Область, к которой относится изобретение
[0001] Настоящее изобретение относится к пиразолопиридиновому производному или его фармакологически приемлемой соли, которое обладает отличным лецитин-холестерин-ацетилтрансфераза(далее указывается как LCAT)-активирующим эффектом (предпочтительно, обратимым LCAT-активирующим эффектом).
Предпосылки изобретения
[0002] Сердечно-сосудистые заболевания (например, сердечное заболевание, цереброваскулярное заболевание и почечное заболевание), вызванные гипертензией, дислипидемией, сахарным диабетом или т.п., являются серьезными проблемами для развитых стран. Антигипертензивные, антидислипидемические и антидиабетические лекарственные средства используют для лечения заболеваний гипертензии, дислипидемии и гипергликемии, соответственно. В клинических условиях α и β блокаторы, диуретики, антагонисты кальция, ингибиторы ACE и A-II антагонисты и т.д. используют в качестве антигипертензивных лекарственных средств; ингибиторы HMG-CoA редуктазы, анионообменные смолы, производные никотиновой кислоты, пробукол и фибраты и т.д. используют в качестве антидислипидемических лекарственных средств; и инсулины, сульфонилмочевинные средства, метформин, глитазоны и ингибиторы DPP4 и т.д. используют в качестве антидиабетических лекарственных средств. Эти лекарственные средства способствуют регуляции кровяного давления или уровней липидов или глюкозы в крови. Тем не менее даже применение этих лекарственных средств не привело к существенному улучшению показателей смертности, связанных с сердечным заболеванием, цереброваскулярным заболеванием и почечным заболеванием. Таким образом, существует потребность в разработке более лучших терапевтических средств для лечения этих заболеваний.
[0003] Непосредственным фактором риска сердечно-сосудистых заболеваний является атеросклероз, связанный с утолщением стенки артерии. Это утолщение вызвано образованием бляшек в результате аккумуляции окисленного холестерина липопротеинов низкой плотности (далее указываются как ЛПНП) в макрофагах и т.п. в артериальной стенке (не-патентные документы 1 и 2). Эти атеросклеротические бляшки ингибируют кровоток и способствуют образованию кровяных сгустков.
[0004] Результаты многих эпидемиологических исследований показывают, что концентрации липопротеинов в сыворотке связаны с заболеваниями, такими как дислипидемия и артериосклероз (например, не-патентный документ 3). Как повышенная концентрация ЛПНП холестерина в крови, так и пониженная концентрация холестерина липопротеинов высокой плотности (далее указывается как ЛПВП) в крови являются факторами риска коронарных заболеваний.
[0005] В периферических тканях ЛПВП способствуют оттоку холестерина, который, в свою очередь, этерифицируется посредством LCAT на ЛПВП с образованием холестерилового сложного эфира. Повышенная активность LCAT способствует оттоку холестерина из макрофагов (например, не-патентные документы 4 и 5). Соответственно, лекарственные средства, которые повышают активность LCAT, считаются полезными в качестве лекарственных средств для лечения или профилактики заболеваний, таких как дислипидемия и артериосклероз.
[0006] Пептидное соединение (например, не-патентный документ 6) и, например, соединение, описанное в патентном документе 1 в виде малой молекулы, известны в качестве таких лекарственных средств, которые повышают активность LCAT.
[0007] Соединение, описанное в патентном документе 2, известно как соединение, имеющее пиразолопиридиновый скелет. Патентный документ 2 не содержит никакого указания относительно LCAT-активирующего эффекта, хотя в этой литературе раскрывается действие, направленное против рецептора LPA.
Перечень цитируемых документов
Патентные документы
[0008] Патентный документ 1: WO2008/002591
Патентный документ 2: WO2012/028243
Не-патентные документы
[0009] Не-патентный документ 1: Ross, R., Annu. Rev. Physiol. 1995, Vol. 57, p. 791-804
Не-патентный документ 2: Steinberg, D., J. Biol. Chem. 1997, Vol. 272, p. 20963-20966
Не-патентный документ 3: Badimon, J. Clin. Invest., 1990, Vol. 85, p. 1234-1241
Не-патентный документ 4: Matsuura, F., J. Clin. Invest. 2006, Vol. 116, p. 1435-1442
Не-патентный документ 5: Yvan-Charvet, L., Arterioscler. Thromb. Vasc. Biol. 2007, Vol. 27, p. 1132-1138
Не-патентный документ 6: Iwata, A., Атеросклероз. 2011, Vol. 218, p. 300-307
Сущность изобретения
Техническая проблема
[0010] Известные в настоящее время соединения, обладающие LCAT-активирующим эффектом, что касается их безопасности и эффективности, являются менее чем удовлетворительными. Таким образом, существует настоятельная потребность в активаторах LCAT, обладающих отличными свойствами безопасности и эффективности.
Решение проблемы
[0011] Авторы настоящего изобретения осуществили различные способы синтеза и исследования с целью получения нового анти-артериосклеротического лекарственного средства, которое обладает отличным LCAT-активирующим эффектом и непосредственно способствует оттоку холестерина из макрофагов. В результате, авторы настоящего изобретения создали настоящее изобретение, обнаружив, что пиразолопиридиновое производное, имеющее определенную структуру, или его фармакологически приемлемая соль обладает отличным LCAT-активирующим эффектом.
[0012] Настоящее изобретение предоставляет пиразолопиридиновое производное или его фармакологически приемлемую соль, которое обладает отличным LCAT-активирующим эффектом (предпочтительно, обратимым LCAT-активирующим эффектом), и лекарственное средство, включающее такое производное.
[0013] Более конкретно, настоящее изобретение относится к:
(1) соединению, представленному общей формулой (I), или его фармакологически приемлемой соли:
[0014]
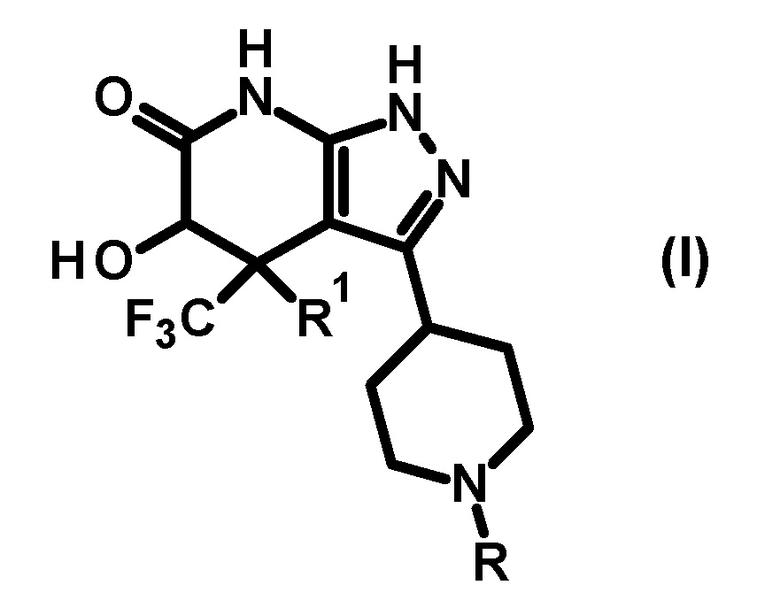
[0015] где R представляет собой необязательно замещенную арильную группу (заместитель(заместители) представляет(представляют) собой 1-3 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из атома галогена, C1-6 алкильной группы, C3-7 циклоалкильной группы, трифторметильной группы, дифторметокси группы, трифторметокси группы, циано группы, C1-6 алкокси группы, C3-7 циклоалкокси группы, фенильной группы, C2-7 алкоксикарбонильной группы, бензилоксикарбонильной группы, ди(C1-6 алкил)аминокарбонильной группы и ди(C1-6 алкил)амино группы) или
необязательно замещенную гетероарильную группу (гетероарил представляет собой 5- или 6-членное кольцо; гетероатом(гетероатомы) на кольце гетероарильной группы представляет(представляют) собой 1 или 2 атома азота, и кольцо необязательно дополнительно содержит один атом азота, атом кислорода или атом серы; и (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из атома галогена, C1-6 алкильной группы, C3-7 циклоалкильной группы, трифторметильной группы, дифторметокси группы, трифторметокси группы, циано группы, C1-6 алкокси группы, C3-7 циклоалкокси группы, фенильной группы, C2-7 алкоксикарбонильной группы, бензилоксикарбонильной группы, ди(C1-6 алкил)аминокарбонильной группы и ди(C1-6 алкил)амино группы), и R1 представляет собой атом водорода или гидрокси группу;
(2) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой необязательно замещенную арильную группу (заместитель(заместители) представляет(представляют) собой 1-3 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из атома галогена, C1-6 алкильной группы, C3-7 циклоалкильной группы, трифторметильной группы, дифторметокси группы, трифторметокси группы, циано группы, C1-6 алкокси группы, C3-7 циклоалкокси группы, фенильной группы, C2-7 алкоксикарбонильной группы, бензилоксикарбонильной группы, ди(C1-6 алкил)аминокарбонильной группы и ди(C1-6 алкил)амино группы);
(3) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой замещенную арильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из атома хлора, атома фтора, C1-3 алкильной группы, трифторметильной группы, дифторметокси группы, трифторметокси группы, циано группы и C1-3 алкокси группы);
(4) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой замещенную фенильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из атома хлора, дифторметокси группы, трифторметокси группы и циано группы);
(5) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой замещенную фенильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из дифторметокси группы, трифторметокси группы и циано группы);
(6) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой необязательно замещенную гетероарильную группу (гетероарил представляет собой 5- или 6-членное кольцо; гетероатом(гетероатомы) на кольце гетероарильной группы представляет(представляют) собой 1 или 2 атома азота, и кольцо необязательно дополнительно содержит один атом азота, атом кислорода или атом серы; и (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из атома галогена, C1-6 алкильной группы, C3-7 циклоалкильной группы, трифторметильной группы, дифторметокси группы, трифторметокси группы, циано группы, C1-6 алкокси группы, C3-7 циклоалкокси группы, фенильной группы, C2-7 алкоксикарбонильной группы, бензилоксикарбонильной группы, ди(C1-6 алкил)аминокарбонильной группы и ди(C1-6 алкил)амино группы);
(7) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой замещенную гетероарильную группу (гетероарил представляет собой 5- или 6-членное кольцо; гетероатом на кольце гетероарильной группы представляет собой один атом азота, и кольцо необязательно дополнительно содержит один атом азота, атом кислорода или атом серы; и (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из атома галогена, C1-3 алкильной группы, C3-6 циклоалкильной группы, трифторметильной группы, дифторметокси группы, трифторметокси группы, циано группы, C1-3 алкокси группы, C2-4 алкоксикарбонильной группы и бензилоксикарбонильной группы);
(8) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой замещенную пиридильную, пиримидильную, пиразинильную, пиридазинильную, тиадиазолильную или тиазолильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из атома хлора, атома фтора, C1-3 алкильной группы, циклопропильной группы, трифторметильной группы, дифторметокси группы, трифторметокси группы, циано группы, C1-3 алкокси группы, C2-4 алкоксикарбонильной группы и бензилоксикарбонильной группы);
(9) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой замещенную пиридильную, пиримидильную, пиразинильную или пиридазинильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из изопропильной группы, трифторметильной группы, дифторметокси группы, циано группы и изопропокси группы);
(10) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой пиридильную, пиримидильную, пиразинильную или тиадиазолильную группу, замещенную трифторметильной группой;
(11) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой пиридильную, пиримидильную или пиразинильную группу, замещенную трифторметильной группой;
(12) соединению в соответствии с любым из пунктов (1)-(11) или его фармакологически приемлемой соли, где R1 представляет собой атом водорода;
(13) соединению в соответствии с пунктом (12) или его фармакологически приемлемой соли, где соединение или соль выбраны из группы, состоящей из следующих:
5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-3-[1-(5-изопропоксипиридин-2-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридазин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-3-{1-[2-изопропил-6-(трифторметил)пиримидин-4-ил]пиперидин-4-ил}-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиразин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-4-(трифторметил)-3-{1-[2-(трифторметил)пиримидин-5-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
6-{4-[5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-4-(трифторметил)пиридин-3-карбонитрил,
3-{1-[3-хлор-5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-4-(трифторметил)-3-{1-[4-(трифторметил)-1,3-тиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-4-(трифторметил)-3-{1-[4-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
3-[1-(5-хлорпиридин-2-ил)пиперидин-4-ил]-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиримидин-4-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-3-[1-(6-изопропоксипиридазин-3-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-3-[1-(6-изопропоксипиридазин-3-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он и
3-[1-(2-циклопропилпиримидин-5-ил)пиперидин-4-ил]-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он;
(14) соединению в соответствии с пунктом (12) или его фармакологически приемлемой соли, где соединение или соль выбраны из группы, состоящей из следующих:
(+)-цис-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридазин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиразин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-5-гидрокси-4-(трифторметил)-3-{1-[2-(трифторметил)пиримидин-5-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-6-{4-[5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-4-(трифторметил)пиридин-3-карбонитрил,
(+)-цис-3-[1-(5-хлорпиридин-2-ил)пиперидин-4-ил]-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиримидин-4-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-5-гидрокси-3-[1-(6-изопропоксипиридазин-3-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-5-гидрокси-3-[1-(6-изопропоксипиридазин-3-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он и
(+)-цис-3-[1-(2-циклопропилпиримидин-5-ил)пиперидин-4-ил]-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он;
(15) соединению в соответствии с любым из пунктов (1)-(11) или его фармакологически приемлемой соли, где R1 представляет собой гидрокси группу;
(16) соединению в соответствии с пунктом (15) или его фармакологически приемлемой соли, где соединение или соль выбраны из группы, состоящей из следующих:
4,5-дигидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он и
4,5-дигидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиразин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он;
(17) соединению в соответствии с пунктом (15) или его фармакологически приемлемой соли, где соединение или соль выбраны из группы, состоящей из следующих:
(+)-4,5-дигидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он и
(+)-4,5-дигидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиразин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он;
(18) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой замещенную фенильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из атома хлора, дифторметокси группы, трифторметокси группы и циано группы), и R1 представляет собой атом водорода;
(19) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой замещенную фенильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из дифторметокси группы, трифторметокси группы и циано группы), и R1 представляет собой атом водорода;
(20) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой замещенную пиридильную, пиримидильную, пиразинильную, пиридазинильную, тиадиазолильную или тиазолильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из атома хлора, атома фтора, C1-3 алкильной группы, циклопропильной группы, трифторметильной группы, дифторметокси группы, трифторметокси группы, циано группы, C1-3 алкокси группы, C2-4 алкоксикарбонильной группы и бензилоксикарбонильной группы), и R1 представляет собой атом водорода;
(21) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой замещенную пиридильную, пиримидильную, пиразинильную или пиридазинильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из изопропильной группы, трифторметильной группы, дифторметокси группы, циано группы и изопропокси группы), и R1 представляет собой атом водорода;
(22) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой пиридильную, пиримидильную или пиразинильную группу, замещенную трифторметильной группой, и R1 представляет собой атом водорода;
(23) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой замещенную фенильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из атома хлора, дифторметокси группы, трифторметокси группы и циано группы), и R1 представляет собой гидрокси группу;
(24) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой замещенную фенильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из дифторметокси группы, трифторметокси группы и циано группы), и R1 представляет собой гидрокси группу;
(25) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой замещенную пиридильную, пиримидильную, пиразинильную, пиридазинильную, тиадиазолильную или тиазолильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из атома хлора, атома фтора, C1-3 алкильной группы, циклопропильной группы, трифторметильной группы, дифторметокси группы, трифторметокси группы, циано группы, C1-3 алкокси группы, C2-4 алкоксикарбонильной группы и бензилоксикарбонильной группы), и R1 представляет собой гидрокси группу;
(26) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой замещенную пиридильную, пиримидильную, пиразинильную или пиридазинильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из изопропильной группы, трифторметильной группы, дифторметокси группы, циано группы и изопропокси группы), и R1 представляет собой гидрокси группу;
(27) соединению в соответствии с пунктом (1) или его фармакологически приемлемой соли, где R представляет собой пиридильную, пиримидильную или пиразинильную группу, замещенную трифторметильной группой, и R1 представляет собой гидрокси группу;
(28) соединению в соответствии с любым из пунктов (1)-(13) и (15)-(27) или его фармакологически приемлемой соли, где трифторметильная группа в 4-положении пиразолопиридинового кольца и гидрокси группа в 5-положении этого кольца находятся в цис-положении относительно друг друга;
(29) соединению в соответствии с любым из пунктов (1)-(13), (15), (16) и (18)-(28) или его фармакологически приемлемой соли, где оптическое вращение представляет собой (+);
(30) фармацевтической композиции, содержащей соединение в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемую соль в качестве активного ингредиента;
(31) фармацевтической композиции для профилактики или лечения артериосклероза, артериосклеротического сердечного заболевания, коронарного сердечного заболевания, цереброваскулярного заболевания, заболевания периферических сосудов, дислипидемии, гипо-ЛПВП-холестеринемии, гипер-ЛПНП-холестеринемии или почечного заболевания, содержащей соединение в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемую соль в качестве активного ингредиента;
(32) профилактическому или терапевтическому средству от артериосклероза, содержащему соединение в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемую соль в качестве активного ингредиента;
(33) профилактическому или терапевтическому средству от дислипидемии, содержащему соединение в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемую соль в качестве активного ингредиента;
(34) профилактическому или терапевтическому средству от заболевания, вызванного повышенной концентрацией ЛПНП холестерина в крови, содержащему соединение в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемую соль в качестве активного ингредиента;
(35) профилактическому или терапевтическому средству от заболевания, вызванного пониженной концентрацией ЛПВП-холестерина в крови, содержащему соединение в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемую соль в качестве активного ингредиента;
(36) активатору LCAT, содержащему соединение в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемую соль в качестве активного ингредиента;
(37) обратимому активатору LCAT, содержащему соединение в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемую соль в качестве активного ингредиента;
(38) анти-артериосклеротическому средству, содержащему соединение в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемую соль в качестве активного ингредиента;
(39) способу активации LCAT, включающему введение человеку эффективного количества соединения в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемой соли;
(40) способу профилактики или лечения заболевания, включающему введение человеку эффективного количества соединения в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемой соли;
(41) способу профилактики или лечения артериосклероза, включающему введение человеку эффективного количества соединения в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемой соли;
(42) способу профилактики или лечения дислипидемии, включающему введение человеку эффективного количества соединения в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемой соли;
(43) способу профилактики или лечения заболевания, вызванного повышенной концентрацией ЛПНП холестерина в крови, включающему введение человеку эффективного количества соединения в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемой соли;
(44) способу профилактики или лечения заболевания, вызванного пониженной концентрацией ЛПВП-холестерина в крови, включающему введение человеку эффективного количества соединения в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемой соли;
(45) соединению в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемой соли для применения в способе лечения или профилактики артериосклероза;
(46) соединению в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемой соли для применения в способе лечения или профилактики дислипидемии;
(47) соединению в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемой соли для применения в способе лечения или профилактики заболевания, вызванного повышенной концентрацией ЛПНП холестерина в крови; и
(48) соединению в соответствии с любым из пунктов (1)-(29) или его фармакологически приемлемой соли для применения в способе лечения или профилактики заболевания, вызванного пониженной концентрацией ЛПВП-холестерина в крови.
[0016] Далее представлено определение заместителей в соединении (I) по настоящему изобретению.
[0017] Соединение (I) по настоящему изобретению охватывает как соединение, представленной формулой (I), так и соединение, представленное формулой, которая представляет его таутомер:
[0018]
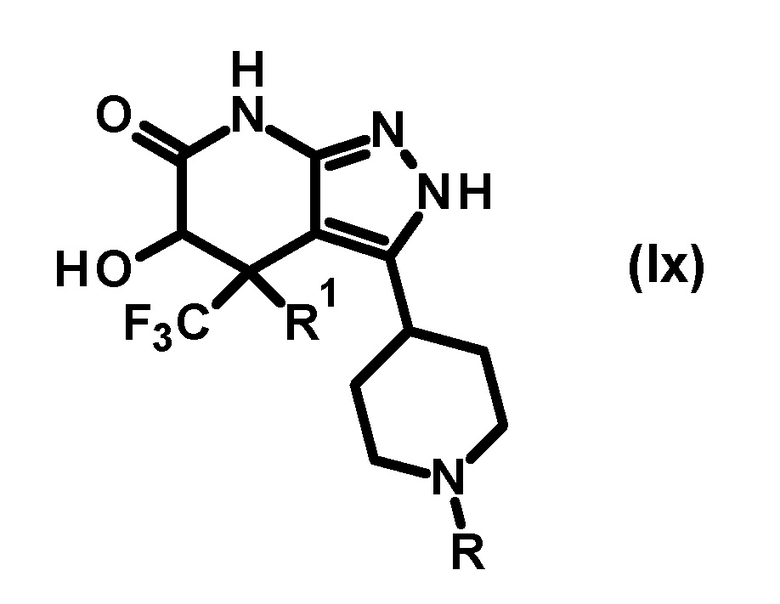
[0019] В настоящей заявке, соединение (I), включая любой такой таутомер, также представлено структурной формулой (I) и его соответствующим химическим названием для удобства, если не указано иное. Соединение (I), раскрытое в настоящей заявке, также охватывает любой изомер дополнительного таутомера (амид-имид кислоты) соединения (I) по настоящему изобретению. В настоящей заявке, соединение (I), включая любой такой изомер, также представлено структурной формулой (I) и его соответствующим химическим названием для удобства.
[0020] В соединении (I) по настоящему изобретению, "арильная группа" представляет собой, например, фенильную группу или нафтильную группу, и предпочтительно представляет собой фенильную группу.
[0021] В соединении (I) по настоящему изобретению, "атом галогена" относится к атому фтора, атому хлора, атому брома или атому иода, и предпочтительно представляет собой атом фтора или атом хлора, более предпочтительно атом хлора.
[0022] В соединении (I) по настоящему изобретению, "C1-6 алкильная группа" относится к линейной или разветвленной насыщенной углеводородной группе, содержащей от 1 до 6 атомов углерода. Примеры такой группы могут включать метильную группу, этильную группу, пропильную группу, изопропильную группу, бутильную группу, втор-бутильную группу, трет-бутильную группу, изобутильную группу, пентильную группу и гексильную группу. C1-6 алкильная группа предпочтительно представляет собой линейную или разветвленную насыщенную углеводородную группу, содержащую от 1 до 3 атомов углерода (C1-3 алкильную группу), более предпочтительно метильную группу.
[0023] В соединении (I) по настоящему изобретению, "C3-7 циклоалкильная группа" относится к циклической насыщенной углеводородной группе, содержащей от 3 до 7 атомов углерода, такой как циклопропильная группа, циклобутильная группа, циклопентильная группа или циклогексильная группа, и предпочтительно представляет собой циклическую насыщенную углеводородную группу, содержащую от 3 до 6 атомов углерода (C3-6 циклоалкильную группу), более предпочтительно циклопропильную группу.
[0024] В соединении (I) по настоящему изобретению, "C1-6 алкокси группа" относится к атому кислорода, связанному указанной выше "C1-6 алкильной группой". Примеры такой группы могут включать метокси группу, этокси группу, пропокси группу, изопропокси группу и бутокси группу. C1-6 алкокси группа предпочтительно представляет собой атом кислорода, связанный вышеуказанной "C1-3 алкильной группой" (C1-3 алкокси группа), более предпочтительно метокси группу.
[0025] В соединении (I) по настоящему изобретению, "C3-7 циклоалкокси группа" относится к атому кислорода, связанному вышеуказанной "C3-7 циклоалкильной группой". Примеры такой группы могут включать циклопропилокси группу, циклобутилокси группу, циклопентилокси группу, циклогексилокси группу и циклогептилокси группу.
[0026] В соединении (I) по настоящему изобретению, "C2-7 алкоксикарбонильная группа" относится к карбонильной группе, связанной вышеуказанной "C1-6 алкокси группой". Примеры такой группы могут включать метоксикарбонильную группу, этоксикарбонильную группу, пропоксикарбонильную группу и бутоксикарбонильную группу. C2-7 Алкоксикарбонильная группа предпочтительно представляет собой карбонильную группу, связанную вышеуказанной "C1-3 алкокси группой" (C2-4 алкоксикарбонильная группа), более предпочтительно метоксикарбонильную группу или этоксикарбонильную группу.
[0027] В соединении (I) по настоящему изобретению, "ди(C1-6 алкил)амино группа" относится к амино группе, связанной двумя одинаковыми или отличными друг от друга вышеуказанными "C1-6 алкильными группами". Ди(C1-6 алкил)амино группа предпочтительно представляет собой диметиламино группу.
[0028] В соединении (I) по настоящему изобретению, "ди(C1-6 алкил)аминокарбонильная группа" относится к карбонильной группе, связанной вышеуказанной "ди(C1-6 алкил)амино группой". Ди(C1-6 алкил)аминокарбонильная группа предпочтительно представляет собой диметиламинокарбонильную группу.
[0029] В соединении (I) по настоящему изобретению, "гетероарильная группа (гетероарил представляет собой 5- или 6-членное кольцо; гетероатом(гетероатомы) на кольце гетероарильной группы представляет(представляют) собой 1 или 2 атома азота, и кольцо необязательно дополнительно содержит один атом азота, атом кислорода или атом серы)" может представлять собой, например, пиридильную группу, пиразинильную группу, пиримидильную группу, пиридазинильную группу, оксазолильную группу, тиазолильную группу, изоксазолильную группу, изотиазолильную группу, пиррольную группу, пиразолильную группу, имидазолильную группу, триазолильную группу или тиадиазолильную группу. Гетероарильная группа предпочтительно представляет собой 5- или 6-членную гетероарильную группу (гетероатом на гетероарильном кольце представляет собой один атом азота; и кольцо необязательно дополнительно содержит один атом азота, атом кислорода или атом серы), более предпочтительно пиридильную группу, пиримидильную группу, пиразинильную группу, пиридазинильную группу, тиадиазолильную группу или тиазолильную группу, еще более предпочтительно пиридильную группу, пиримидильную группу, пиразинильную группу, пиридазинильную группу или тиадиазолильную группу, еще более предпочтительно пиридильную группу, пиримидильную группу, пиразинильную группу или тиадиазолильную группу, особенно предпочтительно пиридильную группу, пиримидильную группу или пиразинильную группу.
[0030] Соединение (I) по настоящему изобретению содержит основную группу и поэтому может образовывать кислотно-аддитивную соль с фармакологически приемлемой кислотой. В настоящем изобретении примеры "его фармакологически приемлемой соли" могут включать: гидрогалогениды, такие как гидрофторид, гидрохлорид, гидробромид и гидроиодид; соли неорганических кислот, такие как нитрат, перхлорат, сульфат и фосфат; низшие алкансульфонаты, такие как метансульфонат, трифторметансульфонат и этансульфонат; арилсульфонаты, такие как бензолсульфонат и п-толуолсульфонат; соли органических кислот, такие как ацетат, малат, фумарат, сукцинат, цитрат, тартрат, оксалат и малеат; и соли аминокислот, такие как соль орнитина, глутамат и аспартат.
[0031] Соединение (I) по настоящему изобретению или его фармакологически приемлемая соль, когда их оставляют на воздухе, могут образовывать гидрат, абсорбируя воду. Такие гидраты также включены в объем настоящего изобретения.
[0032] Соединение (I) по настоящему изобретению или его фармакологически приемлемая соль, когда их оставляют в растворителе, могут образовывать сольват после выделения из растворителя. Такие сольваты также включены в объем настоящего изобретения.
[0033] Соединение (I) по настоящему изобретению включает оптические изомеры на основании асимметрического центра в молекуле. Эти изомеры соединения по настоящему изобретению и смеси этих изомеров все представлены одной формулой, т.е. общей формулой (I), если не указано иное. Таким образом, должно быть понятно, что все такие изомеры и смеси таких изомеров включены в объем настоящего изобретения.
[0034] Соединение (I) по настоящему изобретению включает геометрические изомеры на основании 4-5-положения пиразолопиридинового кольца. Как цис-, так и транс-формы включены в настоящее изобретение, если не указано иное. Например, образуются обе геометрические формы, и их инструментальные данные можно сравнить для определения их соответствующих структур. В настоящем изобретении трифторметильная группа в 4-положении и гидрокси группа в 5-положении предпочтительно находятся в цис-положении относительно друг друга.
[0035] Соединение (I) по настоящему изобретению может содержать изотоп(изотопы) одного или нескольких атомов, образующих такое соединение, в неестественном отношении. Примеры изотопов включают дейтерий (2H), тритий (3H), иод-125 (125I) и углерод-14 (14C). Альтернативно, соединение может быть мечено радиоактивным изотопом, например, тритием (3H), иодом-125 (125I) или углеродом-14 (14C). Такое радиоактивно-меченое соединение является полезным в качестве терапевтического или профилактического средства, реагента для исследований, например, реагента для анализов, и диагностического средства, например, диагностического средства для визуализации in vivo. Должно быть понятно, что все изотопные варианты соединения по настоящему изобретению включены в объем настоящего изобретения, независимо от того являются они радиоактивными или нет.
Выгодные эффекты изобретения
[0036] Соединение, представленное общей формулой (I) по настоящему изобретению, или его фармакологически приемлемая соль обладает отличным LCAT-активирующим эффектом и является полезным в качестве активного ингредиента в терапевтическом или профилактическом средстве от артериосклероза, артериосклеротического сердечного заболевания, коронарного сердечного заболевания (включая сердечную недостаточность, инфаркт миокарда, стенокардию, сердечную ишемию, сердечно-сосудистое расстройство и рестеноз, вызванный ангиогенезом), цереброваскулярного заболевания (включая инсульт и церебральный инфаркт), заболевания периферических сосудов (включая диабетические сосудистые осложнения), дислипидемии, гипо-ЛПВП-холестеринемии, гипер-ЛПНП-холестеринемии или почечного заболевания, в частности, в качестве анти-артериосклеротического средства. Соединение (I) по настоящему изобретению или его фармакологически приемлемая соль имеет высокую концентрацию в крови (AUC, Cmax) при введении животным (человеку, обезьянам и т.д.), и можно ожидать, что оно будет показывать отличную лекарственную эффективность.
Краткое описание чертежа
[0037] [Фиг. 1] Фиг. 1 показывает кривую доза-ответ для определения 50% эффективной концентрации (EC50) активации LCAT в Примерах испытаний 1 и 2 по настоящему изобретению.
Описание вариантов осуществления
[0038] Далее будут описаны типичные способы получения соединения (I) по настоящему изобретению и исходные соединения для использования в получении соединения (I) по настоящему изобретению. Однако настоящее изобретение не ограничивается этими способами.
[0039] Способ получения 1
Способ получения 1 представляет собой способ получения соединения (I) по настоящему изобретению из соединения (II).
[0040]
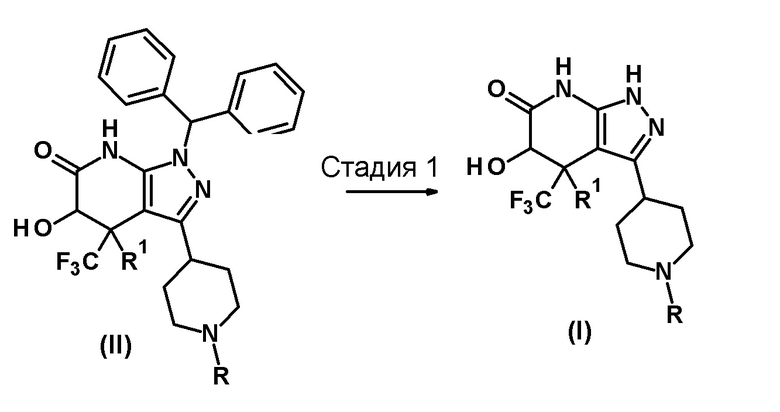
[0041] В этих формулах R и R1 имеют значения, определенные выше.
[0042] (Стадия 1)
Эта стадия включает удаление дифенилметильной группы из соединения (II) в инертном растворителе с образованием соединения (I).
[0043] Примеры реагента, используемого для удаления дифенилметильной группы из соединения (II), включают реагенты, способные удалять тритильную группу, описанные, например, в P.G. Wuts, T.W. Greene, Greene's Protective Groups in Organic Synthesis. Third Edition, 2006, John Wiley & Sons, Inc.
[0044] Растворитель, используемый на этой стадии, предпочтительно представляет собой спирт, такой как метанол или этанол; простой эфир, такой как тетрагидрофуран или 1,4-диоксан; алкилгалогенид, такой как дихлорметан или хлороформ; сложный эфир, такой как этилацетат; ароматический углеводород, такой как толуол; или содержащий их смешанный растворитель, более предпочтительно алкилгалогенид, еще более предпочтительно дихлорметан.
[0045] Реагент, используемый на этой стадии, предпочтительно представляет собой хлористоводородную кислоту или трифторуксусную кислоту, более предпочтительно трифторуксусную кислоту. Соединение, известное как акцептор катионов, такое как триэтилсилан, анизол или тиоанизол, можно использовать в качестве добавки.
[0046] Температура реакции на этой стадии предпочтительно 0°C-100°C, более предпочтительно 0°C-50°C.
[0047] Время реакции на этой стадии предпочтительно составляет от 5 минут до 24 часов, более предпочтительно от 10 минут до 6 часов.
[0048] Способ получения 2
Промежуточное соединение (II) для соединения по настоящему изобретению, где R1 представляет собой атом водорода, также можно получить следующим способом:
[0049]
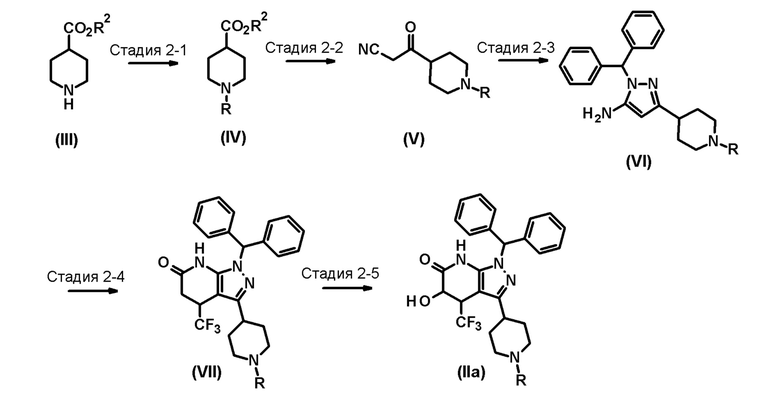
[0050] В этих формулах R имеет значение, определенное выше, и R2 представляет собой метильную группу или этильную группу.
[0051] (Стадия 2-1)
(i) Эта стадия включает взаимодействие соединения (III) с арилирующим агентом или гетероарилирующим агентом через реакцию Бухвальда-Хартвига с использованием палладиевого катализатора, в присутствии лиганда, помимо палладиевого катализатора, и основания в инертном растворителе с образованием соединения (IV).
[0052] Палладиевый катализатор, лиганд, основание и реакционные условия, используемые на этой стадии, конкретно не ограничиваются, при условии, что они представляют собой реагенты и условия для использования в обычных реакциях Бухвальда-Хартвига. Реагенты и условия описаны, например, в A.R. Muci, S.L. Buchwald, Top. Curr. Chem. 2002, Vol. 219, p. 131.
[0053] Растворитель, используемый на этой стадии, представляет собой простой эфир, такой как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или трет-бутилметиловый эфир; или ароматический углеводород, такой как бензол, толуол или ксилол. Растворитель предпочтительно представляет собой толуол или диоксан, более предпочтительно толуол.
[0054] Палладиевый катализатор, используемый на этой стадии, предпочтительно представляет собой ацетат палладия(II) или палладий(0) дибензилиденацетон, более предпочтительно палладий(0) дибензилиденацетон.
[0055] Лиганд, используемый на этой стадии, предпочтительно представляет собой трициклогексилфосфин, 1,3-бис(дифенилфосфино)пропан, 2,2'-бис(дифенилфосфанил)1,1'-бинафтил, 2-(дициклогексилфосфино)бифенил или 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенил, более предпочтительно 2,2'-бис(дифенилфосфанил)1,1'-бинафтил.
[0056] Основание, используемое на этой стадии, предпочтительно представляет собой карбонат натрия, карбонат калия, карбонат цезия, трет-бутоксид натрия или трет-бутоксид калия, более предпочтительно трет-бутоксид натрия.
[0057] Арилирующий агент или гетероарилирующий агент, используемый на этой стадии, относится к соединению, представленному формулой R-Cl, R-Br или R-I, и предпочтительно представляет собой соединение, представленное формулой R-Cl или R-Br (где R имеет значение, определенное выше).
[0058] Температура реакции на этой стадии предпочтительно находится на уровне от 20°C до 150°C, более предпочтительно от 50°C до температуры кипения растворителя.
[0059] Для ускорения реакции на этой стадии, реакционный раствор можно подвергнуть нагреванию и также можно подвергнуть микроволновому облучению.
[0060] Время реакции на этой стадии предпочтительно составляет от 5 минут до 120 часов, более предпочтительно от 10 минут до 96 часов.
[0061] (ii) Альтернативно, эта стадия включает взаимодействие соединения (III) с арилирующим агентом или гетероарилирующим агентом в присутствии основания в инертном растворителе с образованием соединения (IV).
[0062] Растворитель, используемый на этой стадии, может представлять собой галогенированный углеводород, такой как дихлорметан, хлороформ, тетрахлорид углерода, 1,2-дихлорэтан, хлорбензол или дихлорбензол; простой эфир, такой как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или трет-бутилметиловый эфир; ароматический углеводород, такой как бензол, толуол или ксилол; амид, такой как формамид, N,N-диметилформамид, диметилацетамид, N-метил-2-пирролидон или гексаметилфосфортриамид; или сульфоксид, такой как диметилсульфоксид. Растворитель предпочтительно представляет собой амид или сульфоксид, более предпочтительно N,N-диметилформамид или диметилсульфоксид.
[0063] Основание, используемое на этой стадии, может представлять собой органическое основание, такое как триэтиламин, диизопропилэтиламин, 1,8-диазабицикло[5.4.0]-7-ундецен, N-метилморфолин, пиридин, диметиламинопиридин или 2,6-лутидин. Основание предпочтительно представляет собой триэтиламин, диизопропилэтиламин, 1,8-диазабицикло[5.4.0]-7-ундецен, пиридин или диметиламинопиридин.
[0064] Арилирующий агент или гетероарилирующий агент, используемый на этой стадии, относится к соединению, представленному формулой R-F, R-Cl или R-Br, и предпочтительно представляет собой соединение, представленное формулой R-F или R-Cl (где R имеет значение, определенное выше).
[0065] Температура реакции на этой стадии предпочтительно находится на уровне от 20°C до 200°C.
[0066] Для ускорения реакции на этой стадии реакционный раствор можно подвергнуть нагреванию и также можно подвергнуть микроволновому облучению.
[0067] Время реакции на этой стадии предпочтительно составляет от 5 минут до 120 часов, более предпочтительно от 10 минут до 96 часов.
[0068] (Стадия 2-2)
Эта стадия включает взаимодействие соединения (IV) с ацетонитрилом с использованием основания в инертном растворителе с образованием соединения (V).
[0069] Растворитель, используемый на этой стадии, может представлять собой простой эфир, такой как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или трет-бутилметиловый эфир; ароматический углеводород, такой как бензол, толуол или ксилол; алифатический углеводород, такой как гексан; или содержащий их смешанный растворитель. Растворитель предпочтительно представляет собой простой эфир, более предпочтительно тетрагидрофуран.
[0070] Основание, используемое на этой стадии, предпочтительно может представлять собой неорганическое основание, такое как гидрид натрия, карбонат натрия, карбонат калия или карбонат цезия; или металлорганическое основание, такое как трет-бутоксид натрия, трет-бутоксид калия или н-бутиллитий. Основание более предпочтительно представляет собой гидрид натрия или н-бутиллитий.
[0071] Температура реакции на этой стадии предпочтительно находится на уровне от -100°C до 0°C, более предпочтительно от -78°C до -40°C.
[0072] Время реакции на этой стадии предпочтительно составляет от 5 минут до 3 часов, более предпочтительно от 15 минут до 2 часов.
[0073] (Стадия 2-3)
Эта стадия включает взаимодействие соединения (V) с дифенилметилгидразиновым соединением в инертном растворителе с образованием соединения (VI).
[0074] Растворитель, используемый на этой стадии, может представлять собой спирт, такой как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, трет-бутанол, изоамиловый спирт, октанол, циклогексанол, 2-метоксиэтанол, диэтиленгликоль или глицерин; ароматический углеводород, такой как бензол, толуол или ксилол; или содержащий их смешанный растворитель. Растворитель предпочтительно представляет собой спирт, более предпочтительно этанол.
[0075] Дифенилметилгидразиновое соединение, используемое на этой стадии, может представлять собой, например, безводный дифенилметилгидразин, гидрохлорид дифенилметилгидразина или ацетат дифенилметилгидразина. Дифенилметилгидразиновое соединение предпочтительно представляет собой гидрохлорид дифенилметилгидразина или ацетат дифенилметилгидразина.
[0076] Температура реакции на этой стадии предпочтительно находится на уровне от 20°C до 120°C, более предпочтительно от 50°C до температуры кипения растворителя.
[0077] Время реакции на этой стадии предпочтительно составляет от 10 минут до 24 часов, более предпочтительно от 1 часа до 5 часов.
[0078] (Стадия 2-4)
Эта стадия включает взаимодействие соединения (VI) с трифторацетальдегидным эквивалентом и кислотой Мельдрума в инертном растворителе с образованием соединения (VII).
[0079] Растворитель, используемый на этой стадии, может представлять собой спирт, такой как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, трет-бутанол, изоамиловый спирт, октанол, циклогексанол, 2-метоксиэтанол, диэтиленгликоль или глицерин; ароматический углеводород, такой как бензол, толуол или ксилол; или содержащий их смешанный растворитель. Растворитель предпочтительно представляет собой спирт, более предпочтительно этанол.
[0080] Трифторацетальдегидный эквивалент, используемый на этой стадии, может представлять собой, например, трифторацетальдегид алкил гемиацеталь или трифторацетальдегид диалкил ацеталь. Трифторацетальдегидный эквивалент предпочтительно представляет собой трифторацетальдегид этил гемиацеталь.
[0081] Температура реакции на этой стадии предпочтительно находится на уровне от 0°C до 100°C, более предпочтительно от 20°C до температуры кипения растворителя.
[0082] Время реакции на этой стадии предпочтительно составляет 30 минут до 24 часов, более предпочтительно от 1 часа до 6 часов.
[0083] (Стадия 2-5)
Эта стадия включает взаимодействие соединения (VII) с окислителем в присутствии основания в инертном растворителе с образованием соединения (IIa).
[0084] Растворитель, используемый на этой стадии, может представлять собой простой эфир, такой как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или трет-бутилметиловый эфир; ароматический углеводород, такой как бензол, толуол или ксилол; алифатический углеводород, такой как гексан; или содержащий их смешанный растворитель. Растворитель предпочтительно представляет собой простой эфир, более предпочтительно тетрагидрофуран.
[0085] Основание, используемое на этой стадии, предпочтительно может представлять собой неорганическое основание, такое как гидрид натрия, гидрид калия, карбонат натрия, карбонат калия или карбонат цезия; или металлорганическое основание, так как трет-бутоксид натрия, трет-бутоксид калия, н-бутиллитий, диизопропиламид лития, гексаметилдисилазид лития, гексаметилдисилазид калия или 2,2,6,6-тетраметилпиперидид лития. Основание предпочтительно представляет собой металлорганическое основание, более предпочтительно диизопропиламид лития.
[0086] Окислитель, используемый на этой стадии, предпочтительно представляет собой бис(триметилсилил) пероксид, 3-фенил-2-(фенилсульфонил)оксазиридин (реагент Дэйвиса) или (10-камфорсульфонил)оксазиридин, более предпочтительно (10-камфорсульфонил)оксазиридин.
[0087] Температура реакции на этой стадии предпочтительно находится на уровне от -100°C до 100°C, более предпочтительно от -78°C до 30°C.
[0088] Время реакции на этой стадии предпочтительно составляет 1 часа до 10 часов, более предпочтительно от 2 часов до 5 часов.
[0089] Соединение (IIa) имеет цис-транс изомеры. Эти цис-транс изомеры можно разделить известным способом, таким как хроматография. Также, гидрокси группы в смесях цис-транс изомеров соединения (IIa) в произвольном соотношении являются защищенными, и смеси можно перемешивать в присутствии основания в инертном растворителе, с последующим удалением защитной группы с получением смесей, содержащих цис-транс изомеры соединения (IIa) в разных соотношениях.
[0090] Примеры гидрокси-защитной группы, используемой в этой процедуре, включают защитные группы, которые являются стабильными в щелочных условиях, и которые могут быть удалены, как описано, например, в P.G. Wuts, T.W. Greene, Greene's Protective Groups in Organic Synthesis. Third Edition, 2006, John Wiley & Sons, Inc. Защитная группа предпочтительно представляет собой тетрагидропиранильную группу.
[0091] Основание, используемое в этой процедуре, предпочтительно может представлять собой неорганическое основание, такое как гидрид натрия, гидрид калия, карбонат натрия, карбонат калия или карбонат цезия; или металлорганическое основание, такое как трет-бутоксид натрия, трет-бутоксид калия, н-бутиллитий, диизопропиламид лития, гексаметилдисилазид лития, гексаметилдисилазид калия или 2,2,6,6-тетраметилпиперидид лития. Основание предпочтительно представляет собой металлорганическое основание, более предпочтительно диизопропиламид лития.
[0092] Температура реакции предпочтительно находится на уровне от -78°C до 100°C, более предпочтительно от -58°C до 10°C.
[0093] Время реакции предпочтительно составляет от 1 часа до 10 часов, более предпочтительно от 2 часов до 3 часов.
[0094] Способ получения 3
Промежуточное соединение (II) соединения по настоящему изобретению, где R1 представляет собой гидрокси группу, также можно получить следующим способом:
[0095]
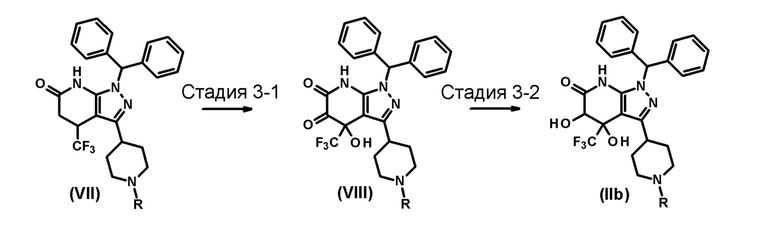
[0096] В этих формулах R имеет значение, определенное выше.
[0097] (Стадия 3-1)
Эта стадия включает взаимодействие соединения (VII) с окислителем в инертном растворителе с образованием соединения (VIII).
[0098] Растворитель, используемый на этой стадии, может представлять собой галогенированный углеводород, такой как дихлорметан, хлороформ, тетрахлорид углерода, 1,2-дихлорэтан, хлорбензол или дихлорбензол; простой эфир, такой как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или трет-бутилметиловый эфир; или ароматический углеводород, такой как бензол, толуол или ксилол. Растворитель предпочтительно представляет собой галогенированный углеводород, более предпочтительно дихлорметан.
[0099] Окислитель, используемый на этой стадии, предпочтительно представляет собой реагент Десса-Мартина.
[0100] Температура реакции на этой стадии предпочтительно находится на уровне от -5°C до 40°C, более предпочтительно от 0°C до 30°C.
[0101] Время реакции на этой стадии предпочтительно составляет 30 минут до 3 часов, более предпочтительно от 1 часа до 2 часов.
[0102] (Стадия 3-2)
Эта стадия включает взаимодействие соединения (VIII) с восстановителем в инертном растворителе с образованием соединения (IIb).
[0103] Растворитель, используемый на этой стадии, может представлять собой спирт, такой как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, трет-бутанол, изоамиловый спирт, октанол, циклогексанол, 2-метоксиэтанол, диэтиленгликоль или глицерин; простой эфир, такой как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или трет-бутилметиловый эфир; ароматический углеводород, такой как бензол, толуол или ксилол; или содержащий их смешанный растворитель. Растворитель предпочтительно представляет собой спирт, более предпочтительно метанол.
[0104] Восстановитель, используемый на этой стадии, предпочтительно представляет собой борогидрид натрия.
[0105] Температура реакции на этой стадии предпочтительно находится на уровне от -5°C до 40°C, более предпочтительно от 0°C до 30°C.
[0106] Время реакции на этой стадии предпочтительно составляет 10 минут до 3 часов, более предпочтительно от 30 минут до 2 часов.
[0107] Способ получения 4
Промежуточное соединение (II) соединения по настоящему изобретению также можно получить следующим способом:
[0108]
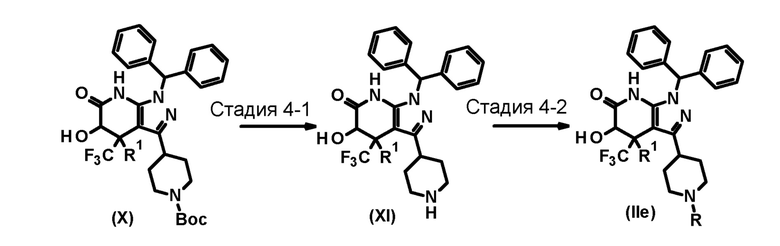
[0109] В этих формулах Boc представляет собой трет-бутоксикарбонильную группу, и R и R1 имеют значения, определенные выше.
[0110] (Стадия 4-1)
Эта стадия включает удаление Boc группы из соединения (X) с образованием соединения (XI).
[0111] Соединение (X) можно получить, например, в соответствии со способами, описанными в Ссылочных примерах 12, 13, 14 и 16 или Ссылочных примерах 25 и 26.
[0112] Примеры реагента, используемого для удаления Boc из соединения (X), включают реагенты, способные удалять Boc, описанные, например, в P.G. Wuts, T.W. Greene, Greene's Protective Groups in Organic Synthesis. Third Edition, 2006, John Wiley & Sons, Inc.
[0113] Растворитель, используемый на этой стадии, предпочтительно представляет собой спирт, такой как метанол или этанол; простой эфир, такой как тетрагидрофуран или 1,4-диоксан; алкилгалогенид, такой как дихлорметан или хлороформ; сложный эфир, такой как этилацетат; ароматический углеводород, такой как толуол; нитрил, такой как ацетонитрил; или содержащий их смешанный растворитель, более предпочтительно алкилгалогенид, нитрил или смешанный растворитель, состоящий из алкилгалогенида и нитрила, еще более предпочтительно смешанный растворитель, состоящий из дихлорметана и ацетонитрила.
[0114] Реагент, используемый на этой стадии, предпочтительно представляет собой комбинацию хлортриметилсилана и иодида натрия.
[0115] Температура реакции на этой стадии предпочтительно находится на уровне от 0°C до 100°C, более предпочтительно от 0°C до 50°C.
[0116] Время реакции на этой стадии предпочтительно составляет от 5 минут до 24 часов, более предпочтительно от 10 минут до 6 часов.
[0117] (Стадия 4-2)
Эта стадия включает взаимодействие соединения (IId) с арилирующим агентом или гетероарилирующим агентом в присутствии основания в инертном растворителе с образованием соединения (II).
[0118] Эту стадию можно осуществить таким же способом, как способ осуществления стадии 2-1(ii).
[0119] Способ получения 5
Промежуточное соединение (II) соединения по настоящему изобретению, где R1 представляет собой гидрокси группу, также можно получить следующим способом:
[0120]
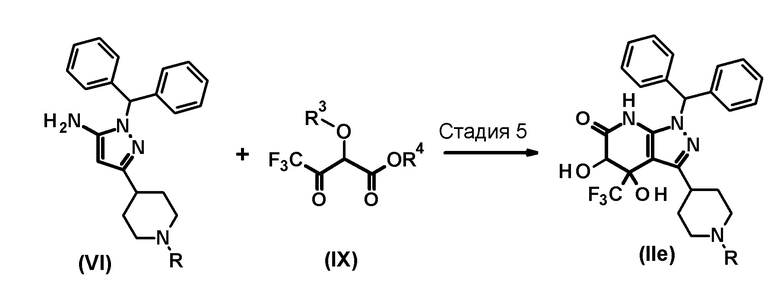
[0121] В этих формулах R имеет значение, определенное выше, R3 представляет собой атом водорода, метильную группу, триэтилсилильную группу, трет-бутилдиметилсилильную группу или трет-бутилдифенилсилильную группу, и R4 представляет собой метильную группу или этильную группу.
[0122] (Стадия 5)
Эта стадия включает конденсацию соединения (VI) и соединения (IX) при нагревании в растворителе, инертном к реакции, или в отсутствие растворителя, с образованием соединения (IIe).
[0123] Растворитель, используемый на этой стадии, может представлять собой органическую кислоту, такую как уксусная кислота, муравьиная кислота, щавелевая кислота, метансульфоновая кислота, п-толуолсульфоновая кислота, камфорсульфоновая кислота, трифторуксусная кислота или трифторметансульфоновая кислота; простой эфир, такой как диэтиловый эфир, диизопропиловый эфир, тетрагидрофуран, диоксан, диметоксиэтан или трет-бутилметиловый эфир; спирт, такой как метанол, этанол, н-пропанол, изопропанол, н-бутанол, изобутанол, трет-бутанол, изоамиловый спирт, октанол, циклогексанол, 2-метоксиэтанол, диэтиленгликоль или глицерин; ароматический углеводород, такой как бензол, толуол или ксилол; или содержащий их смешанный растворитель. Растворитель предпочтительно представляет собой смешанный растворитель, состоящий из этанола и уксусной кислоты.
[0124] Температура реакции на этой стадии обычно находится на уровне от 40°C до 150°C, предпочтительно от 50°C до 130°C, более предпочтительно от 60°C до температуры кипения растворителя.
[0125] Для ускорения реакции на этой стадии реакционный раствор можно подвергнуть нагреванию и также можно подвергнуть микроволновому облучению.
[0126] Время реакции на этой стадии обычно составляет от 5 минут до 72 часов, предпочтительно от 15 минут до 24 часов, более предпочтительно от 30 минут до 3 часов.
[0127] Когда R3 в соединении (IX) представляет собой метильную группу, триэтилсилильную группу, трет-бутилдиметилсилильную группу или трет-бутилдифенилсилильную группу, гидрокси-защитную группу можно удалить из соединения, полученного в результате вышеуказанной реакции, способом, описанным, например, в P.G. Wuts, T.W. Greene, Greene's Protective Groups in Organic Synthesis. Third Edition, 2006, John Wiley & Sons, Inc., с образованием соединения (IIe).
[0128] Если необходимо, продукт каждой стадии, описанной выше, можно выделить из реакционной смеси в виде свободного соединения или его соли после завершения реакции рутинным способом, например, (1) способом непосредственного концентрирования реакционного раствора, (2) способом отфильтровывания нерастворимого вещества, такого как катализатор, и концентрирования фильтрата, (3) способом добавления воды и растворителя, не смешиваемого с водой (например, дихлорэтана, диэтилового эфира, этилацетата или толуола), к реакционному раствору для экстрагирования продукта, или (4) способом сбора кристаллизованного или осажденного продукта фильтрованием. Выделенный продукт можно очистить, если необходимо, рутинным способом, например, путем перекристаллизации, повторного осаждения или различными хроматографическими методами. Альтернативно, продукт каждой стадии можно использовать на следующей стадии без выделения или очистки.
[0129] Соединение (I) по настоящему изобретению выделяют и очищают в виде свободного соединения или его фармакологически приемлемой соли, гидрата или сольвата. Фармакологически приемлемую соль соединения (I) по настоящему изобретению можно получить через солеобразующую реакцию соединения (I) рутинным способом. Выделение и очистку осуществляют с использованием обычных химических процедур, таких как экстракция, концентрирование, дистилляция, кристаллизация, фильтрование, перекристаллизация или различные хроматографические методы.
[0130] Различные изомеры можно разделить на основании различий в физико-химических свойствах между изомерами. Например, рацемическую смесь можно преобразовать в оптически чистый изомер, например, методом фракционированной кристаллизации для получения диастереомерной соли с оптически активным основанием или кислотой, или хроматографией с использованием хиральной колонки. Также, диастереомерную смесь можно разделить, например, методом фракционированной кристаллизации или различными хроматографическими методами. Альтернативно, оптически активное соединение также можно получить с использованием соответствующего оптически активного исходного вещества.
[0131] Примеры лекарственных форм соединения, представленного общей формулой (I) по настоящему изобретению, или его фармакологически приемлемой соли могут включать: формы для перорального введения, такие как таблетки, гранулы, порошки, капсулы и сиропы; и формы для парентерального введения, такие как инъекции и суппозитории. Эти препараты можно вводить системно или местно.
[0132] Примеры пероральных лекарственных форм, включающих соединение, представленное общей формулой (I) по настоящему изобретению, или его фармакологически приемлемую соль, включают таблетки, пилюли, гранулы, порошки, капсулы, растворы, суспензии, эмульсии, сиропы и эликсиры. Примеры парентеральных лекарственных форм, включающих соединение, представленное общей формулой (I) по настоящему изобретению, или его фармакологически приемлемую соль, включают инъекции, мази, гели, кремы, пластыри, аэрозоли, препараты для ингаляции, спреи, глазные капли и суппозитории. Лекарственные средства в этих формах можно получить в соответствии с рутинным способом с использованием добавок, подходящим образом выбранных в соответствии с необходимостью из фармацевтически приемлемых добавок, таких как эксципиенты, связующие, разбавители, стабилизаторы, антисептики, красители, солюбилизирующие вещества, суспендирующие вещества, буферы и смачивающие вещества.
[0133] Доза, при которой вводят соединение, представленное общей формулой (I) по настоящему изобретению, или его фармакологически приемлемую соль, варьирует в зависимости от симптомов, массы тела и возраста реципиента (теплокровного животного, например, человека), способа введения и т.д. Например, в случае перорального введения, одноразовая доза составляет 0,01 мг/кг массы тела (предпочтительно 0,03 мг/кг массы тела) в качестве нижнего предела и 300 мг/кг массы тела (предпочтительно 100 мг/кг массы тела) в качестве верхнего предела, и желательно осуществлять введение от одного до нескольких раз в день, в соответствии с симптомами. В случае внутривенного введения, одноразовая доза составляет 0,01 мг/кг массы тела (предпочтительно 0,03 мг/кг массы тела) в качестве нижнего предела и 300 мг/кг массы тела (предпочтительно 100 мг/кг массы тела) в качестве верхнего предела, и желательно осуществлять введение от одного до нескольких раз в день, в соответствии с симптомами.
[0134] Далее настоящее изобретение будет описано более подробно со ссылкой на Примеры, Примеры испытаний и Примеры формулирования. Однако объем настоящего изобретения не ограничивается этим. В представленных ниже примерах гексан представляет собой н-гексан; THF представляет собой тетрагидрофуран; IPA представляет собой 2-пропанол; DMF представляет собой N,N'-диметилформамид; DMSO представляет собой диметилсульфоксид; и CSA представляет собой (±)-10-камфорсульфоновую кислоту.
Примеры
[0135] (Ссылочный пример 1) 1-(Дифенилметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1H-пиразол-5-амин
[0136]
[0137] н-Бутиллитий (2,69 M раствор в гексане, 17,5 мл, 47,1 ммоль) добавляли по каплям при -78°C к раствору безводного ацетонитрила (2,47 мл, 47,1 ммоль) в безводном THF (70 мл) и смесь перемешивали при указанной выше температуре в течение 10 минут. Добавляли по каплям раствор этил 1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-карбоксилата (соединение, описанное в опубликованной заявке WO2005/40119, 5,69 г, 18,8 ммоль) в THF (30 мл) при указанной выше температуре и смесь перемешивали в течение 30 минут. Затем добавляли уксусную кислоту (6 мл) и температуру смеси повышали до комнатной температуры. К реакционному раствору добавляли этилацетат и Целит(R) и смесь перемешивали приблизительно в течение 10 минут и фильтровали через Целит. Растворитель в фильтрате отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=90/10-50/50 (градиент)] с получением нитрильного промежуточного соединения.
[0138] Дифенилметилгидразин гидрохлорид (4,64 г, 19,8 ммоль) добавляли к раствору нитрильного промежуточного соединения, полученного с использованием процедур, описанных выше, в этаноле (100 мл) и смесь перемешивали при 50°C в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении и полученный остаток разделяли на органический и водный слои путем добавления насыщенного водного раствора бикарбоната натрия и этилацетата. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=90/10-50/50 (градиент)] с получением указанного в заголовке соединения (5,44 г, выход: 61%).
[0139] 1H-ЯМР (400 Гц, CDCl3) δ: 8,38 (1H, с), 7,59 (1H, дд, J=9 Гц, 2 Гц), 7,38-7,27 (6H, м), 7,25-7,18 (4H, м), 6,66 (1H, с), 6,64 (1H, с), 5,41 (1H, с), 4,41 (2H, д, J=13 Гц), 3,23 (2H, с), 3,05-2,98 (2H, м), 2,88-2,81 (1H, м), 2,01 (2H, дд, J=13 Гц, 3 Гц), 1,66 (2H, ддд, J=25 Гц, 13 Гц, 4 Гц).
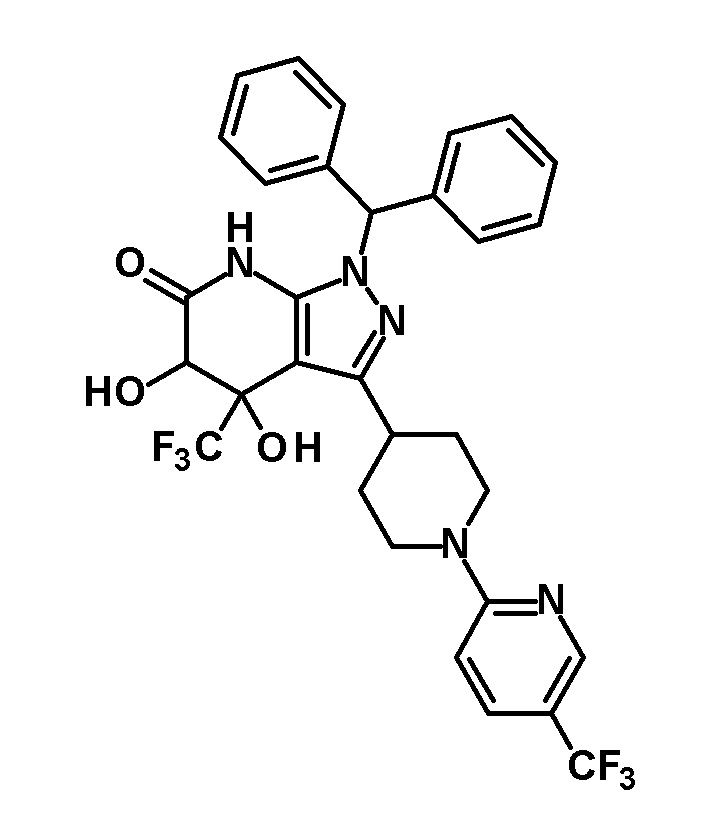
[0140] (Ссылочный пример 2) 1-(Дифенилметил)-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0141]
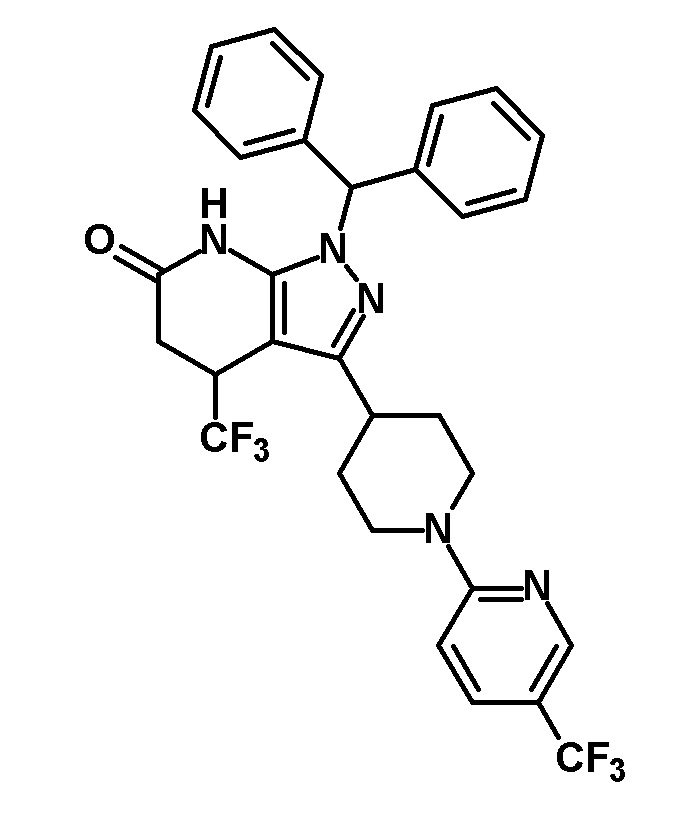
[0142] Трифторацетальдегид этил гемиацеталь (2,68 г, 18,6 ммоль) добавляли к раствору 1-(дифенилметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1H-пиразол-5-амина (4,50 г, 9,42 ммоль), полученного в Ссылочном примере 1, и кислоты Мельдрума (2,65 г, 18,4 ммоль) в этаноле (40 мл) и смесь перемешивали в течение 5 часов при нагревании до температуры кипения с обратным холодильником. Растворитель отгоняли из реакционного раствора при пониженном давлении. Полученный остаток очищали три раза колоночной хроматографией на силикагеле [NH-силикагель, элюент: (i) гексан/дихлорметан=50/50-0/100 (градиент), (ii) дихлорметан/метанол=100/0-90/10 (градиент)] с получением указанного в заголовке соединения (2,85 г, выход: 50%).
[0143] 1H-ЯМР (400Гц, DMSO-d6) δ: 11,18 (1H, с), 8,38 (1H, с), 7,75 (1H, дд, J= 9 Гц, 2 Гц), 7,36-7,16 (10H, м), 6,96 (1H, д, J=9 Гц), 6,81 (1H, с), 4,49-4,41 (2H, м), 4,10-4,00 (1H, м), 3,16-2,89 (5H, м), 1,95-1,46 (4H, м).
[0144] (Ссылочный пример 3) транс-1-(Дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0145]
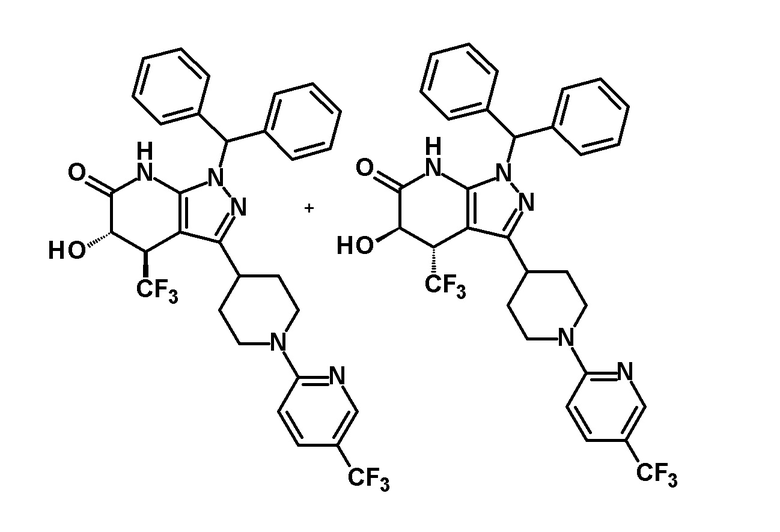
[0146] н-Бутиллитий (2,6 M раствор в гексане, 1,75 мл, 4,70 ммоль) добавляли при -78°C к раствору диизопропиламина (0,65 мл, 4,6 ммоль) в безводном THF (12 мл) и смесь перемешивали при указанной выше температуре в течение 15 минут. Затем добавляли раствор 1-(дифенилметил)-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (980 мг, 1,63 ммоль), полученного в Ссылочном примере 2, в безводном THF (10 мл) и смесь перемешивали в течение 30 минут. К реакционному раствору добавляли при указанной выше температуре раствор (1S)-(+)-(10-камфорсульфонил)оксазиридина (302 мг, 1,32 ммоль) и (1R)-(-)-(10-камфорсульфонил)оксазиридина (302 мг, 1,32 ммоль) в безводном THF (8 мл) и смесь нагревали до комнатной температуры и перемешивали в течение 3 часов. К реакционному раствору добавляли насыщенный водный раствор хлорида аммония, с последующим экстрагированием этилацетатом три раза. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=80/20-50/50]. Полученный частично очищенный продукт очищали с использованием смешанного растворителя дихлорметан-метанол с получением указанного в заголовке соединения (611 мг, выход: 61%).
[0147] 1H-ЯМР (400 Гц, DMSO-d6) δ: 11,26 (1H, с), 8,38 (1H, с), 7,74 (1H, дд, J=9 Гц, 3 Гц), 7,36-7,15 (10H, м), 6,96 (1H, д, J=9 Гц), 6,81 (1H, с), 6,68 (1H, д, J=5 Гц), 4,50-4,39 (2H, м), 4,20 (1H, д, J=5 Гц), 3,99-3,89 (1H, м), 3,11-2,89 (3H, м), 1,92-1,77 (2H, м), 1,71-1,47 (2H, м).
[0148] (Ссылочный пример 4) цис-1-(Дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0149]
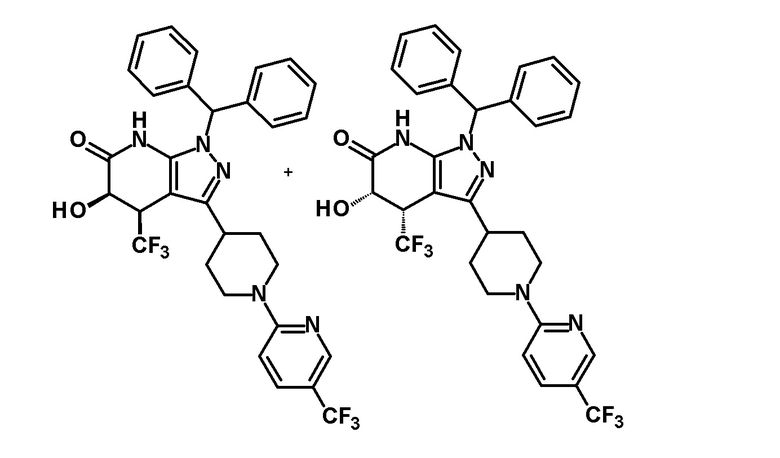
[0150] 3,4-Дигидро-2H-пиран (0,25 мл, 2,7 ммоль) добавляли к раствору транс-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (730 мг, 1,19 ммоль), полученного в Ссылочном примере 3, и CSA (30 мг, 0,129 ммоль) в дихлорметане (10 мл) и смесь перемешивали в течение 8 часов при нагревании до температуры кипения с обратным холодильником. К реакционному раствору добавляли триэтиламин и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=80/20-60/40 (градиент)] с получением защищенного спиртового промежуточного соединения.
[0151] Диизопропиламид лития (1,09 M раствор в гексане и THF (3,50 мл, 3,81 ммоль) добавляли при 0°C к раствору защищенного спиртового промежуточного соединения, полученного с использованием процедур, описанных выше, в безводном THF (20 мл) и смесь перемешивали при указанной выше температуре в течение 2 часов. К смеси добавляли метанол (1 мл) при -40°C и затем температуру смеси повышали до комнатной температуры. К смеси добавляли насыщенный водный раствор хлорида аммония, с последующим экстрагированием этилацетатом три раза. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=100/0-75/25 (градиент)] с получением промежуточного продукта синтеза.
[0152] CSA (60 мг, 0,258 ммоль) добавляли к раствору промежуточного продукта синтеза, полученного с использованием процедур, описанных выше, в метаноле (5 мл) и смесь перемешивали при комнатной температуре в течение 1 часа и затем при 50°C в течение 30 минут. К реакционному раствору добавляли триэтиламин (0,2 мл) и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=80/20-50/50 (градиент)] с получением указанного в заголовке соединения (95 мг, выход: 13%).
[0153] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,21 (1H, с), 8,38 (1H, с), 7,75 (1H, д, J=9 Гц), 7,36-7,23 (8H, м), 7,15 (1H, с), 7,14 (1H, с), 6,96 (1H, д, J=9 Гц), 6,74 (1H, с), 5,80 (1H, д, J=4 Гц), 4,60-4,55 (1H, м), 4,49-4,39 (2H, м), 4,20-4,10 (1H, м), 3,11-2,88 (3H, м), 1,97-1,45 (4H, м).
[0154] (Ссылочный пример 5) 1-(Дифенилметил)-4-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-4,7-дигидро-1H-пиразоло[3,4-b]пиридин-5,6-дион
[0155]
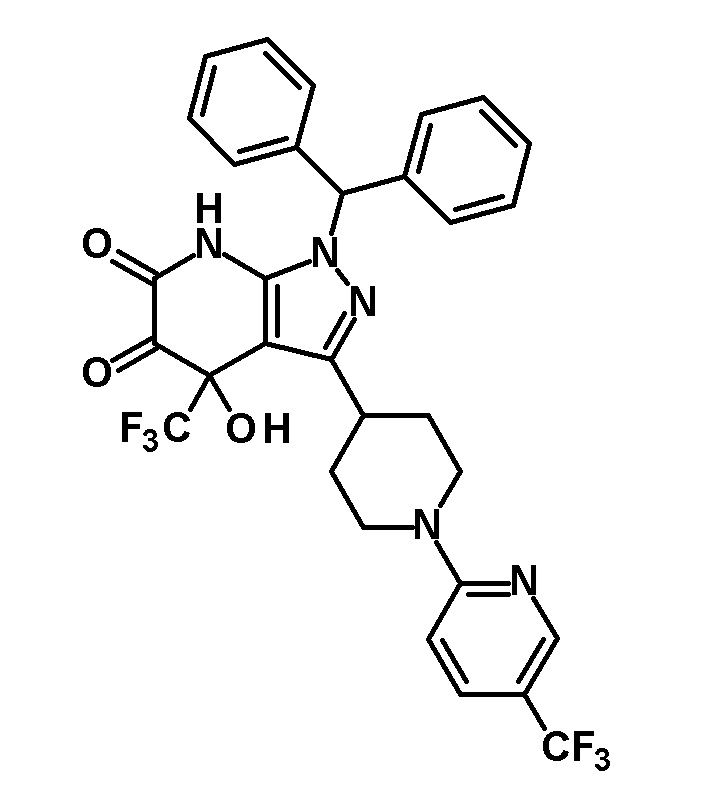
[0156] Реагент Десса-Мартина (210 мг, 0,495 ммоль) добавляли к раствору транс-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (200 мг, 0,325 ммоль), полученного в Ссылочном примере 3, в дихлорметане (5 мл) и смесь перемешивали при комнатной температуре в течение 1 часа. Снова добавляли реагент Десса-Мартина (100 мг, 0,236 ммоль) и смесь перемешивали в течение 15 минут. Опять добавляли реагент Десса-Мартина (100 мг, 0,236 ммоль) и смесь перемешивали в течение 15 минут. К реакционному раствору добавляли насыщенный водный раствор бикарбоната натрия, с последующим экстрагированием этилацетатом три раза. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=80/20-60/40 (градиент)] с получением указанного в заголовке соединения (41 мг, выход: 20%).
[0157] MS (ESI) m/z: 629 (M+H)+.
[0158] (Ссылочный пример 6) 1-(Дифенилметил)-4,5-дигидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0159]
[0160] Борогидрид натрия (10 мг, 0,264 ммоль) добавляли к раствору 1-(дифенилметил)-4-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-4,7-дигидро-1H-пиразоло[3,4-b]пиридин-5,6-диона (41 мг, 0,0651 ммоль), полученного в Ссылочном примере 5, в метаноле (2,0 мл) и смесь перемешивали в течение 1 часа. К реакционному раствору добавляли насыщенный водный раствор хлорида аммония, с последующим экстрагированием этилацетатом три раза. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=70/30-60/40 (градиент)] с получением указанного в заголовке соединения (31 мг, выход: 75%).
[0161] 1H-ЯМР (400 Гц, DMSO-d6) δ: 11,23 (1H, с), 8,37 (1H, с), 7,73 (1H, д, J=9 Гц), 7,36-7,22 (8H, м), 7,16 (2H, д, J=8 Гц), 6,95 (1H, д, J=9 Гц), 6,84 (1H, с), 6,75 (1H, с), 5,97 (1H, д, J=4 Гц), 4,54-4,38 (3H, м), 3,19 (1H, т, J=11 Гц), 3,04-2,92 (2H, м), 2,00 (1H, д, J=12 Гц), 1,79-1,47 (3H, м).
[0162] (Ссылочный пример 7) Этил 1-(5-изопропоксипиридин-2-ил)пиперидин-4-карбоксилат
[0163]
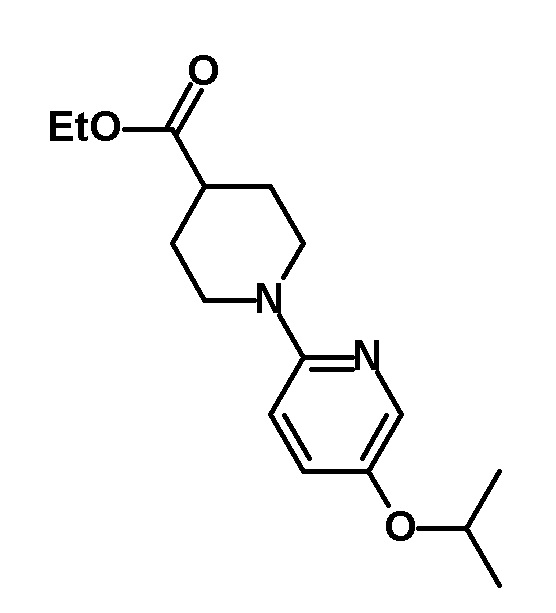
[0164] трет-Бутоксид натрия (0,56 г, 5,79 ммоль), трис(дибензилиденацетон)дипалладий(0) (0,106 г, 0,116 ммоль) и рац.-2,2'-бис(дифенилфосфино)1,1'-бинафтил (0,216 г, 0,347 ммоль) добавляли к раствору 2-бром-5-изопропоксипиридина (соединение, описанное в опубликованной заявке WO2009/81789, 1,00 г, 4,63 ммоль) и этил пиперидин-4-карбоксилата (2,14 мл, 13,9 ммоль) в толуоле (22 мл) и смесь перемешивали при 120°C в течение 1,5 часов. Реакционный раствор охлаждали до комнатной температуры и к раствору добавляли воду, с последующим экстрагированием этилацетатом. Полученный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=100/0-90/10 (градиент)] с получением указанного в заголовке соединения (803 мг, выход: 59%).
[0165] 1H-ЯМР (400 MГц, CDCl3) δ: 7,91 (1H, д, J=3 Гц), 7,18-7,13 (1H, м), 6,66 (1H, д, J=9 Гц), 4,41-4,33 (1H, м), 4,15 (2H, кв., J=7 Гц), 4,10-4,05 (2H, м), 2,95-2,86 (2H, м), 2,52-2,45 (1H, м), 2,04-1,98 (2H, м), 1,85-1,75 (2H, м), 1,30 (6H, д, J=6 Гц), 1,26 (3H, т, J=7 Гц).
[0166] (Ссылочный пример 8) 1-(Дифенилметил)-3-[1-(5-изопропоксипиридин-2-ил)пиперидин-4-ил]-1H-пиразол-5-амин
[0167]
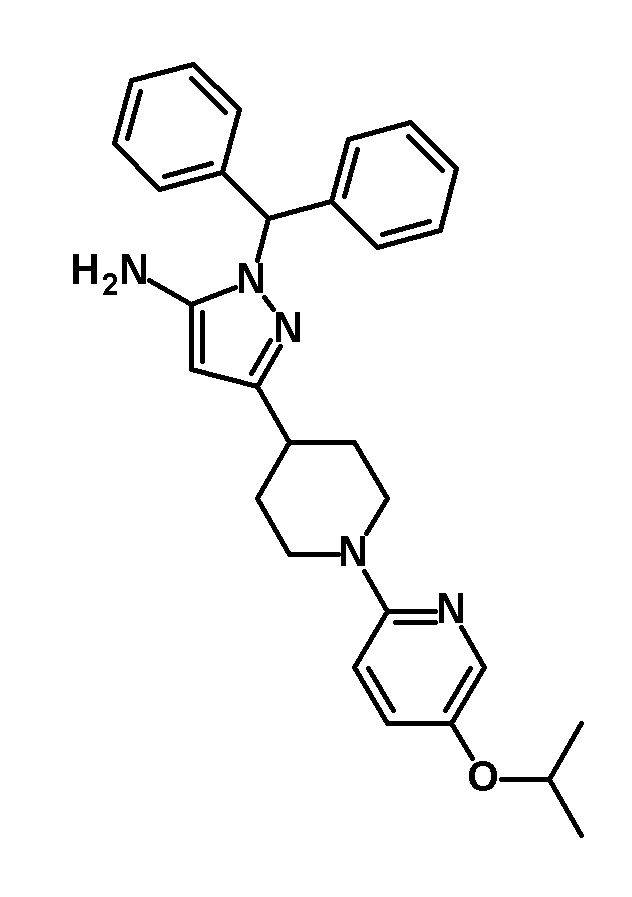
[0168] Указанное в заголовке соединение (1,65 г, выход: 48%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 1, с использованием этил 1-(5-изопропоксипиридин-2-ил)пиперидин-4-карбоксилата (2,14 г, 7,32 ммоль), полученного в Ссылочном примере 7, вместо этил 1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-карбоксилата.
[0169] 1H-ЯМР (400 MГц, CDCl3) δ: 7,92 (1H, д, J=3 Гц), 7,38-7,27 (6H, м), 7,24-7,18 (4H, м), 7,13 (1H, дд, J=9 Гц, 3 Гц), 6,68 (1H, с), 6,66 (1H, д, J=9 Гц), 5,43 (1H, с), 4,41-4,31 (1H, м), 4,19-4,11 (2H, м), 3,27-3,16 (2H, м), 2,89-2,69 (3H, м), 2,05-1,97 (2H, м), 1,79-1,66 (2H, м), 1,30 (6H, д, J=6 Гц).
[0170] (Ссылочный пример 9) 1-(Дифенилметил)-3-[1-(5-изопропоксипиридин-2-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0171]
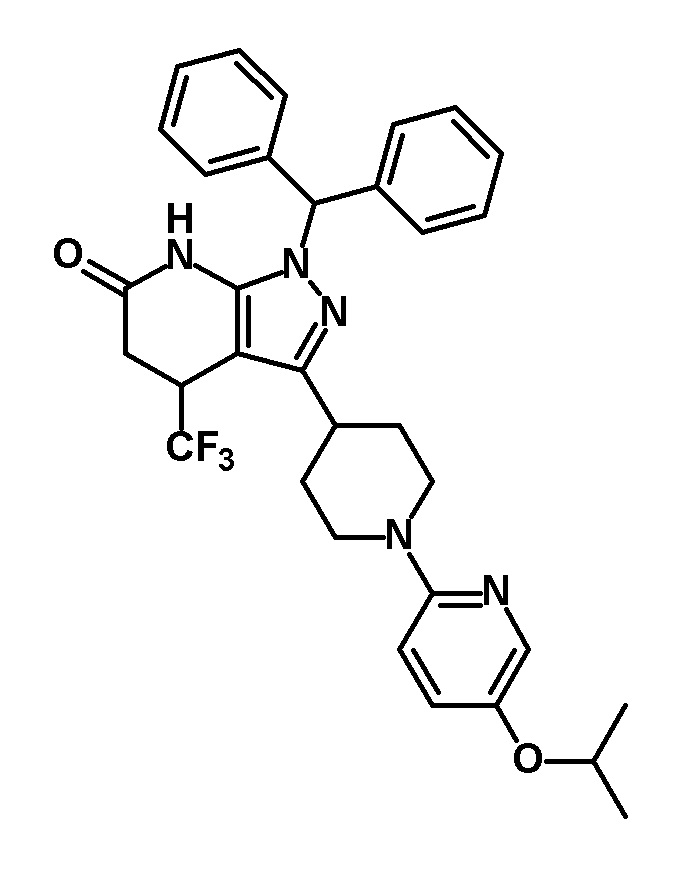
[0172] Указанное в заголовке соединение (1,50 г, выход: 72%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 2, с использованием 1-(дифенилметил)-3-[1-(5-изопропоксипиридин-2-ил)пиперидин-4-ил]-1H-пиразол-5-амина (1,65 г, 3,53 ммоль), полученного в Ссылочном примере 8, вместо 1-(дифенилметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1H-пиразол-5-амина.
[0173] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,17 (1H, с), 7,82 (1H, д, J=3 Гц), 7,37-7,17 (11H, м), 6,82-6,76 (2H, м), 4,47-4,37 (1H, м), 4,21-4,11 (2H, м), 4,09-3,98 (1H, м), 3,11 (1H, дд, J=17 Гц, 8 Гц), 2,85-2,70 (4H, м), 1,95-1,53 (4H, м), 1,22 (6H, д, J=6 Гц).
[0174] (Ссылочный пример 10) транс-1-(Дифенилметил)-5-гидрокси-3-[1-(5-изопропоксипиридин-2-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0175]
[0176] Указанное в заголовке соединение (1,10 г, выход: 71%) получали путем осуществления такой же реакции, как в Ссылочном примере 3, с использованием 1-(дифенилметил)-3-[1-(5-изопропоксипиридин-2-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (1,50 г, 2,54 ммоль), полученного в Ссылочном примере 9, вместо транс-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0177] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,25 (1H, с), 7,82 (1H, д, J=3 Гц), 7,37-7,18 (11H, м), 6,81 (1H, с), 6,79 (1H, д, J=9 Гц), 6,67 (1H, д, J=5 Гц), 4,47-4,37 (1H, м), 4,22-4,10 (3H, м), 3,97-3,87 (1H, м), 2,86-2,72 (3H, м), 1,90-1,52 (4H, м), 1,22 (6H, д, J=6 Гц).
[0178] (Ссылочный пример 11) цис-1-(Дифенилметил)-5-гидрокси-3-[1-(5-изопропоксипиридин-2-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0179]
[0180] Указанное в заголовке соединение (66 мг, выход: 15%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 4, с использованием транс-1-(дифенилметил)-5-гидрокси-3-[1-(5-изопропоксипиридин-2-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (0,78 г, 1,3 ммоль), полученного в Ссылочном примере 10, вместо транс-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0181] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,20 (1H, с), 7,82 (1H, д, J=3 Гц), 7,37-7,19 (9H, м), 7,19-7,14 (2H, м), 6,79 (1H, д, J=9 Гц), 6,74 (1H, с), 5,78 (1H, д, J=4 Гц), 4,60-4,53 (1H, м), 4,47-4,36 (1H, м), 4,22-4,08 (3H, м), 2,86-2,72 (3H, м), 1,95-1,83 (1H, м), 1,80-1,52 (3H, м), 1,22 (6H, д, J=6 Гц).
[0182] (Ссылочный пример 12) трет-Бутил 4-[5-амино-1-(дифенилметил)-1H-пиразол-3-ил]пиперидин-1-карбоксилат
[0183]
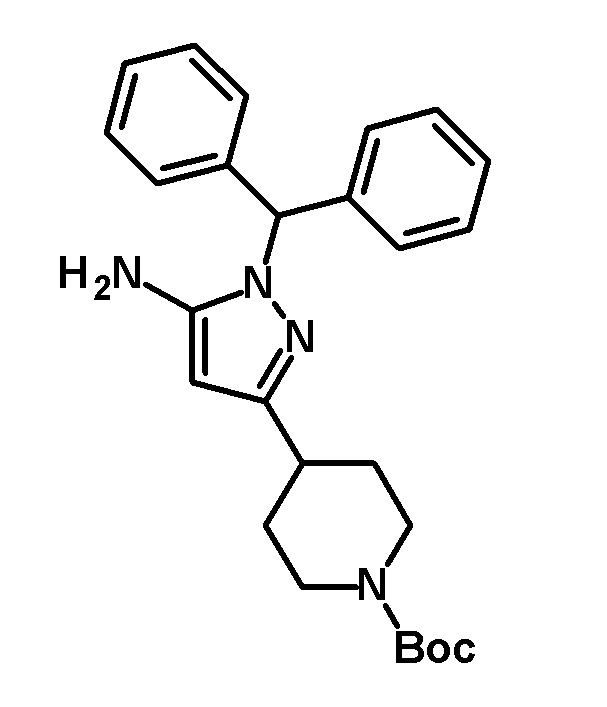
[0184] Дифенилметилгидразин гидрохлорид (8,57 г, 36,5 ммоль) добавляли к раствору трет-бутил 4-(цианоацетил)пиперидин-1-карбоксилата (соединение, описанное в опубликованной заявке WO2004/14910, 7,1 г, 28 ммоль) в этаноле (71 мл) и смесь перемешивали при 50°C в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении и полученный остаток разделяли на органический и водный слои путем добавления насыщенного водного раствора бикарбоната натрия и этилацетата. Органический слой сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=95/5-40/60 (градиент)] с получением указанного в заголовке соединения (7,43 г, выход: 59%).
[0185] 1H-ЯМР (400 Гц, CDCl3) δ: 7,37-7,19 (10H, м), 6,66 (1H, с), 5,40 (1H, с), 4,11 (1H, уш.с), 3,23-3,20 (1H, м), 2,82-2,65 (3H, м), 1,89-1,86 (2H, м), 1,61-1,52 (4H, м), 1,46 (9H, с).
[0186] (Ссылочный пример 13) трет-Бутил 4-[1-(дифенилметил)-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилат
[0187]
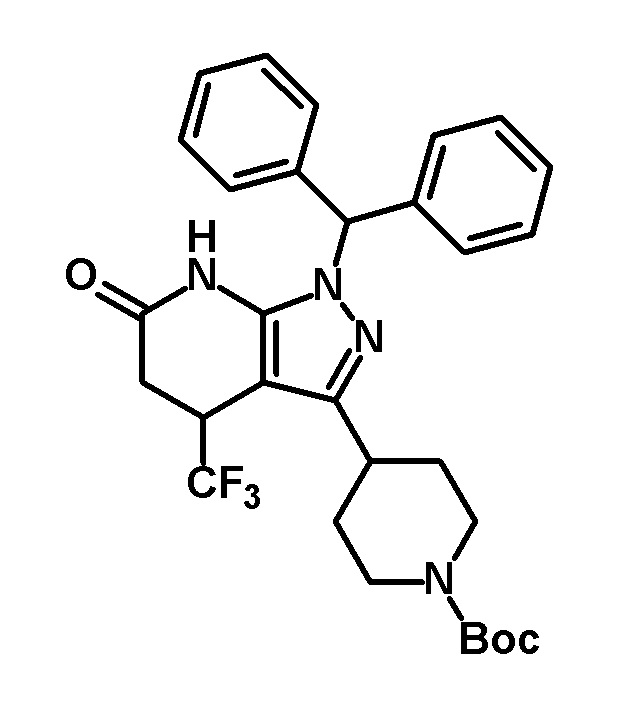
[0188] Указанное в заголовке соединение (1,48 г, выход: 46%) получали путем осуществления такой же реакции, как в Ссылочном примере 2, с использованием трет-бутил 4-[5-амино-1-(дифенилметил)-1H-пиразол-3-ил]пиперидин-1-карбоксилата (2,50 г, 5,78 ммоль), полученного в Ссылочном примере 12, вместо 1-(дифенилметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1H-пиразол-5-амина.
[0189] 1H-ЯМР (400 Гц, CDCl3) δ: 7,44-7,12 (10H, м), 6,69 (1H, с), 4,13 (1H, уш.с), 3,65-3,55 (1H, м), 2,95-2,69 (7H, м), 1,87-1,55 (4H, м), 1,45 (9H, с).
[0190] (Ссылочный пример 14) трет-Бутил 4-[транс-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилат
[0191]
[0192] Указанное в заголовке соединение (450 мг, выход: 35%) получали путем осуществления такой же реакции, как в Ссылочном примере 3, с использованием трет-бутил 4-[1-(дифенилметил)-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилата (1,24 г, 2,24 ммоль), полученного в Ссылочном примере 13, вместо транс-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0193] 1H-ЯМР (400 Гц, DMSO-d6) δ: 11,25 (1H, с), 7,37-7,19 (10H, м), 6,82 (1H, с), 6,66 (1H, д, J=5 Гц), 4,19 (1H, д, J=5 Гц), 3,99-3,87 (2H, м), 2,91-2,75 (3H, м), 1,78-1,40 (4H, м), 1,40 (9H, с).
[0194] (Ссылочный пример 15) транс-1-(Дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридазин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0195]
[0196] Хлортриметилсилан (0,10 мл, 0,79 ммоль) и иодид натрия (120 мг, 0,801 ммоль) добавляли при комнатной температуре к смешанному раствору трет-бутил 4-[транс-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилата (180 мг, 0,79 ммоль), полученного в Ссылочном примере 14, в дихлорметане (2 мл) и ацетонитрила (2 мл) и смесь перемешивали в течение 2 часов. Растворитель отгоняли из реакционного раствора при пониженном давлении с получением промежуточного продукта синтеза.
[0197] 3-Хлор-6-(трифторметил)пиридазин (130 мг, 0,712 ммоль) и N,N-диизопропилэтиламин (0,20 мл, 1,2 ммоль) добавляли к раствору промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, в DMSO (5 мл) и смесь перемешивали при комнатной температуре в течение двух с половиной дней. К реакционному раствору добавляли воду, с последующим экстрагированием этилацетатом три раза. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=90/10-50/50] с получением указанного в заголовке соединения (123 мг, выход: 63%).
[0198] 1H-ЯМР (400Гц, DMSO-d6) δ: 11,27 (1H, с), 7,75 (1H, д, J=10Гц), 7,41 (1H, д, J=10Гц), 7,35-7,17 (10H, м), 6,82 (1H, с), 6,69 (1H, д, J=5 Гц), 4,55-4,50 (2H, м), 4,21-4,20 (1H, м), 4,00-3,93 (1H, м), 3,19-2,97 (3H, м), 1,96-1,53 (4H, м).
[0199] (Ссылочный пример 16) трет-Бутил 4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилат
[0200]
[0201] Указанное в заголовке соединение (115 мг, выход: 15%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 4, с использованием трет-бутил 4-[транс-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилата (765 мг, 1,34 ммоль), полученного в Ссылочном примере 14, вместо транс-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0202] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,21 (1H, с), 7,37-7,15 (10H, м), 6,74 (1H, с), 5,79 (1H, д, J=4 Гц), 4,57-4,54 (1H, м), 4,16-3,90 (3H, м), 2,86-2,39 (3H, м), 1,83-1,35 (4H, м), 1,39 (9H, с).
[0203] (Ссылочный пример 17) цис-1-(Дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридазин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0204]
[0205] Хлортриметилсилан (64,6 мкл, 0,511 ммоль) и иодид натрия (65,3 мг, 0,435 ммоль) добавляли при комнатной температуре к смешанному раствору трет-бутил 4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилата (108 мг, 0,189 ммоль), полученного в Ссылочном примере 16, в дихлорметане (3 мл) и ацетонитрила (1 мл) и смесь перемешивали в течение 4 часов. Растворитель отгоняли из реакционного раствора при пониженном давлении и к остатку добавляли насыщенный водный раствор бикарбоната натрия, с последующим экстрагированием этилацетатом три раза. Полученный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении с получением промежуточного продукта синтеза (94 мг).
[0206] 3-Хлор-6-(трифторметил)пиридазин (21 мг, 0,16 ммоль) и N,N-диизопропилэтиламин (24 мкл, 0,14 ммоль) добавляли к раствору части (45 мг) промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, в DMSO (1 мл) и смесь перемешивали при комнатной температуре в течение 18 часов. К реакционному раствору добавляли воду, с последующим экстрагированием этилацетатом три раза. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=95/5-50/50] с получением указанного в заголовке соединения (34 мг, выход: 58%).
[0207] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,22 (1H, с), 7,75 (1H, д, J=10Гц), 7,41 (1H, д, J=10Гц), 7,34-7,24 (8H, м), 7,17-7,13 (2H, м), 6,75 (1H, с), 5,81 (1H, д, J=4 Гц), 4,61-4,48 (3H, м), 4,24-4,11 (1H, м), 3,22-3,09 (2H, м), 3,05-2,94 (1H, м), 2,03-1,94 (1H, м), 1,87-1,79 (1H, м), 1,76-1,63 (1H, м), 1,62-1,51 (1H, м).
[0208] (Ссылочный пример 18) транс-1-(Дифенилметил)-5-гидрокси-3-{1-[2-изопропил-6-(трифторметил)пиримидин-4-ил]пиперидин-4-ил}-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0209]
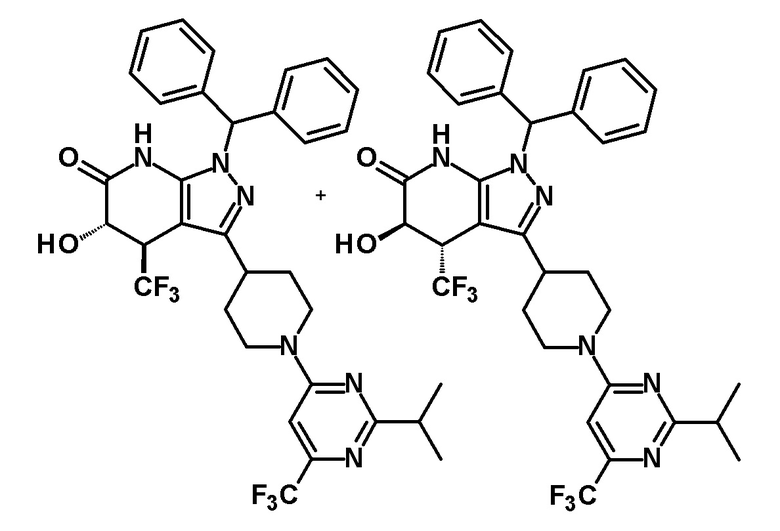
[0210] Указанное в заголовке соединение (300 мг, выход: 96%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 15, с использованием 4-хлор-2-изопропил-6-(трифторметил)пиримидина (соединение, описанное в опубликованной заявке WO2010/134478, 270 мг, 1,20 ммоль), вместо 3-хлор-6-(трифторметил)пиридазина.
[0211] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,27 (1H, с), 7,38-7,23 (9H, м), 7,18 (2H, д, J=7 Гц), 7,07 (1H, с), 6,82 (1H, с), 6,69 (1H, д, J=6 Гц), 4,20 (1H, д, J=6 Гц), 4,03-3,89 (1H, м), 3,19-2,86 (5H, м), 1,98-1,41 (4H, м), 1,21 (6H, д, J=7 Гц).
[0212] (Ссылочный пример 19) транс-1-(Дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиразин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0213]
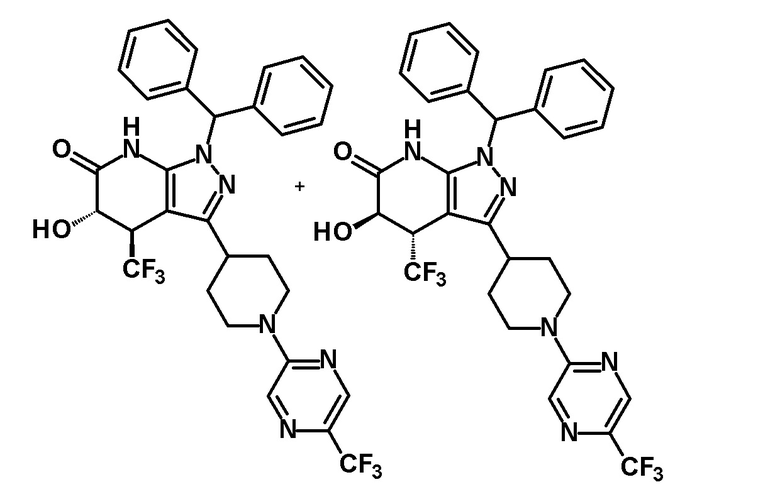
[0214] Указанное в заголовке соединение (106 мг, выход: 62%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 15, с использованием 2-хлор-5-(трифторметил)пиразина (120 мг, 0,657 ммоль) вместо 3-хлор-6-(трифторметил)пиридазина.
[0215] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,27 (1H, с), 8,45 (1H, с), 8,43 (1H, с), 7,35-7,17 (10H, м), 6,82 (1H, с), 6,70 (1H, д, J=5 Гц), 4,52-4,46 (2H, м), 4,21-4,19 (1H, м), 3,99-3,92 (1H, м), 3,19-3,14 (2H, м), 3,02-2,95 (1H, м), 1,94-1,85 (2H, м), 1,72-1,51 (2H, м).
[0216] (Ссылочный пример 20) 6-{4-[цис-1-(Дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-4-(трифторметил)пиридин-3-карбонитрил
[0217]
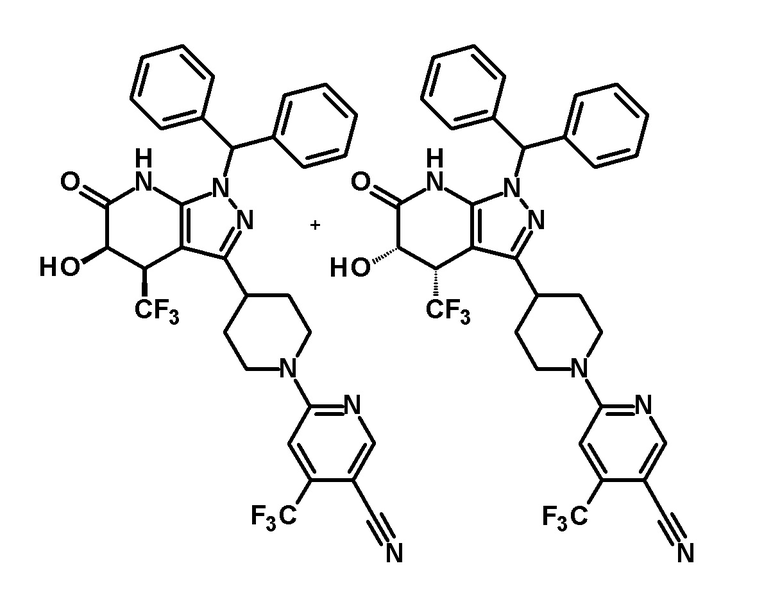
[0218] Указанное в заголовке соединение (47 мг, выход: 70%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 17, с использованием 6-хлор-4-(трифторметил)пиридин-3-карбонитрила (26 мг, 0,12 ммоль) вместо 3-хлор-6-(трифторметил)пиридазина.
[0219] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,23 (1H, с), 8,67 (1H, с), 7,37-7,12 (11H, м), 6,75 (1H, с), 5,82 (1H, д, J=4 Гц), 4,69-4,44 (3H, м), 4,24-4,12 (1H, м), 3,26-3,10 (2H, м), 3,05-2,94 (1H, м), 1,91-1,80 (2H, м), 1,71-1,50 (2H, м).
[0220] (Ссылочный пример 21) цис-3-{1-[3-Хлор-5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0221]
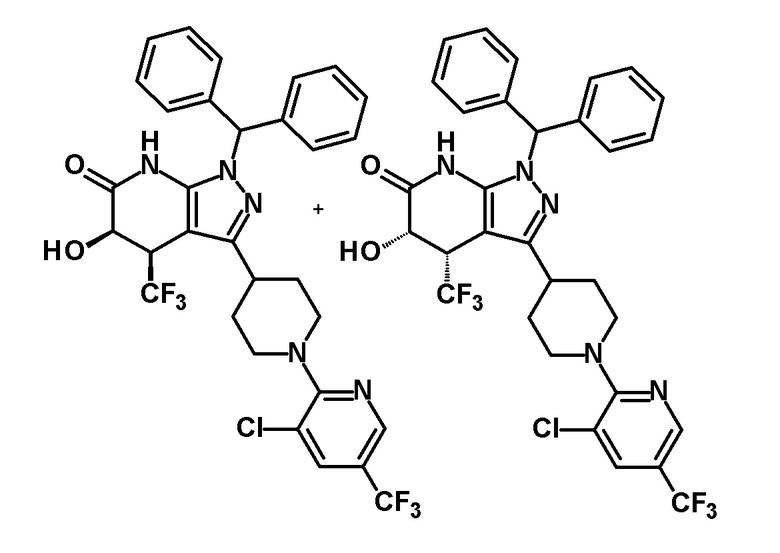
[0222] Указанное в заголовке соединение (40 мг, выход: 58%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 17, с использованием 3-хлор-2-фтор-5-(трифторметил)пиридина (25 мг, 0,13 ммоль) вместо 3-хлор-6-(трифторметил)пиридазина.
[0223] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,22 (1H, с), 8,54 (1H, д, J=2 Гц), 8,16 (1H, д, J=2 Гц), 7,39-7,25 (8H, м), 7,18-7,14 (2H, м), 6,76 (1H, с), 5,81 (1H, д, J=4 Гц), 4,61-4,55 (1H, м), 4,22-4,10 (1H, м), 4,09-3,97 (2H, м), 3,07-2,83 (3H, м), 2,02-1,93 (1H, м), 1,87-1,79 (2H, м), 1,77-1,65 (1H, м).
[0224] (Ссылочный пример 22) цис-1-(Дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[4-(трифторметил)-1,3-тиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0225]
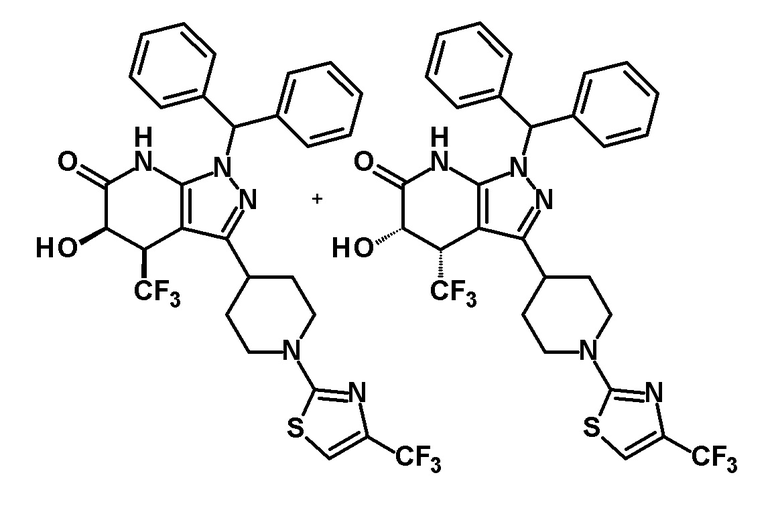
[0226] Указанное в заголовке соединение (12 мг, выход: 15%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 17, с использованием 2-хлор-4-(трифторметил)-1,3-тиазола (45 мг, 0,13 ммоль) вместо 3-хлор-6-(трифторметил)пиридазина.
[0227] 1H-ЯМР (400 MГц, CDCl3) δ: 7,39-7,33 (7H, м), 7,08-7,02 (4H, м), 6,90 (1H, д, J=1 Гц), 6,81 (1H, уш.с), 6,67 (1H, с), 4,49 (1H, д, J=7 Гц), 4,07-3,96 (2H, м), 3,65 (1H, д, J=2 Гц), 3,18-3,09 (2H, м), 2,83-2,74 (1H, м), 1,99-1,76 (4H, м).
[0228]
(Ссылочный пример 23) цис-1-(Дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0229]
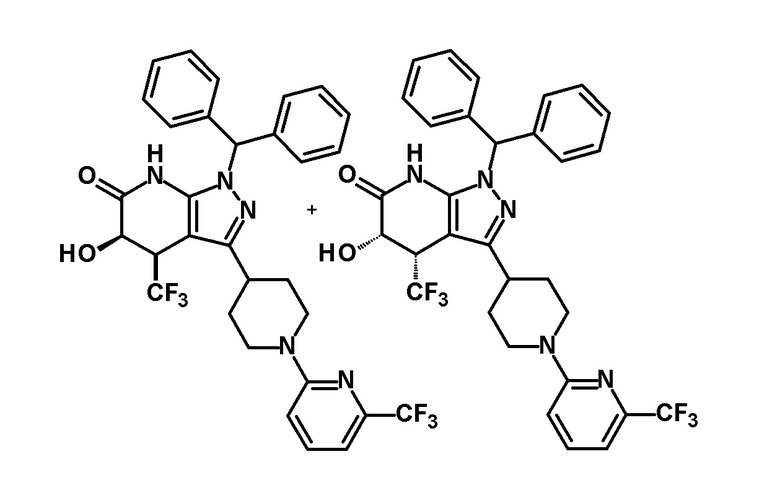
[0230] Указанное в заголовке соединение (18 мг, выход: 24%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 17, с использованием 2-фтор-6-(трифторметил)пиридина (24 мг, 0,15 ммоль) вместо 3-хлор-6-(трифторметил)пиридазина.
[0231] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,21 (1H, с), 7,70 (1H, т, J=8 Гц), 7,37-7,23 (8H, м), 7,18-7,10 (3H, м), 6,97 (1H, д, J=7 Гц), 6,74 (1H, с), 5,80 (1H, д, J=4 Гц), 4,60-4,55 (1H, м), 4,41-4,31 (2H, м), 4,20-4,10 (1H, м), 3,04-2,85 (3H, м), 1,98-1,90 (1H, м), 1,85-1,75 (1H, м), 1,72-1,60 (1H, м), 1,60-1,48 (1H, м).
[0232] (Ссылочный пример 24) цис-1-(Дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[4-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0233]
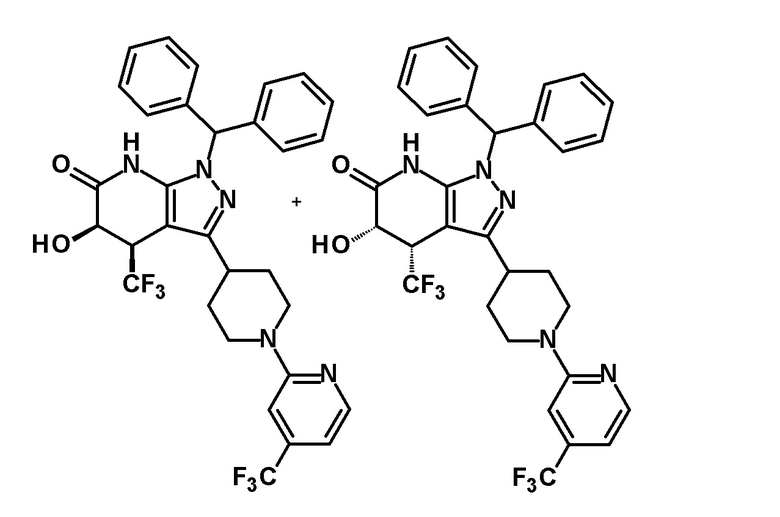
[0234] Указанное в заголовке соединение (29 мг, выход: 36%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 17, с использованием 2-фтор-4-(трифторметил)пиридина (42 мкл, 0,34 ммоль) вместо 3-хлор-6-(трифторметил)пиридазина.
[0235] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,21 (1H, с), 8,29 (1H, д, J=5 Гц), 7,37-7,23 (8H, м), 7,18-7,13 (2H, м), 7,08 (1H, с), 6,81 (1H, д, J=5 Гц), 6,74 (1H, с), 5,80 (1H, д, J=4 Гц), 4,61-4,54 (1H, м), 4,47-4,36 (2H, м), 4,22-4,10 (1H, м), 3,06-2,86 (3H, м), 1,97-1,88 (1H, м), 1,83-1,74 (1H, м), 1,72-1,60 (1H, м), 1,60-1,47 (1H, м).
[0236] (Пример 1) транс-5-Гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0237]
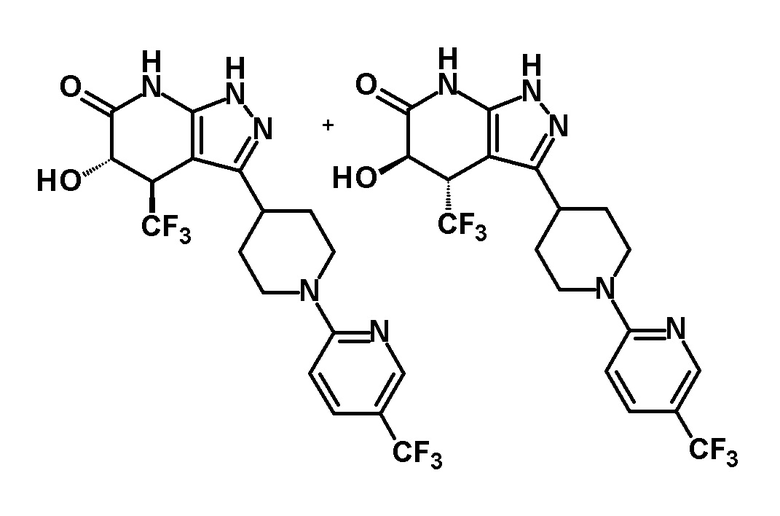
[0238] Триэтилсилан (0,02 мл, 0,13 ммоль) и трифторуксусную кислоту (2 мл) добавляли к раствору транс-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (34 мг, 0,0552 ммоль), полученного в Ссылочном примере 3, в дихлорметане (2 мл) и смесь перемешивали при комнатной температуре в течение 15 минут. К реакционному раствору добавляли насыщенный водный раствор бикарбоната натрия, с последующим экстрагированием этилацетатом три раза. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=60/40-0/100 (градиент)] с получением указанного в заголовке соединения (20 мг, выход: 81%).
[0239] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,13 (1H, с), 10,52 (1H, с), 8,42 (1H, с), 7,79 (1H, т, J=6 Гц), 6,54 (1H, д, J=5 Гц), 4,63-4,52 (2H, м), 4,16 (1H, д, J=5 Гц), 3,94-3,85 (1H, м), 3,14-2,92 (3H, м), 1,83-1,62 (4H, м), 1,29-1,21 (1H, м);
MS (ESI) m/z: 450 (M+H)+.
[0240] (Пример 2) цис-5-Гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0241]
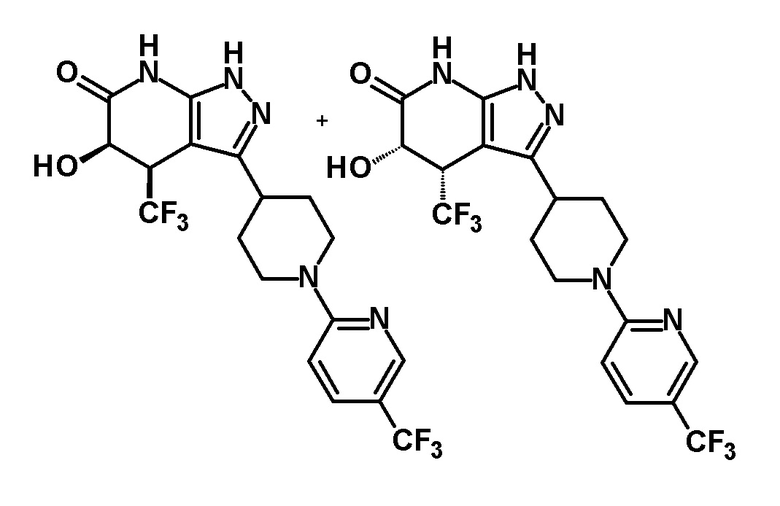
[0242] Триэтилсилан (0,05 мл, 0,3 ммоль) и трифторуксусную кислоту (3 мл) добавляли к раствору цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (95 мг, 0,154 ммоль), полученного в Ссылочном примере 4, в дихлорметане (3 мл) и смесь перемешивали при комнатной температуре в течение 1 часа. К реакционному раствору добавляли насыщенный водный раствор бикарбоната натрия, с последующим экстрагированием этилацетатом три раза. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [NH-силикагель, элюент: этилацетат/метанол=100/0-95/5 (градиент)] с получением указанного в заголовке соединения (45 мг, выход: 65%).
[0243] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,18 (1H, с), 10,53 (1H, с), 8,42 (1H, с), 7,79 (1H, дд, J=9 Гц, 2 Гц), 7,01 (1H, д, J=9 Гц), 5,51 (1H, д, J=4 Гц), 4,62-4,52 (2H, м), 4,46-4,42 (1H, м), 4,20-4,11 (1H, м), 3,13-2,93 (3H, м), 1,89-1,56 (4H, м);
MS (ESI) m/z: 450 (M+H)+.
[0244] (Пример 3) (+)-цис-5-Гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0245]
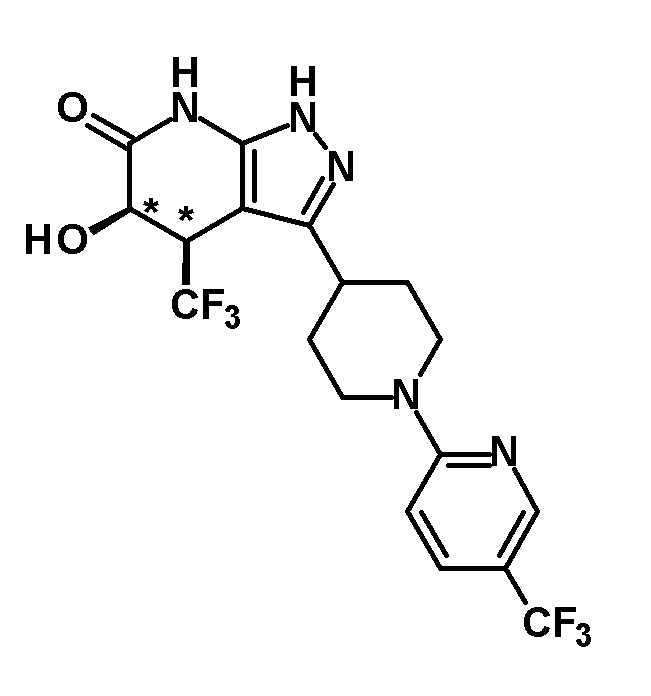
[0246] цис-5-Гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он (0,14 г, 0,31 ммоль), полученный в Примере 2, растворяли в этаноле (30 мл) при нагревании. К раствору затем добавляли гексан (10 мл) и полученный раствор очищали 10 отдельными порциями при помощи ВЭЖХ [колонка: Chiralpak IA (20 мм в.д. ×250 мм); изготовитель Daicel Corporation, элюент: гексан/этанол=60/40, скорость потока: 15 мл/мин] с получением указанного в заголовке соединения (51 мг, выход: 36%, оптически активная форма).
[0247] Оптическую чистоту измеряли с использованием ВЭЖХ [колонка: Chiralpak IA (4,6 мм в.д. ×250 мм); изготовитель Daicel Corporation, элюент: гексан/IPA=60/40, скорость потока: 1,0 мл/мин].
[0248] Оптическая чистота 99% или выше (время удерживания: 10,4 мин);
[α]D25=+12° (DMF, c=1,01).
[0249] (Пример 4) 4,5-Дигидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0250]
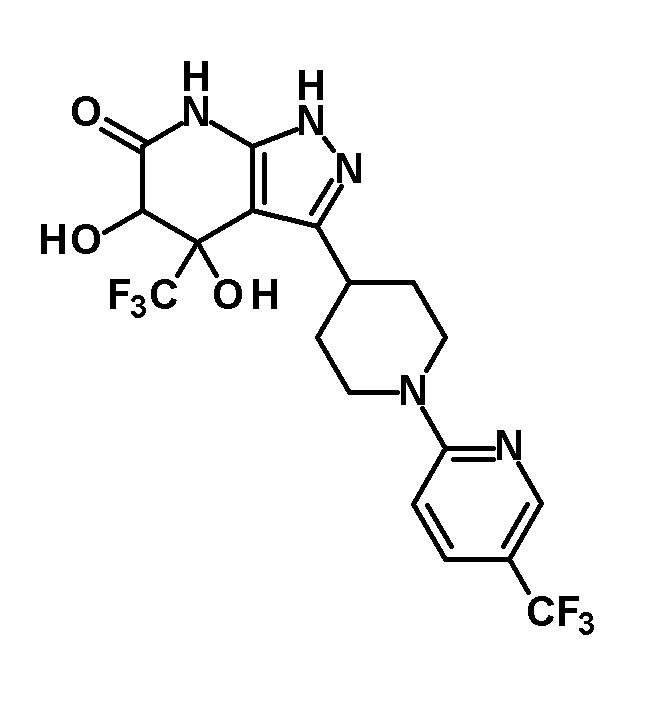
[0251] Триэтилсилан (0,015 мл, 0,094 ммоль) и трифторуксусную кислоту (1 мл) добавляли к раствору 1-(дифенилметил)-4,5-дигидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (31 мг, 0,049 ммоль), полученного в Ссылочном примере 6, в дихлорметане (1 мл) и смесь перемешивали в течение 30 минут. К реакционному раствору добавляли насыщенный водный раствор бикарбоната натрия, с последующим экстрагированием этилацетатом три раза. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=60/40-20/80] с получением указанного в заголовке соединения (15 мг, 0,032 ммоль).
[0252] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,22 (1H, с), 10,55 (1H, с), 8,42 (1H, с), 7,79 (1H, дд, J=9 Гц, 2 Гц), 7,00 (1H, д, J=9 Гц), 6,79 (1H, с), 5,66 (1H, д, J=4 Гц), 4,58 (2H, д, J=13 Гц), 4,35 (1H, с), 3,33-3,24 (1H, м), 2,94 (2H, т, J=12 Гц), 1,90 (1H, д, J=10Гц), 1,76-1,55 (3H, м);
MS (ESI) m/z: 466 (M+H)+.
[0253] (Пример 5) цис-5-Гидрокси-3-[1-(5-изопропоксипиридин-2-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0254]
[0255] Указанное в заголовке соединение (25 мг, выход: 54%) получали путем осуществления такой же реакции, как в способе, описанном в Примере 2, с использованием цис-1-(дифенилметил)-5-гидрокси-3-[1-(5-изопропоксипиридин-2-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (64 мг, 0,11 ммоль), полученного в Ссылочном примере 11, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0256] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,23 (1H, с), 10,54 (1H, с), 7,84 (1H, д, J=3 Гц), 7,25 (1H, дд, J=9 Гц, 3 Гц), 6,83 (1H, д, J=9 Гц), 5,51 (1H, д, J=4 Гц), 4,47-4,41 (2H, м), 4,32-4,21 (2H, м), 4,19-4,08 (1H, м), 2,98-2,87 (1H, м), 2,80-2,69 (2H, м), 1,86-1,62 (4H, м), 1,23 (6H, д, J=6 Гц);
MS (ESI) m/z: 440 (M+H)+.
[0257] (Пример 6) транс-5-Гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридазин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0258]
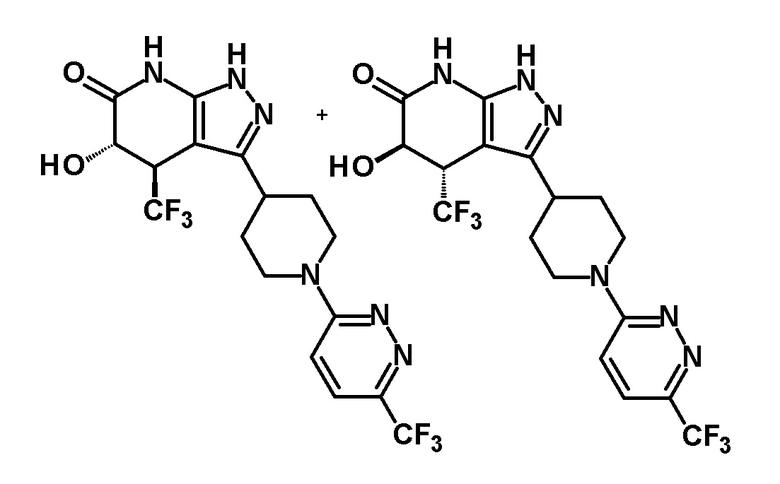
[0259] Указанное в заголовке соединение (14 мг, выход: 96%) получали путем осуществления такой же реакции, как в способе, описанном в Примере 2, с использованием транс-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридазин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (20 мг, 0,0324 ммоль), полученного в Ссылочном примере 15, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0260] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,14 (1H, с), 10,53 (1H, с), 7,82 (1H, д, J=10 Гц), 7,47 (1H, д, J=10 Гц), 6,55 (1H, д, J=5 Гц), 4,69-4,62 (2H, м), 4,17-4,16 (1H, м), 3,94-3,86 (1H, м), 3,17-3,04 (3H, м), 1,86-1,69 (4H, м);
MS (ESI) m/z: 451 (M+H)+.
[0261] (Пример 7) цис-5-Гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридазин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0262]
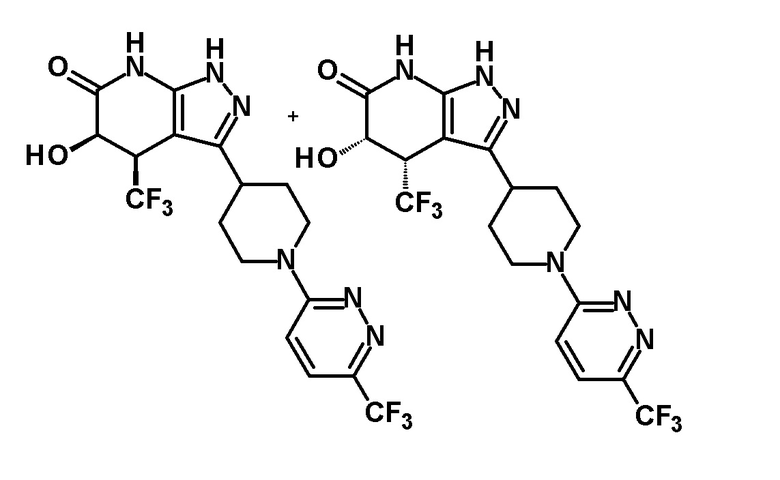
[0263] Указанное в заголовке соединение (17 мг, выход: 73%) получали путем осуществления такой же реакции, как в способе, описанном в Примере 2, с использованием цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридазин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (32 мг, 0,052 ммоль), полученного в Ссылочном примере 17, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0264] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,19 (1H, с), 10,54 (1H, с), 7,81 (1H, д, J=10 Гц), 7,47 (1H, д, J=10 Гц), 5,53 (1H, д, J=4 Гц), 4,73-4,60 (2H, м), 4,48-4,41 (1H, м), 4,23-4,11 (1H, м), 3,19-3,03 (3H, м), 1,93-1,84 (1H, м), 1,82-1,61 (3H, м);
MS (ESI) m/z: 451 (M+H)+.
[0265] (Пример 8) транс-5-Гидрокси-3-{1-[2-изопропил-6-(трифторметил)пиримидин-4-ил]пиперидин-4-ил}-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0266]
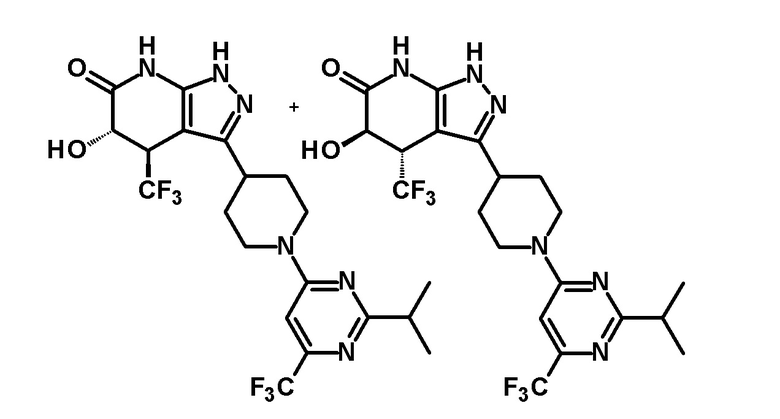
[0267] Указанное в заголовке соединение (52 мг, выход: 93%) получали путем осуществления такой же реакции, как в способе, описанном в Примере 2, с использованием транс-1-(дифенилметил)-5-гидрокси-3-{1-[2-изопропил-6-(трифторметил)пиримидин-4-ил]пиперидин-4-ил}-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (75 мг, 0,114 ммоль), полученного в Ссылочном примере 18, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0268] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,11 (1H, с), 10,53 (1H, с), 7,14 (1H, с), 6,55 (1H, уш.с), 5,34-4,58 (2H, м), 4,16 (1H, с), 3,90 (1H, кв., J=10 Гц), 3,82-3,35 (2H, м), 3,17-2,87 (2H, м), 1,86-1,60 (4H, м), 1,23 (6H, д, J=7 Гц);
MS (ESI) m/z: 493 (M+H)+.
[0269] (Пример 9) цис-5-Гидрокси-3-{1-[2-изопропил-6-(трифторметил)пиримидин-4-ил]пиперидин-4-ил}-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0270]
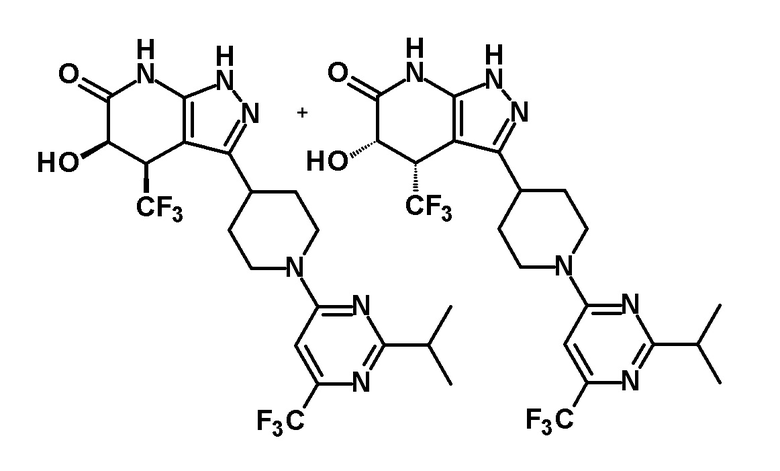
[0271] Защищенную форму дифенилметила получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 4, с использованием транс-1-(дифенилметил)-5-гидрокси-3-{1-[2-изопропил-6-(трифторметил)пиримидин-4-ил]пиперидин-4-ил}-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (300 мг, 0,456 ммоль), полученного в Ссылочном примере 18, вместо транс-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0272] Указанное в заголовке соединение (7 мг, выход: 9%) получали путем осуществления такой же реакции, как в способе, описанном в Примере 2, с использованием защищенной формы дифенилметила, полученной, с использованием процедур, описанных выше, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0273] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,12 (1H, с), 10,50 (1H, с), 7,10 (1H, с), 5,49 (1H, с), 4,41-4,39 (1H, м), 4,17-4,05 (1H, м), 3,10-2,84 (4H, м), 1,86-1,52 (4H, м), 1,19 (6H, д, J=7 Гц);
MS (ESI) m/z: 493 (M+H)+.
[0274] (Пример 10) транс-5-Гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиразин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0275]
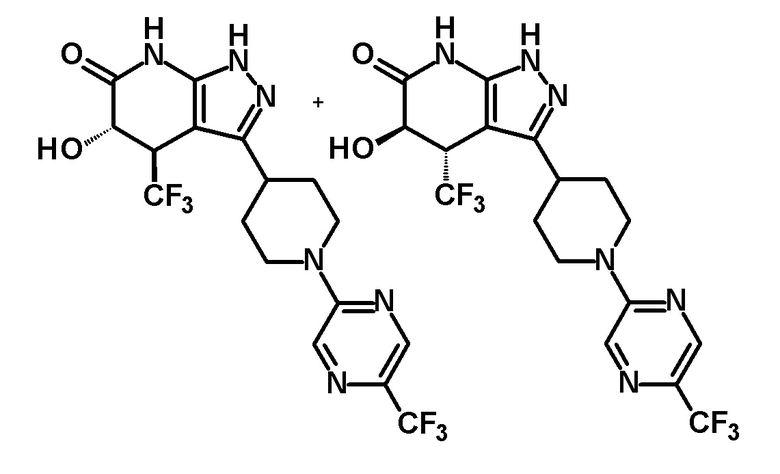
[0276] Указанное в заголовке соединение (70 мг, выход: 90%) получали путем осуществления такой же реакции, как в способе, описанном в Примере 2, с использованием транс-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиразин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (106 мг, 0,172 ммоль), полученного в Ссылочном примере 19, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0277] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,13 (1H, с), 10,54 (1H, с), 8,51 (1H, с), 8,50 (1H, с), 6,59 (1H, д, J=5 Гц), 4,66-4,59 (2H, м), 4,17-4,16 (1H, м), 3,98-3,83 (1H, м), 3,18-2,98 (3H, м), 1,87-1,64 (4H, м);
MS (ESI) m/z: 451 (M+H)+.
[0278] (Пример 11) цис-5-Гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиразин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0279]
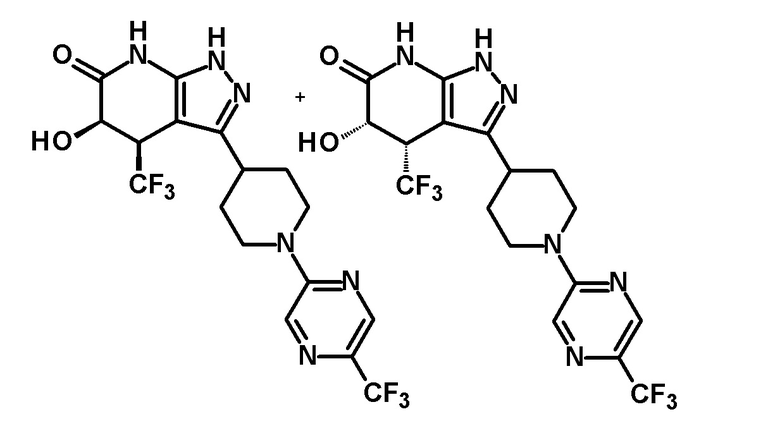
[0280] Промежуточный продукт синтеза получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 15, с использованием трет-бутил 4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилата (115 мг, 0,202 ммоль), полученного в Ссылочном примере 16, вместо трет-бутил 4-[транс-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилата и с использованием 2-хлор-5-(трифторметил)пиразина (90 мг, 0,493 ммоль) вместо 3-хлор-6-(трифторметил)пиридазина.
[0281] Указанное в заголовке соединение (58 мг, выход: 99%) получали путем осуществления такой же реакции, как в способе, описанном в Примере 2, с использованием промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0282] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,18 (1H, с), 10,54 (1H, с), 8,51 (1H, с), 8,50 (1H, с), 5,54 (1H, д, J=4 Гц), 4,65-4,59 (2H, м), 4,46-4,43 (1H, м), 4,22-4,13 (1H, м), 3,16-3,03 (3H, м), 1,90-1,61 (4H, м);
MS (ESI) m/z: 451 (M+H)+.
[0283] (Пример 12) цис-5-Гидрокси-4-(трифторметил)-3-{1-[2-(трифторметил)пиримидин-5-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0284]
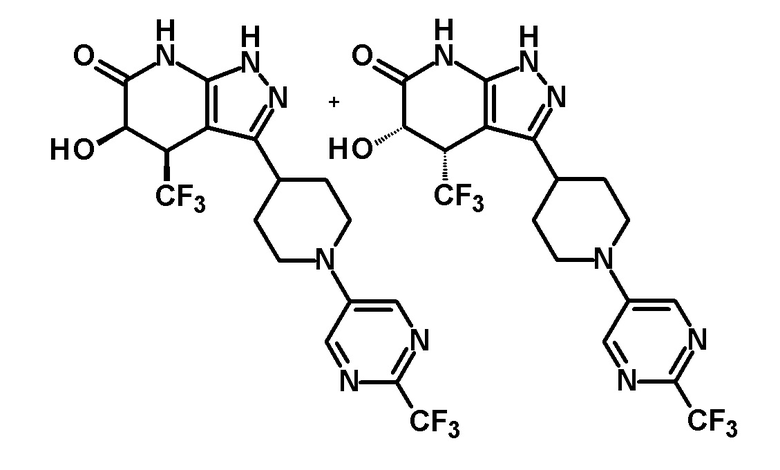
[0285] Хлортриметилсилан (25 мкл, 0,20 ммоль) и иодид натрия (25 мг, 0,17 ммоль) добавляли при комнатной температуре к смешанному раствору трет-бутил 4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилата (42 мг, 0,074 ммоль), полученного в Ссылочном примере 16, в дихлорметане (3 мл) и ацетонитриле (1 мл) и смесь перемешивали в течение 2 часов. Растворитель отгоняли из реакционного раствора при пониженном давлении и к остатку добавляли насыщенный водный раствор бикарбоната натрия, с последующим экстрагированием этилацетатом три раза. Полученный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении с получением промежуточного продукта синтеза.
[0286] 5-Хлор-2-(трифторметил)пиримидин (30 мг, 0,16 ммоль) и 1,8-диазабицикло[5,4,0]-7-ундецен (0,050 мл, 0,33 ммоль) добавляли к раствору промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, в DMSO (3 мл) и смесь перемешивали при 70°C в течение 18 часов. К реакционному раствору добавляли воду, с последующим экстрагированием этилацетатом три раза. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=80/20-0/100] с получением защищенной формы дифенилметила.
[0287] Указанное в заголовке соединение (6,7 мг, выход: 20%) получали путем осуществления такой же реакции, как в способе, описанном в Примере 2, с использованием защищенной формы дифенилметила, полученной с использованием процедур, описанных выше, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0288] 1H-ЯМР (400 MГц, DMSO-d6)δ: 12,22 (1H, с), 10,55 (1H, с), 8,67 (2H, с), 5,53 (1H, д, J=3 Гц), 4,44 (1H, д, J=6 Гц), 4,25-4,10 (3H, м), 3,14-2,95 (3H, м), 1,93-1,38 (4H, м);
MS (ESI) m/z: 451 (M+H)+.
[0289] (Пример 13) 6-{4-[цис-5-Гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-4-(трифторметил)пиридин-3-карбонитрил
[0290]
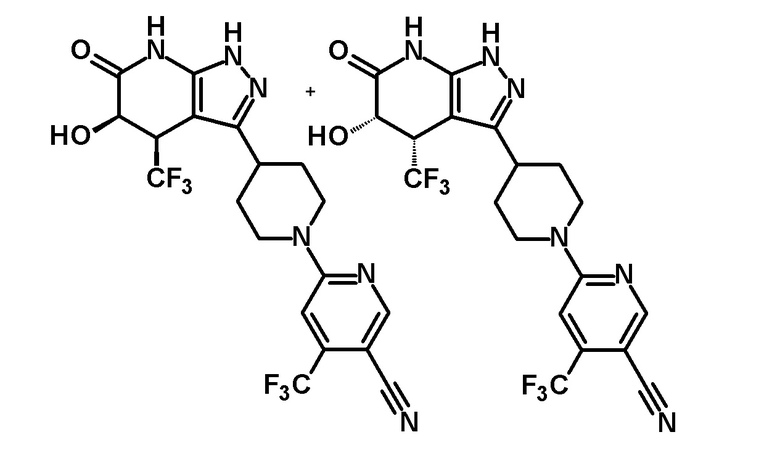
[0291] Указанное в заголовке соединение (23 мг, выход: 69%) получали путем осуществления такой же реакции, как в способе, описанном в Примере 2, с использованием 6-{4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-4-(трифторметил)пиридин-3-карбонитрила (45 мг, 0,070 ммоль), полученного в Ссылочном примере 20, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0292] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,17 (1H, с), 10,54 (1H, с), 8,73 (1H, с), 7,34 (1H, с), 5,54 (1H, д, J=4 Гц), 4,87-4,56 (2H, м), 4,48-4,41 (1H, м), 4,22-4,11 (1H, м), 3,20-3,01 (3H, м), 1,94-1,85 (1H, м), 1,82-1,57 (3H, м);
MS (ESI) m/z: 475 (M+H)+.
[0293] (Пример 14) цис-3-{1-[3-Хлор-5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0294]
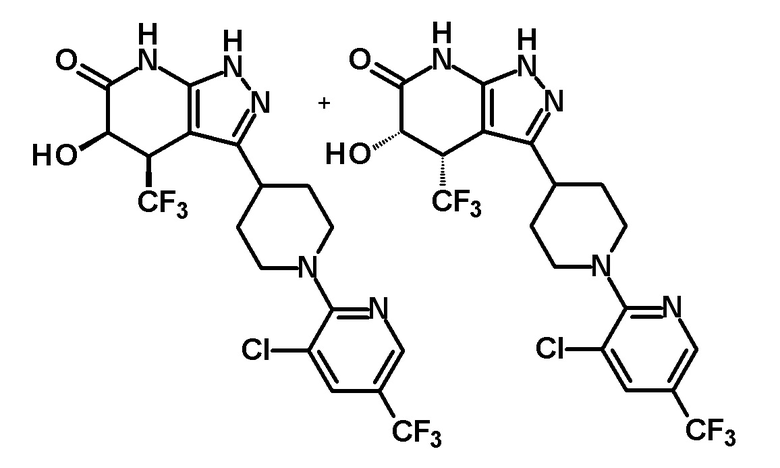
[0295] Указанное в заголовке соединение (21 мг, выход: 78%) получали путем осуществления такой же реакции, как в способе, описанном в Примере 2, с использованием цис-3-{1-[3-хлор-5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (36 мг, 0,055 ммоль), полученного в Ссылочном примере 21, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0296] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,28 (1H, с), 10,54 (1H, с), 8,57 (1H, д, J=2 Гц), 8,20 (1H, д, J=2 Гц), 5,52 (1H, д, J=4 Гц), 4,47-4,42 (1H, м), 4,21-4,07 (3H, м), 3,07-2,91 (3H, м), 1,98-1,74 (4H, м);
MS (ESI) m/z: 484 (M+H)+.
[0297] (Пример 15) цис-5-Гидрокси-4-(трифторметил)-3-{1-[4-(трифторметил)-1,3-тиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0298]
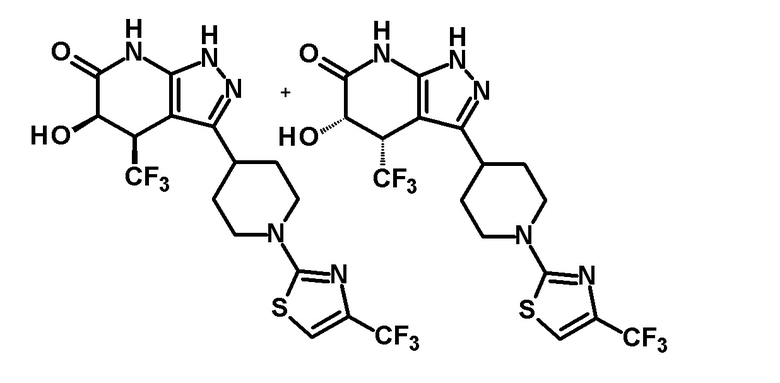
[0299] Указанное в заголовке соединение (6,4 мг, выход: 73%) получали путем осуществления такой же реакции, как в способе, описанном в Примере 2, с использованием цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[4-(трифторметил)-1,3-тиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (12 мг, 0,019 ммоль), полученного в Ссылочном примере 22, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0300] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,26 (1H, с), 10,55 (1H, с), 7,55 (1H, д, J=1 Гц), 5,53 (1H, д, J=3 Гц), 4,45 (1H, дд, J=7 Гц, 4 Гц), 4,20-4,11 (1H, м), 4,04-3,95 (2H, м), 3,21-2,99 (3H, м), 1,92-1,67 (4H, м);
MS (ESI) m/z: 456 (M+H)+.
[0301] (Пример 16) цис-5-Гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0302]
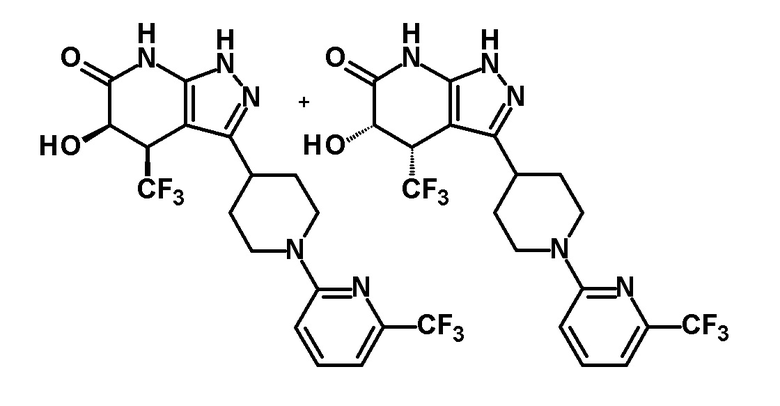
[0303] Указанное в заголовке соединение (16 мг, выход: 71%) получали путем осуществления такой же реакции, как в способе, описанном в Примере 2, с использованием цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (31 мг, 0,050 ммоль), полученного в Ссылочном примере 23, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0304] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,21 (1H, с), 10,54 (1H, с), 7,74 (1H, т, J=9 Гц), 7,17 (1H, д, J=9 Гц), 7,01 (1H, д, J=7 Гц), 5,51 (1H, д, J=4 Гц), 4,54-4,41 (3H, м), 4,22-4,09 (1H, м), 3,09-2,86 (3H, м), 1,92-1,82 (1H, м), 1,81-1,58 (3H, м);
MS (ESI) m/z: 450 (M+H)+.
[0305] (Пример 17) цис-5-Гидрокси-4-(трифторметил)-3-{1-[4-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0306]
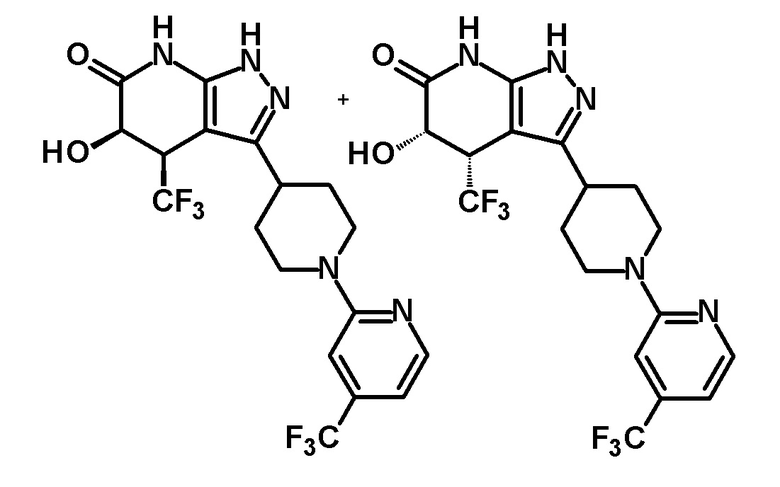
[0307] Указанное в заголовке соединение (14 мг, выход: 74%) получали путем осуществления такой же реакции, как в способе, описанном в Примере 2, с использованием цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[4-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (26 мг, 0,042 ммоль), полученного в Ссылочном примере 24, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0308] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,19 (1H, с), 10,53 (1H, с), 8,33 (1H, д, J=5 Гц), 7,13 (1H, с), 6,85 (1H, д, J=5 Гц), 5,51 (1H, д, J=4 Гц), 4,62-4,49 (2H, м), 4,47-4,41 (1H, м), 4,21-4,10 (1H, м), 3,10-3,00 (1H, м), 3,00-2,86 (2H, м), 1,89-1,80 (1H, м), 1,78-1,57 (3H, м);
MS (ESI) m/z: 450 (M+H)+.
[0309] (Ссылочный пример 25) Метил 3-[1-(трет-бутоксикарбонил)пиперидин-4-ил]-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-5-карбоксилат
[0310]
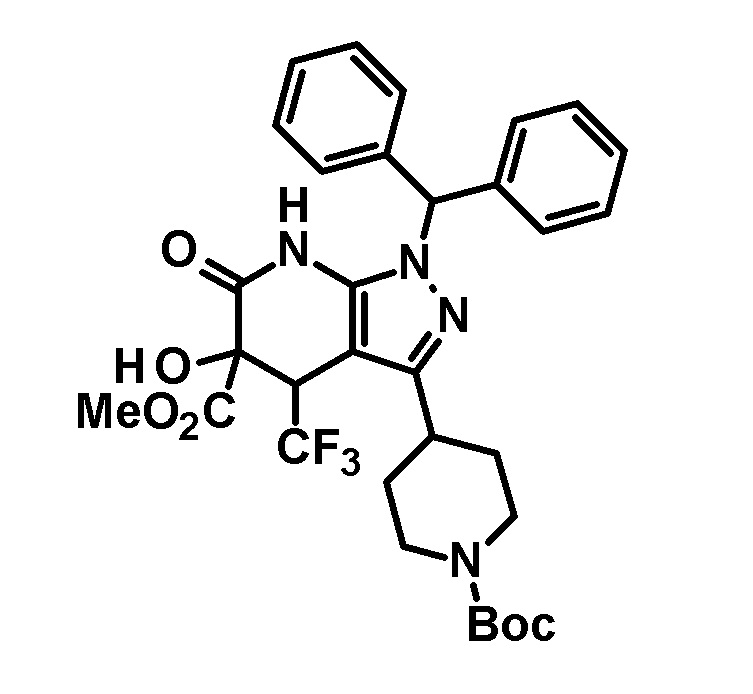
[0311] Диизопропиламид лития (раствор в гексане и THF, 14,5 мл, 15,8 ммоль) добавляли по каплям при -78°C к раствору трет-бутил 4-[1-(дифенилметил)-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилата (2,92 г, 5,27 ммоль), полученного в Ссылочном примере 13, и диметилкарбоната (0,665 мл, 7,90 ммоль) в THF (50 мл). После удаления охлаждающей бани смесь перемешивали в течение 30 минут, при этом температура смеси спонтанно повышалась. К реакционному раствору добавляли насыщенный водный раствор хлорида аммония, с последующим экстрагированием этилацетатом три раза. Полученный органический слой сушили над сульфатом магния и растворитель отгоняли при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле [NH-силикагель, элюент: дихлорметан/метанол=100/0-90/10 (градиент)] с получением промежуточного продукта синтеза.
[0312] 1,8-Диазабицикло[5,4,0]-7-ундецен (далее указывается как DBU; 1,57 мл, 10,5 ммоль), (1S)-(+)-(10-камфорсульфонил)оксазиридин (0,725 г, 3,16 ммоль) и (1R)-(-)-(10-камфорсульфонил)оксазиридин (0,725 г, 3,16 ммоль) добавляли к раствору промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, в THF (50 мл) и смесь перемешивали при комнатной температуре в течение 2 часов. К реакционному раствору добавляли насыщенный водный раствор хлорида аммония, с последующим экстрагированием этилацетатом. Полученный органический слой промывали насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: дихлорметан/метанол=99/1-90/10 (градиент)] с получением указанного в заголовке соединения (2,77 г, выход: 84%).
[0313] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,52 (1H, с), 7,38-7,17 (10H, м), 6,86 (1H, с), 4,12-3,89 (3H, м), 3,73 (3H, с), 2,89-2,67 (3H, м), 1,81-1,29 (4H, м), 1,39 (9H, с).
[0314] (Ссылочный пример 26) трет-Бутил (+)-4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилат
[0315]
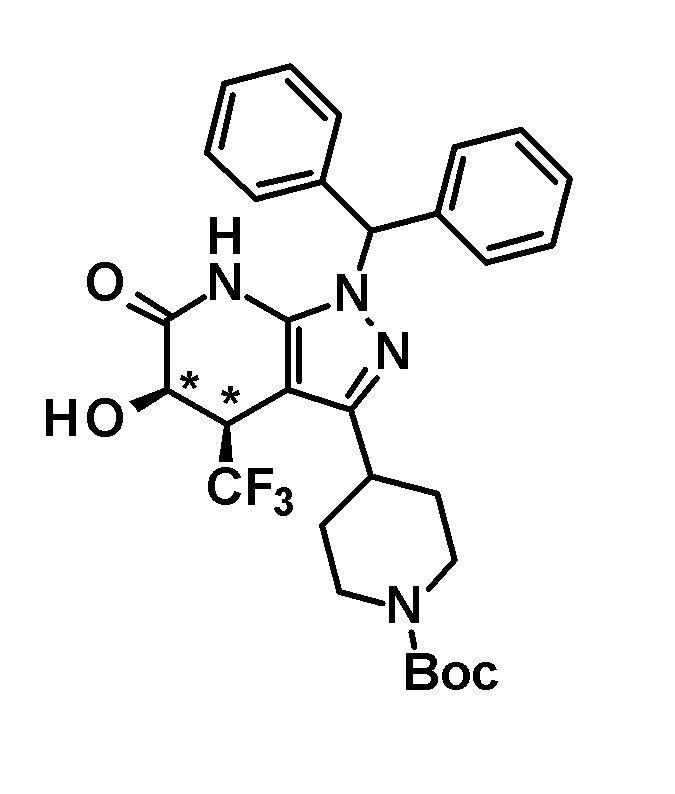
[0316] Гидроксид лития моногидрат (0,873 г, 20,8 ммоль) добавляли к смешанному раствору метил 3-[1-(трет-бутоксикарбонил)пиперидин-4-ил]-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-5-карбоксилата (4,36 г, 6,94 ммоль), полученного в Ссылочном примере 25, в 1,4-диоксане (50 мл) и воде (20 мл) и смесь перемешивали при 50°C в течение 1 часа. Реакционный раствор охлаждали до комнатной температуры и к раствору добавляли насыщенный водный раствор хлорида аммония, с последующим экстрагированием этилацетатом. Полученный органический слой промывали насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=95/5-50/50 (градиент)] с получением промежуточного продукта синтеза.
[0317] Часть (1,23 г) промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, растворяли в этилацетате. К раствору добавляли нейтральный силикагель для адсорбции и растворитель отгоняли при пониженном давлении. Полученный порошок очищали жидкостной флэш-хроматографией [колонка: Chiralflash IC (30 мм в.д. ×100 мм); изготовитель Daicel Corporation, элюент: гексан/этанол=91/9, скорость потока: 12 мл/мин] с получением указанного в заголовке соединения (0,55 г, выход: 27%, оптически активная форма).
[0318] Оптическую чистоту измеряли с использованием ВЭЖХ [колонка: Chiralpak IA (4,6 мм в.д. × 250 мм); изготовитель Daicel Corporation, элюент: гексан/IPA=70/30, скорость потока: 1,0 мл/мин].
[0319] Оптическая чистота 99% или выше (время удерживания: 4,3 мин);
[α]D25=+35° (DMF, c=1,00).
[0320] (Пример 18) (+)-цис-5-Гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиразин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0321]
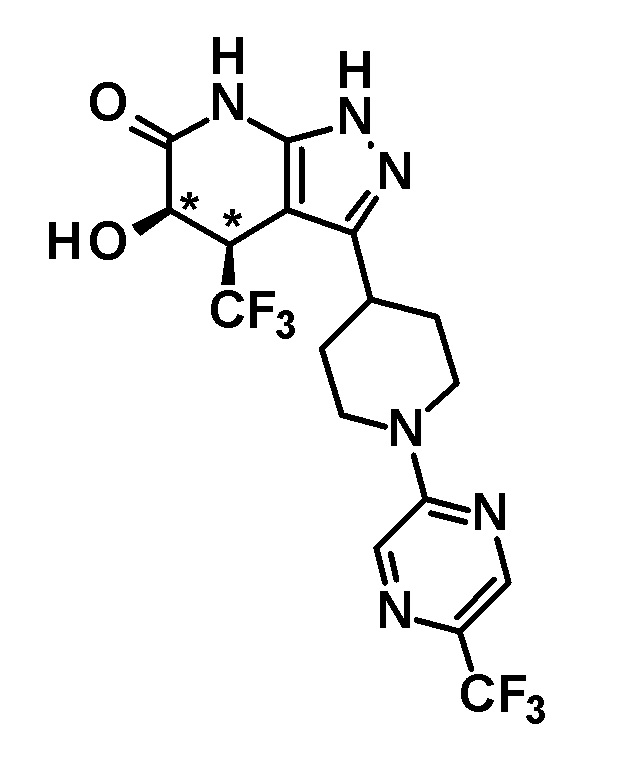
[0322] Хлортриметилсилан (72,3 мкл, 0,573 ммоль) и иодид натрия (73,1 мг, 0,488 ммоль) добавляли к смешанному раствору трет-бутил (+)-4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилата (121 мг, 0,212 ммоль), полученного в Ссылочном примере 26, в дихлорметане (5 мл) и ацетонитриле (2 мл) и смесь перемешивали при комнатной температуре в течение 4 часов. Растворитель отгоняли из реакционного раствора при пониженном давлении и остаток разделяли на органический и водный слои путем добавления насыщенного водного раствора бикарбоната натрия и этилацетата. Полученный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении.
[0323] 2-Хлор-5-(трифторметил)пиразин (31,4 мкл, 0,254 ммоль) и N,N-диизопропилэтиламин (54,1 мкл, 0,318 ммоль) добавляли к раствору полученного остатка в DMSO (2 мл) и смесь перемешивали при комнатной температуре в течение 1 часа и затем оставляли выстаиваться в течение ночи. Реакционный раствор разбавляли этилацетатом, промывали водой и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=95/5-50/50 (градиент)] с получением промежуточного продукта синтеза.
[0324] Триэтилсилан (0,113 мл, 0,707 ммоль) и трифторуксусную кислоту (1,35 мл, 17,7 ммоль) добавляли к раствору промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, в дихлорметане (2 мл) и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель отгоняли из реакционного раствора и остаток разделяли на органический и водный слои путем добавления этилацетата и насыщенного водного раствора бикарбоната натрия. Полученный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: этилацетат/метанол=100/0-95/5 (градиент)] с получением указанного в заголовке соединения (66 мг, выход: 71%, оптически активная форма).
[0325] [α]D25=+13° (DMF, c=1,00).
[0326] (Пример 19) (+)-цис-5-Гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридазин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0327]
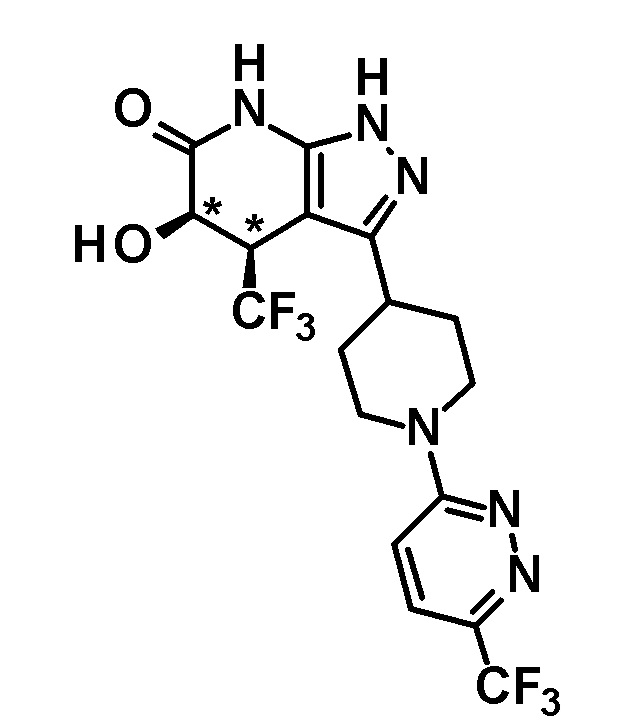
[0328] Указанное в заголовке соединение (70 мг, выход: 69%, оптически активная форма) получали путем осуществления такой же реакции, как в способе, описанном в Примере 18, с использованием 3-хлор-6-(трифторметил)пиридазина (50,6 мг, 0,277 ммоль) вместо 2-хлор-5-(трифторметил)пиразина.
[0329]
[α]D25=+12° (DMF, c=1,00).
[0330] (Пример 20) (+)-6-{4-[цис-5-Гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-4-(трифторметил)пиридин-3-карбонитрил
[0331]
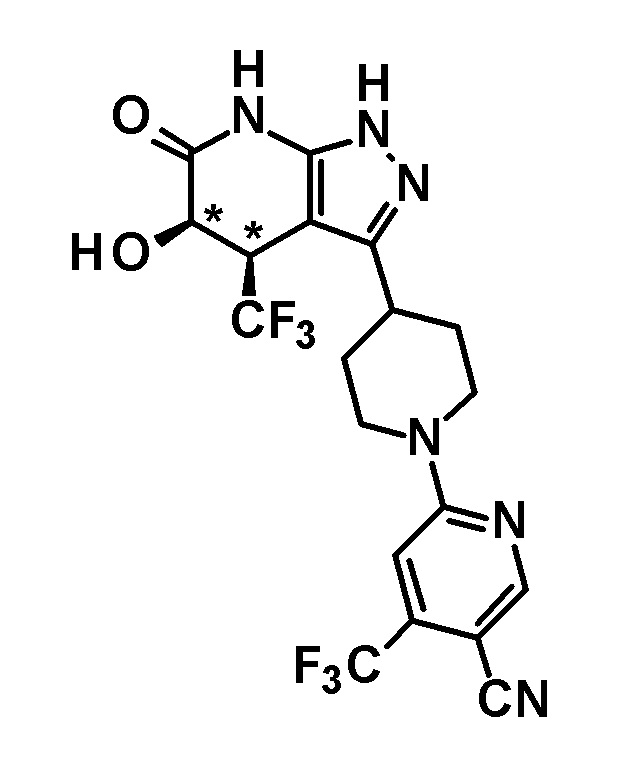
[0332] Указанное в заголовке соединение (56 мг, выход: 68%, оптически активная форма) получали путем осуществления такой же реакции, как в способе, описанном в Примере 18, с использованием 6-хлор-4-(трифторметил)пиридин-3-карбонитрила (43,9 мг, 0,212 ммоль) вместо 2-хлор-5-(трифторметил)пиразина.
[0333] [α]D25=+18° (DMF, c=1,00).
[0334] (Пример 21) (+)-цис-5-Гидрокси-4-(трифторметил)-3-{1-[2-(трифторметил)пиримидин-5-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0335]
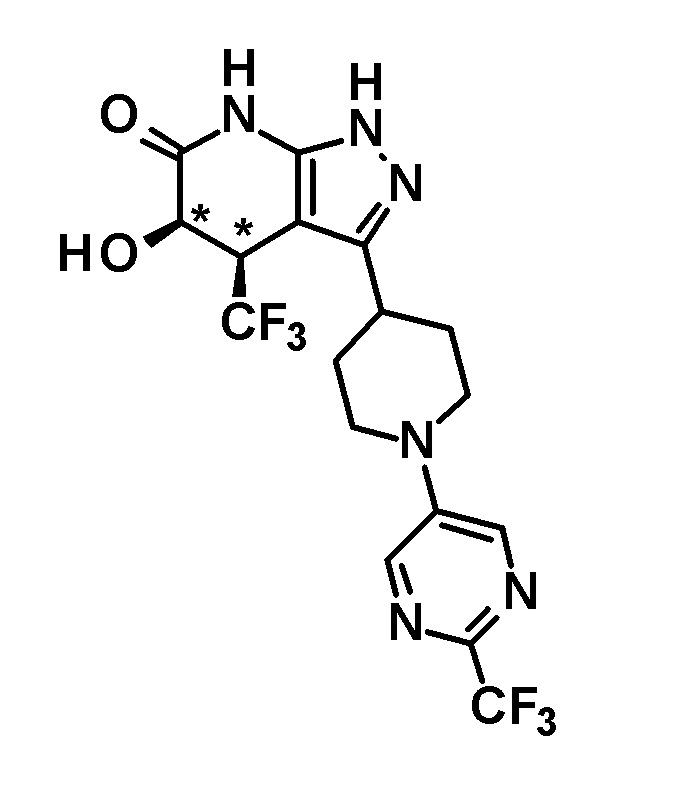
[0336] Хлортриметилсилан (0,31 мл, 2,5 ммоль) и иодид натрия (0,31 г, 2,1 ммоль) добавляли к смешанному раствору трет-бутил (+)-4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилата (0,52 г, 0,91 ммоль), полученного в Ссылочном примере 26, в дихлорметане (25 мл) и ацетонитриле (10 мл) и смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель отгоняли из реакционного раствора при пониженном давлении и остаток разделяли на органический и водный слои путем добавления насыщенного водного раствора бикарбоната натрия и этилацетата. Полученный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении.
[0337] 5-Хлор-2-(трифторметил)пиримидин (0,37 мл, 2,0 ммоль) и DBU (0,62 мл, 4,2 ммоль) добавляли к раствору полученного остатка в DMSO (30 мл) и смесь перемешивали при 70°C в течение 18 часов. Реакционный раствор разбавляли этилацетатом, промывали водой и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=95/5-50/50 (градиент)] с получением промежуточного продукта синтеза.
[0338] Триэтилсилан (0,200 мл, 1,26 ммоль) и трифторуксусную кислоту (1,0 мл, 13 ммоль) добавляли к раствору промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, в дихлорметане (3 мл) и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель отгоняли из реакционного раствора и остаток разделяли на органический и водный слои путем добавления этилацетата и насыщенного водного раствора бикарбоната натрия. Полученный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: этилацетат/метанол=50/50-0/100 (градиент)] с получением указанного в заголовке соединения (0,11 г, выход: 26%, оптически активная форма).
[0339] [α]D25=+7,2° (DMF, c=1,00).
[0340] (Пример 22) (+)-цис-3-[1-(5-Хлорпиридин-2-ил)пиперидин-4-ил]-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0341]
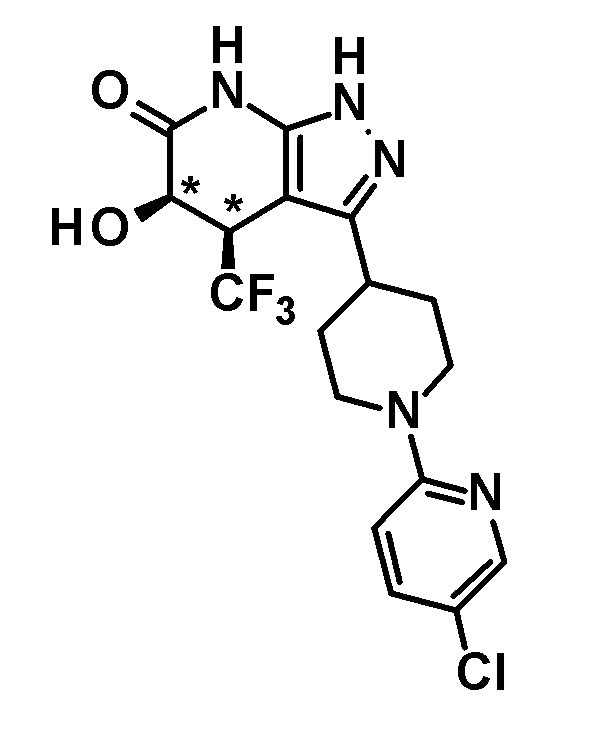
[0342] Указанное в заголовке соединение (39 мг, выход: 44%, оптически активная форма) получали путем осуществления такой же реакции, как в способе, описанном в Примере 21, за исключением того, что использовали 5-хлор-2-фторпиридин (55,8 мкл, 0,556 ммоль) вместо 5-хлор-2-(трифторметил)пиримидина, и смесь перемешивали при комнатной температуре в течение 1 часа и оставляли, вместо перемешивания, при 70°C в течение 18 часов.
[0343] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,20 (1H, с), 10,53 (1H, с), 8,11 (1H, д, J=3 Гц), 7,58 (1H, дд, J=9 Гц, 3 Гц), 6,91 (1H, д, J=9 Гц), 5,51 (1H, уш.с), 4,45-4,37 (3H, м), 4,18-4,10 (1H, м), 3,04-2,97 (1H, м), 2,92-2,81 (2H, м), 1,84-1,58 (4H, м);
MS (ESI) m/z: 416 (M+H)+.
[0344] [α]D25=+12° (DMF, c=1,01).
[0345] (Ссылочный пример 27) Оптически активная форма цис-1-(дифенилметил)-5-гидрокси-3-(пиперидин-4-ил)-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она
[0346]
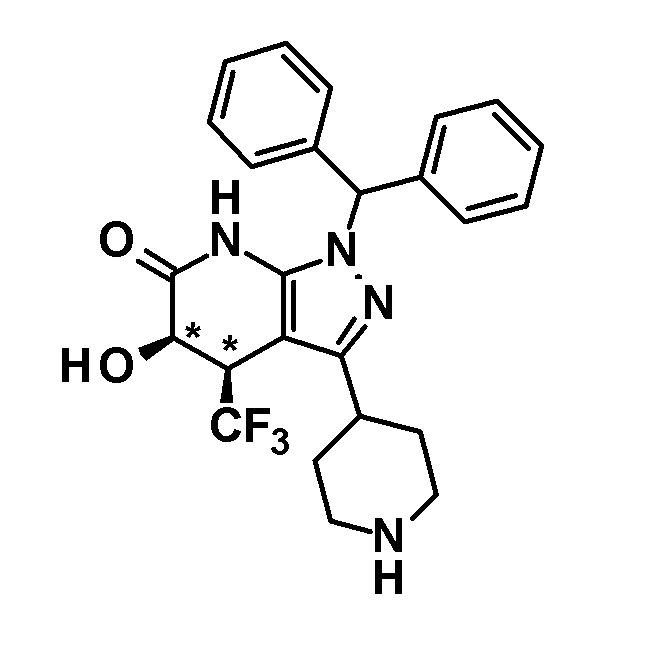
[0347] Хлортриметилсилан (1,42 мл, 11,2 ммоль) и иодид натрия (1,68 г, 11,2 ммоль) добавляли к смешанному раствору трет-бутил (+)-4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилата (3,20 г, 5,61 ммоль), полученного в Ссылочном примере 26, в дихлорметане (100 мл) и ацетонитриле (30 мл) и смесь перемешивали при комнатной температуре в течение 2 часов. К реакционному раствору добавляли N-диизопропилэтиламин (2,86 мл, 16,8 ммоль) и воду (15 мл) и растворитель отгоняли при пониженном давлении. К полученному остатку добавляли дихлорметан (150 мл) и смесь перемешивали. Осадок собирали фильтрованием с получением указанного в заголовке соединения (2,00 г, выход: 76%, оптически активная форма).
[0348] 1H-ЯМР (400 MГц, DMSO-d6) δ: 7,41-7,11 (10H, м), 6,80 (1H, с), 5,86 (1H, д, J=4 Гц), 4,60-4,55 (1H, м), 4,16-4,07 (1H, м), 3,33-3,21 (2H, м), 2,99-2,87 (3H, м), 2,03-1,66 (4H, м).
[0349]
(Ссылочный пример 28) Оптически активная форма цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она
[0350]
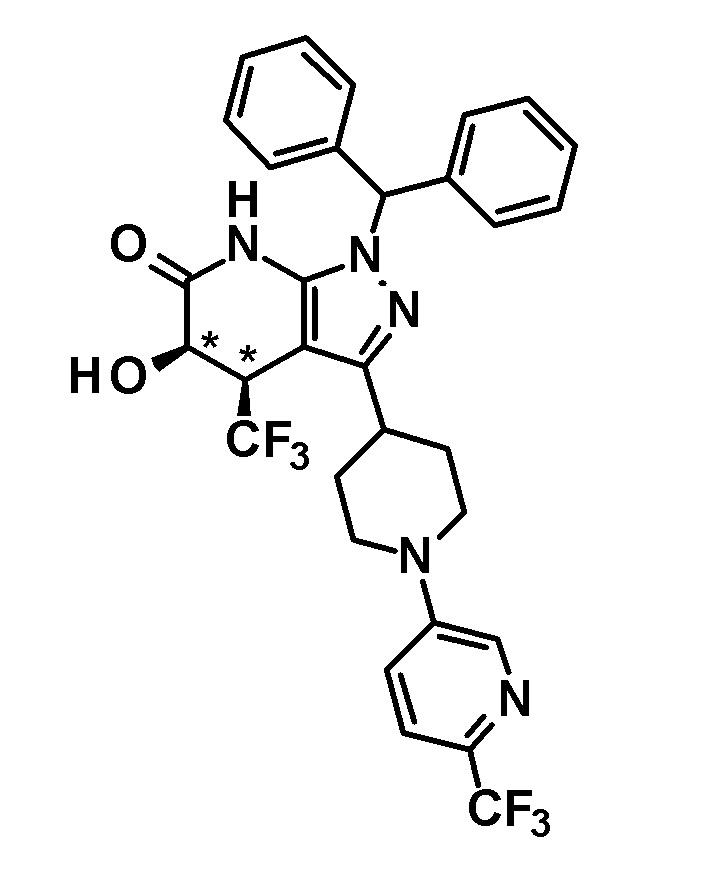
[0351] DBU (87,6 мкл, 0,587 ммоль) и 5-фтор-2-(трифторметил)пиридин (51,9 мкл, 0,440 ммоль) добавляли при комнатной температуре к суспензии оптически активной формы цис-1-(дифенилметил)-5-гидрокси-3-(пиперидин-4-ил)-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (0,138 г, 0,293 ммоль), полученного в Ссылочном примере 27, в DMSO (3 мл) и смесь перемешивали при комнатной температуре в течение 3 часов и затем при 60°C в течение 8 часов на масляной бане. Реакционный раствор снова доводили до комнатной температуры, затем разбавляли этилацетатом и промывали водой и насыщенным солевым раствором, в указанном порядке. Органический слой сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=93/7-55/45 (градиент)] с получением указанного в заголовке соединения (0,135 г, выход: 75%, оптически активная форма).
[0352] 1H-ЯМР (400 MГц, CDCl3) δ: 8,32 (1H, д, J=3 Гц), 7,90 (1H, с), 7,50 (1H, д, J=9 Гц), 7,39-7,34 (6H, м), 7,19 (1H, дд, J=9 Гц, 3 Гц), 7,15-7,10 (4H, м), 6,66 (1H, с), 4,55 (1H, д, J=7 Гц), 3,96-3,81 (3H, м), 3,72 (1H, с), 3,04-2,97 (2H, м), 2,87-2,79 (1H, м), 2,04-1,85 (4H, м).
[0353] (Пример 23) (+)-цис-5-Гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0354]
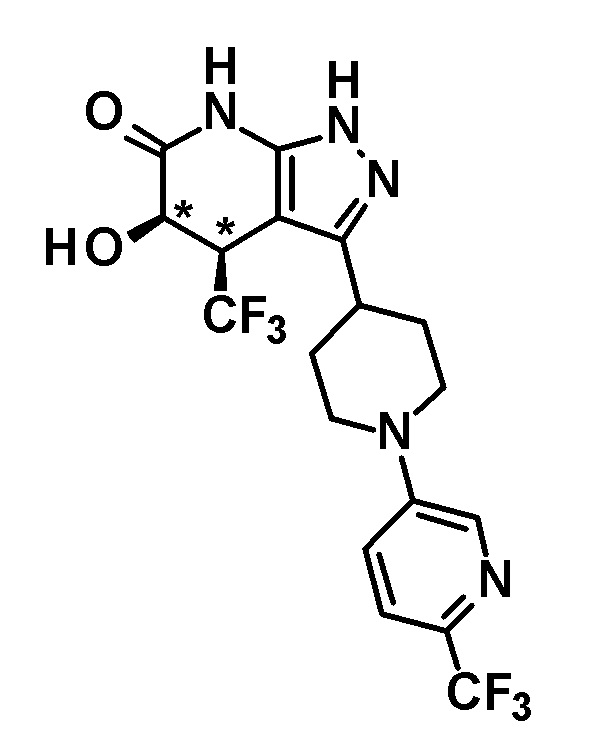
[0355] Трифторуксусную кислоту (0,8 мл, 10 ммоль) добавляли при комнатной температуре к раствору оптически активной формы цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (0,135 г, 0,219 ммоль), полученной в Ссылочном примере 28, и триэтилсилана (0,140 мл, 0,877 ммоль) в дихлорметане (4 мл) и смесь перемешивали в течение 45 минут. Растворитель отгоняли из реакционного раствора при пониженном давлении и полученный остаток разделяли на органический и водный слои путем добавления этилацетата и насыщенного водного раствора бикарбоната натрия. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=83/17-0/100 (градиент)] с получением указанного в заголовке соединения (88,9 мг, выход: 90%, оптически активная форма).
[0356] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,24 (1H, с), 10,54 (1H, с), 8,46 (1H, д, J=3 Гц), 7,64 (1H, д, J=9 Гц), 7,47 (1H, дд, J=9 Гц, 3 Гц), 5,52 (1H, д, J=4 Гц), 4,45 (1H, дд, J=7 Гц, 4 Гц), 4,20-4,05 (3H, м), 3,06-2,91 (3H, м), 1,87-1,73 (4H, м);
MS (ESI) m/z: 450 (M+H)+;
[α]D25=+7,5° (DMF, c=0,958).
[0357] (Пример 24) (+)-3-Фтор-5-{4-[цис-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}пиридин-2-карбонитрил
[0358]
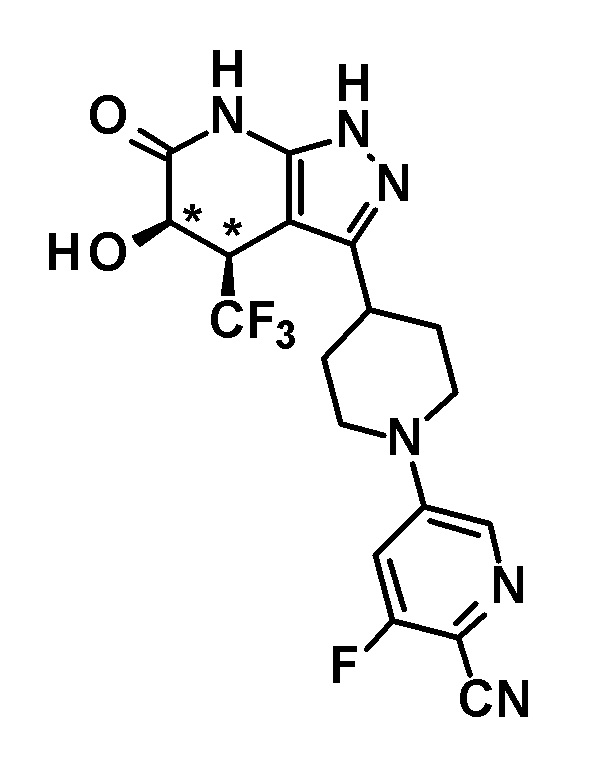
[0359] DBU (0,135 мл, 0,901 ммоль) и 3,5-дифторпиридин-2-карбонитрил (94,7 г, 0,676 ммоль) добавляли при комнатной температуре к суспензии оптически активной формы цис-1-(дифенилметил)-5-гидрокси-3-(пиперидин-4-ил)-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (0,212 г, 0,451 ммоль), полученной в Ссылочном примере 27, в DMSO (2 мл) и смесь перемешивали в течение 66 часов. Реакционный раствор разбавляли этилацетатом и промывали водой и насыщенным солевым раствором, в указанном порядке. Органический слой сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=93/7-50/50 (градиент)] с получением промежуточного продукта синтеза.
[0360] Указанное в заголовке соединение (84,8 г, выход: 44%, оптически активная форма) получали путем осуществления такой же реакции, как в способе, описанном в Примере 23, с использованием промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, вместо оптически активной формы цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0361] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,20 (1H, с), 10,54 (1H, с), 8,36-8,35 (1H, м), 7,45 (1H, дд, J=14 Гц, 2 Гц), 5,53 (1H, д, J=4 Гц), 4,46-4,43 (1H, м), 4,23-4,11 (3H, м), 3,12-3,04 (3H, м), 1,88-1,65 (4H, м);
MS (ESI) m/z: 425 (M+H)+;
[α]D25=+14° (DMF, c=1,01).
[0362] (Пример 25) (+)-цис-5-Гидрокси-4-(трифторметил)-3-{1-[2-(трифторметил)пиримидин-4-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0363]
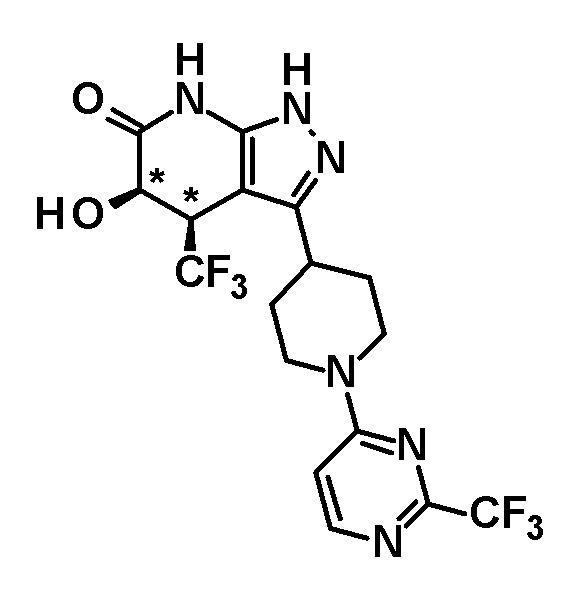
[0364] Указанное в заголовке соединение (0,121 г, выход: 60%, оптически активная форма) получали путем осуществления такой же реакции, как в способе, описанном в Примере 24, с использованием 4-хлор-2-трифторметилпиримидина (0,123 г, 0,676 ммоль) вместо 3,5-дифторпиридин-2-карбонитрила.
[0365] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,17 (1H, с), 10,54 (1H, с), 8,34 (1H, д, J=7 Гц), 7,13 (1H, д, J=7 Гц), 5,52 (1H, д, J=4 Гц), 4,74-4,30 (3H, м), 4,21-4,12 (1H, м), 3,16-2,99 (3H, м), 1,91-1,57 (4H, м);
MS (ESI) m/z: 451 (M+H)+;
[α]D25=+12° (DMF, c=1,01).
[0366] (Ссылочный пример 29) Оптически активная форма цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она
[0367]
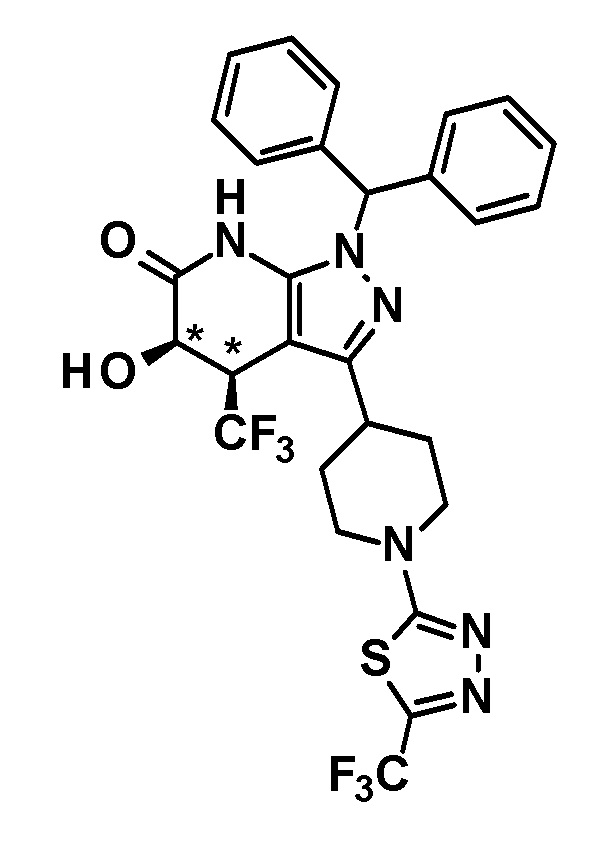
[0368] DBU (0,135 мл, 0,901 ммоль) и 2-хлор-5-трифторметил-1,3,4-тиадиазол (0,127 г, 0,676 ммоль) добавляли при комнатной температуре к суспензии оптически активной формы цис-1-(дифенилметил)-5-гидрокси-3-(пиперидин-4-ил)-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (0,212 г, 0,451 ммоль), полученной в Ссылочном примере 27, в DMSO (1 мл) и смесь перемешивали в течение 18 часов. Реакционный раствор разбавляли этилацетатом и промывали насыщенным солевым раствором три раза. Полученный органический слой сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=95/5-50/50 (градиент)] с получением указанного в заголовке соединения (0,207 г, выход: 74%, оптически активная форма).
[0369] 1H-ЯМР (400 MГц, CDCl3) δ: 7,40-7,37 (6H, м), 7,13-7,06 (5H, м), 6,68 (1H, с), 4,53 (1H, д, J=7 Гц), 4,08-4,00 (2H, м), 3,93-3,85 (1H, м), 3,69 (1H, д, J=3 Гц), 3,39-3,32 (2H, м), 2,92-2,85 (1H, м), 2,04-1,88 (4H, м).
[0370]
(Пример 26) (+)-цис-5-Гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0371]
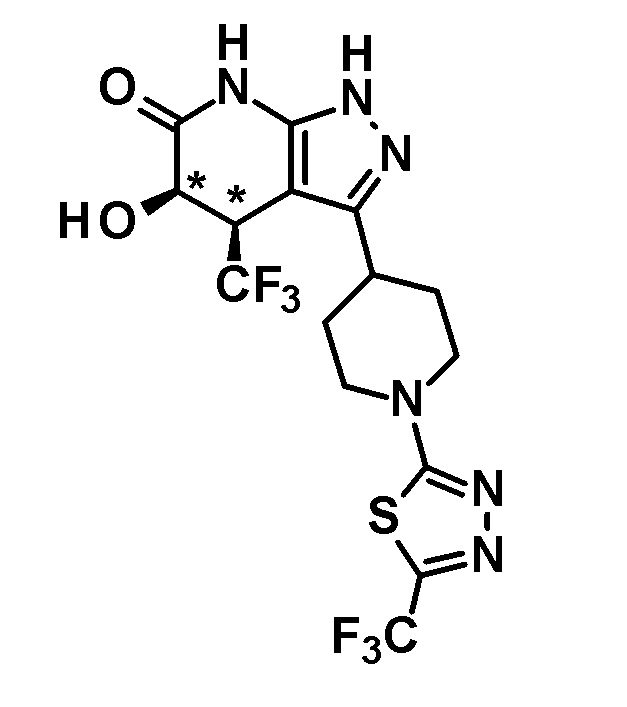
[0372] Трифторуксусную кислоту (0,8 мл, 10 ммоль) добавляли при комнатной температуре к раствору оптически активной формы цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (0,196 г, 0,315 ммоль), полученной в Ссылочном примере 29, и триэтилсилана (0,201 мл, 1,26 ммоль) в дихлорметане (4 мл) и смесь перемешивали в течение 45 минут. Растворитель отгоняли из реакционного раствора при пониженном давлении и полученный остаток разделяли на органический и водный слои путем добавления этилацетата и насыщенного водного раствора бикарбоната натрия. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=80/20-0/100 (градиент)] с получением указанного в заголовке соединения (0,109 г, выход: 76%, оптически активная форма).
[0373] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,25 (1H, с), 10,56 (1H, с), 5,54 (1H, уш.с), 4,45 (1H, д, J=7 Гц), 4,21-4,12 (1H, м), 4,09-4,01 (2H, м), 3,42-3,35 (2H, м), 3,14-3,06 (1H, м), 1,93-1,76 (4H, м);
MS (ESI) m/z: 457 (M+H)+;
[α]D25=+8,7° (DMF, c=1,01).
[0374] (Ссылочный пример 30) Оптически активная форма 5-{4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-3-метилпиридин-2-карбонитрила
[0375]
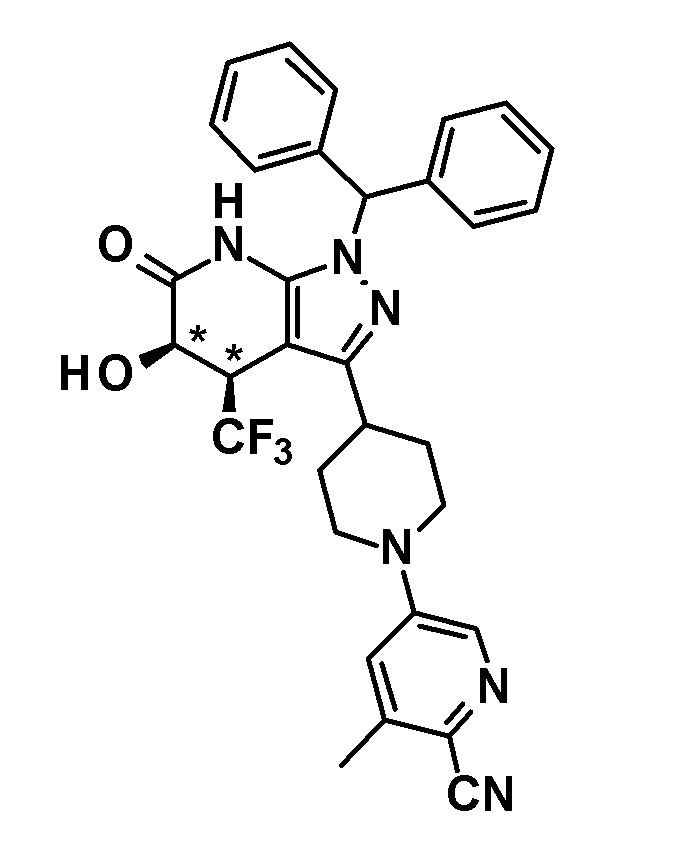
[0376] Карбонат цезия (0,734 г, 2,25 ммоль) и 5-хлор-3-метилпиридин-2-карбонитрил (0,206 г, 1,35 ммоль) добавляли при комнатной температуре к суспензии оптически активной формы цис-1-(дифенилметил)-5-гидрокси-3-(пиперидин-4-ил)-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (0,212 г, 0,451 ммоль), полученной в Ссылочном примере 27, в DMSO (2 мл) и смесь перемешивали при 150°C в течение 2 часов. Реакционную суспензию охлаждали до комнатной температуры, затем разбавляли этилацетатом и промывали насыщенным солевым раствором. Полученный органический слой сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=92/8-50/50 (градиент)] с получением указанного в заголовке соединения (0,226 г, выход: 86%, оптически активная форма).
[0377] 1H-ЯМР (400 MГц, CDCl3) δ: 8,14 (1H, д, J=3 Гц), 7,40-7,36 (6H, м), 7,12-7,06 (5H, м), 6,91 (1H, д, J=3 Гц), 6,69 (1H, с), 4,53 (1H, д, J=7 Гц), 3,94-3,86 (3H, м), 3,69 (1H, д, J=3 Гц), 3,08-3,00 (2H, м), 2,89-2,81 (1H, м), 2,45 (3H, с), 2,02-1,80 (4H, м).
[0378] (Пример 27) (+)-5-{4-[цис-5-Гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-3-метилпиридин-2-карбонитрил
[0379]
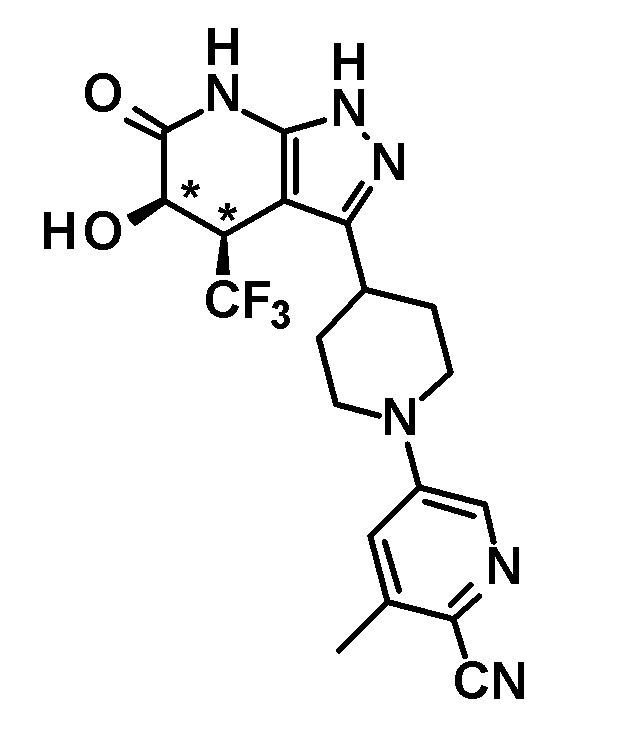
[0380] Указанное в заголовке соединение (0,115 г, выход: 73%, оптически активная форма) получали путем осуществления такой же реакции, как в способе, описанном в Примере 26, с использованием оптически активной формы 5-{4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-3-метилпиридин-2-карбонитрила (0,221 г, 0,377 ммоль), полученной в Ссылочном примере 30, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0381] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,21 (1H, с), 10,53 (1H, с), 8,30 (1H, д, J=3 Гц), 7,33 (1H, д, J=3 Гц), 5,52 (1H, д, J=4 Гц), 4,45-4,43 (1H, м), 4,17-4,11 (3H, м), 3,07-2,94 (3H, м), 2,39 (3H, с), 1,87-1,65 (4H, м);
MS (ESI) m/z: 421 (M+H)+;
[α]D25=+12° (DMF, c=0,984).
[0382] (Ссылочный пример 31) Оптически активная форма 5-{4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}пиразин-2-карбонитрил
[0383]
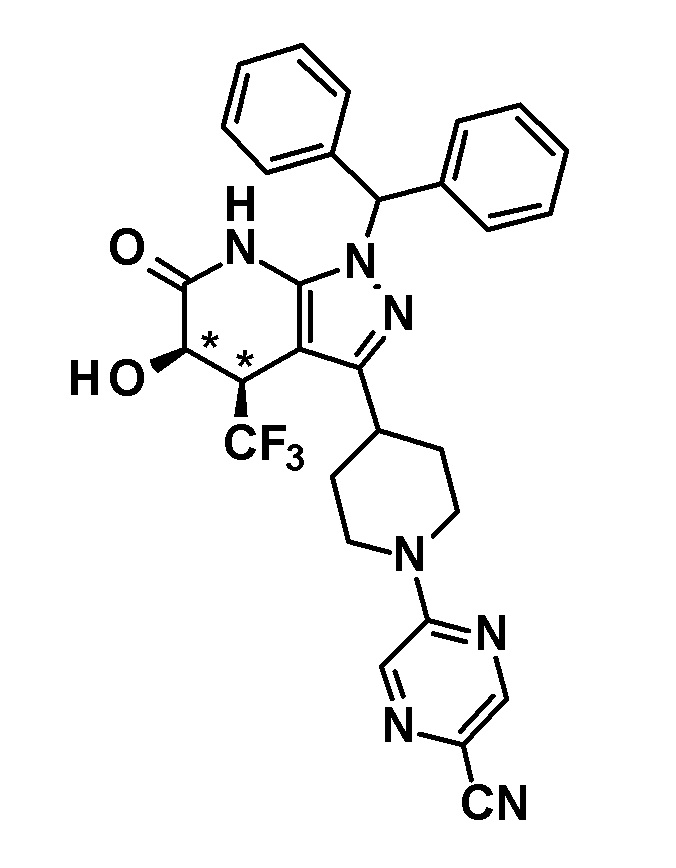
[0384] Указанное в заголовке соединение (132 мг, выход: 66%, оптически активная форма) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 28, с использованием 5-хлор-2-цианопиразина (73,4 мг, 0,526 ммоль) вместо 5-фтор-2-(трифторметил)пиридина.
[0385] 1H-ЯМР (400 MГц, CDCl3) δ: 8,31 (1H, с), 8,12 (1H, с), 7,40-7,36 (6H, м), 7,12-7,06 (4H, м), 6,67 (1H, с), 4,54 (1H, д, J=7 Гц), 4,50-4,45 (2H, м), 3,95-3,86 (1H, м), 3,70 (1H, с), 3,24-3,16 (2H, м), 2,94 (1H, тт, J=11 Гц, 4 Гц), 2,04-1,74 (4H, м).
[0386] (Пример 28) (+)-5-{4-[цис-5-Гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}пиразин-2-карбонитрил
[0387]
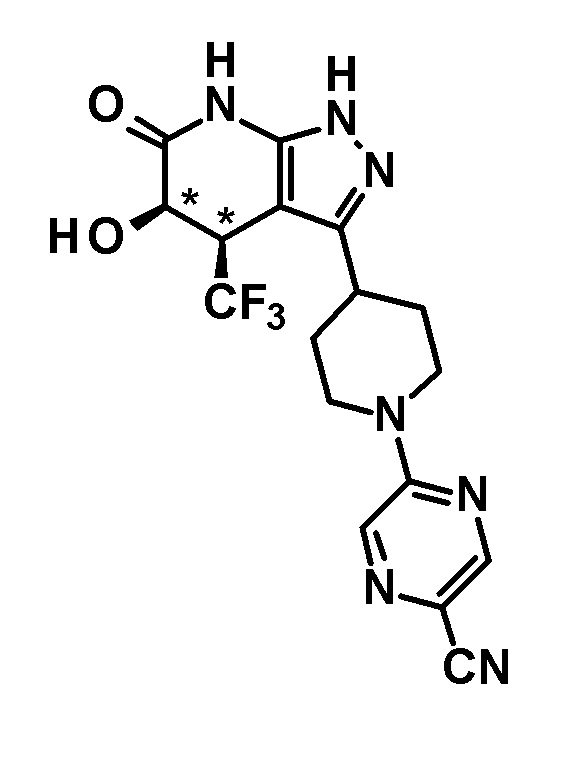
[0388] Указанное в заголовке соединение (78 мг, выход: 86%, оптически активная форма) получали путем осуществления такой же реакции, как в способе, описанном в Примере 26, с использованием оптически активной формы 5-{4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}пиразин-2-карбонитрила (128 мг, 0,223 ммоль), полученной в Ссылочном примере 31, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0389] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,18 (1H, с), 10,54 (1H, с), 8,58 (1H, с), 8,50 (1H, с), 5,53 (1H, уш.с), 4,68-4,61 (2H, м), 4,45-4,42 (1H, м), 4,20-4,11 (1H, м), 3,14-3,05 (3H, м), 1,92-1,61 (4H, м);
MS (ESI) m/z: 408 (M+H)+;
[α]D25=+17° (DMF, c=1,00).
[0390] (Пример 29) Оптически активная форма цис-5-гидрокси-3-[1-(6-фенилпиридазин-3-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она
[0391]
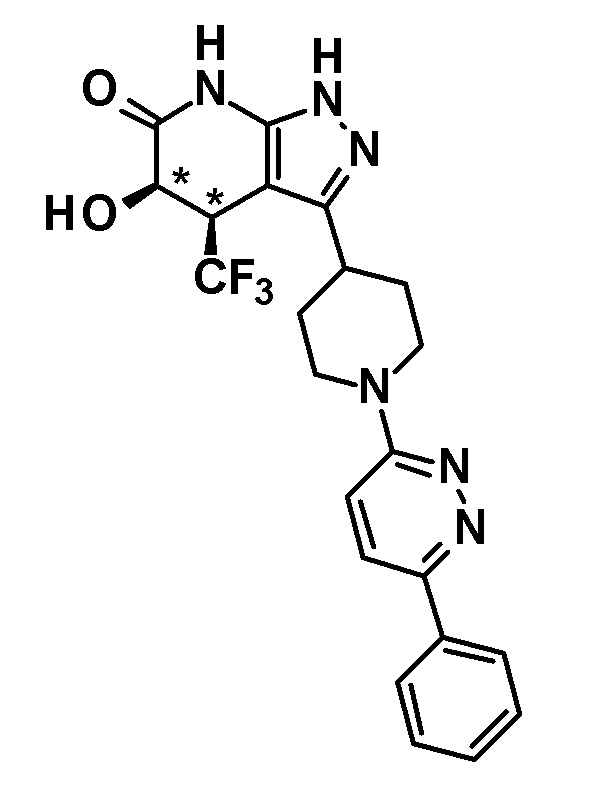
[0392] Промежуточный продукт синтеза получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 28, с использованием 3-хлор-6-фенилпиридазина (0,167 г, 0,876 ммоль) вместо 5-фтор-2-(трифторметил)пиридина.
[0393]
Указанное в заголовке соединение (29 мг, выход: 14%, оптически активная форма) получали путем осуществления такой же реакции, как в способе, описанном в Примере 26, с использованием промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0394] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,22 (1H, с), 10,54 (1H, с), 8,05-7,94 (3H, м), 7,51-7,40 (4H, м), 5,52 (1H, д, J=4 Гц), 4,64-4,57 (2H, м), 4,47-4,44 (1H, м), 4,22-4,13 (1H, м), 3,53-3,51 (4H, м), 3,08-2,98 (3H, м);
MS (ESI) m/z: 459(M+H)+.
[0395] (Пример 30) (+)-цис-5-Гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиримидин-4-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0396]
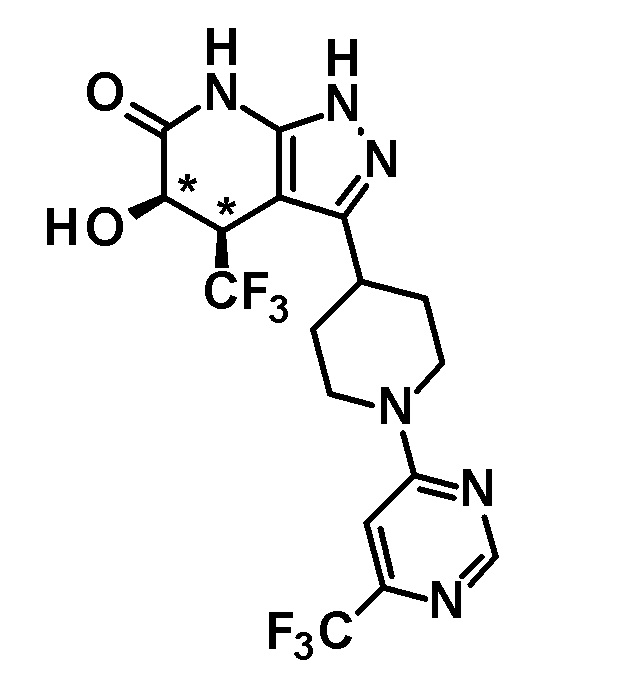
[0397] Карбонат цезия (0,582 г, 1,79 ммоль) и 4-хлор-6-(трифторметил)пиримидин (0,163 г, 0,893 ммоль) добавляли при комнатной температуре к суспензии оптически активной формы цис-1-(дифенилметил)-5-гидрокси-3-(пиперидин-4-ил)-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (210 мг, 0,446 ммоль), полученной в Ссылочном примере 27, в DMSO (5 мл) и смесь перемешивали при 80°C в течение 8 часов. К реакционному раствору добавляли воду, с последующим экстрагированием этилацетатом три раза. Полученный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=100/0-60/40 (градиент)] с получением промежуточного продукта синтеза.
[0398] Трифторуксусную кислоту (2 мл, 26,1 ммоль) добавляли при комнатной температуре к раствору промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, и триэтилсилана (0,116 мл, 0,727 ммоль) в дихлорметане (2 мл) и смесь перемешивали в течение 7,5 часов. Растворитель отгоняли из реакционного раствора при пониженном давлении и к полученному остатку добавляли этилацетат. Органический слой промывали насыщенным водным раствором бикарбоната натрия, водой и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали препаративной тонкослойной хроматографией на силикагеле [элюент: дихлорметан/метанол=10/1] с получением указанного в заголовке соединения (48 мг, выход: 24%, оптически активная форма).
[0399] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,17 (1H, с), 10,55 (1H, с), 8,64 (1H, с), 7,34 (1H, с), 5,54 (1H, д, J=4 Гц), 4,46-4,43 (1H, м), 4,21-4,12 (1H, м), 3,13-2,97 (3H, м), 1,91-1,58 (4H, м);
MS (ESI) m/z: 451(M+H)+;
[α]D25=+10° (DMF, c=1,02).
[0400] (Ссылочный пример 32) Оптически активная форма 5-{4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}пиридин-3-карбонитрила
[0401]
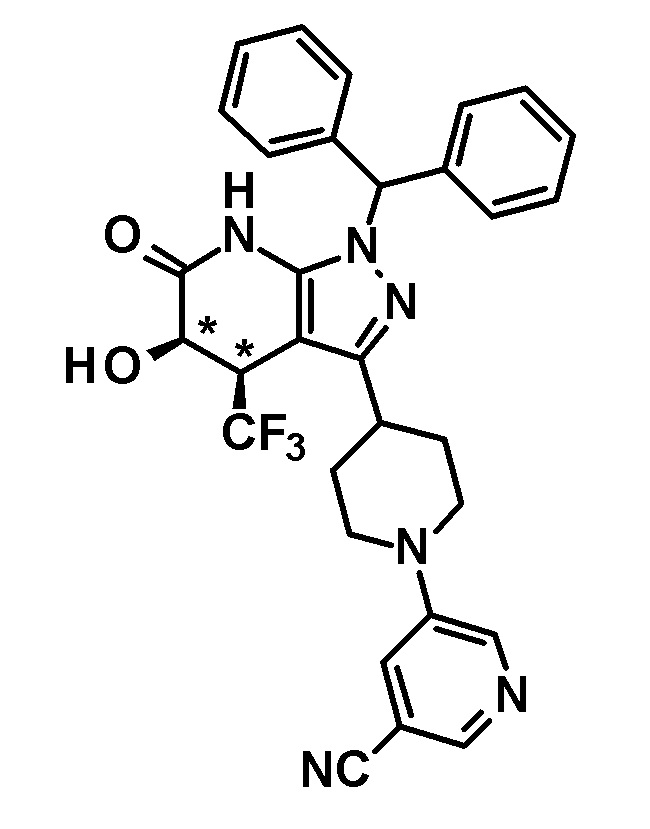
[0402] Указанное в заголовке соединение (42,7 г, выход: 70%, оптически активная форма) получали путем осуществления реакции при 80°C в течение 1 часа и затем при 100°C в течение 5 часов способом, описанным в Ссылочном примере 30, с использованием 5-фторпиридин-3-карбонитрила (26,0 мг, 0,213 ммоль) вместо 5-хлор-3-метилпиридин-2-карбонитрила.
[0403] 1H-ЯМР (400 MГц, CDCl3) δ: 8,46 (1H, д, J=3 Гц), 8,25 (1H, д, J=2 Гц), 7,43-7,04 (10H, м), 6,91 (1H, с), 6,71 (1H, с), 4,56-4,51 (1H, м), 3,96-3,86 (1H, м), 3,83-3,73 (2H, м), 3,72-3,67 (1H, м), 3,02-2,93 (2H, м), 2,87-2,76 (1H, м), 2,08-1,85 (4H, м).
[0404] (Пример 31) (+)-5-{4-[цис-5-Гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}пиридин-3-карбонитрил
[0405]
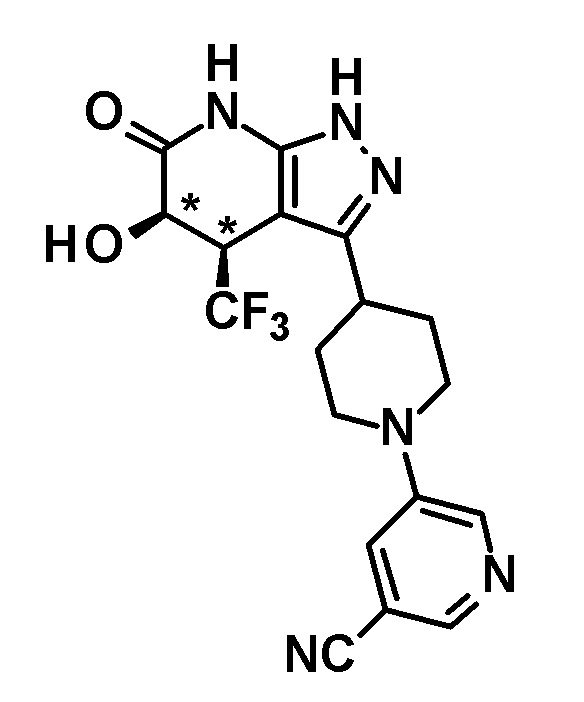
[0406] Указанное в заголовке соединение (0,134 г, выход: 85%, оптически активная форма) получали путем осуществления такой же реакции, как в способе, описанном в Примере 26, с использованием оптически активной формы 5-{4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}пиридин-3-карбонитрила (0,222 г, 0,388 ммоль), полученной в Ссылочном примере 32, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0407] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,24 (1H, с), 10,54 (1H, с), 8,62 (1H, д, J=3 Гц), 8,31 (1H, д, J=2 Гц), 7,81 (1H, дд, J=3 Гц, 2 Гц), 5,65-5,34 (1H, м), 4,48-4,38 (1H, м), 4,21-4,09 (1H, м), 4,07-3,92 (2H, м), 3,06-2,78 (3H, м), 1,90-1,63 (4H, м);
MS (ESI) m/z: 407(M+H)+;
[α]D25=+9,3° (DMF, c=1,01).
[0408] (Ссылочный пример 33) Бензил 2-хлор-5-(трифторметил)пиридин-4-карбоксилат
[0409]
[0410] Бензилбромид (0,634 мл, 3,86 ммоль) добавляли при охлаждении льдом к раствору 2-хлор-5-(трифторметил)пиридин-4-карбоновой кислоты (0,870 г, 3,86 ммоль) и карбоната калия (0,640 г, 4,63 ммоль) в DMF (10 мл) и смесь перемешивали при указанной выше температуре в течение 10 минут и затем при комнатной температуре в течение 3 дней. К реакционному раствору добавляли охлажденный льдом насыщенный водный раствор бикарбоната натрия, с последующим экстрагированием этилацетатом два раза. Полученный органический слой промывали водой и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан:этилацетат=100/0-90/10 (градиент)] с получением указанного в заголовке соединения (1,22 г, выход: количественный).
[0411] 1H-ЯМР (400 MГц, CDCl3) δ: 8,78 (1H, с), 7,68 (1H, с), 7,46-7,36 (5H, м), 5,39 (2H, с).
[0412] (Ссылочный пример 34) Бензил 2-{4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-5-(трифторметил)пиридин-4-карбоксилат
[0413]
[0414] Указанное в заголовке соединение (1,06 г, выход: 73%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 17, с использованием бензил 2-хлор-5-(трифторметил)пиридин-4-карбоксилата (0,913 г, 2,83 ммоль), полученного в Ссылочном примере 33, вместо 3-хлор-6-(трифторметил)пиридазина.
[0415] 1H-ЯМР (400 MГц, CDCl3) δ: 8,43 (1H, с), 7,44-7,33 (11H, м), 7,11-7,06 (5H, м), 6,86 (1H, с), 6,68 (1H, с), 5,34 (2H, с), 4,54-4,51 (1H, м), 4,47-4,41 (2H, м), 3,94-3,86 (1H, м), 3,69 (1H, д, J=3 Гц), 3,12-3,05 (2H, м), 2,89 (1H, тт, J=11 Гц, 4 Гц), 2,01-1,69 (4H, м).
[0416] (Ссылочный пример 35) Оптически активная форма бензил 2-{4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-5-(трифторметил)пиридин-4-карбоксилата
[0417]
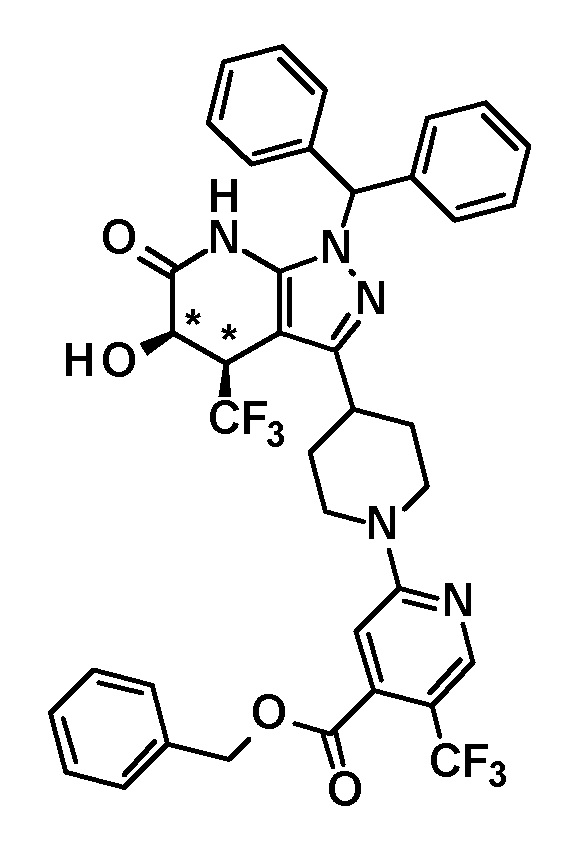
[0418] Смешанный раствор бензил 2-{4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-5-(трифторметил)пиридин-4-карбоксилата (250 мг, 0,333 ммоль), полученного в Ссылочном примере 34, в гексане (4 мл) и IPA (2 мл) очищали при помощи ВЭЖХ [колонка: Chiralpak IA (20 мм в.д. × 250 мм); изготовитель Daicel Corporation, элюент: гексан/IPA=70/30, скорость потока: 18 мл/мин] с получением указанного в заголовке соединения (100 мг, выход: 40%, оптически активная форма).
[0419] Оптическую чистоту измеряли с использованием ВЭЖХ [колонка: Chiralpak IA (4,6 мм в.д. × 150 мм); изготовитель Daicel Corporation, элюент: гексан/IPA=70/30, скорость потока: 1,0 мл/мин].
[0420] Оптическая чистота 99% (время удерживания: 4,4 мин).
[0421]
(Пример 32) Бензил (+)-2-{4-[цис-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-5-(трифторметил)пиридин-4-карбоксилат
[0422]
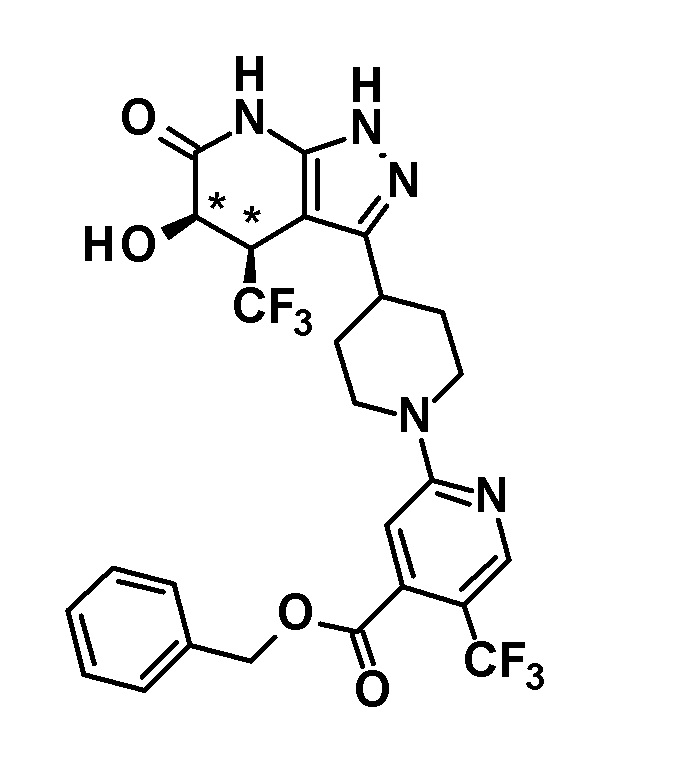
[0423] Указанное в заголовке соединение (60,2 мг, выход: 77%, оптически активная форма) получали путем осуществления такой же реакции, как в способе, описанном в Примере 26, с использованием оптически активной формы бензил 2-{4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-5-(трифторметил)пиридин-4-карбоксилата (100 мг, 0,133 ммоль, оптически активная форма), полученной в Ссылочном примере 35, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она, с последующей очисткой препаративной тонкослойной хроматографией на силикагеле [элюент: дихлорметан/метанол=10/1].
[0424] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,16 (1H, с), 10,53 (1H, с), 8,49 (1H, с), 7,46-7,34 (5H, м), 7,20 (1H, с), 5,52 (1H, д, J=4 Гц), 5,34 (2H, с), 4,63-4,56 (2H, м), 4,45-4,42 (1H, м), 4,20-4,11 (1H, м), 3,13-2,99 (3H, м), 1,87-1,57 (4H, м);
MS (ESI) m/z: 584(M+H)+;
[α]D25=+12° (DMF, c=1,01).
[0425] (Ссылочный пример 36) Бензил 2-{4-[цис-5-{[трет-бутил(диметил)силил]окси}-1-(дифенилметил)-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-5-(трифторметил)пиридин-4-карбоксилат
[0426]
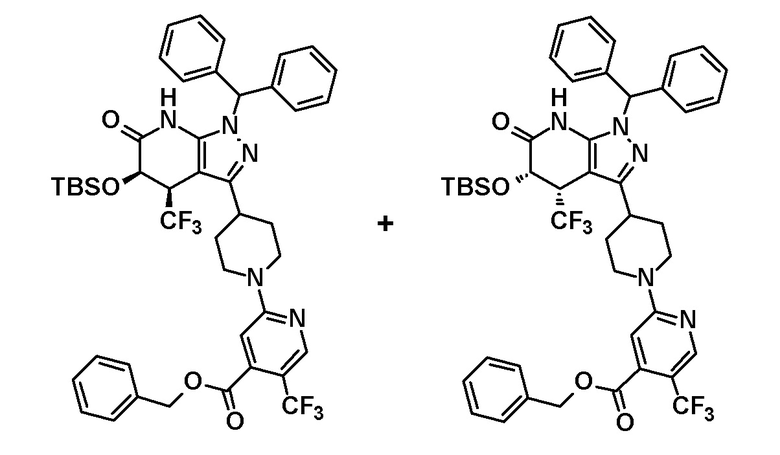
[0427] трет-Бутилдиметилхлорсилан (0,404 г, 2,68 ммоль) и имидазол (0,243 г, 3,57 ммоль) добавляли к раствору бензил 2-{4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-5-(трифторметил)пиридин-4-карбоксилата (670 мг, 0,894 ммоль), полученного в Ссылочном примере 34, в DMF (5 мл) и смесь перемешивали в течение ночи при комнатной температуре. К реакционному раствору добавляли воду, с последующим экстрагированием этилацетатом два раза. Полученный органический слой сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=100/0-85/15 (градиент)] с получением указанного в заголовке соединения (673 мг, выход: 87%).
[0428] 1H-ЯМР (400 MГц, CDCl3) δ: 8,43 (1H, с), 7,74 (1H, уш.с), 7,45-7,32 (11H, м), 7,19-7,16 (2H, м), 7,13-7,10 (2H, м), 6,87 (1H, с), 6,62 (1H, с), 5,35 (2H, с), 4,59 (1H, д, J=7 Гц), 4,49-4,38 (2H, м), 3,68-3,60 (1H, м), 3,14-3,04 (2H, м), 2,86 (1H, тт, J=11 Гц, 4 Гц), 1,99-1,72 (4H, м), 0,91 (9H, с), 0,14 (3H, с), 0,08 (3H, с).
[0429] (Ссылочный пример 37) цис-1-(Дифенилметил)-5-гидрокси-3-{1-[4-(пиперидин-1-илкарбонил)-5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0430]
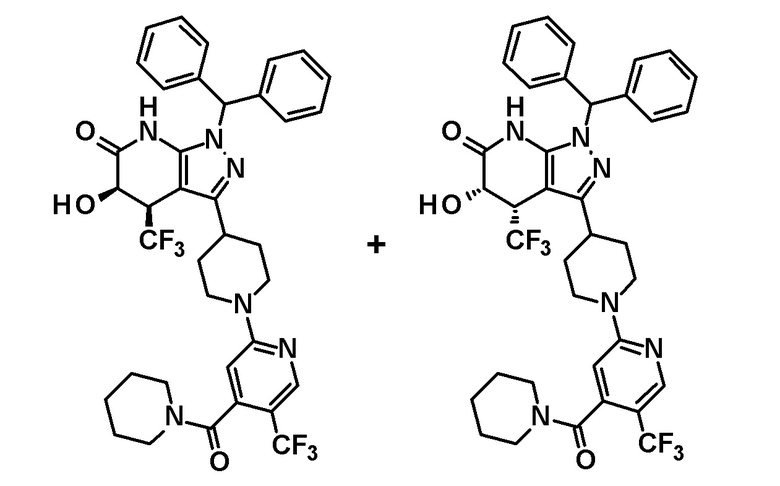
[0431] 10% Палладий на активированном угле (300 мг) добавляли к раствору бензил 2-{4-[цис-5-{[трет-бутил(диметил)силил]окси}-1-(дифенилметил)-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-5-(трифторметил)пиридин-4-карбоксилата (670 мг, 0,775 ммоль), полученного в Ссылочном примере 36, в этилацетате (20 мл) и смесь перемешивали при комнатной температуре в течение 5 часов в атмосфере водорода. После фильтрования через Целит растворитель отгоняли при пониженном давлении с получением неочищенного продукта (600 мг).
[0432] Пиперидин (35,5 мкл, 0,388 ммоль), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (0,147 г, 0,388 ммоль) и N,N-диизопропилэтиламин (0,133 мл, 0,775 ммоль) добавляли при комнатной температуре к раствору части (150 мг) неочищенного продукта, полученного, с использованием процедур, описанных выше, в DMF (3 мл) и смесь перемешивали в течение ночи при комнатной температуре. К реакционному раствору добавляли воду, с последующим экстрагированием этилацетатом три раза. Полученный органический слой сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали препаративной тонкослойной хроматографией на силикагеле [элюент: гексан/этилацетат=2/3] с получением указанного в заголовке соединения (79 мг, выход: 56%).
[0433] 1H-ЯМР (400 MГц, CDCl3) δ: 8,37 (1H, с), 7,44 (1H, д, J=25 Гц, 4 Гц), 7,37-7,32 (6H, м), 7,11-7,06 (4H, м), 6,67 (1H, с), 6,40 (1H, д, J=4 Гц), 4,50 (1H, д, J=7 Гц), 4,45-4,32 (2H, м), 3,92-3,84 (1H, м), 3,77-3,59 (3H, м), 3,16-3,14 (2H, м), 3,09-2,99 (2H, м), 2,89-2,82 (1H, м), 1,98-1,37 (10H, м).
[0434] (Пример 33) цис-5-Гидрокси-3-{1-[4-(пиперидин-1-илкарбонил)-5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0435]
[0436] Указанное в заголовке соединение (44 мг, выход: 51%) получали путем осуществления такой же реакции, как в способе, описанном в Примере 32, с использованием цис-1-(дифенилметил)-5-гидрокси-3-{1-[4-(пиперидин-1-илкарбонил)-5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (112 мг, 0,154 ммоль), полученного в Ссылочном примере 37, вместо оптически активной формы бензил 2-{4-[цис-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-5-(трифторметил)пиридин-4-карбоксилата.
[0437] 1H-ЯМР (400 MГц, CDCl3) δ: 10,72 (1H, уш.с), 8,44 (1H, с), 6,51 (1H, с), 4,74-4,69 (1H, м), 4,55 (1H, д, J=7 Гц), 4,44-4,38 (1H, м), 4,16-4,07 (1H, м), 3,97-3,89 (1H, м), 3,78-3,73 (1H, м), 3,63-3,56 (1H, м), 3,21-3,17 (2H, м), 3,13-2,94 (3H, м), 2,01-1,42 (10H, м);
MS (ESI) m/z: 561(M+H)+.
[0438] (Ссылочный пример 38) трет-Бутил 4-[4,5-дигидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилат
[0439]
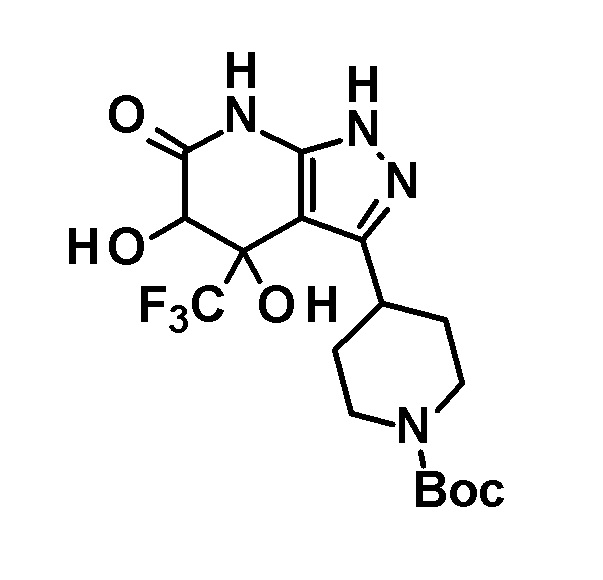
[0440] Раствор этил 2-триэтилсилилоксиацетата (соединение, описанное в литературе J. Org. Chem., 2008, Vol. 73, p. 6268-6278, 18,37 г, 84,13 ммоль) и этанола (0,1474 мл, 2,524 ммоль) в толуоле (40 мл) добавляли при комнатной температуре к суспензии гидрида натрия (63% дисперсия в масле, 5,127 г, 134,6 ммоль) в толуоле (80 мл), затем к смеси добавляли раствор этилтрифторацетата (15,07 мл, 126,2 ммоль) в толуоле (20 мл) и смесь перемешивали в течение 5 минут и затем перемешивали при 80°C в течение 30 минут. К реакционному раствору добавляли насыщенный водный раствор хлорида аммония при охлаждении льдом, с последующим экстрагированием этилацетатом. Полученный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении с получением неочищенного маслянистого продукта (23,6 г).
[0441] Смешанный раствор неочищенного маслянистого продукта (23,6 г), полученного, с использованием процедур, описанных выше, и трет-бутил 4-[5-амино-1-(дифенилметил)-1H-пиразол-3-ил]пиперидин-1-карбоксилата (10,83 г, 25,04 ммоль), полученного в Ссылочном примере 12, в этаноле (150 мл) и уксусной кислоте (50 мл) перемешивали в течение 4 часов при нагревании до температуры кипения с обратным холодильником. Растворитель отгоняли из реакционного раствора при пониженном давлении и к полученному остатку добавляли этилацетат. Органический слой промывали насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. К полученному остатку добавляли этилацетат и гексан и образовавшийся осадок собирали фильтрованием с получением твердого вещества. Растворитель в фильтрате затем отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=90/10-50/50 (градиент)] и объединяли с ранее полученным твердым веществом с получением промежуточного продукта синтеза.
[0442] Триэтилсилан (10,7 мл, 67,4 ммоль) и трифторуксусную кислоту (90 мл, 1176 ммоль) добавляли к суспензии промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, в дихлорметане (300 мл) и смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель отгоняли из реакционного раствора при пониженном давлении. К полученному остатку добавляли диэтиловый эфир и гексан и смесь перемешивали в течение 30 минут. Образовавшийся осадок собирали фильтрованием с получением бесцветного твердого вещества.
[0443] Раствор ди-трет-бутилдикарбоната (5,53 г, 25,4 ммоль) и триэтиламина (4,69 мл, 33,8 ммоль) в этилацетате (30 мл) добавляли при комнатной температуре к смешанной суспензии бесцветного твердого вещества, полученного с использованием процедур, описанных выше, в этилацетате (120 мл) и THF (40 мл) и смесь перемешивали при комнатной температуре в течение 3 часов и оставляли выстаиваться в течение ночи при комнатной температуре. Реакционный раствор промывали насыщенным водным раствором бикарбоната натрия и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=50:50-0:100 (градиент)] с получением указанного в заголовке соединения (3,09 г, выход: 44%).
[0444] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,24 (1H, с), 10,55 (1H, с), 6,76 (1H, с), 5,65 (1H, д, J=4 Гц), 4,33 (1H, уш.с), 4,13-3,99 (2H, м), 3,20-3,09 (1H, м), 2,84-2,59 (2H, м), 1,84-1,76 (1H, м), 1,66-1,46 (3H, м), 1,42 (9H, с).
[0445]
(Ссылочный пример 39) Оптически активная форма трет-бутил 4-[4,5-дигидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилата
[0446]
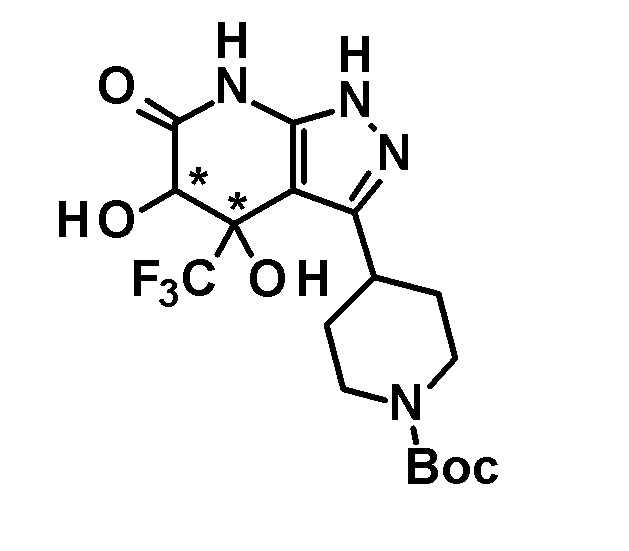
[0447] Смешанный раствор трет-бутил 4-[4,5-дигидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилата (0,79 г, 1,9 ммоль), полученного в Ссылочном примере 38, в этилацетате и метаноле адсорбировали на силикагеле и растворитель отгоняли при пониженном давлении. Полученный порошок очищали жидкостной флэш-хроматографией [колонка: Chiralflash IA (30 мм в.д. ×100 мм); изготовитель Daicel Corporation, элюент: гексан/IPA=90/10, скорость потока: 12 мл/мин] с получением указанного в заголовке соединения (0,34 г, выход: 43%, оптически активная форма).
[0448] Оптическую чистоту измеряли с использованием ВЭЖХ [колонка: Chiralpak IA (4,6 мм в.д. × 250 мм); изготовитель Daicel Corporation, элюент: гексан/IPA=80/20, скорость потока: 1,0 мл/мин].
[0449] Оптическая чистота 99% или выше (время удерживания: 7,7 мин).
[0450] (Пример 34) (+)-4,5-Дигидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиразин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0451]
[0452] Трифторуксусную кислоту (2 мл) добавляли при комнатной температуре к суспензии оптически активной формы трет-бутил 4-[4,5-дигидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-карбоксилата (0,34 г, 0,81 ммоль), полученной в Ссылочном примере 39, в дихлорметане (6 мл) и смесь перемешивали в течение 2 часов. Растворитель отгоняли из реакционного раствора при пониженном давлении и полученный остаток отверждали путем добавления диэтилового эфира и гексана. Растворитель удаляли декантацией и полученное твердое вещество сушили при пониженном давлении с получением промежуточного продукта синтеза.
[0453] 2-Хлор-5-(трифторметил)пиразин (0,15 мл, 1,2 ммоль) и N,N-диизопропилэтиламин (0,41 мл, 2,4 ммоль) добавляли при комнатной температуре к раствору промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, в DMSO (5 мл) и смесь перемешивали в течение 1 часа и затем оставляли выстаиватсья в течение ночи. К реакционному раствору добавляли этилацетат. Органический слой промывали водой и насыщенным солевым раствором, в указанном порядке, и сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=50/50-0/1000 (градиент)] с получением указанного в заголовке соединения (0,31 г, выход: 82%, оптически активная форма).
[0454] Оптическую чистоту измеряли с использованием ВЭЖХ [колонка: Chiralpak IA (4,6 мм в.д. ×250 мм); изготовитель Daicel Corporation, элюент: гексан/IPA=60/40, скорость потока: 1,0 мл/мин].
[0455] Оптическая чистота 99% или выше (время удерживания: 6,7 мин);
1H-ЯМР (400 MГц, DMSO-d6) δ: 12,21 (1H, с), 10,57 (1H, с), 8,51 (1H, с), 8,50 (1H, с), 6,81 (1H, с), 5,68 (1H, д, J=4 Гц), 4,69-4,56 (2H, м), 4,37-4,31 (1H, м), 3,46-3,34 (1H, м), 3,11-2,97 (2H, м), 1,98-1,88 (1H, м), 1,83-1,58 (3H, м);
MS (ESI) m/z: 467 (M+H)+;
[α]D25=+3,9° (DMF, c=0,924).
[0456] (Пример 35) (+)-4,5-Дигидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0457]
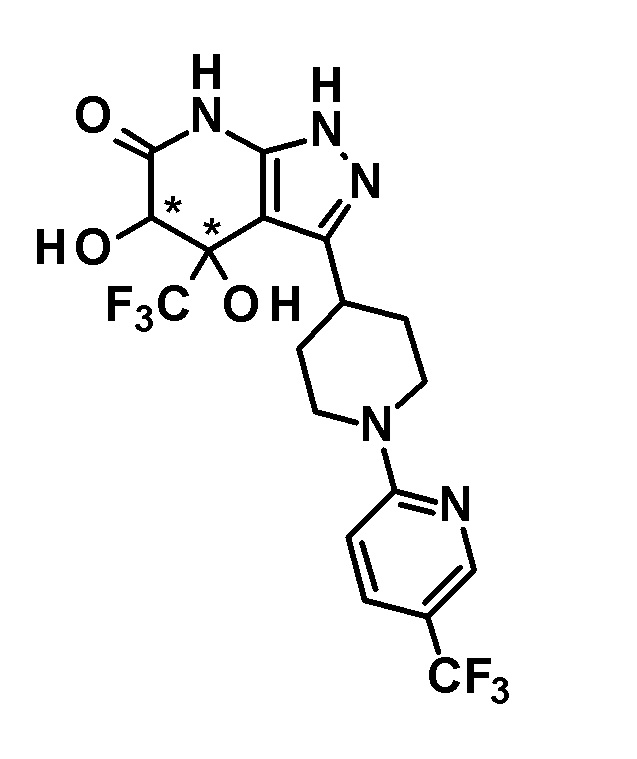
[0458] Указанное в заголовке соединение (890 мг, выход: 89%, оптически активная форма) получали путем осуществления такой же реакции, как в способе, описанном в Примере 34, с использованием 2-фтор-5-(трифторметил)пиридина (0,468 мл, 3,89 ммоль) вместо 2-хлор-5-(трифторметил)пиразина.
[0459] Оптическую чистоту измеряли с использованием ВЭЖХ [колонка: Chiralpak IA (4,6 мм в.д. ×250 мм); изготовитель Daicel Corporation, элюент: гексан/IPA=60/40, скорость потока: 1,0 мл/мин].
[0460] Оптическая чистота 99% или выше (время удерживания: 6,8 мин);
[α]D25=+4,0° (DMF, c=1,00).
[0461] (Пример 36) (+)-цис-5-Гидрокси-3-[1-(5-изопропоксипиридин-2-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0462]
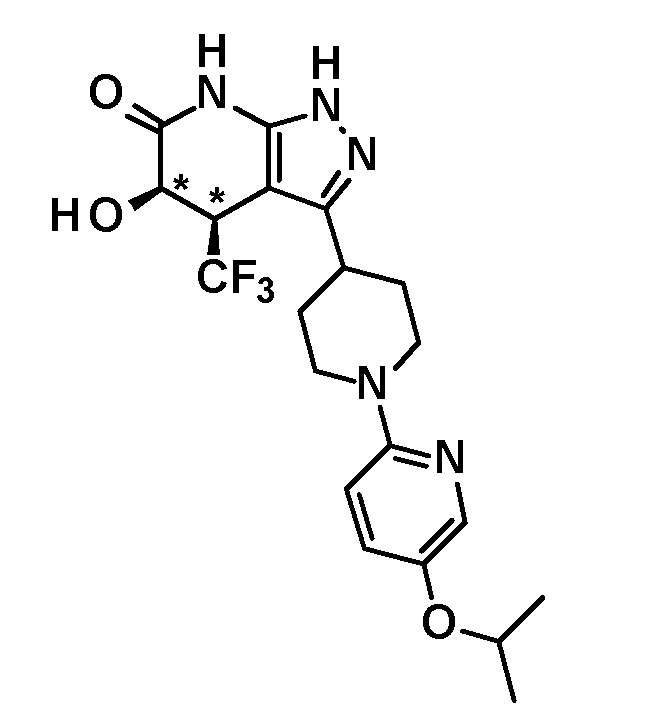
[0463] (+)-Ментилхлорформиат (0,139 мл, 0,655 ммоль) добавляли при 0°C к смешанной суспензии цис-5-гидрокси-3-[1-(5-изопропоксипиридин-2-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (0,240 г, 0,546 ммоль), полученного в Примере 5, и триэтиламина (83,3 мкл, 0,601 ммоль) в THF (24 мл) и этилацетате (6 мл) и смесь перемешивали при 0°C в течение 2 часов и затем при комнатной температуре в течение 16 часов. Добавляли дополнительное количество THF (24 мл), затем дополнительно добавляли триэтиламин (83,3 мкл, 0,601 ммоль) и (+)-ментилхлорформиат (0,139 мл, 0,655 ммоль) при 0°C и смесь перемешивали при комнатной температуре в течение 3 часов. Снова добавляли триэтиламин (41,6 мкл, 0,300 ммоль) и (+)-ментилхлорформиат (69,6 мкл, 0,328 ммоль) и смесь перемешивали при комнатной температуре в течение 2 часов. К реакционной смеси добавляли воду, с последующим экстрагированием этилацетатом. Полученный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=93/7-40/60 (градиент)] с получением каждого из соединения, элюируемого первым (0,156 г), и соединения, элюируемого вторым (0,139 г).
[0464] Морфолин (43,7 мкл, 0,502 ммоль) добавляли при комнатной температуре к раствору соединения, элюируемого первым, полученного, с использованием процедур, описанных выше, в ацетонитриле (4 мл) и смесь перемешивали в течение 16 часов. К смеси добавляли дихлорметан (3 мл) и смесь перемешивали в течение 10 минут. Затем образовавшийся осадок собирали фильтрованием с получением указанного в заголовке соединения (70,2 мг, выход: 29%, оптически активная форма).
[0465] [α]D25=+8,3° (DMF, c=0,922).
[0466] (Ссылочный пример 40) 3-{1-[5-(Дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-1H-пиразол-5-амин
[0467]
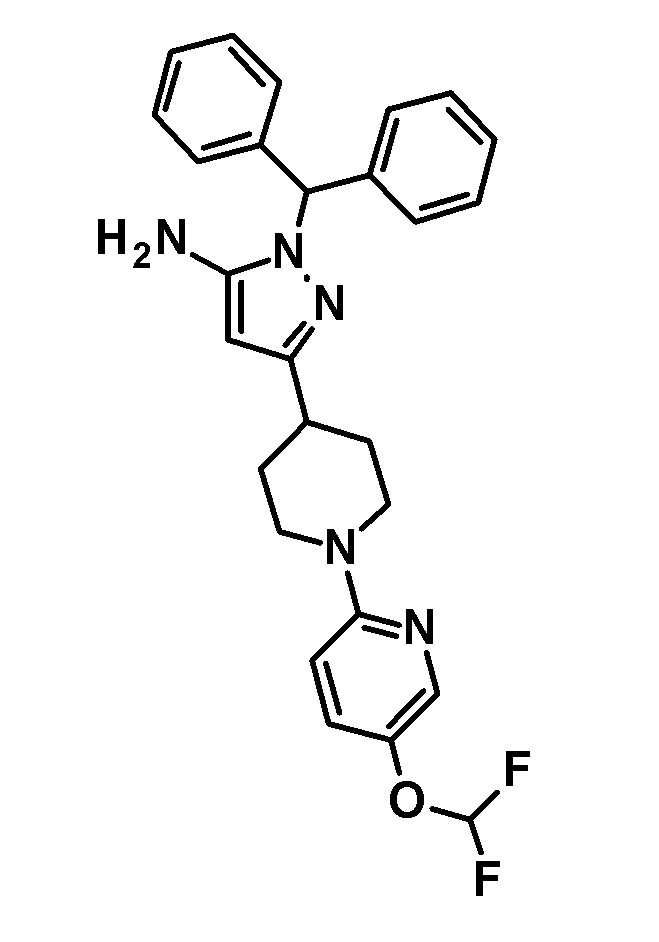
[0468] Указанное в заголовке соединение (2,04 г, выход: 83%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 1, с использованием этил 1-[5-(дифторметокси)пиридин-2-ил]пиперидин-4-карбоксилата (соединение, описанное в опубликованной заявке WO2013/187462, 1,56 г, 5,19 ммоль) вместо этил 1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-карбоксилата и с использованием ацетата дифенилметилгидразина (1,38 г, 5,34 ммоль), вместо гидрохлорида дифенилметилгидразина.
[0469] 1H-ЯМР (400 MГц, DMSO-d6) δ: 7,99 (1H, д, J=3 Гц), 7,41 (1H, дд, J=9 Гц, 3 Гц), 7,33-7,19 (10H, м), 6,95 (1H, д, J=75 Гц), 6,87 (1H, д, J=9 Гц), 6,59 (1H, с), 5,31 (1H, уш.с), 5,17 (1H, с), 4,23-4,20 (2H, м), 2,91-2,84 (2H, м), 2,68-2,58 (1H, м), 1,85-1,80 (2H, м), 1,54-1,44 (2H, м).
[0470]
(Ссылочный пример 41) 3-{1-[5-(Дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0471]
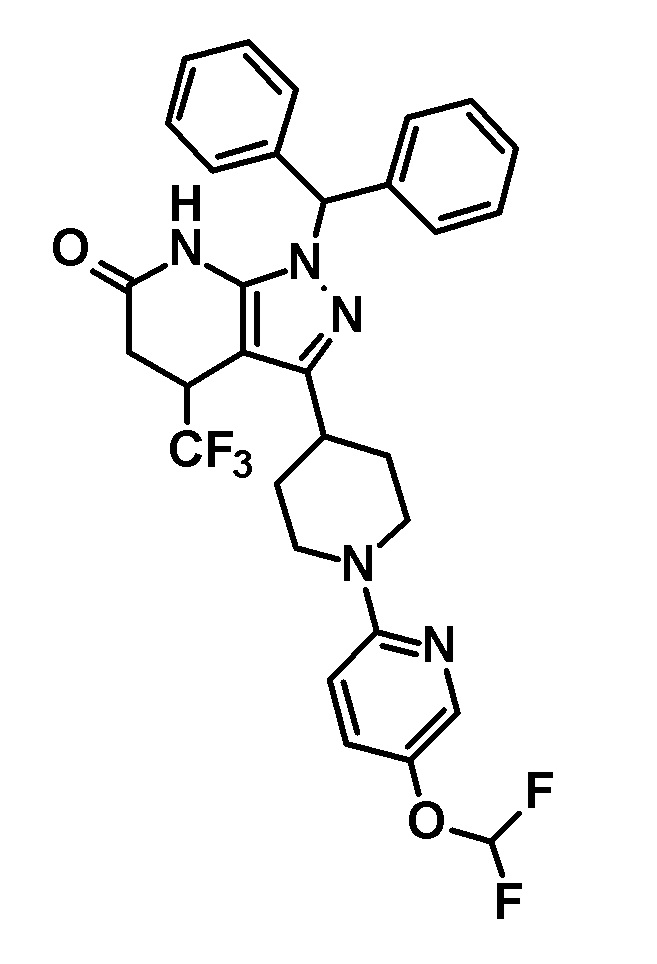
[0472] 1,4-Диазабицикло[2,2,2]октан (530 мг, 4,72 ммоль), кислоту Мельдрума (1,35 г, 9,37 ммоль) и трифторацетальдегид этил гемиацеталь (1,30 г, 9,02 ммоль) добавляли к раствору 3-{1-[5-(дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-1H-пиразол-5-амина (1,17 г, 2,46 ммоль), полученного в Ссылочном примере 40, в этаноле (20 мл) и смесь перемешивали в течение 6 часов при нагревании до температуры кипения с обратным холодильником. Растворитель отгоняли из реакционного раствора при пониженном давлении. К полученному остатку добавляли уксусную кислоту (10 мл) и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель отгоняли при пониженном давлении. К полученному остатку добавляли насыщенный водный раствор бикарбоната натрия, с последующим экстрагированием этилацетатом три раза. Полученный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=100/0-50/50 (градиент)] и снова очищали колоночной хроматографией на силикагеле [NH-силикагель, элюент: гексан/этилацетат=100/0-50/50 (градиент)] с получением указанного в заголовке соединения (1,35 г, выход: 92%).
[0473] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,18 (1H, с), 7,99 (1H, д, J=3 Гц), 7,41 (1H, дд, J=9 Гц, 3 Гц), 7,36-7,17 (10H, м), 6,95 (1H, д, J=74 Гц), 6,88 (1H, д, J=9 Гц), 6,81 (1H, с), 4,30-4,25 (2H, м), 4,09-3,98 (1H, м), 3,15-3,08 (1H, м), 2,94-2,81 (4H, м), 1,91-1,77 (2H, м), 1,72-1,51 (2H, м).
[0474] (Ссылочный пример 42) Метил 3-{1-[5-(дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-5-карбоксилат
[0475]
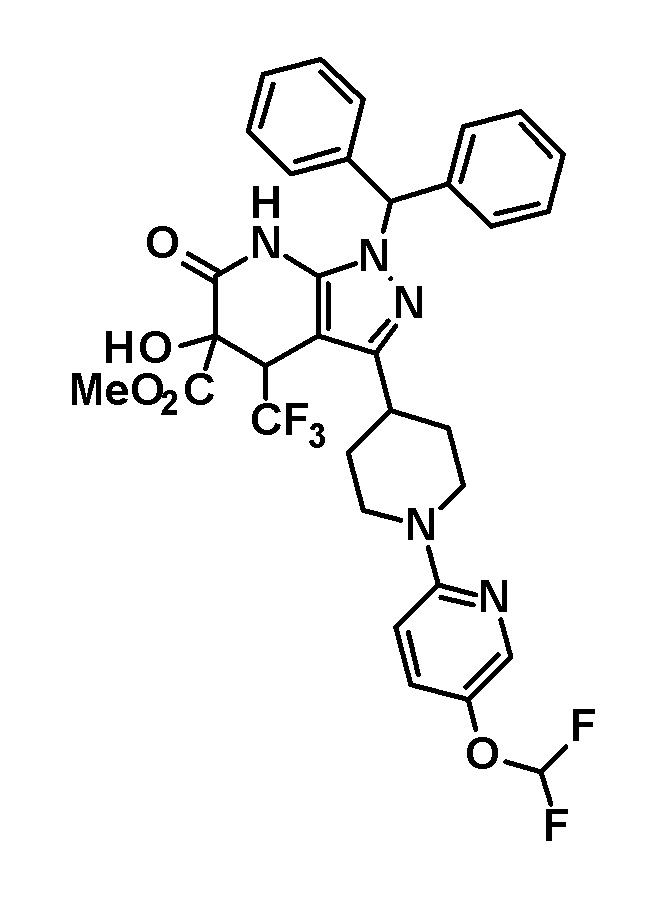
[0476] Диметилкарбонат (0,30 мл, 3,6 ммоль) добавляли к раствору 3-{1-[5-(дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (1,28 г, 2,14 ммоль), полученного в Ссылочном примере 41, в THF (20 мл) и смесь охлаждали до -78°C. Затем добавляли по каплям диизопропиламид лития (раствор в гексане и THF, 5,90 мл, 6,43 ммоль) и смесь нагревали до 0°C и перемешивали при этой температуре в течение 30 минут. К реакционному раствору добавляли насыщенный водный раствор хлорида аммония, с последующим экстрагированием этилацетатом три раза. Полученный органический слой сушили над сульфатом магния и растворитель отгоняли при пониженном давлении. Полученный остаток очищали хроматографией на силикагеле [NH-силикагель, элюент: дихлорметан/метанол=100/0-90/10 (градиент)] с получением промежуточного продукта синтеза.
[0477] DBU (1,23 мл, 8,25 ммоль), (1S)-(+)-(10-камфорсульфонил)оксазиридин (157 мг, 0,684 ммоль) и (1R)-(-)-(10-камфорсульфонил)оксазиридин (155 мг, 0,676 ммоль) добавляли к раствору промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, в THF (10 мл) и смесь перемешивали при комнатной температуре в течение 2 часов. К реакционному раствору добавляли насыщенный водный раствор хлорида аммония, с последующим экстрагированием этилацетатом три раза. Полученный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [NH-силикагель, элюент: дихлорметан/метанол=100/0-90/10 (градиент)] с получением указанного в заголовке соединения (558 мг, выход: 39%).
[0478] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,48 (1H, с), 7,95 (1H, д, J=3 Гц), 7,36 (1H, дд, J=9 Гц, 3 Гц), 7,32-7,12 (10H, м), 6,91 (1H, д, J=75 Гц), 6,85-6,81 (2H, м), 4,28-4,17 (2H, м), 4,10-4,03 (1H, м), 3,69 (3H, с), 2,89-2,77 (3H, м), 1,86-1,82 (2H, м), 1,47-1,35 (2H, м).
[0479] (Ссылочный пример 43) цис-3-{1-[5-(Дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0480]
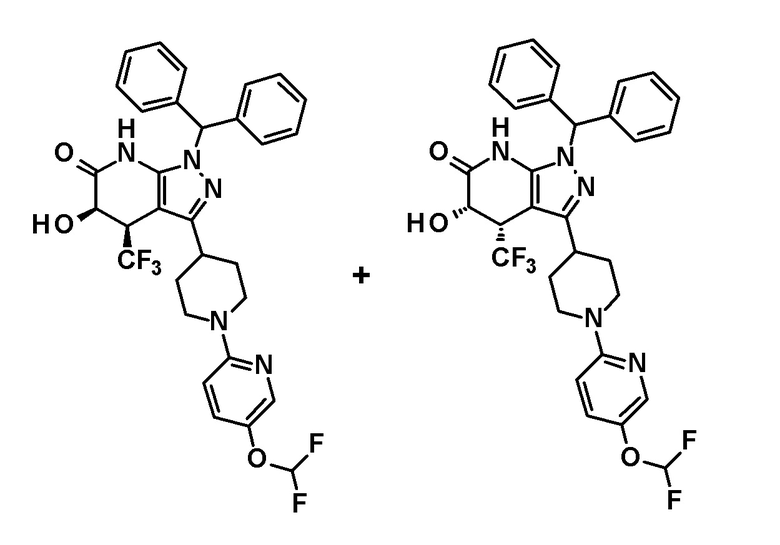
[0481] Гидроксид лития (60 мг, 2,5 ммоль) добавляли к смешанному раствору метил 3-{1-[5-(дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-5-карбоксилата (558 мг, 0,831 ммоль), полученного в Ссылочном примере 42, в этаноле (4 мл) и воде (2 мл) и смесь перемешивали при 50°C в течение 2 часов. Растворитель отгоняли из реакционного раствора при пониженном давлении и к полученному остатку добавляли насыщенный водный раствор хлорида аммония и этилацетат, с последующим экстрагированием этилацетатом три раза. Полученный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом магния и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=100/0-50/50 (градиент)] с получением указанного в заголовке соединения (345 мг, выход: 68%).
[0482] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,21 (1H, с), 7,99 (1H, д, J=3 Гц), 7,41 (1H, дд, J=9 Гц, 3 Гц), 7,36-7,14 (10H, м), 6,95 (1H, д, J=74 Гц), 6,88 (1H, д, J=9 Гц), 6,74 (1H, с), 5,79 (1H, д, J=4 Гц), 4,58-4,54 (1H, м), 4,31-4,24 (2H, м), 4,18-4,10 (1H, м), 2,95-2,82 (3H, м), 1,97-1,49 (4H, м).
[0483] (Пример 37) цис-3-{1-[5-(Дифторметокси)пиридин-2-ил]пиперидин-4-ил}-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0484]
[0485] Указанное в заголовке соединение (103 мг, выход: 87%) получали путем осуществления такой же реакции, как в способе, описанном в Примере 26, с использованием цис-3-{1-[5-(дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (162 мг, 0,264 ммоль), полученного в Ссылочном примере 43, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она, с последующей очисткой полученного остатка колоночной хроматографией на силикагеле [элюент: этилацетат/метанол=100/0-95/5 (градиент)].
[0486] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,22 (1H, с), 10,53 (1H, с), 8,02 (1H, д, J=3 Гц), 7,44 (1H, дд, J=9 Гц, 3 Гц), 7,25 (1H, с), 6,97 (1H, д, J=74 Гц), 6,92 (1H, д, J=9 Гц), 5,51 (1H, д, J=4 Гц), 4,45-4,34 (2H, м), 4,19-4,10 (1H, м), 3,04-2,94 (1H, м), 2,90-2,80 (2H, м), 1,85-1,58 (4H, м);
MS (ESI) m/z: 448 (M+H)+.
[0487] (Ссылочный пример 44) 1-(Дифенилметил)-3-{1-[5-(трифторметил)пиридин-3-ил]пиперидин-4-ил}-1H-пиразол-5-амин
[0488]
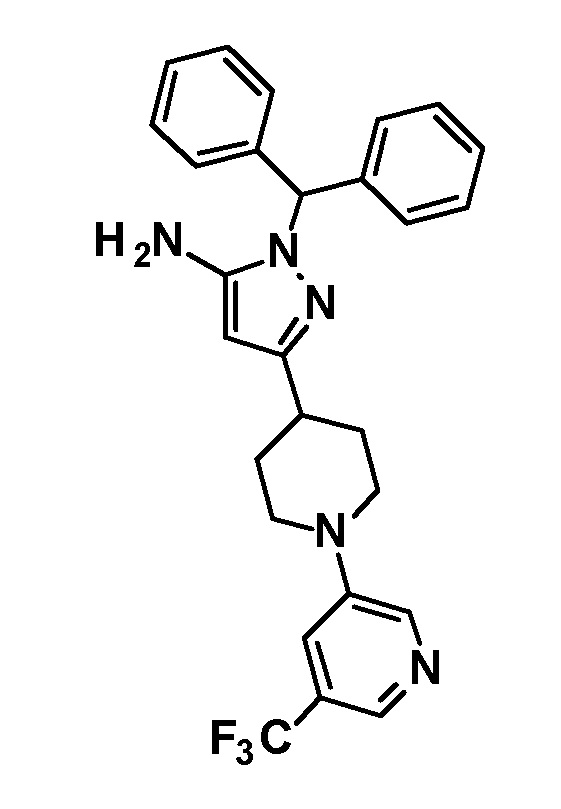
[0489] Указанное в заголовке соединение (2,55 г, выход: 80%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 1, с использованием этил 1-[5-(трифторметил)пиридин-3-ил]пиперидин-4-карбоксилата (соединение, описанное в опубликованной заявке WO2013/187462, 2,02 г, 6,68 ммоль) вместо этил 1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-карбоксилата.
[0490] 1H-ЯМР (400 MГц, CDCl3) δ: 8,45-8,41 (1H, м), 8,25-8,22 (1H, м), 7,37-7,14 (11H, м), 6,64 (1H, с), 3,78-3,69 (2H, м), 3,25-3,16 (2H, м), 2,94-2,84 (2H, м), 2,78-2,69 (1H, м), 2,06-1,98 (2H, м), 1,82-1,69 (2H, м).
[0491] (Ссылочный пример 45) 1-(Дифенилметил)-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0492]
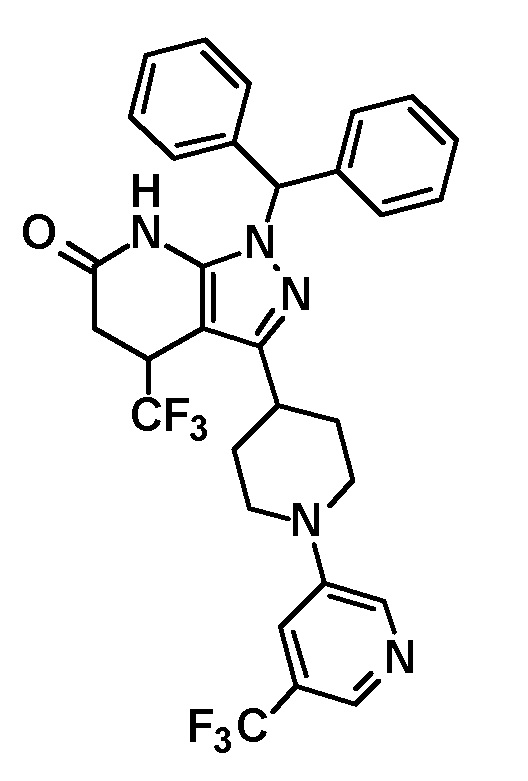
[0493] Осуществляли такую же реакцию, как в способе, описанном в Ссылочном примере 41, с использованием 1-(дифенилметил)-3-{1-[5-(трифторметил)пиридин-3-ил]пиперидин-4-ил}-1H-пиразол-5-амина (2,55 г, 5,34 ммоль), полученного в Ссылочном примере 44, вместо 3-{1-[5-(дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-1H-пиразол-5-амина. К полученному остатку добавляли диизопропиловый эфир и смесь перемешивали. Осадок собирали фильтрованием с получением указанного в заголовке соединения (2,85 г, выход: 89%).
[0494] 1H-ЯМР (400 MГц, CDCl3) δ: 8,45 (1H, д, J=3 Гц), 8,31-8,25 (1H, м), 7,42-7,08 (10H, м), 7,06-6,99 (1H, м), 6,70 (1H, с), 3,86-3,75 (2H, м), 3,66-3,54 (1H, м), 3,01-2,74 (5H, м), 2,08-1,84 (4H, м).
[0495]
(Ссылочный пример 46) Метил 1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-3-ил]пиперидин-4-ил}-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-5-карбоксилат
[0496]
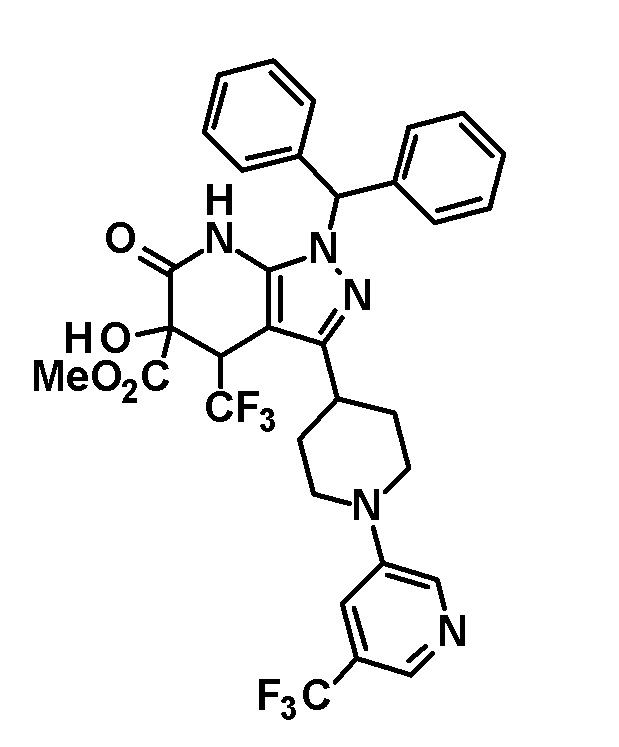
[0497] Осуществляли такую же реакцию, как в способе, описанном в Ссылочном примере 42, с использованием 1-(дифенилметил)-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (2,85 г, 4,7 ммоль), полученного в Ссылочном примере 45, вместо 3-{1-[5-(дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она. К полученному остатку добавляли метанол и смесь перемешивали. Осадок собирали фильтрованием с получением указанного в заголовке соединения (0,980 г, выход: 44%).
[0498] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,54 (1H, с), 8,58 (1H, д, J=3 Гц), 8,26-8,23 (1H, м), 7,57-7,53 (1H, м), 7,37-7,24 (8H, м), 7,22-7,17 (2H, м), 6,88 (1H, с), 4,17-4,07 (1H, м), 3,97-3,84 (2H, м), 3,74 (3H, с), 2,98-2,78 (3H, м), 1,98-1,79 (3H, м), 1,68-1,56 (1H, м).
[0499] (Пример 38) (+)-цис-5-Гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0500]
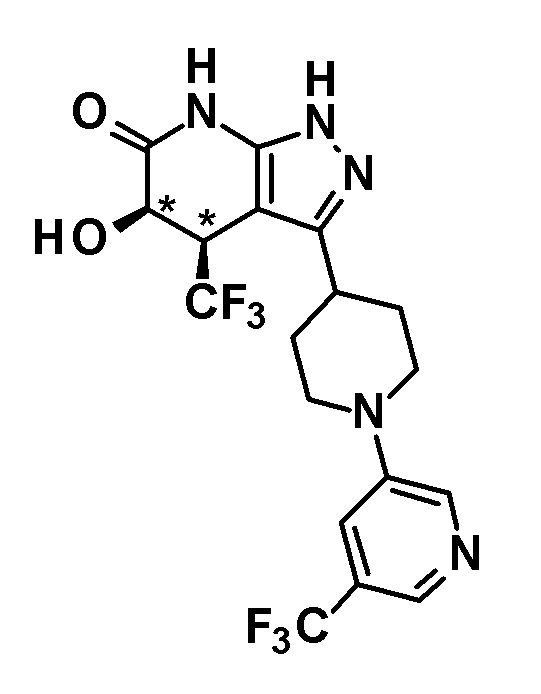
[0501] Гидроксид лития моногидрат (0,183 г, 4,36 ммоль) добавляли к смешанному раствору метил 1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-3-ил]пиперидин-4-ил}-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-5-карбоксилата (0,980 г, 1,45 ммоль), полученного в Ссылочном примере 46, в 1,4-диоксане (15 мл) и воде (5 мл) и смесь перемешивали при 60°C в течение 1 часа. К реакционному раствору добавляли водный раствор хлорида аммония, с последующим экстрагированием этилацетатом. Полученный органический слой сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении с получением неочищенного продукта (0,911 г).
[0502] Промежуточный продукт синтеза получали путем осуществления такой же реакции, как в способе, описанном в Примере 26, с использованием части (0,250 г) неочищенного продукта, полученного, с использованием процедур, описанных выше, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0503] Смешанный раствор промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, в этилацетате и метаноле адсорбировали на силикагеле и растворитель отгоняли при пониженном давлении. Полученный порошок очищали жидкостной флэш-хроматографией [колонка: Chiralflash IA (30 мм в.д. ×100 мм); изготовитель Daicel Corporation, элюент: гексан/этанол=20/80, скорость потока: 10 мл/мин] с получением указанного в заголовке соединения (63,5 мг, выход: 35%, оптически активная форма).
[0504] Оптическую чистоту измеряли с использованием ВЭЖХ [колонка: Chiralpak IA (4,6 мм в.д. ×150 мм); изготовитель Daicel Corporation, элюент: этанол, скорость потока: 2,0 мл/мин].
[0505] Оптическая чистота 98% (время удерживания: 5,0 мин).
[0506] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,26 (1H, с), 10,55 (1H, с), 8,62 (1H, д, J=3 Гц), 8,29-8,26 (1H, м), 7,63-7,59 (1H, м), 5,53 (1H, д, J=4 Гц), 4,47-4,42 (1H, м), 4,21-4,01 (3H, м), 3,03-2,85 (3H, м), 1,90-1,71 (4H, м);
MS (ESI) m/z: 450 (M+H)+;
[α]D25=+5,4° (DMF, c=1,00).
[0507] (Ссылочный пример 47) 1-(Дифенилметил)-3-[1-(6-изопропоксипиридин-3-ил)пиперидин-4-ил]-1H-пиразол-5-амин
[0508]
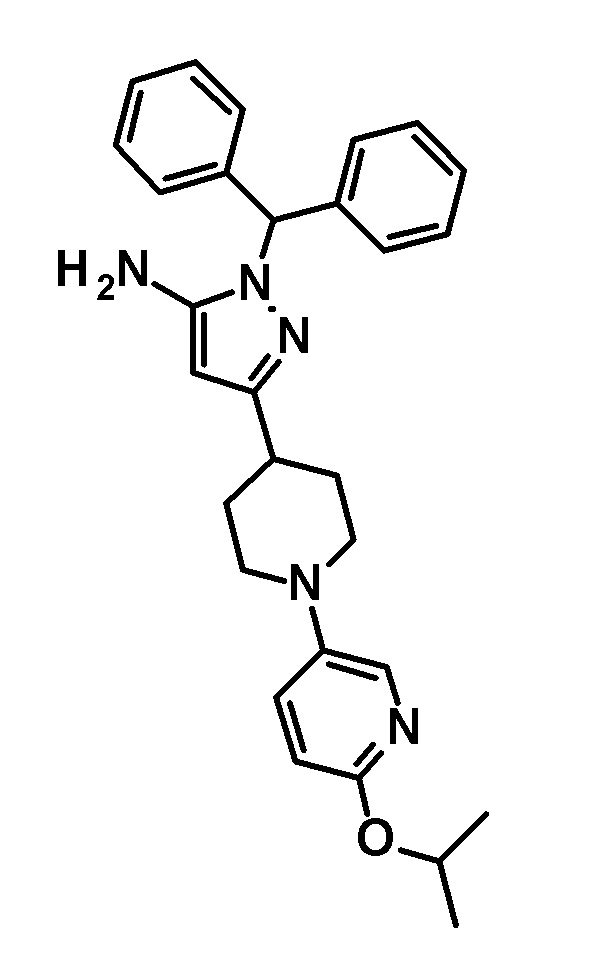
[0509] Указанное в заголовке соединение (531 мг, выход: 34%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 1, с использованием этил 1-(6-изопропоксипиридин-3-ил)пиперидин-4-карбоксилата (соединение, описанное в опубликованной заявке WO2013/187462, 980 мг, 3,35 ммоль), вместо этил 1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-карбоксилата.
[0510] 1H-ЯМР (CDCl3) δ: 7,81 (1H, д, J=3 Гц), 7,37-7,28 (7H, м), 7,23-7,20 (4H, м), 6,67 (1H, с), 6,61 (1H, д, J=9 Гц), 5,46 (1H, с), 5,21-5,15 (1H, м), 3,52-3,47 (2H, м), 3,25 (2H, с), 2,72 (2H, тд, J=12 Гц, 3 Гц), 2,67 (1H, тт, J=12 Гц, 4 Гц), 2,05-2,00 (2H, м), 1,88-1,77 (2H, м), 1,32 (6H, д, J=7 Гц).
[0511] (Ссылочный пример 48) 1-(Дифенилметил)-3-[1-(6-изопропоксипиридин-3-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0512]
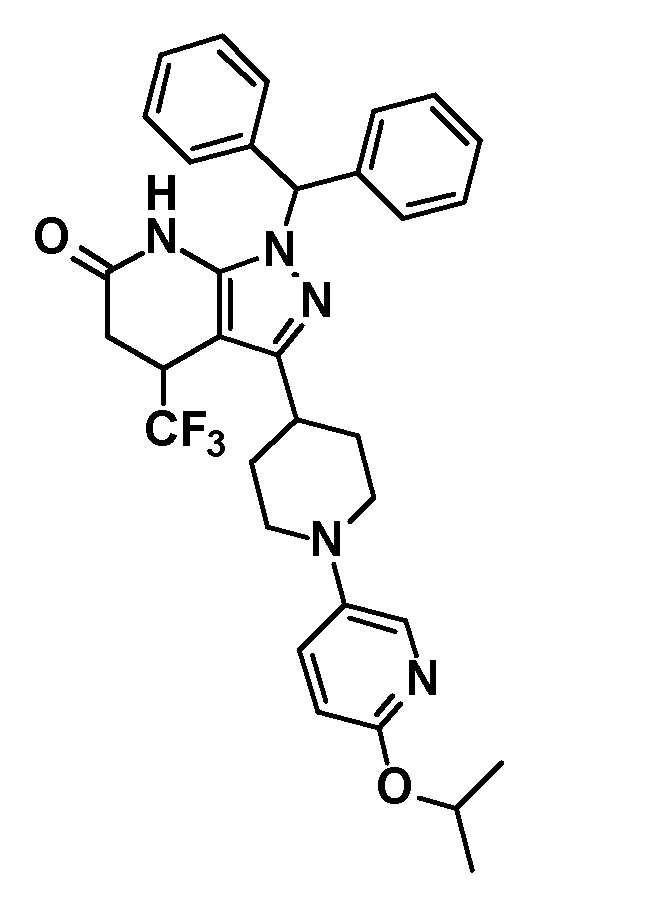
[0513] Указанное в заголовке соединение (512 мг, выход: 81%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 41, с использованием 1-(дифенилметил)-3-[1-(6-изопропоксипиридин-3-ил)пиперидин-4-ил]-1H-пиразол-5-амина (500 мг, 1,07 ммоль), полученного в Ссылочном примере 47, вместо 3-{1-[5-(дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-1H-пиразол-5-амина.
[0514] 1H-ЯМР (CDCl3) δ: 8,32 (1H, уш.с), 7,80 (1H, д, J=3 Гц), 7,38-7,33 (6H, м), 7,28 (1H, дд, J=9 Гц, 3 Гц), 7,23-7,18 (4H, м), 6,67 (1H, с), 6,61 (1H, д, J=9 Гц), 5,21-5,15 (1H, м), 3,66-3,57 (1H, м), 3,56-3,48 (2H, м), 2,90-2,85 (2H, м), 2,77-2,66 (3H, м), 2,12-1,93 (4H, м), 1,33 (6H, д, J=6 Гц).
[0515] (Ссылочный пример 49) Метил 1-(дифенилметил)-5-гидрокси-6-оксо-3-[1-(6-изопропоксипиридин-3-ил)пиперидин-4-ил]-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-5-карбоксилат
[0516]
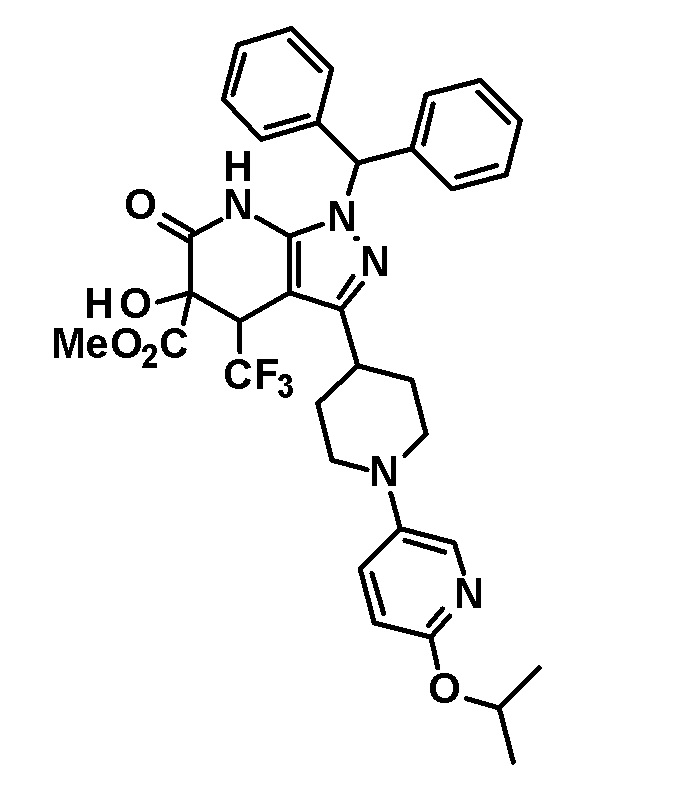
[0517] Указанное в заголовке соединение (285 мг, выход: 52%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 42, с использованием 1-(дифенилметил)-3-[1-(6-изопропоксипиридин-3-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (490 мг, 0,831 ммоль), полученного в Ссылочном примере 48, вместо 3-{1-[5-(дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0518] 1H-ЯМР (CDCl3) δ: 7,78 (1H, д, J=3 Гц), 7,41-7,36 (6H, м), 7,28-7,26 (1H, м), 7,18-7,10 (5H, м), 6,76 (1H, с), 6,61 (1H, д, J=9 Гц), 5,21-5,15 (1H, м), 4,27 (1H, с), 3,89 (3H, с), 3,83-3,76 (1H, м), 3,55-3,46 (2H, м), 2,76-2,66 (3H, м), 2,15-1,82 (4H, м), 1,32 (6H, д, J=6 Гц).
[0519] (Пример 39) Оптически активная форма цис-5-гидрокси-3-[1-(6-изопропоксипиридин-3-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она
[0520]
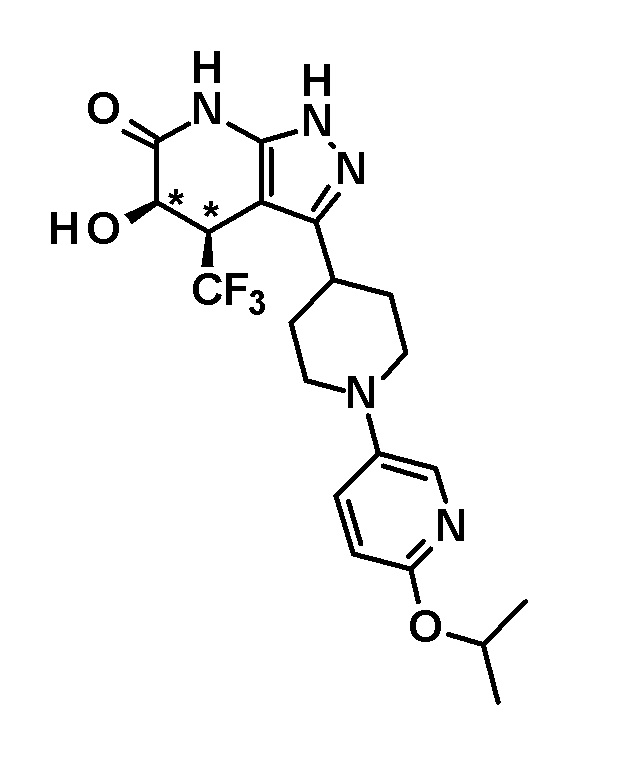
[0521] Неочищенный продукт получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 43, с использованием метил 1-(дифенилметил)-5-гидрокси-6-оксо-3-[1-(6-изопропоксипиридин-3-ил)пиперидин-4-ил]-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-5-карбоксилата (282 мг, 0,425 ммоль), полученного в Ссылочном примере 49, вместо метил 3-{1-[5-(дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-5-карбоксилата.
[0522] Промежуточный продукт синтеза (187 мг) получали путем осуществления такой же реакции, как в способе, описанном в Примере 26, с использованием неочищенного продукта, полученного, с использованием процедур, описанных выше, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0523] Триэтиламин (0,0638 мл, 0,461 ммоль) и (+)-ментилхлорформиат (0,107 мл, 0,502 ммоль) добавляли при 0°C к смешанной суспензии части (184 мг) промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, в THF (15 мл) и этилацетате (5 мл) и смесь перемешивали при 0°C в течение 1 часа и затем при комнатной температуре в течение 4 часов. Снова добавляли триэтиламин (0,0697 мл, 0,502 ммоль) и (+)-ментилхлорформиат (0,133 мл, 0,628 ммоль) и смесь перемешивали при комнатной температуре в течение 3 часов. Опять добавляли триэтиламин (0,0104 мл, 0,754 ммоль) и (+)-ментилхлорформиат (0,178 мл, 0,837 ммоль) и смесь перемешивали в течение ночи. К реакционной смеси добавляли воду, с последующим экстрагированием этилацетатом три раза. Полученный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=100/0-70/30 (градиент)] с получением каждого из соединения, элюируемого первым (70 мг), и соединения, элюируемого вторым (67 мг).
[0524] Указанное в заголовке соединение (39,3 мг, выход: 22%, оптически активная форма) получали путем осуществления такой же реакции, как в способе, описанном в Примере 36, с использованием части (68 мг) соединения, элюируемого первым, полученного, с использованием процедур, описанных выше, вместо цис-5-гидрокси-3-[1-(5-изопропоксипиридин-2-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0525] Оптическую чистоту измеряли с использованием ВЭЖХ [колонка: Chiralpak IA (4,6 мм в.д. × 150 мм); изготовитель Daicel Corporation, элюент: гексан/этанол=50/50, скорость потока: 1,0 мл/мин].
[0526] Оптическая чистота 98% (время удерживания: 8,9 мин);
1H-ЯМР (400 MГц, DMSO-d6) δ: 12,28 (1H, с), 10,55 (1H, с), 7,80 (1H, д, J=3 Гц), 7,44 (1H, дд, J=9 Гц, 3 Гц), 6,63 (1H, д, J=9 Гц), 5,51 (1H, д, J=4 Гц), 5,15-5,09 (1H, м), 4,46-4,43 (1H, м), 4,19-4,10 (1H, м), 3,63-3,58 (2H, м), 2,87-2,79 (1H, м), 2,70-2,62 (2H, м), 1,94-1,75 (4H, м), 1,25 (6H, д, J=6 Гц);
MS (ESI) m/z: 440 (M+H)+.
[0527] (Ссылочный пример 50) 3-[1-(2-Циклопропилпиримидин-5-ил)пиперидин-4-ил]-1-(дифенилметил)-1H-пиразол-5-амин
[0528]
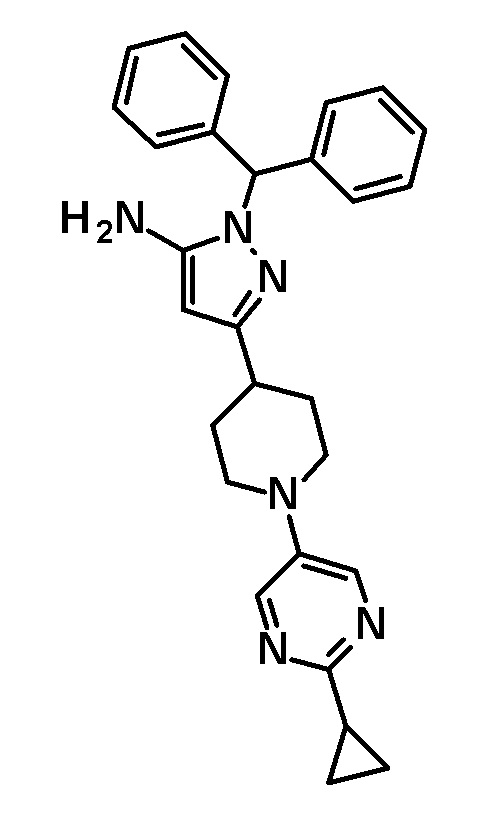
[0529] Указанное в заголовке соединение (0,878 г, выход: 34%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 1, с использованием этил 1-(2-циклопропилпиримидин-5-ил)пиперидин-4-карбоксилата (соединение, описанное в опубликованной заявке WO2013/187462, 1,60 г, 5,81 ммоль), вместо этил 1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-карбоксилата.
[0530] 1H-ЯМР (CDCl3) δ: 8,25 (2H, с), 7,37-7,28 (6H, м), 7,22-7,20 (4H, м), 6,67 (1H, с), 5,44 (1H, с), 3,65-3,60 (2H, м), 3,26 (2H, с), 2,85-2,78 (2H, м), 2,75-2,68 (1H, м), 2,19-2,13 (1H, м), 2,06-2,01 (2H, м), 1,85-1,74 (2H, м), 1,04-0,95 (4H, м).
[0531] (Ссылочный пример 51) 3-[1-(2-Циклопропилпиримидин-5-ил)пиперидин-4-ил]-1-(дифенилметил)-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0532]
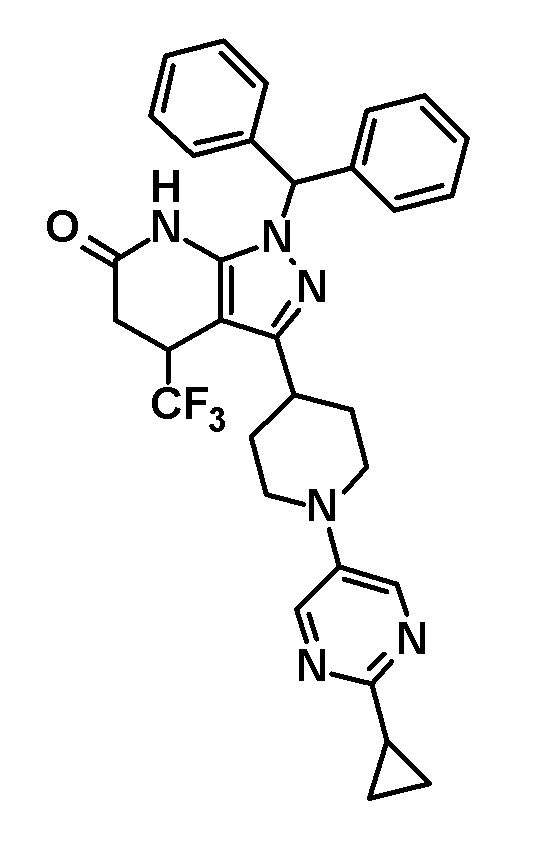
[0533] Указанное в заголовке соединение (0,927 г, выход: 84%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 41, с использованием 3-[1-(2-циклопропилпиримидин-5-ил)пиперидин-4-ил]-1-(дифенилметил)-1H-пиразол-5-амина (0,873 г, 1,94 ммоль), полученного в Ссылочном примере 50, вместо 3-{1-[5-(дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-1H-пиразол-5-амина, с последующей очисткой полученного остатка колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=90/10-20/80 (градиент)].
[0534] 1H-ЯМР (400 MГц, CDCl3) δ: 8,25 (2H, с), 7,78 (1H, с), 7,39-7,34 (6H, м), 7,18-7,14 (4H, м), 6,68 (1H, с), 3,69-3,56 (3H, м), 2,88-2,70 (5H, м), 2,20-2,13 (1H, м), 2,04-1,90 (4H, м), 1,04-0,96 (4H, м).
[0535] (Ссылочный пример 52) Метил 3-[1-(2-циклопропилпиримидин-5-ил)пиперидин-4-ил]-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-5-карбоксилат
[0536]
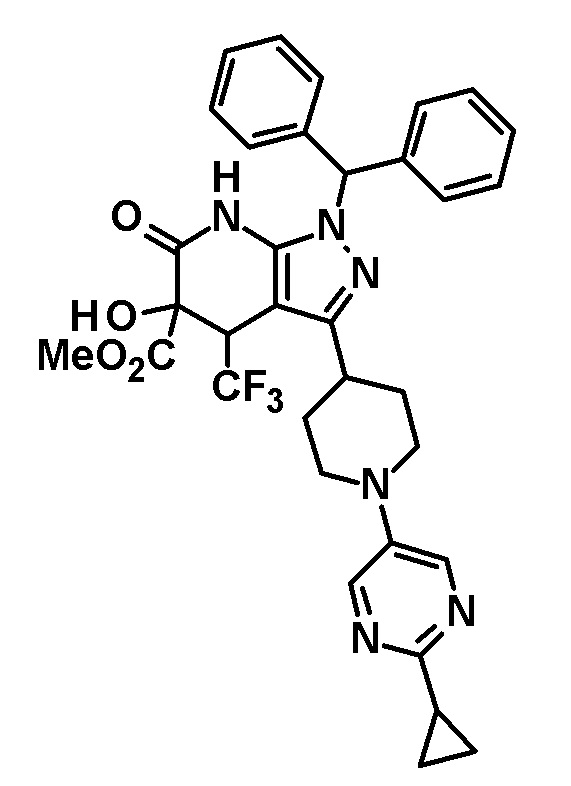
[0537] Осуществляли такую же реакцию, как в способе, описанном в Ссылочном примере 42, с использованием 3-[1-(2-циклопропилпиримидин-5-ил)пиперидин-4-ил]-1-(дифенилметил)-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (0,927 г, 1,62 ммоль), полученного в Ссылочном примере 51, вместо 3-{1-[5-(дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она. К полученному остатку добавляли дихлорметан и смесь перемешивали. Осадок собирали фильтрованием с получением указанного в заголовке соединения (0,404 г, выход: 39%).
[0538] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,54 (1H, с), 8,34 (2H, с), 7,37-7,25 (9H, м), 7,21-7,19 (2H, м), 6,88 (1H, с), 4,11 (1H, кв., J=10Гц), 3,74-3,67 (5H, м), 2,80-2,70 (3H, м), 2,11-2,05 (1H, м), 1,94-1,77 (3H, м), 1,67-1,57 (1H, м), 0,94-0,84 (4H, м).
[0539] (Ссылочный пример 53) цис-3-[1-(2-Циклопропилпиримидин-5-ил)пиперидин-4-ил]-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0540]
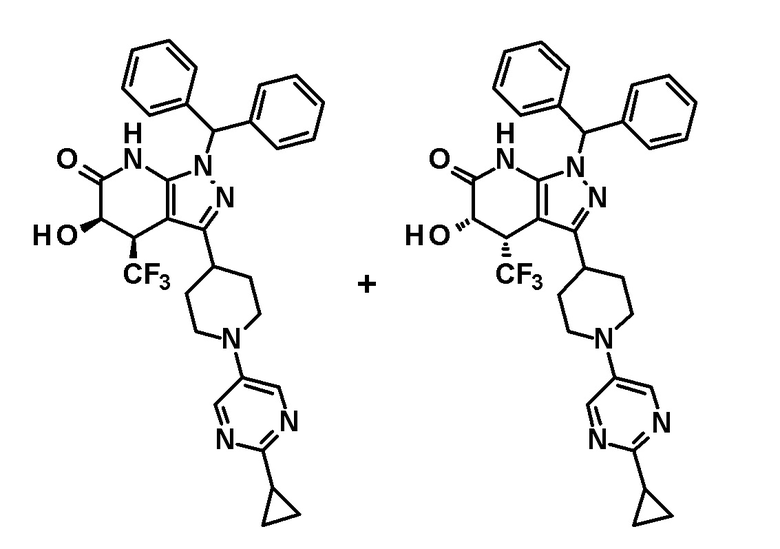
[0541] Указанное в заголовке соединение (0,307 г, выход: 84%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 43, с использованием метил 3-[1-(2-циклопропилпиримидин-5-ил)пиперидин-4-ил]-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-5-карбоксилата (0,400 г, 0,619 ммоль), полученного в Ссылочном примере 52, вместо метил 3-{1-[5-(дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-5-карбоксилата.
[0542] 1H-ЯМР (400 MГц, DMSO-d6) δ: 11,22 (1H, с), 8,35 (2H, с), 7,36-7,17 (10H, м), 6,75 (1H, с), 5,80 (1H, д, J=4 Гц), 4,57 (1H, дд, J=7 Гц, 4 Гц), 4,19-4,10 (1H, м), 3,75-3,68 (2H, м), 2,81-2,71 (3H, м), 2,11-2,05 (1H, м), 1,97-1,91 (1H, м), 1,83-1,65 (3H, м), 0,94-0,84 (4H, м).
[0543] (Пример 40) Оптически активная форма цис-3-[1-(2-циклопропилпиримидин-5-ил)пиперидин-4-ил]-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она
[0544]
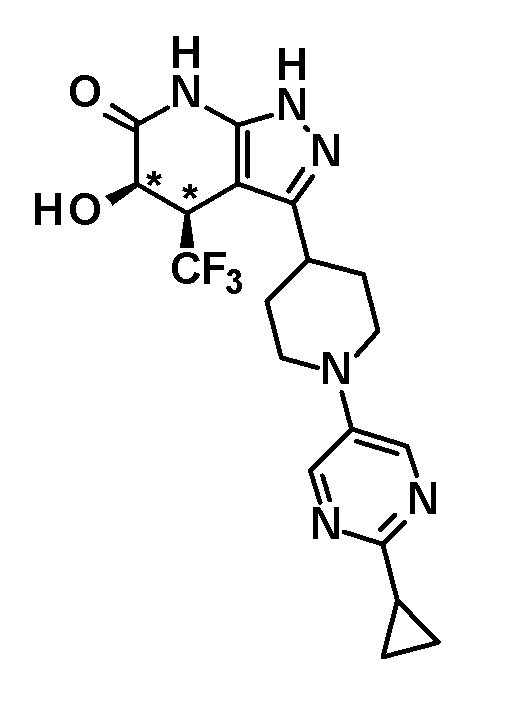
[0545] Промежуточный продукт синтеза получали путем осуществления такой же реакции, как в способе, описанном в Примере 26, с использованием цис-3-[1-(2-циклопропилпиримидин-5-ил)пиперидин-4-ил]-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (0,302 г, 0,513 ммоль), полученного в Ссылочном примере 53, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0546] (+)-Ментилхлорформиат (0,121 мл, 0,571 ммоль) добавляли при 0°C к смешанной суспензии промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, и триэтиламина (72,6 мкл, 0,523 ммоль) в THF (6 мл) и этилацетате (6 мл) и смесь перемешивали при комнатной температуре в течение 2,5 часов. Снова добавляли триэтиламин (19,8 мкл, 0,143 ммоль) и (+)-ментилхлорформиат (40,4 мкл, 0,190 ммоль) при 0°C и смесь перемешивали при комнатной температуре в течение 1 часа. Опять добавляли триэтиламин (13,2 мкл, 0,0952 ммоль) и (+)-ментилхлорформиат (30,3 мкл, 0,143 ммоль) при 0°C и смесь перемешивали при комнатной температуре в течение 1 часа. К реакционной смеси добавляли этилацетат. Органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=93/7-40/60 (градиент)] с получением каждого из соединения, элюируемого первым (111 мг), и соединения, элюируемого вторым (83,3 мг).
[0547] Указанное в заголовке соединение (52,1 мг, выход: 24%, оптически активная форма) получали путем осуществления такой же реакции, как в способе, описанном в Примере 36, с использованием соединени элюируемого первым, полученного, с использованием процедур, описанных выше, вместо цис-5-гидрокси-3-[1-(5-изопропоксипиридин-2-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0548] Оптическую чистоту измеряли с использованием ВЭЖХ [колонка: Chiralpak IA (4,6 мм в.д. ×250 мм); изготовитель Daicel Corporation, элюент: гексан/этанол=10/90, скорость потока: 1,0 мл/мин].
[0549] Оптическая чистота 99% (время удерживания: 12,5 мин);
1H-ЯМР (400 MГц, DMSO-d6) δ: 12,26 (1H, с), 10,54 (1H, с), 8,38 (2H, с), 5,51 (1H, д, J=4 Гц), 4,44 (1H, дд, J=7 Гц, 4 Гц), 4,19-4,10 (1H, м), 3,88-3,79 (2H, м), 2,96-2,86 (1H, м), 2,82-2,72 (2H, м), 2,12-2,06 (1H, м), 1,90-1,73 (4H, м), 0,95-0,85 (4H, м);
MS (ESI) m/z: 423 (M+H)+.
[0550] (Ссылочный пример 54) 1-(Дифенилметил)-3-[1-(6-изопропоксипиридазин-3-ил)пиперидин-4-ил]-1H-пиразол-5-амин
[0551]
[0552] н-Бутиллитий (1,6 M раствор в гексане, 23 мл, 36,6 ммоль) добавляли по каплям при -78°C к раствору безводного ацетонитрила (1,92 мл, 36,6 ммоль) в безводном THF (20 мл) и смесь перемешивали при указанной выше температуре в течение 40 минут. Добавляли по каплям раствор этил 1-[6-(изопропилокси)пиридазин-3-ил]пиперидин-4-карбоксилата (соединение, описанное в опубликованной заявке WO2013/187462, 4,30 г, 14,7 ммоль) в безводном THF (20 мл) при указанной выше температуре и смесь перемешивали в течение 40 минут. Затем добавляли уксусную кислоту (2,52 мл) и температуру смеси повышали до комнатной температуры. Реакционный раствор разделяли на органический и водный слои путем добавления этилацетата и насыщенного солевого раствора и затем подвергали для экстрагированию этилацетатом три раза. Полученный органический слой сушили над безводным сульфатом натрия и растворитель отгоняли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=100/0-60/40 (градиент)] с получением промежуточного продукта синтеза (2,1 г).
[0553] Указанное в заголовке соединение (2,68 г, выход: 41%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 12, с использованием части (2,00 г) промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, вместо трет-бутил 4-(цианоацетил)пиперидин-1-карбоксилата.
[0554] 1H-ЯМР (400 MГц, CDCl3) δ: 7,37-7,28 (6H, м), 7,22-7,20 (4H, м), 7,03 (1H, д, J=10Гц), 6,75 (1H, д, J=10Гц), 6,67 (1H, с), 5,44-5,38 (2H, м), 4,20-4,15 (2H, м), 3,23 (2H, с), 2,97 (2H, тд, J=13 Гц, 3 Гц), 2,79 (1H, тт, J=12 Гц, 4 Гц), 2,04-1,98 (2H, м), 1,78-1,68 (2H, м), 1,37 (6H, д, J=6 Гц).
[0555] (Ссылочный пример 55) 1-(Дифенилметил)-3-[1-(6-изопропоксипиридазин-3-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0556]
[0557] Осуществляли такую же реакцию, как в способе, описанном в Ссылочном примере 41, с использованием 1-(дифенилметил)-3-[1-(6-изопропоксипиридазин-3-ил)пиперидин-4-ил]-1H-пиразол-5-амина (2,28 г, 4,87 ммоль), полученного в Ссылочном примере 54, вместо 3-{1-[5-(дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-1H-пиразол-5-амина. К полученному остатку добавляли диэтиловый эфир и гексан и смесь перемешивали. Осадок собирали фильтрованием с получением указанного в заголовке соединения (2,38 г, выход: 83%).
[0558] 1H-ЯМР (400 MГц, CDCl3) δ: 7,41-7,35 (6H, м), 7,14-7,10 (4H, м), 7,07 (1H, уш.с), 7,02 (1H, д, J=9 Гц), 6,76 (1H, д, J=9 Гц), 6,70 (1H, с), 5,43-5,36 (1H, м), 4,27-4,15 (2H, м), 3,65-3,56 (1H, м), 3,02-2,95 (2H, м), 2,86-2,78 (3H, м), 2,00-1,79 (4H, м), 1,37 (6H, д, J=6 Гц).
[0559] (Ссылочный пример 56) Метил 1-(дифенилметил)-5-гидрокси-6-оксо-3-[1-(6-изопропоксипиридазин-3-ил)пиперидин-4-ил]-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-5-карбоксилат
[0560]
[0561] Осуществляли такую же реакцию, как в способе, описанном в Ссылочном примере 42, с использованием 1-(дифенилметил)-3-[1-(6-изопропоксипиридазин-3-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (2,37 г, 4,01 ммоль), полученного в Ссылочном примере 55, вместо 3-{1-[5-(дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она. К полученному остатку добавляли метанол и смесь перемешивали. Осадок собирали фильтрованием с получением указанного в заголовке соединения (807 мг, выход: 31%).
[0562] 1H-ЯМР (400 MГц, CDCl3) δ: 7,42-7,36 (6H, м), 7,16-7,14 (2H, м), 7,09-7,06 (2H, м), 7,01 (1H, д, J=10Гц), 6,83 (1H, уш.с), 6,79 (1H, с), 6,75 (1H, д, J=9 Гц), 5,43-5,37 (1H, м), 4,26 (1H, с), 4,24-4,18 (2H, м), 3,90 (3H, с), 3,82-3,76 (1H, м), 3,00-2,92 (2H, м), 2,85-2,77 (2H, м), 2,03-1,73 (4H, м), 1,36 (6H, д, J=6 Гц).
[0563]
(Ссылочный пример 57) цис-1-(Дифенилметил)-5-гидрокси-3-[1-(6-изопропоксипиридазин-3-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0564]
[0565] Указанное в заголовке соединение (716 мг, выход: 98%) получали путем осуществления такой же реакции, как в способе, описанном в Ссылочном примере 43, с использованием метил 1-(дифенилметил)-5-гидрокси-6-оксо-3-[1-(6-изопропоксипиридазин-3-ил)пиперидин-4-ил]-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-5-карбоксилата (804 мг, 1,21 ммоль), полученного в Ссылочном примере 56, вместо метил 3-{1-[5-(дифторметокси)пиридин-2-ил]пиперидин-4-ил}-1-(дифенилметил)-5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-5-карбоксилата.
[0566] 1H-ЯМР (400 MГц, CDCl3) δ: 7,42-7,37 (6H, м), 7,12-7,06 (4H, м), 7,02 (1H, д, J=9 Гц), 6,76 (1H, д, J=9 Гц), 6,74-6,72 (2H, м), 5,44-5,37 (1H, м), 4,52 (1H, д, J=7 Гц), 4,28-4,17 (2H, м), 3,97-3,89 (1H, м), 3,69-3,67 (1H, м), 3,03-2,95 (2H, м), 2,86-2,78 (1H, м), 1,91 (4H, ддд, J=45 Гц, 15 Гц, 11 Гц), 1,37 (6H, д, J=6 Гц).
[0567] (Пример 41) (+)-цис-5-Гидрокси-3-[1-(6-изопропоксипиридазин-3-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он
[0568]
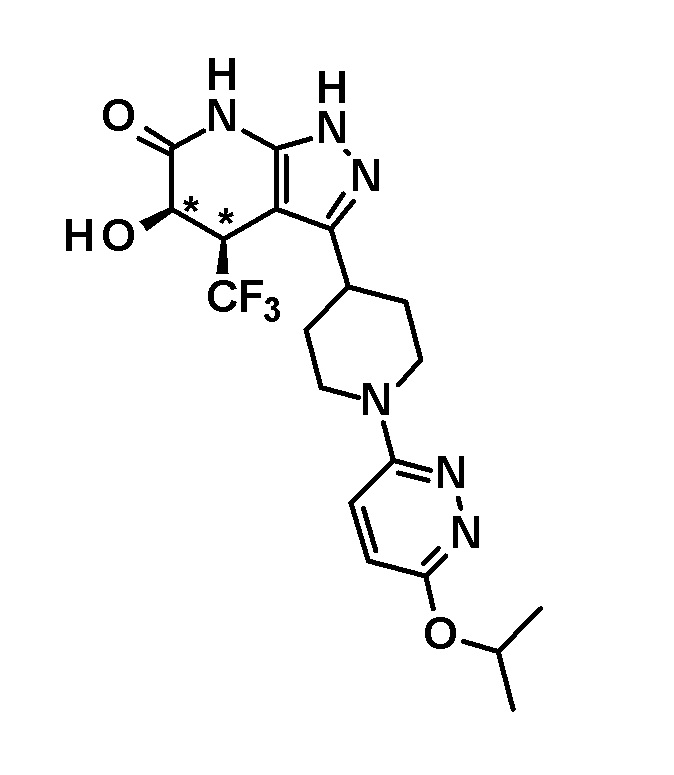
[0569] Промежуточный продукт синтеза (501 мг) получали путем осуществления такой же реакции, как в способе, описанном в Примере 26, с использованием цис-1-(дифенилметил)-5-гидрокси-3-[1-(6-изопропоксипиридазин-3-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она (714 мг, 1,18 ммоль), полученного в Ссылочном примере 57, вместо цис-1-(дифенилметил)-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0570] Триэтиламин (0,172 мл, 1,24 ммоль) и (+)-ментилхлорформиат (0,288 мл, 1,36 ммоль) добавляли при 0°C к смешанной суспензии части (498 мг) промежуточного продукта синтеза, полученного, с использованием процедур, описанных выше, в THF (15 мл) и этилацетате (5 мл) и смесь перемешивали при указанной выше температуре в течение 1 часа и затем при комнатной температуре в течение 3 часов. Снова добавляли триэтиламин (23,5 мкл, 0,170 ммоль) и (+)-ментилхлорформиат (36,0 мкл, 0,170 ммоль) при комнатной температуре и смесь оставляли в течение ночи. К реакционной смеси добавляли воду, с последующим экстрагированием этилацетатом три раза. Полученный органический слой промывали насыщенным солевым раствором и сушили над безводным сульфатом натрия и растворитель концентрировали при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле [элюент: гексан/этилацетат=100/0-60/40 (градиент)] с получением каждого из соединения, элюируемого первым (107 мг), и соединения (119 мг), элюируемого вторым.
[0571] Указанное в заголовке соединение (64,7 мг, выход: 13%, оптически активная форма) получали путем осуществления такой же реакции, как в способе, описанном в Примере 36, с использованием части (105 мг) соединения, элюируемого первым, полученного, с использованием процедур, описанных выше, вместо цис-5-гидрокси-3-[1-(5-изопропоксипиридин-2-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-она.
[0572] 1H-ЯМР (400 MГц, DMSO-d6) δ: 12,23 (1H, с), 10,54 (1H, с), 7,40 (1H, д, J=10Гц), 6,94 (1H, д, J=10Гц), 5,51 (1H, д, J=4 Гц), 5,28-5,21 (1H, м), 4,46-4,43 (1H, м), 4,33-4,26 (2H, м), 4,19-4,11 (1H, м), 3,02-2,95 (1H, м), 2,92-2,81 (2H, м), 1,86-1,65 (4H, м), 1,31 (6H, д, J=6 Гц);
MS (ESI) m/z: 441 (M+H)+;
[α]D25=+10° (DMF, c=1,01).
[0573] (Пример испытания 1) Измерение активности LCAT (in vitro)
Фракцию, состоящую из ЛПВП3 (1,125 < удельный вес < 1,210 г/мл), получали из плазмы здорового субъекта путем центрифугирования в градиенте плотности. Полученную фракцию диализировали против фосфатно-буферного солевого раствора (pH 7,4) и использовали в качестве источника фермента и акцептора для LCAT. Каждое испытываемое лекарственное средство подготавливали путем растворения в диметилсульфоксиде. [14C]Холестерин, содержащий DTNB (реагент Эллмана, конечная концентрация: 0,5 мМ), меркаптоэтанол (конечная концентрация: 12,5 мМ) и 0,6% бычий сывороточный альбумин, добавляли к фосфатно-буферному солевому раствору (pH 7,4), содержащему 1 мг/мл ЛПВП3, и затем добавляли испытываемое лекарственное средство при изменяющихся концентрациях для доведения общего количества до 80 мкл. Эту смесь инкубировали при 37°C приблизительно в течение 16 часов. Затем добавляли смешанный раствор гексана и изопропанола (отношение компонентов смеси=3:2) для остановки реакции. После перемешивания гексановый слой собирали и этот слой упаривали досуха. Добавляли раствор хлороформа (концентрация: 10 мг/мл) и смесь наносили на пластину с тонким слоем силикагеля и проявляли с использованием смешанного раствора гексана, диэтилового эфира и этилацетата (отношение компонентов смеси=85:15:2). Радиоактивность части, соответствующей холестеринолеату, измеряли с использованием анализатора изображений BAS-2500 (изготовитель Fujifilm Corp.). Образец, к которому не добавляли испытываемое лекарственное средство, обрабатывали и анализировали аналогичным образом. EC50 значение активации LCAT рассчитывали в соответствии с формулой, представленной ниже, относительно активности LCAT в образце, к которому не добавляли испытываемое лекарственное средство. Результаты показаны в Таблице 1.
[0574] [Формула 1]
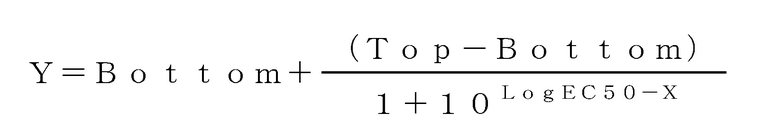
[0575] где X представляет собой логарифм концентрации испытываемого лекарственного средства;
Y представляет собой реактивность (активность LCAT) испытываемого лекарственного средства;
Top представляет собой максимальное значение (максимальное плато);
Bottom представляет собой минимальное значение (минимальное плато); и
EC50 представляет собой 50% эффективную концентрацию.
[0576]
[0577] Как можно видеть из этих результатов, соединение по настоящему изобретению обладает отличным LCAT-активирующим эффектом и является полезным в качестве лекарственного средства для лечения или профилактики заболеваний, таких как дислипидемия и артериосклероз.
[0578] (Пример испытания 2) Измерение активности LCAT (в плазме)
Плазму человека, обезьяны Cynomolgus или трансгенной мыши с человеческим LCAT использовали в качестве источника фермента и акцептора для LCAT. Каждое испытываемое лекарственное средство подготавливали путем растворения в диметилсульфоксиде. [14C]Холестерин, содержащий DTNB (реагент Эллмана, конечная концентрация: 0,5 мМ), меркаптоэтанол (конечная концентрация: 12,5 мМ) и 0,6% бычий сывороточный альбумин, добавляли к 5 мкл каждой плазмы и 45 мкл PBS и затем добавляли испытываемое лекарственное средство при изменяющихся концентрациях для доведения общего количества до 80 мкл. Эту смесь инкубировали при 37°C приблизительно в течение 16 часов. Затем добавляли смешанный раствор гексана и изопропанола (отношение компонентов смеси=3:2) для остановки реакции. После добавления воды и перемешивания гексановый слой собирали и этот слой упаривали досуха. Добавляли раствор хлороформа (концентрация: 10 мг/мл) и смесь наносили на пластину с тонким слоем силикагеля и проявляли с использованием смешанного раствора гексана, диэтилового эфира и этилацетата (отношение компонентов смеси=85:15:2). Радиоактивность части, соответствующей холестеринолеату, измеряли с использованием анализатора изображений BAS-2500 (изготовитель Fujifilm Corp.). Образец, к которому не добавляли испытываемое лекарственное средство, обрабатывали и анализировали аналогичным образом. EC50 значение активации LCAT рассчитывали в соответствии с формулой, представленной ниже, относительно активности LCAT в образце, к которому не добавляли испытываемое лекарственное средство.
[0579] [Формула 2]
[0580] где X представляет собой логарифм концентрации испытываемого лекарственного средства;
Y представляет собой реактивность (активность LCAT) испытываемого лекарственного средства;
Top представляет собой максимальное значение (максимальное плато);
Bottom представляет собой минимальное значение (минимальное плато); и
EC50 представляет собой 50% эффективную концентрацию.
[0581] (Пример испытания 3) Измерение активности LCAT (ex vivo)
Измеряли активность LCAT в плазме обезьяны Cynomolgus или трансгенной мыши с человеческим LCAT, которые получали испытываемое лекарственное средство. [14C]Холестерин, содержащий DTNB (реагент Эллмана, конечная концентрация: 0,26 мМ), меркаптоэтанол (конечная концентрация: 2 мМ) и 0,6% бычий сывороточный альбумин, добавляли к 25 мкл каждой плазмы для доведения общего количества до 40 мкл. Эту смесь инкубировали при 37°C в течение 1 часа. Затем добавляли смешанный раствор гексана и изопропанола (отношение компонентов смеси=3:2) для остановки реакции. После добавления воды и перемешивания гексановый слой собирали и этот слой упаривали досуха. Добавляли раствор хлороформа (концентрация: 10 мг/мл) и смесь наносили на пластину с тонким слоем силикагеля и проявляли с использованием смешанного раствора гексана, диэтилового эфира и этилацетата (отношение компонентов смеси=85:15:2). Радиоактивность части, соответствующей холестеринолеату, измеряли с использованием анализатора изображений BAS-2500 (изготовитель Fujifilm Corp.). Рассчитывали скорость изменения активации LCAT в каждой точке времени по сравнению с активностью LCAT до введения средства.
[0582] (Пример испытания 4) Испытание эффективности лекарственного средства на обезьянах Cynomolgus.
Каждое испытываемое лекарственное средство растворяли в смешанном растворе [4/1 (об./об.)] пропиленгликоля (Sigma-Aldrich Corp.)-Tween 80 (Sigma-Aldrich Corp.) или 0,5% (масс./об.) водном растворе метилцеллюлозы и раствор перорально вводили обезьянам Cynomolgus в течение 1 или 7 дней. В день 1 или 7 периода введения кровь собирали перед введением и после введения и получали плазму. Измеряли содержание холестерина в плазме с использованием коммерчески доступного набора для анализа (Cholesterol-E Wako, Wako Pure Chemical Industries, Ltd.). Профиль липопротеинов анализировали при помощи ВЭЖХ (колонка: LipopropakXL, изготовитель Tosoh Corp.). Содержание ЛПВП-холестерина и не-ЛПВП-холестерина рассчитывали в соответствии со следующей формулой для расчета:
[0583] Содержание ЛПВП-холестерина=содержание холестерина в плазме × (площадь пика ЛПВП-холестерина/общая сумма пиков)
Содержание не-ЛПВП-холестерина=содержание холестерина в плазме × (площадь пика не-ЛПВП-холестерина/общая сумма пиков)
Процент (%) повышения уровня ЛПВП после введения одной дозы 10 мг/кг по сравнению с уровнем перед введением определяли из AUC до введения и через 24 часа после введения. Результаты показаны в Таблице 2.
[0585] (Пример испытания 5) Испытание эффективности лекарственного средства на трансгенных мышах с человеческим LCAT
Каждое испытываемое лекарственное средство растворяли в смешанном растворе [4/1 (об./об.)] пропиленгликоля (Sigma-Aldrich Corp.)-Tween 80 (Sigma-Aldrich Corp.) или 0,5% (масс./об.) водном растворе метилцеллюлозы и раствор перорально вводили трансгенной мыши с человеческим LCAT в течение 1, 4 или 7 дней. В день 1, 4 или 7 периода введения кровь собирали перед введением и после введения и получали плазму. Измеряли содержание холестерина в плазме с использованием коммерчески доступного набора для анализа (Cholesterol-E Wako, Wako Pure Chemical Industries, Ltd.). Профиль липопротеинов анализировали при помощи ВЭЖХ (колонка: LipopropakXL, изготовитель Tosoh Corp.). Содержание ЛПВП-холестерина и не-ЛПВП-холестерина рассчитывали в соответствии со следующей формулой для расчета:
[0586] Содержание ЛПВП-холестерина=содержание холестерина в плазме × (площадь пика ЛПВП-холестерина/общая сумма пиков)
Содержание не-ЛПВП-холестерина=содержание холестерина в плазме × (площадь пика не-ЛПВП-холестерина/общая сумма пиков)
Как можно видеть из этих результатов, соединение по настоящему изобретению демонстрирует отличный LCAT-активирующий эффект и является полезным в качестве лекарственного средства для лечения или профилактики заболеваний, таких как дислипидемия и артериосклероз.
(Пример формулирования 1) Твердая капсула
Каждую стандартную состоящую из двух частей твердую желатиновую капсулу, используемую в качестве оболочки, заполняют 100 мг соединения Примера 1 в форме порошка, 150 мг лактозы, 50 мг целлюлозы и 6 мг стеарата магния с получением стандартной единицы в виде капсулы, которую, в свою очередь, промывают и затем сушат.
[0587] (Пример формулирования 2) Мягкая капсула
Получают смесь соединения Примера 2, добавленного в легко усваемое масло, например, соевое масло, масло семян хлопчатника или оливковое масло, и вводят в желатиновую оболочку с использованием поршневого насоса, с получением мягкой капсулы, содержащей 100 мг активного ингредиента, которую, в свою очередь промывают и затем сушат.
[0588] (Пример формулирования 3) Таблетка
В соответствии с рутинным способом, таблетку получают с использованием 100 мг соединения Примера 3, 0,2 мг коллоидного диоксида кремния, 5 мг стеарата магния, 275 мг микрокристаллической целлюлозы, 11 мг крахмала и 98,8 мг лактозы.
[0589] Если желательно, на таблетку наносят покрытие.
[0590] (Пример формулирования 4) Суспензия
Получают суспензию, содержащую 100 мг соединения Примера 4 в виде тонкодисперсного порошка, 100 мг натрий карбоксиметилцеллюлозы, 5 мг бензоата натрия, 1,0 г раствора сорбита (Japanese Pharmacopoeia) и 0,025 мл ванилина, в 5 мл.
[0591] (Пример формулирования 5) Инъекция
Соединение Примера 6 (1,5% масс.) перемешивают в 10% масс. пропиленгликоля, затем доводят до установленного объема при помощи воды для инъекций и затем стерилизуют с получением инъекции.
Промышленная применимость
[0592] Соединение, представленное общей формулой (I) по настоящему изобретению, или его фармакологически приемлемая соль обладает отличным LCAT-активирующим эффектом и является особенно полезным в качестве активного ингредиента терапевтического или профилактического средства от артериосклероза, артериосклеротического сердечного заболевания, коронарного сердечного заболевания (включая острые коронарные синдромы, сердечную недостаточность, инфаркт миокарда, стенокардию, сердечную ишемию, сердечно-сосудистое расстройство и рестеноз, вызванный ангиогенезом), цереброваскулярного заболевания (включая инсульт и церебральный инфаркт), заболевания
периферических сосудов (включая заболевание периферических артерий и диабетические сосудистые осложнения), дислипидемии, LCAT дефицита, гипо-ЛПВП-холестеринемии, гипер-ЛПНП-холестеринемии, сахарного диабета, гипертензии, метаболического синдрома, болезни Альцгеймера, помутнения роговицы или почечного заболевания, в частности, в качестве анти-артериосклеротического средства.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ СЕРИНА В КАЧЕСТВЕ АГОНИСТОВ ГРЕЛИНОВЫХ РЕЦЕПТОРОВ | 2015 |
|
RU2695649C2 |
| ЦИКЛОАЛКИЛНИТРИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2655380C2 |
| ИНГИБИТОРЫ КИНАЗЫ | 2013 |
|
RU2637944C2 |
| ГЕМИНАЛЬНО-ЗАМЕЩЕННЫЕ ЦИАНОЭТИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ JANUS КИНАЗ | 2014 |
|
RU2664533C2 |
| ИНГИБИТОРЫ СИГМА-РЕЦЕПТОРА | 2005 |
|
RU2404972C2 |
| АМИНОТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2009 |
|
RU2552642C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE10 | 2011 |
|
RU2543386C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| ПРОЛЕКАРСТВА СОПРЯЖЕННО-БИЦИКЛИЧЕСКИХ АНТАГОНИСТОВ C5aR | 2019 |
|
RU2794327C2 |
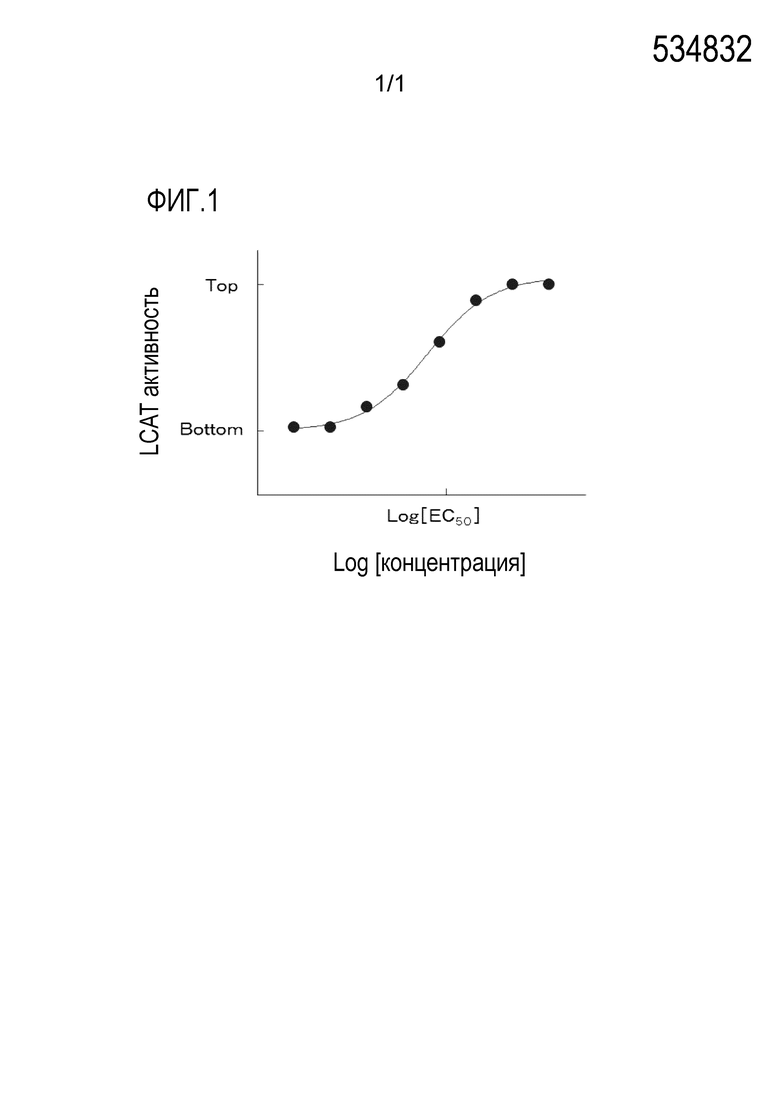
Изобретение относится к соединению, представленному общей формулой (I), или к его фармакологически приемлемой соли, в которой радикалы и символы имеют значения, приведенные в формуле изобретения. Данное соединение обладает отличным LCAT-активирующим эффектом и является полезным в качестве активного ингредиента терапевтического или профилактического средства от артериосклероза, артериосклеротического сердечного заболевания, коронарного сердечного заболевания (включая сердечную недостаточность, инфаркт миокарда, стенокардию, сердечную ишемию, сердечно-сосудистое расстройство и рестеноз, вызванный ангиогенезом), цереброваскулярного заболевания (включая инсульт и церебральный инфаркт), заболевания периферических сосудов (включая диабетические сосудистые осложнения), дислипидемии, гипо-ЛПВП-холестеринемии, гипер-ЛПНП-холестеринемии или почечного заболевания, в частности, в качестве анти-артериосклеротического средства. 16 н. и 25 з.п. ф-лы, 1 ил., 2 табл., 41 пр.
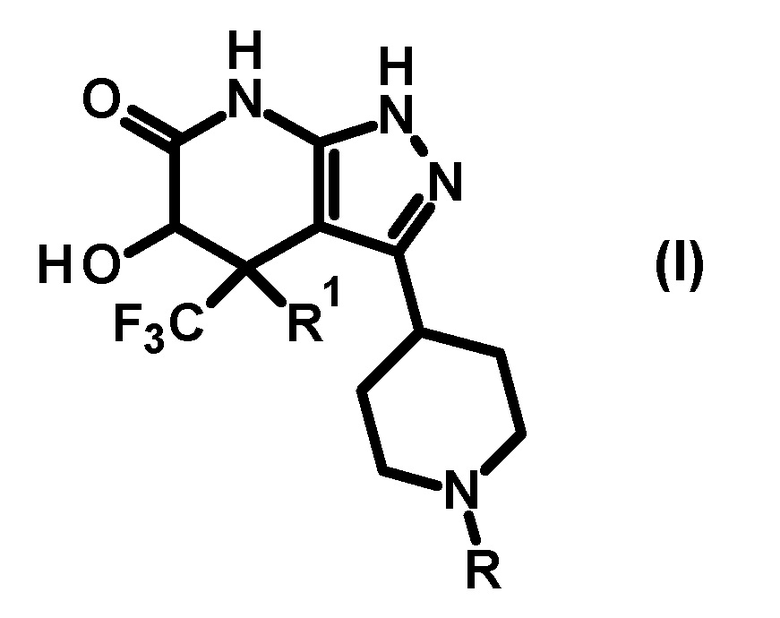
1. Соединение, представленное общей формулой (I), или его фармакологически приемлемая соль:
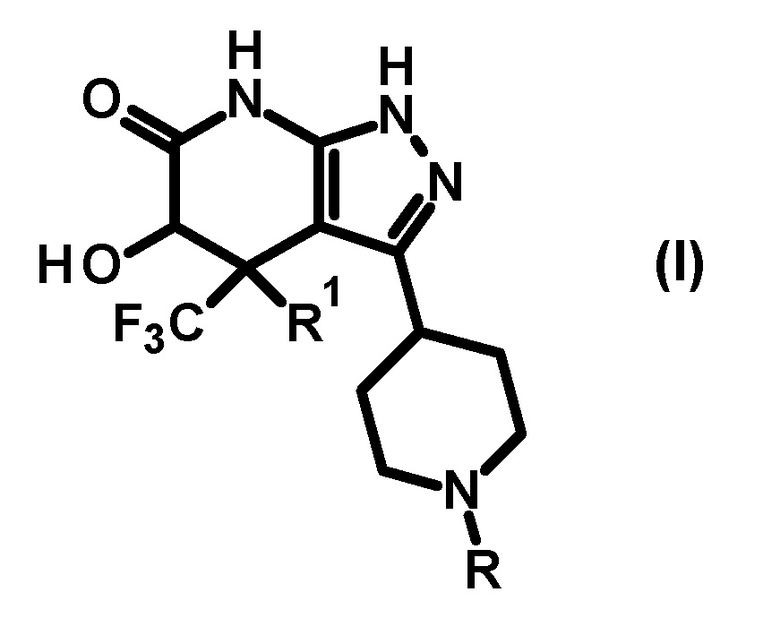
где R представляет собой замещенную пиридильную, пиримидильную, пиразинильную, пиридазинильную, тиадиазолильную или тиазолильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из атома хлора, атома фтора, C1-3 алкильной группы, циклопропильной группы, трифторметильной группы, дифторметокси группы, трифторметокси группы, циано группы, C1-3 алкокси группы, C2-4 алкоксикарбонильной группы и бензилоксикарбонильной группы), и
R1 представляет собой атом водорода или гидрокси группу.
2. Соединение по п. 1 или его фармакологически приемлемая соль, где R представляет собой замещенную пиридильную, пиримидильную, пиразинильную или пиридазинильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из изопропильной группы, трифторметильной группы, дифторметокси группы, циано группы и изопропокси группы).
3. Соединение по п. 1 или его фармакологически приемлемая соль, где R представляет собой пиридильную, пиримидильную, пиразинильную или тиадиазолильную группу, замещенную трифторметильной группой.
4. Соединение по п. 1 или его фармакологически приемлемая соль, где R представляет собой пиридильную, пиримидильную или пиразинильную группу, замещенную трифторметильной группой.
5. Соединение по п. 1 или его фармакологически приемлемая соль, где R1 представляет собой атом водорода.
6. Соединение по п. 5 или его фармакологически приемлемая соль, где соединение или соль выбраны из группы, состоящей из следующих:
5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-3-[1-(5-изопропоксипиридин-2-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридазин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-3-{1-[2-изопропил-6-(трифторметил)пиримидин-4-ил]пиперидин-4-ил}-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиразин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-4-(трифторметил)-3-{1-[2-(трифторметил)пиримидин-5-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
6-{4-[5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-4-(трифторметил)пиридин-3-карбонитрил,
3-{1-[3-хлор-5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-4-(трифторметил)-3-{1-[4-(трифторметил)-1,3-тиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-4-(трифторметил)-3-{1-[4-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
3-[1-(5-хлорпиридин-2-ил)пиперидин-4-ил]-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиримидин-4-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-3-[1-(6-изопропоксипиридазин-3-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
5-гидрокси-3-[1-(6-изопропоксипиридазин-3-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он и
3-[1-(2-циклопропилпиримидин-5-ил)пиперидин-4-ил]-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он.
7. Соединение по п. 5 или его фармакологически приемлемая соль, где соединение или соль выбраны из группы, состоящей из следующих:
(+)-цис-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридазин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-5-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиразин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-5-гидрокси-4-(трифторметил)-3-{1-[2-(трифторметил)пиримидин-5-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-6-{4-[5-гидрокси-6-оксо-4-(трифторметил)-4,5,6,7-тетрагидро-1H-пиразоло[3,4-b]пиридин-3-ил]пиперидин-1-ил}-4-(трифторметил)пиридин-3-карбонитрил,
(+)-цис-3-[1-(5-хлорпиридин-2-ил)пиперидин-4-ил]-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиридин-3-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-гидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-5-гидрокси-4-(трифторметил)-3-{1-[6-(трифторметил)пиримидин-4-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-5-гидрокси-3-[1-(6-изопропоксипиридазин-3-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он,
(+)-цис-5-гидрокси-3-[1-(6-изопропоксипиридазин-3-ил)пиперидин-4-ил]-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он и
(+)-цис-3-[1-(2-циклопропилпиримидин-5-ил)пиперидин-4-ил]-5-гидрокси-4-(трифторметил)-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он.
8. Соединение по п. 1 или его фармакологически приемлемая соль, где R1 представляет собой гидрокси группу.
9. Соединение по п. 8 или его фармакологически приемлемая соль, где соединение или соль выбраны из группы, состоящей из следующих:
4,5-дигидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он и
4,5-дигидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиразин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он.
10. Соединение по п. 8 или его фармакологически приемлемая соль, где соединение или соль выбраны из группы, состоящей из следующих:
(+)-4,5-дигидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиридин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он и
(+)-4,5-дигидрокси-4-(трифторметил)-3-{1-[5-(трифторметил)пиразин-2-ил]пиперидин-4-ил}-1,4,5,7-тетрагидро-6H-пиразоло[3,4-b]пиридин-6-он.
11. Соединение по п. 1 или его фармакологически приемлемая соль, где R представляет собой замещенную фенильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из атома хлора, дифторметокси группы, трифторметокси группы и циано группы), и R1 представляет собой атом водорода.
12. Соединение по п. 1 или его фармакологически приемлемая соль, где R представляет собой замещенную фенильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из дифторметокси группы, трифторметокси группы и циано группы), и R1 представляет собой атом водорода.
13. Соединение по п. 1 или его фармакологически приемлемая соль, где R представляет собой замещенную пиридильную, пиримидильную, пиразинильную, пиридазинильную, тиадиазолильную или тиазолильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из атома хлора, атома фтора, C1-3 алкильной группы, циклопропильной группы, трифторметильной группы, дифторметокси группы, трифторметокси группы, циано группы, C1-3 алкокси группы, C2-4 алкоксикарбонильной группы и бензилоксикарбонильной группы), и R1 представляет собой атом водорода.
14. Соединение по п. 1 или его фармакологически приемлемая соль, где R представляет собой замещенную пиридильную, пиримидильную, пиразинильную или пиридазинильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из изопропильной группы, трифторметильной группы, дифторметокси группы, циано группы и изопропокси группы), и R1 представляет собой атом водорода.
15. Соединение по п. 1 или его фармакологически приемлемая соль, где R представляет собой пиридильную, пиримидильную или пиразинильную группу, замещенную трифторметильной группой, и R1 представляет собой атом водорода.
16. Соединение по п. 1 или его фармакологически приемлемая соль, где R представляет собой замещенную фенильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из атома хлора, дифторметокси группы, трифторметокси группы и циано группы), и R1 представляет собой гидрокси группу.
17. Соединение по п. 1 или его фармакологически приемлемая соль, где R представляет собой замещенную фенильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из дифторметокси группы, трифторметокси группы и циано группы), и R1 представляет собой гидрокси группу.
18. Соединение по п. 1 или его фармакологически приемлемая соль, где R представляет собой замещенную пиридильную, пиримидильную, пиразинильную, пиридазинильную, тиадиазолильную или тиазолильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из атома хлора, атома фтора, C1-3 алкильной группы, циклопропильной группы, трифторметильной группы, дифторметокси группы, трифторметокси группы, циано группы, C1-3 алкокси группы, C2-4 алкоксикарбонильной группы и бензилоксикарбонильной группы), и R1 представляет собой гидрокси группу.
19. Соединение по п. 1 или его фармакологически приемлемая соль, где R представляет собой замещенную пиридильную, пиримидильную, пиразинильную или пиридазинильную группу (заместитель(заместители) представляет(представляют) собой 1 или 2 одинаковые или отличные друг от друга группы, выбранные из группы, состоящей из изопропильной группы, трифторметильной группы, дифторметокси группы, циано группы и изопропокси группы), и R1 представляет собой гидрокси группу.
20. Соединение по п. 1 или его фармакологически приемлемая соль, где R представляет собой пиридильную, пиримидильную или пиразинильную группу, замещенную трифторметильной группой, и R1 представляет собой гидрокси группу.
21. Соединение по любому из пп. 1-6 и 8-20 или его фармакологически приемлемая соль, где трифторметильная группа в 4-положении пиразолопиридинового кольца и гидрокси группа в 5-положении этого кольца находятся в цис-положении относительно друг друга.
22. Соединение по любому из пп. 1-6, 8, 9 и 11-20 или его фармакологически приемлемая соль, где оптическое вращение представляет собой (+).
23. Фармацевтическая композиция, обладающая активирующим действием в отношении лецитин-холестерин-ацетилтрансферазы (LCAT), содержащая соединение по любому из пп. 1-22 или его фармакологически приемлемую соль в качестве активного ингредиента.
24. Фармацевтическая композиция для профилактики или лечения артериосклероза, артериосклеротического сердечного заболевания, коронарного сердечного заболевания, цереброваскулярного заболевания, заболевания периферических сосудов, дислипидемии, гипо-ЛПВП-холестеринемии, гипер-ЛПНП-холестеринемии, LCAT недостаточности, или почечного заболевания, содержащая соединение по любому из пп. 1-22 или его фармакологически приемлемую соль в качестве активного ингредиента.
25. Профилактическое или терапевтическое средство от артериосклероза, содержащее соединение по любому из пп. 1-22 или его фармакологически приемлемую соль в качестве активного ингредиента.
26. Профилактическое или терапевтическое средство от дислипидемии, содержащее соединение по любому из пп. 1-22 или его фармакологически приемлемую соль в качестве активного ингредиента.
27. Профилактическое или терапевтическое средство от заболевания, вызванного повышенной концентрацией ЛПНП холестерина в крови, содержащее соединение по любому из пп. 1-22 или его фармакологически приемлемую соль в качестве активного ингредиента.
28. Профилактическое или терапевтическое средство от заболевания, вызванного пониженной концентрацией ЛПВП-холестерина в крови, содержащее соединение по любому из пп. 1-22 или его фармакологически приемлемую соль в качестве активного ингредиента.
29. Активатор LCAT, содержащий соединение по любому из пп. 1-22 или его фармакологически приемлемую соль в качестве активного ингредиента.
30. Обратимый активатор LCAT, содержащий соединение по любому из пп. 1-22 или его фармакологически приемлемую соль в качестве активного ингредиента.
31. Анти-артериосклеротическое средство, содержащее соединение по любому из пп. 1-22 или его фармакологически приемлемую соль в качестве активного ингредиента.
32. Способ активации LCAT, включающий введение человеку эффективного количества соединения по любому из пп. 1-22 или его фармакологически приемлемой соли.
33. Способ профилактики или лечения артериосклероза, артериосклеротического сердечного заболевания, коронарного сердечного заболевания, цереброваскулярного заболевания, заболевания периферических сосудов, дислипидемии, гипо-ЛПВП-холестеринемии, гипер-ЛПНП-холестеринемии, LCAT недостаточности, или почечного заболевания, включающий введение человеку эффективного количества соединения по любому из пп. 1-22 или его фармакологически приемлемой соли.
34. Способ профилактики или лечения артериосклероза, включающий введение человеку эффективного количества соединения по любому из пп. 1-22 или его фармакологически приемлемой соли.
35. Способ профилактики или лечения дислипидемии, включающий введение человеку эффективного количества соединения по любому из пп. 1-22 или его фармакологически приемлемой соли.
36. Способ профилактики или лечения заболевания, вызванного повышенной концентрацией ЛПНП холестерина в крови, включающий введение человеку эффективного количества соединения по любому из пп. 1-22 или его фармакологически приемлемой соли.
37. Способ профилактики или лечения заболевания, вызванного пониженной концентрацией ЛПВП-холестерина в крови, включающий введение человеку эффективного количества соединения по любому из пп. 1-22 или его фармакологически приемлемой соли.
38. Соединение по любому из пп. 1-22 или его фармакологически приемлемая соль для применения в способе лечения или профилактики артериосклероза.
39. Соединение по любому из пп. 1-22 или его фармакологически приемлемая соль для применения в способе лечения или профилактики дислипидемии.
40. Соединение по любому из пп. 1-22 или его фармакологически приемлемая соль для применения в способе лечения или профилактики заболевания, вызванного повышенной концентрацией ЛПНП холестерина в крови.
41. Соединение по любому из пп. 1-22 или его фармакологически приемлемая соль для применения в способе лечения или профилактики заболевания, вызванного пониженной концентрацией ЛПВП-холестерина в крови.
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| ИНГИБИТОР АТЕРОСКЛЕРОТИЧЕСКОГО УТОЛЩЕНИЯ ВНУТРЕННЕЙ ОБОЛОЧКИ СОСУДОВ | 1992 |
|
RU2114620C1 |

