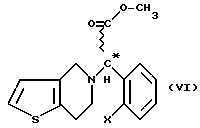
Изобретение относится к новому способу получения соединений общей формулы (VI) (см. в конце описания), где X означает атом галогена.
Известно, что метил(2-галогенофенил)-(6,7-дигидро-4Н-тиено [3,2-c]пиридин-5-ил) ацетаты и их соли могут успешно применяться при лечении, прежде всего, благодаря тому, что они ингибируют агрегацию тромбоцитов и оказывают противотромботическое действие.
Особенно полезным представителем этих соединений, подпадающим под общую формулу VI (где X - атом хлора), является правовращающий метил (+)-[(S)-(2-хлорфенил)-(6,7-дигидро-4Н-тиено [3, 2-c] пиридин-5-ил)ацетат-гидросульфат], имеющий международное незапатентованное название (INN) "clopidogrel" (заявка на Европейский патент, публикация N 099802).
Широкомасштабное получение соединений общей формулы (VI) (где X - атом галогена) ранее осуществлялось только в результате применения производных α- галогенфенилуксусной кислоты, являющихся сильными раздражителями слизистой оболочки и слезоточивыми средствами, применение которых требует особых условий и является небезопасным с точки зрения здоровья и окружающей среды (заявки на Европейский патент, публикации N N 099802, 0420706, 0466569). Более того, выход в результате применения известных способов является довольно низким.
Нашей целью является избежать применения вышеуказанных небезопасных промежуточных соединений (таких как, например, α-бром-(2-хлорфенил)уксусная кислота и ее метиловый эфир) и существенно увеличить выход соединений общей формулы (VI) в результате синтеза.
Поскольку в синтезе в соответствии с данным изобретением каждое промежуточное соединение является хиральным, при получении оптически активного конечного продукта, такого как, например, клопидогрель, имеется возможность применения, начиная с первой стадии, оптически активных соединений в качестве промежуточных соединений. Экономическим преимуществом данного способа является, среди прочих, избежание получения нежелательного изомера.
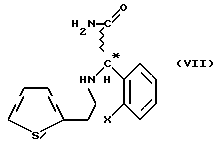
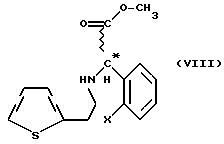
Мы обнаружили, что получение соединений общей формулы (VI) способом, показанным на схеме 1 (см. в конце описания), позволяет избежать применения нежелательных промежуточных соединений и, кроме того, выход при синтезе намного выше. Предметом данного изобретения является третий этап реакционной схемы 1. Оптически активные соединения общей формулы (VI) получают либо из оптически активных соединений общей формулы (VII) (см. в конце описания), либо из оптически активных промежуточных соединений, полученных в результате расщепления промежуточных соединений общей формулы (VIII) (см. в конце описания), или в результате расщепления рацемических соединений общей формулы (VI).
В соответствии с нашим изобретением рацемическое или оптически активное соединение общей формулы (VII) (где X - атом галогена) превращают в рацемическое или оптически активное соединение общей формулы (VIII) (где X - атом галогена) и, при желании, полученное рацемическое соединение общей формулы (VIII) расщепляют на его оптически активные изомеры, а затем в результате замыкания кольца известным способом соединения общей формулы (VIII) превращают в рацемическое или оптически активное соединение общей формулы (VI) и, при желании, рацемические соединения общей формулы (VI) расщепляют на их оптические изомеры и/или их превращают в соли, и/или рацемические или оптически активные соединения выделяют из их солей.
Предпочтительно, соединения общей формулы (VII) подвергают реакции с метанолом в присутствии метилсерной кислоты. Реакцию также проводят под давлением, предпочтительно, составляющем 5-20 бар (500-2000 кПа). Наиболее предпочтительный температурный интервал составляет 50- 150oC. Метилсерную кислоту получают в реакторе посредством кипячения метанола и серной кислоты с обратным холодильником.
Замыкание кольца получаемых соединений общей формулы (VIII) осуществляют известным способом. Расщепление известных рацемических соединений общей формулы (VIII) или рацемических соединений общей формулы (VI) осуществляют при помощи известного способа расщепления, приводящего к получению оптически активных соединений общей формулы (VI).
Получение исходных соединений, применяемых в нашем изобретении, иллюстрируется примерами. Исходные материалы, показанные на схеме 1, могут быть приобретены; синтез соединения формулы (II) описан, например, в заявке на французский патент, публикация N 2608607.
Дальнейшие подробности данного изобретения приведены в нижеследующих примерах, не ограничивающих его объем.
Пример 1
[2-(2-Тиенил)этиламино] (2-хлорфенил)ацетонитрил
104 г (1 моль) бисульфита натрия растворяют в смеси 900 мл воды и 250 мл этанола и к раствору добавляют 140,6 г (1 моль) о-хлорбензальдегида. Через несколько минут осаждается аддукт бисульфита и альдегида в виде белых кристаллов, при этом температура поднимается до 40oC. После 1 часа перемешивания к реакционной смеси добавляют 127,2 г (1 моль) 2-(2- тиенил)этиламина, затем ее перемешивают при 50oC в течение 2 часов. На протяжении этого времени кристаллический бисульфит альдегида превращается в маслянистое вещество. Смесь охлаждают до комнатной температуры и к ней добавляют раствор 49 г (1 моль) цианида натрия в 100 мл воды. При добавлении температура реакционной смеси поднимается до 40oC. Затем смесь перемешивают при 60oC до завершения реакции (1 час). После этого маслянистую органическую фазу экстрагируют 400 мл 1,2-дихлорэтана, промывают 2х200 мл воды до устранения цианида, следы 2-(2-тиенил)этиламина удаляют обработкой 100 мл 3% раствора соляной кислоты. Дихлорэтановую фазу сушат над безводным сульфатом натрия и выпаривают в вакууме. Продуктом является остаточное, быстро кристаллизующееся масло. Вес: 260 г (94%), температура плавления: 40-41oC.
Продукт был идентифицирован с применением элементарного анализа, ИК-спектра и 1H-ЯМР-спектроскопии.
Пример 2
[2-(2-Тиенил)этиламино](2-хлорфенил)ацетонитрил
9,8 г (0,2 моль) цианида натрия растворяют в 70 мл воды и к раствору сначала добавляют 32,8 г (0,2 моль) 2-(2-тиенил)этиламин-гидрохлорида, а затем в течение нескольких минут раствор 28,2 г (0,2 моль) о-хлорбензальдегида в 30 мл этанола. При добавлении температура смеси поднимается до 45oC. Затем реакционную смесь перемешивают при 60oC в течение 2 часов, после этого охлаждают до комнатной температуры и разбавляют 50 мл воды. Полученный маслянистый продукт экстрагируют 100 мл 1,2-дихлорэтана, органическую фазу промывают 2х50 мл воды до устранения цианида, а следы 2-(2-тиенил)этиламина удаляют обработкой 20 мл 3% раствора соляной кислоты, получая в качестве продукта остаточное, быстро кристаллизующееся масло. Вес: 52 г (94%), температура плавления: 40-41oC.
Продукт идентифицировали, как указано в Примере 1. Качество продукта идентично качеству продукта, приготовленного в соответствии с Примером 1.
Пример 3
[2-(2-Тиенил)этиламино] (2-хлорфенил) ацетонитрил-гидрохлорид
276,7 г (1 моль) [2-(2-тиенил) этиламино] (2-хлорфенил) ацетонитрила, полученного в соответствии с Примером 1 или 2, растворяют в 600 мл этанола и к этому раствору добавляют 600 мл 10% водного раствора соляной кислоты. В течение нескольких минут осаждаются белые кристаллы, их собирают, промывают 60 мл 1: 1 смеси 10% соляной кислоты и этанола, а затем ацетоном и сушат. Вес: 305 г (97,4%), температура плавления: 153-154oC.
Продукт был идентифицирован с применением элементного анализа, ИК-спектра и 1H-ЯМР-спектроскопии.
Пример 4
[2-(2-Тиенил)этиламино](2-хлорфенил) ацетонитрил-гидробромид
13,8 г (0,05 моль) [2-(2-тиенил) этиламино] (2-хлорфенил) ацетонитрила, полученного в соответствии с Примером 1 или 2, растворяют в 30 мл этанола и к этому раствору добавляют 40 мл 20% водного раствора бромистого водорода. Продукт, осаждающийся в течение нескольких минут, собирают, промывают этилацетатом, а затем сушат. Вес: 14 г (78,2%), температура плавления: 144-145oC.
Продукт был идентифицирован с применением элементного анализа, ИК-спектра и 1H-ЯМР-спектроскопии.
Пример 5
[2-(2-Тиенил)этиламино] (2-хлорфенил)ацетамид-гидрохлорид
В 1200 мл метилацетата вводят 204 г (5,6 моль) газообразного хлористого водорода при температуре 15-25oC и к раствору добавляют 221,4 г (0,8 моль) [2-(2-тиенил)этиламино] (2-хлорфенил) ацетонитрила формулы (I), полученного, как описано в Примере 1, и 48 мл (1,2 моль) метанола. Смесь перемешивают при 20-25oC в течение 6 часов. В ходе реакции вначале осаждается гидрохлорид исходного "нитрила", а затем постепенно гидрохлорид образующегося "амида кислоты" в виде белых кристаллов. Кристаллы собирают фильтрацией, промывают метилацетатом и сушат. Вес: 249 г (94%), температура плавления: 231-232oC.
Продукт был идентифицирован с применением элементного анализа, ИК-спектра и 1H-ЯМР-спектроскопии.
Пример 6
[2-(2-Тиенил)этиламино] (2-хлорфенил)ацетамид гидрохлорид
В 700 мл метилацетата при 0-10oC вводят 109,8 г (3 моль) газообразного хлористого водорода, к раствору добавляют 83 г (0,3 моль) [2-(2-тиенил)этиламино] (2-хлорфенил) ацетонитрила формулы (I), полученного в соответствии с Примером 1 или 2, и 15 мл (0,37 моль) метанола, и смесь медленно, в течение 20 минут, нагревают до 45-50oC. Затем реакционную смесь перемешивают при 45-50oC в течение 4 часов, кристаллический продукт отфильтровывают при комнатной температуре, промывают этилацетатом и сушат. Вес: 90,4 г (91%), температура плавления: 231-232oC. Качество продукта идентично качеству продукта из Примера 5.
Пример 7
[2-(2-Тиенил)этиламино] (2-хлорфенил)ацетамид
24,8 г (0,075 моль) [2-(2-тиенил)этиламино](2-хлорфенил) ацетамид-гидрохлорида, полученного в соответствии с Примером 5 или 6, смешивают со 170 мл воды, а затем при небольшом охлаждении добавляют 30 мл 10% раствора гидроокиси натрия и 170 мл 1,2-дихлорэтана. Фазы разделяют, водную фазу экстрагируют 2х20 мл 1,2-дихлорэтана, а соединенный органический слой выпаривают в вакууме. Остаток: 22 г, быстро кристаллизующееся масло. Сырой продукт перекристаллизуют из 80 мл изопропилацетата, получая 19,5 г кристаллического основания формулы (VII). Выход: 88,2%, температура плавления: 90-92oC.
Продукт был идентифицирован с применением элементного анализа, ИК-спектра и 1H-ЯМР-спектроскопии.
Пример 8
[2-(2-Тиенил)этиламино] (2-хлорфенил)ацетамид-гидробромид
14,7 г (0,05 моль) [2-(2-тиенил) этиламино] (2-хлорфенил)ацетамида, полученного, как описано в Примере 7, растворяют в 150 мл ацетона. К раствору добавляют 4 мл 60% водного раствора бромистого водорода, и выпавшие в осадок белые кристаллы отфильтровывают, промывают ацетоном и сушат.
Продукт был идентифицирован с применением элементного анализа, ИК-спектра и 1H-ЯМР-спектроскопии.
Пример 9
Метил [2-(2-тиенил)этиламино] (2-хлорфенил)ацетат-гидрохлорид
21,5 мл (0,4 моль) 100% серной кислоты растворяют при охлаждении в 100 мл метанола, раствор нагревают с обратным холодильником в течение 0,5 часа, а затем охлаждают до комнатной температуры; к нему добавляют 33,1 г (0,1 моль) [2-(2-тиенил) этиламино] (2-хлорфенил) ацетамид-гидрохлорида, полученного, как описано в Примере 5, смесь нагревают в условиях дефлегмации с обратным холодильником в течение 10 часов. Затем метанол отгоняют в вакууме, к остатку добавляют 150 мл 1,2-дихлорэтана и 150 мл воды, хорошо встряхивают и разделяют две фазы. Водный слой экстрагируют 2х30 мл 1,2-дихлорэтана, соединенные органические слои промывают 80 мл 5% раствора гидроокиси натрия, затем 100 мл воды, сушат над безводным сульфатом натрия и выпаривают в вакууме. Вес остатка: 28,5 г. Маслянистый продукт, представляющий собой основание формулы (VIII), растворяют в 50 мл изопропилацетата, к нему добавляют 7,3 мл (0,087 моль) концентрированного раствора соляной кислоты и смесь перемешивают при комнатной температуре в течение 1 часа. Осажденный продукт отфильтровывают, промывают 2х10 мл изопропилацетата и сушат. Вес: 28,4 г (82%), температура плавления: 177-178oC (литер. 175oC).
Продукт был идентифицирован с применением элементного анализа, ИК-спектра и 1H-ЯМР-спектроскопии, а также определением температуры плавления.
Пример 10
Метил [2-(2-тиенил)этиламино] (2-хлорфенил)ацетат-гидрохлорид
В 150 мл метанола при охлаждении растворяют 8,5 мл (0,15 моль) 96% серной кислоты, а затем раствор нагревают в условиях дефлегмации с обратным холодильником в течение 0,5 часа. После охлаждения до комнатной температуры к раствору добавляют 20 г (0,0678 моль) [2-(2-тиенил)этиламино] (2-хлорфенил)ацетамида, подпадающего под общую формулу (VII) и полученного, как описано в Примере 7, смесь помещают в закрытый прибор (автоклав) и перемешивают в нем при 130oC и выше в течение 5 часов, в то время как внутреннее давление поднимается до 13 бар (1300 кПа). Затем реакционную смесь охлаждают до комнатной температуры (остаточное давление составляет 1-2 бар (100-200 кПа), метанол отгоняют в вакууме, к остатку добавляют 100 мл изопропилацетата и 100 мл воды, pH смеси доводят до 7,5, добавляя по каплям 60 мл 10% раствора гидроокиси натрия при охлаждении и перемешивании, при этом поддерживая температуру смеси на уровне комнатной. Фазы разделяют, органическую фазу перемешивают с 60 мл 3% водного раствора малеиновой кислоты при 40-50oC в течение 10 минут, а затем две фазы разделяют. После повторного экстрагирования водного раствора малеиновой кислоты 30 мл изопропилацетата органические слои соединяют, сушат над безводным сульфатом натрия и упаривают наполовину по объему. После добавления 5 мл концентрированного раствора соляной кислоты продукт осаждается в виде масла, кристаллизующегося в течение нескольких минут. Его охлаждают до 0-(+5)oC, через 2 часа кристаллы собирают фильтрацией, промывают небольшим количеством изопропилацетата и сушат. Вес: 19,4 г (82,5%), температура плавления: 177-178oC. Качество продукта идентично качеству материала, полученного в Примере 9.
Пример 11
Метил [2-(2-тиенил)этиламино] (2-хлорфенил) ацетат-гидробромид
Проводят такую же процедуру, как и Примере 9, полученный метил [2-(2-тиенил)этиламино] (2-хлорфенил) ацетат растворяют в 50 мл изопропилацетата, к раствору добавляют 8 мл 62% водного раствора бромистого водорода и смесь перемешивают при комнатной температуре в течение 1 часа. В течение этого времени кристаллизуется продукт. Кристаллы собирают, промывают 2х10 мл изопропилацетата и сушат. Вес: 32,5 г (83%), температура плавления: 164-165oC.
Продукт был идентифицирован с применением элементного анализа, ИК-спектра и 1H-ЯМР-спектроскопии.
Пример 12
Гидрат метил (2-хлорфенил) (6,7-дигидро-4Н-тиено [3, 2-е] пиридин-5-ил) ацетат-гидрохлорида
К 28,4 г (0,082 моль) метил [2-(2- тиенил)этиламино] (2-хлорфенил) ацетат-гидрохлорида, полученного в соответствии с Примером 9 или 10, добавляют 50 мл 1,2-дихлорэтана и раствор 7,5 (0,09 моль) гидрокарбоната натрия в 100 мл воды. Смесь хорошо перемешивают, фазы разделяют, водную фазу промывают 2х30 мл 1,2-дихлорэтана, соединенный органический слой сушат над безводным сульфатом натрия и растворитель удаляют в вакууме. Остаток (25 г основания ацетата) растворяют в 90 мл муравьиной кислоты, к раствору добавляют 4 г (0,13 моль) параформальдегида и смесь перемешивают при 50oC в течение 20 минут. Затем большую часть муравьиной кислоты отгоняют в вакууме, остаток растворяют в смеси 100 мл воды и 100 мл 1,2-дихлорэтана, фазы разделяют, водную фазу вновь экстрагируют 30 мл 1,2- дихлорэтана, соединенную органическую фазу хорошо встряхивают со 100 мл 5% раствора гидрокарбоната натрия, фазы разделяют, органическую фазу сушат над безводным сульфатом натрия и выпаривают в вакууме. Остаток растворяют в 45 мл ацетона и к раствору добавляют 6,5 мл (0,077 моль) концентрированной соляной кислоты при 5-10oC при охлаждении. Продукт медленно кристаллизуется. Смесь перемешивают в течение 1 часа при 0-10oC, затем кристаллы отфильтровывают, промывают 2х10 мл ацетона и сушат. Вес: 26,7 г (теоретический: 30,8 г). Выход: 86,6%, температура плавления: 138-140oC (температура плавления, указанная в литературе: 130-140oC).
Продукт был идентифицирован с применением элементного анализа, ИК-спектра и 1Н-ЯМР-спектроскопии, а также определения температуры плавления.
Пример 13
Левовращающий [2-(2-тиенил)этиламино] (2-хлорфенил)- ацетонитрил-гидрохлорид
10 г (0,036 моль) рацемического [2-(2-тиенил) этиламино]-(2-хлорфенил)ацетонитрила (I) растворяют в 15 мл ацетона, к раствору добавляют 10 г (0,043 моль) (1R)-(-)-камфор-10-сульфоновой кислоты и 0,5 мл (0,013 моль) муравьиной кислоты. Смесь нагревают до 50-55oC, затем через 1-2 минуты ее охлаждают до комнатной температуры. Таким образом постепенно осаждается соль, образуемая правовращающим энантиомером исходного материала и (1R)-(-)-камфор-10-сульфоновой кислотой в слегка оптически загрязненной форме. Кристаллы разделяют фильтрацией. К маточному раствору добавляют 7 мл метилацетата, содержащего 10% хлористого водорода, или вводят рассчитанное количество сухого газообразного хлористого водорода, кристаллический осадок отфильтровывают, промывают ацетоном и сушат. Вес: 2,5 г, [α]
После рекристаллизации из этанола: [α]
Продукт был идентифицирован с применением элементного анализа, ИК-спектра и 1Н-ЯМР-спектроскопии.
Пример 14
Правовращающий [2-(2- тиенил)этиламино] (2-хлорфенил) ацетонитрил-гидрохлорид
Проводят такую же процедуру, как и в предыдущем Примере, однако в качестве разрешающей кислоты применяют (1S)-(+)-камфор-10-сульфоновую кислоту. Продукт: вес: 2,5 г, [α]
Продукт был идентифицирован с применением элементного анализа, ИК-спектра и 1Н-ЯМР-спектроскопии.
Пример 15
Правовращающий [2-(2-тиенил)этиламино] (2-хлорфенил)-ацетамид
11,8 г (0,037 моль) левовращающего [2-(2-тиенил)этиламино]- (2-хлорфенил) ацетонитрил-гидрохлорида суспендируют в 100 мл метилацетата и в суспензию вводят 9,6 г сухого газообразного хлористого водорода при комнатной температуре. После этого добавляют 3,6 г (0,113 моль) метанола и смесь перемешивают при комнатной температуре пока через 6 часов реакция не будет завершена. Затем осажденный кристаллический материал - гидрохлоридную соль продукта - отфильтровывают, суспендируют в воде и нейтрализуют двууглекислым натрием при перемешивании.
Осажденный белый кристаллический сырой продукт отфильтровывают, сушат и подвергают перекристаллизации из этанола.
Вес: 5 г, [α]
Продукт был идентифицирован с применением элементного анализа, ИК-спектра и 1H-ЯМР-спектроскопии.
Пример 16
Правовращающий [2-(2-тиенил)этиламино](2-хлорфенил)-ацетамид
38 г (0,129 моль) рацемического [2-(2-тиенил) этиламино] -(2-хлорфенил) ацетамида растворяют при 50oC в 380 мл изопропанола, содержащего 0-0,4%, предпочтительно, 0,2% воды, и к этому раствору добавляют раствор 10,6 г (0,071 моль) L(+)-винной кислоты, имеющий температуру 50oC, в 230 мл изопропанола, содержащего 0-0,4%, предпочтительно, 0,2% воды. Смесь перемешивают при 50oC в течение 30 минут. Образуется густой белый осадок. К смеси добавляют 3,4 мл (0,09 моль) муравьиной кислоты и перемешивание продолжают при 50oC в течение 1 часа. Затем реакционную смесь охлаждают до комнатной температуры, перемешивают в течение еще одного часа и твердую фазу отфильтровывают. Осажденный материал представляет собой соль, образуемую левовращающим энантиомером исходного материала и L(+)-винной кислотой, в слегка оптически загрязненной форме. Вес: 30 г. Температура плавления: 167-169oC после кристаллизации из этанола. Маточный раствор выпаривают в вакууме. Остаток (29 г) поглощают 200 мл воды и 200 мл 1,2-дихлорэтана и нейтрализуют при перемешивании 16 г (0,19 моль) гидрокарбоната натрия. Фазы разделяют, водный слой промывают 2х30 мл 1,2- дихлорэтана, соединенный органический слой экстрагируют 50 мл воды, сушат над безводным сульфатом натрия и выпаривают в вакууме. Вес: 18 г. Сырой продукт подвергают перекристаллизации из 70 мл этанола, промывают небольшим количеством этанола и сушат. Вес: 12,6 г. Температура плавления: 122-124oC, [α]
Продукт был идентифицирован с применением элементного анализа, ИК-спектра и 1H-ЯМР-спектроскопии.
В результате упаривания фильтрата может быть выделено 4 г рацемического исходного материала.
Пример 17
Правовращающий [2-(2-тиенил)этиламино] (2-хлорфенил) ацетамид
76 г (0,257 моль) рацемического [2-(2-тиенил)этиламино] -(2-хлорфенил) ацетамида растворяют при 50oC в 1200 мл изопропанола, содержащего 0,2% воды, к этому раствору добавляют 21,2 г (0,141 моль) L(+)-винной кислоты и 8,3 г (0,18 моль) муравьиной кислоты. Смесь перемешивают при 50oC в течение 1 часа, при этом образуется густой белый осадок. Затем реакционную смесь охлаждают до комнатной температуры в течение 1 часа, перемешивают в течение еще двух часов и твердую фазу отфильтровывают.
Осажденный материал представляет собой соль, образуемую левовращающим энантиомером исходного материала и L(+)-винной кислотой, в слегка оптически загрязненной форме. Вес: 57 г. Температура плавления: 167-169oC после кристаллизации их этанола.
После фильтрации первого твердого материала 5,2 г (0,141 моль) газообразной соляной кислоты вводят в фильтрат для осаждения гидрохлорида продукта. Образующийся белый выкристаллизовавшийся материал фильтруют и сушат. Вес: 41,7 г.
Полученную слегка оптически загрязненную соль поглощают 100 мл этанола и к этому раствору постепенно добавляют 5,3 г (0,13 моль) гидроокиси натрия, растворенной в 70 мл этанола, для получения свободного основания. Образующийся продукт, содержащий некоторое количество хлорида натрия, отфильтровывают и промывают дистиллированной водой. После сушки его количество составляет 27,7 г, 73% от содержания правовращающего энантиомера в исходном материале. Температура плавления: 122-124oC, [α]
После выпаривания этанольного фильтрата в вакууме и поглощения остатков водой выделяют 9 г рацемического исходного материала.
Пример 18
Правовращающий метил[2-(2-тиенил)этиламино] (2-хлорфенил)- ацетат-гидрохлорид
В 40 мл метанола при охлаждении растворяют 11,5 мл (0,215 моль) 100% серной кислоты, раствор нагревают в условиях дефлегмации в течение 30 минут, затем после охлаждения до комнатной температуры добавляют 12,4 г (0,042 моль) правовращающего [2-(2-тиенил)этиламино] (2-хлорфенил)ацетамида и смесь нагревают при дефлегмации в течение 6-7 часов до окончания реакции. Метанол отгоняют в вакууме, к остатку добавляют 75 мл 1,2-дихлорэтана и 75 мл воды, смесь хорошо встряхивают и фазы разделяют. Водную фазу экстрагируют 2х20 мл 1,2-дихлорэтана, соединенную органическую фазу экстрагируют 50 мл 5% раствора гидроокиси натрия, затем 50 мл воды и сушат над безводным сульфатом натрия. Сушильный материал отфильтровывают и в раствор при охлаждении вводят 1,5 г (0,041 моль) сухого газообразного хлористого водорода. Осажденный кристаллический продукт отфильтровывают, промывают 1,2-дихлорэтаном и сушат. Вес: 12,1 г, температура плавления: 185-186oC (разложение), [α]
Продукт был идентифицирован с применением элементного анализа, ИК-спектра и 1H-ЯМР-спектроскопии.
Пример 19
Правовращающий метил α-(2-тиенил)этиламино) (2-хлорфенил) ацетат через расщепление рацемата
а) 175 г гидрохлоридной соли соединения общей формулы (VIII) (где X - атом хлора) растворяют в смеси 0,75 л дихлорметана и 0,25 л воды и к раствору медленно добавляют 45 г гидрокарбоната натрия. После смешивания органическую фазу отделяют декантацией. После обычной процедуры обработки получают аминоэфир, который затем растворяют в 850 мл ацетона, и к раствору добавляют 87 г (+)-камфор-10-сульфоновой кислоты. Смесь выдерживают при комнатной температуре в течение 12 часов и полученный осадок отделяют. Таким образом получают 146,5 г камфорсульфоната, [α]
b) 33,5 г гидрохлоридной соли соединения общей формулы (VIII) (где X - атом хлора) и 14,6 г (+)-винной кислоты смешивают в 500 мл изопропанола, нагревают до 50oC, а затем оставляют при комнатной температуре. Полученный осадок отделяют и 4 раза кристаллизуют из изопропанола. Таким образом получают (+)-тартрат желаемого правовращающего продукта. Температура плавления: 105oC. Удельное вращение амина [α]
Пример 20
Левовращающий метиловый эфир α -(2-тиенилэтиламино) (2- хлорфенил)уксусной кислоты через расщепление рацемата
100 г гидрохлорида рацемата соединения общей формулы (VIII) (где X - атом хлора) и 30 г гидрокарбоната натрия смешивают в 500 мл дихлорметана и 200 мл воды. После перемешивания органическую фазу отделяют декантацией, а растворитель отгоняют в вакууме. Остаток растворяют в 800 мл ацетона и к этому раствору добавляют 53,3 г (-)-камфор-10-сульфоновой кислоты. Смесь оставляют при комнатной температуре в течение 12 часов. Полученный осадок отделяют и суспендируют в 300 мл ацетона. Нерастворимый твердый остаток кристаллизуют из смеси 600 мл ацетона и 160 мл метилэтилкетона, получая 52,5 г (-)-камфорсульфоната желаемого продукта, температура плавления: 95oC, [α]
Пример 21
Гидрохлоридная соль метилового эфира (+)-(S)-(2- хлорфенил) (6,7-дигидро-4H-тиено [3, 2-c] пиридин-5-ил) уксусной кислоты
6 г (0,017 моль) правовращающего метил [2-(2-тиенил) этиламино] (2-хлорфенил) ацетат-гидрохлорида суспендируют в 6,7 мл 38% водного раствора формалина и нагревают до 60oC при перемешивании. Исходный материал растворяется при 60oC, полученный раствор перемешивают при этой температуре в течение 30 минут до завершения реакции. Затем реакционную смесь разбавляют 100 мл 1,2-дихлорэтана и 150 мл воды, после энергичного встряхивания фазы разделяют. Водную фазу экстрагируют 2х30 мл 1,2-дихлорэтана, соединенную органическую фазу экстрагируют 100 мл воды, сушат над безводным сульфатом натрия, фильтруют и выпаривают в вакууме. Оставшиеся 6 г материала растворяют в 30 мл диэтилового эфира и, при охлаждении реакционной смеси, в раствор при комнатной температуре добавляют 0,6 г сухого газообразного хлористого водорода. Осажденный кристаллический материал отфильтровывают, промывают эфиром и сушат. Вес: 5,5 г. Температура плавления: 130-132oC, [α]
Пример 22
a) Соль метилового эфира (+)-(2-хлорфенил)-(6,7-дигидро-4H-тиено [3, 2-c] пиридин-5-ил) уксусной кислоты с (-)-камфорсульфоновой кислотой
32 г (0,0994 моль) метилового эфира (2-хлорфенил) (6,7-дигидро-4H-тиено [3, 2-c] пиридин-5-ил) уксусной кислоты растворяют в 150 мл ацетона и к раствору добавляют 9,95 г (0,0397 моль) левовращающего моногидрата 10-камфорсульфоновой кислоты. Гомогенную реакционную смесь оставляют при комнатной температуре. Через 48 часов появляется небольшое количество кристаллов. Смесь концентрируют выпариванием до 50 мл и оставляют при комнатной температуре в течение 24 часов. Полученные кристаллы отфильтровывают, промывают ацетоном и сушат. Полученные таким образом кристаллы вновь растворяют в очень небольшом количестве (50 мл) горячего ацетона и после охлаждения кристаллы отфильтровывают, промывают ацетоном и сушат. Таким образом получают целевое соединение. Выход: 88%. Температура плавления: 165oC, [α]
b) Метиловый эфир (+)-(2-хлорфенил)-(6,7- дигидро-4H- тиено [3, 2-c] пиридин-5-ил) уксусной кислоты
К суспензии, полученной из 200 г соли, образованной (-)-камфорсульфоновой кислотой с метиловым эфиром (+)-(2-хлорфенил) (6, 7-дигидро-4H-тиено [3, 2-с] пиридин-5-ил) уксусной кислоты и 800 мл дихлорметана добавляют 800 мл раствора гидрокарбоната натрия. После перемешивания органическую фазу отделяют декантацией, сушат над сульфатом натрия и растворитель удаляют в вакууме. Получают метиловый эфир (+)-(2-хлорфенил) (6, 7-дигидро-4H-тиено [3, 2-c]-пиридин-5-ил) уксусной кислоты в виде раствора в 800 г дихлорметана. После перемешивания органическую фазу отделяют декантацией, сушат над сульфатом натрия и растворитель удаляют в вакууме.
Метиловый эфир (+)-(2-хлорфенил) (6,7-дигидро-4H-тиено [3,2-c]пиридин-5-ил) уксусной кислоты получают в виде бесцветного масла.
с) Кислая сернокислая соль метилового эфира (+)-(2-хлорфенил) (6, 7-дигидро-4H-тиено [3, 2-c]пиридин-5-ил) уксусной кислоты
Остаток, полученный в предыдущем примере, растворяют в 500 мл ледяного ацетона, к этому раствору по каплям добавляют 20,7 мл концентрированной серной кислоты (93,64%; плотность 1,83). Полученный осадок отделяют фильтрацией, промывают 1000 мл ацетона и сушат в вакуумной печи при 50oC. Таким образом получают 139 г целевой соли в виде белых кристаллов. Температура плавления: 184oC, [α]
 н
н
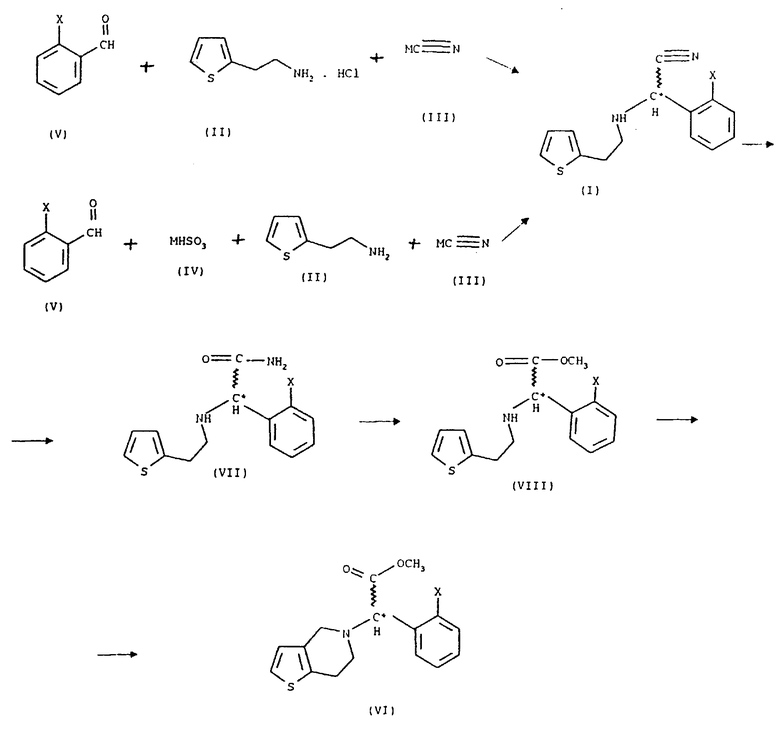
Изобретение относится к способу получения рацемических или оптически активных метил(2-галогенофенил)(6,7-дигидро-4Н-тиено[3,2-с] пиридин-5-ил)ацетатов формулы VI или их солей путем превращения рацемического или оптического активного соединения формулы VII в соединение VIII с последующей циклизацией последнего. Указанные соединения применяются при лечении благодаря тому, что они ингибируют агрегацию тромбоцитов. Способ позволяет увеличить выход целевого продукта и избежать использования токсичного промежуточного соединения. 3 з.п.ф-лы.
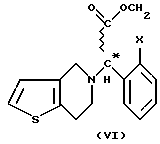
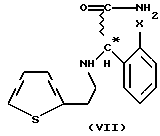
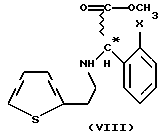
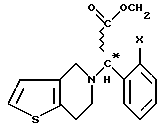
где Х - галоген,
или их солей, отличающийся тем, что рацемическое или оптически активное соединение общей формулы VII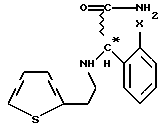
где Х - галоген,
превращают в рацемическое или оптически активное соединение общей формулы VIII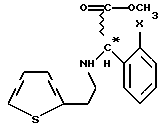
где Х - галоген,
при необходимости расщепляют полученное рацемическое соединение общей формулы VIII на его оптически активные изомеры, а затем рацемическое или оптически активное соединение общей формулы VIII подвергают циклизации известным способом с получением рацемического или оптически активного соединения общей формулы VI и, при необходимости, рацемическое соединение общей формулы VI расщепляют на его оптические изомеры, и/или превращают их в соли, и/или выделяют рацемическое или оптически активное соединение общей формулы VI из его солей.
| СПОСОБ ПОЛУЧЕНИЯ ТИАВИЦИКЛАНОВ ИЛИ МОНОТИ-АЦИКЛАНОВ | 0 |
|
SU281459A1 |
| Способ получения производных тиено-/3,2- @ / пиридина или их солей | 1983 |
|
SU1272994A3 |
| Способ получения в - феиилэтиламина и его замещенных | 1951 |
|
SU99802A2 |

