Настоящее изобретение относится к производным секо-баккатина III.
Дитерпены, имеющие таксановый скелет, в частности, паклитаксел и доцетаксел, в настоящее время используются в медицине для лечения опухолей различного происхождения.
Однако существующие в настоящее время производные таксана обладают значительными побочными действиями, а также быстро индуцируют резистенцию, аналогично другим противоопухолевым лекарственным средствам.
Настоящее изобретение относится к производным секо-баккатина III, который раскрыт в US 5756776, характеризующемся биодоступностью при пероральном введении, пониженной токсичностью и чрезвычайно высокой антиангиогенной активностью.
Соединения по настоящему изобретению имеют общую формулу (I):
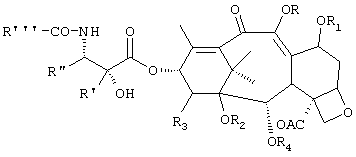
где:
- R и R1, которые могут быть одинаковыми или различными, представляют собой водород, C1-C18 ацильную группу, необязательно замещенную ароильную группу или группу -CONR6R7, в которой R6 и R7, которые могут быть одинаковыми или различными, представляют собой C1-C4 алкильную, бензильную или фенильную группу;
- R2 представляет собой водород или вместе с R3 образует карбонатный или тиокарбонатный остаток;
- R3 представляет собой водород или группу -OR5, где R5 представляет собой водород, или вместе с R2 образует карбонатный или тиокарбонатный остаток;
- R4 представляет собой бензоильную группу, необязательно замещенную в мета-положении, или гетароильную группу;
R' представляет собой водород или C1-C4 алкил;
R" представляет собой C1-C4 алкил, C2-C6 алкенил, арил или гетарил;
R"' представляет собой C1-C4 алкил, C1-C18 ацил, арил или трет-бутокси группу;
при условии, что R и R1 не могут оба представлять водород.
C1-C18 ацильная группа предпочтительно представляет такие группы, как формил, ацетил, н-пропаноил, н-гексаноил.
Необязательно замещенная ароильная группа предпочтительно представляет бензоил, необязательно замещенный одним или тремя заместителями, выбранными из атомов галогена или C1-C4 алкила, C1-C4 алкокси, C1-C4 галогеналкила, C1-C4 галогеналкокси, циано и нитро групп.
Мета-замещенная бензоильная группа предпочтительно представляет собой 3-галогенбензильную или 3-метоксибензоильную группу.
Гетароильная группа предпочтительно представляет собой 5- или 6-членный гетероарил, содержащий в кольце один или два атома кислорода, азота или серы и замещенный карбонильной группой, например, 2- или 3-теноил, никотиноил или 2- или 3-фуроил.
Арил предпочтительно представляет собой фенил, и гетарил предпочтительно представляет собой 2- или 3-фурил, 2- или 3-тиенил, 2-, 3- или 4-пиридил.
Предпочтительная группа соединений формулы (I) представляет собой такую группу, в которой:
R и R1, которые являются одинаковыми, представляют собой C1-C18 ацильную группу, необязательно замещенную бензоильную группу, как определено выше, или группу -CONR6R7, более предпочтительно R и R1 представляют собой ацетил или 3,4,5-триметоксибензоил;
R2 представляет собой водород;
R3 представляет собой водород;
R4 представляет собой бензоил;
R' представляет собой водород или метил;
R" представляет собой C1-C4 алкил или C1-C6 алкенил, более предпочтительно, изобутил или изобутенил;
R''' представляет собой трет-бутокси группу.
Следующей группой предпочтительных соединений является группа, в которой R представляет собой водород и R1 представляет собой ацил, ароил или группу CONR6R7, как определено выше, R2 и R3 представляют собой водород, R4 представляет собой бензоил, R1 представляет собой водород или метил, R" представляет собой C1-C4 алкил или C2-C6 алкенил, и R"' представляет собой трет-бутокси.
Этерификация гидроксильных групп в положениях С-7 и С-9 индуцирует, в сравнении с известными соединениями, повышение цитотоксического действия на резистентные клеточные линии, а также улучшенную абсорбцию при пероральном введении. Соединения по изобретению являются менее активными по сравнению с паклитакселом, взятым в качестве сравнительного лекарственного средства, в связывании с тубулином, но при этом обладают сопоставимой цитотоксичностью в отношении чувствительных опухолевых клеток. Эти соединения в основном отличаются от соединений предшествующего уровня техники по антиангиогенной активности. Таблица показывает активность in vivo некоторых производных С-секо-10-дегидро-10-деацетил-7,9-бис-ацетил-баккатина III и С-секо-10-дегидро-10-деацетил-7,9-бис-ацетил-1,14-карбонат-баккатина III, имеющих одинаковую изосериновую цепь.
Антиангиогенную активность оценивали в испытании Matrigel, в котором ангиогенез индуцировали при помощи FGF-2 (150 мг/гранула), абсорбированном на грануле Matrigel (12,5 мг/мл, 0,5 мл), который подкожно инъецировали мышам C57BL6N.
Испытываемое соединение вводили перорально ежедневно или внутрибрюшинно через день, в концентрациях, указанных в таблице. Через 7 дней ангиогенный ответ оценивали путем измерения содержания гемоглобина в гранулах по методу Drabkin.
Антиангиогенная активность in vivo соединения примера II.
Соединения по изобретению получают взаимодействием С-секо-10-дегидро-10-деацетил-7,9-гидрокси баккатина III, описанного в US 5756776, с реакционноспособным производным карбоновой кислоты (хлорангидрид или ангидрид), в соответствии с известными способами ацилирования.
Сложные диэфиры С7 и С9 можно получить, используя, по меньшей мере, два эквивалента реакционноспособного производного. Карбаматные группы могут быть введены традиционными способами, например путем взаимодействия фосгена и амина формулы R6R7NH.
Полученные соединения затем подвергают взаимодействию, в соответствии с известными методами, с изосериновым производным, обычно оксазолидиновым производным, которое при обработке кислотой в мягких условиях дает соединения (I).
Соединения по изобретению характеризуются низкой системной токсичностью: при дозах, эффективных для ингибирования роста опухоли, они не вызывают ни потери веса, ни очевидной нейротоксичности; у "голых" мышей, которым были трансплантированы опухолевые клетки человека, доза паклитаксела, который использовали в качестве сравнительного лекарственного средства, вызывающая такую же противоопухолевую активность, также вызывала тремор и потерю веса до 20%.
Соединения по настоящему изобретению, благодаря их высокой растворимости в воде, можно легко формулировать в виде препаратов для инъекций.
Соединения (I) можно также формулировать в виде обычной лекарственной формы для перорального введения (капсулы или таблетки).
Поскольку они обладают низкой токсичностью, соединения (I) можно вводить внутривенно при дозах вплоть до 600 мг/м2 и перорально при дозах вплоть до 1000 мг/м2. Дозы можно уменьшить до 50 мг/м2 при лечении ревматоидного артрита.
Приведенные ниже примеры более подробно иллюстрируют изобретение, не ограничивая его объем.
Пример I - получение С-секо-10-дегидро-10-деацетил-7,9-бис-ацетил-баккатина III.
К раствору 300 мг 10-дегидро-10-деацетилбаккатина III в 5 мл метанола добавляют 1 экв. CeCl3·3Н2О и реакционную смесь перемешивают в течение 10 мин. После полного растворения добавляют небольшими порциями 80 мг NaBH4. Через 10 мин раствор обрабатывают равным объемом водного раствора NH4Cl и экстрагируют СН2Cl2. Хлорированный растворитель удаляют, остаток переносят в 1 мл пиридина, охлаждают до 0°С в течение 1 часа, затем добавляют 150 мг уксусного ангидрида. Раствор оставляют на 2 часа при 0°С, затем разбавляют 10 мл воды и подвергают обратной экстракции CH2Cl2. Хлорированный растворитель отгоняют в вакууме и остаток хроматографируют на силикагеле, элюируя смесью н-гексан/этилацетат, с получением 260 мг С-секо-10-дегидро-10-деацетил-7,9-бис-ацетил-баккатина III (m/z 630).
Пример II - получение 13-[(2R, 3S)-3-изобутил-2-гидрокси-3-трет-бутоксикарбониламинопропаноил]-С-секо-10-дегидро-10-деацетил-7,9-бис-ацетил-баккатина III.
630 мг С-секо-10-дегидро-10-деацетил-7,9-бис-ацетил-баккатина III растворяют в 5 мл толуола и добавляют 335 мг дициклогексилкарбодиимида (DCC), 50 мг (4S,5R)-N-Вос-2-(2,4-диметоксифенил)-4-изобутил-5-оксазолидинкарбоновой кислоты и 20 мг 4-диметиламинопиридина. Раствор нагревают при 60°С в течение 24 часов, затем обрабатывают этилацетатом и насыщенным раствором NaHCO3. Органическую фазу сушат и фильтруют через силикагель для удаления мочевины. Растворитель выпаривают в вакууме до сухости и остаток переносят в раствор метанола/хлористоводородной кислоты, поддерживая температуру на уровне 0°С в течение 1 часа. Раствор нейтрализуют до рН 5, затем разбавляют водой и желаемое соединение обратно экстрагируют CH2Cl2. Растворитель выпаривают с получением 700 мг 13-[(2R, 3S)-3-изобутил-2-гидрокси-3-трет-бутоксикарбониламинопропаноил]-С-секо-10-дегидро-10-деацетил-7,9-бис-ацетил-баккатина III.
Белый порошок, т.пл. 91°С; 1H-ЯМР (300 М Гц, CDCl3 60°С) δ 8,03 (2Н, д, AA'-Bz), 7,61 (1Н, т, C-Bz), 7,48 (2Н, т, ВВ'-Bz), 6,17 (1H, шир. т, J=7 Гц, Н-13), 5,63 (1H, д, J=8 Гц, H-2), 5,27 (1H, д, J=10 Гц, H-5), 5,16 (1H, шир. д, J=8 Гц, H-20a), 4,65 (1H, д, J=9,5 Гц, NH), 4,39 (1H, д, J=8 Гц, H-3), 4,32 (1H, шир. д, J=8 Гц, H-20b), 4,31 (1H, м, Н-7а), 4,25 (1H, д, J=3 Гц, Н-2'), 4,18 (1H, м, Н-3'), 4,12 (1H, м, H-7b), 2,89 (1H, м, Н-14а), 2,46 (2Н, м, Н-6а b H-14b), 2,28 (3Н, с, ОАс), 2,22 (3Н, с, ОАс), 2,10 (1H, м, Н-6b), 1,94 (3Н, с, Н-19), 1,85 (3Н, шир. с, Н-18), 1,60 (1H, м, Н-4'а), 1,42 (1H, м, H-4'b), 1,30 (9Н, с, ВОС), 1,26 (3Н, с, Н-17), 1,19 (3Н, с, Н-16), 0,99 (6Н, д, J=7 Гц, Н-6' и Н-7'); HREIMS м/z [М]+(871,3999) (вычислено для C45H61NO16, 871,3990).
Пример III - получение 13-[(2R, 3S)-3-фенил-2-гидрокси-3-трет-бутоксикарбониламинопропаноил]-С-секо-10-дегидро-10-деацетил-7,9-бис-ацетил-баккатина III.
630 мг С-секо-10-дегидро-10-деацетил-7,9-бис-ацетил-баккатина III растворяют в 5 мл толуола и добавляют 335 мг DCC, 525 мг (4S, 5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-5-оксазолидинкарбоновой кислоты и 20 мг 4-диметиламинопиридина.
Раствор нагревают при 60°С в течение 24 часов, затем обрабатывают этилацетатом и насыщенным раствором NaHCO3. Органическую фазу сушат и фильтруют через силикагель для удаления мочевины. Растворитель выпаривают в вакууме до сухости и остаток переносят в раствор метанола/хлористоводородной кислоты, поддерживая температуру на уровне 0°С в течение 1 часа. Раствор нейтрализуют до рН 5, затем разбавляют водой и желаемое соединение обратно экстрагируют СН2Cl2. Растворитель выпаривают с получением 700 мг 13-[(2R, 3S)-3-изобутил-2-гидрокси-3-трет-бутоксикарбониламинопропаноил] -С-секо-10-дегидро-10-деацетил-7,9-бис-ацетил-баккатина III, который кристаллизуют из этилацетата с получением 645 мг чистого соединения.
Белый порошок, т.пл. 110°С; 1H-ЯМР (300 МГц, CDCl3 60°С) δ 8,03 (2Н, д, AA'-Bz), 7,61 (1Н, т, C-Bz), 7,48 (2Н, т, ВВ'-Bz), 6,17 (1H, шир. т, J=7 Гц, Н-13), 5-63 (1H, д, J=8 Гц, H-2), 5-27 (1H, д, J=10 Гц, H-5), 5,16 (1H, шир. д, J=8 Гц, H-20a), 4,65 (1H, д, J=9,5 Гц, NH), 4,39 (1H, д, J=8 Гц, H-3), 4,32 (1H, шир. д, J=8 Гц, H-20b), 4,31 (1H, м, Н-7а), 5,29 (1H, д, J=9,4 Гц, H), 4,67 (1H, шир. с, H-2'), 4,12 (1H, м, Н-7b), 2,89 (1H, м, Н-14а), 2,46 (2Н, м, Н-6а и Н-14b), 2,28 (3Н, с, ОАс), 2,22 (3Н, с, ОАс), 2,10 (1H, м, Н-6b), 1,94 (3Н, с, Н-19), 1,85 (3Н, шир. с, Н-18), 7,42 (5Н, с, 3'Ph), 1,39 (9Н, с, tBu), 1,26 (3Н, с, Н-17), 1,19 (3Н, с, Н-16).
Пример IV - получение С-секо-10-дегидро-10-деацетил-7,9-бис-триметоксибензоил-баккатина III.
К раствору 546 мг С-секо-10-дегидро-10-деацетил-баккатина III в 3 мл пиридина добавляют небольшими порциями 575 мг триметоксибензоилхлорида. Через 3 часа раствор выливают в 30 мл воды и экстрагируют СН2Cl2; органическую фазу промывают кислотами до полного удаления пиридина. Растворитель выпаривают с получением 905 мг С-секо-10-дегидро-10-деацетил-7,9-бис-триметоксибензоил-баккатина III (m/z 936).
Пример V - получение 13-[(2R, 3S)-3-фенил-2-гидрокси-3-трет-бутоксикарбониламинопропаноил]-С-секо-10-дегидро-10-деацетил-7,9-бис-триметоксибензоил-баккатина III.
930 мг 13-[(2R, 3S)-С-секо-10-дегидро-10-деацетил-7,9-бис-триметоксибензоил-баккатина III растворяют в 15 мл толуола и добавляют 335 мг DCC, 525 мг (4S, 5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-5-оксазолидинкарбоновой кислоты и 20 мг 4-диметиламинопиридина. Раствор нагревают при 60°С в течение 24 часов, затем обрабатывают этилацетатом и насыщенным раствором NaHCO3. Органическую фазу сушат и фильтруют через силикагель для удаления мочевины. Растворитель выпаривают в вакууме до сухости и остаток переносят в раствор метанола/хлористоводородной кислоты, поддерживая температуру на уровне 0°С в течение 1 часа. Раствор нейтрализуют до рН 5, затем разбавляют водой и желаемое соединение обратно экстрагируют CH2Cl2. Растворитель выпаривают с получением 940 мг 13-[(2R, 3S)-3-изобутил-2-гидрокси-3-трет-бутоксикарбониламинопропаноил]-С-секо-10-дегидро-10-деацетил-7,9-бис-триметоксибензоил-баккатина III, который кристаллизуют из этилацетата с получением 878 мг чистого соединения.
Пример VI - получение 1,14-карбоната С-секо-10-дегидро-10-деацетил-7,9-бис-ацетил-баккатина III.
1 г 10-деацетил-14β-гидроксибаккатина III, полученного как описано в US 5698712, растворяют в метаноле и обрабатывают 6 г Cu(ОАс)2 и реакционную смесь перемешивают в течение 120 часов. Соль отфильтровывают, растворитель удаляют и остаток хроматографируют на колонке с силикагелем, элюируя смесью 6:4 гексан/этилацетат, с получением 0,9 г 1,14-карбоната 10-дегидро-10-деацетил-14β-гидроксибаккатина III (M+568). 300 мг этого соединения растворяют в метаноле и обрабатывают 1 экв. CeCl3·3Н2О и реакционную смесь перемешивают в течение 10 минут. После полного растворения добавляют небольшими порциями 80 мг NaBH4. Через 10 минут раствор обрабатывают равным объемом водного раствора NH4Cl и экстрагируют CH2Cl2. Хлорированный растворитель удаляют, остаток переносят в 1 мл пиридина, охлаждают до 0°С в течение 1 часа, затем добавляют при перемешивании 150 мг уксусного ангидрида. Раствор оставляют на 2 часа при температуре 0°С, затем разбавляют 10 мл воды и обратно экстрагируют при помощи CH2Cl2. Хлорированный растворитель отгоняют в вакууме и остаток хроматографируют на силикагеле, элюируя смесью н-гексан/этилацетат с получением 250 мг 1,14-карбоната С-секо-10-дегидро-10-деацетил-7,9-бис-ацетил-баккатина III (m/z 658).
Пример VII - получение 1,14-карбоната 13-[(2R, 3S)-3-изобутил-2-гидрокси-3-трет-бутоксикарбониламинопропаноил]-С-секо-10-дегидро-10-деацетил-7,9-бис-ацетил-баккатина III.
600 мг 1,14-карбоната С-секо-10-дегидро-10-деацетил-7,9-бис-ацетил-баккатина III обрабатывают, как описано в Примере II, с получением 680 мг указанного в заголовке соединения.
Белый порошок, т.пл. 121°С; 1H ЯМР (300 МГц, CDCl3 60°С) δ 8,03 (2Н, д, AA'-Bz), 7,61 (1Н, т, C-Bz), 7,48 (2Н, т, ВВ'-Bz), 6,44 (1H, шир. д, J=7 Гц, Н-13), 5,63 (1H, д, J=8 Гц, H-2), 5,27 (1H, д, J=10 Гц, H-5), 5,16 (1H, шир. д, J=8 Гц, Н-20а), 4,65 (1H, д, J=9,5 Гц, NH), 4,39 (1H, д, J=8 Гц, H-3), 4,32 (1H, шир. д, J=8 Гц, H-20b), 4,31 (1H, м, Н-7а), 4,30 (1H, д, J=3 Гц, Н-2'), 4,08 (1H, м, Н-3'), 4,12 (1H, м, Н-7b), 4,84 (1H, д, Н-14а, 7 Гц), 2,46 (1H, м, Н-6а), 2-28 (3Н, с, ОАс), 2,22 (3Н, с, ОАс), 2,10 (1H, м, Н-6b), 1,94 (3Н, с, Н-19), 1,85 (3Н, шир. с, Н-18), 1,60 (1H, м, Н-4'а), 1,42 (1H, м, H-4'b), 1,30 (9Н, с, ВОС), 1,26 (3Н, с, Н-17), 1,19 (3Н, с, Н-16), 0,99 (6Н, д, J=7 Гц, Н-6' и Н-7').
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПАКЛИТАКСЕЛА | 2001 |
|
RU2276147C2 |
| ПОЛУСИНТЕТИЧЕСКИЙ ТАКСАН, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2134688C1 |
| ПРОИЗВОДНЫЕ ТАКСОЛА С ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2007 |
|
RU2419622C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТАКСАНА И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ПРИ ИХ ПОЛУЧЕНИИ | 1993 |
|
RU2116302C1 |
| ПРОИЗВОДНЫЕ ТАКСАНА, ФУНКЦИОНАЛИЗИРОВАННЫЕ ПО 14-ПОЛОЖЕНИЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2320652C2 |
| ПРОИЗВОДНЫЕ ТАКСАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2137764C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТАКСАНА | 2001 |
|
RU2275365C2 |
| АНАЛОГИ Δ-ИЗОТАКСОЛА, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2142950C1 |
| С-4 КАРБОНАТСОДЕРЖАЩИЕ ТАКСАНЫ | 2000 |
|
RU2243223C2 |
| ПРОИЗВОДНЫЕ ТАКСАНА | 1993 |
|
RU2188198C2 |
Настоящее изобретение относится к новым полусинтетическим таксанам формулы 1:
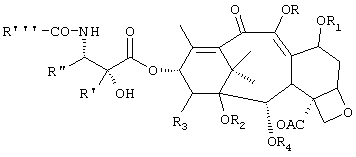 (1),
(1),
где R и R1, которые могут быть одинаковыми или различными, представляют собой водород, C1-C18 ацильную группу, бензоильную группу; R2 и R3 представляет собой водород или вместе R2 и R3 образует карбонатный или тиокарбонатный остаток; R4 представляет собой бензоильную группу, необязательно замещенную в мета-положении; R' представляет собой водород или C1-C4 алкил; R" представляет собой C1-C4алкил или фенил; R''' представляет собой трет-бутокси группу; при условии, что R и R1 не могут оба представлять водород. Изобретение также относится к фармацевтической композиции на основе соединений формулы 1, обладающей противоопухолевым, антиангиогенным и антиартрозным действием. Технический результат - получение новых соединений, проявляющих цитотоксичность, сравнимую с цитотоксичностью других таксанов, но с пониженной системной токсичностью, которые можно вводить внутривенно или перорально. 2 н. и 4 з.п. ф-лы, 1 табл.
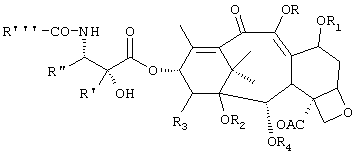
где R и R1, которые могут быть одинаковыми или различными, представляют собой водород, C1-C18 ацильную группу, бензоильную группу;
R2 и R3 представляют собой водород или вместе R2 и R3 образуют карбонатный или тиокарбонатный остаток;
R4 представляет собой бензоильную группу, необязательно замещенную в мета- положении;
R' представляет собой водород или C1-C4 алкил;
R" представляет собой C1-C4 алкил или фенил;
R''' представляет собой трет-бутокси группу;
при условии, что R и R1 не могут оба представлять водород.
R и R1, которые являются одинаковыми, представляют собой C1-C18 ацильную группу, более предпочтительно R и R1 представляют собой ацетил, или бензоильную группу;
R2 представляет собой водород;
R3 представляет собой водород;
R4 представляет собой бензоил;
R' представляет собой водород или метил;
R" представляет собой C1-C4 алкил, более предпочтительно, изобутил;
R''' представляет собой трет-бутокси группу.
13-[(2R,3S)-3-изобутил-2-гидрокси-3-трет-бутоксикарбониламинопропаноил]-С-секо-10-дегидро-10-деацетил-7,9-бис-ацетил-баккатина III,
13-[(2R,3S)-3-фенил-2-гидрокси-3-трет-бутоксикарбониламинопропаноил]-С-секо-10-дегидро-10-деацетил-7,9-бис-ацетил-баккатина III,
1,14 карбоната 13-[(2R,3S)-3-изобутил-2-гидрокси-3-трет-бутоксикарбониламинопропаноил]-С-секо-10-дегидро-10-деацетил-7,9-бис-ацетил-баккатина III.
| US 5756776 A, 26.03.1996 | |||
| RU 93043106 A, 20.03.1996 | |||
| RU 2059631 C1, 10.05.1996. |

