Изобретение касается новых таксанов формулы 1, способов их получения и их фармацевтического использования: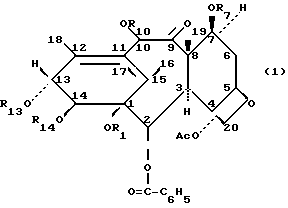
В общей формуле 1 R1, взятый отдельно, является водородом и R14, взятый отдельно, является водородом, три-/низший алкил/-силилом или алканоилом с 2-9 углеродными атомами, незамещенным или моно-, ди- или тризамещенным атомом галогена или монозамещенным фенилом, который является незамещенным или моно-, ди- или тризамещенным атомом галогена, низшим алкокси или низшим галоалкокси; или
R1 и R14, вместе взятые, являются карбонилом или низшим алкилиденом, незамещенным или замещенным фенилом;
каждый из R7 и R10 является водородом, три-/низший алкил/-силилом или алканоилом с 2-9 углеродными атомами, незамещенным или моно-, ди- или тризамещенным атомом галогена или монозамещенным фенилом, который незамещен или моно-, ди- или тризамещен атомом галогена, низшим алкокси или низшим галоалкокси, и
R13 представляет собой водород, триалкилсилил, -COCHOHCH-/C6H5/NHCOC6H5, -COCHOHCH-/C6H5/NHCOОС/CH3/3, алканоил с 2-9 углеродными атомами, незамещенный или моно-, ди- или тризамещенный атомом галогена или монозамещенный фенилом, который незамещен или моно-, ди- или тризамещен атомами галогена, низшим алкокси или низшим галоалкокси;
Ac является ацетилом.
Термин "низший алкил" означает одновалентную насыщенную разветвленную или прямую углеводородную цепь, содержащую от 1 до 8 углеродных атомов. Представителями таких алкильных групп являются метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, неопентил, трет-пентил, гексил, изогексил, гептил, октил и тому подобные.
Термин "низший алкокси" означает низшую алкильную группу, присоединенную к остальной молекуле через эфирную кислородную связь. Представителями таких алкоксигрупп являются метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси, пентокси, изопентокси, неопентокси, трет-пентокси, гексокси, изогексокси, гептокси, октокси и тому подобные.
Термин "низший алкилиден" означает изначально двухвалентную насыщенную разветвленную или прямую углеводородную цепь, содержащую от 1 до 8 углеродных атомов. Представителями таких алкилиденовых групп являются метилиден, этилиден, пропилиден, изопропилиден, бутилиден, изобутилиден, пентилиден, неопентилиден, гексилиден, гептилиден, октилиден и тому подобные.
Представителями низших галоалкоксигрупп являются дифторметокси, трифторметокси.
Соединение формулы 1, в которой R1=R7=R10=R13=R14=H /соединение формулы 1а/, является новым таксаном, который может быть выделен по-существу в чистой форме из растительного материала рода Taxus, главным образом из надземных частей растения Taxus Wallichiana. Этот вид распространен в Азии, в частности в отрогах Гималаев. Структура соединения формулы 1а отнесена к новому таксану посредством ядерного магнитного резонанса /на ядрах 1H и 13C/ и рентгеноструктурного анализа. Оно содержит дополнительные оксигруппы в положении 14 β в сравнении со структурой 10-деацетилбаккатина III, известного синтонического вещества, используемого в синтезе противоопухолевых средств таксола и таксотера. Присутствие этих дополнительных оксигрупп придает молекуле более высокую гидрофильность, чем гидрофильность самого 10-деацетилбаккатина III, с вытекающими отсюда преимуществами введения перфузией больным противоопухолевых средств, содержащих этот вид дитерпеновых ядер.
Новый таксан формулы 1а может быть выделен из растительных материалов, предпочтительно из иголок, двумя различными способами экстракции.
Можно экстрагировать растительный материал либо смешивающимся с водой растворителем, или несмешивающимся с водой растворителем. В первом случае /метод A/ может быть использован апротонный растворитель, такой как метанол или этанол, он может быть использован один или в сочетании с водой. Апротонный растворитель средней полярности, такой как ацетон, один или в смеси с водой может быть также использован. Соединение формулы 1а экстрагируют из растительного материала этими растворителями при комнатной температуре. Полученный экстракт концентрируют до удаления органического растворителя, отфильтровывают от образовавшегося нерастворимого материала и затем обрабатывают несмешивающимся с водой растворителем, например этилацетатом, или хлорированным растворителем, предпочтительно хлористым метиленом. Органический экстракт, содержащий соединение 1а, затем выпаривают досуха и полученный остаток очищают колоночной хроматографией. Если используют несмешивающийся с водой растворитель /метод B/, то может быть применен ароматический углеводород, например бензол или толуол, или хлорированный углеводород, например хлористый метилен, хлороформ, 1,2-дихлорэтан, 1,1-трихлорэтан. В этом случае экстракцию растительного материала проводят при комнатной температуре или при температуре образования флегмы выбранного растворителя.
Полученный экстракт концентрируют, очищают от полярных примесей обработкой водно-спиртовой смесью, например 30% водным метанолом, и выпаривают досуха.
Остаток аналогично остатку, полученному методом A, подвергают затем дальнейшей очистке колоночной хроматографией.
В обоих способах остаток, полученный двумя различными экстракциями растительного материала, очищают колоночной хроматографией на силикагеле, элюируя смесью растворителей, такой как циклогексан-ацетон, хлористый метилен-этилацетат или хлористый метилен-метанол, регулируя отношения растворителей в этих смесях так, чтобы выделить максимально возможное количество соединения 1а. В зависимости от качества исходного растительного материала получают выходы соединения 1а в пределах от 0,1 до 0,01%. Хроматографические фракции, содержащие соединение 1а, накапливают, выпаривают досуха и остаток кристаллизуют из метанола, этилацетата или ацетонитрила, чтобы получить чистое соединение 1а.
Соединение формулы 1 может быть получено из соединения 1а известными методами.
Из соединения 1а могут быть получены частично дериватизированные продукты, так как оксигруппы имеют следующий порядок реакционной способности: 7-OH > 10-OH > 14-OH > 13-OH.
Поэтому, либо используя соответствующие количества реактивов, или регулируя соответственно температуры и время реакции, можно получить частичные дериваты.
Соединения, в которых все или некоторые из R7, R10, R13, R14 являются алканоильными группами с 2-9 углеродными атомами, как определено выше, могут быть получены взаимодействием соединения 1а или его производного с ангидридом карбоновой кислоты формулы R5-COO-CO-R5, где R5 представляет собой алкильную группу с 1-8 углеродными атомами, незамещенную или моно-, ди- или тризамещенную атомом галогена или монозамещенную фенилом, который является незамещенным или моно-, ди- или тризамещенный атомом галогена, низшим алкокси или низшим галоалкокси, или с эквивалентным производным соединения 1а в присутствии 4-аминозамещенного аминопиридина и в подходящем растворителе.
Реакция ацилирования может быть проведена при комнатной температуре или необязательно при нагревании реакционной смеси с обратным холодильником.
Альтернативно соединение 1а или его производное может быть образовано кислотой формулы R5COOH в хлорированном растворителе, таком как хлористый метилен или хлороформ в присутствии карбодиимида, такого как дициклогексилкарбодиимид или 1-/3-диметиламинопропил/-3-этилкарбодиимид, и катализатора, такого как 4-/N,N-диметиламино/-пиридин или 4-/пирролидино/-пиридин при комнатной температуре или при температуре образования флегмы растворителя /температуре рефлекса/.
Соединение 1, в котором R7, R10, R13 и R14 представляют собой три-/низший алкил/-силильную группу, может быть получено из соединения 1a или его производного обработкой силилирующим агентом в соответствующем растворителе и в присутствии подходящего катализатора. Например, может быть использован трет-бутилдиметилхлорсилан в количестве 1,1 эквивалента на каждую оксигруппу, участвующую в реакции, в присутствии 2,2 эквивалента имидазола или 4-/N, N-диметиламино/-пиридина в диметилформамиде и при комнатной температуре.
Соединения формулы 1, в которых группы R1 и R14, вместе взятые, образуют CO-группу, могут быть получены взаимодействием соединения 1a и подходящего хлорформиата в основном растворителе, например пиридине. Например, при взаимодействии соединения 1a с избытком трихлорэтилового эфира хлормуравьиной кислоты в пиридине при 80oC в течение нескольких минут достигают ацилирования группой -COOCH2CCl3 оксигрупп в положениях 7, 10 и 14.
Однако в реакционной среде имеет место внутримолекулярное нуклеофильное замещение между заместителем в 14-положении и оксигруппой в 1-положении, приводящее к получению карбоната формулы 1b.
Производные формулы 1, в которых R1 и R14, вместе взятые, образуют низший алкилиден, незамещенный или замещенный фенилом, могут быть получены взаимодействием соединения 1a с альдегидом или кетоном в подходящих катализированных условиях. Например, при обработке соединения 1a 2,2-диметоксипропаном в ацетоновых растворах и в присутствии пара-толуолсульфоната пиридина, получают циклический кетон формулы 1c, вовлекающий в свое образование оксигруппы в положениях 14 и 1 соединения формулы 1a.
Соединения формулы 1 согласно изобретению обладают антимитотической активностью, сравнимой с активностью известных таксанов, таких как таксол или его производные, и противоопухолевой активностью in vivo.
In vitro они проявляют активность по отношению к тубулину головного мозга /The lanski, Roc. Natl. Acaol Sci, USA 70, 765, 1973/ и к культуре клеток лейкоцитов человека. Соединения согласно изобретению проявляют активность по отношению к тубулину, превышающую в два раза таковую соответствующих производных баккатина III.
Соединения могут быть введены перрорально или парентерально, одни или в сочетании с другими терапевтическими средствами, включая противоопухолевые средства, стероиды и так далее, больному, нуждающемуся в таком лечении. Парентеральные пути введения включают внутримышечный, внутриполостной, внутривенный и внутриартериальный. Что касается любого лекарства этого типа, то режимы доз должны быть рассчитаны в зависимости от конкретной неоплазмы, состояния больного и наблюдаемой реакции, но обычно дозы находятся в пределах от 10 до 30 мг/м2 в день в течение пяти дней или от 150 до 250 мг/м2, однократно вводимые каждые три недели. Имея низкую токсичность в сравнении с другими, используемыми в настоящее время средствами, они позволяют избежать токсической реакции либо снижением дневной дозы, или введением соединения в альтернативные дни, или при более длительных интервалах, таких как через каждые три-пять дней. Пероральные дозированные формы включают таблетки и капсулы, содержащие 1-10 мг лекарственного средства на дозированную единицу. Изотонические солевые растворы, содержащие 20-100 мг/мл, могут быть использованы для парентерального введения.
Следующие ниже примеры поясняют далее настоящее изобретение.
Пример 1. Выделение 14-окси-10-деацетилбаккатина III, соединение формулы 1a /соединение 1, в котором R1=R7=R10=R13=H/ из листьев Taxus Wallichiana. Метод A.
1 кг листьев Taxus Wallichiana, прошедших вакуумную сушку при 35oC. тонко мололи и экстрагировали 6 порциями метанола по 3 литра каждая при перемешивании при комнатной температуре, при этом каждая экстракция продолжалась шесть часов. Собранные экстракты концентрировали при пониженном давлении до объема 1 литра, выдерживали 24 часа и нерастворимый материал отфильтровывали, фильтрат экстрагировали 6 раз по 500 мл хлористого метилена. Собранные органические фазы концентрировали в вакууме до 500 мл, и полученный раствор очищали на хроматографической колонке, содержащей 300 г силикагеля, используя хлористый метилен в качестве элюента для полного удаления аполярных соединений с последующим использование смеси хлористый метилен-этилацетат в отношении 85:15, элюируя таким образом чистое соединение 1a. Собранные фракции концентрировали досуха. Остаток кристаллизовали из семи объемов метанола. Кристаллизованное твердое вещество фильтровали с напорным насосом, промывали небольшим количеством метанола и сушили в вакууме при 40oC. Получали 800 мг соединения 1a, точка плавления 215-217oC, M+ при м/2560.
Элементный анализ для C29H36O11:
вычислено: C 62,13; H 6,47%,
найдено: C 62,07; H 6,53%.
Пример 2. Выделение 14-окси-10-деацетилбаккатина III, соединение 1a /соединение 1, в котором R1=R7=R10=R13=R14=H/ из листьев Taxus Wallichiana.
1 кг листьев Taxus Wallichiana, высушенных в вакууме при 35oC, подвергали тонкому помолу и экстрагировали 6 порциями толуола по 3 литра каждая при перемешивании при комнатной температуре, при этом каждая экстракция длилась шесть часов. Собранные экстракты концентрировали при пониженном давлении до объема 500 мл и обрабатывали 3 частями 20% водного метанола по 70 мл каждая. Толуоловую фазу концентрировали в вакууме досуха и остаток растворяли в 500 мл хлористого метилена. Полученный раствор очищали колоночной хроматографией, как описано в примере 1, и получали чистое соединение 1a, точка плавления 215-217oC.
Пример 3. Получение соединения 1b /соединение 1, в котором R7=R10 - -COCH2CCl3, R13 - H, R1 и R14, вместе взятые, образуют CO/.
300 мг соединения 1a /0,53 ммоля/ растворяли в 6 мл безводного пиридина и обрабатывали пять минут при 80oC 0,49 мл /3,41 ммоля/ трихлорацетилхлорформиата. Реакционную смесь охлаждали до комнатной температуры, затем добавляли несколько капель метанола для разложения избыточного реактива. Реакционную смесь разбавляли водой и экрагировали хлористым метиленом. Органическую фазу отделяли, сушили над сульфатом натрия и концентрировали досуха. Остаток очищали через хроматографическую колонку, содержащую 7 г силикагеля, и элюировали смесью гексан-этилацетат в отношении 1:1. Получали 295 мг /50%/ соединения 1b. M+ /масс-спектр/ при m/z 955 /937 + H4/.
Элементный анализ для C36H35O16Cl6:
вычислено: C 46,10; H 3,84; Cl 22,73%,
найдено: C 45,98; H 3,91; Cl 22,67%.
Пример 4. Получение соединения 1c / соединение 1, в котором R7=R10=R13 - H; R14 и R1, вместе взятые, являются C/CH3/2/.
100 мг соединения 1a растворяли в 17 мл ацетона, предварительно отогнанного через сульфат меди, и обрабатывали 7 мл 2,2-диметоксипропана и 150 мг пара-толуолсульфоната пиридина при помешивании при комнатной температуре. Затем реакционную смесь выпаривали досуха и остаток извлекали хлористым метиленом. Органический раствор промывали водой для удаления соли пиридиния, сушили над сульфатом магния и растворитель отгоняли при пониженном давлении. Остаток очищали хроматографией на колонке с 5 г силикагеля, используя в качестве элюента этиловый эфир. Получали 82 мг /76%/ соединения 1c, точка плавления 166-170oC, масс-спектр M+ при m/z 618/600 + NH4/.
Элементный анализ для C32H40O11:
вычислено: C 64,00; H 6,67%,
найдено: C 63,89; H 6,71%.
Пример 5. Получение 14 β-окси-10-дезацетил-7,10-дихлорацетата баккатина III/ соединение 1, R1=R13=R14 - H; R7=R10 - COCHCl2/.
200 мг соединения 1a растворяли в 3 мл безводного пиридина и обрабатывали при комнатной температуре в течение 24 часов 160 микролитрами дихлоруксусного ангидрида и 10 мг 4-/N,N-диметиламино/-пиридина.
Реакционную смесь разбавляли водой со льдом и экстрагировали хлороформом. Органическую фазу промывали разбавленной хлористоводородной кислотой затем водой, сушили над сульфатом натрия и выпаривали досуха.
Остаток очищали колоночной хроматографией на силикагеле, элюируя смесью гексан-этилацетат в отношении 3:7. Получали 217 мг 7,10-дихлорацетата, который кристаллизовали из хлороформа, M+792.
Элементный анализ для C33H36O13Cl4:
вычислено: C, 50,00; H 4,54; Cl 17,93%;
найдено: C 49,86; H 4,71; Cl 17,01%.
Пример 6. Получение 10-деацетил-14β-гидроксибаккатин III-1,14-карбоната (R1 и R14 вместе образуют  R7 = R10 = R13 - H).
R7 = R10 = R13 - H).
Безводный пиридин (8,3 мл, 8,1 г, 20 мол. экв.) добавляют к суспензии 10-деацетил-14β-гидроксибаккатина III (2,8 г, 5,14 ммоль) в CH2Cl2. Получают прозрачный раствор, который охлаждают до -10oC. После этого при перемешивании добавляют 26,6 мл 1,9 М раствора фосгена в толуоле. Спустя 15 мин после окончания добавления, смесь разбавляют в насыщенном растворе NaHCO3, а затем в воде (20 мл). Ее экстрагируют CH2Cl2, и органическую фазу промывают разбавленной HCl и водой. После сушки MgSO4, выпаривания растворителя и колоночной хроматографии (с использованием 10 г силикагеля и смеси гексанэтилацетата 3 : 7 как элюента) получено 2,8 г продукта, имеющего M+ 568.
Пример 7. Получение 10-деацетил-14β-гидрокси-7-O-триэтилсилил-баккатин III-1,14-карбоната (R1 и R14 вместе образуют  R10 = R13 - H; R7 - триэтилсилил).
R10 = R13 - H; R7 - триэтилсилил).
10 г 1,14-карбоната 10-деацетил-14β-гидроксибаккартина III разбавляют в 50 мл диметилформамида и добавляют к 5 мл триэтилсилилхлорида в присутствии имидазола. Реакционную смесь оставляют на 2 часа при комнатной температуре, как только реакция завершилась, о чем судят по TCX, раствор пиридина выливают на 500 г льда, и суспензию экстрагируют оттуда метилхлоридом. Дегидратированную на MgSO4 органическую фазу концентрируют досуха. Остаток (11,8 г) кристаллизуют из смеси ацетон : гексан. Получено 10 г продукта, имеющего M+ 702.
Пример 8. Получение 14β-гидрокси-7-O-триэтилсилилбаккатин III-1,14-карбоната (R1 и R4 вместе образуют 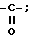 R10 - Ac; R7 - триэтилсилил, R13 - H).
R10 - Ac; R7 - триэтилсилил, R13 - H).
10 г 10-деацетил-14β-гидрокси-7-O-триэтилсилилбаккатин III-1,14-карбоната разбавляют в метилхлориде и экстрагируют в течение 12 часов двумя эквивалентами ацетилхлорида. Реакционную смесь промывают H2O, и органическую фазу концентрируют досуха в вакууме. Остаток снова помещают в метанол, и отделившееся твердое вещество отфильтровывают и кристаллизуют из смеси ацетон : гексан. Получают 10 г требуемого продукта, имеющего M+ 744.
Пример 9. Получение 13-[(2R, 3S)-3-бензоиламино-2-гидрокси-3-фенилпропаноил]-14β-гидроксибаккатин III-1,14 карбоната.
0,5 г 7-O-триэтилсилил-14β-гидроксибаккатин III-1,14-карбоната разбавляют в 60 мл толуола; 800 мг (4S, 5R)-N-бензоил-2,2-диметил-4-фенил-5-оксазолидинкарбоновой кислоты; 400 г циклогексилкарбодиимида и 40 г N,N-диметиламинопиридина. Реакционную смесь выдерживают при температуре 80oC в течение двух часов, после чего ее фильтруют и промывают водой, затем органическую фазу концентрируют досуха. Остаток обрабатывают метанолом, содержащим 0,1% H2SO4, при 10oC. Метанольный раствор разбавляют в воде, и продукт экстрагируют этилацетатом; органическую фазу концентрируют досуха, и остаток очищают хроматографией на силикагеле со смесью ацетон : гексан 4 : 6. Получают 580 г продукта с M+ m/z 897.
Пример 10. Получение 13-[(2R, 3S)-3-бутоксилкарбониламино-2-гидрокси-3-фенилпропаноил] 14β-гидроксибаккатин III 1,14-карбоната.
0,5 г 7-O-триэтилсилил-14β-гидроксибаккатина III разбавляют в 60 мл толуола; 800 мг (4S, 5R)-N-(третбутоксикарбонил)-2,2-диметил-4-фенил-5-оксазолидинкарбоновой кислоты, 400 г циклогексилкарбодиимида и до 40 мг N,N-диметиламинопиридина. Реакционную смесь выдерживают при 80oC в течение двух часов, после этого ее фильтруют и промывают водой; органическую фазу после этого концентрируют досуха. Остаток обрабатывают метанолом, содержащим 1% H2SO4, при 10oC. Метанольный раствор разбавляют водой и продукт экстрагируют этилацетатом; органическую фазу концентрируют досуха, и остаток очищают колоночной хроматографией на силикагеле путем элюирования смесью ацетон : гексан 4 : 6. Получают 580 мг продукта, M+ m/z 851.
Результаты биологических испытаний
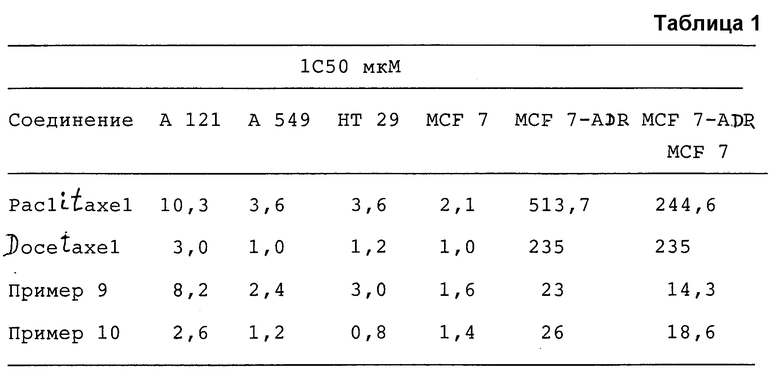
Соединения, полученные по примерам 9 и 10, подвергали испытаниям in vitro на цитотоксичность и противоопухолевую активность в сравнении со стандартными продуктами паклитаксел (paclitaxel) и доцетаксел (docetaxel).
Цитотоксическая активность.
Продукты испытывали по обычно известным специалистам методикам на следующим клеточных линиях:
A 121 : карцинома яичников человека
A 159 : (CCL70) рак легких человека
HT 29 : (HT B-38) аденокарцинома ободочной кишки человека
MCF 7 : аденокарцинома молочной железы человека
MCF 7-ADR : устойчивая в адриамицину аденокарцинома молочной железы
Приведенные в таблице 1 результаты указывают на то, что соединения, являющиеся предметом настоящего изобретения, обладают значительной цитотоксической активностью и отличаются от соединений, обычно известных как удивительно активные в отношении клеток, устойчивых к адриамицину, что можно видеть из соотношения между клетками MCF 7-ADR и клетками MCF 7.
Данные, полученные in vitro.
Противоопухолевая активность соединений в примерах 9 и 10 была рассчитана по следующей экспериментальной модели.
"Атимичные (лишенные тимуса) мыши Balb/c nu/nu возраста шесть недель (самки) получены от Harlan/Sprague-Dawley и использованы как реципиенты опухолевой молочной железы человека (MCF 7) после периода карантина 2 недели. Мышам имплантируют подкожно примерно 50 мг фрагментов неомертвевшей опухоли с использованием обычной техники трансплантации троакаром (12 калибра). Хемотерапию начинают через 4-10 дней после имплантации опухоли, когда опухоль является осязаемой (размера 100 мм3). Рост опухоли отслеживают два раза в неделю путем измерения циркулем в двух направлениях. Для определения противоопухолевой активности аналогов паклитаксела рассчитывают величину ингибирования роста опухоли (величину O/K). Одновременно для подвергнутой обработке и контрольной групп определяют вес опухоли. Когда вес опухоли в контрольной группе (K) достигает 1-1,5 г, определяют вес опухоли в каждой группе, подвергнутой обработке (O), включая животных без видимых опухолей. Величину O/K рассчитывают как отношение веса опухоли обработанных животных к весу опухоли контроля.
Веса опухолей оценивают из размера опухолей на основе предположения, что 1 мм3 = 1 мг. Полагают, что O/K ≤ 0,4 является минимальным уровнем для активности (стандарты Национального института рака (NCJ)).
Представленные в таблице 2 результаты показывают, что соединения, являющиеся предметом настоящего изобретения, как оказалось, обладают значительной противоопухолевой активностью, которая превосходит активность ранее известных продуктов.
Примеры фармацевтических композиций.
Пример 11. Инъекционный раствор для парентерального введения содержит
Соединение примера 9 - 9 мг
Кремофор EL - 175 мг
Спирт абсолютн. - g.s. до 0,4 мг
Пример 12. Таблетки для орального применения содержат
Соединение примера 10 - 50 мг
Поперечно-сшитая натрий-карбоксиметилцеллюлоза - 12,5 мг
Лактоза (высушенная распылением) - 133,75 мг
Микрокристаллическая целлюлоза - 50 мг
Коллоидная двуокись кремния - 1,25 мг
Стеарат магния - 2,5 мгп

Изобретение относится к новым производным таксана III формулы 1, в которой R1 - H; R14 - H, C2-C9-алканоил, незамещенный атомом галогена, или R1 и R14, взятые вместе, представляют собой карбонил или низший алкилиден; R7= R10 - H, три(низший алкил)силил или C2-C9-алканоил, незамещенный или моно-, ди-, тризамещенный атомом галогена; R13 - H, -COCHOHCH-(C6H5) NHCOC6H5 или -COCHOHCH (C6H5)NHCOOC(CH3)3-группа; Ас - ацетил, эти соединения обладают противоопухолевой активностью. Изобретение включает также способы получения соединений формулы 1, где R1=R7=R10=R13=R14 - H из надземных частей растений Taxus Walliсhiana, а также способы соединений формулы 1 с различными значениями радикалов R1, R7, R10, R13 и R14, а также фармацевтическую композицию на основе производных таксана формулы 1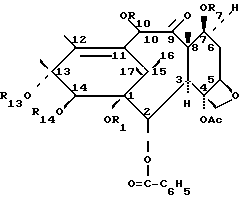
8 с. и 4 з.п.ф-лы, 2 табл.
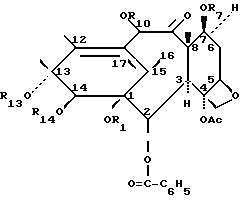
в которой R1, взятый в отдельности, является атомом водорода;
R14, взятый в отдельности, представляет собой атом водорода или алканоил с 2 - 9 углеродными атомами, незамещенный или моно-, ди- или тризамещенный атомом галогена,
или R1 и R14, вместе взятые, представляют собой карбонил или низший алкилиден;
R7 представляет собой атом водорода, три(низший алкил)-силил или алканоил с 2 - 9 углеродными атомами, незамещенный или моно-, ди- или тризамещенный атом галогена;
R10 - атом водорода, алканоил с 2 - 9 углеродными атомами, незамещенный или моно-, ди- или тризамещенный атомом галогена,
R13 - атом водорода, -COCHOHCH- (C6H5) NHCOC6H5 или -COCHOHCH(C6H5) NHCOOC(CH3)3 группа,
AC - ацетил.
R5COOH,
где R5 представляет собой алкильную группу с 1 - 8 углеродными атомами, незамешенную или моно-, ди- или тризамещенную атомом галогена,
или с ее активированным производным.
| EP, 0253738, C 07 D 305/14, 1988 | |||
| EP, 0473326, C 07D 305/14, 1992 | |||
| PCT, 92 09582, C 07 D 305/14, 1992. |

