Изобретение относится к конъюгатам терапевтически применяемых антрациклинов с носителями, такими, как поликлональные и моноклональные антитела, или белки, или пептиды природного или искусственного происхождения, со способами их получения, с фармацевтическими композициями, содержащими эти конъюгаты, и с использованием этих композиций при лечении некоторых опухолей млекопитающих. Это изобретение также относится к новым производным антрациклина и к способам их получения.
В последние годы было синтезировано много цитотоксических антрациклинов. Например, антрациклины, имеющие морфолиновое кольцо или замещенное морфолиновое кольцо, связанные по положению C-3' фрагмента сахарозы, демонстрировали многообещающую противоопухолевую активность при воздействии на опухоли подопытных мурен (Bioactive molecules, 55 - 101, т. 6. Под ред. J. William Lown, Elveiser, 1983).
Изобретение связано с новыми связующими агентами для выделения терапевтически применяемых лекарственных веществ из биоактивных препаратов с целью улучшения терапевтического действия этих лекарств и уменьшения их токсического действия при введении в человеческий организм. Более конкретно эти биоактивные препараты включают в себя лекарственные вещества, имеющие первичные или вторичные гидроксильные группы и относящиеся к терапевтическим классам, таким, как антибиотики, противоопухолевые или противовирусные соединения, связанные с носителями, такими, как антитела, реакционноспособные относительно выбранной популяции клеток, или с белками, пептидами или полимерами природного или искусственного происхождения, реакционноспособными относительно рецепторных тканей.
Каждое лекарственное вещество, содержащее по крайней мере первичную или вторичную гидроксильную группу, ковалентно связано с носителем через отросток связующего агента и связано с этим отростком связующего агента через ацетальную связь, чувствительную к кислоте и находящуюся в положении его первичной или вторичной гидроксильной группы. Чувствительная к кислоте ацетальная связь биоактивного препарата изобретения делает возможным выделение активного лекарственного вещества в кислой внешней или внутренней окружающей среде "ткани-мишени".
В соответствии с этим изобретение представляет конъюгаты общей формулы I
[A - O - W - Z]a - T
где фрагмент A-O - остаток лекарственного вещества формулы A-O-H, в которой - O-H - первичная или вторичная гидроксильная группа; а - целое число от 1 до 30; W - группа общей формулы II:
где
b - целое число от 1 до 4, B - алкилен C1-C3, а R1 и R2 независимо - водород, галоген, алкил, фенил или замещенный фенил; Z - промежуточная группа, а T - фрагмент носителя.
Предпочтительным значением a являются целые числа от 1 до 5. Приемлемым значением для B является группа -/CH2/2-. При определении R1 и R2 типичным галогеном являются хлор, бром и йод, а алкилом может быть C1-C4 - алкил, такой, как метил или этил. Замещенным фенилом может быть галоген- или алкилзамещенный фенил, и в этом случае галоген и алкил могут быть такими же, как определенные выше. Предпочтительными группами Z являются:
(i) -NH);
(ii) -NH-/CH2/c-S-S-, где c - целое число от 1 до 4;
(iii) -NH-[C]d -N=CH-, где
a) d равно 0,
b) d равно 1, а [C] представляет собой -NH-CO/CH2/e-O-/CH2/e-, в котором e - целое число от 2 до 4,
c) d - целое число от 1 до 4, а [C] представляет собой -CH2/f-O-/CH2/f-, в котором f равно 1 или 2, или
d) d - целое число от 2 до 6, а [C] является CH2;
(iv) -NH-[C]d-NH-CO-, где [C] и d имеют значения, определенные выше;
(v) -[D]-NH-, где [D] представляет собой -NH-(CH2)g - CO-, в котором g - целое число от 2 до 6;
(vi) -[E]-CO-, где [E] представляет собой -NH-/CH2/g-NH-, в котором g - целое число от 2 до 6, или
(vii) пиперазинилкарбонильный фрагмент формулы: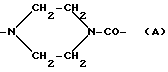
В формуле I лекарственное вещество A-O-H предпочтительно представляет собой противоопухолевый препарат, относящийся к классу антрациклинов, как, например, доксорубицин, его 3'-демино-3'-морфолинил-производное, в которых морфолиновое кольцо возможно замещено в положении 2'' алкоксильными остатками типа C1-C4-алкоксильных групп; аналоги пиримидина типа 5-фтордеоксиуридина или арабинофуранозилцитозина (цитарабина); производные винка алкалоидов типа 4-дезацетилвинилбластина; или другие противоопухолевые препараты типа подофиллотоксина или иллюдинов. С другой стороны, лекарственное вещество A-O-H может быть противовирусным препаратом, например, 3'-азидо-3'деокситимидин (AZT), бромвинилдеоксиуридин (BVDU), 9-[/2-оксиэтокси/метил] гуанин(ацикловир) или 9-[(1,3-диокси-2-пропокси)метил]гуанин(ганцикловир), или антибиотиком, например, тиенамицин или новые производные пенема, такие, как (5R, 6S)-2-карбамоилоксиметил-6-[(IR)-оксиэтил]-2-пенем, -3-карбоновая кислота и ацетоксиметил (5R, 6S)-2-карбомолилоксиметил-6-[/IR/-оксиэтил]-2-пенем-3-карбоксилат.
В тех случаях, когда фрагмент A-O- происходит из антрациклина A-O-H, то типичный фрагмент A-O- изображается формулой: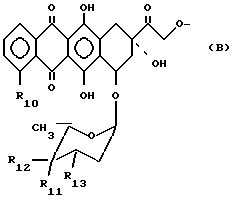
где
R10 - атом водорода или гидрокси- или метоксигрупа, один из R11 и R12 - атом водорода, а другой - гидроксильная группа, или R11 - атом водорода или йода, а R12 - атом водорода, R13 - аминогруппа или атом азота, заключенный в морфолиновом кольце. Морфолиновым кольцом может быть морфолиновое (MO), 3-циано-4-морфолиновое (CM) или 2-метокси-4-морфолиновое (MM) кольцо, в котором атом азота связан с C-3 следующим образом: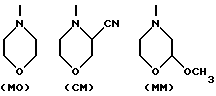
Фрагмент носителя T обычно выбирают из поликлонального антитела или его фрагмента, включающего в себя центр закрепления антигена, способный связывать опухолевоспецифический антиген; моноклонального антитела или его фрагмента, включающего в себя центр закрепления антигена, способный связывать антиген, предпочтительно или селективно выраженный в популяциях опухолевых клеток; пептида или белка, способного предпочтительного или селективно связываться с опухолевой клеткой; и полимерного носителя.
Фрагмент носителя, предпочтительно фрагмент носителя, происходящий из веществ, которое может быть представлено в виде T-[NH2]x, где x представляет собой число аминогрупп, пригодных для конденсации, может быть выбран из поликлональных антител, выращенных на фоне опухолевоспецифических антигенов, или из моноклональных антител, связанных с антигенами, предпочтительно или селективно выраженными в популяциях опухолевых клеток; или из природных или рекомбинантных пептидов, или белков, или факторов роста, предпочтительно или селективно связывающихся с клетками опухоли, или из природных или синтетических полимерных носителей, таких, как, например, полилизин. Часть носителя может быть также образована из частей указанных носителей, таких, как Fab или F(ab')2 фрагментов антител, или из частей вышеуказанных пептидов или белков, полученных посредством методов рекомбинантной ДНК.
Типичными примерами вышеупомянутых антител и соответствующими терапевтическими возможными применениями являются:
антитело к Т-клетке, такое, как антитело Т101 [Royston I и др., J. Immunol. 1980, 125, 725];
антитело к CD5, такое, как антитело ОКТ1 (Орто) ATCC CRL 8000 (хронические лимфоцитарные лейкозы);
антитело к трансферриновому рецептору, такое, как антитело ОКТЭ (Орто) АТСС CRL 8021 (опухоль яичника и другие опухоли);
антитело к меланоме, такое, антитело MAb 9.2.27 Bumol, T.F. и др., Proc. Natl. Acad. USA, 1982, 79, 1245 (меланомы);
антитела к сигнальному гену карциномы, такие, как: анти-CEA 116 NS-3 ATCC CRL 8019, анти альфа-фетопротеин ОМ 3-1.1 ATCC HB 134 (также гепатомы), 791T/36[Embleton, M. J. и др., Br. J. Cancer 1981, 43, 582] (также остеогенная саркома), B 72.3 [US-A-4 522 918 /1985/] (колоректальные карциномы и другие опухоли);
антитело к овариальной карциноме, такое, как антитело OVB 3 ATCC H B 9147;
антитело к карциноме молочной железы, такое, как антитело (HMGF антиген) [Aboud-Pirak, E. и др., Cancer Res. 1988, 48, 3188];
антитело к карциноме мочевого пузыря, такое, как антитело IG3.10 [Yu, D. S. и др., Eur. J. Urol. 1987, 13, 198.
Типичными примерами вышеупомянутых факторов роста или белков природного или рекомбинантного происхождения являются FGF, EGF, PDGF, TCF-α , α-MS , интерлейкины, интерфероны, TNF меланотропин (MSH) и т.д.
Фрагмент носителя, происходящий из вещества, которое может быть представлено в виде T-[SH]x1, где x1 обозначает число тионильных групп, доступных для конденсации, предпочтительно производят из тиолированных поликлональных или моноклональных антител, полученных с использованием, например, N-сукцинимидил-S-ацетилтиоацетата (SATA), N-сукцинимидил-3-(2-пиридилтио)пропионата (SPDP), D,L-гомоцитеин тиолактона, N-ацетил-D,L-гомоцистеин тиолактона или 2-аминотиолактона.
Фрагмент носителя, происходящий из вещества, которое может быть представлено в виде T-[CHO] x2, где x2 обозначает общее число формильных групп, доступных для конденсации, предпочтительно производят из моноклональных или поликлональных антител, содержащих углеводный фрагмент, предпочтительно находящийся в Fc-области, селективно окисленный до альдегидных групп, используя ферментативные или химические методы, как описано в US-A-4 671958. Носитель T-[CHO] x2 может быть также получен при формилировании или при окислении подходящих полимерных носителей, или при окислении до альдегидных групп углеводных остатков подходящих гликопротеинов.
Фрагмент носителя, происходящий из вещества, которое может быть представлено в виде T-[COOH]x3, где x3 обозначает общее число карбоксильных групп, доступных для конденсации, предпочтительно производят из полиаспаргиновой или полиглутаминовой кислоты или из растворимых или биоразлагаемых синтетических сополимеров типа тех, что образуются из N-(2-оксипропил)метакриламида (HMPA), имеющего следующее строение: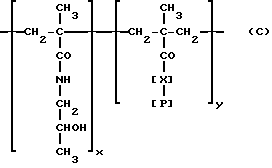
[X]: -Gly-Phe-Leu-Gly; -HN-(CH2)n-CO; n = 1 - 5.
[P]: OH, O-C6H4-n-NO2.
x/y /90/10 + 95/5 мол/мол.%; Mw 10000 - 40000, предпочтительно 12000 - 20000 (J., Kopecek. Biodegradation of polymers for biomedical use. B: IUPAC Macromolecules, H. Benoit and P. Rempp. Ed.: 505 - 520 (1982) Pergamon Press, Oxford, England] или из сополимеров полиаминокислот типа поли(GluNa, Ala, Tyr), или Mw 25000 - 50000 дальтонов, которые используют в качестве способных быть мишенью лекарств-носителей для тканей легкого (приводимые ниже) (R. Duncan и др. Journal of Bioactive and Compatible Polymers, т. 4, 1989), или из карбоксильных групп, естественно присутствующих в моноклональных или поликлональных антителах, или химически введенных обработкой антитела бифункциональными ангидридами (например, малеиновым ангидридом).
Структура поли(Glu Na, Ala, Tyr)сополимера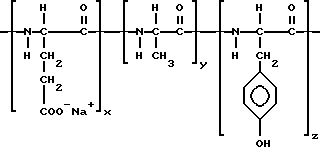
x : y : z = 1 : 1 : 1.
Кроме того, изобретение предлагает способ получения коньюгата формулы I, который включает в себя конденсацию производного формулы III,
A-O-W-OH
где
A-O- и W определены выше, с веществом или веществами, которое (которые) способно (способны) предоставить указанную промежуточную группу Z и фрагмент носителя T в указанный конъюгат формулы I, при этом образуется указанный конъюгат формулы I.
Конденсация может быть проведена через активированное производное, такое, как смешанный ангидрид, азид или активированный сложный эфир производного формулы III, или прямой реакцией в присутствии конденсирующего агента, такого, как дициклогексилкарбодиимид. Соответствующий способ включает в себя превращение производного формулы III в активированное производное формулы IV
A-O-W-L
где
A-O- и W имеют значения, указанные выше, а L представляет собой активированную группу для создания амидной связи, группу типа N-оксисукционоимидной, ее водорастворимого производного-N-оксисульфосукцинимидной, или является 2,4-динитрофенокси-, 2,3,4,5,6-пентафторфенокси- или третбутоксикарбонилокси-группой; и
(i') конденсацию полученного соединения формулы IV с веществом формулы T-[NH2] x, определенным выше, с получением биоактивного препарата формулы I, имеющего амидную связь (связи) или
(ii') конденсацию соединения формулы IV с тиопроизводным формулы NH2-(CH2)-SH, таким, как 2-аминоэтантиол, в котором с = 2, и конденсацию получающегося соединения формулы V
A-O-W-NH-(CH2)c - SH
где
A-O-, W и с определены выше, с веществом формулы T-[SH]x1, определенным выше, с получением конъюгата формулы I, имеющего дисульфидную связь (связи); или
(iii') взаимодействие соединения формулы IV с гидразином, сукциновым или адипиновым производным дигидразида, или с диаминосоединением и конденсацию получающегося соединения формулы VI
A-O-W-NH[C]d-NH2
где
A-O-, W, C и d определены выше, с веществом формулы T-[CHO]x2, определенным выше, с получением биоактивного препарата формулы I, имеющего оксимную связь (связи).
приемлемый способ (iv') включает конденсацию соединения общей формулы VI, описанного выше, с веществом формулы VII
T[CO-L']y
которое получается в результате реакции, затрагивающей носитель формулы T-[COOH] x3, определенный выше, и в котором у является целым числом от 1 до 30 и представляет собой общее количество активированных карбоксильных групп, T является фрагментом носителя в получающемся конъюгате формулы I, а L' представляет собой гидроксил или активированную группу для создания амидной связи, возможно в присутствии конденсирующего агента с получением конъюгата формулы I, в котором возможно присутствующие непрореагировавшие активированные карбоксильные группы могут быть подавлены фармацевтически приемлемым амином, таким, как 1-амино-2-пропанол.
Другие приемлемые способы включают (v') конденсацию соединения формулы IV с аминопроизводным формулы H2N-[CH2]g-COOH, в котором g определено выше, при этом образуется производное формулы VIII
A-O-W-[D]-OH
где
A-O-, W и [D] определены выше; возможно превращение соединения формулы VIII в активированное производное формулы IX
A-O-W-[D]-L
где
A-O-, W, [D] и L определены выше, и конденсацию получающегося соединения IX или соединения формулы VIII в присутствии конденсирующего агента с веществом формулы T-[Nh2] x, определенным ранее, с образованием биоактивного препарата формулы I.
Другие способы включают в себя (vi') взаимодействие соединения формулы IV с аминопроизводным формулы H2N-(CH2)g-NH2, в котором g определено выше, при этом получают производное формулы X
A-O-W-[E]-H
где
[E] определен выше, или
(VII') взаимодействие соединения формулы IV с пиперазином с образованием производного формулы XI
A-O-W[пиперазин]-H
и конденсацию соединения формулы X или XI, приведенных выше, с ранее описанным соединением формулы VII с получением конъюгата формулы I.
Например, способ активации или превращения соединения формулы III или VIII в производное IV или IX заключается во взаимодействии соединения формулы III или VIII с N-оксисукцинимидом или с его водорастворимой солью - 3-замещенным сульфонатом натрия - в присутствии N, N'дициклогексилкарбодиимида в среде растворителя, такого, как этилацетат или N, N'диметилформамид. В таком случае в формулах IV и IX L представляет собой остаток: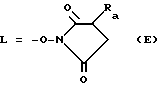
где
Ra представляет собой водород или натрийсульфатную группу.
Способ активации для превращения носителей вида T-[COOH]x3 в соединения общей формулы VII заключается во взаимодействии такого носителя с пара-нитрофенолом в присутствии N,N'дициклогексилкарбодиимида в среде растворителя, такого, как диметилформамид, в этом случае L' представляет собой пара-NO2-C6H5O-; или с N-этоксикарбонил-2-этокси-1,2-дигидрониноном (EEDQ) (B. Belleau и др., JACS, 90, 1651 (1968) в среде растворителя, такого, как тетрагидрофуран или диметилформамид, в этом случае в формуле VII L' представляет собой остаток:
L'= -OCOOC2H5 (F)
Другая методика включает взаимодействие щелочной соли носителя T-[COOH] x3, типа натриевой соли, с аклкилгалоид-карбонатом, таким, как (C1-C4)алкил-галоид-карбонат предпочтительно с этилхлоркарбонатом (C2H5O-COCl), в среде растворителя, таком, как вода или диметилформамид. В этом случае в формуле VII остаток L' представляет собой -OCOOC2H5.
Методики конденсаций для получения конъюгатов формулы I, исходя из производного формулы III или VIII и носителя формулы T-[NH2]x, осуществляют в условиях, допускающих образование ковалентных связей амидного типа и совместимых со структурой этого носителя. В соответствии с химической стабильностью носителя в водных или органических растворителях для проведения реакции сочетания с промежуточными соединениями общих формул IV, V, VI, IX, X и XI, могут быть использованы различные методики. Для носителей, чувствительных к органическим растворителям, предпочтительные условия заключаются в использовании водных растворов, содержащих буфер, при pH 7 - 9,5, реакцию проводят при 4 - 37oC в течение нескольких часов или в течение нескольких дней.
Методики получения конъюгатов формулы I конденсацией производного 5 с носителем формулы T-[SH]x1 осуществляют в условиях, включающих использование содержащих буфер водных растворов при pH 6-7, при температуре 4oC и времени реакции от нескольких часов до нескольких дней.
Методики получения конъюгатов формулы I конденсацией производного VI с носителем формулы T-[CHO]x2 осуществляют в условиях, допускающих образование ковалентных связей оксимного или гидразонного типа и совместимых со структурой носителя. Предпочтительные условия включают в себя использование содержащих буфер водных растворов при pH 4 - 7,5, температура реакции составляет 4 - 37oC, а время реакции - от нескольких часов до нескольких дней.
Методики получения конъюгатов формулы I конденсацией производного VI, X или XI с носителем формулы T-[CO-L']y осуществляют в условиях, допускающих образование ковалентных связей амидного типа и совместимых со структурой носителя. Предпочтительные условия включают использование содержащих буфер водных растворов при pH 7 - 9,5, реакцию проводят при 4 - 37oC в течение нескольких часов или нескольких дней.
Другие условия включают использование обезвоженных диметилсульфоксида или диметилформамида, реакцию проводят при комнатной температуре в течение 1 - 3 ч.
В данном случае промежуток времени "от нескольких часов до нескольких дней" может составлять от 4 часов до 5 дней.
Например, подходящими условиями для конденсации соединений формулы IV и IX и антител T-[NH2]x являются: водный 0,1 М раствор фосфата натрия и водный 0,1 М раствор хлорида натрия при pH 8, содержащие моноклональное тело (концентрация 1 мг/мл), обрабатываемые при 20oC в течение 24 ч 30-кратным мольным избытком 10 мас.% об. раствора IV или IX в N, N-диметилформамиде. Конъюгат очищают иль-фильтрацией на колонке Sephadex G-25 Pnarmacia Fine Chemical, Piscataway, N, H), элюируя PBS (солевым раствором, содержащим фосфатный буфер).
Подходящими условиями для сочетания соединений формулы V и функционализированных антител, содержащих тиогруппы, являются: водный 0,1 М раствор ацетата натрия и водный 0,1 М раствор хлорида натрия при pH 6, содержащие моноклональное антитело (концентрация 1 мг/мл), обрабатываемые 30-кратным мольным избытком 5 мас.%/об. раствора соединения V в том же буфере в течение 24 ч при 4oC. Конъюгат очищают гель-фильтрацией, как описано выше.
Подходящими условиями для сочетания соединений формулы VI и носителей типа T-[CHO] x2 являются: новый 0,1 М раствор ацетата натрия и водный 0,1 М раствор хлорида натрия при pH 6-7, содержащие моноклональное антитело (концентрация 1 мг/мл), обрабатываемые при 20oC в течение 24 ч 30-кратным мольным избытком 5 мас.%/об. раствора соединения VI в том же буфере. Конъюгат очищают гель-фильтрацией, как описано выше.
Подходящими условиями для конденсации соединений формул VI, X и XI и активированного носителя формулы VII являются: безводный полярный растворитель, такой, как диметилформамид, содержащий 5 - 50 мг/мл соединения VI, X или XI, обрабатываемый при 20oC в течение 1 - 24 ч эквивалентным количеством соединения VII.
Соединения общей формулы III, а также активированные соединения формулы IV, V, VI, IX, X и XI являются новыми соединениями и, следовательно, относятся к сфере притязаний изобретения.
Соединения III, V, VI, X и XI являются как полезными промежуточными соединениями, так и/или терапевтически активными препаратами. Более конкретно промежуточные соединения общей формулы III являются полезными пролекарствами соединений общей формулы A-O-H, описанных ранее. Производные формулы III могут быть получены способом, проиллюстрированным на схеме I. Этот способ включает концентрацию лекарственного вещества формулы A-O-H с подходящими энолэфирным производным, имеющим скрытую карбоксильную группу, таким, как производное формулы VII: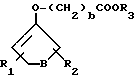
где
B, b, R1 и R2 определены ранее, а R3 представляет собой защитную группу, такую, как метил или этил; и удаление защитной группы из получающегося соединения.
Схема 1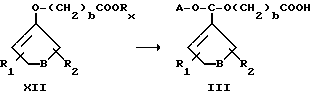 .
.
Не ограничивая изобретение, противоопухолевые антрациклины представляют собой гидроксилированные лекарственные вещества (A-OH), пригодные для изготовления биоактивных препаратов общей формулы I.
Из класса антрациклинов 3'-деамино-3'-(2-метокси-4-морфолинил) -доксорубицин (XIII b) или 3'-деамино-3'-(4-морфолинил)доксорубицин (XIII) (E.M. Acton и др. Morpholinyl Anthracyclines и Bioactuive Molecules т. 6. /Под ред. J. W. Lown, Elsevier, 1988, представляют собой лучшие соединения.
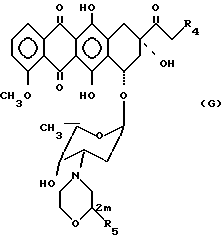
A-OH=XIIIa: R4=OH, R5=H A-O-=XIIIa': R4=O-, R5=H.
A-OH=XIIIb: R4=OH, R5=OCH3 A-O-=XIIIb': R4=O-, R5=OCH.
Превращение антрациклинов формулы XIIIa, b в новые чувствительные к кислоте производные общей формулы III, определенные выше, можно осуществить обработкой соединением формулы XII, проиллюстрированным ранее, в котором Rx представляет собой остаток, такой, как этильная группа, полученным по методике, описанной J. A. Landgrebe и др. в J. Org. Chem. 43, 1244 (1975), взаимодействием подходящего соединения, имеющего кетогруппу, с этилдиазоацетатом. Предпочтительными условиями для проведения реакции антрациклинов, а также и других соединений, имеющих первичные или вторичные гидроксильные группы, с соединением XII являются использование сульфокислотного катализатора, такого, как пара-толуолсульфокислота или камфарсульфокислота, в полярном растворителе, таком, как диметилформамид, реакцию проводят при комнатной температуре, время реакции составляет от нескольких часов до одного дня. Скрытую карбоксильную группу кислотно-чувствительного фрагмента, связанного с антрациклинами в соединениях формулы III, превращают в активированную карбонильную группу соединений формулы IV, из которых путем соединения с подходящими носителями получают новые биоактивные препараты. Возможным вариантом является превращение производных антрациклина общей формулы IV в производные общей формулы V, VI, VIII, IX, X и XI и их взаимодействие с подходящими носителями с получением новых биоактивных препаратов общей формулы I.
Терапевтически применяемые лекарственные соединения типа тех, что писаны ранее, могут реагировать таким же образом, как описано для антрациклинов, с соединениями формулы XII и превращаться в биоактивные препараты для лечения некоторых заболеваний млекопитающих.
Биоактивные препараты формулы I изобретения являются терапевтически полезными препаратами, поскольку они содержат ацетальную связь, которая при гидролизе, катализируемым ионом оксония, или при расщеплении in vivo под действием ферментов выделяет исходное лекарственное соединение A-OH. Например, хорошо известно, что в злокачественных опухолях наблюдается по сравнению с обычной тканью высокая скорость гликолиза. Это приводит к повышенному образованию лактата и, следовательно, к уменьшению pH в опухоли (H. M. Rauen и др., Z. Naturforsch, Teil B, 23 (1968) 1461).
Конъюгаты, полученные по описанным способам, были охарактризованы различными химико-физическими методами. Например, сохранение первоначального молекулярного веса и отсутствие образования агрегатов оценивали методами хроматографической гель-фильтрации (Yu, D. S. и др., J. Urol. 1430, 145, 1988) с одновременным и независимым определением антрациклина и антитела при различных длинах волн и методами электрофореза в геле. Общее распределение заряда полученных соединений оценивали методами ионообменной хроматографии. Концентрацию антрациклина определяли спектрофотометрическим титрованием на фоне стандартной калибровочной кривой, полученной с исходным антрациклином. Концентрацию белка определяли колориметрическими исследованиями, такими, как анализ бицинконовой кислотой (Smith P. K. и др., Anal. Biochem. 150, 76, 1985) или анализ красителем Bradford (Bradford, M.M., Anal. Biochem. 72, 243, 1976). Сохранение активности связывания антигенов антител после процессов соединения оценивали методом ELISA (Yu, D.S. и др., J. Urol. 140, 415, 1988) и цитофлюориметрическими методами (Calledo J. и др., Int. J. Cancer 33. 737, 1984). Оценку сохранения цитотоксичности конъюгатов по сравнению с исходным лекарственным соединением проводили исследованием ингибирования "клетками-мишенью" 3H-тимидина после инкубационного периода, достаточно длительного для проявления максимального цитотоксического эффекта (Dillmann, R.O. и др., Cancer Res. 48, 6097, 1988).
Оценку селективной цитотоксичности конъюгатов по отношению к позитивному антигену по сравнению с отношением к клеточной линии негативного антигена проводили, исследуя ингибирование поглощения 3H-тимидина позитивным антигеном относительно клеточных линий негативного антигена после короткого инкубационного периода (Dillmann R.O. и др., Cancer Res. 48, 6096, 1988).
Чувствительность конъюгата к кислоте оценивали вышеупомянутыми хроматографическими методами после инкубации соединений в подходящих буферных растворах.
С другой стороны, для оценки стабильности в плазме конъюгаты метили радиоактивным изотопом в фрагменте антитела (225I) и/или в фрагменте антрациклина (14C) и использовали аналитические методы жидкостной хроматографии высокого давления (HPLC).
Биологическая активность.
Воздействие соединения III' на клетки Lovo (аденокарциномы толстой кишки человека) и на Lovo-Doxo-резистентные (Lovo) (DX) клетки исследовали in vitro в сравнении с доксорубицином и 3'-деамино-3'[2(S)-метокси-4-морфолинил] доксорубицином (XIII), используя метод пластинчатого посева отдельных изолированных клеток через 4 ч после обработки (анализ колонии клеток). Концентрацию 50% ингибирования (IC50) вычисляли по кривым концентрация - восприимчивость. Данные, представленные в табл. 1, свидетельствуют о том, что соединение III' более цитотоксично, чем доксорубицин, как по отношению к чувствительным, так и к резистентным клеточным линиям, но в 15 - 20 раз менее цитотоксично, чем его исходное соединение XIIIb. Соединение III было испытано in vivo на CDP-1 мышах с P388-доксорубицин-резистивным лейкозом (P388/DX Johnson) в сравнении с доксорубицином и соединением XIIIb. Данные, представленные в табл. 2, свидетельствуют о том, что соединение III, введенное внутрибрюшинно через 1 день после заражения опухолью (доза 0,22 мг/кг), является очень активным препаратом (T/C% 184).
Конъюгаты формулы Ii, Iii, Iv, Ivi и Ivii (полученные соответственно в примерах 5, 6, 9, 11 и 12), в которых остаток цитотоксичного лекарственно вещества 3'-деамино-3'-[2(S)-метокси-4-морфолинил]доксорубицин привязан через кислотно-чувствительную связь, испытывали in vivo при воздействии на P388/DX Johnson лейкоз в сравнении с доксорубицином и несвязанным лекарственным веществом. Данные, приведенные в табл. 3, свидетельствуют о том, что все новые соединения, введенные внутривенно через 1 день после заражения опухолью, сохраняют активность, равную активности несвязанного лекарственного вещества XIIIb. Терапевтический индекс, выраженный как отношение между LD50-100 и оптимальной дозой, для всех конъюгантов выше, чем для свободного лекарственного вещества XIIIb'.
В табл. 4 представлены t50% выделения 3'-деамино-3'-[2(S)-метокси-4-морфолин] доксорубицина XIIIb из конъюгантов при 37oC и pH 3 (ацетатный буфер).
Терапевтическое действие этих соединений и улучшение их терапевтического действия по сравнению с исходным соединением оценивали на образцах животных с трансплантированными опухолями человека. Лишенных волос мышей с ксенотрансплантантами человеческих опухолей лечили подходящими эквивалентными дозами конъюгатов, свободного лекарственно соединения, антител и физической смесью лекарственного соединения и антител, регистрировали рост опухоли и проводили сравнение между различными группами, которые были подвергнуты лечению.
Конъюгаты приготовляли в виде фармацевтических композиций вместе с фармацевтически приемлемым носителем или разбавителем. Может быть использован любой подходящий носитель или разбавитель. Приемлемые носители и разбавители включают физиологический солевой раствор и декстрозный раствор Ringer.
Следующие примеры иллюстрируют изобретение, но не ограничивают его.
Пример 1. Получение 14-0-(1-карбоксиметилокси-циклогексил)-3'-диамино-3'-[2(S)-метокси- -4-морфолинил]доксорубицина [III; A-O- = XIIIb, b = 1, B = -(CH2)2-, R1 = R2 = H].
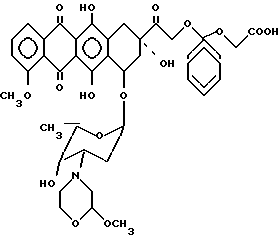
3'-деамино-3'-[2(S)-метокси-4-морфолинил] -доксорубицин (XIIIb) (0,68 г, 1 ммоль), полученный по методике, описанной в заявке Великобритании N 8928654.6, растворяли в 20 мл безводного диметилформамида и обрабатывали в присутствии 50 мг расплавленной пара-толуолсульфокислоты 6 г (32 ммоль) этил 2-(циклогексен-1-ил)-оксиацетата [XII'; b = 1, R3 = C2H5, B = -(CH2)2-, R1 = R2 = H] . Эту смесь перемешивали при комнатной температуре в течение двух часов, после чего выливали в водный раствор бикарбоната натрия и экстрагировали хлористым метиленом. Органический слой промывали водой, сушили над безводным cульфатом натрия и фильтровали. Растворитель удаляли при пониженном давлении, а остаток хроматографировали на испарительной колонке с кремневой кислотой, используя в качестве элюирующей системы смесь хлористого метилена/метанола (98/2 по об. ) и получали эфирное производное (R3 = C2H5) соединения III', обозначенного в заглавии.
Это производное обрабатывали в течение 1 ч при температуре 0oC и при перемешивании в присутствии азота 200 мл 0,1 N водного раствора гидроокиси натрия. После чего pH смеси доводили уксусной кислотой до 8,2 и экстрагировали н-бутанолом. Органический слой промывали водой, концентрировали при пониженном давлении до малого объема и хроматографировали на колонке с кремневой кислотой, используя в качестве элюирующей системы смесь хлористого метилена/метанола (8-/20 по об.), с получением соединения III', обозначенного в заглавии, в виде свободнокислотного производного (0,35 г, выход 43%), которое обрабатывали эквивалентным количеством водного раствора соляной кислоты и лиофилизировали.
Тонкослойная хроматография на пластинке с кизельгуром (Merck) F254, элюирующая система хлористый метилен/метанол (95/5 по об.),
Rf = 0,13, MS (масс-спектрометрия) - FD : m/e 783 (M+).
1ЯМР (200 МГц, DMCO d6 δ :
1,12 (дублет, J = 6,4 Гц, 3H, CH3 - 5'); 1,3 - 1,9 (мультиплент, 12H, 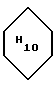 , CH2 - 2');2,0 - 2,7 (мультиплет, 7H, CH2-8, O-CH2CH2-N, O-CH-CH2-N, H-3'); 2,94 (синглет, 2H, CH2-10); 3,24 (синглет, 3H, CH-OCH3); 3,40, 3,73 (два мультиплета, 2H, NCH2CH2O); 3,57 (мультиплет, 1H, H-4'); 3,91 (синглет, 2H, OCH2COOH); 3,97 (синглет, 3H, OCH3-4); 4,04 (двойной квадруплет, J = 6,4, 1,0 Гц, 1H, H-5'); 4,35 (двойной дуплет, J =2,2, 5,2 Гц, 1H, OCH-OCH3); 4,61 (синглет, 2H, CH2- 14); 4,92 (мультиплет, 1H, H - 7); 5,24 (мультиплет, 1H - 1); 7,6 - 7,9 (мультиплет, 3H, H-1, H-2, H - 3); 13,20 (широкий синглет, 1H, OH-11); 14,02 (синглет, 1H, OH - 6).
, CH2 - 2');2,0 - 2,7 (мультиплет, 7H, CH2-8, O-CH2CH2-N, O-CH-CH2-N, H-3'); 2,94 (синглет, 2H, CH2-10); 3,24 (синглет, 3H, CH-OCH3); 3,40, 3,73 (два мультиплета, 2H, NCH2CH2O); 3,57 (мультиплет, 1H, H-4'); 3,91 (синглет, 2H, OCH2COOH); 3,97 (синглет, 3H, OCH3-4); 4,04 (двойной квадруплет, J = 6,4, 1,0 Гц, 1H, H-5'); 4,35 (двойной дуплет, J =2,2, 5,2 Гц, 1H, OCH-OCH3); 4,61 (синглет, 2H, CH2- 14); 4,92 (мультиплет, 1H, H - 7); 5,24 (мультиплет, 1H - 1); 7,6 - 7,9 (мультиплет, 3H, H-1, H-2, H - 3); 13,20 (широкий синглет, 1H, OH-11); 14,02 (синглет, 1H, OH - 6).
Пример 2. Получение N - оксисукцинимидпроизводного 14-0-(1-карбоксиметилокси-циклогексил)-3'-деамино-3'-[2(S)- метокси-4-морфолинил]доксорубицина [IV; A-O-=XIIIb, b=1, B=-(CH2)2-, R1 = R2 = H,L= 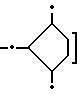
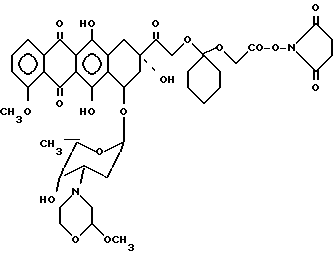
Соединение III (0,2 г 0,25 ммоль), полученное по методике, описанной в примере 1, растворяли в 8 мл безводного диметилформамида, охлаждали до 0oC и обрабатывали 0,2 г N-оксисукцинимида и 0,4 г дициклогексилкарбодиимида. Смесь выдерживали при 0oC в течение двух часов, а затем оставляли на ночь при комнатной температуре. После этого смесь концентрировали при пониженном давлении до малого объема и хроматографировали на испарительной колонке с кремневой кислотой, использяуя в качестве элюирующей системы смесь хлористого метилена/метанола (97/3 по об.). Элюат, содержащий соединение, указанное в заглавии, концентрировали досуха при пониженном давлении, затем остаток поглощали этилацетатом, фильтровали и концентрировали при пониженном давлении до малого объема. В заключение добавляли этиловый эфир и 0,130 г соединения IV', указанного в заглавии, собирали на воронке из спекшего стекла, промывали этиловым эфиром и хранили в атмосфере азота.
Тонкослойная хроматография на пластинке с кизельгуром (Merck)F254, элюирующая система хлористый метилен/метанол (95/5 по об.), Rf = 0,37, MS - FD: m/e 864 (M+).
Пример 3. Получение 14-0-(1-гидразинокарбонилметилокси-циклогексил)-3'-деамино-[2(S)- метокси-4-морфолинил]доксорубицина [VI, A-O-=XIIIb', b=1, B=-(CH2)2-, R1=R2=H,C=0].
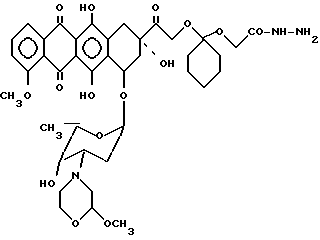
20 мг соединения IV', полученного по методике, описанной в примере 2, растворяли в 5 мл тетрагидрофурана и добавляли к 1 М раствору гидразингидрата в изопропаноле. Смесь выдерживали при 0oC в течение 70 мин. После этого добавляли хлористый метилен и смесь трижды промывали холодной водой. Органический слой отделяли, сушили над безводным сульфатом натрия, фильтровали и отгоняли в вакууме растворитель. Остаток хроматографировали на колонке с кремневой кислотой, используя в качестве элюирующей системы смесь хлористого метилена/метанола (99/1 по об.). Соединение VI', 20 мг, осаждали этиловым эфиром.
Тонкослойная хроматография на пластинке с кизельгуром (Merck) F254, элюирующая система хлористый метилен/метанол (95/5/ по об.).
Rf=0,25; MS - PO : m/e 797 (M+).
1ЯМР (200 МГц, DMCO d6) δ :
1,36 (дублет, J = 6,6 Гц, 3H, CH3 - 5'); 1,4 - 1,7 (мультиплет, 1OH,  ; 1,76 (мультиплет, 2H, CP2 - 2'); 2,30 (мультиплет. 2H, CH2 - 8); 2,50 (мультиплет. 1H, H-3'); 2,55 (мультиплет, 4H, N - CH2 - CH(OCH3), N-CH2CH2-0); 2,93 (дублет, J =18,8 Гц, 1H, H-10 ах); 3,22 (двойной дублет, J= 1,0, 18,8 Гц, 1H, H-10e); 3,39(синглет, 3H, CH/OCH3); 3,55, 3,95 (два мультиплета, 2H, NCH2CH2O); 3,70 (мультиплет, 1H, H -4); 3,95 (мультиплет. 1H, H - 5'); 4,09 (синглет, 3H, CH3 - O - 4); 4,10 (мультиплет, 2H, O-CH2 CONH); 4,50 (мультиплет, 1H, NCH2-CH(OCH3)), 4,67 (синглет, 2H,CH2-14); 5,28 (мультиплет, 1H, H-7); 5,54 (мультиплет, 1H, H - 1); 7,64 (широкий синглет, 1H, CONHNH2); 7/78 (триплет, J = 7,9 Гц, 1H, H - 2); 8,04 (дублет, J = 7,9 Гц, 1H, H-1); 13,32 (синглет, 1H, OH -II); 13,98 (синглет, 1H, OH - 6).
; 1,76 (мультиплет, 2H, CP2 - 2'); 2,30 (мультиплет. 2H, CH2 - 8); 2,50 (мультиплет. 1H, H-3'); 2,55 (мультиплет, 4H, N - CH2 - CH(OCH3), N-CH2CH2-0); 2,93 (дублет, J =18,8 Гц, 1H, H-10 ах); 3,22 (двойной дублет, J= 1,0, 18,8 Гц, 1H, H-10e); 3,39(синглет, 3H, CH/OCH3); 3,55, 3,95 (два мультиплета, 2H, NCH2CH2O); 3,70 (мультиплет, 1H, H -4); 3,95 (мультиплет. 1H, H - 5'); 4,09 (синглет, 3H, CH3 - O - 4); 4,10 (мультиплет, 2H, O-CH2 CONH); 4,50 (мультиплет, 1H, NCH2-CH(OCH3)), 4,67 (синглет, 2H,CH2-14); 5,28 (мультиплет, 1H, H-7); 5,54 (мультиплет, 1H, H - 1); 7,64 (широкий синглет, 1H, CONHNH2); 7/78 (триплет, J = 7,9 Гц, 1H, H - 2); 8,04 (дублет, J = 7,9 Гц, 1H, H-1); 13,32 (синглет, 1H, OH -II); 13,98 (синглет, 1H, OH - 6).
Пример 4. Получение 14-0-[1-4/4-карбокси-1-н-бутил/карбамоилметил-окси-1-циклогексил/] -3'- деамино-3'-[2/S/-метокси-4-морфолинил]доксорубицина [VIII, A-O-=XIIIb', b=1, B=-(CH2)2-, R1=R2=H, g=4].
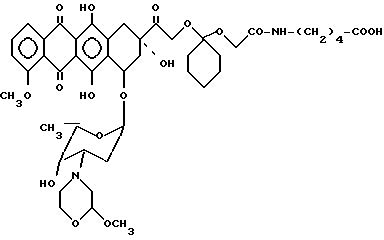
15 мг (0,15 ммоль) γ - аминомасляной кислоты растворяли в 2,5 мл 0,05 М фосфатного буфера с pH 7,6 и добавляли к соединению IV (30 мг 0,033 ммоль), полученному по методике, описанной в примере 2, растворенному в 3 мл ацетонитрила. Смесь 0выдерживали при комнатной температуре в течение ночи, затем доводили ее pH до 6, добавляя уксусную кислоту, и экстрагировали н-бутанолом. Органический слой отделяли и испаряли в вакууме. Остаток хроматографировали на колонке с кремневой кислотой, используя в качестве элюирующей системы смесь хлористого мтилена/метанола (95/5 по об.) и получали 20 мг соединения VIII', указанного в заглавии.
Тонкослойная хроматография на пластинке с кизельгуром (Merck) F254, элюирующая система хлористый метилен/метанол (95/1 по об.), Rf = 0,55, MS - FD:m/e 884 (M+).
Пример 5. Gолучение конъюгата формулы I', содержащего полиглутаминовую кислоту [I': A-O- = XIIIb, B = -(CH2)2-, R1=R2=H, Z = -NH-NH-CO-, a = 2, T = поли-L-глутаминовая кислота, b = 1.
85 мг поли-L-глуутаминовой кислоты, молекулярный вес 2000 - 15000 (Sigma) и 20 мг соединения VI', полученного по методике, описанной в примере 3, растворяли в 2 мл диметилформамида и перемешивали в течение трех часов. После этого добавили 25 мг N-этоксикарбонил-2-этокси-1,2-дигидрохинолина. Смесь перемешивали в течение ночи, а затем выливали в смесь, состоящую из этилового и петролейного эфира. Осадок собирали на фильтре из спекшегося стекла, промывали этиленовым эфиром и растворяли в 8 мл 2,5-ного водного раствора бикарбоната натрия. Раствор пропускали через колонку с обращенной фазой RP-8, 40 - 63 мкм (Merck) (30•1,8 см) и элюировали смесью воды и ацетонитрила, содержащей 0 - 20% ацетонитрила. Элюат, содержащий конъюгат, лиофилизировали, затем собирали на фильтре из спекшегося стекла, промывали метанолом и этиловым эфиром и получали 45 мг соединения I', обозначенного в заглавии. По данным метода спектроскопии этот конъюгат содержит 13% метоксиморфолинового соединения формулы XIIIb.
Пример 6. Получение конъюгата формулы I'', содержащего поли (GluNa, Ala, Tyr) [I'' : A-O- = XIIIb', b = 1, B = -(CH2)2-, R1=R2=H, Z = -NH-NH-CO-, a = 2, T = поли (GluNa, Ala, Tyr)].
По методике, описанной в примере 5, 60 мг поли (GluNa, Ala, Tyr) (1 : 1 : 1), MW 25000 - 50000 (Sigma) и 20 мг соединения VI', полученного в примере 3, растворяли в 2 мл диметилформамида, перемешивали в течение трех часов, обрабатывали 25 мг N-этоксикарбонил-2-этокси-1,2-дигидрохинолина и получали после очистки на колонке с обращенной фазой 35 мг соединения I'', указанного в заглавии.
Пример 7. Получение противомолекулярного конъюгата формулы I''' [I''' : A-O- = XIIIb', b = 1, B = -(CH2)2-, R1 = R2 = H, Z = -NH-, T = EpI].
10-2 М раствор соединения IV', описанного в примере 2, в N, N- диметилформамиде (37,5 моль) постепенно добавляли при комнатной температуре и перемешивали к 1 мл раствора (концентрация 2 мг/мл) очищенного мышиного моноклонального антитела к человеческой меланоме EpI (Giacomini, P. и др., Int. J. Cancer. 39, 729 (1978)) в 0,1 М NaH2PO4 и 0,1 М NaCl, pH 8. Реакционную смесь перемешивали в темноте при комнатной температуре в течение ночи и центрифугировали. Конъюгат выделяли гель-фильтрационной хроматографией на колонке Sephadex - G25 (PD-10, Pharmacia), элюируя PBS (Gibco, 10X, Cat. N. 042.04200 М) pH 7,3. Единственный пик собирали (1,5 мл) и спектрофотометрически оценивали при 480 нм содержание антрациклина. Содержание белка определяли колориметрическим анализом белка (BCA, Pierce). Конъюгат содержал 1,27 мг/мл антитела, соотношение антрациклин/антитело составляло 11,4/1,0. Физико-химические показатели продукта оценивали методами жидкостной хроматографии высокого давления - гель-фильтрации (колонка BioSil SEC-250, 0,1 М NaH2PO4, 0,1 м NaCl, pH 70) с двойным детектированием длин волн (280 и 480 нм) и методом SDS - PAGE (электрофорез в полиакриламидном геле). По данным метода жидкостной хроматографии высокого давления было обнаружено образование агрегатов у 50% белка путем элюирования из свободного объема колонки.
По данным метода SDS-PAGE как абсорбция антрциклина, так и реакция белка с красителем Coomasie происходили при молекулярном весе 160 kD, тем самым подтверждая образование ковалентных связей и указывая на образование агрегатов, обусловленное нековалентным взаимодействием.
Пример 8. Получение конъюгата формулы Iiv против карциномы толстой кишки {Iiv:A-O- = XIIIb', b = 1, B = -(CH2)2-, R1=R2=H, Z = -NH-NH-, T = B 72.3].
1 мл раствора B 72.3 антитела (US-A- 4 522 918) в 0,1 м NaH2PO4, pH буфера 6 (концентрация антитела в растворе 2,6 мг/мл) обрабатывали в темноте при 4oC мл водного 0,1 М раствора NaIO4. Через час продукт очищали гель-фильтрационной хроматографией на колонке Sephadex G25 (PD-10 Pharmacia), элюируя 0,1 М NaH2PO4 буфером с pH 6. Фракцию, содержащую белок (1,7 мг, 2 мл), обрабатывали в том же буфере 30 молярным эквивалентом 10 мас.%/об.% раствора соединения VI', описанного в примере 3. Смесь выдерживали в темноте при 37oC в течение 24 ч, после чего очищали, как описано в примере 7.
Конъюгат содержал 0,48 мг/мл антитела с соотношением антрациклин/белок 2, 4/1 и был на 85% мономерным.
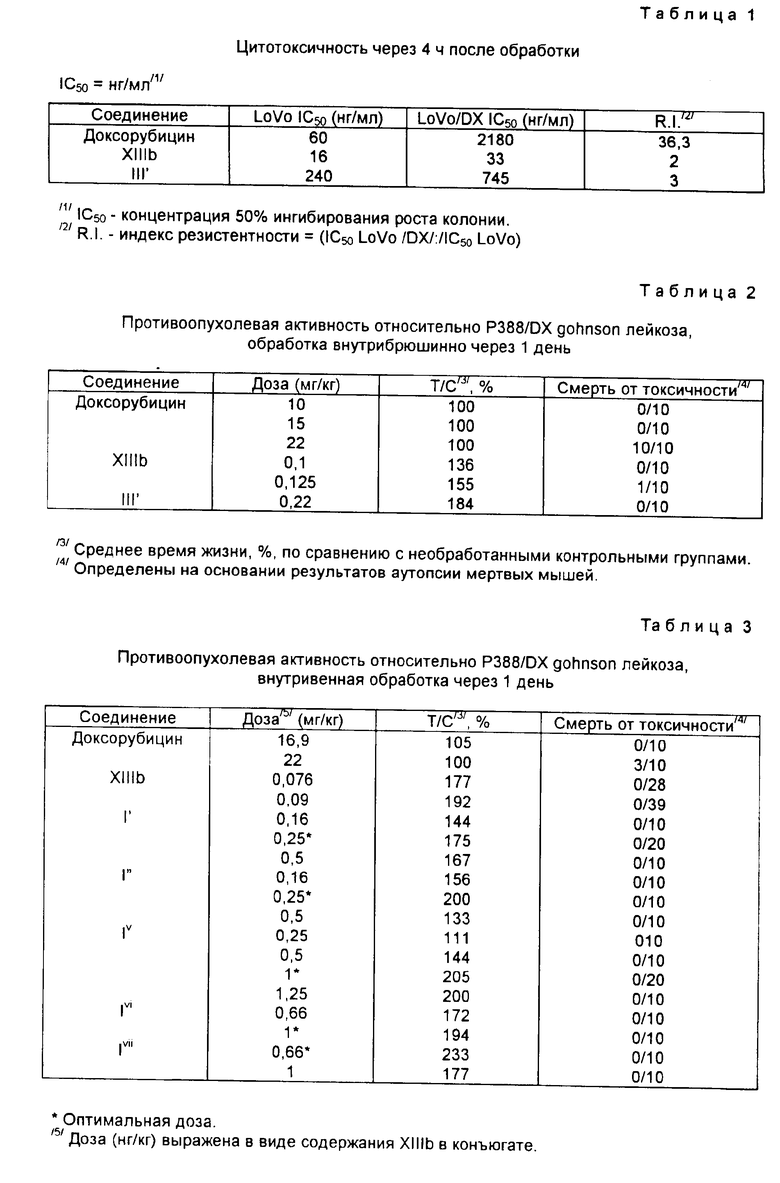
Пример 9. Получение сополимера 3-метакрилоиламино-2-оксипропана и 14-0-{ 1-[4-/N-метакрилоилглицилфенилаланил-лейцилглицил/гидразино] - карбонилметилокси-циклогексил}-3'-деамино-3'-[2/S/-метокси-4-морфолинил] доксорубицина (IV)
[IV:A-O- = XIIIb', b = 1, B = -(CH2)2-, R1=R2=H, Z = -NHNH-CO-, T = HPMA X = Gly - Phe - Leu - Gly] (см. фиг. 1).
0,58 г сополимера HPMA [X = Gly-Phe-Leu-Gly], содержание P = OC6H4-пара-NO2 4,2% (мол(мол%)), полученного по методике, описанной J. Kopecek и др. , Macromol. Chem. 177, 2833 - 2848 (1976), радикальной полимеризацией 3-мтакрилоиламино- 2-оксипропана и 4-нитрофенилового эфира N-метакрилоилглицилфенилаланил -лейцилглицина, растворяли в 15 мл осушенного диметилсульфоксида, содержащих 0,1 г производного VI', полученного по методике, описанной в примере 3. Раствор выдерживали при комнатной температуре в течение двух дней, затем добавляли к нему 0,4 мл 1-амино-2-пропанола и выливали реакционную смесь в 300 мл смеси, состоящей из ацетона и этилового эфира (1: 1 об./об.). Осадок собирали и повторно растворяли его в 10 мл 95%-ного этанола, после чего из раствора, добавляя 80 мл ацетона, осаждали соединение Iv, указанное в заглавии, собирали его и промывали этиловым эфиром. Было получено 0,51 г.
Содержание антрациклина (мас. /мас. %), рассчитанное в виде содержания хлоргидрата 3'-деамино-3'-[2(S)-метокси-4-морфолинил]- доксорубицина (XIIa), составляло 3,3%. % молярное соотношение в формуле IV (r:s:t) = (95,7:0,79: 3,52).
Пример 10. Получение 14-0-/1-пиперазинкарбонилметилокси-циклогексил/ -3'-деамино-[2/S/-метокси-4-морфолинил] доксорубицина [XI', A-O- = XIIIb', b = 1, B = -(CH2)2-, R1=R2=H].
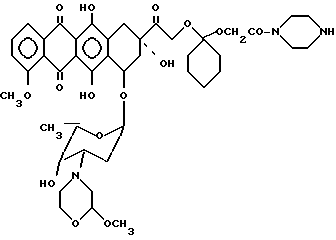
100 мг соединения IV, полученного по методике, описанной в примере 2, растворяли в 20 мл безводного тетрагидрофурана, охлаждали до 0oC и добавляли к раствору 50 мг пиперазина в 2 мл безводного тетрагидрофурана. Реакционную смесь перемешивали при 0oC в течение 15 мин, затем разбавляли 100 мл хлористого метилена и промывали водой (3 • 50 мл). Органический слой отделяли, сушили над безводным сульфатом натрия и при пониженном давлении удаляли растворитель. Неочищенный продукт хроматографировали на колонке с кремниевой кислотой, используя в качестве элюирующей системы смесь хлористого метилена/метанола (80/20 по об.). Соединение IX', 80 мг, осаждали этиловым эфиром.
Тонкослойная хроматография на пластинке с кизельгуром (Merck) F254, элюирующая система хлористый метилен/метанол (70/30 по об.)
Rf = 0,60.
FD - MS : m/z 868 (100, [M + H]+).
1ЯМР (200 МГц, CDCl3) δ :
1,35 (дублет, J = 6,6 Гц, 3H, 5'-CH3); 1,3 - 1,9 (мультиплет, 12H, 2'-CH2, циклогексан); 2,11 (двойной дублет, J = 4,2, 14,6 Гц, 1H, 8 ax-H); 2,3 - 2,5 (мультиплет, 5H, 8 eq-H, 3'-H, 3'' ax-H, 5''-CH2''); 2,60 (двойной дублет, J = 3,9, 11,2 Гц, 1H, 3'' eg-H); 2,86 (мультиплет, 4Н, CH2-NH-CH2); 3,00 (дублет J = 18,9 Гц, 1H, 10ax-H); 3,23 (двойной дублет, J = 1,2, 18,9 Гц, 1H, 10 eq-H); 3,37 (синглет, 3H, 2'' -OCH3); 3,4 - 3,6 (мультиплет, 5H, CON(CH2)2, 6'' ax-H); 3,90 (мультиплет, 1H, 6'' eq-H); 3,98 (квадруплет, J = 6,6 Гц, 1H, 5'-H); 4,07 (синглет, 3H, 4-OCH3); 4,18 (синглет, 2H, OCH2CON); 4,48 (двойной дублет, J = 2,5, 3,9 Гц, 1H, 2'' eq-H); 4,79 (мультиплет, 2H, 14-CH2); 5,24 (мультиплет, 1H, 7-H); 5,53 (мультиплет, 1H, 1'-H); 7,37 (дублет, J = 7,7 Гц, 1H, 3-H); 7,76 (двойной дублет, J =7,7, 6,8 Гц, 1H, 2-H); 8,02 (дублет, J = 6,8 Гц, 1H, 1-H); 13,30 (широкий синглет, 1H, 11-OH); 13,98 (синглет, 6-OH).
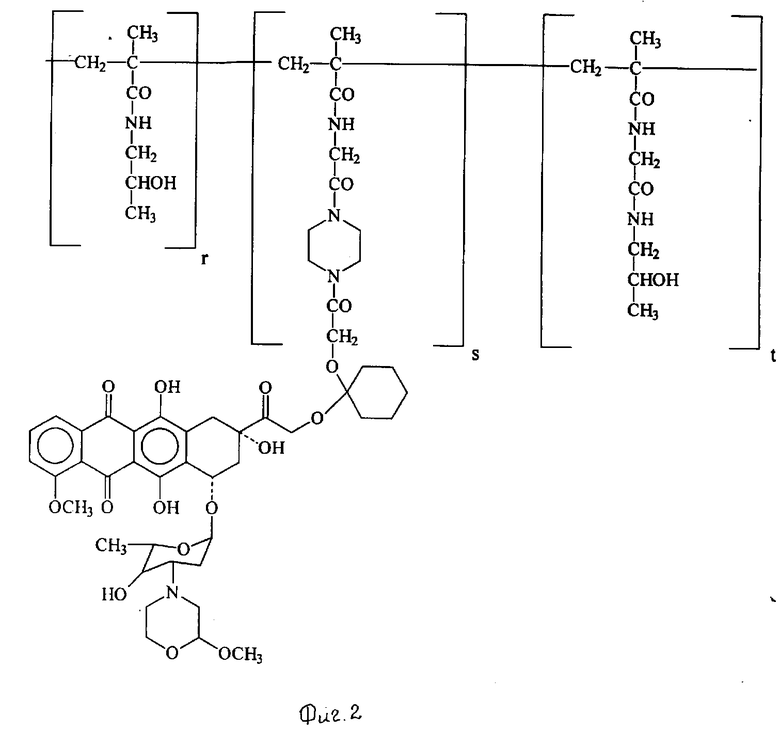
Пример 11. Получение сополимера 3-метакрилоиламино-2-оксипропана и 14-0-{ 1-[4-/N-метакрилоилглицил/пиперазин-1ил] -карбонил-метилокси-циглокгексил}-3'-деамино-3'- [2/S/-метокси-4-морфолин] доксорубицина (Ivi) [Ivi: A-O- = XIIIb', b=1, B = -(CH2)2-, R1=R2 = H, Z = [1,4-пиперазил]-CO-, T = HPMA и X = HN-CH2-CO-] (см. фиг. 2).
0,42 г сополимера HPMA [X = HN-CH2-CO-, содержание P = OC6H4-пара-NO2 7,6% мол/мол/] , полученного по методике, описанной G.Kopecek и др., Macromol. Chem. 177, 2833-2848 (1976), радикальной полимеризацией 3-метакрилоиламино-2-оксипропана и 4-нитрофенилового эфира N-метариклоилглицина, растворяли в 2,2 мл безводного диметилмсульфоксида и проводили реакцию с 0,07 г производного XI', полученного по методике, описанной в примере 10. Раствор выдерживали при комнатной температуре в течение двух часов, затем добавляли к нему 0,05 мл 1-амино-2-пропанола и выливали в 200 мл смеси, состоящей из ацетона и этилового эфира (1:1 об./об.). Осадок собирали, повторно растворяли в 10 мл 95%-ного этанола, из раствора осаждали 80 мл ацетона соединение Ivi, указанное в заглавии, собирали его и промывали этиловым эфиром. Получали 0,39 г соединения Ivi.
Содержание антрациклина (мас. /мас. %), рассчитанное в виде содержания хлоргидрата 3'-деамино-3'-[2/S/-метокси-4-морфолинил] доксорубицина /XIII/, составляло 9,74%. %молярное соотношение в формуле Ivi (r:s:t) = (92,4 : 2,15 : 5,45).
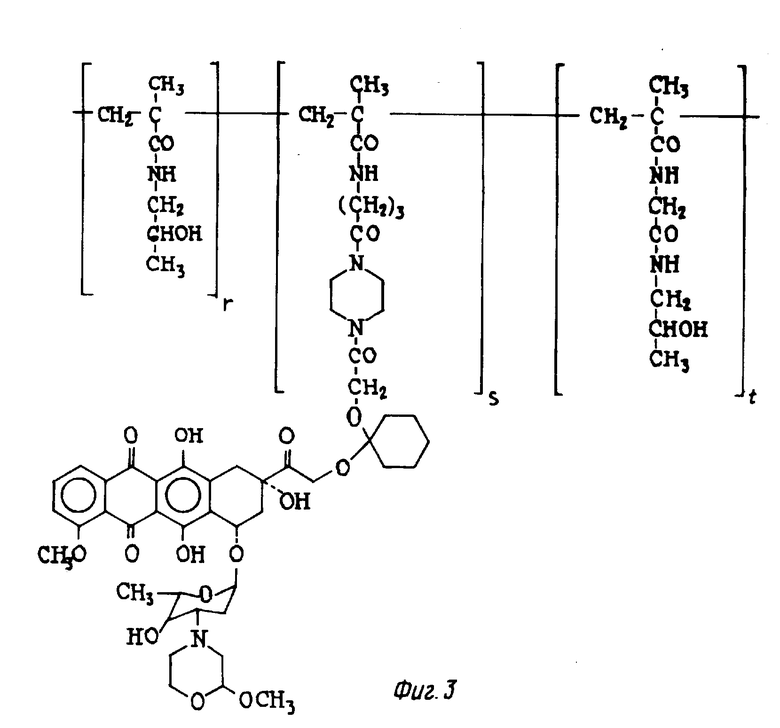
Пример 12. Полученное сополимера 3-метакрилоиламина-2-оксипропана и 14-O-{ 1-[4-/6-метакрилоиламинокапроил/пиперазин-1ил] -карбонилметилоксициклогексил} -3'-деамино-3'-[2 /S/-метокси-4-морфолинил]доксорубцина (Ivii) [Ivii : A-O- =XIIIb', b = 1, B = -/CH2/2-, R1 = R2 = H, Z = [1,4-пиперазинил]-CO-, T = HPMA и X = HN-/CH2/5-CO-] (см. фиг. 3).
0,6 г сополимер HPMA X = HN-/CH2/5-CO-, содержание P = PC6H4-пара-NO2 5,3% (мол/мол), полученного про методике, описанной J. Kopecek и др. Macromol. Chem. 1977, 2833 - 2848 (1976), радикальной полимеризацией 3-метакрилоиламино-2-оксипропана и 4-нитрофенилового эфира N-метакрилоиламинокапроната, растворяли в 3 мл безводного диметилсульфоксида и проводили реакцию с 0,1 г производного XI, полученного по методике описанной в примере 10. Раствор выдерживали при комнатной температуре в течение двух часов, добавляли к нему 0,1 мл 1-амино-2-пропанола и выливали в 200 мл смеси, состоящей из ацетона и этилового эфира (1:1 об./об.). Следуя методике, описанной в примере 11, было получено 0,58 г соединения Ivii, указанного в заглавии.
Содержание антрациклина (мас.мас.%), рассчитанное в виде содержания хлоргидрата 3'-деамино-3'-[2/S/-метокси-4-морфолинил] доксорубцина (XIII), составляло 9,2%. % молярное соотношение в формуле Ivii (r : s : t) = (94,7 : 2,05 : 3,31).
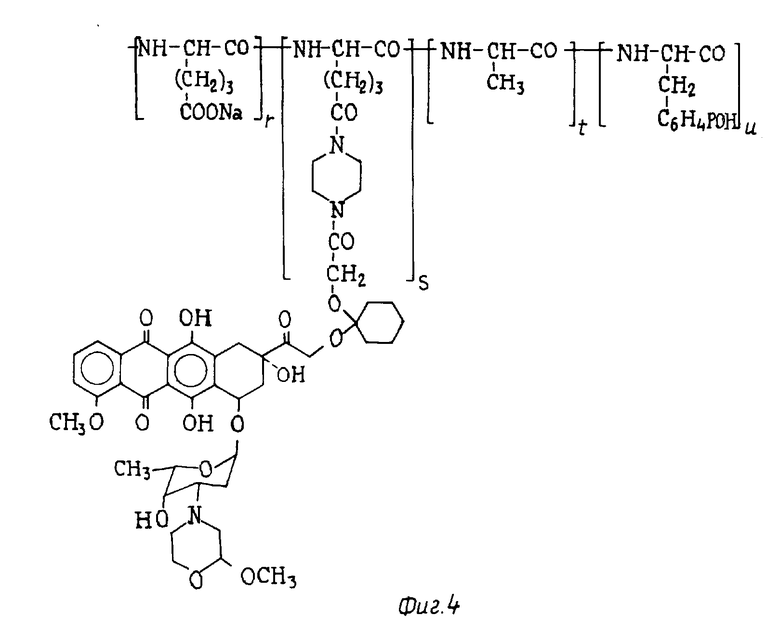
Пример 13. Получение конъюгата поли(Glu-Na, Ala, Tyr) ( 1:1:1) и 14-O-(1-пиперазинокарбонилметилокси-циклогексил)-3'-деамино- [2/S/-метокси-4-морфолинил] доксорубцина (Iviii) [Iviii, A-O- = XIIIb', b = 1, B = -(CH2(2-, R1 = R2 = H; Z = [1,4-пиперазинил]-CO-, T = поли (Glu-Na, Ala, Tyr)] (см. фиг. 4).
0,2 г поли(Glu-Na, Ala, Tyr) (1:1:1), Mw25000 - 40000 (Sigma) растворяли при перемешивании и комнатной температуре в 5 мл воды. Подкисляя водный раствор до pH 3 0,1 N HCl, осаждали соответствующую свободную кислоту. 0,17 г, поли (Glu-OH, Ala, Tyr), выделенных и высушенных в вакууме, растворяли в 10 мл безводного диметилформамида и добавляли к 0,035 г 14-O-(1-пиперизинокарбонилметилокси-циклогексил)-3'-деамино -[2/S/-метокси-4-морфолинил] -доксорубцина (XI'), полученного по методике, описанной в примере 10, и к 0,08 г N-этоксикарбонил-2-этокси-1,2-дигидрохинолина (EEDQ). Другую порцию EEDQ (0,08 г) добавляли через три часа. Реакционную смесь перемешивали в течение ночи при комнатной температуре, а затем выливали в 300 мл этилового эфира. Осадок суспендировали в 10 мл воды и обрабатывали 14 мл 0,1 N NaOH; pH раствора доводили до 8,5, добавляли 0,1 N HCl, после чего раствор пропускали через колонку Sephadex G10. Водный раствор лиофилизировали и получали 0,16 г соединения Iviii, указанного в названии.
Содержание антрациклина (мас. мас. %), рассчитанное в виде содержания хлоргидрата 3'-деамино-3'-[2(S)метокси-4-морфолинил] -доксорубцина (XIII), составляло 10%. % молярное соотношение в соединении формулы Iviii (r:s:t:u) = 31,33 : 2,01 : 33,33 : 33,33).
| название | год | авторы | номер документа |
|---|---|---|---|
| КОНЪЮГАТЫ АНТРАЦИКЛИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 1992 |
|
RU2107690C1 |
| ПРОИЗВОДНЫЕ 3'-АЗИРИДИНО-АНТРАЦИКЛИНА, СПОСОБЫ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2149163C1 |
| БИОЛОГИЧЕСКИ АКТИВНЫЕ ПОЛИМЕРСВЯЗАННЫЕ АНТРАЦИКЛИНЫ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2145965C1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ АНТРАЦИКЛИНОВЫЕ ГЛИКОЗИДЫ, СВЯЗАННЫЕ С ПОЛИМЕРНЫМ ПРОИЗВОДНЫМ | 1992 |
|
RU2118171C1 |
| АНТРАЦИКЛИНОВЫЙ ГЛИКОЗИД, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1992 |
|
RU2118328C1 |
| ПОЛИМЕРНЫЙ КОНЪЮГАТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2149646C1 |
| ПРОИЗВОДНЫЕ АЗА-АНТРАЦИКЛИНОНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2159245C2 |
| АНТРАЦИКЛИНОВЫЕ ГЛИКОЗИДЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ КИСЛОТНО-АДДИТИВНЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ДИИОДОПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1991 |
|
RU2100366C1 |
| ПРОИЗВОДНЫЕ АНТРАЦИКЛИНА | 1995 |
|
RU2159619C2 |
| УРЕИДОПРОИЗВОДНЫЕ НАФТАЛИНФОСФОНОВЫХ КИСЛОТ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2136692C1 |

Изобретение может быть использовано при лечении некоторых опухолей млекопитающих. Конъюгат общей формулы [A - O - W - Z]a - T, где T - фрагмент носителя, A - O - W - остаток терапевтически приемлемого антрациклина, Z - спейсерная группа. Конъюгат получают конденсированием соединения формулы A - O - W - OH или его активированного производного с веществом (или их смесью), которое является спейсерной группой Z, и с фрагментом носителя T. Конденсация может быть осуществлена в присутствии конденсирующего агента. T - T-клеточные антитела, антитела против CD5, против рецептора трансферрина и др. Заявлено соединение формулы A - O - W - [пиперазин]-H для получения конъюгата. Задача изобретения - улучшение терапевтического действия лекарств и уменьшение их токсического действия при введении в организм. 3 с. и., 10 з. п. ф-лы, 4 табл.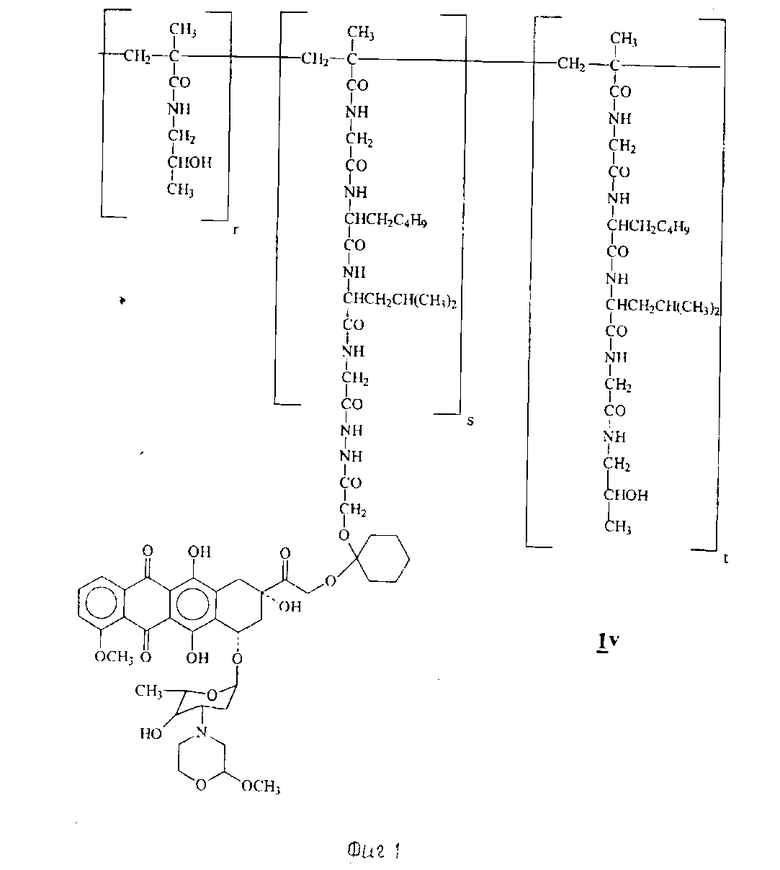
[A-O-W-Z]a-T,
где фрагмент A-O-W представляет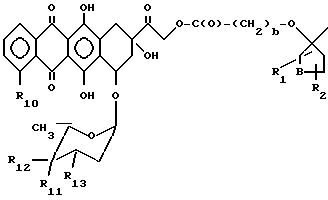
в которой R10 - водород или гидрокси- или метоксигруппа;
один из R11 и R12 - водород, а другой - гидроксигруппа или R11 - водород или йод, а R12 - водород;
R13 - аминогруппа или азот, присутствующий в морфолиновом кольце, где b = 1;
В - -(СН2)n-, где n = 1 - 3;
R1 и R2 независимо - водород;
Z - спейсерная группа формулы -NHNHCO-, -NH-, -NHNH- или фрагмент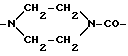
T - фрагмент носителя, выбранного из поликлонального антитела или его части, содержащей антигенсвязывающий сайт, способный связываться с ассоциированным с опухолью антигеном; моноклонального антитела или его части, содержащей антигенсвязывающий центр, способный связываться с антигеном, предпочтительно или избирательно экспрессированным на популяциях опухолевых клеток; пептида или белка, способного предпочтительно или избирательно связываться с опухолевыми клетками, и полимерного носителя.
A-O-W-OH,
или его активированного производного с веществом или смесью веществ, которое (которая) является указанной спейсерной группой Z и фрагментом носителя T по п.1, либо которое (которая) способно (способна) предоставить указанную спейсерную группу Z и фрагмент носителя T по п.1.
A-O-W-L
где A-O-W определено в п.1;
L - группа для создания амидной связи,
а вещество, способное предоставить спейсерную группу Z и фрагмент носителя T, имеет формулу
T-[NH2]x,
в которой x - число аминогрупп, способных к конденсации.
A-O-W-NH-NH2,
где A-O-W определено в п.1,
а указанным веществом, способным предоставить спейсерную группу Z и фрагмент носителя Т, является вещество формулы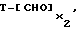
где x2 - число формильных групп, способных к конденсированию.
T-[CO-L']y,
где T - фрагмент носителя по п.1;
y представляет целое число от 1 до 30 и целое число карбоксильных групп на фрагменте носителя;
L' - гидроксильная или активирующая группа для создания амидной связи, необязательно в присутствии конденсирующего агента.
H2N-[CH2]g-COOH,
в которой g = 1 - 6, целое число,
с образованием производного общей формулы VIII
A-O-W-[D]-OH,
где A-O-W определен в п.1;
[D] представляет -HN-[CH2]g-CO-, где g имеет указанное значение,
причем конденсирование указанного соединения формулы VIII с веществом формулы
T-[NH2]x,
осуществляют в присутствии конденсирующего агента.
A-O-W-[D]-L,
полученное из соединения формулы VIII, где L - активирующая группа для создания амидной связи, и конденсирование полученного соединения формулы IX с веществом формулы T-[NH2] x, осуществляют в присутствии конденсирующего агента.
A-O-W-[пиперазин]-H,
где A-O-W определено в п.1,
и указанное соединение формулы II конденсируют с веществом формулы VII.
| Способ размножения копий рисунков, текста и т.п. | 1921 |
|
SU89A1 |
| ЭПОКСИДНАЯ КОМПОЗИЦИЯ | 0 |
|
SU328147A1 |
