Настоящее изобретение относится к новым промежуточным продуктам, используемым в синтезе производных 14β-гидрокси-1,14-карбонат-дезацетилбаккатина III и к способам их получения. Промежуточные соединения, полученные способом по данному изобретению, могут быть использованы при получении новых производных таксана, обладающих противоопухолевой активностью.
Таксаны представляют собой один из наиболее важных классов противоопухолевых агентов, разработанных в последние годы. Паклитаксел представляет собой сложный дитерпен, выделенный из коры Taxus brevifolia и рассматривается как «ведущее соединение» для лечения рака. В настоящее время идет интенсивный поиск производных таксана, обладающих более высокой терапевтической активностью и улучшенным фармакокинетическим профилем. Конкретные разработки относятся к производным баккатина III, модифицированным различным образом по отношению к основной структуре. Примерами указанных соединений являются производные 14β-гидрокси-баккатина III, описанные в US 5705508, WO 97/43291, WO 96/36622. В настоящее время производные 14β-гидрокси-1,14-карбонат-деацетилбаккатина III получают из предшественника 14β-гидрокси-деацетилбаккатина III, который является природным соединением, получаемым в малых количествах экстракцией из листьев Taxus wallichiana, как описано в ЕР 559019. Существует большая потребность в новых промежуточных соединениях или широко используемых альтернативных способах, которые позволяют просто и эффективно получать производные 14β-гидрокси-1,14-карбонат-деацетилбаккатина III.
В настоящее время обнаружено, что 14β-гидрокси-1,14-карбонат-деацетилбаккатин III может быть получен по способу с использованием 10-дезацетилбаккатина III в качестве исходного соединения, которое, в отличие от 14β-гидрокси-баккатина III, может быть легко выделено в больших количествах из листьев Taxus baccata.
Таким образом, настоящее изобретение относится к способу получения 14β-гидрокси-1,14-карбонат-дезацетилбаккатина III, включающему следующие стадии:
1) защита гидроксигрупп в положениях 7 и 10 10-дезацетилбаккатина III

где R и R1 выбраны из водорода, С1-С10 алкила или арила, С1-С10 алкил- или арил-карбонила, трихлорацетила, С1-С4 триалкилсилила; предпочтительно, когда R и R1 одинаковы, они представляют собой трихлорацетил, тогда как, когда они различны, предпочтительно R является трихлорацетилом и R1 является ацетилом, или R является триэтилом или триметилсилилом и R1 представляет собой ацетил;
2) двустадийное окисление с получением производного, окисленного в положении 13- и гидроксилированного в положении 14

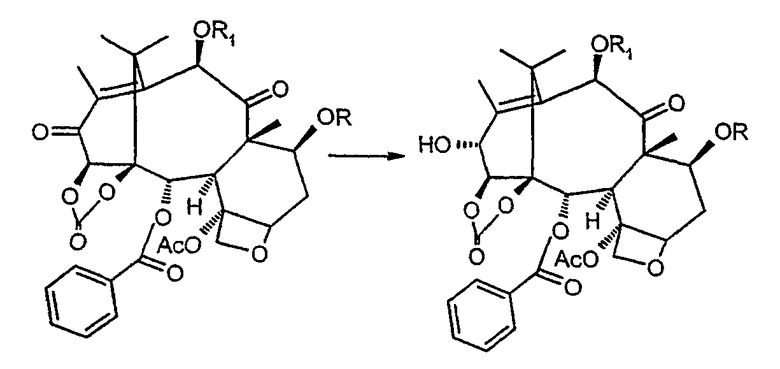
3) карбоксилирование вицинальных гидроксилов в положениях 1 и 14 с получением 1,14-карбонатного производного
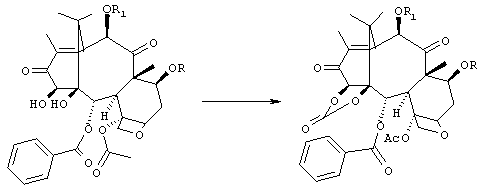
4) восстановление карбонила в положении 13

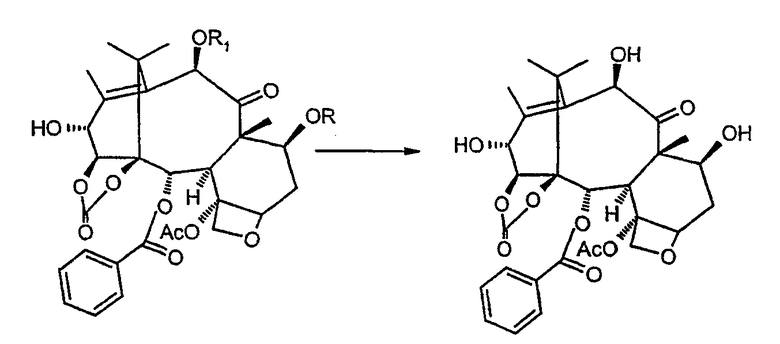
5) удаление защитных групп в положениях 7 и 10

Способы защиты гидроксилов в положениях 7 и 10 описаны Holton et al., Tetrahedron Letters 39 (1998), 2883-2886. Селективная защита гидроксилов исходного соединения дезацетилбаккатина III возможна благодаря их различной реакционной способности. В частности, обнаружено, что реакционная способность по отношению к ацилирующим, алкилирующим или силилирующим агентам изменяется в порядке C(7)-OH>C(10)-OH>C(13)-OH>C(1)-OH, поэтому группы в положениях 7 и 10 могут быть селективно защищены, тогда как гидроксилы в положениях 1 и 13 могут оставаться свободными.
Кроме того, путем изменения условий проведения реакции возможно полностью изменить порядок реакционности гидроксилов в положениях 7 и 10, допуская, таким образом, их различное замещение. Примеры реагентов и реакционных условий, используемых при защите гидроксилов в положении 10 и 7, описаны в публикациях, цитируемых выше.
Стадия окисления гидроксила в положении 13 осуществляется с помощью диоксида магния или диоксида висмута в растворителе, выбранном из ацетонитрила, ацетона или смеси 9:1 этилацетата/метиленхлорида, при интенсивном перемешивании предпочтительно с помощью диоксида магния в ацетонитриле или ацетоне. Реакция протекает быстро с образованием производного, окисленного в положении 13, которое может быть выделено из реакционной среды, тогда как более продолжительное взаимодействие дает производное, окисленное в положении 13 и гидроксилированное в положении 14.
Следующую стадию карбоксилирования гидроксилов в положениях 1 и 14 обычно осуществляют с помощью фосгена или трифосгена в смеси метиленхлорид/толуол в присутствии пиридина. Затем полученное 1,14-карбонатное производное легко может быть восстановлено в положении 13 с образованием соответствующего 13-гидроксипроизводного. Указанное восстановление происходит региоселективно на карбониле в положении 13, тогда как карбонил в положении 9 остается неизмененнным, и стереоселективно, почти исключительно давая 13-α изомер. Эту реакцию обычно осуществляют с помощью борогидрида натрия в метаноле и достигают высоких выходов. Последняя стадия представляет собой удаление защитных групп гидроксилов в положениях 7 и 10 с получением конечного продукта 14β-гидрокси-1,14-карбонат-дезацетилбаккатина III. Условия и реагенты, которые могут быть использованы для селективного удаления защитных групп гидроксилов в положении 7 и 10, описаны Zheng et al., Tetrahedron Lett., 1995, 36, 2001, и Datta et al., J. Org. Chem., 1995, 60, 761. Полученный конечный продукт представляет собой чрезвычайно полезное промежуточное соединение для синтеза разнообразных производных таксана. Как указано выше, это промежуточное соединение до сих пор получали исходя из 14β-гидрокси-баккатина III, экстрагируемого из листьев Taxus wallichiana с низкими выходами. Способ по настоящему изобретению позволяет получать те же промежуточные соединения с высокими выходами исходя из соединения, доступного в больших количествах. Примеры соединений, обладающих противоопухолевой активностью, которые могут быть получены исходя из 14β-гидрокси-1,14-карбонат дезацетилбаккатина III, представлены в US 5705508, WO 97/43291, WO 96/36622.
В соответствии с предпочтительным вариантом выполнения способа по настоящему изобретению дезацетилбаккатин III подвергают взаимодействию с трихлорацетилхлоридом в метиленхлориде в присутствии триэтиламина и с использованием N,N-диметиламинопиридина (ДМАП) в каталитических количествах. Оказалось, что использование трихлорацетата в качестве защитной группы при окислении, карбоксилировании и восстановлении очень удобно в соответствии со способом по данному изобретению. В частности, производное 7,10-бистрихлорацетата, полученное с количественным выходом из исходного соединения, после окисления и карбоксилирования легко восстанавливается в положении 13 с одновременным удалением защитных трихлорацетатных групп с получением 14β-гидрокси-1,14-карбонат-дезацетилбаккатина III. Использование ДМАП в каталитических количествах обеспечивает определенные преимущества с точки зрения промышленности и окружающей среды, принимая во внимание то, что до сих пор ацилирование этого субстрата проводили в пиридине с последующими проблемами слива оставшегося растворителя.
Следующие промежуточные соединения, полученные в соответствии с предпочтительным вариантом выполнения, описанным выше, являются частью настоящего изобретения.

Следующие примеры иллюстрируют данное изобретение более подробно.
Пример I
Получение 7,10-бистрихлорацетил-10-дезацетилбаккатина III
Первый вариант
4,77 мл ангидрида трихлоруксусной кислоты (42,32 ммоль) добавляют по каплям к раствору 10 г 10-деацетилбаккатина III (18,4 ммоль) в 125 мл сухого метиленхлорида и 42 мл пиридина. Реакционную смесь перемешивают в течении трех часов или до завершения реакции, контролируя с помощью тонкослойной хроматографии (ТСХ) на силикагеле с использованием в качестве элюента смеси 1:1 н-гексан/этилацетат. После завершения реакции добавляют 5 мл метанола для разложения избытка ангидрида трихлоруксусной кислоты, затем добавляют воду. Органическую фазу тщательно промывают подкисленной (HCl) водой для удаления пиридина, тогда как оставшуюся органическую фазу сушат над MgSO4 и концентрируют досуха в вакууме с получением бледно-желтого твердого вещества (17 г), которое кристаллизуют из хлороформа, [α]D -34° (CH2Cl2, C 5,8). ИК (KBr): 3517, 1771, 1728, 1240, 981, 819, 787, 675 см-1;
1Н-ЯМР (200 МГц): δ 8,11 (Bz C), 7,46 (Bz, BB'), 6,50 (с, H-10), 5,72 (м, H-7, H-29), 5,02 (д, J=8 Гц, H-5), 4,95 (8м, H-13), 4,37 (д, J=8 Гц, H-20а), 4,18 (д, J=8 Гц, H-20b), 4,02 (д, J=6 Гц, H-3), 2,32 (с, 4-Ac), 2,22 (с, H-18), 1,91 (с, H-19), 1,25 и 1,11 (с, H-16, H-17), 1,94 (м, H 14α), 1,89 (м, H 14β).
Второй вариант
10-Дезацетилбаккатин III (10 г, 18,38 ммоль) суспендируют в CH2Cl2 (120 мл), добавляют ДМАП (220 мг, 1,4 ммоль, 0,1 экв.) и охлаждают до 0°C на ледяной бане. Добавляют Et3N (10,26 мл, 73,6 ммоль, 4 экв.) и сразу же после этого Cl3CCOCl (4,12 мл, 36,8 ммоль, 2 экв.) в токе азота в течение 5 мин, поддерживая температуру ниже 10°C. После завершения добавления смесь оставляют перемешиваться в ледяной бане в течение 15 мин, затем баню оставляют и реакционную смесь перемешивают при комнатной температуре в течение 1 ч. Через 1 ч реакционную смесь анализируют с помощью ТСХ (AcOEt 2/н-гексан 3, Rf 10-DAB III=0,05, Rf 7,10-бистрихлорацетил-10-DAB III=0,26) и добавляют Cl3CCOCl (1 мл, 0,5 экв.). Перемешивание продолжают при комнатной температуре в течение 10 мин, затем реакционную смесь выливают в химический стакан, содержащий 160 г растертого льда и оставляют при перемешивании до установления равновесного состояния при комнатной температуре (около 1 ч). Затем отделяют водную фазу и экстрагируют СН2Cl2 (3×40 мл). Объединенные органические фазы промывают 1 н. HCl (20 мл), затем насыщенным раствором NaHCO3 (20 мл), сушат над Na2SO4 и выпаривают растворитель. Масса неочищенного продукта 16,5 г. После кристаллизации из хлороформа ИК, 1Н-ЯМР и [α]D спектры согласуются с теми, которые получены для соединений с использованием пиридина и ангидрида трихлоруксусной кислоты.
Пример II
Окисление по положению 13 и гидроксилирование по положению 14 7,10-бистрихлорацетата 10-дезацетилбаккатина III
30 г активированного MnO2 добавляют к раствору 7,10-бистрихлорацетата 10-дезацетилбаккатина III (3 г) в ацетонитриле (40 мл), перемешивают суспензию магнитной мешалкой при комнатной температуре и наблюдают за ходом реакции с помощью ТСХ (смесь петролейный эфир-этилацетат 5:5; Rf исходного сырья примерно 0,31). Примерно через один час завершается образование 13-дегидропроизводного (ТСХ анализ, Rf 13-дегидропроизводного примерно 0,50). Затем перемешивание продолжают в течение приблизительно 72 часов, на протяжении этого времени 13-дегидропроизводное медленно окисляется до соответственно 14β-гидроксипроизводного (Rf примерно 0,36). Реакционную смесь фильтруют через целит и осадок повторно промывают этилацетатом. Растворитель выпаривают и остаток очищают колоночной хроматографией на силикагеле (100 мл, элюент - смесь петролейный эфир-этилацетат 7:3) с получением 170 мг 13-дегидропроизводного и 2,38 г 14β-гидрокси-13-дегидропроизводного.
13-Дегидро-14β-гидрокси-10-дезацетилбаккатин III, 7,10-бистрихлорацетат, белый порошок, т. пл. 97°C. ИК (KBr диск): 3440, 1780, 1767, 1736, 1686, 1267, 1232, 1103, 1010, 854 см-1;
1Н-ЯМР (200 МГц, CDCl3): δ 8,07 (Bz AA'), 7,60 (Bz, C), 7,49 (Bz, BB'), 6,52 (c, H-10), 5,92 (д, J=6,7 Гц, H-2), 5,70 (ушир. т, J=8,0 Гц, H-7), 4,95 (ушир. д, J=8,2 Гц, H-5), 4,37 (д, J=8,2 Гц, H-20a), 4,31 (д, J=8,2 Гц, H-20b), 4,17 (с, H14), 4,02 (д, J=6,7 Гц, H-3), 2,71 (м, H-6), 2,29 (с, OAc), 2,17 (с, OAc), 1,96 (с, H-18), 1,27, 1,01 (с, H-16, H-17 и H-19).
Пример III
Окисление/гидроксилирование 7-триэтилсилилбаккатина III
10 г активированного MnO2 добавляют к раствору 7-триэтилсилилбаккатина III (1,0 г) в ацетонитриле (10 мл), перемешивают суспензию магнитной мешалкой при комнатной температуре и наблюдают за ходом реакции с помощью ТСХ (смесь 6:4 петролейный эфир-этилацетат; Rf исходного сырья примерно 0,25). Примерно через два часа завершается образование 13-дегидропроизводного (ТСХ анализ, Rf 13-дегидропроизводного приблизительно 0,45). Затем продолжают перемешивание примерно в течение 188 часов, в процессе этого добавляют еще MnO2 (10 г). 13-Дегидропроизводное медленно окисляется до, соответственно, 14β-гидроксипроизводного (Rf примерно 0,38). Реакционную смесь фильтруют через целит и остаток промывают этилацетатом. Выпаривают растворитель и остаток очищают колоночной хроматографией на силикагеле (40 мл, элюент - смесь 7:3 петролейный эфир-этилацетат) с получением 126 мг 13-дегидропроизводного, 479 мг (46%) 14β-гидрокси-13-дегидропроизводного и 189 мг их смеси. 13-Дегидро-7-триэтилсилилбаккатин III, белый порошок, т. пл. 168°C, [α]D 25 -35 (CH2Cl2, C 0,67). ИК (KBr): 3488, 1726, 1711, 1676, 1373, 1269, 1244, 1230, 1105 см-1. 1Н-ЯМР (200 МГц CDCl3): δ 8,07 (Bz AA'), 7,60 (Bz, C), 7,49 (Bz, BB'), 6,59 (c, H-10), 5,69 (д, J=6,9 Гц, H-2), 4,92 (д, J=8,2 Гц, H-5), 4,48 (дд, J=10,6 Гц, H-7), 4,33 (д, J=8,0 Гц, H-20a), 4,12 (д, J=8,0 Гц, H-20b), 3,91, (д, J=6,9 Гц, H-3), 2,96 (д, J=20 Гц, H-14a), 2,65 (д, J=20 Гц, H-20b), 2,50 (м, H-6α), 2,23 (c, OAc), 2,19 (c, OAc + H-18), 1,67, 1,28, 1,19 (c, H-16, H-17 и H-19), 0,19 (м, TES).
13-Дегидро-14β-гидрокси-10-дезацетилбаккатин III, 7,10-бистрихлорацетат, белый порошок, т. пл. 153°C, [α]D 25 +20 (CH2Cl2, C 0,75). ИК (KBr): 3431, 1723, 1692, 1371, 1269, 1242, 1223, 1096 см-1. 1Н-ЯМР (500 МГц CDCl3): δ 8,06 (Bz AA'), 7,60 (Bz, C), 7,48 (Bz, BB'), 6,51 (c, H-10), 5,88 (д, J=6,9 Гц, H-2), 4,90 (д, J=8,2 Гц, H-5), 4,47 (дд, J=10,67 Гц, H-7), 4,30 (д, J=8 Гц, H-20a), 4,28 (д, J=8,2 Гц, H-20b), 4,13 (ушир. д, J=2 Гц, H-14), 3,84 (д, J=6,9 Гц, H-3), 3,69 (ушир. д, J=2 Гц, 14-OH), 3,62 (c, 1-OH), 2,52 (м, H-6α), 2,24 (c, OAc), 2,21 (c, OAc), 2,11 (c, H-18), 1,92 (м, H-6β), 1,74, 1,56, 1,28 (c, Н-16, H-17 и H-19), 0,94 (м, TES), 0,59 (м, TES). HRNS: 714,3092 (вычислено для C37H50O12Si 714,3092).
Пример IV
Окисление/гидроксилирование 7-триэтилсилилбаккатина III
10 г активированного MnO2 добавляют к раствору 7-триэтилсилилбаккатина III (1,0 г) в ацетонитриле (10 мл), перемешивают при комнатной температуре и наблюдают за ходом реакции с помощью ТСХ (смесь петролейный эфир-этилацетат 6:4; Rf исходного сырья примерно 0,25). Примерно через два часа завершается образование 13-дегидропроизводного (ТСХ анализ, Rf 13-дегидропроизводного примерно 0,45). Затем продолжают перемешивание в течение приблизительно 188 часов, в течение которых добавляют еще MnO2 (10 г). 13-Дегидропроизводное медленно окисляется до, соответственно, 14β-гидроксипроизводного (Rf примерно 0,38). Реакционную смесь фильтруют через целит и остаток промывают этилацетатом. Растворитель выпаривают и остаток очищают колоночной хроматографией на силикагеле (40 мл, элюент - смесь 7:3 петролейный эфир-этилацетат) с получением 126 мг 13-дегидропроизводного, 479 мг (46%) 14β-гидрокси-13-дегидропроизводного и 189 мг их смеси.
13-Дегидро-7-триэтилсилилбаккатин III, белый порошок, т. пл. 210°C, [α]D 25 -48 (CH2Cl2, C 0,50). ИК (KBr): 3478, 1728, 1676, 1373, 1271, 1240, 1071, 1026 см-1. 1Н-ЯМР (200 МГц, CDCl3): δ 8,07 (Bz AA'), 7,64 (Bz, C), 7,50 (Bz, BB'), 6,46 (c, H-10), 5,70 (д, J=6,9 Гц, H-2), 4,95 (д, J=8,2 Гц, H-5), 4,51 (дд, J=10,7 Гц, H-7), 4,32 (д, J=8,4 Гц, H-20a), 4,14 (д, J=8,4 Гц, H-20b), 3,92, (д, J=6,9 Гц, H-3), 2,99 (д, J=20 Гц, H-14a), 2,68 (д, J=20 Гц, H-14b), 2,56 (м, H-6α), 2,29 (c, OAc), 2,18 (c, OAc), 2,08 (c, H-18), 1,68, 1,29, 1,20 (c, H-16,H-17 и H-19), 0,19.
13-Дегидро-14β-гидрокси-7-триэтилсилилбаккатин III, белый порошок, т. пл. 220°C, [α]D 25 +19 (CH2Cl2, C 0,42). ИК (KBr): 3568, 1710, 1719, 1686, 1372, 1282, 1240, 1219, 1073 см-1. 1Н-ЯМР (200 МГц, CDCl3): δ 8,09 (Bz AA'), 7,60 (Bz, C), 7,51 (Bz, BB'), 6,39 (c, H-10), 5,89 (д, J=6,9 Гц, H-2), 4,94 (д, J=8,2 Гц, H-5), 4,47 (дд, J=10,7 Гц, H-7), 4,31 (ушир. c, -H-20a + H-20b), 4,15 (c, H-14), 3,69 (д, J=6,9 Гц, H-3), 2,29 (c, OAc), 2,16 (c, H-18), 2,14 (c, OAc), 1,74, 1,21, 1,10 (c, H-16, H-17 и H-19). HRMS: 600,6112 0,19 (вычислено для C31H36O12Si 600,6103).
Пример V
Получение 1,14-карбонат-13-дегидро-7-тес-баккатина III
Раствор 13-дегидро-14β-гидрокси-7-триэтилсилилбаккатина III (124 мг, 1,17 ммоль) в СН2Cl2 (1 мл) и пиридин (0,56 мл, 6,8 ммоль, 20 моль эквив.) добавляют по каплям в течение 5 мин к раствору фосгена (1,8 мл 20% раствора в толуоле, 3,4 ммоль, 20 моль эквив.) в СН2Cl2 (2 мл). Смесь перемешивают при комнатной температуре в течение 1 часа и затем избыток фосгена нейтрализуют насыщенным раствором NaHCO3 и экстрагируют СН2Cl2. Органическую фазу промывают насыщенным раствором NaHCO3, насыщенным солевым раствором и сушат (Na2SO4). Растворитель выпаривают с получением красноватого остатка, который очищают на небольшой колонке с силикагелем (около 5 мл, элюент - гексан/этилацетат 8:2) с получением 118 мг (92%) карбоната. В том случае, когда реакцию проводят с триэтиламином в качестве основания без обратного добавления, получают смесь 1,14-карбоната и 2-дебензоил-1,2-карбонат-14 бензоата (примерно 1:15).
13-Дегидро-14β-гидрокси-7-триэтилсилилбаккатин III 1,14-карбонат, белый порошок, т. пл. 153°C, [α]D 25 +23 (CH2Cl2, C 0,75). ИК (KBr): № полосы OH 1834, 1734, 1709, 1373, 1242, 1225, 1088, 1057 см-1. 1Н-ЯМР (200 МГц CDCl3): δ 7,99 (Bz AA'), 7,60 (Bz, C), 7,48 (Bz, BB'), 6,51 (c, H-10), 6,12 (д, J=6,9 Гц, H-2), 4,90 (д, J=8,2 Гц, H-5), 4,78 (c, H-14), 4,44 (дд, J=10,7 Гц, H-7), 4,34 (д, J=8 Гц, H-20a), 4,19 (д, J=8,2 Гц, H-20b), 3,80 (д, J=6,9 Гц, H-3), 2,50 (м, H-6α), 2,23 (c, OAc), 2,22 (c, OAc), 2,19 (c, H-18), 1,92 (м, H-6β), 1,72, 1,39, 1,26 (c, -H-16, H-17 и H-19), 0,90 (м, TES), 0,56 (м, TES). HRNS: 740,2851 (вычислено для C38H48O13Si 740, 2864).
13-Дегидро-14β-гидроксибаккатин III 1,14-карбонат, белый порошок, 240°C, [α]D 25 -2,5 (CH2Cl2, C 0,4). ИК (KBr): 3539, 1831, 1736, 1240, 1088, 1068, 1057, 1024 см-1. 1Н-ЯМР (200 МГц CDCl3): δ 7,98 (Bz AA'), 7,61 (Bz, C), 7,50 (Bz, BB'), 6,39 (c, H-10), 6,14 (д, J=6,9 Гц, H-2), 4,98 (д, J=8,2 Гц, H-5), 4,80 (c, H-14), 4,43 (дд, J=10,7 Гц, H-7), 4,35 (д, J=8 Гц, H-20a), 4,24 (д, J=8,2 Гц, H-20b), 3,80 (д, J=6,9 Гц, H-3), 2,50 (м, H-6α), 2,30 (c, OAc), 2,20 (c, OAc), 2,15 (c, H-18), 1,90 (м, H-6β), 1,74, 1,34, 1,25 (c, H-16, H-17 и H-19). HRMS: 626,2005 (вычислено для C33H34О1 626,1999).
Пример VI
Получение 1,14-карбонат-7-О-триэтилсилилбаккатина III
Избыток NaBH4 (примерно 20 мг) добавляют малыми порциями к раствору 13-дегидро-14β-гидрокси-7-триэтилсилилбаккатин III 1,14-карбоната (50 мг) в метаноле (5 мл). Через 30 мин добавляют в реакционную смесь насыщенный NH4Cl, экстрагируют этилацетатом, промывают солевым раствором, высушивают над Na2SO4 и удаляют растворитель с получением остатка, который очищают колоночной хроматографией на силикагеле (примерно 5 мл, элюирование гексан-этилацетатом 8:2) с получением 35 мг 13α-гидроксипроизводного и 9 мг 13β-гидроксипроизводного.
14β-Гидрокси-7-триэтилсилилбаккатин III 1,14-карбонат, [α]D 25 -35 (CH2Cl2, C 0,60). ИК (KBr): 3054, 1819, 1736, 1603, 1371, 1261, 1238, 1090, 1069 см-1. 1Н-ЯМР (200 МГц, CDCl3): δ 8,06 (Bz AA'), 7,65 (Bz, C), 7,50 (Bz, BB'), 6,47 (c, H-10), 6,12 (д, J=6,9 Гц, H-2), 5,05 (ушир. д, J=5,5 Гц, H-13), 4,98 (ушир. д, J=9 Гц, H-5), 4,83 (д, J=5 Гц, H-14), 4,50 (дд, J=10,7 Гц, H-7), 4,34 (д, J=8 Гц, H-20a), 4,23 (д, J=8 Гц, H-20b), 3,75 (д, J=6,9 Гц, H-3), 2,56 (м, H-6α), 2,34 (c, OAc), 2,22 (c, OAc), 1,78 (м, H-6β), 1,35 (c, H-18), 1,75, 1,18, 0,95 (c, H-16, H-17 и H-19), 0,90 (м, TES), 0,62 (м, TES).
14β-Гидрокси-7-триэтилсилил-13-эпибаккатин III 1,14-карбонат, некристаллический, [α]D 25 -13 (CH2Cl2, C 0,60). ИК (KBr): 3630, 1825, 1734, 1603, 1375, 1262, 1091, 1071, 1049 см-1. 1Н-ЯМР (200 МГц, CDCl3): δ 8,01 (Bz AA'), 7,63 (Bz, C), 7,48 (Bz, BB'), 6,44 (c, H-10), 6,12 (д, J=7,2 Гц, H-2), 4,90 (ушир. д, J=9 Гц, H-5), 4,81 (д, J=8 Гц, H-14), 4,48 (ушир., J=8, H-13), 4,50 (дд, J=10, 7 Гц, H-7), 4,41 (д, J=8 Гц, H-20a), 4,31 (д, J=8 Гц, H-20b), 3,68 (д, J=7,2 Гц, H-3), 2,60 (м, H-6α), 2,32 (c, OAc), 2,26 (c, H-18), 2,21 (c, OAc), 1,80 (м, H-6β), 1,72, 1,43, 1,27 (c, H-16, H-17 и H-19), 0,93 (м, TES), 0,61 (м, TES).
Пример VII
Получение 13-дегидро-14β-гидрокси-7,10-бистрихлорацетилбаккатин III 1,14-карбоната
К раствору 13-дегидро-14β-гидрокси-7,10-бистрихлорацетилбаккатина III (200 мг) в СН2Cl2 (2 мл) и пиридине (1,12 мл, 20 эквив.) добавляют в течение 5 мин раствор фосгена (20% в толуоле, 3,6 мл, 20 эквив.) в СН2Cl2 (2 мл). Смесь перемешивают при комнатной температуре в течение 1 ч, затем нейтрализуют избыток фосгена насыщенным раствором NaHCO3 (3 мл). Смесь экстрагируют СН2Cl2, органическую фазу промывают насыщенным раствором NaHCO3, затем насыщенным раствором NaCl и высушивают над Na2SO4. После удаления растворителя остаток очищают колоночной хроматографией на силикагеле (элюент - гексан/ AcOEt 9:1) с получением 175 мг(89%) карбоната.
13-Дегидро-14β-гидрокси-7,10-бистрихлорацетилбаккатин III 1,14-карбонат, твердое белое некристаллическое вещество. ИК (KBr): 1834, 1771, 1735, 1709, 1232, 1103, 1010, 854 см-1.
1Н-ЯМР (200 МГц, CDCl3): δ 8,03 (Bz AA'), 7,60 (Bz, C), 7,50 (Bz, BB'), 6,52 (c, H-10), 5,92 (д, J=6,7 Гц, H-2), 5,70 (ушир. т, J=8,0 Гц, H-7), 4,95 (ушир. д, J=8,2 Гц, H-20b), 4,77 (c, H-14), 4,02 (д, J=6,7 Гц, H-3), 2,71 (м, H-6), 2,29 (c, OAc), 1,96 (c, H-18), 1,27-1,01 (м, H-16, H-17, H-19).
Пример VIII
Получение 14β-гидрокси-10-деацетилбаккатин III 1,14-карбоната
Раствор 13-дегидро-14β-гидрокси-7,10-бистрихлорацетилбаккатин III 1,14-карбоната (500 мг) в MeOH (8 мл) охлаждают до 0°C на ледяной бане и в течение 5 мин к нему добавляют твердый NaBH4 (44 мг). Смесь перемешивают при комнатной температуре в течение 1 ч затем охлаждают до 0°C. В течение 5 мин добавляют ацетон (2 мл), смесь концентрируют, затем добавляют AcOEt (10 мл) и фильтруют через целит. Чистый раствор промывают насыщенным раствором NaCl и высушивают над Na2SO4. Растворитель выпаривают с получением остатка (4,5:1 смесь эпимеров С13), который очищают колоночной хроматографией на силикагеле (элюент - гексан/AcOEt 1:1) с получением 251 мг 13β эпимера и 55 мг 13α эпимера (88% всего) карбоната с удаленной защитной группой.
13α-14β-Гидрокси-10-дезацетилбаккатин III 1,14-карбонат, некристаллическое твердое белое вещество. ИК (KBr): 3520 (OH), 1834, 1709,1232, 1103, 1010, 854 см-1.
1Н-ЯМР (200 МГц, CDCl3): δ 8,03 (Bz AA'), 7,60 (Bz, C), 7,50 (Bz, BB'), 6,27 (c, H-10), 5,92 (д, J=6,7 Гц, H-2), 4,95 (ушир. д, J=8,2 Гц, H-20b), 4,85 (м, H-13), 4,77 (c, H-14), 4,42 (ушир. т, J=8,0 Гц, H-7), 4,02 (д, J=6,7 Гц, H-3), 2,71 (м, H-6), 2,29 (c, OAc), 1,96 (c, H-18), 1,27-1,01 (м, H-16, H-17, H-19).
13α-14β-Гидрокси-10-дезацетилбаккатин III 1,14-карбонат, некристаллическое твердое белое вещество. ИК (KBr): 3520 (OH), 1834, 1709, 1232, 1103, 1010, 854 см-1.
1Н-ЯМР (200 МГц, CDCl3): δ 8,03 (Bz AA'), 7,60 (Bz, C), 7,50 (Bz, BB'), 6,27 (c, H-10), 5,92 (д, J=6,7 Гц, H-2), 4,95 (ушир. д, J=8,2 Гц, H-20b), 4,80 (м, H-13), 4,77 (c, H-14), 4,42 (ушир. т, J=8,0 Гц, H-7), 4,02 (д, J=6,7 Гц, H-3), 2,71 (м, H-6), 2,29 (c, OAc), 1,96 (c, H-18), 1,27 - 1,01 (м, H-16, H-17, H-19).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТАКСАНА | 2001 |
|
RU2275365C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПАКЛИТАКСЕЛА | 2001 |
|
RU2276147C2 |
| ПОЛУСИНТЕТИЧЕСКИЕ ТАКСАНЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2001 |
|
RU2259363C2 |
| ПОЛУСИНТЕТИЧЕСКИЙ ТАКСАН, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2134688C1 |
| ПРОИЗВОДНЫЕ ТАКСАНА, ФУНКЦИОНАЛИЗИРОВАННЫЕ ПО 14-ПОЛОЖЕНИЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2320652C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ТАКСАНА | 2007 |
|
RU2434014C2 |
| ПРОИЗВОДНЫЕ ТАКСАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2168513C2 |
| ПРОИЗВОДНЫЕ ТАКСАНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2245882C2 |
| СПОСОБ ПОЛУЧЕНИЯ 14БЕТА-ГИДРОКСИБАККАТИН III-1,14-КАРБОНАТА | 2002 |
|
RU2291866C2 |
| ПРОИЗВОДНЫЕ 10-ДЕАЦЕТИЛБАККАТИНА III И 10-ДЕАЦЕТИЛ-14β-ГИДРОКСИБАККАТИНА III, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2152936C1 |
Изобретение относится к новому способу получения 14β-гидрокси-1,4-карбонат-дезацетилбаккатина III и промежуточных продуктов, используемых при получении новых производных таксана, обладающих противоопухолевой активностью. Способ включает следующие стадии: a) защиту гидроксилов в положениях 7 и 10 10-дезацетилбаккатина III, где R и R1 выбраны из водорода, C1-C10 алкила или арила, C1-C10 алкил- или арилкарбонила, трихлорацетила, C1-C4 триалкилсилила; предпочтительно, когда R и R1 одинаковые, они представляют собой трихлорацетил, тогда как, когда они различны, предпочтительно R представляет собой трихлорацетил и R1 представляет собой ацетил, или R представляет собой триэтил или триметилсилил и R1 представляет собой ацетил; b) двустадийное окисление с получением производного, окисленного до карбонила в положении 13 и гидроксилированного в положении 14; c) карбоксилирование вицинальных гидроксилов в положениях 1 и 14 с получением 1,14-карбонатного производного; d) восстановление карбонила в положении 13; e) удаление защитных групп в положениях 7 и 10. Изобретение также относится к промежуточным продуктам. Технический результат - получения промежуточных соединений для синтеза таксана. 2 н. и 6 з.п. ф-лы.
a. защиту гидроксилов в положениях 7 и 10 10-дезацетилбаккатина III:

где R и R1 выбраны из водорода, С1-С10 алкила или арила, С1-С10 алкил- или арил-карбонила, трихлорацетила, С1-С4 триалкилсилила; предпочтительно, когда R и R1 одинаковые, они представляют собой трихлорацетил, тогда как, когда они различны, предпочтительно, R представляет собой трихлорацетил и R1 представляет собой ацетил, или R представляет собой триэтил или триметилсилил и R1 представляет собой ацетил;
b. двухстадийное окисление с получением производного, окисленного до карбонила в положении 13 и гидроксилированного в положении 14:
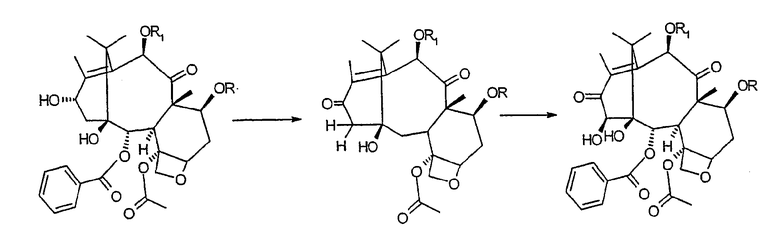
с. карбоксилирование вицинальных гидроксилов в положениях 1 и 14 с получением 1,14-карбонатного производного:

d. восстановление карбонила в положении 13:
е. удаление защитных групп в положениях 7 и 10:
13-дегидро-14β-гидрокси-10-дезацетилбаккатин III,
1,14-карбонат-13-дегидро-7-триэтилсилилбаккатин III,
13-дегидро-14β-гидрокси-7,10-бистрихлорацетилбаккатин III
1,14-карбонат.
| US 5698712 A, 16.12.1997.US 5750562 A, 12.05.1998.EP 0559019 A1, 08.09.1993.US 5705508 A, 06.01.1998.RU 2144920 C1, 27.01.2000.RU 2115649 C1, 20.07.1998. |

