Данное изобретение относится к новым антрациклиновым гликозидам, способам их получения и фармацевтическим композициям, содержащим их.
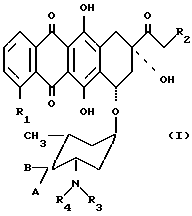
Изобретение предлагает антрациклиновые гликозиды общей формулы 1, в которых аминогруппа сахарной части несет моно- или бис-алкилзамещенные цепи
где
R1 есть водород или метоксигруппа; R2 есть водород или гидроксигруппа, A и B оба являются водородом или одна из A и B есть водород и другая есть гидрокси или группа формулы -OSO2R5, в которой R5 есть C1-C4 алкил или арил, произвольно замещенный C1-C4 алкилом, нитро, амино, метокси или галогеном; R3 есть атом водорода или группа формулы 2 и R4 есть группа формулы 2
-(CH2)n-X,
в которой
n есть 2 или 3 и X есть гидроксигруппа, галоген или группа формулы OSO2R5, в которой R5 есть, как определено выше и при условии, если R2, X и A являются гидроксигруппой и R3 = H, то должно быть 3, или фармацевтически приемлемую их соль.
Соединения данного изобретения демонстрируют противоопухолевую активность.
Изобретение включает следующие соединения:
1a: N-(3-гидроксипропил) даунорубицин
(R1 = OCH3, R2 = R3 = H, A = OH, B = H, R4 = (CH2)3-OH);
1b: N-(3-гидроксипропил) доксорубицин
(R1 = OCH3, R2 = OH, R3 = H, A = OH, B = H, R4 = (CH2)3-OH);
1c: N,N-бис(3-гидроксипропил) даунорубицин
(R1 = OCH3, R2 = H, A = OH, B = H, R3 = R4 = (CH2)3-OH);
1d: 4-деметокси-N-(2-гидроксиэтил)даунорубицин
(R1 = R2 = R3 = H, A = OH, B = H, R4 = C(H2)2-OH);
1e: N,N-бис(2-гидроксиэтил) даунорубицин
(R1 = OCH3, R2 = H, A = OH, B = H, R3 = R4 = (CH2)2-OH);
1f: N,N-бис-(2-гидроксиэтил) доксорубицин
(R1 = OCH3, R2 = OH, A = OH, B = H, R3 = R4 = (CH2)2-OH);
1g: 4-деметокси-N,N-бис-(2-гидроксиэтил)даунорубицин
(R1 = R2 = H, A = OH, B = H, R3 = R4 = (CH2)2-OH);
1h: 4-деметокси-4'-O-метансульфонил-N,N-бис-(2-хлорэтил) даунорубицин
(R1 = R2 = H, A = OSO2CH3, B = H, R3 = R4 = (CH2)2-Cl);
1i: 4-деметокси-N,N-бис-(2-хлорэтил) даунорубицин
(R1 = R2 = H, A = OH, B = H, R3 = R4 = (CH2)2-Cl)
1j: 4'-O-метансульфонил-N,N-бис-(2-хлорэтил) даунорубицин
(R1 = OCH3, R2 = H, A = OSO2CH3, B = H, R3 = R4 = (CH2)2-Cl);
1k: N,N-бис-(2-хлорэтил) даунорубицин
(R1 = OCH3, R2 = H, A = OH, B = H, R3 = R4(CH2)2-Cl);
1l: 4'-эпи-4'-O-метансульфонил-N,N-бис-(2-хлорэтил) даунорубицин
(R1 = OCH3, R2 = H, A = H, B = OSO2CH3, R3 = R4(CH2)2-Cl);
(R1 = OCH3, R2 = H, A = H, B = OH, R3 = R4 = (CH2)2-Cl);
1n: 4-деметокси-4'-эпи-4'-O-метансульфонил-N, N-бис(2-хлорэтил) даунорубицин
[R1 = R2 = H, A = H, B = OSO2CH3, R3 = R4 = (CH2)2-Cl];
1o: N,N-бис-(2-хлорэтил) доксорубицин
[R1 = OCH3, R2 = OH, A = OH, B = H, R3 = R4 = (CH2)3-Cl]
и фармацевтически приемлемые их соли, такие как гидрохлоридная соль.
Соединения данного изобретения могут быть получены несколькими методами, исходя из соединений формулы 3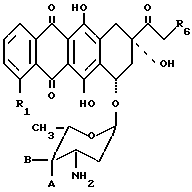
где
R1, A и B есть такие, как определено выше для формулы 1, R6 есть водород, гидроксильная группа или кислотно-чувствительная маскирующая группа для гидроксигруппы. Примеры подходящих защищающих групп включают группы, описанные в Международной заявке на патент PCT/EP91/01449, опубликованной как W092/02255, которая основана на заявке на Пат. Великобритании 9017024,2, поданной 3.8.1990 под названием "New Linker for Bioactive Agents".
Примеры исходных соединений формулы 3 включают даунорубицин [3a : R1 = OCH3, R2 = H, A = OH, B = H], 4-деметокси-даунорубицин [3b : R1 = OCH3, R2 = H, A = OH, B = H], доксорубицин [3c : R1 = OCH3, R2 = OH, A = OH, B = H], 4-деметокси-4'-эпи-даунорубицин [3d : R1 = R2 = H, A = H, B = OH], 4'-эпи-даунорубицин [3e : R1 = OCH3, R2 = H, A = H, B = OH] или маскированное производное доксорубицина при C-14.
Соединения общей формулы 1 могут быть получены алкилированием аминогруппы сахарной части, используя обычные методы. Например, моно 2- или 3-гидркосиалкиламино производные, которые представляют собой соединения формулы 1, определенные выше, в которых R1 и R4 такие, как определено выше, где X есть гидрокси группа, могут быть получены способом, включающим
(i) взаимодействие соединения формулы 3, определенного выше, причем соединение формулы 3 растворяют в полярном апротонном растворителе, с алкилирующим агентом общей формулы 4
X - (CH2)n - Гал
где
n есть 2 или 3; X - гидрокси и Гал является галогеном,
и при необходимости (ii) очистку полученных антрациклиновых гликозидов формулы 1 на хроматографической колонке; и/или, при необходимости,
(iii) превращение антрациклинового гликозида формулы 1 в его фармацевтически пригодную кислую соль присоединения. Антрациклиновый гликозид формулы 1 может быть превращен в его гидрохлоридную соль, например, обработкой безводным хлористым водородом.
Предпочтительно галогеном в формуле 4 является иод или бром. Полярный апротонный растворитель является предпочтительно сухим и представляет собой, например, диметилформамид или ацетонитрил. Реакцию обычно проводят при температуре от 20 до 30oC, обычно в течение периода времени от четырех до двадцати четырех часов. Стадию очистки (ii) обычно проводят на колонке с силикагелем, используя в качестве элюента смесь метиленхлорид : метанол (80 : 20 об/об).
Бис-2- или 3-гидрокси-алкиламино производные, которые представляют собой соединения формулы 1, определенные выше, в которых R3 и R4 являются группой формулы 2, как определено выше, где X есть гидрокси группа, могут быть получены вышеописанным способом.
Бис-2-гидроксиэтиламино соединения общей формулы 1 могут быть также получены способом, включающем (i) взаимодействие соединения формулы 3, описанного выше, предпочтительно в виде свободного основания, с этиленоксидом, и при необходимости (ii) очистку образовавшегося антрациклинового гликозида формулы 1, на хроматографической колонке; и/или при необходимости (iii) превращение антрациклинового гликозида формулы 1 в его фармацевтически пригодную кислую соль присоединения. Предпочтительно соединение формулы 3 сначала растворяют в растворителе, включающем метанол и метиленхлорид. Обычно стадию (i) проводят в темноте, и при начальной температуре около -40oC. Затем температуру обычно медленно повышают до комнатной температуры. Обычно реакционную смесь оставляют при комнатной температуре в течение 3 дней.
Антрациклиновые гликозиды общей формулы 1, определенные выше, в которых R2 есть водород и один или оба R3 и R4 представляют собой группу формулы (CH2)n-OSO2R5, определенную выше, могут быть получены способом, включающем обработку соответствующих моно- или бис-гидроксиаминопроизводных формулы 1 сульфонилхлоридом формулы 5
Cl-SO2R5,
где
R5 представляет собой остаток, определенный выше;
и при необходимости (ii) очистку образовавшегося антрациклинового гликозида формулы 1 на хроматографической колонке. Обычно стадию (i) проводят в апротонном растворителе, таком как метиленхлорид. Обычно ее проводят в присутствии третичного амина, такого как триэтиламин или пиридин.
Также можно получить антрациклиновые гликозидные производные общей формулы 1, в которых R2 является гидроксигруппой из соответствующих моно или бис-гидроксиамино соединений формулы 1, и эту C-14 гидрокси группу, R2 = OH, маскируют кислотно-чувствительной защищающей группой. Путем кислотной обработки последнего в мягких условиях получают желаемый сульфонилалкиламиноантрациклин.
Соединения общей формулы 1, в которых X есть галоид, можно получить по способу, включающему (i) растворение антрациклинового гликозида формулы 1, определенного выше, в котором один или оба R3 и R4 являются группой формулы OSO2R5, где R5 есть алкильная или арильная группа, в апротонном растворителе, и взаимодействие образовавшегося раствора с соответствующей солью галогенида и при необходимости (ii) очистку полученного антрациклинового гликозида на хроматографической колонке; и/или, при необходимости (iii) выделение требуемого соединения в виде соответствующего гидрогалогенида. Когда X есть хлор, соответствующая хлорная соль может быть, например, хлоридом пиридиния или хлоридом н-тетрабутиламмония. Апротонным растворителем, например, является ацетон или диметилформамид. Реакцию обычно проводят при комнатной температуре.
Антрациклиновые гликозиды общей формулы 1, определенные выше, в которых X есть хлор, R2 есть водород, оба A и B являются водородом или один из A или B является группой - OSO2R5, в которой R5 такая, как описана выше, могут быть получены способом, включающим (i) обработку соответствующего моно или би-гидроксиалкиламино производного формулы 1, описанного выше, в сухом пиридине, сульфонилхлоридом формулы 5; и при необходимости (ii) очистку образовавшегося антрациклинового гликозида на хроматографической колонке; и/или при необходимости (iii) выделение требуемого соединения в виде соответствующего гидрогалогенида. Обычно стадию (i) проводят в темноте и при температуре 0oC. Обычно ее проводят в атмосфере азота. Температуру поддерживают при 0oC в течение вплоть до 16 часов.
Можно также получить антрациклиновые гликозидные производные общей формулы 1, в которых R2 является гидроксильной группой из соответствующих моно или бис-гидрoксиалкиламино соединений формулы 1, и эту C-14 гидроксигруппу, R2 = OH, маскируют кислотно-чувствительной защищающей группой. Путем кислотной обработки в мягких условиях последнего получают требуемое хлоралкиламино антрациклиновое производное.
Соединения данного изобретения имеют активность в качестве противоопухолевых агентов. Млекопитающее, например человека, можно лечить способом, включающем введение ему оральным или парентеральным путем фармацевтически эффективного количества антрациклинового гликозида формулы 1, определенного выше, или его фармацевтически пригодной кислой соли присоединения.
Изобретение обеспечивает также фармацевтическую композицию, включающую фармацевтически приемлемый разбавитель или носитель и в качестве активного начала антрациклиновый гликозид формулы 1, или его фармацевтчиески пригодную соль. Могут использоваться обычные носители или разбавители. Композиция может быть сформулирована и применяться, например, внутривенно обычным способом.
Следующие примеры иллюстрируют данное изобретение, не ограничивая его.
Пример 1. Получение: N-(3-гидроксипропил)даунорубицина
[R1 = OCH3, R2OR3 = H, A = OH, B = H, R4(CH2)3-OH] (1a)
К раствору даунорубицина (3a) (0,2 г, 0,38 ммоль) в безводном диметилформамиде (2 мл) добавляют 3-бром-1-пропанол [4 : X = OH, Гал = Br, n = 3] (200 мкл, 2,16 ммоль) при комнатной температуре в атмосфере азота и перемешивание продолжают в течение пяти дней.
После этого растворитель удаляют в вакууме; неочищенное масло растворяют в метиленхлориде (10 мл) и добавляют ангидрид трифторуксусной кислоты (425 мкл, 3 ммоль). Смесь перемешивают в течение одного часа при 0oC и затем выливают в насыщенный водный раствор бикарбоната натрия и экстрагируют метиленхлоридом. Объединенные органические экстракты промывают водой и органический растворитель удаляют при пониженном давлении. Неочищенное масло растворяют в метаноле (50 мл) и перемешивают в течение одного часа при 40oC, концентрируют до небольшого объема и очищают с помощью флеш-хроматографии на колонке с кремниевой кислотой, используя в качестве элюирующей системы смесь метанола и метиленхлорида (10/90 по объему), получая после обработки метанольной безводной соляной кислотой названное соединение 1a (0,12 г, выход 54%) в виде гидрохлоридной соли.
ТСХ на кизельгуровой пластине F254 (Merck), элюирующая система: метиленхлорид, метанол, уксусная кислота, вода (80:20, 7:3 по объему)
Rf = 0,42
FD - MC : м/e 569 (M+)
1ПМР (200 МГц, DMCOd6) δ : 1,16 (д, J = 6,4 Гц, 3H, CH3-5'); 1,5 - 1,8 (м, 4H, CH2CH2, CH2-2'); 2,15 (м, 2H, CH2-8); 2,25 (с, 3H, COCH3); 2,6 - 2,9 (м, 2H, CH2NH); 2,99, 2,89 (ABк, J = 18,0 Гц, 2H, CH2-10); 3,31 (м, 1H, H-3'); 3,41 (м, 2H, CH2-OH); 3,63 (м, 1H, H-4'); 3,92 (с, 3H, OCH3); 4,14 (к, J = 6,4 Гц, 1H, H-5'); 4,96 (м, 1H, H-7); 5,29 (м, 1H, H-1); 5,49 (с, 1H, OH-9); 7,6 - 7,9 (м, 3H, ароматические H-ы).
Пример 2. Получение: N,N-бис (3-гидроксипропил)даунорубицина
[R1 = OCH3, R2 = H, A = OH, B = H, R3 = R4 = (CH2)2-OH] (1c)
Названное соединение получают, выдерживая даунорубицин (3a) (0,2 г, 0,38 ммоль) и 3- бром-1-пропанол (200 мкл, 2,16 ммоль) в безводном диметилформамиде в течение трех недель в атмосфере азота. После этого растворитель удаляют в вакууме и неочищенное масло очищают с помощью флеш-хроматографии на колонке с кремниевой кислотой, используя в качестве элюирующей системы смесь метанола и метиленхлорида (10/90 по объему), получая после обработки метанольной безводной соляной кислотой N,N-бис (3-гидроксипропил)-даунорубицин (1c) (0,10 г, выход 50%) в виде гидрохлоридной соли.
ТСХ на кизельгуровой пластине F254 (Merck), элюирующая система : метиленхлорид, метанол, уксусная кислота, вода (30:4:1:0,5 по объему) Rf = 0,14.
FD - MC : м/e 627 (M+)
1ПМР (200 МГц, CDCl3) δ : 1,29 (д, J = 6,4 Гц, 3H, CH3-5'); 1,8 - 1,5 (М, 5H: 2xNHCH2CH2, H-2' экв); 2,04 (м, 1H, H-2', акс); 2,09 (М, 1H, H-8 акт); 2,34 (м, 1H, H-8 экв); 2,41 (с, 3H, COCH3); 2,5 - 2,9 (М, 5H, 2xNHCH2, H-3'); 2,95 (д, J = 19,0 Гц, 1H, H-10 акс); 3,21 (дд, J = 1,5 19,0 Гц, 1H, H-10 экв.); 3,8 - 3,6 (м, 5H, 2xCH2OH, H-4'); 4,07 (с, 3H, OCH3; 4,09 (к, J = 6,4 Гц, 1H, H-5'); 5,29 (м, 1H, H-7); 5,57 (д, J = 3,3 Гц, 1H, H-1'); 7,38 (д, J = 8,4 Гц, 1H, H-3); 7,77 (дд, J = 7,4, 8,4 Гц, 1H, H-2); 8,01 (д, J = 7,4 Гц, 1H, H-1); 14,0, 13,0 (широкие сигналы, 2H, 2x фенольные - OH).
Пример 3. Получение N,N-бис-(2-гидроксиэтил) доксорубицина
[R1 = OCH3, R2 = OH, A = OH, B = H, R3 = R4 = (CH2)2-OH] (1f)
Смесь доксорубина (3c) (0,15 г, 0,258 ммоль), метанола и метиленхлорида (25 мл, 1:1 по объему) выливают в хорошо закрытый круглодонный сосуд, охлаждают при - 40oC и добавляют этиленоксид (15 мл). Реакционную смесь медленно доводят до комнатной температуры и выдерживают в течение двух дней в темноте. После этого удаляют растворители при пониженном давлении и остаток очищают с помощью флеш-хроматографии на колонке с кремниевой кислотой, используя в качестве элюирующей системы смесь метиленхлорида и метанола (80:20 по объему). Названное соединение 1f (0,1 г, выход 60%) превращают в гидрохлоридную соль путем обработки метанольной безводной соляной кислотой. ТСХ на кизельгуровой пластине F254 (Merck), элюирующая система : метиленхлорид, метанол, уксусная кислота, вода (80:20:7:3 по объему)
Rf=0,36.
FD = MC : м/е 631 (М+)
1ПМР (200 МГц, DMCOd6) δ : 1,13 (д, J=6,6 Гц, 3H, CH3-5'); 1,53 (м, 1H, H-2' экв); 1,94 (м, 1H, H -2'акс); 2,16 (м, 2Н CH2-8); 2,65 (м, 4H, N(CH2CH2OH)2; 2,82 (м, 1H, H-3'); 2,98 (м, 2H, CH2-10); 3,33 (м, 4H, N(CH2CH2OH)2; 3,57 (м, 1H, H-4'); 4,00 (c, 3H, OCH3-4); 4,02 (дд, J = <2, 6,6 Гц, 1H, H-5'); 4,35 (шм, 3H, OH-4', N(CH2CH2OH)2); 4,56 (м, 2H, CH2-14); 4,84 (т, J = 6,2 Гц, 1H, OH-14); 4,98 (м, 1H, H-7); 5,30 (м, 1H, H-1'); 5,40 (с, 1H, OH-9); 7,67 (м, 1H, H-2); 7,93 (м, 2H, H-1, H-3); 13,28 (шс, 1H, OH-11); 14,06 (с, 1H, OH-6).
Пример 4. Получение 4-деметокси-N,N-бис-(2-гидроксиэтил) даунорубицина
[R1 = R2 = H, A = OH, B = H, R3 = R4 = (CH2)2 - OH (1g)
Свободное основание 4-деметоксидаунорубицин (3b) (0,23 г, 0,5 ммоль) превращают в названное соединение 1g, следуя методике, описанной в примере 3. Выход: 0,15 г в виде гидрохлоридной соли после обработки метанольной безводной соляной кислотой.
ТСХ на кизельгуровой пластине F254 (Merck), элюирующая система: метиленхлорид, метанол, уксусная кислота, вода (80:20:7:3 по объему)
Rf = 0,28
FD = MC : м/е 585 (M+)
1ПМР (200 МГц, DMCOd6) δ :
1611 (д, J = 6,6 Гц, 3H, CH3-5'); 1,55 (м, 1H, H-2'ед) : 1,93 (м, 1H, H-2 акс); 2,17 (м, 2H, CH2-8); 2,26 (c, 3H, COCH3); 2,65 (м, 4H, N(CH2CH2OH)2); 2,82 (м, 1H, H-3'); 2,99 (м, 2H, CH2-10); 3,30 (м, 4H, (CH2CH2OH)2; 3,59 (м, 1H, H-4'); 4,06 (дк, J =< 2, 6,6 Гц, 1H, H-5'); 4,35 [шм, 3H, OH-4', N(CH2CH2OH)2]; 4,98 (м, 1H, H-7); 5,30 (М: 1H, OH-9); 8,00 (м, 2H, H-2, H-3); 8,30 (м, 2H, H-1, H-4); 13,35 (c, 1H, OH-11); 13,55 (c, OH-6).
Пример 5. Получение N,N-бис (2-гидроксиэтил) даунорубицина
[R1 = OCH3, R2 = H, A = OH, B = H, R3 = R4 = (CH2)2 - OH] (1e)
Названное соединение 1e получают из даунорубицина (3а), следуя такой же методике, как описано в примере 4.
ТСХ на кизельгуровой пластине F254 (Merck), элюирующая система:
метиленхлорид, метанол, уксусная кислота, вода (80:20:7:3 по объему)
Rf = 0,28
MC : м/е 615 (M+)
Пример 6. Получение 4-деметокси-4'-O-метансульфонил-N, N-бис(2-хлорэтил)-даунорубицина
[R1 = R2 = H, A = OSO2CH3, B = H, R3 = R4 = (CH2)2-Cl] (1h)
4-деметокси-бис(2-гидроксиэтил) даунорубицин (1g) (0,3 г, 0,5 ммоль), полученный, как описано в примере 4, растворяют в сухом пиридине (15 мл), охлажденном до 0o, и добавляют метансульфонил хлорид (1 мл) и выдерживают в течение ночи при 0oC при перемешивании в атмосфере азота. После этого реакционную смесь выливают в воду со льдом и экстрагируют метиленхлоридом. Органический слой промывают холодной водой, сушат над безводным сульфатом натрия и концентрируют под пониженным давлением. Неочищенное вещество очищают с помощью флеш-хроматографии на колонке с кремниевой кислотой, используя в качестве элюирующей системы смесь метиленхлорида и ацетона (97:3 по объему), получая названное соединение 1 h (0,1 г, выход 30%).
ТСХ на кизельгуровой пластине F254 (Merck), элюирующая система:
метиленхлорид, ацетон (20:1) по объему) Rf = 0,48
MC : м/е 680 (М+)
1ПМР (200 МГц, CDC I3) δ : 1,35 (д, J = 6,5 Гц, 3H, CH3-5'); 1,8 - 2,4 (м, 4H, H-2'экв H-2'акс, H-8акс, H-8экв; 2,42 (c, 3H, CH3-CO); 3,06 (т, 4H, J = 7,1 Гц, CH2-N-CH2); 3,17 (м, 1H, H-3); 3,43 (м, 4H, 2xCH2-Cl); 4,16 (м, 1H, H-5'); 4,40 (c, 1H, OH-9); 4,91 (c, 1H, H-4'); 5,30 (дд, J = 2,1, 3,7 Гц, 1Н, H-7); 5,60 (д, 1H, J = 3,4 Гц, H-1'); 7,85 (м, 2H, H-2, H-3); 8,37 (м, 2H, H-1, H-4); 13,55 (c, 1H, OH-11); 13,64 (c, 1H, OH-6).
Пример 7. Биологические испытания 4-деметокси-4'-O-метансульфонил-N,N-бис (2-хлорэтил) даунорубицин (соединение 1h) испытывают "in vitro" как ингибитор роста колоний двух линий человеческих клеток: LoVo (колония аденокарцинома) и LoXo/DX (колония аденокарцинома, устойчивая к доксорубицину) в сравнении с 4-деметокси-даунорубицином (3b) и доксорубицином (3с) (таблица 1). При сравнении с 4-деметоксидаунорубицином и доксорубицином для соединения Ih наблюдают поразительно более высокую активность по линии доксорубицин-резистентных клеток.
Соединение 1h также оценивают "in vivo" против Р388 мышиных лейкозов, чувствительных (таблица 2) и резистентных к доксорубицину (gohnson). При испытании при стойком лейкозе соединение 1 показывает высокую активность (таблица 3).
Примеры на композицию
Пример 8. 4-деметокси-4'-O-метансульфонил-N,N-бис(2-хлорэтил)-даунорубицин (1h), полученный по примеру 6, растворяют в смеси Кремофора фирмы Басф (Cremofor EL) и этанола в объемном соотношении 6,5:3,5. Концентрация активного вещества составляет 1 мг/мл. Полученная композиция проявляет стабильность, равную 95,1% при 4oC в течение 7 суток.
Пример 9. 4-деметокси-4'-O-метансульфонил-N,N-бис(2-хлорэтил)-даунорубицин (1h), полученный по примеру 6, растворяют в N,N-диметилацетамиде; концентрация активного вещества составляет 5 мг/мл. Полученная композиция проявляет стабильность, равную 95,1% при 4oC в течение 15 суток.
Эксперимент
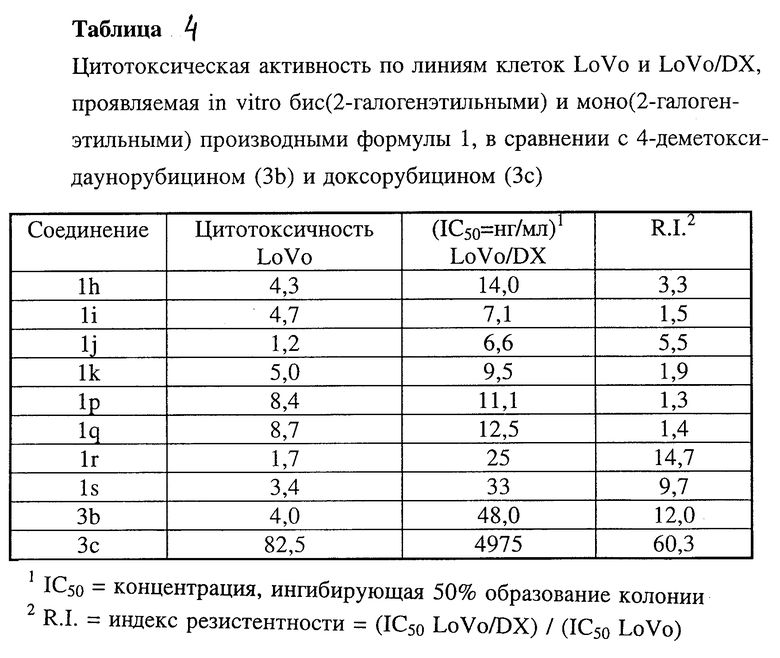
Производные антрациклина формулы 1 по изобретению, в которых NR3R4 является бис(2-галогеналкильной) группой и которые представляют собой 4-деметокси-3'-N, N-бис(2-хлорэтил)даунорубицин (1i), 3'-N, N-бис(2-хлорэтил)даунорубицин (1k) и соответствующие 4'-O-метансульфонильные производные, в частности 4-деметокси-4'-O-метансульфонил-3'-N,N-бис(2-хлорэтил)даунорубицин (1h) и 4'-O-метансульфонил-3'-N,N-бис(2-хлорэтил)даунорубицин (1j), испытывали in vitro в качестве ингибитора роста колоний двух линий клеток человека: LoVo (аденокарцинома) и LoVo/DX (соответствующая сублиния, устойчивая к доксорубицину) в сравнении с 4-деметокси-даунорубицином (3b) и доксорубицином (3c) (таблица 4 и 4a)
У бис(2-галогеналкил)антрациклина были отмечены более высокая активность в отношении доксорубицин-резистентной линии клеток и низкий индекс резистентности по сравнению с соединениями 3b и 3c (таблицы 4).
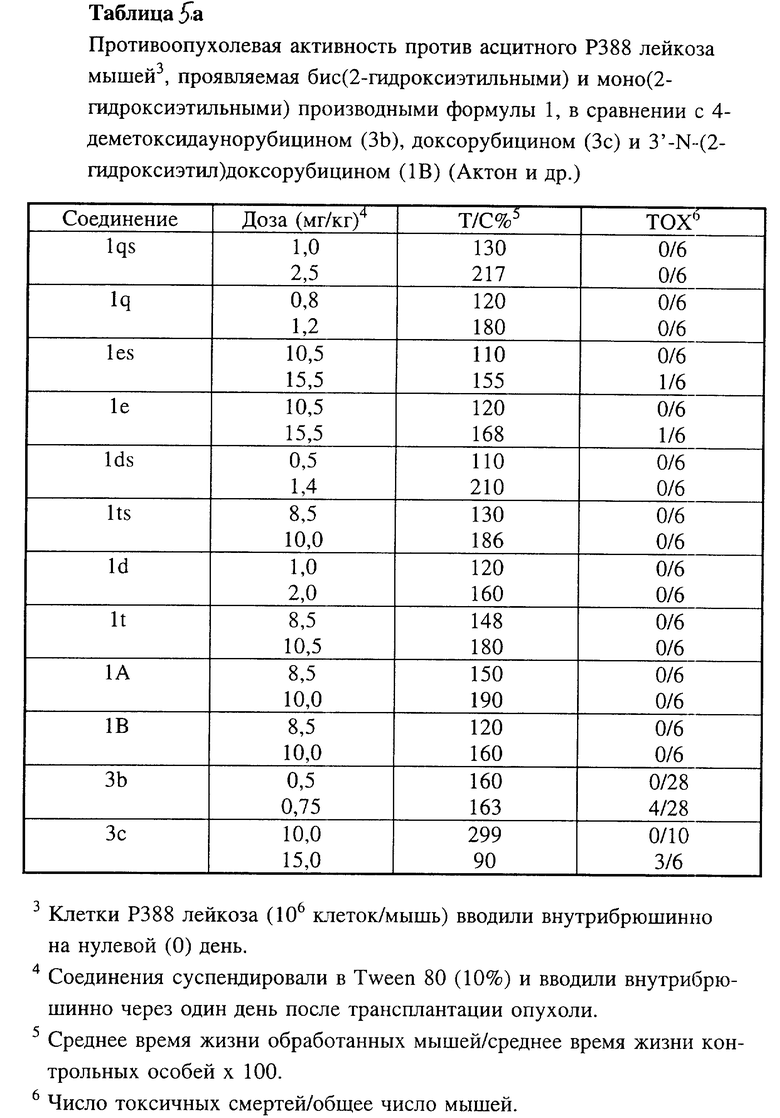
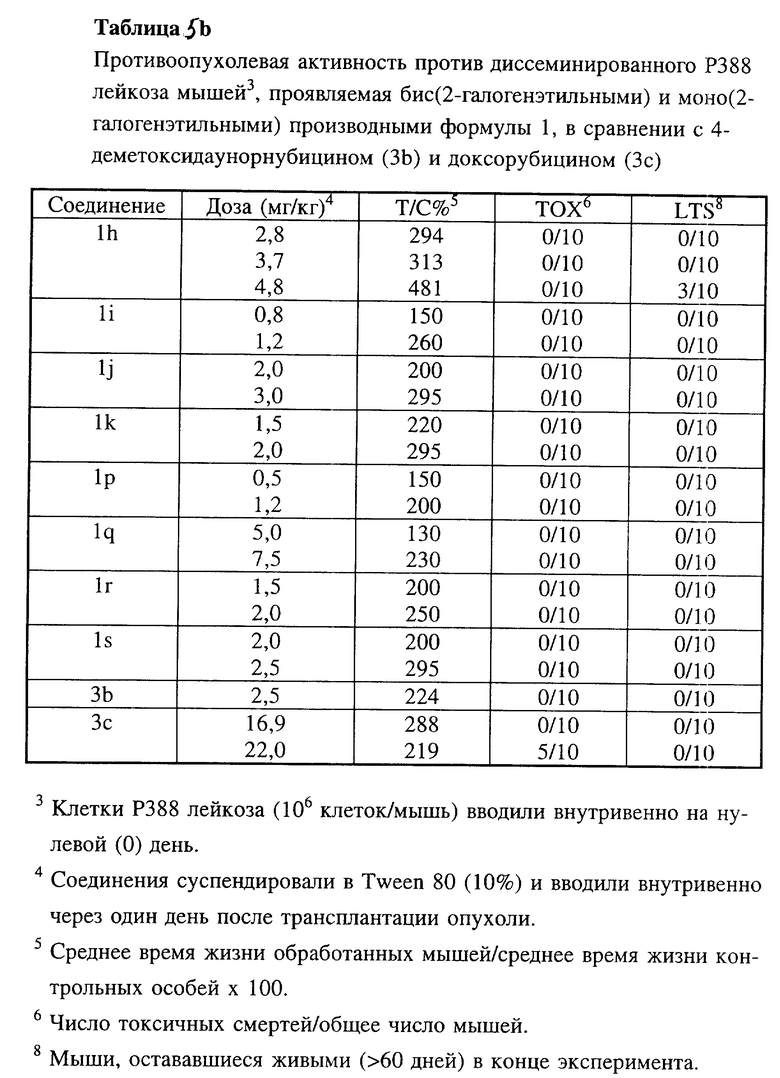
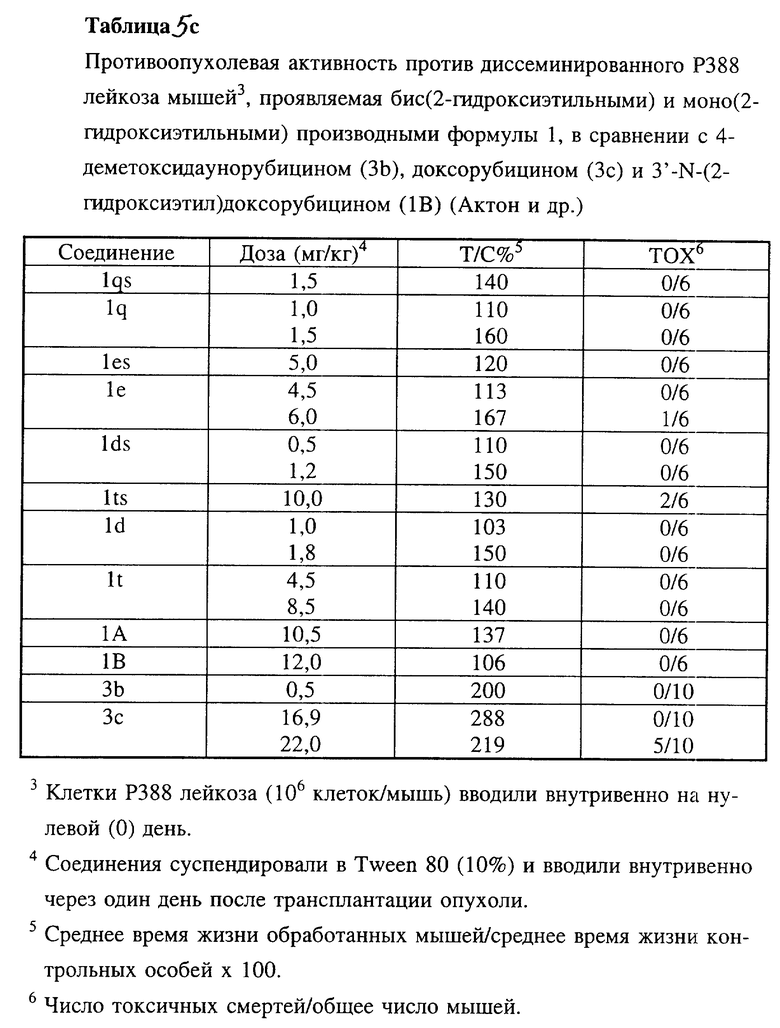
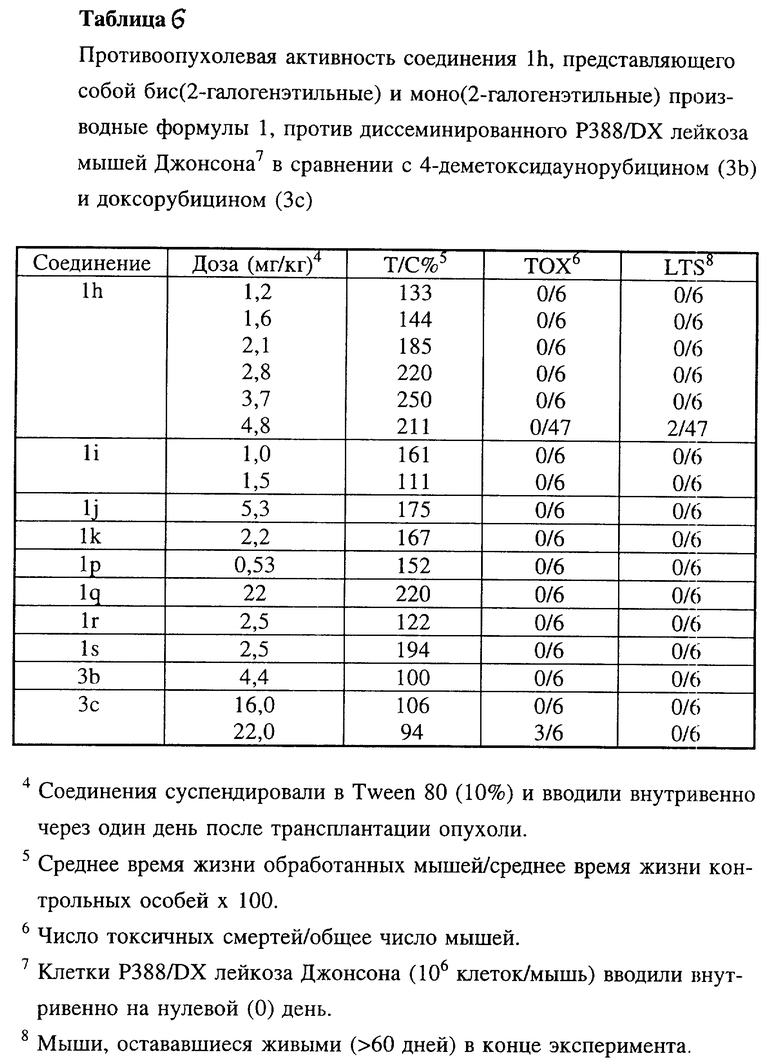
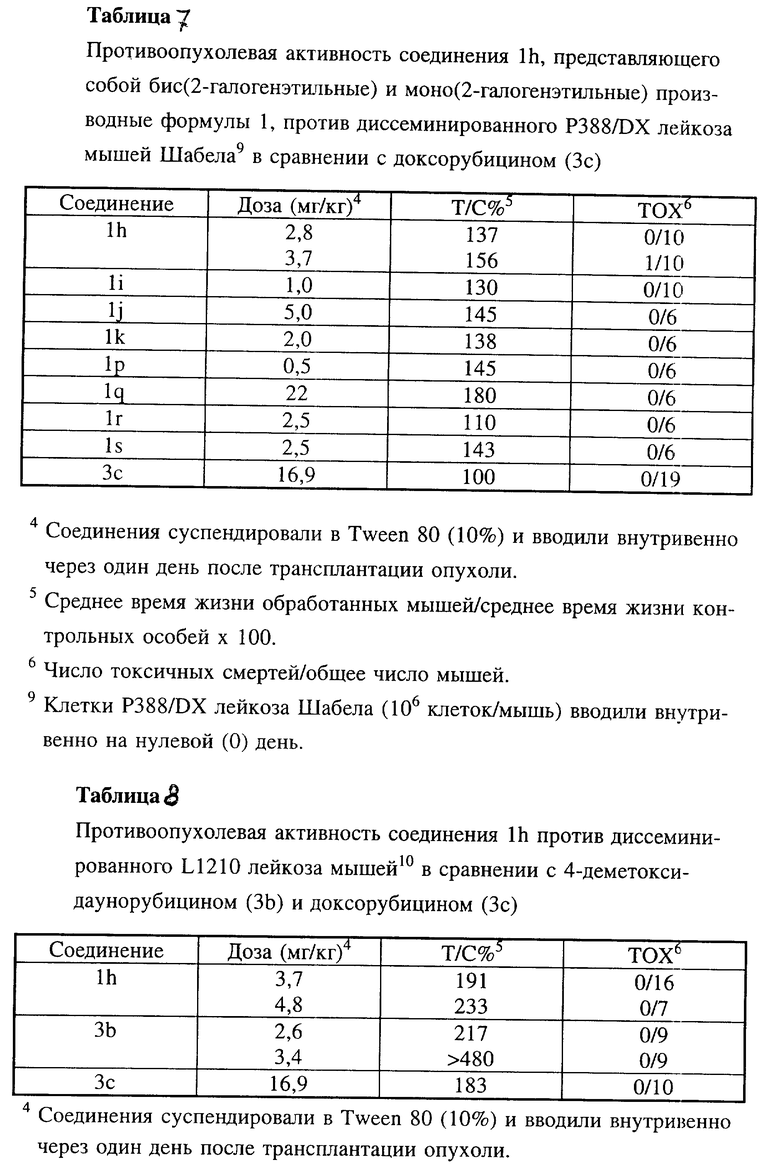
Соединения 1h, 1i, 1j и 1k по настоящему изобретению испытывали in vivo против асцитного Р388 лейкоза мышей (таблицы 5, 5a, 5c), диссеминированного Р388 лейкоза мышей (таблица 5b), диссеминированного P388/DX лейкоза Джонсона (Johnson) и P388/DX лейкоза мышей Шабела (Schabel) (таблицы 6 и 7) в сравнении с соединениями 3b и 3c.
В этих экспериментальных лейкозах мышей производные бис(2-галогеналкил)антрациклина проявили аналогичную или более высокую активность в случае чувствительных к лекарственным средствам моделей Р388 лейкоза, причем исключительно высокой эта активность была в отношении доксорубицин-резистентных Р388 лейкозов.
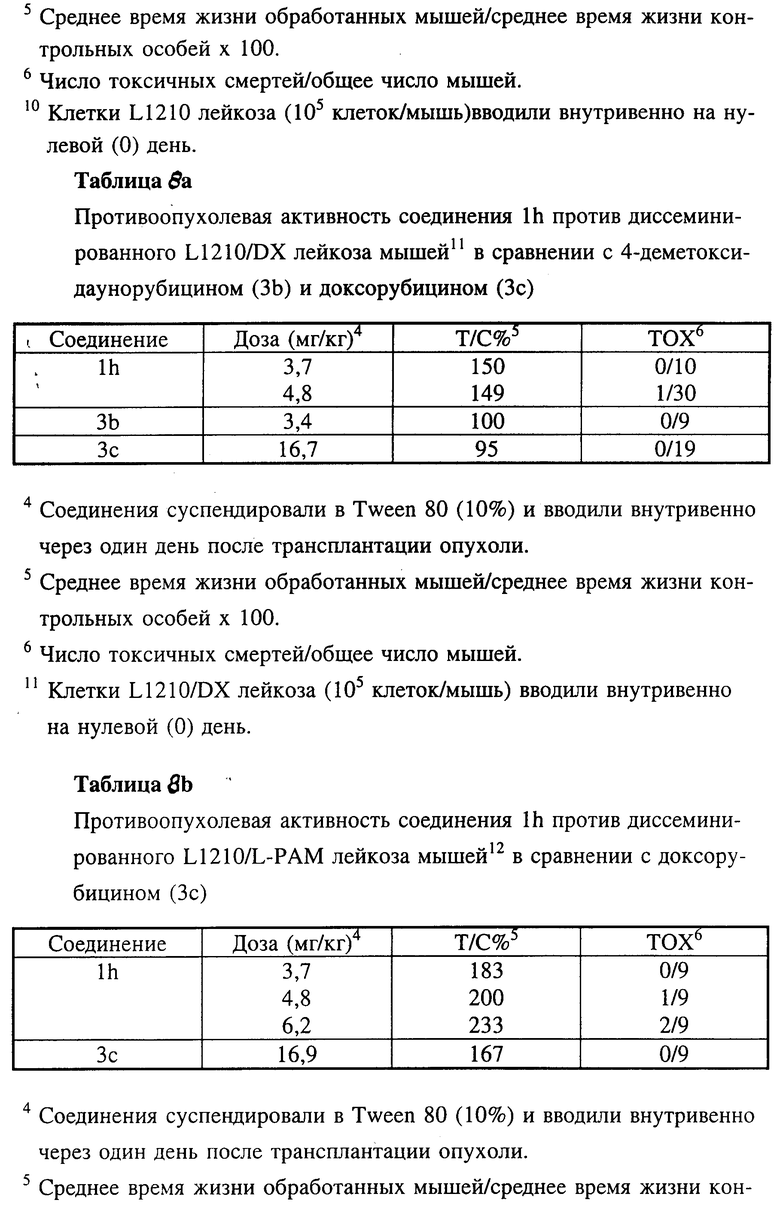
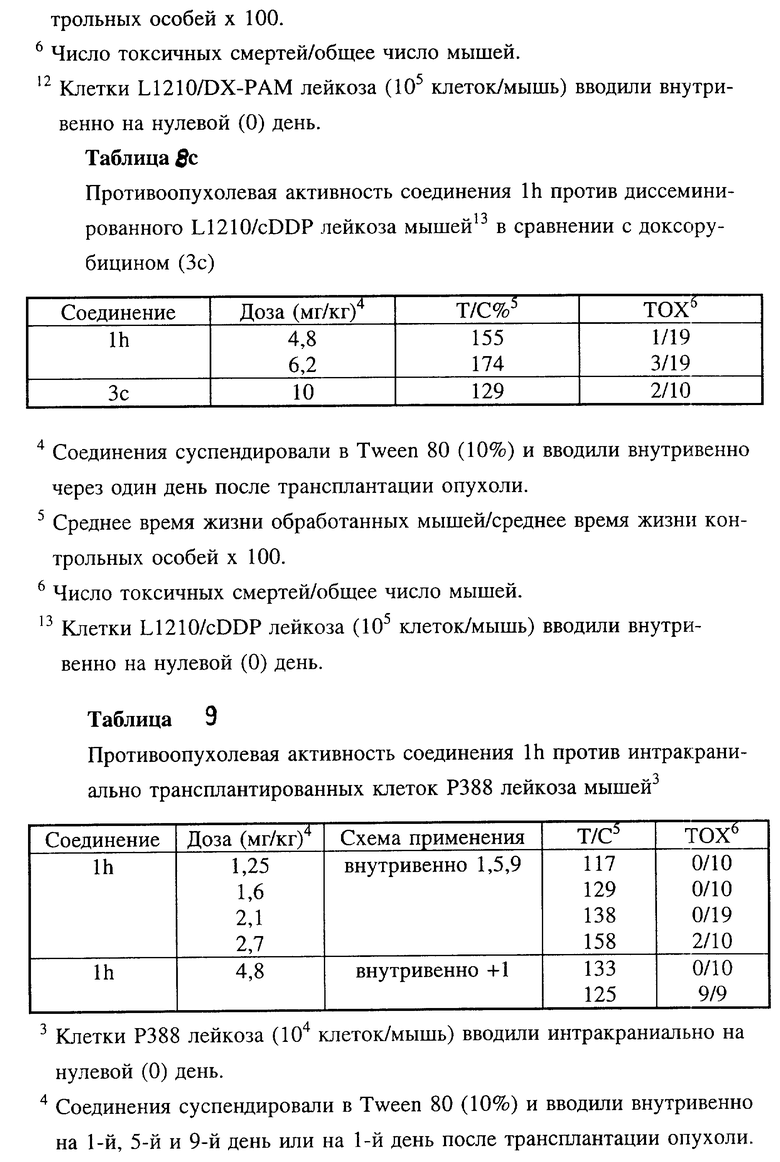
Противоопухолевая активность соединения 1h по настоящему изобретению оценивали также in vivo в отношении диссеминированного L1210 лейкоза мышей (таблица 8) и диссеминированных L1210 лейкозов, устойчивых к разным противоопухолевым средствам, таких как доксорубицин-резистентный L1210 лейкоз, L1210/DX (таблица 8а), мелфалан-резистентный L1210 лейкоз, L1210/L-PAM (таблица 8b) и цисплатин-резистентный L1210 лейкоз, L1210/cDDP (таблица 8c) в сравнении с соединениями 3b и 3c.
Во всех этих экспериментальных лейкозах мышей соединение 1h характеризовалось более высокой противоопухолевой активностью.
Кроме того, было установлено, что соединение 1h также проявляет активность против интракраниально трансплантированного P388 лейкоза мышей (таблица 9), в то время как у соединений 3b и 3c отсутствовала активность при значении T/C%, равном 90-100.
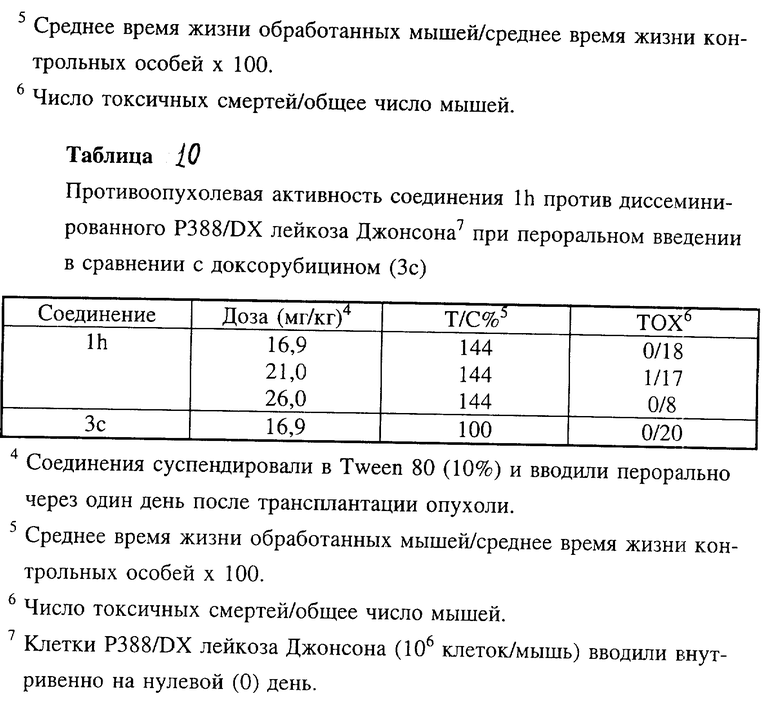
Соединение lh оказалось активным против доксорубицин-резистентного Р388 лейкоза мышей (лейкоз Джонсона) при пероральном введении (таблице 10).
Производные антрациклина формулы 1 по настоящему изобретению, в которых NR3R4 является N,N-бис(2-гидроксиалкильной) группой и которые представляют собой 4-деметокси-3'-N, N-бис(2-гидроксиэтил)даунорубицин (1g), 3'-N,N-бис(2-гидроксиэтил)даунорубицин (1e) и соответствующие 4'-О-метансульфонильные производные формулы 1, в которой A = CH3SO2O, в частности 4-деметокси-4'-О-метансульфонил-3'-N, N-бис(2-гидроксиэтил)даунорубицин (1qs) и 4'-O-метансульфонил-3'-N,N-бис(2-гидроксиэтил)даунорубицин (1es), испытывали in vitro в отношении линий клеток LoVo и LoVo/DX (таблица 4а) и in vivi в отношении асцитного и диссеминированного Р388 лейкозов (таблицы 5a и 5c).
Было установлено, что эти соединения являются в 2х103 - 8х103 раз менее цитотоксическими, чем соответствующие N,N-бис(2-хлоралкильные) производные при испытании in vitro на клетках LoVo и LoVo/DX.
4'-O-метансульфонильные производные 3'-N,N-бис(2-гидроксиэтильных) производных оказались более активными, чем соответствующие производные, содержащие гидроксильную группу в положении C-4', но менее активными в отношении чувствительных к лекарственным средствам Р388 лейкоза и совершенно инертными к доксорубицинрезистентному Р388 лейкозу (лейкоз Джонсона) T/C% = 100) по сравнению с соответствующими 3'-N,N-бис(2-галогеналкильными) производными.
Производные антрациклинов формулы 1 по настоящему изобретению, в которых NR3R4 является N(2-галогенэтильной) группой и которые представляют собой 4-деметокси-4'-O-метансульфонил-3'-N-(2-хлорэтил)даунорубицин (1p: R1 = R2 = B = H, A = CH3SO2O), 4'-O-метансульфонил-3'-N-)2-хлорэтил)даунорубицин (1q: R1 = OCH3, R2 = B = H, A = CH3SO2O), 4'-O-метансульфонил-3'-N-(2-хлорэтил)доксорубицин (1r: R1 = OCH3, R2 = OH, B = H, A = CH3SO2O) и 4'-O-метансульфонил-3'-N-(2-бромэтил)доксорубицин (1s: R1 = OCH3, R2 = OH, B = H, A = CH3SO2O), проявляли удивительно высокую активность при испытании in vitro на LoVo и LoVo/DX клетках (таблица 4) и оказывали противоопухолевое действие при испытании in vivi против асцитного Р388 лейкоза мышей (таблица 5), диссеминированного Р388 лейкоза мышей (таблица 5b), диссеминированного Р388/DX лейкоза мышей Джонсона и Р388/DX лейкоза мышей Шабела (табл. 6 и 7).
N-монозамещенные производные формулы 1 по настоящему изобретению, имеющие в алкильной цепи гидроксильную группу, которые представляют собой 4-деметокси-3'-N-)2-гидроксиэтил)даунорубицин (1d), 3'-N-(2-гидроксиэтил)даунорубицин (1t: R1 = OCH3, R2 = B = H, A = OH), соответствующие 4'-O-метансульфонильные производные, в частности 4-деметокси-4'-O-метансульфонил-3'-N-(2-гидроксиэтил)даунорубицин (lds: R1=R2=B=H, A=CH3SO2O), 4'-O-метансульфонил-3'-N-(2-гидроксиэтил)даунорубицин (1ts: R1=OCH3, R2=B=H, A= CH3SO2O) и 4'-O-метансульфонил-3'-N-(2-гидроксиэтил)доксорубицин (1A: R1= OCH3, R2= OH, B=H, A=CH3SO2O), испытывали in vitro по линиям клеток LoVo и LoVo/DX (таблица 4a) и in vivo в отношении асцитного и диссеминированного Р388 лейкозов (таблицы 5a и 5c) также в сравнении с 3'-N-(2-гидроксиэтил)доксорубицином (1B: R1= OCH3, R2=OH, B=H), описанным Актоном (Acton) и др. в журнале (Journal of Medicinal Chemictry), которое было выбрано в качестве ближайшего прототипа для соединения lA по настоящему изобретению, поскольку единственным различием между этими двумя соединениямии является наличие в соединении lA метансульфонильной группы в положении 4'-оксигруппы, в то время как соединение lB, представленного Актоном и др., имеет гидроксильную группу.
Было установлено, что 4'-O-метансульфонильные производные 3'-N-(2-гидроксиэтильных) производных являются более активными, чем соответствующие 3'-N-(2-гидроксиэтильные) производные, имеющие гидроксильную группу в положении C-4', но менее активными в отношении чувствительных к лекарственным средствам Р388 лейкозов и инертными к доксорубицин-резистентному Р388 лейкозу (лейкоз Джонсона) (T/C%=100) в сравнении с соответствующими 3'-N-(2-галогеналкильными производными).
Получение 4-деметокси-N-(2-гидроксиэтил)даунорубицина [R1=R2=R3=H, A=OH, B=H, R4=(CH2)2-OH] (1d)
4-Деметоксидаунорубицин (3b) в виде свободного основания (0,23 г, 0,5 ммоль) растворяли в сухом диметилформамиде (10 мл) и в течение 2-х дней при комнатной температуре обрабатывали 1-бромэтанолом (2 мл). После этого реакционную смесь разбавляли водой (200 мл) и экстрагировали метиленхлоридом (100 мл х 3). Указанное в заголовке соединение 1d (0,3 г) выделяли посредством испарительной хроматографии на колонках из силикагеля, производя элюирование смесью метиленхлорида и метанола (с объемным отношением 98:2), и после обработки безводным раствором хлористо-водородной кислоты в метаноле превращали его в соответствующую хлористо-водородную соль.
Тонкослойная хроматография на кизельгуровой пластине F254 (Merck) с использованием в качестве элюирующей системы смеси метиленхлорида, метанола, уксусной кислоты и воды (с объемным отношением 30/4/1/0,5) Rf = 0,32.
Масс-спектр с частотным разделением: m/e 541.
Спектр 1H ЯМР (400 МГц, DMSO-D6), δ : 1,17 (д, J=6,4 Гц, 3H, CH3-5'); 1,84 (м, 2H, CH2-2'); 2,17 (д, J=4,7 Гц, 2H, CH2-8); 2,25 (с, 3H, COCH3); 2,90 (м, 2H, NH+ CH2CH2OH); 3,69 (м, 1H, H-4'); 4,14 (к, J=6,4 Гц, 1H, H-5'); 4,54, 5,20 (два широких сигнала, 2H, OHCH2 + OH-4'); 5,01 (т, J=4,7 Гц, 1H, H-7); 5,31 (м, 1H, H-1'); 5,42 (с, 1H, OH-9); 8,00 (м, 2H, Н2, H-2 + H-3); 8,30 (м, 2H, H-1 + H-4).
Получение 4-деметокси-N-N-бис(2-хлорэтил)даунорубицина [R1=R2=H, A=OH, B=H, R3=R4=(CH2)2-Cl] (1i)
4-Деметокси-N, N-бис(2-гидроксиэтил)даунорубицин (1 г) (0,52 г, 0,9 ммоль), полученный в соответствии с процедурой, описанной в примере 4, растворяли в смеси метиленхлорида (12 мл) и пиридина (2,2 мл). Реакционную смесь охлаждали при 0oC, промывали аргоном и добавляли мезилхлорид (0,49 мл, 6,3 ммоль). Через 1 час эту смесь выливали в лед, содержащий 0,1 н. водный раствор HCl (200 мл), и экстрагировали метиленхлоридом (300 мл). Органическую фазу концентрировали до небольшого объема (10 мл) при пониженном давлении и обрабатывали ацетоном (20 мл) и хлоридом лития (20 мг). После выдерживания в течение 4 часов при температуре 35oC эту смесь разбавляли метиленхлоридом (200 мл) и промывали водой (100 мл x 2). Указанное в заголовке соединение li (0,2 г) выделяли посредством испарительной хроматографии на колонках из силикагеля, производя элюирование смесью метиленхлорида и ацетона (с объемным отношением 98:2).
Тонкослойная хроматография на кизельгуровой пластине F254 (Merck) с использованием в качестве элюирующей системы смеси метиленхлорида и ацетона (с объемным отношением 95/5) Rf = 0,42.
Масс-спектр с частотным разделением: m/e 622.
Спектр 1H ЯМР (200 МГц, CDCl3), δ : 1,38 (д, J=6,6 Гц, 3H, CH3-5'); 1,80 (двойной дублет, J=5,0, 12,5 Гц, 1H, H-2'экв); 2,00 (дважды двойной дублет, J= 3,7, 12,5, 12,5 Гц, 1H, H-2'акс); 2,14 (двойной дублет, J=4,1, 14,7 Гц, 1H, H-8 акс), 2,35 (дважды двойной дублет, J=1,7, 2,1, 14,7 Гц, 1H, H-8 экв); 2,43 (с, 3H, COCH3); 2,8-3,1 [м, 5H, (N-CH2CH2Cl)2 + H-3']; 3,03 (д, J= 19,0 Гц, 1H, H-10 акс); 3,26 (двойной дублет, J=1,7, 19,0 Гц, 1H, H-10 экв); 3,50 [м, 4H, (NCH2CH2Cl)2]; 3,53 (м, 1H, H-4'); 4,08 (к, J=6,6 Гц, 1H, H-5'); 4,63 (с, 1H, OH-9); 5,30 (двойной дублет, J=2,1, 4,1 Гц, 1N, H-7); 5,58 (д, J= 3,7 Гц, 1H, H-1'); 7,88 (м, 2H, H-1 + H-4); 8,34 (м, 2H, H-2 + H-3); 13,35 (с, 1H, OH-11); 13,62 (с, 1H, OH-6).
Получение 4'-O-метансульфонил-N,N-бис(2-хлорэтил)даунорубицина [R1=OCH3, R2=H, A=OSO2CH3, B=H, R3=R4=(CH2)2-Cl] (1j) и N,N-бис(2-хлорэтил)даунорубицина [R1=OCH3, R2=H, A=OH, B=H, R3=R4=(CH2)2-Cl] (1k)
N, N-бис(2-гидроксиэтил)даунорубицин [R1= OCH3, R2=H, A=OH, B=H, R3=R4= (CH2)2-OH] (0,155 г, 0,252 ммоль), полученный из даунорубицина (3a) в соответствии с процедурой, описанной в примере 4, растворяли в пиридине (3 мл) при температуре 0oC под слоем аргона и обрабатывали мезилхлоридом (0,15 мл, 1,94 ммоль). Через 15 минут реакционную смесь выливали в лед, содержащий 0,1 н.водный раствор HCl (50 мл), и экстрагировали метиленхлоридом (100 мл x 2). Органическую фазу промывали водой, сушили над безводным сульфатом натрия и концентрировали до небольшого объема при пониженном давлении. Эту смесь разбавляли ацетоном (10 мл), добавляли хлорид лития (10 мг) и в течение 4 часов выдерживали при температуре 35oC. После этого реакционную смесь экстрагировали метиленхлоридом и промывали водой. Органическую фазу сушили над безводным сульфатом натрия, концентрировали до небольшого объема и очищали посредством испарительной хроматографии на колонках из силикагеля, производя элюирование смесью метиленхлорида и ацетона (с объемным отношением 100/1), в результате чего были получены указанные в заголовке соединения 1j (45 мг) и 1k (53 мг).
Соединение 1j:
Тонкослойная хроматография на кизельгуровой пластине F254 (Merck) с использованием в качестве элюирующей системы смеси метиленхлорида и ацетона (с объемным отношением 95/5) Rf = 0,52.
Масс-спектр с частотным разделением: m/e (lj) 730.
Спектр 1H ЯМР (400 МГц, CDCl3), δ: 1,34 (д, J=6,4 Гц, 3H, CH3-5'); 1,74 (двойной дублет, J=4,7, 13,2 Гц, 1H, H=2'экв); 1,93 (дважды двойной дублет, J= 4,0, 13,2, 13,2 Гц, 1H, H-2'акс); 2,10 (двойной дублет, J=4,3, 15,0 Гц, 1H, H-8 акс); 2,34 (дважды двойной дублет, J=1,7, 2,1, 15,0 Гц, 1H, H-8 экв); 2,41 (с, 3H, COCH3); 2,8-3,1 [м, 5H, (NCH2CH2Cl)2 + H-3']; 2,98 (д, J= 18,8 Гц, 1H, H-10 акс); 3,22 (двойной дублет, J=1,7, 18,8 Гц, 1H, H-10 экв); 3,47 [m, 4H, (NCH2CH2Cl)2]; 3,64 (м, 1H, H-4'); 4,04 (к, J=6,4 Гц, 1H, H-5'); 4,08 (с, 3H, 4-OCH3); 4,59 (с, 1H, OH-9); 5,29 (двойной дублет, J=2,1, 4,3 Гц, 1H, H-7); 5,56 (д, J=4,0 Гц, 1H, H-1'); 7,39 (двойной дублет, J=1,3, 8,5 Гц, 1H, H-3); 7,78 (двойной дублет, J=8,1, 8,5 Гц, 1H, H-2); 8,03 (двойной дублет, J= 1,3, 8,1 Гц, 1H, H-1); 13,30 (с, 1H, OH-11); 14,01 (с, 1H, OH-6).
Соединение 1k:
Тонкослойная хроматография на кизельгуровой пластине F254 (Merck) с использованием в качестве элюирующей системы смеси метиленхлорида и ацетона (с объемным отношением 95/5) Rf = 0,26.
Масс-спектр с частотным разделением: m/e 652.
Спектр 1H ЯМР (400 МГц, CDCl3), δ : 1,33 (д, J=6,4 Гц, 3H, CH3-5'); 1,82 (двойной дублет, J=4,3, 13,2 Гц, 1H, H-2'экв); 2,06 (дважды двойной дублет, J= 3,8, 13,2, 13,2 Гц, 1H, H-2'акс); 2,14 (двойной дублет, J=3,8, 15,0 Гц, 1H, H-8 акс); 2,27 (дважды двойной дублет, J=1,7, 2,1, 15,0 Гц, 1H, H-8 экв); 2,40 (с, 3H, COCH3); 2,96 (д, J=19,2 Гц, 1H, H-10 акс); 3,04 [м, 4H, (NCH2CH2Cl)2] ; 3,15 (м, 1H, H-3'); 3,15 (с, 3H, CH3SO2); 3,24 (двойной дублет, J=1,7, 19,2 Гц, 1H, H-10 экв); 3,41 [м, 4H, (NCH2CH2Cl)2]; 4,09 (с, 3H, OCH3-4); 4,14 (к, J=6,4 Гц, 1H, H-5'); 4,37 (с, 1H, OH-9); 4,89 (м, 1H, H-4'); 5,29 (двойной дублет, J=2,1, 3,8 Гц, 1H, H-7); 5,59 (д, J=3,8 Гц, 1H, H-1'); 7,40 (двойной дублет, J=1,0, 7,7 Гц, 1H, H-3); 7,79 (двойной дублет, J= 7,7, 7,7 Гц, 1H, H-2); 8,04 (двойной дублет, J=1,0, 7,7 Гц, 1H, H-1); 13,29 (с, 1H, OH-11), 14,02 (с, 1H, OH-6).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 3'-АЗИРИДИНО-АНТРАЦИКЛИНА, СПОСОБЫ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2149163C1 |
| БИОЛОГИЧЕСКИ АКТИВНЫЕ ПОЛИМЕРСВЯЗАННЫЕ АНТРАЦИКЛИНЫ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2145965C1 |
| ПРОИЗВОДНЫЕ АЗА-АНТРАЦИКЛИНОНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2159245C2 |
| ПРОИЗВОДНЫЕ АНТРАЦИКЛИНОНА И ИХ ИСПОЛЬЗОВАНИЕ ПРИ АМИЛОИДОЗЕ | 1995 |
|
RU2167661C2 |
| СВЯЗУЮЩИЙ АГЕНТ ДЛЯ БИОАКТИВНЫХ ПРЕПАРАТОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1991 |
|
RU2116087C1 |
| АНТРАЦИКЛИН ГЛИКОЗИД И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1992 |
|
RU2081878C1 |
| АНТРАЦИКЛИНОВЫЙ ГЛИКОЗИД И СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 1990 |
|
RU2073681C1 |
| ПРОИЗВОДНЫЕ АНТРАЦИКЛИНА | 1995 |
|
RU2159619C2 |
| ФТОРИРОВАННЫЕ ПРОИЗВОДНЫЕ 17β-ЗАМЕЩЕННОГО -4-АЗА-5α-АНДРОСТАН-3-ОНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2125061C1 |
| АНТРАЦИКЛИНОВЫЕ ГЛИКОЗИДЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ДИИОДОПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1991 |
|
RU2100366C1 |



Антрациклиновый гликозид общей формулы I, в которой R1 - водород или метоксигруппа; R2 - водород или гидроксигруппа; A и B - оба водород или один из A или B - водород, а другой - гидроксигруппа или группа формулы - OSO2R5, где R5-C1-C4 - алкил, произвольно замещенный C1-C4-алкилом, нитро, амино, метокси или галоидом; R3 - атом водорода или группа формулы II и R4 - группа формулы II: -(СH2)n-X, где n = 2 или 3 и Х - гидроксигруппа, галоид или группа формулы - OSO2R5, где R5 такой, как определено выше, и при условии, что если R2, Х и А - гидроксигруппа и R3 - H, то n должно быть 3, или его фармацевтическая пригодная соль. Соединения изобретения проявляют противоопухолевую активность. Раскрываются способы их получения и фармацевтическая композиция, содержащая их. 3 с. и 5 з.п. ф-лы, 17 табл.
\ \ \ 1 1. Антрациклиновый гликозид общей формулы I \\\6 $$$ \\\1 где R<Mv>1<D> - водород или метоксигруппа; \\\4 R<Mv>2<D> - водород или гидроксигруппа; \\\4 A и B - один водород, а другой гидроксигруппа или группа общей формулы OSO<Mv>2<D>R<Mv>5<D>, где R<Mv>5<D> - C<Mv>1<D> - C<Mv>4<D>-алкил, необязательно замещенный C<Mv>1<D> - C<Mv>4<D>- алкилом, нитро, амино, метокси или галоидом; \\\4 R<Mv>3<D> - водород или группа формулы II \\\6 -(CH<Mv>2<D>)<Mv>n<D> - X; \\\1 где n = 2 или 3 и X - гидроксигруппа, галоид или группа формулы OSO<Mv>2<D>R<Mv>5<D>, где R<Mv>5<D> определен выше и при условии, что если R<Mv>2<D>, X и A являются гидроксигруппой и R<Mv>3<D> - водород, то n = 3; \\\4 R<Mv>4<D> - группа общей формулы II \\\1 или его фармацевтически приемлемая соль. \\\2 2. Соединение по п.1, где в формуле II X - хлор. \ \\2 3. Соединение по п.1, которое выбирают из (3-гидроксипропил)даунорубицина, N, N-бис-(3-гидроксипропил) даунорубицина, 4-деметокси-N-(2-гидроксиэтил)даунорубицина, N, N-бис-(2-гидроксиэтил)даунорубицина, N, N-бис-(2-гидроксиэтил) доксорубицина, 4-деметокси-N, N-бис(2-гидроксиэтил) даунорубицина, 4-деметокси-4'-O-метансульфонил-N, N-бис(2-хлорэтил) даунорубицина, 4-деметокси-N, N-бис(2-хлорэтил) даунорубицина, 4'-O-метансульфонил-N, N- бис(2-хлорэтил)-даунорубицина, N,N-бис(2-хлорэтил)даунорубицина, или их фармацевтически приемлемых солей. \\\2 4. Соединение по пп.1 - 3, которое представляет собой гидрохлоридную соль. \\\2 5. Способ получения антрациклинового гликозида формулы I по п.1, где R<Mv>3<D> и R<Mv>4<D> определены в п. 1, X - гидроксигруппа или оба R<Mv>3<D> и R<Mv>4<D> - группа формулы II, где X - гидроксигруппа или его соли, отличающийся тем, что включает (i) взаимодействие соединения формулы III \ \ \6 $$$ \\\1 где R<Mv>1<D> - водород или метоксигруппа; \\\4 R<Mv>6<D> - водород, гидрокси- или кислотно-чувствительная группа, \ \ \1 причем соединение формулы III растворяют в диметилформамиде с алкилирующим агентом общей формулы IV \\\6 X - (CH<Mv>2<D>)<Mv>n<D> Gаl, \\\1 где n = 2 или 3; \\\4 X - гидроксигруппа; \ \ \ 4 Gаl - бром, \\\1 при 20 - 30<198>C и при необходимости (ii) очистку образующихся антрациклиновых гликозидов формулы I на колонке с силикагелем с использованием в качестве элюента смеси хлористый метилен : метанол (80 : 20) и/или, при необходимости (iii) превращение антрациклинового гликозида формулы I в его хлористоводородную соль путем обработки посредством безводного хлористого водорода. \\\2 6. Способ получения антрациклинового гликозида формулы I по п.1, где оба R<Mv>3<D> и R<Mv>4<D> являются группой формулы II, где n = 2, X - гидроксигруппа, или его соли, отличающийся тем, что включает (i) растворение соединения общей формулы III по п.5 в смеси хлористый метилен - метанол и взаимодействие соединения формулы III с этиленоксидом в темноте при исходной температуре -40<198>C, последовательно повышая температуру до комнатной и поддерживая ее в течение 3 суток, и при необходимости (ii) очистку образующегося антрациклинового гликозида формулы I на хроматографической колонке и/или при необходимости (iii) превращение антрациклинового гликозида формулы I в фармацевтически приемлемую его кислотно-аддитивную соль. \\\2 7. Способ получения антрациклинового гликозида I формулы I по п.1, в котором один или оба R<Mv>3<D> и R<Mv>4<D> представляют собой группу формулы II, в которой X - хлор, A и B - водород или один из A или B является группой формулы OSO<Mv>2<D>R<Mv>5<D>, в которой R<Mv>5<D> - C<Mv>1<D> - C<Mv>4<D>-алкил или необязательно замещенный арил, или его соли, отличающийся тем, что включает (i) растворение антрациклинового гликозида формулы I по п.1, в котором один или оба R<Mv>3<D> и R<Mv>4<D> являются группой формулы II по п.1, в которой X - гидроксигруппа, в сухом пиридине и взаимодействие полученного раствора с сульфонилхлоридом общей формулы V \\\6 Cl-SO<Mv>2<D>R<Mv>5<D>, \\\1 в которой R<Mv>5<D> определен выше, \\\1 в темноте при 0<198>C и при поддержании этой температуры в течение 16 ч, и при необходимости (ii) очистку образующегося антрациклинового гликозида на хроматографической колонке и/или при необходимости (iii) выделение требуемого соединения в виде соответствующего гидрогалогенида. \\\2 8. Фармацевтическая композиция, обладающая противоопухолевой активностью, включающая ингредиент и фармацевтически пригодный разбавитель или носитель, отличающаяся тем, что в качестве активного ингредиента она содержит антрациклиновый гликозид формулы I или его фармацевтически приемлемую соль по п.1 в эффективном количестве.
| Машковский М.Д | |||
| Лекарственные средства | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| - М.: Медицина, 1993, с.458 - 460 | |||
| J.Med Chem | |||
| Пневматический водоподъемный аппарат-двигатель | 1917 |
|
SU1986A1 |
| EP, 0226173 A, C 07 H 15/252, 1987. | |||
