных соединений, их синтеза, их производных, их солей и сольватов, фармацевтических композиций, содержащих их, и применения таких соединений и композиций в медицине и терапии, в частности, в качестве релаксантов основных мышц.
Основными ограничивающими побочными действиями многих клинически эффективных центральных мышечных релаксантов и противосудорожных средств являются вызывание седативного эффекта и нарушение согласованности движений у реципиента, которые серьезным образом ограничивают их применимость. Подобные побочные действия обнаружены у лекарственных средств, применяемых при лечении тревоги, таких как бензодиазепины. Хотя эти эффекты могут быть кратковременными, пациенты при такой терапии часто не могут водить машину или участвовать в определенного рода занятиях.
В настоящее время неожиданно обнаружено, что амиды формулы (I) являются сильными центральными миорелаксантами и обладают значительно пониженной способностью вызывать седативный эффект и нарушение координации по сравнению с известными средствами.
В одном аспекте данное изобретение относится к новым соединениям формулы (I)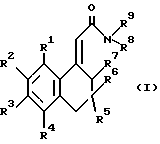
где R1, R2, R3 и R4 выбраны, каждый, из водорода и фтора, и, по меньшей мере, один и не более двух из них представляют собой фтор,
R5 выбран из водорода и C1-C4-алкила,
R6 выбран из водорода, C1-C4-алкила и гидрокси, или
R5 и R6 вместе с углеродом кольца образуют карбонильную группу,
R7 выбран из водорода и гидрокси,
R8 и R9 выбраны (каждый) из водорода, C1-C4-алкила и цикло(C3 или C4) алкила или вместе с азотом образуют морфолиногруппу, и X выбран из связи, метилена и -O- и всегда является связью или -О-, если любой из R5, R6 и R7 не является водородом, и всегда является связью, если R5 и R6 вместе с углеродом кольца образуют карбонильную группу,
и к их солям и сольватам.
Как здесь используется, "C1-C4-алкил" обозначает линейную или разветвленную алкильную группу, имеющую 1, 2, 3 или 4 атома углерода,
"цикло(C3 или C4)алкил" обозначает циклоалкильную группу, имеющую 3 или 4 атома углерода,
"соли" обозначают основные соли, образуемые в случае, когда в формуле (I) один из радикалов R8 и R9 является водородом, и "сольваты" обозначают сочетание в определенных соотношениях соединения формулы (I) и его растворителя.
Необходимо учесть, что соединения формулы (I) могут существовать в различных геометрически изомерных формах и в виде их смесей в любых соотношениях. Настоящее изобретение включает в свой объем такие геометрически изомерные формы или смеси геометрических изомеров, в том числе отдельные E и Z изомеры соединений формулы (I), а также смеси таких изомеров в любых соотношениях.
В формулу (I) включены соединения, где один или несколько углеродных центров являются хиральными. Настоящее изобретение включает в свой объем каждый возможный оптический изомер, практически не содержащий, т.е. имеющий менее 5%, какой-либо другой оптический изомер (изомеры), а также смеси одного или более оптических изомеров в любых соотношениях, в том числе их рацемические смеси.
Специалисту понятно, что некоторые соединения формулы (I) могут существовать в энантиомерных формах в соответствии с направлением вращения плоскости поляризованного света, проходящего через образец этого соединения. Отдельные оптические изомеры, а также их смеси в любых соотношениях входят в объем данного изобретения.
Необходимо учесть, что структурная формула (I) представляет собой лишь двухмерное изображение этих соединений. Отдельные группы соединений формулы (I) включают такие, где:
(i) один из R1, R2, R3 и R4 обозначает фтор,
(ii) два из R1, R2, R3 и R4 обозначают фтор,
(iii) R1 обозначает фтор,
(iv) R2 обозначает фтор,
(v) R3 обозначает фтор,
(vi) R4 обозначает фтор,
(vii) R5 обозначает водород,
(viii) R5 обозначает C1-C4-алкил, предпочтительно алкил, имеющий 1, 2 или 3 атома углерода, и, более предпочтительно, метил или этил,
(ix) R6 обозначает водород,
(x) R6 обозначает C1-C4-алкил, предпочтительно алкил, имеющий 1, 2 или 3 атома углерода, и, более предпочтительно, метил или этил,
(xi) R6 обозначает гидроксигруппу,
(xii) R5 и R6 вместе с углеродом кольца образуют карбонильную группу,
(xiii) R7 обозначает водород,
(xiv) R7 обозначает гидрокси,
(xv) R8 обозначает водород,
(xvi) R8 обозначает C1-C4-алкил, предпочтительно алкил, имеющий 1, 2 или 3 атома углерода, и, более предпочтительно, метил, этил или изопропил,
(xvii) R8 обозначает цикло(C3 или C4) алкил, предпочтительно циклопропил,
(xviii) R9 обозначает водород,
(xix) R9 обозначает C1-C4-алкил, предпочтительно алкил, имеющий 1, 2 или 3 атома углерода, и, более предпочтительно, метил, этил или изопропил,
(xx) R9 обозначает цикло(C3 или C4) алкил, предпочтительно циклопропил,
(xxi) R8 и R9 вместе с азотом образуют морфолиногруппу,
(xxii) X обозначает связь,
(xxiii) X обозначает метилен,
(xxiv) X обозначает -О-,
и их соли и сольваты.
Предпочтительными как класс являются соединения, где >C=O группа и бензольное кольцо находятся на противоположных сторонах экзо-двойной связи, и их соли и сольваты.
Отдельными предпочтительными соединениями формулы (I) являются
(E)-2-(6-фтор-3-метил-1-инданилиден)ацетамид,
(E)-N-циклопропил-2-(6-фтор-3-метил-1-инданилиден)ацетамид,
(E)-2-(6-фтор-3,3-диметил-1-инданилиден)-N-метилацетамид,
(E)-N-циклопропил-2-(6-фтор-3-этил-1-инданилиден)ацетамид,
(E)-N-циклопропил-2-(5,6-дифтор-1-инданилиден)ацетамид,
(E)-2-(5,6-дифтор-1-инданилиден)-N-метилацетамид,
(E)-2-(5,6-дифтор-1-инданилиден)ацетамид,
(E)-2-(5,7-дифтор-1-инданилиден)ацетамид,
(E)-N-циклопропил-2-(4,6-дифтор-1-инданилиден)ацетамид,
(E)-2-(4,6-дифтор-1-инданилиден)-N-изопропилацетамид,
(E)-2-(4,6-дифтор-1-инданилиден)-N,N-диметилацетамид,
(Z)-2-(4,6-дифтор-2-гидрокси-1-инданилиден)ацетамид,
(E)-2-(7-фтор-1,2,3,4-тетрагидро-1-нафтилиден)ацетамид,
(E)-N-циклопропил-2-(7-фтор-1,2,3,4-тетрагидро-1-нафтилиден)-ацетамид,
(E)-N-циклопропил-2-(6-фтор-3,4-дигидро-2H-1-бензопиран-4- илиден)ацетамид,
(E)-2-(4,6-дифтор-1-инданилиден)ацетамид,
(E)-2-(6-фтор-1-инданилиден)ацетамид,
(Z)-2-(6-фтор-2-гидрокси-1-инданилиден)ацетамид,
(E)-2-(6-фтор-3,3-диметил-1-инданилиден)ацетамид,
(E)-2-(6-фтор-3-этил-1-инданилиден)-N,N-диметилацетамид,
(E)-2-(6-фтор-3-гидрокси-1-инданилиден)ацетамид
и их соли и сольваты.
Особенно предпочтительным является (E)-2-(4,6-дифтор-1- инданилиден)ацетамид, вместе с его солями и сольватами.
Предпочтительны соли и сольваты, являющиеся фармацевтически приемлемыми.
Кроме того, настоящее изобретение относится к соединению формулы (I) или его фармацевтически приемлемой соли или сольвату для применения при лечении млекопитающего, в том числе человека.
Данное изобретение относится также к применению соединения формулы (I) или его фармацевтически приемлемой соли или сольвата для получения лекарственного препарата для лечения млекопитающего, в том числе человека.
Фармацевтически приемлемые соли включают соли аммония, соли щелочных металлов, например, соли натрия и калия, и соли щелочноземельных металлов, например, соли магния и кальция.
Соли, которые не являются фармацевтически приемлемыми, находят применение при получении и/или очистке самих соединений и/или таких их солей, которые являются полезными и/или при нетерапевтическом использовании, например, in vitro.
Соединения формулы (I) вместе с их фармацевтически приемлемыми солями и сольватами применимы в медицине в качестве центральных миорелаксантов и, следовательно, могут быть использованы при лечении состояний, ассоциированных с патологически повышенным тонусом скелетных мышц.
Они особенно ценны при релаксации скелетных мышц в спастических, гипертензивных и гиперкинетических состояниях. В частности, их можно применять при лечении и ослаблении симптомов вызванного физическим усилием спазма скелетных мышц, например, в случае боли в нижней части спины. Их можно также применять в таких состояниях, как повреждение спинного мозга, паркинсонизм, хорея, артрит, атетоз, состояние эпилепсии и столбняк, и особенно для ослабления мышечного спазма при таких состояниях, как мышечная спастичность, миозит, спондилит, церебральный (корковый) паралич, цереброваскулярное заболевание и рассеянный склероз. Их можно также применять в качестве предоперационных миорелаксантов.
Соединения формулы (I) вместе с их фармацевтически приемлемыми солями и сольватами можно также применять при лечении в случаях, связанных с конвульсивным состоянием, например, являющимся последствием большого эпилептического, малого эпилептического припадка, психомоторного эпилептического припадка или фокального эпилептического припадка.
Соединения формулы (I) вместе с их фармацевтически приемлемыми солями и сольватами применимы также при лечении тревоги, как здесь используется этот термин, следует понимать как расстройства с симптомом тревоги, страха.
Тревожные расстройства определены в Diagnostic and Statistical Manual of Mental Disorders (Third Edition - Revised, 1987, опублик. The American Psychiatric Association, Washington, D.C., USA, см. стр. 235-253) как психиатрические состояния, имеющие симптомы тревоги и реакции избегания в качестве характерных признаков. В такие состояния включают генерализованное состояние тревоги, простую фобию и состояние паники.
Тревога также наблюдается как симптом, ассоциированный с другими психиатрическими нарушениями, например, навязчивыми компульсивными состояниями, посттравматическим стрессовым состоянием, шизофреническими депрессивными состояниями и основными депрессивными состояниями, и с органическими клиническими состояниями, такими как болезнь Паркинсона, рассеянный склероз, и другими нарушающими физическую трудоспособность расстройствами.
Соединения формулы (I) вместе с их фармацевтически приемлемыми солями и сольватами применимы также при лечении боли, например, связанной с воспалением и/или травмой, и находят применение в качестве мягких и сильных аналгетиков.
Соединения формулы (I) вместе с их фармацевтически приемлемыми солями и сольватами применимы также в лечении воспалительных состояний, например ревматоидного артрита, ревматоидного спондилита, остеоартрита и подагрического артрита, и несуставных воспалительных состояний, например, синдрома образовавшего грыжу (разорванного) выпавшего межпозвоночного диска, бурсита, тендинита, тендосиновита, синдрома фибромиалгии и других воспалительных состояний, связанных с растяжением связок и региональной деформацией скелетных мышц. Особенно следует отметить, что эти соединения менее ульцергенны, чем другие противовоспалительные средства, такие как ибупрофен, напроксе и т.п.
Таким образом, настоящее изобретение относится также к способу лечения
a) состояния, связанного с патологически повышенным тонусом скелетных мышц,
b) состояния, связанного с конвульсиями,
c) тревоги,
d) боли, или
e) воспалительного состояния
у млекопитающего, в том числе человека, предусматривающему введение ему терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемых соли или сольвата.
Соединения формулы (I) и их фармацевтически приемлемые соли и сольваты могут вводиться с сопутствующими другими терапевтическими средствами для лечения перечисленных выше состояний. Для состояний, связанных с патологически повышенным тонусом скелетных мышц, такими другими средствами могут быть аналгетики, такие как кодеин, ацетаминофен, фенацетин и ибупрофен. Для воспалительных состояний (например, артрита) и/или боли, такими другими средствами являются аналгетики, такие как кодеин, оксикодон, ацетаминофен, фенацетин и ибупрофен, антиартритные средства, такие как метотрексат и азатиоприн, и противозастойные средства, такие как эфедрин и псевдоэфедрин.
Соединение, соль или сольват (далее называемые активными ингредиентами) могут вводиться любым путем, в том числе пероральным, ректальным, назальным, наружно (в том числе щечным и подъязычным), вагинальным, парентеральным (в том числе подкожным, внутримышечным, внутривенным и внутрикожным) и чрескожным. Должно быть понятно, что предпочтительный путь введения будет определяться, например, состоянием и возрастом реципиента и видом состояния, которое подлежит лечению.
Количество требуемого активного ингредиента зависит от ряда факторов, в том числе от вида состояния и его тяжести, идентичности реципиента и способа введения, и определяется в конечном счете лечащим врачом.
Как правило, для каждого из указанных состояний подходящая доза активного ингредиента (оцененная в расчете на исходное соединение) находится в диапазоне 0,05-100 мг на кг веса тела реципиента в день, предпочтительно в пределах 0,1-50 мг на кг веса тела в день, наиболее предпочтительно в пределах 0,5-20 мг на кг веса тела в день и оптимально 1-10 мг на кг веса тела в день. Желаемую дозу предпочтительно представляют в виде двух, трех, четырех, пяти, шести или более субдоз, вводимых с соответствующими интервалами в течение дня.
Хотя активный ингредиент можно вводить индивидуально, предпочтительно предоставлять его в виде фармацевтической композиции, содержащей активный ингредиент, указанный выше, вместе с приемлемым носителем. Каждый носитель должен быть "приемлемым" в смысле его совместимости с другими ингредиентами композиции и безвредным для реципиента.
Композиции включают в себя композиции, пригодные для перорального, ректального, назального, наружного (в том числе щечного и подъязычного), вагинального, парентерального (в том числе подкожного, внутримышечного, внутривенного и внутрикожного) или чрескожного введения. Композиции могут быть представлены для удобства в форме стандартных единичных доз и могут быть приготовлены любыми способами, хорошо известными специалисту в области фармации. Такие способы предусматривают стадию соединения активного ингредиента с носителем, состоящим из одного или более дополнительных ингредиентов. Обычно эти композиции готовят однородным и тесным связыванием активного ингредиента с жидкими носителями или тонкоизмельченными твердыми носителями или с обоими типами носителей, после чего продукт формуют, если это необходимо.
Композиции по настоящему изобретению, пригодные для перорального введения, могут быть представлены в виде таких отдельных единиц, как капсулы, крахмальные облатки или таблетки, каждая из которых содержит заданное количество активного ингредиента, в виде порошка или гранул, в виде раствора или суспензии в водной или неводной жидкости, или в виде жидкой эмульсии типа масло в воде или жидкой эмульсии типа вода в масле. Активный ингредиент может быть также представлен в виде болюса, пасты или электуария (лекарственной кашки).
Таблетки могут быть приготовлены прессованием или формованием, иногда с одним или несколькими дополнительными ингредиентами. Прессованные таблетки могут быть приготовлены прессованием в подходящей таблетировочной машине активного ингредиента в свободно-сыпучем виде, таком как порошок или гранулы, иногда смешанные со связывающим веществом (например, повидоном, желатином, гидроксипропилметилцеллюлозой), смазывающим веществом, инертным разбавителем, консервантом, разрыхляющим средством (например, натриевой солью гликолата крахмала, поперечно-сшитым повидоном, натриевой солью поперечно-сшитой карбоксиметилцеллюлозы), поверхностно-активным или диспергирующим агентом. Формованные таблетки могут быть изготовлены формованием в подходящей машине смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Таблетки иногда могут быть покрыты или могут иметь бороздки и могут быть сформованы таким образом, чтобы обеспечить медленное или контролируемое выделение содержащегося в них активного ингредиента, например, с применением гидроксипропилметилцеллюлозы в варьируемых соотношениях, для обеспечения желаемого профиля высвобождения. Таблетки могут быть иногда защищены энтеросолюбильным покрытием для обеспечения выделения в частях кишечника, иных, чем желудок.
Композиции, пригодные для перорального применения, описанные выше, могут также содержать буферирующие агенты, предназначенные для нейтрализации кислотности в желудке. Такие буферы могут быть выбраны из множества органических и неорганических агентов, таких как слабые кислоты или основания, смешанные с их конъюгированными солями.
Композиции для наружного введения в рот включают таблетки, содержащие активный ингредиент на ароматизированной основе, обычно сахарозе и аравийской камеди или трагаканте, пастилки, содержащие активный ингредиент на инертной основе, такой как желатин и глицерин, или сахароза и аравийская камедь, и жидкости для полоскания рта, содержащие активный ингредиент и подходящий жидкий носитель.
Композиции для ректального введения могут быть представлены в виде суппозитория с подходящей основой, содержащей, например, какао-масло или салицилат.
Композиции для вагинального введения могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или спреев, содержащих, кроме активного ингредиента, такие носители, о которых известно, что они пригодны для такого применения.
Композиции, пригодные для парентерального введения, включают водные и неводные изотонические стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатические агенты и растворенные вещества, которые делают эти композиции изотоническими в крови реципиента, и водные и неводные стерильные суспензии, которые могут содержать суспендирующие агенты и загустители, такие как липосомы или другие состоящие из микрочастиц системы, предназначенные для направления этих соединений к компонентам крови или к одному или нескольким органам. Композиции могут быть представлены в запечатанных контейнерах для одной дозы или для множества доз, например, в ампулах или флаконах, и могут храниться в лиофилизированном состоянии, требующем только добавления стерильного жидкого носителя, например, воды для инъекций, непосредственно перед использованием.
Не подготовленные заранее растворы и суспензии для инъекций могут быть получены из стерильных порошков, гранул и таблеток описанного ранее типа.
Композиции, пригодные для чрескожного введения, могут быть представлены в виде отдельных пластырей, приспособленных для того, чтобы оставаться в тесном контакте с эпидермисом реципиента в течение длительного периода времени. Такие пластыри для удобства содержат активный ингредиент в виде, иногда забуференного, водного раствора, например, с концентрацией 0,1-0,2 М активного соединения. В одном частном варианте активный ингредиент может доставляться из пластыря при помощи ионтофореза, как описано в общих чертах в Pharmaceutical Research, 3(6), 318 (1986).
Предпочтительные композиции со стандартными дозами содержат дневную дозу или единицу, дневную разделенную дозу или их удобные части активного ингредиента, например, 1-1500 мг, предпочтительно, 5-1000 мг и, наиболее предпочтительно, 10-700 мг активного ингредиента, в расчете на исходное соединение.
Должно быть понятно, что, кроме конкретно упомянутых ингредиентов, композиции этого изобретения могут содержать другие агенты, общепринятые в этой области, соответствующие типу композиции, например, композиции для перорального введения могут содержать такие дополнительные агенты, как подслащивающие вещества, загустители и ароматизаторы.
Таким образом, данное изобретение относится также к фармацевтической композиции, содержащей соединение формулы (I), или его фармацевтически приемлемую соль, или сольват вместе с приемлемым носителем.
Соединения, соли и сольваты могут быть получены способом, включающим взаимодействие соединения формулы (II)
где R1-R7 и X имеют указанные выше значения и Z обозначает отщепляемую группу, с амином NR8R9 или его подходящим производным. Подходящие отщепляемые группы включают атомы галогенов, такие как хлор или бром, активированные эфиры (например, N-гидроксисукцинимид, пентафторфенил, нитрофенил, 1-гидроксибензотриазол), смешанные ангидриды (например, этоксикарбонилокси) или C1-C6-алкокси (например, этокси). Реакцию удобно проводить в инертном органическом растворителе (например, дихлорметане) при температуре приблизительно -20-120oC и удобно при приблизительно 0-25oC. Пригодными производными амина являются гидратированные или гидрохлоридные производные, например, NH4OH, NH4Cl.
Когда R8 и R9 обозначают H, соединения формулы (I) могут быть получены взаимодействием соединений формулы (II), где X обозначает атом галогена, такой как хлор или бром, с амином в гидратированной форме, например, с NH4OH, в подходящем органическом растворителе (например, дихлорметане) при температуре приблизительно 0-25oC.
Соединения формулы (I), где R6 или R7 обозначают гидроксигруппу, могут быть получены взаимодействием соединений формулы (II), в которой X обозначает C1-C6-алкокси, например, этоксигруппу, а гидроксигруппа защищена, например, SiMe2-трет-Bu с амином, представленным в виде гидрохлорида, например, NH4Cl, в присутствии Me3Al в нейтральных условиях, с последующим удалением защитной группы в нейтральных условиях, например, п-толуолсульфонатом пиридиния (ППТС).
Альтернативно, соединения формулы (I), где R6 или R7 обозначают гидроксигруппу, могут быть получены из соединений формулы (I), где R6 или R7 обозначают H, галогенированием, например, N-бромсуксинимидом (NBS) с последующим гидролизом, например, карбонатом серебра (Ag2CO3). Соединения формулы (I), где R6 или R7 обозначают аллильную гидроксигруппу, могут быть получены из соединений формулы (I), в которой R6 или R7 обозначают H, путем окисления, например, диоксидом селена.
Соединения формулы (II), где Z представляет собой атом галогена, могут быть получены из соединений формулы (III)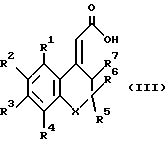
в которой R1 - R7 и X имеют указанные выше значения, путем взаимодействия с галогенирующим агентом (например, оксалилхлоридом или тионилхлоридом) в подходящем органическом растворителе (например, бензоле, толуоле, дихлорметане) иногда в присутствии катализатора (например, ДМФ) при температуре от приблизительно -20oC до температуры кипения с обратным холодильником.
Соединения формулы (II), где Z представляет собой алкоксигруппу (например, этоксигруппу), могут быть получены из соединений формулы (III) взаимодействием с подходящим полярным растворителем (например, органическим спиртом, таким как этанол) иногда в присутствии каталитического количества кислоты (например, п-толуолсульфокислоты) при температуре приблизительно 0oC до температуры перегонки.
Соединения формулы (II), где Z представляет собой активированный эфир (описанный выше), могут быть получены из соединений формулы (III) взаимодействием с фенолом или - N-гидроксисоединением и карбодиимидом (например, дициклогексилкарбодиимидом или 1-(3-диметиламинопропил)-3- этилкарбодиимидом) в растворителе, таком как диметилформамид (ДМФ) или дихлорметан, при 0-50oC.
Соединения формулы (III) могут быть получены дегидратацией соединений формулы (IV)
где R1 - R7 и X имеют указанные выше значения, взаимодействием с подходящим дегидратирующим агентом (например, кислотой, такой как трифторуксусная кислота) в подходящем органическом растворителе (например, дихлорметане) при температуре от приблизительно -20oC до температуры перегонки.
Соединения формулы (IV) могут быть получены омылением соответствующего C1-C6-алкилового эфира основанием (например, гидроксидом натрия) в подходящем полярном растворителе (например, этаноле) при температуре от приблизительно 0oC до температуры перегонки или водной кислотой (например, соляной кислотой) при температуре от приблизительно 0oC до температуры перегонки.
Эфиры соединений формулы (IV), имеющие R6 или R7 в виде защищенных гидроксигрупп, например, защищенных SiMe2-трет-Bu, могут быть дегидратированы в нейтральных условиях (например, Сульфураном Мартина, биc [α,α- биc(тpифтopмeтил)- бензолметанолато] дифенилсера) с получением соответствующих защищенных гидроксисоединений формулы (II), где Z представляет собой C1-C6-алкокси (например, этокси).
Эфиры соединений формулы (IV) могут быть получены из соединений формулы (V)
где R1-R7 и X имеют указанные выше значения взаимодействием с halCH2CO2R, где hal обозначает атом галогена, такой как хлор, бром или иод (предпочтительно бром), a R представляет собой C1-6-алкил (например, этил), в присутствии металла (например, цинка, предпочтительно активированного цинка) и каталитического количества галогена (например, иода) в подходящем органическом растворителе (например, этиловом эфире, бензоле) при температуре от 0oC до температуры перегонки или взаимодействием с литиевой солью этилацетата в подходящем растворителе (например, тетрагидрофуране) при температуре между -100oC и комнатной температурой (например, от -80 до -70oC).
Соединения формулы (V), имеющие R6 или R7 в виде защищенных гидроксигрупп, как описано выше, могут быть получены из соответствующего незащищенного гидроксисоединения формулы (V) путем подходящей защиты при нейтральных условиях, например, т-бутилдиметилсилилхлоридом, в присутствии основания, такого как имидазол.
Соединения формулы (V), имеющие R6 или R7 в виде гидроксигрупп, могут быть получены из соответствующего галоген- (например, бром-) соединения гидролизом в нейтральных условиях, например, карбонатом серебра (Ag2CO3).
Соединения формулы (V), имеющие R6 или R7 в виде аллильного алкила (например, метила), могут быть получены из соответствующих соединений формулы (V), в которых R6 и/или R7 представляют собой H, взаимодействием с основанием (например, гидридом натрия) с последующим алкилированием, например, метилиодидом (MeI).
Соединения формулы (V) могут быть получены из соединения формулы (VI).

где R1-R7, X и Z имеют указанные выше значения, предпочтительно Z представляет собой атом галогена, такой как хлор, циклизацией в присутствии льюисовой кислоты (например, хлорида алюминия в подходящем растворителе (например, дихлорметане) при температуре от приблизительно 0oC до температуры перегонки.
Соединения формулы (VI), где Z представляет собой атом галогена (например, хлор или бром), могут быть получены из соответствующей карбоновой кислоты взаимодействием с галогенирующим агентом (например, оксалилхлоридом или тионилхлоридом) либо в чистом виде, либо в подходящем органическом растворителе (например, метиленхлориде или N,N-диметилформамиде) при температуре от приблизительно 0oC до температуры перегонки.
Соединения формулы (VI), где Z представляет собой алкоксигруппу (например, этоксигруппу), могут быть получены из соединений формулы (VII)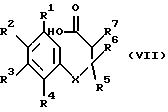
где R1-R7 и X имеют указанные выше значения взаимодействием с подходящим органическим спиртом (например, этанолом) иногда в присутствии каталитического количества кислоты (например, п-толуолсульфокислоты) при температуре от приблизительно 0oC до температуры перегонки.
Карбоновые кислоты могут быть получены омылением соответствующих C1-6-алкиловых эфиров основанием (например, гидроксидом натрия) в подходящем полярном растворителе, например, воде или этаноле) при температуре от приблизительно 0oC до температуры перегонки или водной кислотой (например, соляной кислотой) при температуре от приблизительно 0oC до температуры перегонки.
Карбоновые кислоты, где X не является кислородом, могут быть получены из соединений формулы (VIII)
где R и R1-R6 имеют указанные выше значения, а n обозначает 0 или 1, моно-деэтерификацией сильным основанием (например, водным гидроксидом калия) при температуре перегонки.
Соединения формулы (VIII) могут быть получены реакцией соединения формулы (IX) с соединением формулы (X)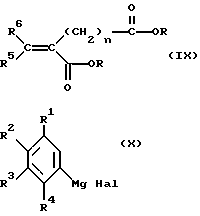
где R, R1-R6 и n имеют указанные выше значения, и Hal обозначает Cl, Br или I (предпочтительно Br), в органическом растворителе (например, безводном диэтиловом эфире) и иногда в присутствии галогенида меди (например, иодида меди (I)) при температуре между -50oC и температурой перегонки.
Соединения формулы (IX) могут быть получены реакцией соединения формулы (XI) с соединением формулы (XII)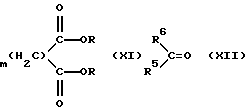
где R, R5 и R6 имеют указанные выше значения, и m обозначает 1 или 2, в органическом растворителе (например, этиловом эфире или дихлорметане) при температуре между комнатной температурой и температурой перегонки.
Соединения формулы (X) могут быть получены из соответствующих галогенсодержащих соединений (например, содержащих бром, хлор) стандартными способами, хорошо известными специалистам в этой области. Сами галогенсоединения могут быть получены коммерчески доступными способами, хорошо известными специалистам в этой области или взятыми из химической литературы.
Альтернативно, соединения формулы (IX) могут быть получены согласно способу E. Z. Eliel, R.O. Hutchins, and Sr.M. Knoeber, Organis Synthesis Coll. Vol. VI, 442, 1988 с модификациями, легко очевидными для специалистов в этой области.
Соединения формул (XI) и (XII) могут быть коммерчески доступными или получены способами, хорошо известными специалистам в этой области или взятыми из химической литературы.
Альтернативно эти эфиры могут быть получены из соединений формулы (XIII)
где R, R1-R4, R6 и R7 имеют указанные выше значения, путем восстановления двойной связи, например, каталитическим восстановлением, например, с оксидом платины (PtO2) и водородом в подходящем органическом растворителе (например, этаноле) при температуре приблизительно 20-60oC.
Если X представляет собой кислород, эфиры могут быть получены реакцией соединения формулы (XIV) с соединением формулы (XV)
где R, R1-R6 и Z имеют указанные выше значения, в подходящем органическом растворителе в присутствии основания (например, гидрида натрия).
Соединения формул (XIV) и (XV) могут быть коммерческим доступными или получены способами, хорошо известными специалистам в этой области или взятыми из химической литературы.
Соединения формулы (XIII) могут быть получены из соединений формулы (XVI)
где R1-R4, R6 и R7 имеют указанные выше значения, путем этерификации подходящим органическим спиртом (например, этанолом), необязательно в присутствии каталитического количества кислоты (например, HCl, п-толуолсульфокислоты, тионилхлорида) при температуре приблизительно 20-60oC.
Соединения формулы (XVI) могут быть также использованы для прямого получения соответствующей ненасыщенной кислоты путем восстановления двойной связи, т. е. каталитическим восстановлением, например, палладием или оксидом платины (PtO2) и водородом, в подходящем органическом растворителе (например, этаноле) при температуре приблизительно от 0oC до температуры перегонки.
Соединения формулы (XVI) могут быть получены из соединений формулы (XVII)
где R1-R4 и R6 имеют указанные выше значения, взаимодействием с HOOCCHR7COOH в органическом основании (например, пиридине), необязательно в органическом растворителе (например, дихлорметане), необязательно с каталитическим количеством основания (например, пиперидина) при температуре приблизительно от 0oC до температуры перегонки.
Соединения формулы (XVII) и HOOCCHR7COOH могут быть коммерчески доступны или получены способами, хорошо известными специалистам в этой области или описанными в химической литературе.
Альтернативно, соединения формулы (I) могут быть получены взаимодействием R8R9NCOCH2PO(OR)2 (где R, R8, R9 имеют указанные выше значения) с основанием (например, NaH) в подходящем органическом растворителе (например, ТГФ или ДМСО) и взаимодействием полученных анионных образцов с соединением формулы (VI) или формулы (VIa), соответственно, при температуре приблизительно от 0oC до температуры перегонки. Реакции способствует добавление анионного стабилизирующего реагента (например, гексаметилдисилазана калия или краун-эфира (например, 15-краун-5).
Соединение R8R9NCOCH2PO(OR)2 может, в зависимости от R, R8 и R9, быть коммерчески доступным или получено способами, хорошо известными специалистам или описанными в химической литературе. Альтернативно, эти соединения могут быть получены взаимодействием подходящего R8R9NCOCH2Z (где Z имеет указанное выше значение) с подходящим P(OR)3 в органическом растворителе (например, ТГФ) при температуре приблизительно 0-50oC.
Соединение R8R9NCOCH2Z может быть получено взаимодействием соответствующего R8R9NH с ZCH2COZ в подходящем органическом растворителе (например, диэтиловом эфире) при температуре приблизительно от 0oC до температуры перегонки.
Соединение R8R9NH может быть доступно коммерчески или получено способами, хорошо известными специалистам в области получения аминов или описанными в химической литературе. Соединение ZCH2COZ может быть получено коммерческим путем или способами, хорошо известными специалистам в области получения таких соединений или описанными в химической литературе.
Альтернативно, соединения формулы (I) могут быть получены взаимодействием R8R9NCOCH2P(+)(Ph))3Cl(-) (где R8, R9 и Z имеют указанные выше значения) с подходящим основанием (например, NaH) в подходящем органическом растворителе (например, диметоксиэтане) при температуре приблизительно 0-50oC и взаимодействием полученной анионной продукта с соединением формулы (V) при температуре приблизительно от 0oC до температуры перегонки.
Соединение R8R9NCOCH2P(+)(Ph)3Cl(-) может быть получено реакцией подходящего R8R9NCOCH2Z с 50% молярным избытком P(Ph)3 (трифенилфосфина) в подходящем органическом растворителе (например, ТГФ) при температуре приблизительно от 20oC до температуры перегонки.
R8R9NCOCH2Z может быть получено, как описано ранее.
Альтернативно, соединения формулы (I) могут быть также получены непосредственно из соединений формулы (III) взаимодействием подходящего связывающего агента (например, дициклогексилкарбодиимида (ДЦК) или этилхлорформиата) с последующим взаимодействием образованного таким образом активированного эфира с подходящим амином, HNR8R9.
Альтернативно, соединения формулы (I), где R5 представляет собой водород, a R6 представляет собой гидроксигруппу, могут быть получены из соединений формулы (I), в которых R5 и R6 вместе образуют карбонильную группу, путем восстановления этой карбонильной группы при помощи подходящего восстанавливающего агента, например, боргидрида натрия, в подходящем растворителе, таком как алканол (например, этанол).
Соединения формулы (I), так же как и любые из промежуточных продуктов, применяемых при получении данных соединений, могут быть подвергнуты одному или нескольким следующим необязательным преобразованиям:
(i) преобразование соединения формулы (I) или его промежуточных продуктов в их соли,
(ii) при образовании соли соединения формулы (I) или его промежуточного продукта преобразования этой соли в соединение формулы (I) или его промежуточный продукт.
Следующие далее примеры иллюстрируют данное изобретение, но не ограничивают его.
Пример 1
Получение (E)-2-(6-фтор-1-инданилиден)ацетамида
а) Получение 3-(4-фторфенил)пропионовой кислоты
Смесь 4-фторкоричной кислоты (300,0 г, 1,8 моля, Aldrich) и 5% палладия на угле (9,0 г) в этаноле (3 л) гидрировали при атмосферном давлении и комнатной температуре в течение 4,5 ч. Смесь фильтровали через целит (диатомовую землю) и фильтрат концентрировали в вакууме, получая 275,1 г (91%) 3-(4-фторфенил)пропионовой кислоты в виде белого твердого вещества, т.пл. 86-88oC.
b) Получение 3-(4-фторфенил)пропионилхлорида
Смесь 3-(4-фторфенил)пропионовой кислоты (275,1 г, 1,6 моля) и тионилхлорида (300 мл, 4,1 моля) нагревали с обратным холодильником в течение 3 часов, охлаждали до комнатной температуры и перегоняли в вакууме водоструйного насоса, получая 287,6 г (96%) 3-(4-фторфенил)пропионилхлорида в виде бледно-розового масла, т. кип. 120-122oC/15 мм Hg.
с) Получение 6-фтор-1-инданона
Раствор 3-(4-фторфенил)пропионилхлорида (287,6 г, 1,5 моля) в дихлорметане (1,4 л) добавляли по каплям в течение 3 ч к охлажденной на льду механически перемешиваемой суспензии хлорида алюминия (226,0 г, 1,7 моля, Aldrich) в дихлорметане (2,2 л) в атмосфере азота. Полученный желтовато-коричневый раствор кипятили с обратным холодильником в течение 5 ч и давали ему остыть до комнатной температуры.
Раствор промывали последовательно водой (2 л), 1 н. гидроксидом натрия (2 л), водой (2 л) и солевым раствором (2 л). Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали до рыжевато-коричневой жидкости (229,1 г, 99%). Жидкость перекристаллизовывали из смеси дихлорметан-гексан, получая 215,7 г (93%) 6-фтор-1-инданона в виде кристаллов нестандартного белого цвета, т.пл. 57-59oC.
d) Получение этил-2-(6-фтор-1-гидрокси-1-инданил)ацетата
(i) Смесь 6-фтор-1-инданона (5,0 г, 33,3 ммоля), этилбромацетата (8,3 г, 50,0 ммолей, Aldrich), активированного порошка цинка (3,2 г, 50,0 ммолей, Mallinckrodt; Org. Synth., Coll. Vol. 6, 290, 1988) и несколько кристаллов иода в смеси диэтиловый эфир-бензол (1:1, 100 мл) нагревали с обратным холодильником в атмосфере азота в течение 24 часов. Смесь фильтровали и фильтрат концентрировали в вакууме. Остаток в диэтиловом эфире интенсивно перемешивали с избытком разбавленного гидроксида алюминия, сушили и концентрировали, получая этил-2-(6-фтор-1-инданил)ацетат в виде янтарного масла (7,6 г, 97%).
(ii) Этилацетат (1,8 г, 20 ммолей добавляли каплями к перемешиваемому охлажденному (на бане ацетона с сухим льдом) 1 н. раствору бис(триметилсилил)амида лития в тетрагидрофуране (20 мл, Aldrich) под азотом. После 15 мин каплями добавляли раствор 6-фтор-1-инданона (3,0 г, 20 ммолей) в тетрагидрофуране (20 мл) и полученную смесь перемешивали в течение 1 часа (на бане с сухим льдом-ацетоном). Добавляли 1н раствор соляной кислоты (20 мл) и смеси давали нагреться до комнатной температуры. Органическую фазу отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали до бледно-желтого масла (5,3 г). Смесь хроматографировали на силикагеле 60 с применением линейного градиента дихлорметан-гексан (1:1) - дихлорметан в качестве элюента. Фракции, содержащие только этил-2-(6-фтор-1-гидрокси-1-инданил)ацетат, объединяли и концентрировали в вакууме, получая 3,1 г (65%) бесцветного масла.
e) Получение 2-(6-фтор-1-гидрокси-1-инданил)уксусной кислоты
Смесь этил-2-(6-фтор-1-гидрокси-1-инданил)ацетата (44,0 г, 0,18 моля), 1 н. гидроксида натрия (180 мл) и абсолютного этанола (280 мл) перемешивали в течение 18 часов при комнатной температуре. Смесь концентрировали в вакууме, добавляли H2O и экстрагировали диэтиловым эфиром. Слой диэтилового эфира промывали солевым раствором, сушили над сульфатом натрия и концентрировали в вакууме, получая 2-(6-фтор-1-гидрокси-1-инданил)уксусную кислоту в виде янтарного масла (37,7 г, 100%, примечание: это соединение самопроизвольно дегидратируется при стоянии при комнатной температуре до смеси олефинов, если оно сразу не подвергается взаимодействию с трифторуксусной кислотой).
f) Получение 2-(6-фтор-1-гидрокси-1-инданил)ацетата лития
Смесь этил-2-(6-фтор-1-гидрокси-1-инданил)ацетата (2,0 г, 8,4 ммоля) 1 н. гидроксида лития (8,4 мл) и абсолютного этанола (13,0 мл) перемешивали в течение 18 часов при комнатной температуре. Смесь концентрировали в вакууме, разбавляли H2O и экстрагировали диэтиловым эфиром. Водную фазу концентрировали в вакууме, разбавляли толуолом (100 мл) и концентрировали в вакууме, получая 2-(6-фтор-1-гидрокси-1-инданил)ацетат лития в виде белого твердого вещества (1,4 г, 77%)
g) Получение (E)-2-(6-фтор-1-инданилиден)уксусной кислоты
Трифторуксусную кислоту (1,5 мл) добавляли к перемешиваемой охлажденной (баня со льдом и метанолом) суспензии 2-(6-фтор-1- гидрокси-1-инданил)ацетата лития (0,5 г, 2,3 ммоля) в дихлорметане (13,5 мл). Через 15 минут смесь концентрировали в вакууме и полученное белое твердое вещество перекристаллизовывали из водного ацетона, получая (E)-2-(6-фтор-1-инданилиден)уксусную кислоту в виде белых кристаллов (0,32 г, 73%), идентичных соединению примера 1i по т.пл. смеси 203-205oC и ЯМР.
h) Получение (E)-2-(6-фтор-1-инданилиден)уксусной кислоты
Трифторуксусную кислоту (100 мл) добавляли к перемешиваемому охлажденному (баня со льдом и метанолом) раствору 2-(6-фтор-1-гидрокси-1-инданил)уксусной кислоты (37,5 г, 0,18 моля) в дихлорметане (900 мл). После 15 минут смесь концентрировали в вакууме, получая (E)-2-(6-фтор-1-инданилиден)уксусную кислоту в виде желтовато-рыжевато-коричневого твердого вещества (33,0 г, 95%), т.пл. 203-205oC.
i) Получение (E)-2-(6-фтор-1-инданилиден)ацетилхлорида
Охлажденную на льду перемешиваемую суспензию (E)-2-(6-фтор-1-инданилиден)уксусной кислоты (384 мг, 2 ммоля) в бензоле обрабатывали оксалилхлоридом (761 мг, 6 ммолей) и давали ей нагреться до комнатной температуры в течение 1,5 часов. Полученный желтый раствор концентрировали в вакууме, получая (E)-2-(6-фтор-1-инданилиден)ацетилхлорид в виде бледно-желтого твердого вещества (421 мг, 100%), т.пл. 97-99oC.
j) Получение (E)-2-(6-фтор-1-инданилиден)ацетамида
29,6% водный раствор гидроксида аммония (17,6 мл, 134 ммоля) добавляли по каплям к перемешиваемому, охлажденному (баня со льдом) раствору (E)-2-(6-фтор-1-инданилиден)ацетилхлорида (14,1 г, 67 ммолей) в дихлорметане (165 мл). После 1 часа полученный белый осадок собирали фильтрованием, растворяли в этилацетате (600 мл) и промывали водой (3х300 мл). Слой этилацетата сушили над сульфатом натрия и концентрировали в вакууме. Полученное твердое вещество не совсем белого цвета промывали гексаном, получая 11,6 г (91%) (E)-2-(6-фтор-1-инданилиден)ацетамида, т.пл. 180-183oC.
Пример 2
Получение (E)-2-(6-фтор-1-инданилиден)ацетамида
Перемешиваемую суспензию (E)-2-(6-фтор-1-инданилиден)уксусной кислоты (0,5 г, 2,6 ммоля) в дихлорметане (10 мл) при -20oC последовательно обрабатывали по каплям этилхлороформиатом (0,3 г, 2,6 ммоля, Aldrich) и триэтиламином (0,3 г, 2,6 ммоля, Eastman). Смесь перемешивали при -20oC в течение 2 ч. Добавляли безводный аммиак в дихлорметане (0,8 М, 12 мл) (Примечание: при применении водного гидроксида аммония смешанный ангидрид частично гидролизовался до кислоты), смесь перемешивали в течение 16 часов при комнатной температуре и затем промывали последовательно водой, раствором бикарбоната натрия, водой и солевым раствором. Слой дихлорметана сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая 0,18 г смеси 6: 1 (E)-2-(6-фтор-1-инданилиден)ацетамида и 2-(5-фтор-1H-инден-3-ил)ацетамида.
Пример 3
Получение (E)-N-этил-2-(6-фтор-1-инданилиден)ацетамида
Это соединение получали аналогично получению в примере 5 с заменой применяемого в примере 5 циклопропиламина этиламином (70 мас.% в воде). Полученные при хроматографии растворы, содержание (E)-N-этил-2-(6-фтор-1-инданилиден)ацетамид, концентрировали на роторном испарителе в вакууме. Перекристаллизация остатка из смеси дихлорметан-гексаны дала 1,7 г (68%) (E)-N-этил-2-(6-фтор-1-индалиниден)ацетамида, т.пл. 125-126oC.
Пример 4
Получение (E)-N-циклопропил-2-(6-фтор-1-инданилиден)ацетамида
К охлажденному на льду перемешиваемому раствору (E)-2-(6-фтор-1-инданилиден)ацетилхлорида в дихлорметане (50 мл) добавляли циклопропиламин (1,65 г, 28,86 ммоля) и реакционную смесь нагревали до комнатной температуры в течение ночи. Смесь выпаривали в вакууме до твердого остатка. Остаток растворяли в этилацетате (300 мл), промывали водой (75 мл) и органический слой концентрировали путем роторного испарения в вакууме. Остаток хроматографировали на силикагеле с применением смеси этилацетатгексаны (0:1 - 1:1 - градиент) в качестве элюента. Фракции, содержащие только продукт реакции, объединяли и концентрировали роторным упариванием в вакууме. Перекристаллизация остатка из смеси дихлорметан-гексаны дала 1,6 г (76%) (E)-N-циклопропил-2-(6-фтор-1-инданилиден)ацетамида в виде белого порошкообразного твердого вещества, т.пл. 124-127oC.
(E)-N-этил-2-(6-фтор-1-инданилиден)-N-метилацетамид получали аналогичным образом с заменой циклопропиламина N-этилметиламином (3,5 мл, 0,025 моля, Aldrich). Остаток хроматографировали на силикагеле с применением смеси этилацетат-гексаны (градиент 1: 5 - 1:2) в качестве элюента. Хроматографические фракции, содержащие (E)-N-этил-2-(6-фтор-1-инданилиден)-N- метилацетамид, концентрировали роторным испарением в вакууме. Перекристаллизация остатка из смеси этилацетат-гексаны дала 1,32 г (61%) (E)-N-этил-2-(6-фтор-1-инданилиден)-N-метилацетамида в виде белого твердого вещества, т.пл. 74-77oC.
Пример 5
Получение (E)-2-(4,6-дифтор-1-инданилиден)ацетамида
а) Получение 3-(2,4-дифторфенил)пропановой кислоты
Смесь 2,4-дифторкоричной кислоты (30,0 г, 0,16 моля, Aldrich) и гидрата оксида платины (0,5 г, EM Scientific ) в 95% этаноле (140 мл) помещали в аппарат Парра для гидрогенизации. После поглощения определенного количества водорода катализатор отфильтровывали и фильтрат концентрировали в вакууме, получая 29,7 г (98%) 3-(2,4-дифторфенил)пропановой кислоты в виде белого твердого вещества. Перекристаллизация 1,0 г из смесей ацетонитрил:вода дала 0.61 г 3-(2,4-дифторфенил)пропановой кислоты в виде белого твердого вещества, т.пл. 104-106oC, ЯМР (DMSO-d6): d 12,2 (шир., 1H), 6,98-7,40 (м, 3H), 2,81 (т, 2H), 2,51 (т, 2H).
Анал.рассчитано для C9H8F2O2 (мол. масса 186,15): C 58,06, H 4,38.
Найдено: C 57,94, H 4,36.
b) Получение 4,6-дифтор-1-инданона
К смеси 3-(2,4-дифторфенил)пропановой кислоты (28,7 г, 0,15 моля) и диметилформамида (5 капель) при температуре окружающей среды добавляли по каплям оксалилхлорид (50 мл, Aldrich). Смесь перемешивали при температуре окружающей среды в течение 18 часов. Избыток оксалилхлорида удаляли перегонкой в вакууме, получая 3-(2,4-дифторфенил)пропионилхлорид. Раствор 3-(2,4-дифторфенил) пропионилхлорида в дихлорметане (300 мл) добавляли по каплям к смеси хлорида алюминия (23,4 г, 0,18 моля, Aldrich) в дихлорметане (300 мл) при температуре бани со льдом. После завершения добавления смесь кипятили с обратным холодильником в течение 3,5 часов и давали ей нагреться до температуры окружающей среды в течение ночи. Реакционную смесь выливали в ледяную воду (1500 мл), две фазы разделяли и водную фазу экстрагировали дихлорметаном. Объединенную органическую фазу промывали последовательно 0,1 н. водным гидроксидом натрия и насыщенным раствором хлорида натрия, сушили над сульфатом натрия и концентрировали в вакууме, получая 21,7 г неочищенного 4,6-дифтор-1-инданона. Хроматография на силикагеле с применением смеси гексаны: дихлорметан (3: 1) в качестве элюента дала 10,1 г светло-желтого твердого вещества. Перекристаллизация 0,5 г из смесей ацетон:вода дала 0,2 г 4,6-дифтор-1-инданона в виде белого твердого вещества, т.пл. 97-99oC, ЯМР (CDCl3) d 7,02-7,27 (м, 2H), 3,12 (т, 2H), 2,76 (м, 2H).
Анал. рассчитано для C9H6F2O (мол. масса 168,14): C 64,29, H 3,60.
Найдено: C 64,18, H 3.61.
c) Получение этил-2-(4,6-дифтор-1-гидрокси-1-инданил)ацетата
Смесь 4,6-дифтор-1-инданона (12,6 г, 0,08 моля), этил-бромацетата (19,0 г, 0,11 моля, Aldrich) активированного цинкового порошка (7,5 г, 0,11 моля, Aldrich, Org. Syn. Coll. Vol. 6, 290, 1988) и несколько кристаллов иода в диэтиловом эфире: толуоле (1: 1, 300 мл) нагревали при 30-35oC в атмосфере азота в течение 24 часов. Добавляли еще немного кристаллов иода, температуру доводили до 40-45oC и смесь выдерживали при этой температуре еще 24 часа. Реакционную смесь фильтровали и концентрировали в вакууме. Остаток обрабатывали смесью диэтилового эфира (450 мл), концентрированного гидроксида аммония (135 мл) и воды (135 мл). Водную фазу отделяли и экстрагировали диэтиловым эфиром. Объединенную органическую фазу промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая 22,7 г неочищенного этил-2-(4,6-дифтор-1-гидрокси-1-инданил)ацетата, который хроматографировали на силикагеле с применением смеси дихлорметан:гексан (9:1) в качестве элюента, получая 12,7 г (66%) желтого масла, ЯМР (CDCl3); d 6,67-6,88 (м, 2H), 4,22 (к, 2H), 3,02 (м, 1H), 2,75 (2м, 3H), 2,31 (м, 2H), 1,28 (т, 3H).
Анал. рассчитано для C13H14F2O3 (мол. масса 256,24): C 60,93, H 5,51.
Найдено: C 60,68, H 5,50.
d) Получение 2-(4,6-дифтор-1-гидрокси-1-инданил)уксусной кислоты
Смесь этил-2-(4,6-дифтор-1-гидрокси-1-инданил)ацетата (12,0 г, 0,147 моля) и 1,0 н. гидроксида натрия (48 мл, 0,048 моля, Universal Scientific Supply Co. ) в этаноле (75 мл) перемешивали в течение 18 часов при температуре окружающей среды. Реакционную смесь концентрировали в вакууме, разбавляли водой и промывали диэтиловым эфиром. Водную фазу нейтрализовали 1,0 н. соляной кислотой (48 мл, 0,048 моля, Universal Scientific Supply Co.) и экстрагировали диэтиловым эфиром. Экстракт диэтилового эфира сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая количественный выход неочищенной 2-(4,6-дифтор-1-гидрокси-1-инданил)уксусной кислоты. Этот материал использовали сразу без дальнейшей очистки.
e) Получение (E)-2-(4,6-дифтор-1-инданилиден)уксусной кислоты
Трифторуксусную кислоту (39,9 г, 0,35 моля) добавляли по каплям к перемешиваемой, охлажденной (баня метанола со льдом) смеси 2-(4,6-дифтор-1-гидрокси-1-инданил)уксусной кислоты (11,3 г, 0,05 моля) в дихлорметане (250 мл). После 35 минут смесь концентрировали в вакууме. К остатку добавляли дихлорметан и смесь концентрировали в вакууме. Эту процедуру повторяли еще раз, получая 6,4 г неочищенной (E)-2-(4,6-дифтор-1-инданилиден)уксусной кислоты. Перекристаллизация 0,9 г из смеси ацетон:вода дала 0,15 г (E)-2-(4,6-дифтор-1-инданилиден)уксусной кислоты в виде белого твердого вещества, т. пл. 238-239oC, ЯМР (DMSO-d6): d 12,25 (шир., 1H), 7,23-7,65 (м, 2H), 6,46 (т, 1H), 3,20-3,28, 2,97-3,20 (2м, 4H), ЯЭО (ядерный эффект Оверхаузера) в стационарном состоянии: освещ. при 6,46d наблюдаемый ЯЭО 21,6% при 7,63d.
Анал. рассчитано для C11H8F2O2 (мол. масса 210,17): C 62,86, H 3,84.
Найдено: С 62,76, H 3,86.
f) Получение (E)-2-(4,6-дифтор-1-инданилиден)ацетилхлорида
Суспензию (E)-2-(4,6-дифтор-1-инданилиден)уксусной кислоты (5,49 г, 0,026 моля) в смеси дихлорметан:диметилформамид (50 мл: 5 капель) обрабатывали оксалилхлоридом (6,6 г, 0,052 моля, Aldrich) и перемешивали при температуре окружающей среды в течение 18 часов. Полученный раствор концентрировали в вакууме и остаток использовали без дальнейшей очистки.
g) Получение (E)-2-(4,6-дифтор-1-инданилиден)ацетамида
30% водный раствор гидроксида аммония (1,7 мл, 0,026 моля) добавляли по каплям к перемешиваемому, охлажденному (ледяная баня) раствору (E)-2-(4,6-дифтор-1-инданилиден)ацетилхлорида (2,97 г, 0,013 моля) в дихлорметане (50 мл). После 4,5 ч. смесь концентрировали в вакууме и остаток распределяли между 5% раствором в воде бикарбоната натрия и этилацетатом. Этилацетат промывали насыщенным водным хлоридом натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле с применением смеси этилацетат: гексаны (7:3) в качестве элюента и растирание полученного твердого вещества с пентаном дали 1,63 г (60%) (E)-2-(4,6-дифтор-1-инданилиден)ацетамида в виде белого твердого вещества, т.пл. 178-180oC, ЯМР (DMSO-d6): d 6,04-7,45 (м, 4H), 6,46 (с, 1H), 2,94-3,00, 3,21-3,27 (2м, 4H): ЯЭО стационарного состояния: освещ. при 6,46 d, наблюдаемый ЯЭО 19% при 7,26d.
Анал. рассчитано для C11H9F2NO (мол. масса 209,19): C 63,15, H 4,34, N 6,70.
Найдено: C 63,07, H 4,36, N 6,67.
h) Получение (E)-2-(4-хлор-1-инданилиден)уксусной кислоты
Трифторуксусную кислоту (25,1 мл) добавляли к перемешиваемому, охлажденному (баня с метанолом и льдом) раствору 2-(4-хлор-1-гидрокси-1-инданил)уксусной кислоты (10,4 г, 0,05 моля) в дихлорметане (230 мл). После 30 минут смесь концентрировали в вакууме. К остатку добавляли дихлорметан и смесь концентрировали в вакууме, получая 6,5 г (68%) белого твердого вещества. Перекристаллизация 0,98 г из смесей ацетонитрил: 2-пропанол дала 0,62 г белого твердого вещества, т.пл. 233-234oC, ЯМР (DMSO-d6): d 12,15 (шир., 1H, COOH), 7,30-6,81 (м, 3H, A), 6,41 (с, 1H, =CH), 3,00-3,06, 3,19-3,22 (2м, 4H, 2хCH2), ЯЭО стационарного состояния: освещ. при 6,41d, наблюдаемый ЯЭО 19,7% при 7,79d.
i) Получение (E)-2-(4-хлор-1-инданилиден)ацетилхлорида
Суспензию (E)-2-(4-хлор-1-инданилиден)уксусной кислоты (5,5 г, 0,03 моля) в смеси диметилформамид: дихлорметан (5 капель: 50 мл) обрабатывали оксалилхлоридом (6,6 г, 0,05 моля) и перемешивали при комнатной температуре в течение 16 часов. Полученный раствор концентрировали в вакууме и остаток использовали без дальнейшей очистки.
j) Получение (E)-2-(4-хлор-1-инданилиден)-N-циклопропилацетамида
Охлажденный на льду раствор (E)-2-(4-хлор-1-инданилиден)ацетилхлорида (2,95 г, 0,013 моля) в дихлорметане (30 мл) обрабатывали циклопропиламином (1,48 г, 0,026 моля, Aldrich) и смесь перемешивали в течение 4 часов. Смесь концентрировали в вакууме и остаток собирали смесью этилацетата и 5% водного бикарбоната натрия. Фазу этилацетата промывали 5% водным бикарбонатом натрия, насыщенным водным NaCl и сушили над сульфатом натрия. Фильтрование и концентрирование дали 3,5 г неочищенного масляного продукта. Хроматография на силикагеле с применением смеси этилацетат:гексан (1:1) в качестве элюента и растирание полученного твердого вещества с гексаном дали 2.46 г (76%) (E)-2-(4-хлор-1-инданилиден)-N-циклопропилацетамида в виде твердого вещества нестандартного белого цвета: т.пл. 140-142oC, ЯМР (DMSO-d6): d 8,16 (д, 1H, NH), 7,29-7,51 (м, 3H, Ar), 6,34 (т, 1H, = CH), 2,93-3,07, 3,18-3,31 (2м, 4H, 2хCH2), 2,72 (м, 1H, CH), 0,62-0,72, 0,39-0,47 (2м, 2хCH2), ЯЭО стационарного состояния: освещ. при 6,34d, наблюдаемый 20,3% ЯЭО при 7,46d.
Анал. рассчитано для C14H14ClNO (мол. масса 247,72): C 67,88, H 5,70, N 5,65.
Найдено: C 67,86, H 5,74, N 5,58.
Пример 6
Получение (E)-N-циклопропил-2-(4,6-дифтор-1-инданилиден)- ацетамида
Охлажденный на льду раствор (E)-2-(4,6-дифтор-1-инданилиден)- ацетилхлорида (2,97 г, 0,013 моля) в дихлорметане (30 мл) обрабатывали циклопропиламином (1,48 г, 0,026 моля, Aldrich) и смесь перемешивали в течение 4 часов. Смесь концентрировали в вакууме и остаток собирали смесью этилацетата и 5% водного бикарбоната натрия. Этилацетатную фазу промывали 5% водным бикарбонатом натрия, насыщенным водным NaCl и сушили над сульфатом натрия. Хроматография на силикагеле с применением смеси этилацетат:гексан (1:1) в качестве элюента и растирание полученного твердого вещества с пентаном дали 1,92 г (59%) (E)-N-циклопропил-2-(4,6-дифтор-1-инданилиден)ацетамида в виде белого твердого вещества: т.пл. 156-158oC.
Пример 7
Получение (E)-2-(4-фтор-1-инданилиден)ацетамида
а) Получение этил-2-фторциннамата
Раствор 2-фторкоричной кислоты (48,4 г, 0,29 моля, Aldrich) и тионилхлорида (5 мл) в этаноле (650 мл) нагревали с обратным холодильником в течение 48 часов. Смесь концентрировали в вакууме. Остаток поглощали этилацетатом, промывали последовательно 5% водным раствором бикарбоната натрия, водой и солевым раствором и сушили над сульфатом натрия. Фильтрование и концентрирование дали 54,25 г (96%) неочищенного этил-2-фторциннамата. Этот материал использовали без дальнейшей очистки.
b) Получение этил-3-(2-фторфенил)пропионата
Смесь этил-2-фторциннамата (29,25 г, 0,176 моля) и гидрата оксида палладия (0,25 г, EM Scientific) в 95% этаноле (150 мл) помещали в аппарат Парра для гидрогенизации и качали под давлением 2-4 атм водорода. После потребления достаточного количества водорода катализатор удаляли фильтрованием и фильтрат концентрировали в вакууме, получая 29,39 г (99%) неочищенного этил-3-(2-фторфенил)пропионата. Этот материал использовали без дальнейшей очистки.
c) Получение 3-(2-фторфенил)пропионовой кислоты
Смесь этил-3-(2-фторфенилпропионата (25,54 г, 0,130 моля) и 50% водного раствора гидроксида натрия (30 мл) в воде (130 мл) нагревали с обратным холодильником в течение 2 часов. После охлаждения смесь промывали диэтиловым эфиром (2х100 мл). Водную фазу охлаждали в бане со льдом и pH устанавливали равной 3 с помощью соляной кислоты. Образовавшийся белый осадок собирали фильтрованием, промывали несколько раз водой и сушили в вакууме при 60oC в течение 18 часов, получая 18,66 г (85%) 3-(2-фторфенил)пропионовой кислоты в виде белого твердого вещества, т.пл. 72-74oC. Этот материал использовали без дальнейшей очистки.
d) Получение 4-фтор-1-инданона
К смеси 3-(2-фторфенил)пропионовой кислоты (18,64 г, 0,111 моля) и диметилформамида (5 капель) при комнатной температуре добавляли по каплям оксалилхлорид (60 мл). Смесь перемешивали при комнатной температуре до прекращения выделения газа. Избыток оксалилхлорида удаляли перегонкой, получая 3-(2-фторфенил)пропионилхлорид. Раствор 3-(2-фторфенил)пропионилхлорида в дихлорметане (230 мл) добавляли по каплям к смеси хлорида алюминия (16,25 г, 0,12 моля) в дихлорметане (230 мл) и эту смесь дефлегмировали в течение 3,5 часов. Реакционную смесь выливали в ледяную воду (1200 мл) и разделяли две фазы. Дихлорметановую фазу промывали последовательно 0,1 н. водным гидроксидом натрия (2х100 мл), водой (200 мл) и солевым раствором (200 мл), сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаточное масло хроматографировали на силикагеле, элюируя смесью гексан:дихлорметан (9:1) и получая 11,0 г неочищенного 4-фтор-1-инданона в виде желтого твердого вещества. Перекристаллизация из смесей ацетон:вода дала 8,02 г (48%) 4-фтор-1-инданона в виде бледно-желтого твердого вещества: т.пл. 71-72oC, ЯМР (DMSO-d6): d 7,51 (м, 3H, Ar), 3,13 (т, 2H, CH2), 2,74 (т, 2H, CH2).
Анал. рассчитано для C9H7FO (мол. масса 150, 152): C 71,99, H 4,70.
Найдено: C 71,86, H 4,79.
e) Получение этил-2-(4-фтор-1-гидрокси-1-инданил)ацетата
Это соединение получали способом, аналогичным описанному в примере 5с для этил-2-(4,6-дифтор-1-гидрокси-1-инданил)ацетата, заменяя 4-фтор-1-инданоном (15,53 г, 0,103 моля) 4,6-дифтор-1- инданон. Хроматография на силикагеле со смесью этилацетат:гексан (19:1) в качестве элюента дала 19,11 г (78%) этил-2-(4-фтор-1- гидрокси-1-инданил)ацетата, который использовали без дальнейшей очистки.
f) Получение 2-(4-фтор-1-гидрокси-1-инданил)уксусной кислоты
Это соединение получали способом, сходным с описанным для 2-(4,6-дифтор-1-гидрокси-1-инданил)уксусной кислоты в примере 5d, заменяя этил-2-(4-фтор-1-гидрокси-1-инданил) ацетатом (17,35 г, 0,0728 моля) этил-2-(4,6-дифтор-1-гидрокси-1- инданил)ацетат и получая количественный выход неочищенной 2-(4-фтор-1-гидрокси-1-инданил)уксусной кислоты. Этот материал использовали непосредственно без дальнейшей очистки.
g) Получение (E)-2-(4-фтор-1-инданилиден)уксусной кислоты
Это соединение получали способом, аналогичным описанному для (E)-2-(4,6-дифтор-1-инданилиден)уксусной кислоты в примере 5e, заменяя 2-(4-фтор-1-гидрокси-1-инданил)уксусной кислотой (14,6 г, 0,069 моля) 2-(4,6-фтор-1-гидрокси-1-инданил)уксусную кислоту и получая неочищенную (E)-2-(4-фтор-1-инданилиден)уксусную кислоту. Перекристаллизация из смесей ацетонитрил: 2-пропанол дала 6,85 г (52%) (E)-2-(4-фтор-1-инданилиден)уксусной кислоты в виде белого твердого вещества, т.пл. 249-251oC.
h) Получение (E)-2-(4-фтор-1-инданилиден)ацетилхлорида
Это соединение получали способом, сходным с описанным для (E)-2-(4,6-дифтор-1-инданилиден)ацетилхлорида в примере 5f, заменяя (E)-2-(4-фтор-1-инданилиден)уксусной кислотой (5,77 г, 0,03 моля) (E)-2-(4,6-дифтор-1-инданилиден)уксусную кислоту. Полученный раствор концентрировали в вакууме и остаток использовали без дальнейшей очистки.
i) Получение (E)-2-(4-фтор-1-инданилиден)ацетамида
Охлажденный на льду раствор (E)-2-(4-фтор-1-инданилен) ацетилхлорида (2,11 г, 0,01 моля) в дихлорметане (65 мл) обрабатывали 30% водным раствором гидроксида аммония (2,63 мл, 0,02 моля) и смесь перемешивали в течение 18 часов. Гексан добавляли к смеси и твердый материал собирали фильтрованием, получая 1,63 г неочищенного продукта. Перекристаллизация из смесей ацетонитрил: вода дала 1,11 г (58%) (E)-2-(4-фтор-1-инданилиден)ацетамида в виде белого твердого вещества, т.пл. 198-200oC.
Пример 8
Получение (E)-2-N-циклопропил-(4-фтор-1-инданилиден)ацетамида
Охлажденный на льду раствор (E)-2-(4-фтор-1-инданилиден) ацетилхлорида (2,11 г, 0,010 моля) в дихлорметане (65 мл) обрабатывали циклопропиламином (1,39 мл, 0,02 моля) и смесь перемешивали в течение 18 часов. Гексан добавляли к смеси и твердый материал собирали фильтрованием и промывали последовательно водой и гексаном, получая 1,22 г неочищенного продукта. Перекристаллизация из смесей ацетонитрил:вода дала 0,83 г (36%) (E)-2-N-циклопропил-(4-фтор-1-инданилиден)ацетамида в виде белого твердого вещества, т.пл. 121-122oC.
Пример 9
Получение (E)-2-(5-фтор-1-инданилиден)ацетамида
а) Получение этил-2-(5-фтор-1-гидрокси-1-инданил)ацетата
Это соединение получали способом, аналогичным описанному в примере 5с для этил-2-(4,6-дифтор-1-гидрокси-1-инданил)ацетата, заменяя 5-фтор-1-инданоном (14,77 г, 0,098 моля, Fairfield) 4,6-дифтор-1-инданон. Хроматография на силикагеле со смесью этилацетат:гексаны (9:1) в качестве элюента дала 19,56 г (83%) аналитически чистого этил-2-(5-фтор-1-гидрокси-1-инданил)ацетата в виде бледно-желтого масла, ЯМР (CDCl3): d 6,88-7,30 (м, 3H, Ar), 5,30 (с, 1H, OH), 4,20 (к, 2H, CH2CH3), 2,66-3,08 (м, 4H, 2CH2), 2,30 (т, 2H, CH2Ar), 1,28 (т, 3H, CH3).
Анал. рассчитано для C13H15FO3 (мол. масса 238,25): C 65,54, H 6,35.
Найдено: C 65,39, H 6,38.
b) Получение 2-(5-фтор-1-гидрокси-1-инданил)уксусной кислоты
Это соединение получали способом, аналогичным описанному для 2-(4,6-дифтор-1-гидрокси-1-инданил)уксусной кислоты в примере 5d, заменяя этил-2-(5-фтор-1-гидрокси-1-инданил)ацетатом (19,55 г, 0,082 моля) этил-2-(4,6-дифтор-1-гидрокси-1-инданил)ацетат и получая 14,70 г (84%) неочищенной 2-(5-фтор-1-гидрокси-1-инданил)уксусной кислоты в виде белого твердого вещества. Этот материал использовали сразу без дальнейшей очистки.
с) Получение (E)-2-(5-фтор-1-инданилиден)уксусной кислоты
Это соединение получали способом, аналогичным описанному для (E)-2-(4,6-дифтор-1-инданилиден)уксусной кислоты в примере 5е, заменяя 2-(5-фтор-1-гидрокси-1-инданил)уксусной кислотой (14,70 г, 0,069 моля) 2-(4,6-дифтор-1-гидрокси-1-инданил)уксусную кислоту. Перекристаллизация из смесей ацетонитрил: 2-пропанол дала 9,05 г (68%) (E)-2-(5-фтор-1-инданилиден)уксусной кислоты в виде белого твердого вещества, т.пл. 240-242oC.
d) Получение (E)-2-(5-фтор-1-инданилиден)ацетилхлорида
Это соединение получали способом, аналогичным описанному для (E)-2-(4,6-дифтор-1-инданилиден)ацетилхлорида в примере 5f, заменяя (E)-2-(5-фтор-1-инданилиден)уксусной кислотой (5,77 г, 0,03 моля) (E)-2-(4,6-дифтор-1-инданилиден)уксусную кислоту. Полученный раствор концентрировали в вакууме и остаток использовали без дальнейшей очистки.
e) Получение (E)-2-(5-фтор-1-инданилиден)ацетамида
Это соединение получали способом, аналогичным описанному для (E)-2-(6-фтор-1-инданилиден)ацетамида в примере 1k, заменяя (E)-2-(5-фтор-1-инданилиден)ацетилхлоридом (3,16 г, 0,015 моля) (E)-2-(6-фтор-1-инданилиден)ацетилхлорид. Перекристаллизация из смесей ацетонитрил:вода дала 1,28 г (44%) (E)-2-(5-фтор-1-инданилиден)ацетамида в виде белого твердого вещества, т.пл. 191-193oC.
Пример 10
Получение (E)-N-циклопропил-2-(5-фтор-1-инданилиден)ацетамида
Раствор (E)-2-(5-фтор-1-инданилиден)уксусной кислоты (0,97 г, 0,005 моля), 1-гидроксибензотриазола (0,68 г, 0,005 моля, Fluka), циклопропиламина (0,35 мл, 0,005 моля, Aldrich) и 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорида (0,96 г, 0,005 моля, Sigma), который добавляли последним, в диметилформамиде (15 мл) перемешивали при комнатной температуре в течение 18 часов и раствор концентрировали в вакууме. Остаток растворяли в этилацетате, промывали последовательно 5% водным раствором лимонной кислоты (3х50 мл), насыщенным водным раствором бикарбоната натрия (2х50 мл) и солевым раствором и сушили над сульфатом натрия. Раствор концентрировали в вакууме, получая неочищенный (E)-N-циклопропил-2-(5-фтор-1-инданилиден)ацетамид. Хроматография на силикагеле с элюцией смесью гексан:этилацетат (1:1) дала 0,52 г (44%) (E)-N-циклопропил-2-(5-фтор-1-инданилиден)ацетамида в виде белого твердого вещества, т.пл.137-138oC.
Пример 11
Альтернативное получение (E)-2-(6-фтор-1-инданилиден)ацетамида
К охлажденной льдом перемешиваемой суспензии NaH (60% дисперсии в минеральном масле, 12,41 г, 60,25 ммоля, Aldrich) в тетрагидрофуране (30 мл) с 15-краун-5 (3,96 г, 17,98 ммоля, Aldrich) добавляли под азотом диэтилкарбамоилметилфосфонат (11,7 г, 59,97 ммоля, K&K-IC) и 6-фтор-1-инданон (9,0 г, 59,96 ммоля), соответственно, в тетрагидрофуране (80 мл). Эту смесь выливали в 200 мл ледяной воды и экстрагировали 3 раза 600 мл диэтилового эфира. Одну органическую фазу промывали последовательно порциями 200 мл водного бисульфита натрия (10%) и насыщенным раствором хлорида натрия. Органическую фазу сушили над карбонатом калия, фильтровали, концентрировали на роторном испарителе в вакууме и повторно подвергали роторному испарению с 200 мл дихлорметана, получая липкий твердый остаток. Этот остаток хроматографировали на Силикагеле 60 со смесью этилацетат:гексан (2:1) в качестве элюента. Фракции, содержащие (E)-2-(6-фтор-1-инданилиден)ацетамид, объединяли и концентрировали роторным испарением в вакууме, получая 2,38 г желтого твердого вещества. Разведение раствора в дихлорметане неочищенного материала гексаном дало 2,16 г (18,8%) (E)-2-(6-фтор-1-инданилиден)ацетамида, т.пл. 178-182oC, ЯМР (DMSO-d6); d 7,4-7,1 (м, 4H, Ar и NH), 6,88 (шир. с, 1H, H), 6,37 (т, 1H, J = 2,54 Гц, =CH), 3,22-3,14 (м, 2H, CH2), 2,95-2,89 (м, 2H, CH2). ЯЭО стационарного состояния: освещ. при d 6,37, значимый наблюдаемый ЯЭО при d 7,33-7,28.
Анал. рассчитано для C11H10FNO: C 69,10, H 5,27, N 7,33.
Найдено: C 69,01, H 5,29, N 7,28.
Пример 12
Получение (E)-N-циклопропил-2-(5-фтор-1,2,3,4-тетрагидро-1- нафтилиден)ацетамида
а) Получение 2-(2-фторфенил)этилбромида
К смеси водной бромистоводородной кислоты (48%, 360 мл) и концентрированной серной кислоты (103,6 мл) при комнатной температуре добавляли по каплям 2-(2-фторфенил)этанол (250 мл, 1,78 моля, Aldrich). Реакционную смесь кипятили с обратным холодильником в течение 7 часов, выливали на 600 мл льда и смесь экстрагировали диэтиловым эфиром. Экстракты в диэтиловом эфире промывали последовательно насыщенным раствором карбоната натрия и солевым раствором. Органическую фазу сушили (MgSO4) и концентрировали в вакууме, получая 359,9 г (99%) 2-(2-фторфенил)этилбромида в виде коричневого масла. Этот продукт использовали без дальнейшей очистки. ЯМР (CDCl3): d 7,6-6,9 (м, 4H, ArH), 3,6 (т, 2H, CH2), 3,2 (т, 2H, CH2).
b) Получение диэтил-2-(2-(2-фторфенил)этилмалоната
К абсолютному этанолу (1,5 л) при комнатной температуре в атмосфере азота добавляли металлический натрий (61,1 г, 2,66 моля) в виде малых кусочков в течение нескольких часов. После перемешивания в течение 24 часов при комнатной температуре смесь нагревали до 40oC и добавляли по каплям диметилмалонат (525,4 г, 3,28 моля) с последующим добавлением 2-(2-фторфенил)этилбромида (359,9 г, 1,77 моля). Смесь дефлегмировали в течение 8 часов. Неочищенный материал очищали вакуумной перегонкой при 85-130oC и 0,60 мм Hg, получая 312,6 г (62%) диэтил-2-(2-(2-фторфенил)этил) малоната в виде прозрачного масла. ЯМР (CDCl3): d 7,6-7,0 (м, 4H, ArH), 4,2 (к, 4H, 2хCH2), 3,4 (т, 1H, CH), 2,6 (к 2H, CH2), 2,2 (т, 2H, CH2), 1,2 (т, 6H, 2хCH3).
c) Получение 2-[2-(2-фторфенил)этил]малоновой кислоты
Смесь диэтил-2-(2-(2-фторфенил) этил) малоната (381,8 г, 1,35 моля) и гидроксида калия (227,3 г, 4,1 моля) в этаноле (500 мл) и воде (500 мл) нагревали с обратным холодильником в течение 24 часов. Реакционную смесь помещали на ледяную баню и добавляли соляную кислоту (6N, 442 мл). Этанол удаляли в вакууме и водный остаток экстрагировали диэтиловым эфиром. Экстракты высушивали (MgSO4) и концентрировали в вакууме, получая 309,3 г (100 %) 2-[2-(2-фторфенил)этил]малоновой кислоты в виде твердого вещества нестандартного белого цвета. Этот продукт использовали без дальнейшей очистки. ЯМР (DMSO-d6): d 6,8-6,2 (м, 4H, ArH), 2,4 (т, 1H, CH), 1,9 (к, 2H, CH2), 1,3 (т, 2H, CH2), ИК (KBr) 1691 см-1.
d) Получение 4-(2-фторфенил) масляной кислоты
2-[2-(2-фторфенил)этил] малоновую кислоту (147 г, 0,65 моля) нагревали в масляной бане при 170oC в течение 2,5 часов. При охлаждении 4-(2-фторфенил) масляная кислота (117,3 г, 99%) кристаллизовалась в виде рыжевато-коричневого твердого вещества. Этот продукт использовали без дальнейшей очистки. ЯМР (CDCl3): d 7,6-7,0 (м, 4H, ArH), 3,0-1,8 (м, 6H, 3хCH2). ИК (чистый) 1709 см-1.
е) Получение 5-фтортетралона
Смесь 4-(2-фторфенил)масляной кислоты (100 г, 0,55 моля) и тионилхлорида (418,8 г, 3,51 моля) нагревали с обратным холодильником в течение 3 часов. Избыток тионилхлорида удаляли в вакууме, получая 110,1 г (100%) 4-(2-фторфенил)бутирилхлорида.
К 4-(2-фторфенил)бутирилхлориду в сероуглероде (1,0 л) при -78oC добавляли хлорид алюминия (93,2 г, 0,7 моля) порциями в течение периода 30 минут. Смесь нагревали до комнатной температуры в течение 30 минут, затем дефлегмировали в течение 2 часов. Реакционную смесь выливали в смесь льда (500 мл) и HCl (6 н. , 500 мл). Сероуглеродный слой отделяли, промывали насыщенным раствором бикарбоната натрия и экстрагировали этилацетатом. Водную фазу экстрагировали этилацетатом. Объединенные экстракты сушили (MgSO4) и концентрировали в вакууме, получая 84,2 г (93%) 5-фтортетралона в виде твердого вещества рыжевато-коричневого цвета. ЯМР (CDCl3): d 7,7 (м, 1H, ArH), 7,1 (м, 2H, ArH), 2,9 (т, 2H, CH2), 2,6 (т, 2H, CH2), 2,1 (к, 2H, CH2).
f) Получение (E)-N-циклопропил-2-(5-фтор-1,2,3,4- тетрагидро-1-нафталиден)ацетамида
К перемешиваемой суспензии NaH (60% дисперсии в минеральном масле, Aldrich) в диметилсульфоксиде при комнатной температуре под азотом добавляли диэтил(циклопропилкарбамоилметил)фосфонат (21,5 г, 0,09 моля). Реакция была слегка экзотермической. К полученному раствору добавляли 5-фтортетралон (13,7 г, 0,08 моля) в диметилсульфоксиде. Смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь выливали в охлажденную льдом воду (800 мл) и экстрагировали 4 раза порциями по 500 мл водой, фильтровали и выпаривали в роторном испарителе в вакууме.
Хроматография на силикагеле с применением смеси этилацетат:гексан (35% : 50%) в качестве элюента с последующим растиранием полученного твердого вещества с пентаном при комнатной температуре дала 5,07 г (25%) (E)-N-циклопропил-2-(5-фтор- 1,2,3,4-тетрагидро-1-нафтилиден)ацетамида, т.пл. 148-149oC.
Пример 13
Получение (E)-N-циклопропил-2-(7-фтор-1,2,3,4-тетрагидро-1- нафтилиден)ацетамида
а) Получение 3-(4-фторбензоил)пропионовой кислоты
Смесь фторбензола (104,4 г, 1,09 моля, Aldrich) и янтарного ангидрида (93,5 г, 0,93 моля) в 1,2-дихлорбензоле (530 мл) нагревали до 50oC. Добавляли порциями хлорид алюминия (245 г, 1,84 моля), поддерживая температуру ниже 60oC. После 4 часов при 60oC с последующими 5 часами при 80oC реакционную смесь выливали в смесь концентрированной HCl (200 мл) и ледяной воды (2 л). Органический слой отделяли и водную фазу экстрагировали дихлорметаном. Объединенные органические фазы сушили и концентрировали в вакууме. Остаток выливали в гексан (2 л) и полученное твердое вещество отфильтровывали и промывали пентаном, получая 164,1 г (89%) 3-(4-фторбензоил)пропионовой кислоты в виде белого твердого вещества. Т. пл. 102-104,5oC (в литературе, J. Org. Chem. 26, 2667, 1961, т.пл. 102,5-103,5oC).
b) Получение 4-(4-фторфенил)масляной кислоты
Смесь 3-(4-фторбензоил)пропановой кислоты (42,3 г, 0,22 моля) и палладия на угле (10%, 3 г) в уксусной кислоте (250 мл) гидрировали при 50 psi (344737,85 пА) и 25oC в течение 6 часов. Смесь фильтровали и концентрировали в вакууме. Остаток перегоняли при 0,02 мм Hg и продукт кристаллизовали, получая 4-(4-фторфенил)масляную кислоту в виде белого твердого вещества (97%), т.пл. 44-46,2oC (в литературе, J. Am. Chem. Soc. 89, 386, 1967, т.пл. 45,5-46,5oC).
с) Получение 7-фтор-1-тетралона
Смесь 4-(4-фторфенил)масляной кислоты (68,2 г, 0,37 моля) и тионилхлорида (155 г, 1,3 моля) нагревали с обратным холодильником в течение 1,25 часа. Смесь концентрировали в вакууме, получая 75,3 г (100%) 4-(4-фторфенил)бутирилхлорида.
К смеси хлорида алюминия (66 г, 0,5% моля) в сероуглероде (600 мл) добавляли по каплям раствор 4-(4-фторфенил)бутирилхлорида (75,3 г, 0,37 моля) в сероуглероде (260 мл), поддерживая внутреннюю температуру ниже 10oC. После нагревания с обратным холодильником в течение 0,5 часа реакционную смесь выливали в смесь концентрированной HCl (50 мл) и ледяной воды (800 мл). Смесь фильтровали и экстрагировали диэтиловым эфиром. Экстракты в диэтиловом эфире сушили и концентрировали в вакууме, получая неочищенный 7-фтор-1-тетралон. Вакуумная перегонка дала чистый 7-фтор-1-тетралон, т. кип. 83oC при 0,3 мм Hg, который отверждался до белого твердого вещества (94%), т.пл. 62-64oC (в литературе, J. Am. Chem. Soc., 89, 386, 1967, т.пл. 63,5-65,0oC.
d) Получение (E)-N-циклопропил-2-(7-фтор-1,2,3,4- тетрагидро-1-нафтилиден)ацетамида
Это соединение получали способом, аналогичным описанному в примере 12f с заменой 5-фтор-1-тетралона и диэтил(циклопропилкарбамоил)метилфосфоната 7-фтор-1-тетралоном (7,76 г, 0,05 моля) и диэтил(циклопропилкарбамоил)метилфосфонатом (11,1 г, 0,05 моля). Хроматография на силикагеле с применением смеси этилацетат:гексан (1:2) в качестве элюента дала 4,38 г (37%) (E)-N-циклопропил-2-(7-фтор-1,2,3,4-тетрагидро-1-нафтилиден)ацетамида, т.пл. 122,8-123,3oC, ЯМР (DMSO-d6): d 8,00 (д, J = 4,0 Гц, 1H), 7,32 (дд, J = 11,2 Гц, 1H), 7,04-7,23 (м, 2H), 6,33 (с, 1H), 3,06 (м, 2H), 2,69 (м, 3H), 1,70 (м, 2H), 0,66 (м, 2H), 0,40 (м, 2H), ЯЭО стационарного состояния: освещ. при 6,39d, наблюдаемый значимый ЯЭО при 7,32d.
Анал. рассчитано для C15H16FNO (мол. масса 245,30): C 73,45, H 6,57, N 5,71.
Найдено: C 73,38, H 6,64, N 5,67.
Пример 14
Получение (E)-2-(7-фтор-1,2,3,4-тетрагидро-1-нафтилиден) -N, N-диметилацетамида
а) Получение этил-2-(7-фтор-1,2,3,4-тетрагидро-1-гидрокси-1- нафтил)ацетата
Этилацетат (5,4 г, 61 ммоль) добавляли по каплям к перемешиваемому охлажденному (баня с ацетоном - сухим льдом) раствору 1 М бис(триметилсилил)амида лития в тетрагидрофуране (61 мл, 0,061 моля, Aldrich) под азотом. После 15 минут добавляли по каплям раствор 7-фтор-1-тетралона (10,0 г, 61 ммоль) в тетрагидрофуране (25 мл) и полученную смесь перемешивали в течение 1 часа (на бане с сухим льдом и ацетоном). Добавляли 1 н. раствор соляной кислоты (61 мл) и смеси давали нагреться до комнатной температуры. Органическую фазу отделяли, сушили над безводным сульфатом натрия, фильтровали и концентрировали до бледно-желтого масла (15,0 г, 100%). Аналитическую пробу получали при помощи хроматографии порции 1,5 г на силикагеле 60 с применением смеси дихлорметан-гексан (1:1) в качестве элюента. Фракции, содержащие только этил-2-(7-фтор-1,2,3,4-тетрагидро-1-гидрокси-1-нафтил)ацетат, объединяли и концентрировали в вакууме, получая 1,2 г (80%) бесцветного масла, ЯМР (DMSO-d6): d 6,93-7,31 (м, 3H, Ar), 5,28 (с, 1H, OH), 3,98 (м, 2H, CH2OOC), 2,60-2,87 (м, 4H, CH2CO, CH2), 2,12-2,28 (м, 1H, CH), 1,78-1,86 (м, 3H, CH, CH2), 1,09 (т, 3H, CH3).
b) Получение этил-2-(7-фтор-1,2,3,4-тетрагидро-1-нафтил)ацетата
Трифторуксусную кислоту (20 мл) добавляли к перемешиваемому охлажденному (баня с сухим льдом и метанолом) раствору неочищенного этил-2-(7-фтор-1,2,3,4-тетрагидро-1-гидрокси-1-нафтил)ацетата (10,0 г, 35,8 моля) в дихлорметане (180 мл). После 4 часов смесь концентрировали в вакууме до прозрачного масла (8,3 г, 100%). ЯМР (DMSO-d6): d 6,94-7,65 (м, 3H, Ar), 6,45 (шир. с, 0,2H, =CH/E), 6,10 (т, 0,8H, =CH/эндо), 4,08 (м, 2H, CH2OOC), 3,67, 3,51 (сс, 2, 2H, H2O, CH2/эндо), 3,08, 2,70, 2,25, 1,77 (м, 4,4H, 5хCH2), 1,26 (т, 0,6H, CH3/E), 1,17 (т, 2,4H, CH3/эндо).
Часть полученной выше смеси E и эндо-эфиров (2,3 г, 10 ммолей), гидрат гипофосфита натрия (1,8 г, 20 ммолей, Aldrich) и 10% палладий на угле (0,2 г) в 75% водном этаноле (20 мл) нагревали с обратным холодильником в течение 2 часов. Смесь фильтровали через целит и фильтрат концентрировали в вакууме. Остаток в дихлорметане промывали последовательно водой (100 мл) и солевым раствором (50 мл), сушили над безводным сульфатом натрия, концентрировали в вакууме и хроматографировали на силикагеле 60 с применением смеси этилацетат-гексан (3: 97) в качестве элюента. Фракции, содержащие только этил-2-(7-фтор-1,2,3,4-тетрагидро-1-нафтил)ацетат, объединяли и концентрировали в вакууме, получая 1,9 г (78%) бледно-желтого масла, ЯМР (DMSO-d6): d 6,89-7,14 (м, 3H, Ar), 4,11 (к, 2H, CH2OOC), 3,15-3,27 (м, 1H, CH), 2,44-2,82 (м, 4H, 2хCH2), 1,52-1,90 (м, 4H, 2xCH2), 1,20 (т, 3H, CH3).
c) Получение этил-2-бром-2-(7-фтор-1,2,3,4- тетрагидро-1-нафтил)ацетата
К перемешиваемому охлажденному (баня с сухим льдом и ацетоном) раствору диизопропиламина (0,3 мл, 1,9 ммоля, Aldrich) в тетрагидрофуране (3 мл) в азоте последовательно добавляли 2,5 н-бутиллитий в гексане (0,8 мл, Aldrich) хлортриметилсилан (0,2 мл, 1,8 ммоля, Aldrich) и этил-2-(7-фтор-1,2,3,4-тетрагидро-1-нафтил)ацетат (236 мг, 1,0 ммоль). Полученный прозрачный раствор перемешивали в течение 1 часа, обрабатывали N-бром-сукциниимидом (180 мг, 1,0 ммоль, Aldrich) и перемешивали еще в течение 0,5 часа перед удалением бани с сухим льдом и ацетоном. Красноватый мутный раствор перемешивали в течение 2 часов при комнатной температуре, обрабатывали разбавленной водной соляной кислотой (4 мэкв) и экстрагировали диэтиловым эфиром (30 мл). Эфирный слой сушили над безводным сульфатом натрия, фильтровали, концентрировали в вакууме и хроматографировали на силикагеле 60 с применением смеси дихлорметан-гексан (1:9) в качестве элюента. Фракции, содержащие только этил-2-бром-2-(7-фтор- 1,2,3,4-тетрагидро-1-нафтил)ацетат в виде 1:4 изомерной смеси объединяли и концентрировали в вакууме до прозрачного масла (171 мг, 54%), ЯМР (DMSO-d6): d 7,00-7,18 (м, 3H, Ar), 5,20 (д, J = 6,2 Гц, 0,8H, В CHCO), 5,17 (д, J = 6,2 Гц, 0,2H, В CHCO), 4,19 (с, 1,6H, CH2OOC), 4,14 (к, 0,4H, CH2OOC), 3,49 (м, 1H, ArCH), 2,69 (м, 2H, ArCH2), 1,81-1,97 (м, 3H, CH, CH2), 1,61-1,67 (м, 1H, CH), 1,21 (т, 0,4H, CH3), 1,07 (т, 0,6H, CH3).
g) Получение (E)-2-(7-фтор-1,2,3,4-тетрагидро-1- нафтилиден)уксусной кислоты
Смесь этил-2-бром-2-(7-фтор-1,2,3,4-тетрагидро-1- нафтил)ацетата (2,2 г, 7,0 ммолей), 1 н. трет-бутоксида калия в тетрагидрофуране (14 мл, Aldrich) и трет-бутаноле (140 мл) перемешивали в течение 5 часов при комнатной температуре. Полученную суспензию концентрировали в вакууме, разбавляли водой (200 мл) и промывали диэтиловым эфиром. Водный слой подкисляли добавлением 1 н. соляной кислоты (14 мл) и экстрагировали диэтиловым эфиром. Эфирный экстракт сушили над безводным сульфатом натрия, фильтровали, концентрировали в вакууме и хроматографировали на Силикагеле 60 с применением смеси этилацетат-гексан (1:1) в качестве элюента. Фракции, содержащие только (E)-2-(7-фтор-1,2,3,4- тетрагидро-1-нафтилиден)уксусную кислоту, объединяли и концентрировали в вакууме, получая белое твердое вещество (0,8 г, 55%), ЯМР (DMSO-d6): d 12,22 (шир.с., 1H, COOH), 7,57 (дд, Jm = 2,6 Гц), Jo = 11,0 Гц, 1H, Ar), 7,12-7,28 (м, 2H, Ar), 6,36 (с, 1H, = CH/E), 3,04 (т, 2H, ArCH2), 2,74 (т, 2H, CH2), 1,74 (м, 2H, CH2), ЯЭО стационарного состояния: освещ. при 6,36, наблюдаемый 25% ЯЭО при 7,57.
e) Получение (E)-2-(7-фтор-1,2,3,4-тетрагидро-1-нафтилиден)- ацетилхлорида
Оксалилхлорид (3 мл, 0,034 моля, Aldrich) добавляли к суспензии (E)-2-(7-фтор-1,2,3,4-тетрагидро-1-нафтилиден)уксусной кислоты (2,03 г, 0,01 моля) в толуоле (35 мл) при 0oC, защищая от влажности атмосферой азота. Перемешиваемой смеси давали нагреться до 25oC и смесь перемешивали в течение 1,5 часа. Концентрирование в вакууме дало (E)-2-(7-фтор-1,2,3,4-тетрагидро- 1-нафтилиден)ацетилхлорид, который растворяли в дихлорметане и использовали без очистки.
f) Получение (E)-2-(7-фтор-1,2,3,4-тетрагидро-1-нафтилиден)- N,N-диметилацетамида
Диметиламин (3 мл, 0,045 моля, Kodak) добавляли к охлажденному (на ледяной бане) раствору (E)-2-(7-фтор-1,2,3,4-тетрагидро-1- нафтилиден)ацетилхлорида (2,3 г, 0,010 моля) в дихлорметане (35 мл). Реакционную смесь нагревали до 25oC и перемешивали в течение 1,5 часа. Летучие компоненты удаляли роторным испарением в вакууме, получая бежевый остаток. Его растворяли в этилацетате (200 мл), промывали деионизованной водой (50 мл) и органический слой концентрировали роторным испарением в вакууме. Остаток хроматографировали на Силикагеле 60 с применением ступенчатого градиента от этилацетат-гексаны/1: 5 до этилацетата. Фракции, содержащие (E)-2-(7-фтор-1,2,3,4-тетрагидро-1-нафтилиден)-N, N- диметилацетамид, объединяли и концентрировали роторным испарением в вакууме до белого твердого вещества. Перекристаллизация из смеси дихлорметан-гексаны дала 1,62 г (70%) (E)-2-(7-фтор-1,2,3,4- тетрагидро-1-нафтилиден)-N, N-диметилацетамида в виде белого кристаллического твердого вещества, т.пл. 66-70oC.
Анал. рассчитано для C14H16FNO (мол. масса 233,278): C 72,08, H 6,91, N 6,00.
Найдено: C 71,97, H 6,94, N 5,95.
Пример 15
Получение (E)-2-(6-фтор-3,3-диметил-1-инданилиден)ацетамида
а) Получение диэтилизопропилиденмалоната
Диэтилизопропилиденмалонат получали по способу E.Z. Eliel, R.O. Hutehins and Sr. M. Knoeber, Organic Synthesis Coll. Vol. VI, 442, 1988, со следующими модификациями. Смесь ацетона (54 г, 0,93 моля, Mallinckrodt), диэтилмалоната (100 г, 0,62 моля, Aldrich), уксусного ангидрида (80 г, 0,78 моля, Mallinckrodt) и хлорида цинка (12,5 г, 0,78 моля, Aldrich) дефлегмировали (90oC, масляная баня) в течение 18 часов, защищая от влажности. Реакционный раствор разбавляли дихлорметаном (500 мл) и промывали холодной водой (3х50 мл). Водные промывки объединяли и экстрагировали дихлорметаном. Все слои дихлорметана объединяли и концентрировали роторным испарением в вакууме. Оставшееся масло перегоняли под вакуумом и фракции, кипящие при 102-138oC при 12 Top, объединяли с осадком резервуара и нагревали в течение 6 часов при 200oC в масляной бане. Темное масло перегоняли, получая 40,1 г (32%) диэтилизопропилиденмалоната в виде прозрачного масла, т. кип. 110-115oC/12 мм Hg.
b) Получение диэтил-2-(2-(4-фторфенил)-2-метилэтил)малоната
Смесь 2-фторфенилмагнийбромида (82 мл 2 н. раствора в этиловом эфире, 0,164 моля, Aldrich) и иодида меди (I) (0,310 мг, 1,63 ммоля, Aldrich) перемешивали в течение 15 минут при -10oC под азотом. К этой смеси добавляли раствор диэтилизопропилиденмалоната (29,6 г, 0,148 моля) в безводном диэтиловом эфире (250 мл) в виде тонкой струи с быстрым перемешиванием. Полученный раствор перемешивали при -10oC в течение 2 часов, при 25oC в течение 30 минут и затем выливали при быстром перемешивании в 0,5 кг раздробленного льда, содержащего 30 мл 12 н. соляной кислоты. Разделяли слои и водный слой экстрагировали этиловым эфиром (3х400 мл). Все эфирные слои объединяли и промывали деионизованной водой (2х25 мл), насыщенным раствором в воде бикарбоната натрия (25 мл) и деионизованной водой (25 мл). Этот эфирный слой концентрировали роторным испарением в вакууме и остаток перегоняли, получая 25,9 г (59%) диэтил-2-(2-(4-фторфенил)-2-метилэтил)малоната в виде прозрачного масла (т. кип. 140-145oC/0,01 мм Hg).
с) Получение 3-(4-фторфенил)-3-метилмасляной кислоты
Раствор диэтил-2-(2-(4-фторфенил)-2-метилэтил)малоната (41 г, 0,138 моля) в гидроксиде калия (85%, 18,25 г, 0,277 моля, Mallinckrodt) в 250 мл деионизованной воды интенсивно дефлегмировали в течение 4 часов с масляной баней при 150oC. После охлаждения на ледяной бане раствор нейтрализовали 18 н. серной кислотой (23 мл, 0,414 моля, Mallinckrodt) и экстрагировали дихлорметаном (4х250 мл). Дихлорметановые экстракты объединяли, промывали водой и концентрировали роторным испарением в вакууме. Остаток суспендировали с водой и кристаллический продукт собирали фильтрованием, получая 24,3 г (90%) 3-(4-фторфенил)-3-метилмасляной кислоты, т.пл. 45-47oC.
d) Получение 3-(4-фторфенил)-3-метилбутирилхлорида
Оксалилхлорид (46,5 г, 0,367 моля, Aldrich) добавляли к раствору 3-(4-фторфенил)-3-метилмасляной кислоты (24 г, 0,122 моля) при -10oC, защищая от влаги атмосферой азота. Перемешиваемой смеси давали нагреться до 25oC и смесь перемешивали в течение 2 часов. Фракционная перегонка дала 26,6 г (76%) 3-(4-фторфенил)-3- метилбутирилхлорида в виде прозрачного масла, т. кип. 132-138oC.
е) Получение 6-фтор-3,3-диметил-1-инданона
Раствор 3-(4-фторфенил)-3-метилбутирилхлорида (19,0 г, 0,0815 моля) в дихлорметане (100 мл) добавляли по каплям в течение 2,5 часов к перемешивающейся смеси хлорида алюминия (13,57 г, 0,102 моля, Aldrich) в дихлорметане (200 мл), защищая от влаги атмосферой азота. После перемешивания в течение 18 часов при 25oC реакционный раствор выливали на лед (400 г) и полученный раствор экстрагировали дихлорметаном (2х200 мл). Слои дихлорметана объединяли, промывали деионизованной водой (50 мл) и концентрировали роторным испарением в вакууме. Остаток растворяли в этилацетате и промывали через подушку силикагеля. Подушку промывали дополнительной порцией этилацетата. Удаление летучих компонентов из объединенных промывок роторным испарением в вакууме дало 15,2 г (99%) 6-фтор-3,3-диметил-1-инданона в виде светло-желтого масла, кристаллизующегося при стоянии, т.пл. 57-62oC.
f) Получение этил-2-(6-фтор-1-гидрокси-3,3-диметил-1- инданил)ацетата
Это соединение получали способом, сходным с описанным для этил-2-(6-фтор-1-гидрокси-1-инданил)ацетата в примере 1d, заменяя 6-фтор-3,3-диметил-1-инданоном 6-фтор-1-инданон и получая активированный цинк нагреванием цинковой пыли (Aldrich) с иодом (Aldrich) без растворителя. Удаление летучих компонентов из рабочего раствора роторным испарением в вакууме дало 16,2 г (82%) этил-2-(6-фтор-1-гидрокси-3,3-диметил-1-инданил)ацетата в виде светло-желтого масла.
g) Получение (E)-2-(6-фтор-3,3-диметил-1-инданилиден)уксусной кислоты
Раствор этил-2-(6-фтор-1-гидрокси-3,3-диметил-1-инданил)ацетата (16 г, 0,0601 моля) в 1 н. гидроксиде натрия (60,1 мл, 0,0601 моля) в этаноле (60 мл) перемешивали в течение 20 часов. Раствор концентрировали до небольшого объема роторным испарением в вакууме, разводили деионизованной водой (100 мл) и подкисляли до pH 3 1 н. соляной кислотой. Этот двухфазный раствор экстрагировали дихлорметаном (2х100 м). Экстракты объединяли, промывали деионизованной водой (20 мл), сушили с сульфатом магния (Mallinckrodt) и концентрировали роторным испарением в вакууме. Остаток растворяли в дихлорметане (30 мл), охлаждали до 0oC и разводили 400 мл холодного (0oC) раствора трифторуксусной кислоты (45 г, Aldrich) в дихлорметане (400 мл). После 15 минут раствор концентрировали роторным испарением в вакууме и остаток кристаллизовали путем добавления гексана, получая 9,23 г (70%) (E)-2-(6-фтор-3,3-диметил-1-инданилиден)уксусной кислоты в виде белого кристаллического твердого вещества, т.пл. 202-203oC.
h) Получение (E)-2-(6-фтор-3,3-диметил-1-инданилиден)ацетилхлорида
К охлажденной на льду перемешиваемой суспензии (E)-2-(6-фтор-3,3-диметил-1-инданилиден)уксусной кислоты (9,0 г, 0,0409 моля) в дихлорметане (200 мл) добавляли оксалилхлорид (15,6 г, 0,123 моля, Aldrich). Перемешиваемой суспензии давали нагреться до 25oC в течение 2 часов. Полученный раствор концентрировали роторным испарением в вакууме с добавлением дихлорметана (4х75 мл), получая (E)-2-(6-фтор-3,3-диметил-1-инданилиден)ацетилхлорид в виде неохарактеризованного масла. Дихлорметан (приблизительно 70 г) добавляли для растворения этого оставшегося масла и полученный раствор разделяли поровну и использовали без дальнейшей очистки в примерах 15i, 16 и 17.
i) Получение (E)-2-(6-фтор-3,3-диметил-1-инданилиден)ацетамида
Раствор 30% водного гидроксида аммония (10 мл, 76 ммолей, Mallinckrodt) добавляли к раствору (E)-2-(6-фтор-З,3-диметил-1-инданилиден)ацетилхлорида (0,01363 моля), полученного из примера 41h, разбавляли дихлорметаном (200 мл) и охлаждали до 0oC. Двухфазный раствор быстро перемешивали и давали ему нагреться до комнатной температуры в течение 18 часов. Реакционный раствор концентрировали роторным испарением в вакууме, разбавляли дихлорметаном (200 мл) и промывали 1 н. водной соляной кислотой (Mejntosh) раствором 5% водного бикарбоната натрия (Mallinckrodt), сушили с сульфатом магния (Mallinckrodt) и концентрировали роторным испарением в вакууме. Остаток хроматографировали на Силикагеле 60 с применением смеси этилацетат-гексаны (1:1) и затем этилацетата. Фракции, содержащие (E)-2-(6-фтор-3,3-диметил-1-инданилиден)ацетамид, объединяли и концентрировали роторным испарением в вакууме. Перекристаллизация из смеси дихлорметан-гексаны дала 2,85 г (95%) (E)-2-(6-фтор-3,3-диметил-1-инданилиден)ацетамида в виде белого кристаллического твердого вещества, т.пл. 167-168oC.
Пример 16
Получение (E)-2-(6-фтор-3,3-диметил-1-инданилиден)-N-метилацетамида
Это соединение получали способом, аналогичным описанному в примере 15i, с заменой раствора 30% водного гидроксида аммония 40% водным раствором метиламина (10 мл, Aldrich). Перекристаллизация из смеси дихлорметан-гексаны дала 2,89 г (91%) (E)-2-(6-фтор-3,3-диметил-1-инданилиден)-N-метилацетамида в виде белого кристаллического твердого вещества, т.пл. 157-158oC.
Пример 17
Получение (E)-N-циклопропил-2-(6-фтор-3,3-диметил-1- инданилиден)ацетамида
Это соединение получали способом, аналогичным описанному в примере 15i, с заменой раствора 30% водного гидроксида аммония циклопропиламином (4 мл, Aldrich). Перекристаллизация из смеси дихлорметан-гексаны дала 2,86 г (81%) (E)-N-циклопропил-2-(6-фтор-3,3-диметил-1-инданилиден)ацетамида в виде белого кристаллического твердого вещества, т.пл. 149-150oC.
Пример 18
Получение (E)-2-(6-фтор-3-метил-1-инданилиден)ацетамида
а) Получение этил-4-фторциннамата
Раствор бутиллития, 2,5 М в гексанах (159 мл, 0,3975 моля, Aldrich) добавляли по каплям в течение 0,25 часа с быстрым перемешиванием к раствору триэтилфосфоноацетата (89,2 г, 0,389 моля, Aldrich) в тетрагидрофуране (800 мл, безводный, Aldrich) при менее 5oC в атмосфере азота. Этот раствор перемешивали еще в течение 0,25 часов и охлаждали до 0oC в бане со льдом, после чего добавляли раствор 4-фторацетофенона (50 г, 0,362 моля, Aldrich) в тетрагидрофуране (50 мл) в виде одной порции. Перемешивание продолжали в течение 18 часов без дополнительного охлаждения. Затем этот раствор концентрировали до приблизительно 100 мл роторным испарением в вакууме и разводили до 500 мл этилацетатом. После промывания деионизованной водой (3х50 мл) этот раствор концентрировали роторным испарением в вакууме. Перегонка при пониженном давлении дала 48 г (63%) этил-4-фторциннамата в виде смеси (E) - (Z) изомеров (соотношение 3:1) с примесью 16% триэтилфосфоноацетата в виде прозрачного масла, т.кип. 138-143oC при 14 мм Hg.
b) Получение этил-3-(4-фторфенил)бутирата
Смесь этил-4-фторциннамата (47,5 г, 0,228 моля) и 10% палладия на угле (0,85 г, Aldrich) в 95% этаноле встряхивали в гидрогенизаторе Парра при 2-3 атм H2-давлении в течение 1 часа. Смесь фильтровали и концентрировали роторным испарением в вакууме. Фракционная перегонка дала 46,5 г (97%) этил-3-(4-фторфенил)бутирата в виде прозрачного масла, т. кип. 122-128oC.
с) Получение 3-(4-фторфенил)бутановой кислоты
Раствор этил-3-(4-фторфенил)бутирата (45,3 г, 0,215 моля), 85% гидроксида калия (14,22 г, 0,215 моля, Mallinckrodt) в 200 мл деионизованной воды дефлегмировали в течение 2 часов, концентрировали роторным испарением в вакууме, подкисляли до pH 3 12 N соляной кислотой (Mallinckrodt) и экстрагировали дихлорметаном (4х200 мл). Слои дихлорметана объединяли, промывали деионизованной водой (50 мл) и концентрировали роторным испарением в вакууме. Остаток кристаллизовали из смеси дихлорметана-гексана, получая 34,5 г (88%) 3-(4-фторфенил)масляной кислоты в виде белого кристаллического твердого вещества.
d) Получение 3-(4-фторфенил)бутирилхлорида
Оксалилхлорид (71 г, 48,8 мл, 0,560 моля, Aldrich) добавляли к смеси 3-(4-фторфенил)масляной кислоте (34 г, 0,187 моля) в 200 мл дихлорметана при 5oC. После перемешивания в течение 20 минут при этой температуре раствору давали нагреться до 25oC и перемешивание продолжали в течение 2 часов. Летучие компоненты удаляли роторным испарением в вакууме с добавлением дихлорметана (4х) во время концентрирования, получая 35,1 г (94%) 3-(4-фторфенил)бутирилхлорида в виде светло-желтого масла.
е) Получение 6-фтор-3-метил-1-инданона
Это соединение получали способом, аналогичным описанному в примере 15е, с заменой 3-(4-фторфенил)-3-метилбутирилхлорида 3-(4-фторфенил)бутирилхлоридом (35,1 г, 174,9 ммоля). Удаление летучих компонентов из объединенных промывок роторным испарением в вакууме дало 26,3 г (92%) 6-фтор-3-метил-1-инданона в виде масла, образующего плавящиеся при низкой температуре кристаллы при стоянии.
f) Получение этил-2-(6-фтор-1-гидрокси-3-метил-1-инданил)ацетата
Это соединение получали способом, аналогичным описанному в примере 15f, с заменой 6-фтор-3,3-диметил-1-инданона 6-фтор-3-метил-1-инданоном (25 г, 140 ммолей). Удаление летучих компонентов из обработанного раствора дало 15,0 г (45%) этил-2-(6-фтор-1-гидрокси-3-метил-1-инданил)ацетата в виде светлого рыжевато-коричневого масла.
g) Получение (E)-2-(6-фтор-3-метил-1-инданилиден)уксусной кислоты
Это соединение получали способом, аналогичным описанному в примере 15g, с заменой этил-2-(6-фтор-1-гидрокси-3,3- диметил-1-инданил)ацетата этил-2-(6-фтор-1-гидрокси-3-метил- 1-инданил)ацетатом (15 г, 59,5 ммоля). Удаление летучих компонентов из обработанного раствора дало 9,3 г (76%) (E)-2-(6-фтор-3-метил-1-инданилиден)уксусной кислоты в виде рыжевато-коричневого твердого вещества, т.пл. 175-177oC.
h) Получение (E)-2-(6-фтор-3-метил-1-инданилиден)ацетилхлорида
Это соединение получали способом, аналогичным описанному в примере 15h , с заменой (E)-2-(6-фтор-3,3-диметил-1-инданилиден)уксусной кислоты (E)-2-(6-фтор-3-метил-1-инданилиден)уксусной кислотой (9 г, 43,6 ммоля). Остаток-продукт растворяли в дихлорметане и использовали без очистки в примерах 17, 18 и 19.
i) Получение (E)-2-(6-фтор-3-метил-1-инданилиден)ацетамида
Это соединение получали способом, аналогичным примеру 15i, с заменой (E)-2-(6-фтор-3,3-диметил-1-инданилиден)ацетилхлорида (E)-2-(6-фтор-3-метил-1-инданилиден)ацетилхлоридом (3,26 г, 14,5 ммоля). Перекристаллизация из смеси дихлорметан-гексаны дала 2,39 г (77%) (E)-2-(6-фтор-3-метил-1-инданилиден)ацетамида в виде белого кристаллического твердого вещества, т.пл. 149-151oC.
Пример 19
Получение (E)-2-(6-фтор-3-метил-1-инданилиден)-N-метилацетамида
Это соединение получали способом, аналогичным описанному в примере 16, с заменой (E)-2-(6-фтор-3,3-диметил-1-инданилиден)ацетилхлорида (E)-2-(6-фтор-3-метил-1-инданилиден)ацетилхлоридом (3,26 г, 14,5 ммоля). Перекристаллизация из смеси дихлорметан-гексаны дала (2,27 г (71%) (E)-2-(6-фтор-3-метил-1-инданилиден)-N-метилацетамида в виде белого кристаллического твердого вещества, т.пл. 168-169oC.
Пример 20
Получение (E)-N-циклопропил-2-(6-фтор-3-метил-1- инданилиден)ацетамида
Это соединение получали способом, аналогичным описанному в примере 17, с заменой (E)-2-(6-фтор-3,3-диметил-1-инданилиден) ацетилхлорида (E)-2-(6-фтор-3-метил-1-инданилиден)ацетилхлоридом (3,26 г, 14,5 ммоля). Перекристаллизация из смеси дихлорметан-гексан дала 2,30 г (67%) (E)-N-циклопропил-2-(6-фтор-3-метил-1- инданилиден)ацетамида в виде белого кристаллического твердого вещества, т.пл. 132-134oC.
Пример 21
Получение (Z)-2-(6-фтор-2-гидрокси-1-инданилиден)ацетамида (Способ A)
а) Получение (Z)-2-(2-бром-6-фтор-1-инданилиден)ацетамида
N-бромсукцинимид (22,57 г, 126,8 ммоля, Aldrich) и бензоилпероксид (1,89 г, 7,8 моля, Aldrich) добавляли к суспензии (E)-2-(6-фтор-1-инданилиден)ацетамида (21,00 г, 109,8 ммоля) в тетрахлориде углерода (400 мл) и бензоле (400 мл). Смесь нагревали с обратным холодильником под сушильной трубкой с хлоридом кальция с освещенной инфракрасной лампой на ней в течение 2 часов, после чего образовался оранжевый раствор. Тепло и свет удаляли и раствор перемешивали при температуре окружающей среды в течение 18 часов. Смесь фильтровали и твердые вещества промывали этилацетатом. Промывки и фильтрат объединяли и упаривали в вакууме. Остаток растворяли в этилацетате (800 мл) и промывали водой (3х200 мл) и солевым раствором (200 мл), сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя сначала смесью гексан:этилацетат (2:1) с постепенным увеличением полярности до гексан:этилацетат (1:1). Фракции, содержащие основное пятно, объединяли и упаривали в вакууме, получая желтое твердое вещество, которое сушили в вакууме при 70oC в течение 18 часов, получая 1,022 г (3%) (Z)-2-(2-бром-6-фтор-1-инданилиден)ацетамида в виде желтого твердого вещества, т.пл. 162-163oC. 1H-ЯМР.
b) Смесь (Z)-2-(2-бром/6-фтор-1-инданилиден)ацетамида (5,30 г, 19,25 ммоля) и нитрата серебра (10,40 г, 61,18 ммоля, Aldrich) в диметоксиэтане (265 мл) и воде (100 мл) нагревали с обратным холодильником в течение 18 часов. Смесь фильтровали и фильтрат разбавляли водой (700 мл) и экстрагировали этилацетатом (6х100 мл). Объединенные экстракты промывали водой (200 мл) и солевым раствором (200 мл), сушили над сульфатом магния, фильтровали и упаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью гексан-этилацетат (2: 1), постепенно увеличивая полярность до отношения гексан: этилацетат 1:1. Фракции, содержащие соединение с Rf=0,18, объединяли и упаривали в вакууме, получая 1,13 г (28%) неочищенного (Z)-2-(6-фтор-2-гидрокси-1- инданилиден)ацетамида в виде оранжевого твердого вещества. Перекристаллизация из смесей этилацетат:гексан дала 0,49 г (12%) (Z)-2-(6-фтор-2-гидрокси-1-инданилиден)ацетамида, в виде твердого вещества нестандартного белого цвета, т.пл. 201-202oC.
c) Получение (Z)-2-(6-фтор-2-гидрокси-1-инданилиден)ацетамида (Способ B)
Суспензию (E)-2-(6-фтор-1-инданилиден)ацетамида (12,00 г, 62,8 ммоля) в дихлорметане (250 мл) добавляли к раствору диоксида селена (5,20 г, 46,9 ммоля, Aldrich) и трет-бутилгидропероксида (25 мл, 260,8 ммоля, Aldrich) в дихлорметане (500 мл). Суспензию перемешивали при температуре окружающей среды в течение 3 дней. Добавляли дополнительное количество трет-бутилгидроксипероксида (10 мл, 104,3 ммоля) и смесь перемешивали при температуре окружающей среды в течение 18 часов. Добавляли дополнительное количество диоксида селена (5,00 г, 45,1 ммоля) и смесь перемешивали при температуре окружающей среды в течение 18 часов. Добавляли дополнительное количество трет-бутилгидроксипероксида (15 мл, 156,5 ммоля) и смесь перемешивали при температуре окружающей среды в течение 18 часов. Смесь фильтровали для удаления приблизительно 1 грамма неочищенного продукта и фильтрат сушили над сульфатом магния, фильтровали и упаривали в вакууме. Добавляли дополнительное количество диоксида селена (5,00 г, 45,1 ммоля) и смесь перемешивали при температуре окружающей среды в течение 18 часов. Смесь концентрировали в вакууме до 300 мл, добавляли гексан и осадок собирали фильтрованием, промывали гексаном и объединяли с твердым материалом, собранным ранее. Объединенный твердый материал растворяли в этилацетате (700 мл), промывали последовательно водой (3х100 мл), солевым раствором (100 мл), концентрировали в вакууме до 100 мл и охлаждали в ледяной бане. Твердое вещество собирали фильтрованием и фильтрат концентрировали в вакууме, получая вторую часть твердых веществ. Все твердые вещества объединяли и хроматографировали на силикагеле, элюируя смесью гексан:этилацетат (1:1). Фракции, содержащие основное пятно, объединяли и упаривали в вакууме, получая 5,80 г твердого вещества нестандартного белого цвета, которое промывали хлороформом (3х50 мл), получая 5,43 г (42%) (Z)-2-(6-фтор-2-гидрокси-1- инданилиден)ацетамида в виде белого твердого вещества, т.пл. 202-204oC.
Пример 22
Получение (Z)-2-(4,6-дифтор-2-гидрокси-1-инданилиден)ацетамида
Суспензию (E)-2-(4,6-дифтор-1-инданилиден)ацетамида (10,0 г, 0,05 моля) в дихлорметане (250 мл), полученную как описано в примере 5g, добавляли порциями в течение 10 минут к смеси 70% водного трет-бутилгидропероксида (19,8 мл, 0,15 моля, Aldrich), и диоксида селена (3,7 г, 0,03 моля, Aldrich) в дихлорметане (500 мл) при температуре окружающей среды. После 18 часов добавляли дополнительно трет-бутилгидропероксид (10 мл, 5,0 М раствора в 2,2,4-триметилпентане, 0,05 моля, Aldrich) и диоксид селена (1,8 г, 0,02 моля) и смесь перемешивали при комнатной температуре. После 18 часов добавляли дополнительно трет-бутилгидропероксид (10 мл 70% водного раствора, 0,08 моля) и диоксид селена (3,7 г, 0,05 моля) и смесь перемешивали при температуре окружающей среды в течение 8 дней. Полученный твердый материал фильтровали и промывали дихлорметаном, получая 5,85 г неочищенного (Z)-2-(4,6-дифтор-2- гидрокси-1-инданилиден)ацетамида. После 7 дней при окружающей температуре вторую часть неочищенного (Z)-2-(4,6-дифтор-2- гидрокси-1-инданилиден)ацетамида получали из фильтрата. Колоночная хроматография на силикагеле с применением этилацетата в качестве элюента с последующей второй колоночной хроматографией на силикагеле с применением смеси этилацетат:гексан (3: 2) в качестве элюента и растирание полученного твердого вещества с пентаном дали 2,38 г (Z)-2-(4,6-дифтор-2-гидрокси-1- инданилиден)ацетамида с виде розового твердого вещества, т.пл. 235-237oC.
Пример 23
Получение (E)-2-(6-фтор-3-гидрокси-1-инданилиден)ацетамида
а) Получение 3-бром-6-фтор-1-инданона
Смесь N-бромсукцинимида (2,76 г, 15,51 ммoля, Aldrich), бензоилпероксида (0,01 г, 0,04 ммоля, Aldrich) и 6-фтор-1-инданона (2,29 г, 15,25 ммоля) в тетрахлориде углерода (20 мл) нагревали с обратным холодильником в атмосфере азота в течение 2 часов. Смесь охлаждали до температуры окружающей среды, фильтровали и твердые вещества промывали дихлорметаном. Промывки и фильтрат объединяли, промывали последовательно 1,0 н. гидроксидом натрия (2х30 мл), водой (2х30 мл) и солевым раствором (30 мл) и упаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя сначала гексаном с постепенным увеличением полярности до гексан:этилацетат (95:5). Фракции, содержащие основное пятно, объединяли и упаривали в вакууме, получая 2,30 г (66%) 3-бром-6- фтор-1-инданона в виде желтого масла, которое использовали без дальнейшей очистки.
b) Получение 3-гидрокси-6-фтор-1-инданона
Смесь 3-бром-6-фтор-1-инданона (2,50 г, 10,0 ммоля) и карбоната серебра (4,19 г, 15,2 ммоля, Aldrich) в диметоксиэтане (85 мл) и воде (65 мл) перемешивали в течение ночи при температуре окружающей среды. Смесь фильтровали через подушку целита и фильтрат разбавляли водой (500 мл) и экстрагировали этилацетатом (4х100 мл). Объединенные экстракты промывали водой (100 мл) и солевым раствором (75 мл), сушили над сульфатом натрия, фильтровали и упаривали в вакууме, получая 2,80 г (количественный выход) неочищенного 3-гидрокси-6-фтор-1-инданона, который использовали без дальнейшей очистки. Хроматография 0,41 г на силикагеле с элюцией смесью гексан:этилацетат (3:4) дала 0,050 г аналитически чистого 3-гидрокси-6-фтор-1-инданона в виде рыжевато-коричневого твердого вещества, т.пл. 73-76oC.
c) Получение 3-((трет-бутилдиметилсилил)окси)-6-фтор-1-инданона
Раствор 3-гидрокси-6-фтор-1-инданона (4,09 г, 24,6 ммоля) в диметилформамиде (10 мл) добавляли к раствору трет-бутилдиметилсилихлорида (4,60 г, 30,5 ммоля, Aldrich) и имидазола (4,22 г, 62,0 ммоля, Aldrich) в диметилформамиде (20 мл). Раствор перемешивали при температуре окружающей среды в течение 18 часов и упаривали в вакууме. Остаток растворяли в дихлорметане (200 мл) и промывали водой (6х75 мл) и солевым раствором (100 мл), сушили над сульфатом магния, фильтровали и упаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью гексан:этилацетат (95:5). Фракции, содержащие основное пятно, объединяли и упаривали в вакууме, и остаток сушили в вакууме при температуре окружающей среды в течение 18 часов, получая 4,14 г (60%) 3-((трет-бутилдиметилсилил)окси)-6-фтор-1-инданона в виде белого твердого вещества, т.пл. 56-58oC.
d) Получение этил-2-(3-((трет-бутилдиметилсилил)окси) -6-фтор-1-гидрокси-1-инданил)ацетата
Раствор этилацетата (1,00 мл, 10,3 ммоля) и диизопропиламина лития (Эту соль получали из диизопропиламина (1,41 мл, 10,0 ммолей, Aldrich) и н-бутиллития (4,00 мл, 1,5 М раствора гексана, 10,0 ммолей, Aldrich) в тетрагидрофуране (15 мл) перемешивали при -78oC под азотом в течение 15 минут. Раствор 3-((трет-бутилдиметилсилил)окси)-6-фтор-1-инданона (2,80 г, 10,0 ммолей) в тетрагидрофуране (15 мл) добавляли по каплям в течение 7 минут и раствор перемешивали при -78oC под азотом в течение 1,5 часа. Раствор хлорида аммония (1,60 г, 30,0 ммолей) в воде (9 мл) добавляли и полученной суспензии давали нагреваться до температуры окружающей среды. Слои разделяли и водный слой экстрагировали эфиром (2х100 мл). Органические экстракты объединяли и промывали последовательно водой (100 мл) и солевым раствором (100 мл), сушили над сульфатом магния, фильтровали и упаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью гексан-этилацетат (98:2), постепенно увеличивая полярность до гексан:этилацетат (4:1). Фракции, содержащие основное пятно, объединяли и упаривали в вакууме. Остаток сушили в вакууме при температуре окружающей среды в течение 18 часов при 60oC, получая 2,86 г (78%) этил-2-(3-((3- трет-бутилдиметилсилил)окси)-6-фтор-1-гидрокси-1-инданил)ацетат в виде прозрачного масла.
е) Получение (E)-этил-2-(3-((трет-бутилдиметилсилил)окси)-6- фтор-1-инданилиден)ацетата
Раствор этил-2-(3-((трет-бутилдиметилсилил)окси)-6-фтор-1- гидрокси-1-инданил)ацетата (2,80 г, 7,6 ммоля) добавляли к раствору бис[2,2,2-трифтор-1-фенил-1-(трифторметил)-этокси] - дифенилсульфурана (6,30 г, 9,4 ммоля), Fluka) дихлорметане (50 мл) под атмосферой азота. Раствор перемешивали при температуре окружающей среды в течение 35 минут и выливали в воду (500 мл). Органический слой отделяли, промывали солевым раствором (250 мл), сушили над сульфатом магния, фильтровали и упаривали в вакууме. Остаток хроматографировали на силикагеле с элюцией смесью гексан:этилацетат (99:1). Фракции, содержащие основное пятно (и также малые примеси), объединяли и выпаривали в вакууме, получая 2,68 г (количественный выход) неочищенного (E)-этил-2-(3-((трет- бутилдиметилсилил)окси)-6-фтор-1-инданилиден)ацетата в виде желтого масла, которое использовали без дальнейшей очистки.
f) Получение (E)-2-(3-((трет-бутилдиметилсилил)окси)-6-фтор- 1-инданилиден)ацетамида
Раствор амида диметилалюминия получали добавлением триметилалюминия (6,5 мл, 1,0 М раствора в толуоле, 13,0 ммолей, Aldrich) к раствору хлорида алюминия (0,695 г, 13,0 ммолей) в дихлорметане (25 мл) под атмосферой азота и перемешиванием в течение 45 минут при температуре окружающий среды. Этот раствор амида диметилалюминия (13,0 ммолей) добавляли к раствору (E)-этил-2-(3-((трет-бутилдиметилсилил)окси)-6-фтор-1-инданилиден)ацетата (1,190 г, 3,4 ммоля) в дихлорметане (60 мл) под азотом. Смесь перемешивали при температуре окружающей среды в течение 30 минут и дефлегмировали в течение 18 часов. После охлаждения при температуре окружающей среды и затем в ледяной бане смесь гасили добавлением по каплям 0,5 н. соляной кислоты до прекращения выделения газа. Раствор разбавляли водой (50 мл), слои разделяли и водный слой экстрагировали дихлорметаном (75 мл). Органические слои объединяли, промывали последовательно водой (75 мл) и солевым раствором (75 мл), сушили над сульфатом магния, фильтровали и упаривали в вакууме. Остаток перекристаллизовывали из смесей дихлорметан:гексан, получая 0,321 г (29%) (E)-2-(3-((трет- бутилдиметилсилил)окси)-6-фтор-1-инданилиден)ацетамида в виде белого твердого вещества, т.пл. 160-165oC.
g) Получение (E)-2-(6-фтор-3-гидрокси-1-инданилиден)ацетамида
Раствор (E)-2-(3-((трет-бутилдиметилсилил)окси)-6-фтор- 1-инданилиден)ацетамида (1,80 г, 5,6 ммоля) и п-толуолсульфоната пиридиния (0,85 г, 3,4 ммоля, Aldrich) в этаноле (65 мл) нагревали при 55-68oC в течение 3,5 часов в атмосфере азота и выпаривали в вакууме. Остаток растворяли в этилацетате (150 мл) и промывали последовательно водой (2х150 мл) и солевым раствором (150 мл), сушили над сульфатом магния, фильтровали и выпаривали в вакууме. Остаток хроматографировали на силикагеле с элюцией этилацетатом, постепенно увеличивая полярность до этилацетат:этанол (95:5). Фракции, содержащие основное пятно, объединяли и выпаривали в вакууме. Остаток сушили в вакууме при 80oC в течение 18 часов, получая 0,72 г (62%) (E)-2-(6-фтор-3-гидрокси-1-инданилиден)ацетамида в виде белого твердого вещества, т.пл. 166-168oC.
Пример 24
Получение (Z)-2-(2,3-дигидрокси-6-фтор-1-инданилиден) ацетамида
а) Получение (Z)-2-(2,3-дибром-6-фтор-1-инданилиден)ацетамида
N-бромсукцинимид (49,37 г, 277,4 ммоля, Aldrich) и бензоилпероксид (1,60 г, 6,6 ммоля, Aldrich) добавляли к суспензии (E)-2-(6-фтор-1-инданилиден)ацетамида (17,68 г, 92,5 ммоля) в четыреххлористом углероде (335 мл) и бензоле (335 мл). Смесь нагревали с обратным холодильником под сушильной трубкой с хлоридом кальция в течение 4 часов, после чего образовался оранжевый раствор. Нагревание удаляли и раствор перемешивали при температуре окружающей среды в течение 18 часов. Смесь фильтровали и твердые вещества промывали этилацетатом. Промывки и фильтрат объединяли и выпаривали в вакууме. Остаток растворяли в этилацетате (800 мл) и промывали водой (3х200 мл) и солевым раствором (200 мл), сушили над сульфатом натрия, фильтровали и выпаривали в вакууме. Остаток хроматографировали на силикагеле с элюцией смесью гексан:этилацетат (2:1). Фракции, содержащие соединение с Rf = 0,4 в смеси гексан: этилацетат (1: 1), объединяли и упаривали в вакууме, получая твердое вещество, которое промывали гексаном и сушили в вакууме при 50oC в течение 18 часов, получая 1,35 г (4%) (Z)-2-(2,3-дибром-6-фтор-1-инданилиден)ацетамида в виде желтого твердого вещества, т.пл. 158-163oC.
b) Смесь (Z)-2-(2,3-дибром-6-фтор-1-инданилиден)ацетамида (0,54 г, 1,55 ммоля) и карбоната серебра (0,56 г, 2,03 ммоля, Aldrich) в диметоксиэтане (15 мл) и воде (30 мл) дефлегмировали в течение 6 часов. Смесь перемешивали в течение ночи при температуре окружающей среды и опять дефлегмировали в течение 6 часов. Смесь разбавляли водой (100 мл) и экстрагировали этилацетатом (6х30 мл). Объединенные экстракты промывали водой (100 мл) и солевым раствором (100 мл) и выпаривали в вакууме. Остаток хроматографировали на силикагеле с элюцией смесью гексан:этилацетат (2:1). Фракции, содержащие соединение с Rf = 0,15, при элюции этилацетатом, объединяли и выпаривали в вакууме, получая 0,12 г (35%) неочищенного (Z)-2-(2,3-дигидрокси-6-фтор-1-инданилиден)ацетамида в виде бежевого твердого вещества. Перекристаллизация из смесей этилацетат: гексан дала 0,037 г (11%) (Z)-2-(2,3-дигидрокси-6-фтор-1-инданилен)ацетамида в виде твердого вещества нестандартного белого цвета, которое, как было показано при помощи 1H-ЯМР, было смесью (85:15) диастереомеров, т.пл. 212-220oC. 1H-ЯМР (DMSO-d6): d 7,82 (д, 2H), 7,28-7,76 (м, 3H), 6,96 (с, 1H), 6,80 (с, 0,15H), 6,54 (с, 0,85H), 6,51 и 6,12 (м, 0,3H), 5,92 (д, 1H), 4,81 (м, 1,7H), ЯЭО стационарного состояния: освещ. при 6,47d, наблюдаемый 20% ЯЭО при 7,38d.
Пример 25
Получение (E)-2-(6-фтор-3-оксо-1-инданилиден)ацетамида.
а) Получение (E)-этил-3-фторциннамата
Это соединение получали способом, аналогичным описанному в примере 18а, с заменой 4'-фторацетофенона 3-фторбензальдегидом (33,6 г, 0,3 ммоля, Aldrich). Перегонка дала 32,85 г (56%) (E)-этил-3-фторциннамата в 5 фракциях (т. кип. 140-155oC при 15 Top), содержащих одинаковое количество, приблизительно 13%, триэтилфосфоноацетата в виде примеси. Этот материал использовали без дополнительной очистки. 1H-ЯМР (DMSO-d6); d 7,66-7,47 (м, 2H), 7,45-7,40 (м, 1H), 7,27-7,20 (м, 1H), 6,70 (д, 1H, JHH = 16 Гц), 4,18 (к, 2H, JHH = 7,2 Гц), 4,10-3,96 (м, 0,78H), 3,77 (д, 0,26H, JPH = 21,3 Гц), 1,24 (т, 3H, JHH = 7,0 Гц), 1,24-1,14 (м, 1,17H).
b) Получение диэтил-2-карбэтокси-3-(3-фторфенил)глутарата
Металлический натрий (0,388 г, 0,0169 моля) перемешивали в диэтилмалонате (15,28 г, 0,0953 моля, Aldrich) в атмосфере азота при 120oC в течение 0,33 часа. К полученному раствору добавляли (E)-этил-3-фторциннамат (16,4 г, 0,0845 моля) и перемешивание продолжали в течение 7 часов при той же температуре. Темный раствор охлаждали, растворяли в дихлорметане (500 мл) и подкисляли 30 мл 1 н. водной соляной кислотой (Macintosh). Летучие компоненты удаляли из полученной пены роторным испарением в вакууме и остаток растворяли в этилацетате. Этот раствор промывали 5% водным бикарбонатом натрия до нейтральной реакции, водой и летучие компоненты удаляли роторным испарением в вакууме. Перегонка дала 20 г материала с т. кипения между 130 и 185oC при 0,150 Top. Повторная перегонка дала 14,72 г (44%) диэтил-2-карбэтокси-3-(3- фторфенил)глутарата в виде прозрачной жидкости, т. кип. 155-160oC при 0,1 Top.
с) Получение 3-(3-фторфенил)глутаровой кислоты
К горячему раствору гидроксида натрия (19,15 г, 0,479 моля) в воде (50 мл) добавляли раствор диэтил-2-карбэтокси- 3-(3-фторфенил)глутарата (18,8 г, 0,0532 моля) и этаноле (36 мл). Полученную суспензию нагревали с обратным холодильником в течение 5 часов. Смесь выливали в ледяную воду и этанол удаляли роторным испарением в вакууме. Оставшийся водный раствор подкисляли концентрированной соляной кислотой (12 н.) и раствор (200 мл) экстрагировали этилацетатом (3х300 мл). Этилацетатные слои объединяли, промывали водой (50 мл) и летучие компоненты удаляли роторным испарением в вакууме, получая твердое вещество, которое перекристаллизовывали из дихлорметана и гексана, получая 9,3 г (77%) 3-(3-фторфенил)глутаровую кислоту в виде белого твердого вещества, т.пл. 126-127,5oC.
d) Получение 2-(6-фтор-3-оксо-1-инданил)уксусной кислоты
Соединяли полифосфорную кислоту (39,6 г, Aldrich) и 3-(3-фторфенил)глутаровую кислоту (6,6 г, 0,0292 моля) и эту смесь нагревали с масляной бане при 120oC в течение 10 минут. Ставший красным раствор охлаждали приблизительно до 60oC и приблизительно 100 мл воды добавляли по каплям с интенсивным перемешиванием. Полученный осадок собирали и промывали водой. Перекристаллизация из дихлорметана и гексана дала 5,3 г (87%) 2-(6-фтор-3-оксо-1-инданил)уксусной кислоты, т.пл. 150-151oC.
е) Получение 2-(6-фтор-3-оксо-1-инданил)ацетилхлорида
Оксалилхлорид (4,5 г, 0,035 моля, Aldrich) добавляли к охлажденной на льду перемешивающейся смеси 2-(6-фтор-3-оксо-1- инданил)уксусной кислоты (5,0 г, 0,024 моля) в дихлорметане (200 мл) в атмосфере азота. Смеси давали нагреться до комнатной температуры и перемешивание продолжали в течение 48 часов. Летучие компоненты удаляли из раствора роторным испарением в вакууме с добавлением дихлорметана (3х50 мл), получая 2-(6-фтор-3-оксо-1-инданил)ацетилхлорид, который использовали без очистки или анализа.
f) Получение 2-(6-фтор-3-оксо-1-инданил)ацетамида
Раствор 2-(6-фтор-3-оксо-1-инданил)ацетилхлорида (полученного из 0,024 моля 2-(6-фтор-3-оксо-1-инданил)уксусной кислоты) в дихлорметане (150 мл) охлаждали до 0oC и перемешивали быстро, добавляя 50 мл гидроксида аммония (28-30%). Полученной смеси давали нагреться до комнатной температуры и перемешивание продолжали в течение 18 часов. Летучие компоненты из этой смеси удаляли роторным испарением в вакууме и остаток растворяли в дихлорметане (250 мл) и промывали водой (3х50 мл). Дихлорметановую фазу затем суспендировали с Силикагелем 60 и летучие компоненты удаляли роторным испарением в вакууме. Эту суспензию затем наносили на колонку Силикагеля 60 (51х400 мм), смоченную дихлорметаном, и продукт получали элюированием смесью метанол:дихлорметан (3:97), получая, после перекристаллизации из метанола, 2,4 г (48%) 2-(6-фтор-3-оксо-1-инданил)ацетамида в виде желтого твердого вещества, т.пл. 150-152oC.
g) Получение (E)-2-(6-фтор-3-оксо-1-инданилиден)ацетамида
Смесь 2-(6-фтор-3-оксо-1-инданил)ацетамида (0,750 г, 0,0036 моля), N-бромсукцинимида (0,750 г, 0,0042 моля, Aldrich), бензоилпероксида (0,270 г, 0,0011 моля, Aldrich) в тетрахлорметане (37 мл) и бензоле (37 мл) перемешивали при нагревании на масляной бане при 120oC в течение 20 минут. Эту реакцию сочетали с подобным образом идущей реакцией (за исключением масштаба реакции - 0.0024 моля). Полученный раствор суспендировали с Силикагелем 60 и летучие компоненты удаляли роторным испарением в вакууме. Этот силикагель затем наносили на колонку Силикагеля 60 (51х450 мм), смоченную дихлорметаном, и продукт получали элюцией смесью метанол:дихлорметан (3:97). После удаления летучих компонентов из объединенных фракций, содержащих продукт, роторным испарением в вакууме остаток перекристаллизовывали из метанола, получая 0,810 г (58%) (E)-2-(6-фтор-3-оксо-1-инданилиден)ацетамида, т.пл. 235oC (разложение).
Пример 26
Получение (E)-N-циклопропил-2-(6-фтор-3-этил-1-инданилиден)ацетамида
а) Получение этил-3-(4-фторфенил)пентеноата
Раствор бутиллития, 1,6 М в гексане (230 мл, 0,368 моля, Aldrich) добавляли по каплям в течение 0,5 часа при быстром перемешивании к раствору триэтилфосфоноацетата (78,9 г, 0,351 моля, Aldrich) в тетрагидрофуране (800 мл безводного, Aldrich) при температуре ниже 5oC в атмосфере азота. Этот раствор перемешивали еще в течение 0,25 часа и охлаждали до -5oC на бане с метанолом и льдом, а затем в виде одной порции добавляли раствор 4'-фторпропиофенона (50 г, 0,328 моля, Aldrich) в тетрагидрофуране (50 мл). Перемешивание продолжали в течение 18 часов без дополнительного охлаждения. Раствор концентрировали до золотисто-желтого шлама роторным испарением в вакууме и шлам разводили до 1000 мл этилацетатом. После промывания деионизованной водой (3х100 мл) этот раствор концентрировали роторным испарением в вакууме. Перегонка при пониженном давлении дала 45,65 г (63%) этил-3-(4-фторфенил)пентеноата в виде смеси (E) и (Z) изомеров (соотношение 1:1) с 30% примеси в виде триэтилфосфоноацетата в виде прозрачной жидкости, т. кип. 140-146oC при давлении отсасывающего устройства.
b) Получение этил-3-(4-фторфенил)валерата
Смесь этил-3-(4-фторфенил)пентеноата (45,65 г, 0,137 моля) и 10% палладия на угле (0,86 г, Aldrich) в 95% этаноле встряхивали при 4 атм давлении водорода в гидрогенизаторе Парра в течение 1,5 часа. Смесь фильтровали и концентрировали роторным испарением в вакууме. Фракционная перегонка дала 38,95 г (63%) этил-3-(4- фторфенил)валерата в виде прозрачного масла с примесью 29% триэтилфосфоноацетата, т.кип. 133-142oC при 17 мм Hg.
с) Получение 3-(4-фторфенил)валериановой кислоты
Это соединение получали способом, аналогичным описанному в примере 18с, с заменой 3-(4-фторфенил)бутирата этил-3-(4-фторфенил)валератом (38,05 г, 0,144 моля, содержащим 29% триэтилфосфоноацетата) и применением избытка 85% гидроксида калия (18,05, 0,273 моля, Mallinckrodt). Дихлорметановые слои объединяли, промывали деионизованной водой (50 мл) и концентрировали роторным испарением в вакууме. Остаток кристаллизовали из гексанов, получая 23,47 г (83%) 3-(4-фторфенил)валериановой кислоты в виде белого кристаллического твердого вещества; ЯМР (DMSO-d6); d 12 (с, 1H), 7,28-7,24 (м, 2H), 7,14-7,08 (м, 2H), 2,91-2,89 (м, 1H), 2,64-2,41 (м, 2H), 1,66-1,62 (м, 1H), 1,56-1,51 (м, 1H), 0,71 (т, 3H, J = 7,3 Гц).
а) Получение 3-(4-фторфенил)валероилхлорида
Это соединение получали способом, аналогичным описанному в примере 18d, с заменой 3-(4-фторфенил)бутирата 3-(4-фторфенил)- валериановой кислотой (23,47 г, 0,120 моля). Летучие компоненты удаляли роторным испарением в вакууме с добавлением дихлорметана (6х250 мл) во время концентрирования, получая 25,25 г (98%) 3-(4-фторфенил)валероилхлорида в виде золотисто-желтой жидкости : ЯМР (DMSO-d6): d 7,3-7,22 (м, 2H), 7,15-7,06 (м, 2H), 2,98-2,73 (м, 1H), 2,66-2,38 (м, 2H), 1,74-1,37 (м, 2H), 0,70 (т, 3H, J= 7,2 Гц).
е) Получение 3-этил-6-фтор-1-инданона
Это соединение получали способом, аналогичным описанному в примере 18е, с заменой 3-(4-фторфенил)бутирилхлорида 3-(4-фторфенил)валероилхлоридом (25,27 г, 0,118 моля). Дихлорметановые экстракты объединяли, промывали деионизованной водой (100 мл) и концентрировали роторным испарением в вакууме. Остаток хроматографировали на Силикагеле 60 с применением ступенчатого градиента, идущего от гексанов до смеси этилацетат:гексаны (1:1). Фракции, содержащие 3-этил-6-фтор-1-инданон, объединяли и концентрировали роторным испарением в вакууме с дихлорметаном (2х150), добавляемым во время концентрирования, получая 17,48 г (83%) 3-этил-6-фтор-1-инданона в виде канареечно-желтого сиропа: ЯМР (DMSO-d6): d 7,74-7,7 (дд, 1H, JHF = 8,4 Гц, JHH = 4,8 Гц), 7,6-7,53 (ддд, 1H, JHF = 9,0 Гц, JHH = 9,0 Гц и 2,7 Гц), 7,37 (дд, 1H, JHF = 7,8 Гц, JHH = 2,4 Гц), ~ 3,3 (м, 1H, частично затемнено водой), 2,88 (дд, 1H, Jгем = 19,2 Гц), 2,39 (дд, 1H, Jгем = 19,2 Гц, J = 2,4 Гц), 1,98-1,90 (м, 1H), 1,54-1,44 (м, 1H), 0,90 (т, 3H, J = 7,3 Гц).
f) Получение цис и транс этил-2-(3-этил-6-фтор-1-гидрокси-1- инданил)ацетата
Это соединение получали способом, аналогичным описанному в примере 18f, с заменой 6-фтор-3-метил-1-инданона 3-этил-6-фтор-1-инданоном (17,3 г, 0,097 моля). Удаление летучих компонентов из обработанного раствора дало 25,17 г (97%) цис и транс этил-2-(3-этил-6-фтор-1-гидрокси-1-инданил)ацетата в виде золотисто-желтого масла: ЯМР (DMSO-d6): d 7,23-7,21 (м, 1H), 7,13-7,06 (м, 2H), 5,48 (с, 1H), 4,0 (к, 2H, J = 7,2 Гц), 2,90-2,82 (м, 1H), 2,80-2,73 (м, 1H), 2,70-2,55 (м, 2H), 2,04-1,9 (м, 1H), 1,83-1,67 (м, 1H), 1,46-1,28 (м, 1H), 1,11 (т, 3H, J = 7,1 Гц), 0,95 (т, 3H, J = 7,3 Гц).
g) Получение (E)-2-(3-этил-6-фтор-1-инданилиден)уксусной кислоты
Это соединение получали способом, аналогичным описанному в примере 18g, с заменой этил-2-(6-фтор-1-гидрокси-3-метил-1-инданил)ацетата этил-2-(3-этил-6-фтор-1-гидрокси-1-инданил)ацетатом (24,85 г, 0,093 моля). Удаление летучих компонентов из обработанного раствора дало бежевый остаток. Перекристаллизация из смеси дихлорметан-гексаны дала 12,91 г (63%) (E)-2-(3-этил-6-фтор-1-инданилиден)уксусной кислоты в виде белого кристаллического твердого вещества, т.пл. 145-148oC.
h) Получение (E)-2-(3-этил-6-фтор-1-инданилиден)ацетилхлорида
Это соединение получали способом, аналогичным описанному в примере 18h, с заменой (E)-2-(6-фтор-3-метил-1-инданилиден) уксусной кислоты (E)-2-(3-этил-6-фтор-1-инданилиден)уксусной кислотой (5,7 г, 25,88 ммоля). Продукт-остаток растворяли в дихлорметане и использовали без очистки в примере 26.
i) Получение (E)-N-циклопропил-2-(6-фтор-3-этил-1- инданилиден)ацетамида
Это соединение получали способом, аналогичным описанному в примере 20, с заменой (E)-2-(6-фтор-3-метил-1-инданилиден)- ацетилхлорида (E)-2-(3-этил-6-фтор-1-инданилиден)ацетилхлоридом (3,18 г, 13,3 ммоля). Перекристаллизация из смеси дихлорметан-гексаны дала 2,24 г (65%) (E)-N-циклопропил-2-(6-фтор-3-этил-1- инданилиден)ацетамида в виде белого кристаллического твердого вещества, т.пл. 143-147oC.
Пример 27
Получение (E)-N-циклопропил-2-(6-фтор-3-пропил-1- инданилиден)ацетамида
Хлорид алюминия (139 г, 1,04 моля) добавляли к раствору бутирилхлорида (55,45, 0,520 моля, Aldrich) в дихлорметане (500 мл) с перемешиванием в атмосфере азота при 25oC. Раствор фторбензола (50,1 г, 0,521 моля, Aldrich) в дихлорметане добавляли и перемешивание продолжали в течение 18 часов.
Реакционный раствор выливали на лед и экстрагировали дихлорметаном (3х400 мл). Комбинированные дихлорметановые экстракты промывали деионизованной водой (2х260 мл), 1,0 н. соляной кислотой (500 мл), насыщенным раствором бикарбоната натрия (2х500 мл) и деионизованной водой (4х250 мл), концентрировали роторным испарением в вакууме. Этот материал объединяли с материалом из подобного получения (с применением 0,26 моля фторбензола) для перегонки. Перегонка при пониженном давлении дала 69,27 г (53%) 4'-фторбутирофенона в виде бледно-желтой жидкости, которая позднее частично кристаллизовалась, т. кип. 108-112oC при 30 миллитор, ЯМР (DMSO-d6): d 8,03 (к, 2H, J = 9,0 Гц и 5,6 Гц), 7,31 (т, 2H, J = 8,9 Гц), 2,97 (т, 2H, J = 7,0 Гц), 1,65-1,55 (м, 2H), 0,91 (т, 3H, J = 7,3 Гц).
b) Получение этил-3-(4-фторфенил)гексаноата
Раствор бутиллития, 2,5 М в гексане (166 мл, 0,416 моля, Aldrich), добавляли по каплям в течение 0,5 часа при быстром механическом перемешивании к раствору триэтилфосфоноацетата (93,2 г, 0,416 моля, Aldrich) в тетрагидрофуране (700 мл, безводный, Aldrich) при температуре ниже 5oC в атмосфере азота. Этот раствор перемешивали еще в течение 0,25 часа и охлаждали до -5oC в бане с метанолом и льдом и затем добавляли в виде одной порции раствор 4'-фторбутирофенона (69 г, 0,415 моля, Aldrich) в тетрагидрофуране (150 мл). Перемешивание продолжали в течение 18 часов без дополнительного охлаждения. Раствор концентрировали до шлама темно-верблюжьего цвета роторным испарением в вакууме и разбавляли до 600 мл деионизованной водой. Водный раствор экстрагировали дихлорметаном (5х500 мл) и дихлорметан концентрировали роторным испарением в вакууме. Перегонка при пониженном давлении дала 58,5 г (60%) этил-3-(4- фторфенил)гексаноата в виде смеси (E) и (Z) изомеров (соединение 1: 1) в виде прозрачной жидкости, т. кип. 140-150oC при давлении отсасывающего устройства.
с) Получение этил-3-(4-фторфенил)гексаноата
Смесь этил-3-(4-фторфенил)гексаноата (58, 12 г, 0,246 моля) и 10% палладия на угле (1,1 г, Aldrich) в 95% этаноле встряхивали в гидрогенизаторе Парра при давлении 2-4 атм водорода в течение 0,75 часа. Смесь фильтровали и концентрировали роторным испарением в вакууме, получая 58,4 г (99,6%) этил-3-(4-фторфенил)гексаноата в виде прозрачной жидкости, ЯМР (DMSO-d6): 7,27-7,20 (м, 2H), 7,12-7,04 (м, 2H), 3,91 (к, 2H, J = 7,2 Гц), 2,99 (м, 1H), 2,66-2,59 (м, 1H), 2,52-2,44 (м, 1H, частично затенено DMSO), 1,59-1,46 (м, 2H), 1,13-1,07 (м, 2H), 1,02 (т, 3H, J = 7,2 Гц), 0,78 (т, 3H, J = 7,5 Гц).
d) Получение 3-(4-фторфенил)гексановой кислоты
Это соединение получали способом, аналогичным описанному в примере 65с, с заменой 3-(4-фторфенил)валерата этил-3-(4- фторфенилгексаноатом (58 г, 0,243 моля). Дихлорметановые слои объединяли, промывали деионизованной водой (250 мл) и концентрировали роторным испарением в вакууме. Остаток выпаривали вместе с гексаном (200 мл), получая 46,81 г (92%) 3-(4-фторфенил)гексановой кислоты в виде желтого масла.
е) Получение 3-(4-фторфенил)гексаноилхлорида
Это соединение получали способом, аналогичным описанному в примере 18d, с заменой 3-(4-фторфенил)масляной кислоты 3-(4-фторфенил)гексановой кислотой (46,5 г, 0,222 моля). Летучие компоненты удаляли роторным испарением в вакууме с добавлением дихлорметана (5х250 мл) во время концентрирования, получая 50,01 г (99%) 3-(4-фторфенил)гексаноилхлорида в виде золотисто-желтой жидкости.
f) Получение 6-фтор-3-пропил-1-инданона
Это соединение получали способом, аналогичным описанному в примере 18е, с заменой 3-(4-фторфенил)бутирилхлорида 3-(4-фторфенил)гексаноилхлоридом (49,95 г, 0,218 моля). Дихлорметановые экстракты объединяли, промывали деионизованной водой (250 мл) и концентрировали роторным испарением в вакууме. Остаток упаривали с дихлорметаном (100 мл), получая 41,26 г (98%) 6-фтор-3-пропил-1- инданона в виде золотисто-желтого сиропа.
g) Получение цис и транс этил-2-(6-фтор-1-гидрокси-3-пропил-1- инданил)ацетата
Это соединение получали способом, аналогичным описанному в примере 18f, с заменой 6-фтор-3-метил 1-инданона 6-фтор-3-пропил- 1-инданоном (40,75 г, 0,212 моля). Удаление летучих компонентов из обработанного раствора дало 57,48 г (97%) цис и транс этил-2-(6-фтор-1-гидрокси-3-пропил-1-инданил)ацетата в виде золотисто-желтого масла.
h) Получение (E)-2-(6-фтор-3-пропил-1-инданилиден)уксусной кислоты
Это соединение получали способом, аналогичным описанному в примере 18g, с заменой этил-2-(6-фтор-1-гидрокси-3-метил- 1-инданил)ацетата этил-2-(6-фтор-1-гидрокси-3-пропил-1- инданил)ацетатом (57,12 г, 0,204 моля). Удаление летучих компонентов из рабочего раствора дало золотисто-желтый остаток. Этот остаток суспендировали в гексанах, получая 24,49 г (51%) (E)-2-(6-фтор-3-пропил-1-инданилиден)уксусной кислоты в виде кристаллического белого твердого вещества, т.пл. 141-144oC, ЯМР (DMSO-d6).
i) Получение (E)-2-(6-фтор-3-пропил-1-инданилиден)ацетилхлорида
Это соединение получали способом, аналогичным описанному в примере 18h, с заменой (E)-2-(6-фтор-3-метил-1-инданилиден)- уксусной кислоты (E)-2-(6-фтор-3-пропил-1-инданилиден)уксусной кислотой (15,01 г, 64,07 ммоля). Остаток-продукт растворяли в дихлорметане и использовали без дальнейшей очистки в примере 27j.
j) Получение (E)-N-циклопропил-2-(6-фтор-3-пропил-1- инданилиден)ацетамида
Это соединение получали способом, аналогичным описанному в примере 20, с заменой (E)-2-(6-фтор-3-метил-1-инданилиден)- ацетилхлорида (E)-2-(6-фтор-3-пропил-1-инданилиден)ацетилхлоридом (3,26 г, 0,013 моля). Летучие компоненты удаляли роторным испарением в вакууме, получая золотисто-желтое масло. Масло хроматографировали на Силикагеле 60 со ступенчатым градиентом гексаны - этилацетат-гексаны (1: 1). Фракции, содержащие (E)-N-циклопропил-2-(6-фтор-3- пропил-1-инданилиден)ацетамид, объединяли и концентрировали роторным испарением в вакууме с гексанами (4х250 мл), добавленными во время концентрирования, получая 2,09 г (59%) (E)-N-циклопропил-2-(6-фтор-3-пропил-1-инданилиден)ацетамида в виде белого порошкообразного твердого вещества, т. пл. 94-97oC.
Пример 28
Получение (Z)-2-(6-фтор-1-инданилиден)ацетамида
Раствор (E)-2-(6-фтор-1-инданилиден)ацетамида (20 г, 104,6 ммоля) в смеси дихлорметан:метанол (3:1) (1000 мл) освещали кварцевой фотохимической иммерсионной лампой с парами ртути Canrad-Hanovia, 450 Вт (Ace Jlass, 7825-35) в течение 0,5 часа. Летучие компоненты удаляли роторным испарением в вакууме, получая бежевый остаток. Остаток хроматографировали на Силикагеле 60 с применением ступенчатого градиента от смеси этилацетат:гексан (1:1) до смеси этилацетат: этанол (1:1). Фракции, содержащие (Z)-2-(6-фтор-1-инданилиден)ацетамид, объединяли и концентрировали роторным испарением в вакууме. Полученное твердое вещество суспендировали в гексане, получая 7,52 г (37%) (Z)-2-(6-фтор-1- инданилиден)ацетамида в виде белого кристаллического твердого вещества, т.пл. 175-177oC.
Пример 29
Получение (E)-2-(4,6-дифтор-3-оксо-1-инданилиден)ацетамида
а) Получение 2,4-дикарбэтокси-3-(3,5-дифторфенил)-5- гидрокси-5-метил-1-циклогексанона
Слегка теплые жидкий 3,5-дифторбензальдегид (5,0 г, 0,0352 моля, Aldrich), 95% этанол (1,75 мл) и пиперидин (0,7 мл) добавляли к этилацетоацетату (9,2 г, 0,0704 моля, Aldrich). Раствор перемешивали до гомогенности и затем помещали в водяную баню для контролирования слегка экзотермической реакции. После 4 часов кристаллическую массу растворяли в теплом дихлорметане (100 мл). При разбавлении гексаном (300 мл) образуется мутный раствор. После стояния в течение 24 часов кристаллический продукт собирали фильтрованием и промывали гексаном, получая 8,0 г (59%) 2,4-дикарбэтокси-3-(3,5-дифторфенил)-5-гидрокси-5-метил-1- циклогексанона, т.пл. 185-186oC.
b) Получение 3-(3,5-дифторфенил)глутаровой кислоты
К горячему (95oC) раствору, приготовленному из гидроксида натрия (322 г, 8,1 моля) и деионизованной воды (322 мл) добавляли смесь 2,4-диацетил-3-(3,5-дифторфенил)-5-гидрокси-5-метил-1- циклогексанона (43 г, 0,112 моля) в этаноле (322 мл) с быстрым перемешиванием. Полученную смесь нагревали с обратным холодильником в течение 4 часов с применением масляной бани при 140oC. Этанол удаляли роторным испарением в вакууме и полученную суспензию охлаждали на ледяной бане и концентрированную соляную кислоту (12 н.) добавляли для доведения pH приблизительно до 1. Осадок растворяли добавлением воды и водный раствор экстрагировали этилацетатом (общий объем 1500 мл). Этилацетатные экстракты объединяли, промывали водой, сушили с MgSO4 и летучие компоненты удаляли роторным испарением в вакууме. Перекристаллизация остатка из дихлорметана дала 9,10 г (33%) 3-(3,5-дифторфенил)глутаровой кислоты, т. пл. 170-172oC.
с) Получение 2-(4,6-дифтор-3-оксо-1-инданил)уксусной кислоты
Это соединение получали способом, аналогичным описанному в примере 25d, с заменой 3-(3-фторфенил)глутаровой кислоты 3-(3,5-дифторфенил)глутаровой кислотой (9,02 г, 36,9 ммоля) и увеличением времени нагревания с 10 минут до 30 минут. Хроматография собранного продукта на колонке Силикагеля 60 (51х450 мм) с элюцией смесью метанол:дихлорметан (4:96) дала материал, который перекристаллизовывали из воды, получая 1,93 г (23%) 2-(4,6-дифтор-3-оксо-1-инданил)уксусной кислоты, т.пл. 170-172oC.
d) Получение 2-(4,6-дифтор-3-оксо-1-инданил)ацетилхлорида
Это соединение получали способом, аналогичным описанному в примере 25е, с заменой 2-(6-фтор-3-оксо-1-инданил)уксусной кислоты 2-(4,6-дифтор-3-оксо-1-инданил)уксусной кислотой (3,85 г, 0,017 моля). Полученный таким образом 2-(4,6-дифтор-3-оксо-1-инданил) ацетилхлорид использовали без дополнительной очистки или анализа.
е) Получение 2-(4,6-дифтор-3-оксо-1-инданил)ацетамида
Это соединение получали способом, аналогичным описанному в примере 25f, с заменой 2-(3-фтор-3-оксо-1-инданил)ацетилхлорида 2-(4,6-дифтор-3-оксо-1-инданил)уксусной кислотой (4,2 г, 17 ммолей). После хроматографии перекристаллизация дважды из смеси дихлорметан:гексан дала 2,8 г (77%) 2-(4,6-дифтор-3-оксо-1-инданил)ацетамида, т.пл. 155-157oC.
f) Получение (E)-2-(4,6-дифтор-3-оксо-1-инданилиден)ацетамида
Смесь 2-(4,6-дифтор-3-оксо-1-инданил)ацетамида (1,0 г, 0,0044 моля), N-бромсукцинимида (0,950 г, 0,00533 моля), 2,2-азобис(2- метилпропионитрила) (0,350 г, 0,00213 моля, Kodak), тетрахлорметана (50 мл) и бензола (50 мл) нагревали с масляной баней при 120oC в течение 1 часа. Реакционный раствор разбавляли дихлорметаном, суспендировали с Силикагелем 60 и летучие компоненты удаляли роторным испарением в вакууме. Этот диоксид кремния затем наносили на колонку Силикагеля 60 (51х450 мм), смоченного дихлорметаном, и продукт удаляли элюированием смесью метанол:дихлорметан (2:98). Летучие компоненты удаляли роторным испарением в вакууме, получая 0,613 г остатка. Остаток перекристаллизовывали из метанола, получая 0,302 г (31%) (E)-2-(4,6-дифтор-3-оксо-1-инданилиден)ацетамида, т.пл. 250oC (разложение).
Пример 30
Получение (E)-N-этил-2-(7-фтор-1,2,3,4-тетрагидро-1- нафтилиден)ацетамида
а) Получение 2-хлор-N-этилацетамида
Хлорацетилхлорид (45 г, 398 ммолей, Aldrich) добавляли по каплям к водному этиламину (70%, 30,7 г, 0,48 моля) в 100 мл деионизованной воды, исходно при -20oC, с перемешиванием. Температуру реакции повышали до 0oC и перемешивание продолжали, пока реакция не переставала быть экзотермической. Полученный раствор подкисляли концентрированной соляной кислотой (7 мл) и экстрагировали дихлорметаном (4х250 мл). Органический слой промывали деионизованной водой (150 мл), фильтровали через стекловату и концентрировали роторным испарением в вакууме, получая бледно-желтый жидкий остаток. Остаток концентрировали с гексаном (200 мл) и дихлорметаном (600 мл), получая 16,01 г (59%) 2-хлор-N-этилацетамида. Спектр этого соединения согласовался с предложенной структурой и соединение использовали без дальнейшей очистки.
b) Получение диэтил-N-этилкарбамоил)метил)фосфоната
2-хлор-N-этилацетамид (15,5 г, 127,5 ммоля) добавляли порциями с перемешиванием к триэтилфосфиту (28 г, 0,17 моля, Aldrich) при 110oC. Затем раствор нагревали до 155oC в течение 30 минут, охлаждали до 125oC и летучие компоненты удаляли перегонкой в вакууме водоструйного насоса (15 мм Hg) при этой температуре. Фракционная перегонка дала 25,06 г (88%) диэтил((N-этилкарбамоил)метил)фосфоната, т. кип. 135-147oC при 0,50 мм Hg. Это соединение использовали без дальнейшего анализа.
с) Получение (E)-N-этил-2-(7-фтор-1,2,3,4-тетрагидро-1- нафтилиден)ацетамида
К перемешиваемой суспензии NaH (80% дисперсия в минеральном масле, 0,91 г, 0,030 моля, Aldrich) в диметилсульфоксиде (150 мл) при комнатной температуре при атмосфере азота добавляли раствор диэтил((N-этилкарбамоил)метил)фосфоната (6,8 г, 0,030 моля) в диметилсульфоксиде (50 мл). Реакция была слегка экзотермической. Реакционную смесь перемешивали в течение 0,75 часов. Раствор 7-фтор-1-тетралона (5,00 г, 0,030 моля) в диметилсульфоксиде (50 мл) добавляли и реакцию перемешивали в течение 1 часа. Реакцию выливали в ледяную воду (300 мл) и экстрагировали диэтиловым эфиром (3х250 мл). Объединенные эфирные фазы промывали водой (100 мл) и концентрировали роторным испарением в вакууме, получая золотисто-желтый сироп. Этот остаток хроматографировали на Силикагеле 60 с применением ступенчатого градиента, идущего от смеси этилацетат: гексаны/1: 3 до смеси этилацетат:гексан 1:1. Фракции, содержащие (E)-N-этил-2-(7- фтор-1,2,3,4-тетрагидро-1-нафтилиден)ацетамид, объединяли и концентрировали роторным испарением в вакууме, получая 3,74 г белого твердого вещества. Перекристаллизация из смеси дихлорметан:гексан дала 3,28 г (46%) (E)-N-этил-2-(7-фтор- 1,2,3,4-тетрагидро-1-нафтилиден)ацетамида в виде пушистого белого твердого вещества, т.пл. 87-89oC.
Анал. рассчитано для C14H16FNO (мол. масса 233,278): С 72,08, H 6,91, N 6,00.
Найдено: C 72,05, H 6,91, N 6,05.
Пример 31
Получение (Z)-N-этил-2-(7-фтор-1,2,3,4-тетрагидро-1- нафталиден)ацетамида
Фракции из хроматографической очистки, описанной в примере 30, содержащие (Z)-N-этил-2-(7-фтор-1,2,3,4-тетрагидро- 1-нафтилиден)ацетамид, объединяли и концентрировали в вакууме, получая 1,07 г (20%) (Z)-N-этил-2-(7-фтор-1,2,3,4-тетрагидро- 1-нафтилиден)ацетамида в виде белого твердого вещества, т.пл. 117-118oC.
Анал. рассчитано для C14H16FNO (мол. масса 233,278): С 72,08, H 6,91, N 6,00.
Найдено: C 71,99, H 6,89, N 6,01.
Пример 32
Получение (E)-2-(4,6-дифтор-3-гидрокси-1-инданилиден)ацетамида
Суспензию (E)-2-(4,6-дифтор-3-оксо-1-инданилиден)ацетамида (0,100 г, 0,45 ммоля) и боргидрида натрия (0,017 г, 0,45 ммоля) в 95% этаноле перемешивали при температуре окружающей среды в течение 2 часов. Смесь охлаждали в ледяной бане и гасили 0,1 н. соляной кислотой (3 мл). Этанол выпаривали в вакууме и остаток растворяли в этилацетате (50 мл), промывали последовательно водой (2х30 мл) и солевым раствором (30 мл), сушили над сульфатом натрия, фильтровали и упаривали в вакууме. Остаток промывали последовательно холодным этилацетатом, гексаном, диэтиловым эфиром, получая 0,046 г (45%) (E)-2-(4,6-дифтор-3-гидрокси-1-инданилиден)ацетамида в виде белого твердого вещества, т. пл. 232-237oC (разложение). 1H-ЯМР (DMSO-d6): δ 7,06-7,42 (м, 4H), 6,49 (с, 1H), 5,52 (д, 1H), 5,30 (м, 1H), 3,44 (м, 1H), 3,03 (1H).
Соединения (см. табл. 1) получали способами, аналогичными описанным в указанных примерах.
Фармацевтические композиции
В следующих примерах 73-78 "Активным ингредиентом" является соединение формулы (I) или фармацевтически приемлемые соль или сольват.
Пример 73
Композиции для таблеток
Композиции A (см. табл. 2) B (см. табл. 3) и C готовили влажным гранулированием ингредиентов с раствором повидона с последующим добавлением стеарат магния и прессованием.
Композиция C - мг/таблетка
Активный ингредиент - 100
Лактоза - 200
Крахмал - 50
Повидон - 5
Стеарат магния - 4 - 359
Следующие композиции D и E готовили прямым прессованием смешанных ингредиентов. Лактоза в композиции E является лактозой типа прямого прессования (Dairy-Crest - "Zeparox").
Композиция D - мг/таблетка
Активный ингредиент - 250
Предварительно желатинированный крахмал NF15 - 150 - 400
Композиция E - мг/таблетка
Активный ингредиент - 250
Лактоза - 150
Avriel - 100 - 500
Композиция F (препарата с контролируемым выделением активного ингредиента)
Композицию готовили влажным гранулированием ингредиентов (см. ниже) с раствором повидона с последующим добавлением стеарата магния и прессованием. - мг/таблетка
(а) Активный ингредиент - 500
(b) Гидроксипропилметилцеллюлоза (Methocel K4M Premium) - 112
(с) Лактоза В.Р. - 53
(d) Повидон В.Р. - 28
(е) Стеарат магния - 7 - 700
Пример 74
Композиция для капсул
Композиция A
Композицию для капсул готовили смешиванием ингредиентов композиции D по примеру 73 и заполнением смесью твердой желатиновой капсулы. Композицию В (nifra) готовят подобным способом.
Композиция В - мг/капсула
(а) Активный ингредиент - 250
(b) Лактоза В.Р. - 143
(с) Натриевая соль гликоллата крахмала - 25
(а) Стеарат магния - 2 - 420
Композиция C - мг/капсула
(а) Активный ингредиент - 250
(b) Macrogol 4000 В.Р. - 350 - 600
Композиция D - мг/капсула
Активный ингредиент - 250
Лецитин - 100
Арахисовое масло - 100 - 450
Капсулы композиций D готовили диспергированием активного ингредиента в лецитине и арахисовом масле и заполнением этой дисперсией мягких эластичных желатиновых капсул.
Композиция E - мг/капсула
(а) Активный ингредиент - 100
(b) Лактоза - 300
(с) Стеарат магния - 2
(d) Лаурилсульфат натрия - 2
(е) Натриевая соль гликоллата крахмала - 50
(f) Тальк, USP - 25 - 479
Композицию для капсулы готовили микронизаций (микроизмельчением) активного ингредиента при помощи GEM-T Type 1047 Jet Mill и смешиванием с оставшимися ингредиентами композиции E. Полученной смесью заполняли твердые с двумя частями желатиновые капсулы.
Композиция F (капсула с контролируемым выделением)
Следующую композицию для капсул с контролируемым выделением активного ингредиента готовили экструдированием ингредиентов a, b и c при помощи экструдера с последующим образованием шарообразных частиц из экструдата и высушиванием. Высушенные осадки затем покрывали контролирующей выделение мембраной (d) и заполняли ими состоящую из двух частей твердую желатиновую капсулу. - мг/капсула
(а) Активный ингредиент - 250
(b) Микрокристаллическая целлюлоза - 125
(с) Лактоза В.Р. - 125
(а) Этилцеллюлоза - 13 - 513
Пример 75
Композиция для инъекций
Активный ингредиент - 0,200 г
95% этанол и PEG 400, 1:1
Стерильная вода - q.s. до 10 мл
Активный ингредиент растворяли в 95% этаноле и полиэтиленгликоле (PEG 400) (1:1). Раствор доводили до указанного объема водой и фильтровали через стерильный микропористый фильтр в стерильный флакон (типа 1) из янтарного стекла на 10 мл, который закрывали стерильным перекрытием и наружной крышкой.
Пример 76
Сироп
Активный ингредиент - 0,25 г
Раствор сорбита - 1,50 г
Глицерин - 2,00 г
Бензоат натрия - 0,005 г
Ароматизатор, Peach 17,42.3169 - 0,0125 мл
Очищенная вода - q.s до 5,00 мл
Активный ингредиент растворяли в смеси глицерина и большей части очищенной воды. Водный раствор бензоата натрия добавляли к раствору с последующим добавлением раствора сорбита и, наконец, ароматизатора. Объем доводили очищенной водой и хорошо перемешивали.
Пример 77
Суппозиторий - мг/суппозиторий
Активный ингредиент - 250
Твердый жир, В.Р. (Witepsol H15-Dynamit Nobel) - 1770 - 2020
1/5 Witepsol H15 расплавляли в резервуаре с паровой рубашкой при 45oC (максимум). Активный ингредиент просеивали через сито 200 мкМ и добавляли к расплавленной основе при смешивании с применением Silverson, снабженного режущей головкой, пока не образовывалась гладкая дисперсия. При поддержании смеси при 45oC остальной Witepsol H15 добавляли к суспензии и перемешивали, обеспечивая гомогенную смесь. Всю суспензию пропускали через сито из нержавеющей стали 250 мкМ и при непрерывном перемешивании давали ей остыть до 40oC. При температуре 38-40oC 2,02 г этой смеси вносили в подходящие пластиковые формы на 2 мл. Суппозиториям давали остыть до комнатной температуры.
Пример 78
Пессарии - мг/пессарий
Активный ингредиент - 250
Безводная декстроза - 380
Картофельный крахмал - 363
Стеарат магния - 7 - 1000
Указанные ингредиенты смешивали непосредственно друг с другом и пессарии готовили прямым прессованием полученной смеси.
Пример 79
Центральная миорелаксирующая активность
Центральную миорелаксирующую активность соединений формулы (I) определяли при помощи теста Straub-хвостов на основе способа, описанного K.O. Ellis, J.F. Carpenter, Neuropharm. 13, 211 (1974).
Результат теста Straub-хвостов выращивают в виде ED50 в мг/кг. ED50 определяют как дозу введенного соединения, предотвращающую Straub-хвосты у 50% мышей. Соединение вводят зондированием за 60 минут до оценки.
Возможное побочное действие этих соединений определяли при помощи rodorod-теста на мышах (с вращаемым цилиндром), описанного H.D. Novak и J.M. Zwolshei, J. Pharmacologocоl Methods, 10, 175 (1983). Результат rodorod-теста на мышах выражали в виде ED50 в мг/кг. ED50 является дозой, которая приводит к тому, что 50% животных не могут удержаться на цилиндре, вращающемся при 11 об/мин.
Антагонизм по отношению к индуцируемым морфином Straub-хвостам отражает эффективность релаксации основных мышц, в то время как неблагоприятный исход в rodorod-тесте отражает седативный эффект и нарушение координации.
Определение отношения неблагоприятного исхода в rodorod-тесте к антагонизму в отношении индуцируемых морфином Straub-хвостов является средством оценки способности к побочным эффектам релаксантов основных мышц (J.D. Novak, Drug Del. Res. 2, 383 (1982) (см. табл. 4).
Пример 80
Противосудорожная активность
Противосудорожную активность соединений формулы (I) определяли при помощи способа, описанного Mehta et al., J. Med. Chem., 24, 465 (1981).
Противосудорожную активность выражали как ED50 в мг/кг. ED50 для защиты против максимальных индуцируемых электрошоком судорог была дозой, которая предотвращала выпрямление задней конечности у 50% животных. ED50 для защиты против индуцируемых Метразолом судорог была дозой, которая предотвращала судороги у 50% животных (см. табл. 5).
Пример 81
Анксиолитическая активность
Анксиолитическую активность соединений формулы (I) измеряли при помощи способа Geller and Seifter, J. Psycho-pharmacologia, 1, 482 (1960) в модификации Pollard and Howard, Psychopharmacology, 62, 117 (1979). Клинически эффективные анксиолитики увеличивают подавленную ответную реакцию. Анксиолитическая активность соединения выражается как самая низкая доза, необходимая для значительного увеличения подавленной ответной реакции в крысах (MED).
Соединение примера N - р. о. MED мг/кг
13 - 3,13 р.о.
MED - минимальная эффективная доза.
Пример 82
Противовоспалительная активность
Соединения формулы (I) обладают противовоспалительной активностью, как показано при помощи модификации стандартного теста индуцированного каррагенаном плеврита, описанного R. Vinegar, J.F. Traux and J.Z. Selph (Pro. Soc. Exp. Biol. Med. 143:711-714, 1973). В этих экспериментах использовали Zewis самцов крыс, весом 160-180 г, разделенных на группы, состоящие из 5 животных. Тестируемые соединения давали голодающим крысам кормлением через желудочный зонд за 0,5 часа перед интраплевральной инъекцией 50 мг каррагенана. После 4 часов экссудат плевры собирали и определяли объем отека и число клеток. ED50 величины определяли линейным регрессионным анализом и эти величины представляли дозы, при которых введенное лекарственное средство давало 50% ингибирование индуцированного каррагенаном накопления клеток и образования отека в плевральной полости крысы (см. табл. 6).
Пример 83
Вызванный адъювантом артрит
Соединения формулы (I) проявляют также хроническую противовоспалительную активность, как показано ингибированием вызываемого адъювантом полиартрита в крысах. Процедура для этого теста была детально описана R. Vinegar, J.F. Truax, J.Z. Selph, A Zea and P.R. Johnston (J. Immunopharmacol. 1: 497-520, 1979). В этих экспериментах использовали самок Zewis крыс с исходным весом 190±10 г. Артритных крыс разделяли на группы, состоящие из 6 животных. Получающим пищу крысам дозировали при помощи зондирования через рот тест-соединения, начиная с 21-го дня после инъекции адъюванта, терапию продолжали до 28-го дня. Частоту и тяжесть артритных повреждений оценивали при помощи модификации процедуры оценки, описанной H.Z.F. Currey and M. Ziff (J. Exp. Med. 121: 185-203, 1968). Вкратце, билатеральные суставы оценивали на отек, эритему и анкилоз, как представлено в табл. 7.
Максимальный возможный балл на крысу был 40. Экспериментальные результаты анализировали одно-факторным ANOVA с последующими post hoc сравнениями терапевтических эффектов по сравнению с необработанными артритными контролями с применением теста Newman-Keuls. Процентное ингибирование каждой обрабатываемой лекарственными средствами группы рассчитывали из среднего значения по отношению к артритному контролю. Соединения примера N 5 значимо (p < 0,01) понижали баллы артрита на 22-ой, 25-ый и 27-ой дни у крыс с вызванным адъювантом артритом при дозах b.i.d.c 50 мг/кг. Массу селезенки и фибриноген плазмы измеряли после смерти на 27-ой день, и эти показатели были значимо понижены (p < 0,01).
Пример 84
Легкая аналгезия
Соединения формулы (I) обладают легкой аналгетической активностью, как показано при помощи модификации теста индуцируемой трипсином гипералгезии задних конечностей крыс, описанного R. Vinegar, J.F. Truax, J. Z. Selph and P. R. Johuston (J. Pharmacol. Meth. 23: 51-61, 1990). В этих исследованиях использовали Zewis самцов весом 160-180 г, которых делили на группы, состоящие из 5-6 животных. Тестируемые соединения давали получавшим пищу крысам зондированием через рот за 0,5 часа перед субплантарной инъекцией 250 мг трипсина в одну заднюю конечность. Спустя час крыс оценивали на гипералгезию при помощи F-образного зажима на мататарзальной зоне инъецированной задней конечности. Определяли латентное состояние (секунды) по болевой ответной реакции (голос или обращение в бегство), причем максимальный латентный период был 4 секунды. Величины ED50 оценивали при помощи линейного регрессионного анализа, и они представляли дозу, при которой данное лекарственное средство давало 50% ингибирование, с применением формулы: (4 сек - контрольный латентный период) - (4 сек - тестируемый латентный период)/4 сек - контрольный латентный период х 100.
Соединение примера N - р.о. ED50, мг/кг
5 - 4,0
Пример 85
Сильная аналгезия
Соединения формулы (I) обладают сильной аналгетической активностью, как показано при помощи болевого теста для фаланг (модификации теста индуцируемой трипсином гипералгезии задних конечностей, описанного R.Vinegar, J.F. Truax, J.Z. Selph and P.R. Johnston (J. Pharmacol. Meth. 23: 51-61, 1990)). В этих исследованиях использовали самцов Zewis крыс весом 160-180 г, которых делили на группы из 5-6 животных. Болевой тест для фаланг является болевым тестом (без гипералгезии), в котором тестируемые соединения дают получающим пищу крысам зондированием через рот. Спустя 1 час F-образные механические зажимы надевали на фаланги одной задней конечности, которые инициировали болевую ответную реакцию (голосовую или обращение в бегство). Латентный период (секунды) до болевой реакции определяли с 3 секундами максимально дозволенного периода времени. ED50 величины определяли линейным регрессионным анализом, и они представляли дозу, при которой данное соединение увеличивало латентный период, давая 50% ингибирование, с применением формулы: (3 сек - контрольный латентный период) - (3 сек - тестируемый латентный период)/3 сек - контрольный латентный период х 100.
Соединение примера N - р.о. ED50, мг/кг
5 - 20
Пример 86
Данные о токсичности
(i) Соединение примера 1
Однократные дозы (15, 45 или 250 мг/кг) вводили зондированием через рот группам 4 получающих пищу самцов CD-1 мышей (Charles River). Максимальная переносимая доза была более 250 мг/кг, так как не наблюдали смерти животных в течение 7 дней после дозирования.
(ii) Соединение примера 5
Однократные дозы (5, 15, 40, 100, 250, 500 или 1000 мг/кг) вводили зондированием через рот группам 4 получавших пищу самцов CD-1 мышей (Charles River). Максимальная переносимая доза была более 1000 мг/кг, так как не было смерти животных в течение 7 дней после дозирования.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ КАРБОЦИКЛИЧЕСКИЕ АМИДЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОИЗВОДНЫЕ ИНДАНА, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ И СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ | 1995 |
|
RU2145954C1 |
| ЭНАНТИОМЕРНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1992 |
|
RU2091386C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНЫХ СОЕДИНЕНИЙ ИЛИ ИХ ПРОИЗВОДНЫХ | 1990 |
|
RU2068849C1 |
| СПОСОБ ПОЛУЧЕНИЯ АМИДОВ НЕНАСЫЩЕННЫХ КИСЛОТ | 1989 |
|
RU2010025C1 |
| ЗАМЕЩЕННЫЕ БЕНЗИМИДАЗОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ОБЛАДАЮЩАЯ АНТИВИРУСНЫМ ДЕЙСТВИЕМ | 1993 |
|
RU2141952C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИАРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 1989 |
|
RU2036901C1 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМКОМПОЗИЦИЯ | 1989 |
|
RU2121998C1 |
| МОНО-, ДИ- ИЛИ ТРИСЛОЖНЫЕ ЭФИРЫ 2-АМИНО-6-(С - С-АЛКОКСИ-9-(βD-АРАБИНОФУРАНОЗИЛ)-9Н-ПУРИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2114860C1 |
| Способ получения амидов ненасыщенных кислот | 1987 |
|
SU1695825A3 |
| ПРИМЕНЕНИЕ 5-ЭТИНИЛУРАЦИЛА В КАЧЕСТВЕ ИНАКТИВАТОРА УРАЦИЛРЕДУКТАЗЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЕЕ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ УСИЛЕНИЯ ДЕЙСТВИЯ ИЛИ СНИЖЕНИЯ ТОКСИЧНОСТИ 5-ФТОРУРАЦИЛА | 1991 |
|
RU2194511C2 |








Описываются новые производные бициклических амидов общей формулы I, где R1, R2, R3 и R4 выбраны (каждый) из водорода и фтора и, по меньшей мере, один и не более двух из них представляют собой фтор, R5 выбран из водорода и С1-С4-алкила, R6 выбран из водорода, С1-С4-алкила и гидроксигруппы или R5 и R6 вместе с углеродом кольца образуют карбонильную группу, R7 выбран из водорода и гидроксигруппы, R8 и R9 выбраны, каждый, из водорода С1-С4-алкила и цикло (С3 или С4)алкила или вместе с азотом образуют морфолиногруппу, их соли и сольваты. Описываются также фармацевтические композиции на основе соединений формулы I, которые находят применение в медицине, в частности, в качестве релаксантов основных мышц. Описывается также способ лечения патологических и воспалительных состояний. Соединения формулы I обладают значительно пониженной способностью вызывать седативный эффект и нарушение координации по сравнению с известными средствами. 10 с. и 21 з.п. ф-лы, 7 табл.


где R1, R2, R3 и R4 выбраны, каждый, из водорода и фтора, и, по меньшей мере, один и не более двух из них представляют собой фтор;
R5 выбран из водорода и C1-C4-алкила;
R6 выбран из водорода, C1-C4-алкила и гидроксигруппы
или R5 и R6 вместе с углеродом кольца образуют карбонильную группу;
R7 выбран из водорода и гидроксигруппы;
R8 и R9 выбраны, каждый, из водорода, C1-C4-алкила и цикло(C3 или C4)алкила или вместе с азотом образуют морфолиногруппу.
1. (E)-2-(6-фтор-3-метил-1-инданилиден)ацетамид,
2. (E)-N-циклопропил-2-(6-фтор-3-метил-1-инданилиден) ацетамид,
3. (E)-2-(6-фтор-3,3-диметил-1-инданилиден)-N-метилацетамид,
4. (E)-N-циклопропил-2-(6-фтор-3-этил-1-инданилиден)ацетамид,
5. (E)-N-циклопропил-2-(5,6-дифтор-1-инданилиден)ацетамид,
6. (E)-2-(5,6-дифтор-1-инданилиден)-N-метилацетамид,
7. (E)-2-(5,7-дифтор-1-инданилиден)-ацетамид,
8. (E)-N-циклопропил-2-(4,6-дифтор-1-инданилиден)ацетамид,
9. (E)-2-(4,6-дифтор-1-инданилиден)-N-изопропилацетамид,
10. (E)-2-(4,6-дифтор-1-инданилиден)-N,N-диметилацетамид,
11. (Z)-2-(4,6-дифтор-2-гидрокси-1-инданилиден)ацетамид,
12. (E)-2-(4,6-дифтор-1-инданилиден)ацетамид,
13. (E)-2-(6-фтор-1-инданилиден)ацетамид,
14. (Z)-2-(6-фтор-2-гидрокси-1-инданилиден)ацетамид,
15. (E)-2-(6-фтор-3,3-диметил-1-инданилиден)ацетамид,
16. (E)-2-(6-фтор-3-этил-1-инданилиден)-N,N-диметилацетамид,
17. (E)-2-(6-фтор-3-гидрокси-1-инданилиден)ацетамид или их соли или сольваты.
| СПОСОБ ОЛИГОМЕРИЗАЦИИ НИЗШИХ ОЛЕФИНОВ В ГАЗОЖИДКОСТНОЙ ФАЗЕ | 1999 |
|
RU2177929C2 |
| Формирователь прямоугольных импульсов | 1983 |
|
SU1241438A1 |
| СПОСОБ ПОЛУЧЕНИЯ МОДИФИЦИРОВАННЫХ ИЗДЕЛИЙ ИЗ ПОЛИАКРИЛОНИТРИЛА | 0 |
|
SU218373A1 |
| Устройство для сушки керамических и т.п. изделий | 1956 |
|
SU114374A3 |
| Способ получения замещенных 2-алкилиден-7-диэтиламино-2Н-1-бензопиранов | 1987 |
|
SU1505942A1 |
| Способ получения амидов ненасыщенных кислот | 1987 |
|
SU1695825A3 |
| US 5075337 A, 24.12.91. | |||

