Изобретение относится к новым псевдополиморфным кристаллическим формам дигидрохлорида 2-[2-[4-(бис-(4-фторфенил)метил)-1-пиперазинил]этокси]уксусной кислоты, способам их получения и к фармацевтическим композициям, их содержащим.
2-[2-[4-(бис-(4-фторфенил)метил)1-пиперазинил] этокси] уксусная кислота, известная как и называемая далее эфлетиризин (INN: Международное несобственное название), представляет собой соединение следующей формулы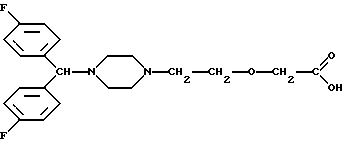
Эфлетиризин подпадает под общую формулу I в европейском патенте 58146 и относится к замещенным производным бензгидрилпиперазина.
Подобно 2-[2-[4-[(4-хлорфенил)фенилметил]-1-пиперазинил]этокси]уксусной кислоте, известной как и называемый ниже цетиризин (INN), эфлетиризин, как было установлено, обладает превосходными антигистаминными свойствами. Он относится к фармакологическому классу антагонистов рецептора гистамина Н1 второго поколения и обнаруживает in vitro высокое сродство и селективность для H1-рецепторов. Как цетиризин, он пригоден в качестве антиаллергенного, антигистаминного, бронходилаторного и антиспазматического агента. Последние клинические исследования показали полезность эфлетиризина при его введении в виде назального аэрозоля для лечения аллергического ринита и рино-конъюктивита (J.-F. Dessanges et.al, Allergy and Clin.Immunol. News (1994), Suppl. 2, abstract 1864; С. De Vos et.al., Allergy and Clin.Immunol. News (1994), Suppl. 2, abstract 428).
Другие недавние клинические фармакологические исследования показали, что эфлетиризин дает неожиданно хорошие результаты при лечении крапивницы, атонического дерматита и чесотки.
Эфлетиризин является аморфным твердым веществом. Однако очень желательно получать продукт с воспроизводимыми характеристиками, который во время приготовления препаративных форм ведет себя всегда одинаково, особенно в отношении предъявляемых требований. По этим причинам заявитель пытался получить кристаллические формы эфлетиризина. Хотя изучалось терапевтическое применение эфлетиризина, таким кристаллическим формам внимание пока не уделялось.
Данное изобретение вытекает из неожиданного обнаружения двух псевдополиморфных кристаллических форм дигидрохлорида эфлетиризина, а именно безводного дигидрохлорида эфлетиризина и моногидрата дигидрохлорида эфлетиризина. Для удобства идентификации безводный дигидрохлорид эфлетиризина далее обозначается как "Форма А", а моногидрат дигидрохлорида эфлетиризина обозначается как "Форма В".
Согласно другому аспекту данное изобретение предусматривает способы получения этих новых псевдополиморфных форм, а также способы конверсии Формы А в Форму В и Формы В в Форму А.
Данное изобретение основано также на установлении того факта, что эти две новые псевдополиморфные формы имеют различные свойства. В частности, установлено, что твердые фармацевтические композиции, содержащие дигидрохлорид эфлетиризина в Форме А, более стабильны во времени при хранении, чем твердые фармкомпозиции, содержащие Форму В. Эта большая стабильность при хранении обусловлена лучшей совместимостью с твердыми носителями и разбавителями, обычно применяемыми в твердых фармкомпозициях.
Соответственно, данное изобретение относится также к фармкомпозициям, содержащим Форму А или Форму В в сочетании с подходящими фармацевтическими эксципиентами, носителями или разбавителями, предпочтительно, к твердым фармкомпозициям, содержащим Форму А.
Что касается способов получения этих псевдополиморфных форм дигидрохлорида эфлетиризина, то Форму В можно получить путем гидролиза в водной среде 2-[4-бис-(4-фторфенил) метил]-1-пиперазинил] этоксиацетамида в присутствии соляной кислоты при температуре, лежащей между 40oС и температурой нагревания реакционной смеси с обратным холодильником. Форма В затем может быть перекристаллизована в водном растворе кислоты или в смеси растворителей, содержащей воду и соляную кислоту.
Форму В затем можно перевести в Форму А путем нагревания до температуры кипения в растворителе, таком как ацетон или метилэтилкетон. Форму А можно опять перевести в Форму В путем перекристаллизации в среде водной соляной кислоты. Следующие примеры иллюстрируют способы получения дигидрохлорида эфлетиризина в Форме А и Форме В в соответствии с настоящим изобретением. В этих примерах дифференциальные термограммы были получены на дифференциальном сканирующем калориметре PERKIN ELMER DSC 7 с температурным градиентом 20oС/мин.
1. Получение 2-[4-[бис-(4-фторфенил)метил]-1-пиперазинил]этоксиацетамида и его дигидрохлорида.
Суспензию 11,0 г (0,038 мол) 1-[бис-(4-фторфенил)метил]пиперазина, 10,5 г (0,076 мол) 2-(2-хлорэтокси)ацетамида и 8,1 г безводного карбоната натрия в 40 мл ксилола нагревают с обратным холодильником при 140oС в течение 4 часов. Образовавшийся осадок отфильтровывают и затем промывают толуолом. Фильтрат и толуол, использованный для промывки, соединяют. Полученную органическую фазу экстрагируют 80 мл 1N водного раствора соляной кислоты и водную фазу дважды промывают толуолом. К полученной водной фазе добавляют толуол, затем добавляют 80 мл 1N водного раствора гидроокиси натрия и экстрагируют один раз толуолом. Органическую фазу промывают один раз водой, сушат над безводным сульфатом натрия и испаряют растворители в ротационном испарителе досуха. На этой стадии остаток после выпаривания представляет собой 2-[4-[бис-(4-фторфенил)метил] -1-пиперазинил] этоксиацетамид, который можно превратить в дигидрохлорид следующим образом: остаток после испарения растворяют в 50 мл изопропанола и отфильтровывают; к изопропанольному раствору добавляют 4,38 N спиртовой раствор соляной кислоты (17,5 мл) и оставляют смесь кристаллизоваться. Осадок отфильтровывают, промывают изопропанолом и диэтиловым эфиром, затем сушат в вакууме. Таким образом получают 15,8 г (90%) дигидрохлорида 2-[4-[бис-(4-фторфенил)метил] -1-пиперазинил]этоксиацетамида.
Точка плавления: 229,51oС
Масс-спектр: 389 (свободное основание М+), 345 и 203.
2. Получение эфлетиризина.
В круглодонной колбе, снабженной механической мешалкой, трубкой для ввода азота и конденсатором, 30 г (0,065 мол) 2-[4-[бис-(4-фторфенил)метил] -1-пиперазинил] этоксиацетамида дихлорида добавляют к смеси 325 мл этанола, 130 мл 1N водного раствора гидроокиси натрия и 62 мл 6,3 N водного раствора гидроокиси натрия. Смесь нагревают с обратным холодильником в атмосфере азота в течение 1,5 часов. Затем реакционную смесь охлаждают до комнатной температуры и рН доводят до 5 при помощи 78 мл 5 N водного раствора соляной кислоты. Добавляют воду и под вакуумом в ротационном испарителе выпаривают этанол. Полученную водную фазу экстрагируют дихлорметаном. Органическую фазу высушивают над безводным сульфатом натрия и выпаривают досуха с получением 25 г сырого эфлетиризина в виде аморфного твердого вещества, 5 г которого перекристаллизовывают из ацетонитрила.
Элементный анализ для C21H24F2N2O3:
Вычислено: С 64,60; Н 6,19; N 7,17; F 9,73.
Найдено: С 64,45; Н 6,27; N 2,24; F 9,44.
3. Получение моногидрата дигидрохлорида эфлетиризина (Форма В).
37%-ный (вес./вес.) водный раствор соляной кислоты (6,38 л) добавляют к суспензии 2,76 кг (7,1 мол) 2-[4-[бис-(4-фторфенил)метил]-1-пиперазинил] этоксиацетамида в 6,4 л воды. Реакционную смесь нагреваю при 65oС в течение часа. Затем ее охлаждают до примерно 0oС и оставляют кристаллизоваться. Образующийся осадок отфильтровывают при 0oС, промывают соляной кислотой 6N (1,5 л) и получают сырой моногидрат дигидрохлорида эфлетиризина.
Сырой продукт затем растворяют при нагревании при 60oС в 13,5 л воды и раствор дважды промывают 1 л толуола. Водную фазу подкисляют 16 л 37% (вес. /вес. ) водного раствора соляной кислоты и охлаждают до 0oС. Образующийся остаток отфильтровывают при 0oС, промывают 6N соляной кислотой (2,4 л) и сушат полученный продукт при 50oС в течение 4 дней. Получают моногидрат дигидрохлорида эфлетиризина в виде твердого вещества белого цвета (выход 3,14 кг; 92%).
На дифференциальной сканирующей термограмме Формы В виден первый эндотермический пик между 155 и 170oС и второй эндотермический пик между 210 и 235oС.
4. Получение безводного дигидрохлорида эфлетиризина (Форма А):
Превращение Формы В в Форму А.
Получают суспензию 3,143 кг (6,53 мол) Формы В, синтезированной в Примере 3, в 35 л метилэтилкетона. Смесь нагревают с обратным холодильником в течение 2 часов. Воду удаляют при нагревании с обратным холодильником в течение 2 часов и 50 минут при добавлении 5 л метилэтилкетона. Полученную смесь охлаждают до 25oС, перемешивают в течение ночи, отфильтровывают и промывают метилэтилкетоном (10 л). Получают безводный дигидрохлорид эфлетиризина (Форма А), который сушат при 50oС в вакууме (выход 98,6%, 2983 г).
Элементный анализ для C21H24F2N2O3 • 2HCl:
Вычислено: С 54,44; Н 5,66; N 6,05; Cl 15,30; F 8,19.
Найдено: С 54,80; Н 5,68; N 5,86; Cl 15,50; F 8,21.
Дифференциальная сканирующая термограмма Формы А показывает эндотермический пик между 220 и 235oС.
5. Превращение Формы А в Форму В.
Готовят суспензию 699 г Формы А, полученной в Примере 4, в 3 л воды. Смесь нагревают при 60oС до полного растворения и сразу же фильтруют. К полученному раствору в течение 30 минут добавляют при 50oС 37% (вес./вес.) водный раствор соляной кислоты (3 л). Кристаллизацию инициируют несколькими кристаллами Формы В. Смесь охлаждают, перемешивают 1 час при комнатной температуре, затем 2 часа при 0oС. Образовавшееся твердое вещество отфильтровывают, промывают 0,6 л 6 N водного раствора соляной кислоты и сушат под вакуумом при 50oС (выход: 676 г, 93%).
Элементный анализ для C21H25F2N2O3 • 2HCl • Н2О:
Вычислено: С 52,41; Н 5,86; N 5,82; Cl 14,73; F 7,89.
Найдено: С 52,08; Н 6,03; N 5,44; Cl 14,55; F 7,83.
Псевдополиморфные Формы А и В дигидрохлорида эфлетиризина характеризуются далее рентгеновскими (Х-лучи) дифракционными спектрами и скоростью растворения.
I. Рентгенограммы порошков.
Рентгеновские исследования проводят на дифрактометре PHILIPS PW 1710 с использованием в качестве источника излучения Сu Кa. Образцы порошка, подвергаемого анализу, помещают в держатель образца без измельчения или перемешивания. Спектры снимали при комнатной температуре при величине угла рассеяния от 2q=4o до 2q=50o со скоростью сканирования 1o/мин.
Для Формы А характерные дифракционные пики наблюдаются при величинах 2q:
13,7o±0,5; 13,9o±0,5; 16,3o±0,5; 18,0o±0,5; 18,6o±0,5; 19,1o±0,5;
23,1o±0,5; 24,1o±0,5; 25,6o±0,5 и 30,2o±0,5.
Для Формы В характерные дифракционные пики наблюдаются при величинах: 7,5o±0,5; 9,7o±0,5; 10,5o±0,5; 10,7o±0,5; 15,7o±0,5; 18,9o±0,5; 19,6o±0,5; 19,9o±05; 20,4o±0,5; 20,9o±0,5; 22,2o±0,5; 22,5o±0,5; 24,6o±0,5; 24,7o±0,5; 25,9o±0,5 и 29,3o±0,5.
II. Скорость растворения.
Исследование биодоступности лекарственных средств показали, что скорость растворения (IDR) менее 0,1 мг/см2/мин часто может свидетельствовать об ограниченной скорости растворения абсорбции лекарства в человеческом организме.
Таким образом IDR является показателем биодоступности. Этот параметр зависит от различных физико-химических свойств, включая химическую форму (соль, сольват), форму кристаллов, растворимость и смачиваемость.
Определение IDR осуществляют следующим образом. Форму, подвергающуюся испытанию, измельчают и смешивают с микрокристаллической целлюлозой AVICEL РН 102 (сухое связующее, улучшающее таблетируемость). Соотношение вещество : эксципиент при смешении составляет 70:30 (вес./вес.). Аликвотные порции (500 мг) прессуют в таблетки при конечной нагрузке 10 т, чтобы получить постоянную известную площадь поверхности с нулевой пористостью.
Определение растворимости проводят при 37oС с использованием 500 мл водной среды при трех различных значениях рН, воспроизводящих ожидаемый интервал значений рН в желудочно-кишечном тракте человека, а именно 1,2, 4,0 и 7,5.
Одинаковые и воспроизводимые гемодинамические условия получали, осуществляя определение в приборе USP XXII 2 (USP-Фармакопея США, 1990), в котором используется лопастная мешалка (50 об/мин), и набор таблеток помещают на дно сосуда (метод статического диска). Для последующего определения IDR выбирают статически доступные (p<0,05) линейные участки всех кривых растворения.
Результаты представлены в таблице 1, иллюстрирующей IDR для Формы А и Формы В, выраженные в мг/см2/мин при трех значениях рН.
Результаты таблицы 1 показывают, что у Формы А и Формы В IDR больше 0,1 мг/см2/мин при трех значениях рН. Это показывает, что растворение, вероятно, не является стадией, лимитирующей скорость в in vivo процессе абсорбции для твердых фармацевтических композиций, содержащих или Форму А, или Форму В. Однако, Форма В характеризуется значительно более низким IDR, чем Форма А. Это означает, что если с терапевтической точки зрения желательна самая высокая скорость растворения твердой дозированной формы, Форма А является предпочтительной кристаллической формой для применения в твердой фармацевтической композиции.
Настоящее изобретение также касается фармацевтических композиций, содержащих Форму А или Форму В в сочетании с подходящими фармацевтическими эксципиентами. Неожиданно оказалось, что в случае твердых фармацевтических композиций лучше использовать Форму А, чем Форму В. Мы установили, что твердые фармацевтические композиции, содержащие Форму А в сочетании с обычными носителями и разбавителями, например, сорбитом, обладает лучшей стабильностью при хранении, чем композиции, содержащие Форму В. Это показывают результаты следующего исследования, направленного на определение стабильности двух псевдополиморфных форм в присутствии D-сорбита при хранении в экстремальных температурных условиях, то есть в герметичных флаконах при 40oС или 60oС.
В этом опыте проводят ЖХВР нескольких образцов: Формы А, Формы В, D-сорбита и 1:1 (вес./вес.) смеси Формы А или Формы В с D-сорбитом.
Каждый образец измельчают и перемешивают. Аликвотные порции (500 мг) прессуют с получением таблеток диаметром 13 мм при нагрузке 1 т, которая является обычной силой сжатия в случае фармацевтических таблеток. Каждая таблетка немедленно измельчается в тонкий порошок и аликвот хранят в тщательно герметизируемом стеклянном флаконе при 40oС или 60oС. Образцы анализируют методом ЖХВР через 0, 4, 16 и 24 недели.
В первой серии определений снимают ЖХВР/УФ спектры с использованием системы Kontron HPLC type 300, снабженной УФ-детектором. Используют колонку Supercosil RP ABZ 250X 4,6 мм вн. диам., частицы размером 5 мкм, используемая мобильная фаза состоит из смеси ацетонитрил/вода 25:75 (об/об), воды, содержащей 770 мг/л ацетата аммония.
Образцы растворяют в смеси ацетонитрил/вода 25:75 (об/об) до концентрации 2 мг/л и 10 мкл этих растворов подвергают ЖХВР.
При сравнении данных ЖХВР для индивидуальных компонентов и для двойных смесей видно, что имеются новые пики, указывающие на взаимодействие с D-сорбитом. Имеющиеся новые пики оценивают количественно и идентифицируют методом ЖХВР/МС, с использованием спектрометра VG Quattro, соединенного с системой Kontron HPLC.
При ЖХВР/УФ в случае чистой Формы А и Формы В после хранения при 40oС или 60oС в течение 4, 16 или 24 недель на спектрограммах нет изменений. Чистый D-сорбит не обнаруживается ЖХВР/УФ. В случае двойных смесей по сравнению со спектрами чистой Формы А или Формы В на спектрограммах появляются новые пики. Эти новые пики особенно значительны в случае Формы В, которая хранилась при 60oС.
При ЖХВР/МС новые пики, наблюдаемые для двойных смесей, идентифицируются для моноэфира сорбита и эфлетиризина и дигидратированных форм моноэфира сорбита и эфлетиризина. Последние появляются после более длительного хранения, чем первый.
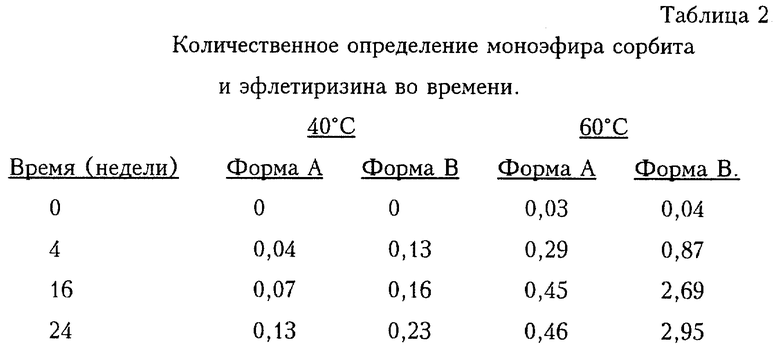
В таблице 2 приведены результаты количественного определения моноэфира сорбита и эфлетиризина, образующегося в бинарных смесях при хранении при 40oС или 60oС. Определение относительных количеств моноэфира проводят методом ЖХВР-УФ.
Таблица 2 показывает, что когда Форму А или Форму В прессуют с получением таблетки вместе с D-сорбитом и хранят при 40oС или 60oС, моноэфир сорбита и эфлетиризина образуется в количестве, увеличивающемся со временем. Далее, она показывает, что образование моноэфира совсем не велико для Формы А при хранении при 40oС или 60oС и для Формы В при хранении при 40oС. Однако, моноэфир образуется в значительном количестве для Формы В при хранении при 60oС. Эти результаты показывают, что дигидрохлорид эфлетиризина в Форме А реагирует с гидроксилированными эксципиентами, носителями или разбавителями, обычно используемыми в твердых фармацевтических композициях, в меньшей степени и что поэтому Форма А более пригодна, чем Форма В для приготовления таких композиций.
Данное изобретение также относится к фармацевтической композиции, содержащей Форму А или Форму В, или смесь Формы А с Формой В в сочетании с подходящими эксципиентами, разбавителями или носителями. Фармацевтические композиции по изобретению могут быть в различных формах. Препаративные формы с непрерывным выделением представляют особенный интерес и даже предпочтительные композиции содержат эксципиенты медленного высвобождения в сочетании с циклодекстрином.
Пример такой композиции: 30 мг безводного 2-[2-[4-[бис(4-фторфенил)метил] -1-пиперазинил] -этокси] уксусной кислоты дигидрохлорида; 14,7 мг Encompress®; 82,3 мг циклодекстрина; 70 мг Methocel® K15MCR; 1 мг Aerosil® 200; 2 мг стеарата магния.

Изобретение относится к новым псевдополиморфным формам дигидрохлорида 2-[2-[4-[бис(4-фторфенил)метил] -1-пиперазинил] этокси] уксусной кислоты, а именно безводному дигидрохлориду 2-[2-[4-[бис-(4-фторфенил)метил]-1-пиперазинил] этокси] уксусной кислоты и моногидрату дигидрохлорида 2-[2-[4-[бис(4-фторфенил)метил] -1-пиперазинил] этокси] уксусной кислоты. Оно также относится к способам получения этих псевдополиморфных форм и к фармкомпозициям, их содержащим. Соединения обладают превосходными антигистаминными свойствами и пригодны в качестве антиаллергенного и антиспазматического агента. 8 с. и 4 з.п. ф-лы, 2 табл.
| 2- @ 4-(Дифенилметил)-1-пиперазинил @ -уксусные кислоты или их амиды,или их нетоксичные фармацевтически приемлемые соли,проявляющие спазмолитическую и антигистаминную активность | 1982 |
|
SU1310397A1 |
| WO 9737982, 16.10.1997 | |||
| Нитевидный носитель магнитнойзАпиСи | 1979 |
|
SU801064A1 |

