Область техники, к которой относится изобретение
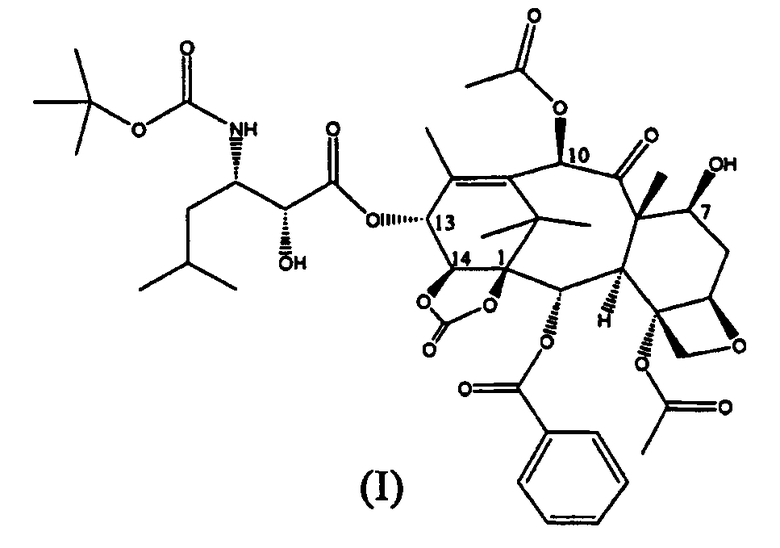
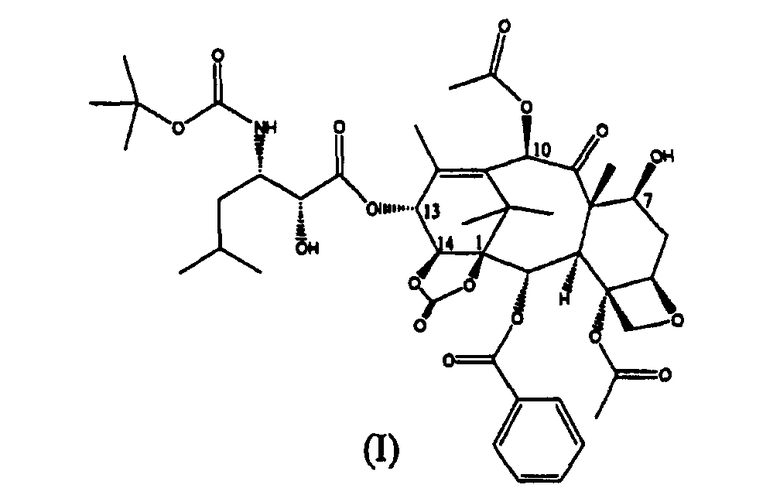
Настоящее изобретение относится к производным таксана, в частности к способу получения 13-N-(Boc-β-изобутилсеринил)-14-β-гидроксибаккатин III-1,14-карбоната (I):
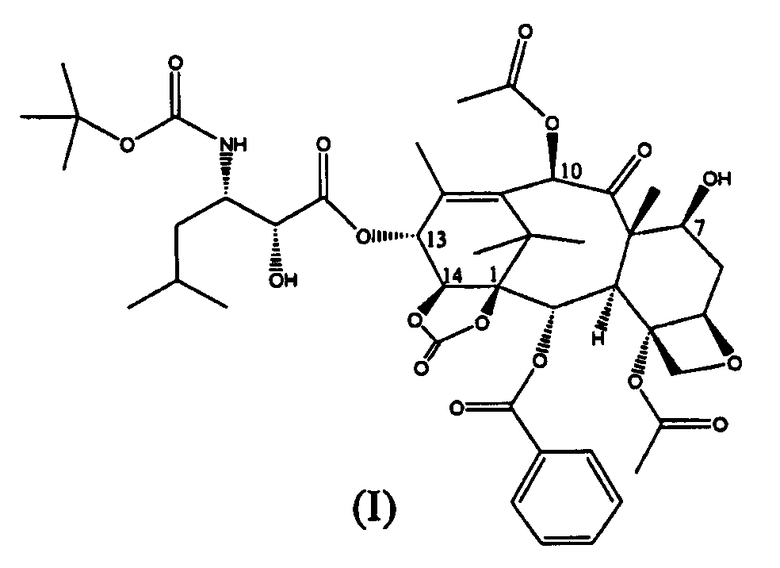
Уровень техники
Соединение (I), впервые раскрытое в WO 01/02407, является высокоактивным в отношении опухолей молочной железы, легкого, яичников, кишечника, простаты, почек и поджелудочной железы, а также, в случае устойчивости, в отношении к известным противоопухолевым агентам, таким как адриамицин, винбластин и некоторые производные Pt.
В литературе также описан ряд синтетических методов получения (I), которые включают использование оксазолидин-защищенной боковой цепи. В патенте US 6737534 10-дезацетилбаккатин III, исходное вещество, легкодоступное из листьев Taxus baccata, сначала защищают по 7- и 10-му положениям, окисляют по 13-положению и затем гидроксилируют по 14-му положению. После этого осуществляется карбонилирование вицинальных 1,14 гидроксигрупп фосгеном с получением 1,14-карбонатного производного, затем следует восстановление 13-кето группы до гидроксигруппы и удаление защитных групп с 7- и 10-го положений с получением 10-дезацетил-14β-гидроксибаккатин III-1,14-карбоната, который селективно ацетилируют по 10-гидроксигруппе, превращают в 7-триэтилсилильное производное и вводят в реакцию с (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислотой. Удаление триэтилсилильной и диметоксибензилиденовой защитных групп приводит к соединению (I).
WO 01/02407 раскрывает два синтетических пути к соединению (I), оба исходя из 14β-гидрокси-10-дезацетилбаккатина III, компонента листьев Taxus wallichiana. Первый способ, описываемый как способ (А), включает следующие стадии:
(а) превращение 14β-гидрокси-10-дезацетилбаккатина III в 7-триэтилсилильное производное;
(б) карбонилирование 1, 14 гидроксигрупп;
(в) ацетилирование 10-гидроксигруппы;
(г) реакцию продукта стадии (в) с (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислотой;
(д) удаление триэтилсилильной и диметоксибензилиденовой групп из продукта стадии (г).
Второй способ, описываемый как способ (Б), включает следующие стадии:
(а') ацетилирование 10-гидроксигруппы в 14β-гидрокси-10-дезацетилбаккатине III;
(б') карбонилирование 1, 14 гидроксигрупп;
(в') силилирование 7-гидроксигруппы;
(г') реакцию продукта стадии (в') с (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислотой;
(д') удаление триэтилсилильной и диметоксибензилиденовой групп из продукта стадии (г').
В способе Б проведение ацетилирования 10-гидроксигруппы до введения защиты по 7-му положению позволяет избежать образования смеси региоизомеров по 7- и 10-положениям, которое происходит в способе А, в котором ацетилирование проводится после защиты 7-гидроксигруппы. Поэтому способ Б является предпочтительным по сравнению со способом А, поскольку он является высокорегиоселективным. Однако масштабирование способа Б до мультикилограммовых количеств представляет собой проблему, поскольку большие количества фосгена не могут быть загружены в реактор из соображений безопасности, и, соответственно, стадия (б') не может быть осуществлена путем добавления 14β-гидрокси-10-дезацетилбаккатина III к фосгену. Если вместо этого фосген барботируют через раствор 14β-гидрокси-10-дезацетилбаккатина III, образуется значительное количество (около 7%) примеси (II).
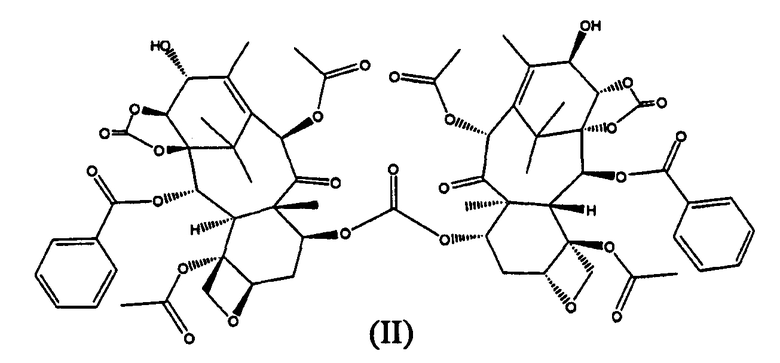
Образование (II) протекает из-за того, что 7-гидроксигруппа также реакционноспособна в отношении фосгена, что приводит к образованию соединения (III).
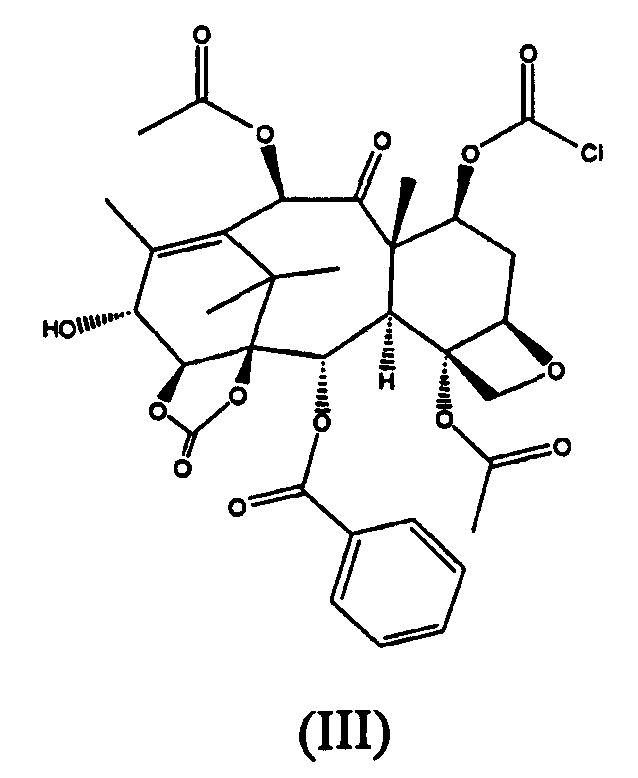
Таким образом, когда карбонилирование проводится в большом масштабе, и фосген барботируют в реактор, соединение (III) реагирует с 14β-гидрокси-10-дезацетилбаккатином III, с образованием (II).
Эта примесь образуется также, когда способ Б проводят в меньшем масштабе, однако ее количество составляет менее 0,4%.
Из-за близкого структурного сходства с 14β-гидроксибаккатин III-1,14-карбонатом, соединение (II) может быть удалено только колоночной хроматографией, что снижает выход и увеличивает стоимость способа, особенно в промышленном масштабе.
Еще одним недостатком способа Б является тот факт, что триэтилсилилфторид, образующийся после удаления ТЭС (триэтилсилильной) группы, не может быть полностью удален кристаллизацией, и для получения конечного продукта, соответствующего по чистоте требованиям, предъявляемым к фармацевтическим продуктам, необходима колоночная хроматография низкого давления. Однако широко известно, что колоночная хроматография низкого давления в промышленных масштабах является затруднительной, дорогостоящей и приводит к проблемам, связанным с транспортировкой и уничтожением окиси кремния, загрязненной токсичными материалами.
Сущность изобретения
В настоящее время было обнаружено, что вышеупомянутые недостатки могут быть преодолены проведением стадии (б') с бис(трихлорметил)карбонатом вместо фосгена и проведением стадии (в') с трихлороацетилхлоридом вместо триэтилсилилхлорида.
Таким образом, изобретение относится к способу получения соединения формулы (I):
который включает следующие стадии:
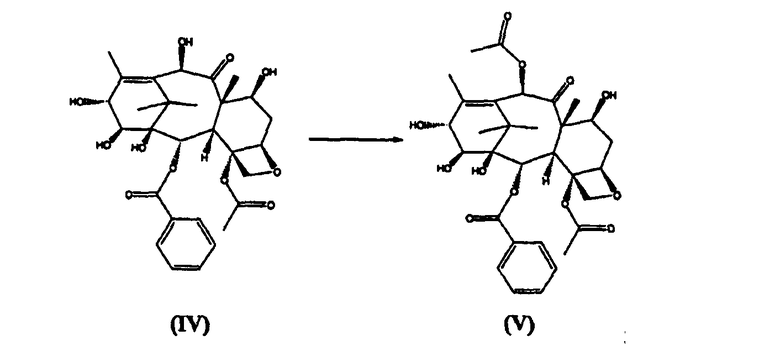
а) ацетилирование 10-гидроксигруппы 14β-гидрокси-10-дезацетилбаккатина III (IV)
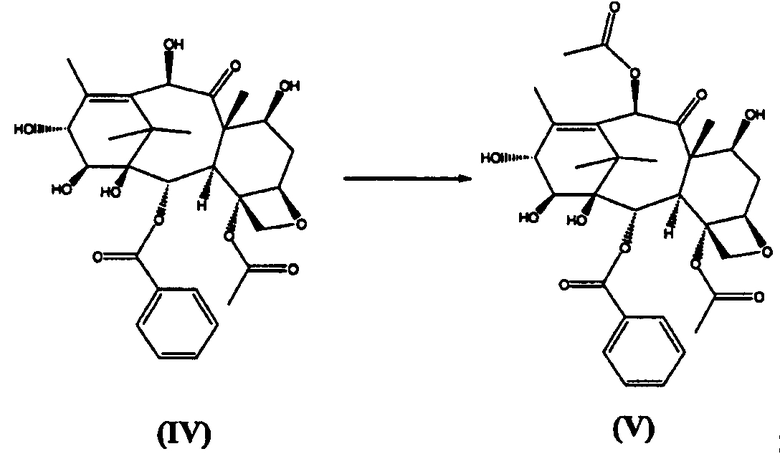
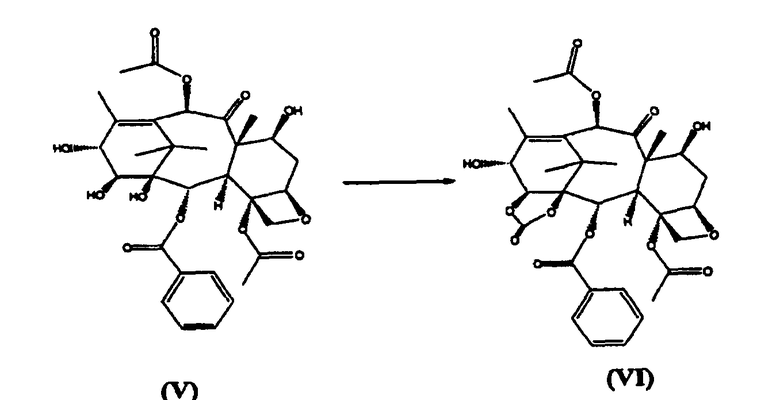
б) реакцию (V) с бис(трихлорметил)карбонатом с получением 1,14-карбонатного производного (VI)
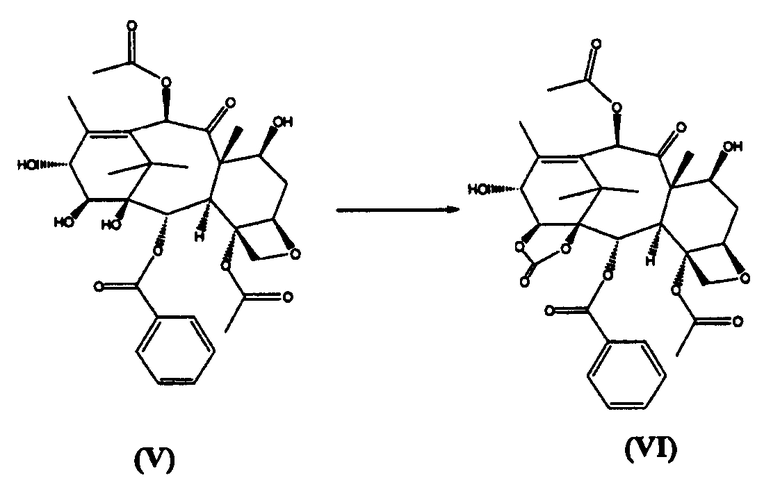
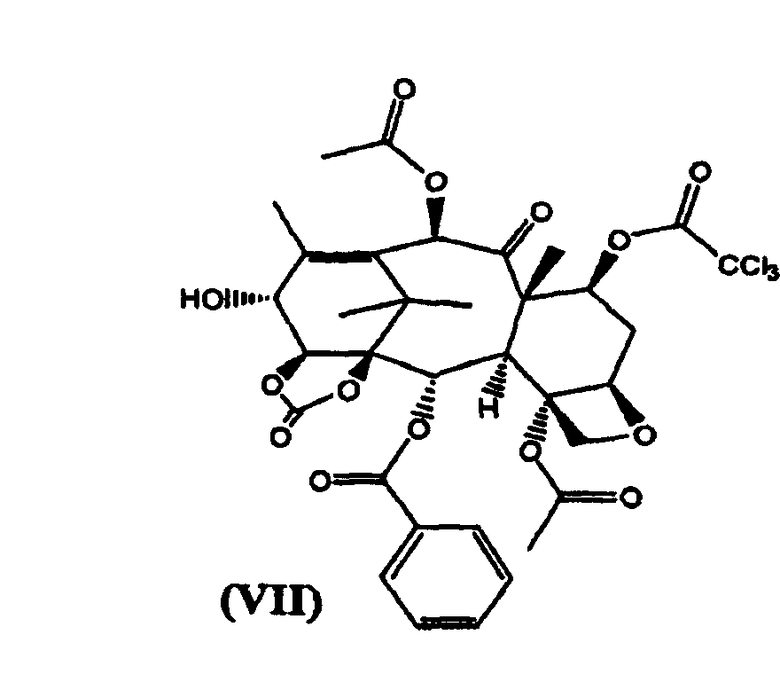
в) реакцию (VI) с трихлорацетилхлоридом с получением (VII)
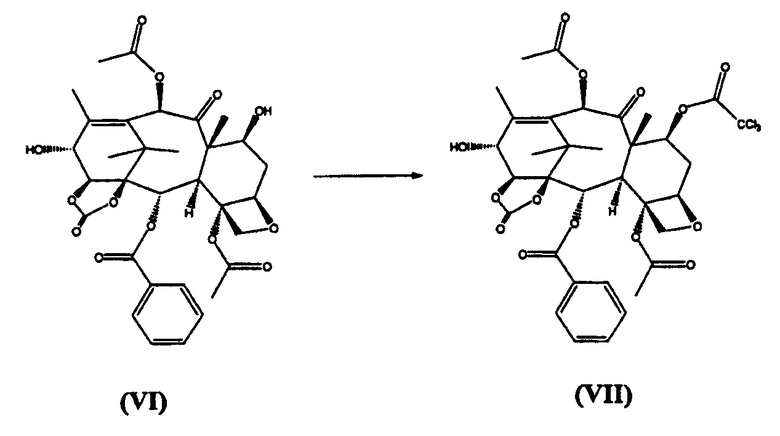
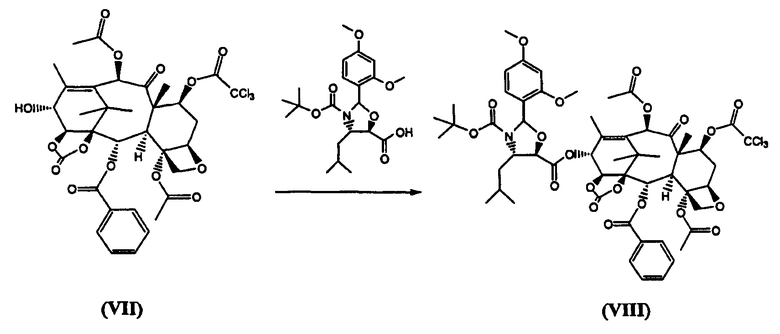
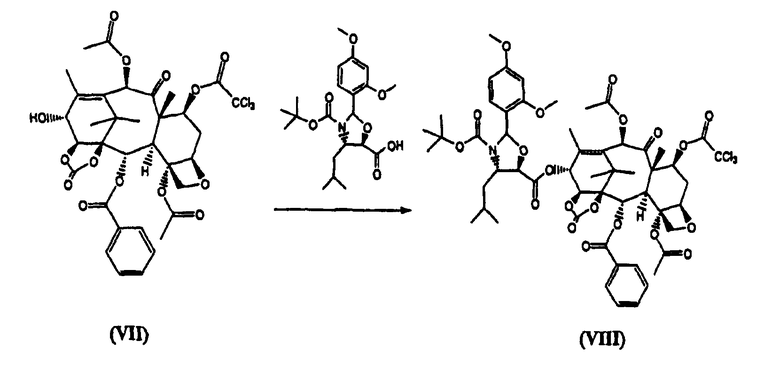
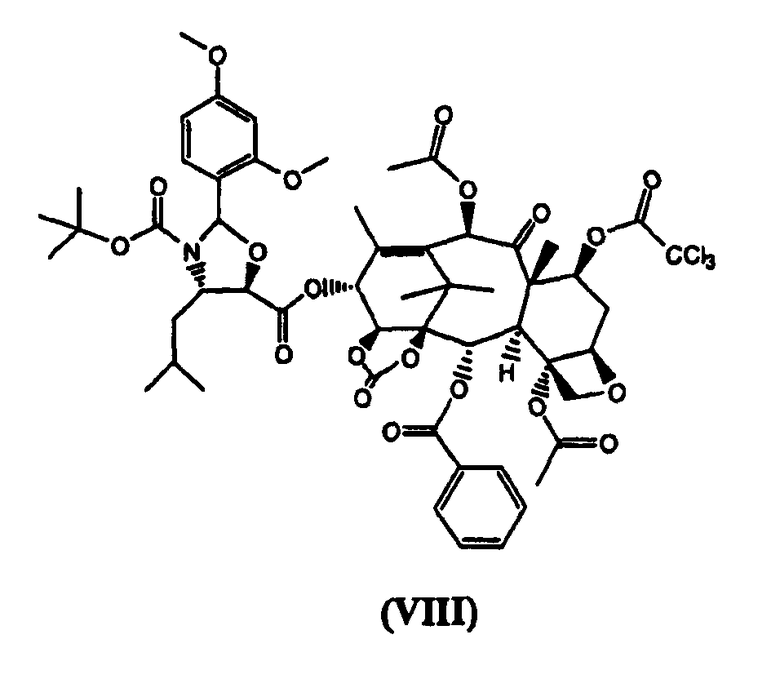
г) реакцию (VII) с (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислотой с получением (VIII)
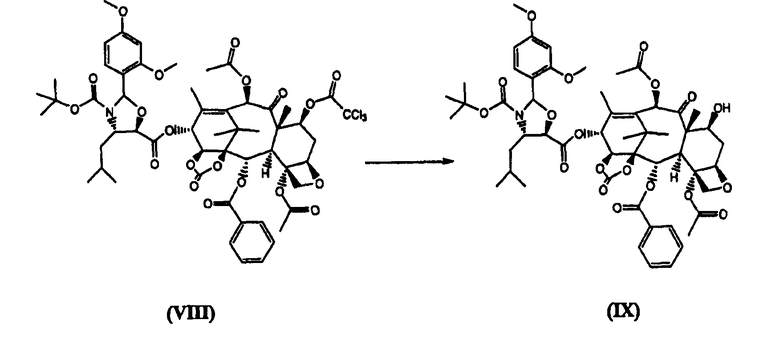
д) удаление из соединения (VIII) защитной трихлорацетильной группы щелочью, предпочтительно, гидроксидом аммония
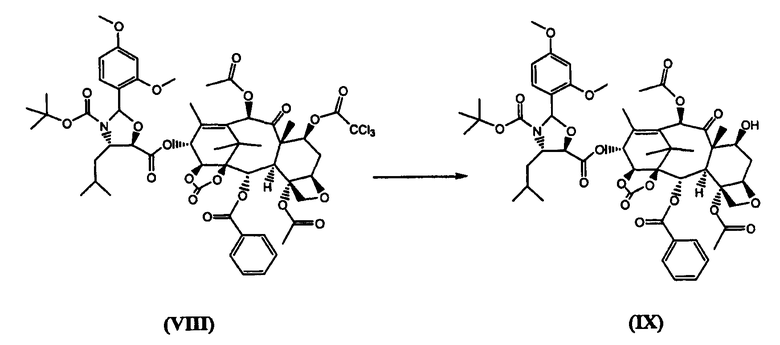
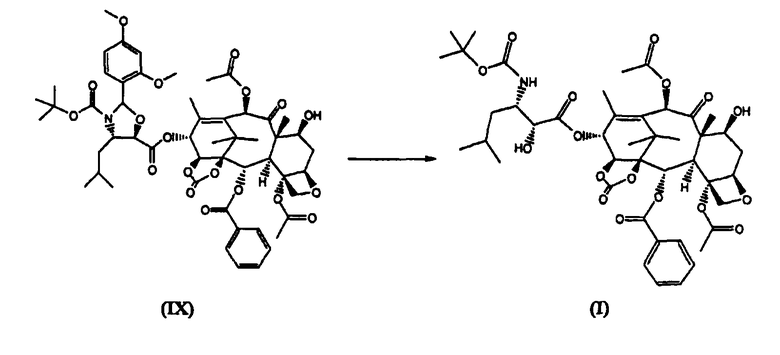
е) удаление из соединения (IX) диметоксибензилиденовой защитной группы
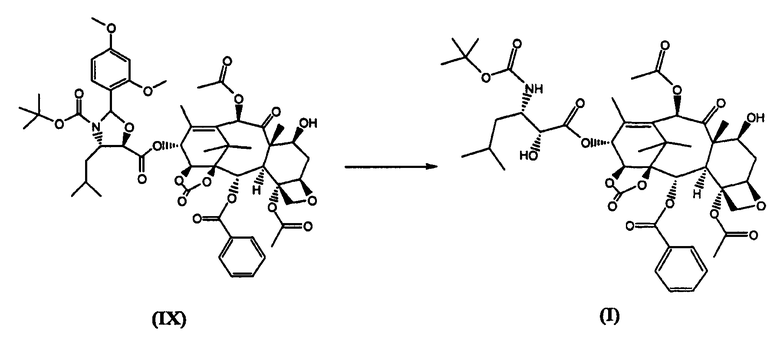
Согласно предпочтительному варианту осуществления изобретения, ацетилирование 10-го положения (стадия а) проводится уксусным ангидридом в присутствии солей церия, скандия или иттербия, предпочтительно, CeCl3×7H2O. Стадия б) проводится с бис(трихлорметил)карбонатом в дихлорометане при 0°C в присутствии основания, предпочтительно, пиридина. Стадия с) проводится с использованием трихлорацетилхлорида в подходящем растворителе, таком как дихлорметан, в присутствии основания, предпочтительно, пиридина, при -10°C. (4S,5R)-N-Boc-2-(2,4-Диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновая кислота для использования на стадии (г') может быть получена как описано в WO 01/02407. Стадия г) проводится в безводном неполярном растворителе, предпочтительно, дихлорметане, в присутствии основания, предпочтительно, 4-диметиламинопиридина (ДМАП), и конденсирующего агента, такого как дициклогексилкарбодиимид (ДЦК), приводя к продукту, который после кристаллизации имеет чистоту выше 98,5%. Трихлорацетильная группа по 7-му положению может быть удалена гидроксидом аммония в апротонном биполярном растворителе, таком как ацетонитрил или N-метилпирролидон, выделение проводят осаждением в воде, получая продукт с чистотой не менее 98,5%. В заключение, продукт стадии д) обрабатывают раствором HCl в метаноле. Соединение (I) затем кристаллизуют из этилацетата и затем из смеси ацетон/гексан, получая твердое вещество с чистотой не менее 99,9%.
Таким образом, использование бис(трихлорметил)карбоната на стадии б) является выгодным, поскольку оно предотвращает образование примеси (II). Использование трихлорацетилхлорида в качестве защитной группы в промежуточном соединении (VII) позволяет получить соединение формулы (VIII), которое легко кристаллизуется из метанола с чистотой более 98,5%, в то время как 7-триэтилсилильный аналог не удалось закристаллизовать из различных растворителей. Еще более важным является то, что трихлорацетамид, который образуется после удаления защиты из 7-го положения, благодаря своей растворимости в смеси воды и либо ацетонитрила, либо N-метилпирролидона, эффективно отделяется от соединения (IX) обработкой гидроксидом аммония. Таким образом, после удаления диметоксибензилиденовой группы и кристаллизации, соединение (I) получается с чистотой не менее 99,9%.
Следующие примеры иллюстрируют изобретение более детально.
Примеры
Пример 1
14β-Гидроксибаккатин III (V) (стадия а)
14β-Гидрокси-10-дезацетилбаккатин III (IV) (10 кг) суспендировали в ТГФ (45 л) и добавляли CeCl3×7H2O (0,5 кг). Добавляли уксусный ангидрид (6,6 кг) в течение 20 минут, и реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем разбавляли добавлением воды (10 л). ТГФ удаляли дистилляцией под вакуумом, остаток высушивали до содержания в нем воды менее 10%, затем кристаллизовали из этилацетата, получая целевое соединение в виде белого порошка (8,2 кг, выход 85%).
1H ЯМР (300 МГц, CDCl3): 1,02 (с, 3Н), 1,08 (с, 3 Н), 1,62 (с, 3Н), 1,78 (ддд, 1Н), 1,99 (д, 3Н), 2,16 (с, 3Н), 2,24 (с, 3Н), 2,46 (ддд, 1Н), 3,43 (ОН, с), 3,73 (д, 1Н), 3,89 (д, 1Н), 4,18 (с, 2Н), 4,35 (дд, 1Н), 4,60 (дд, 1Н), 4,91 (дд, 1Н), 5,73 (д, 1Н), 6,28 (с, 1Н), 7,39 (т, 1Н), 7,52 (дт, 2Н), 8,06 (д, 2Н).
Пример 2
14β-Гидроксибаккатин III-1,14-карбонат (VI) (стадия б)
14β-Гидроксибаккатин III (V) (5 кг) растворяли в смеси дихлорометана (48,0 л) и пиридина (8,0 кг). Реакционную смесь охлаждали до -10°С и добавляли раствор бис(трихлорметилкарбоната) (5,4 кг) в дихлорметане (32,0 л) в течение 30 минут. Реакцию разбавляли добавлением раствора карбоната натрия (11,9 кг) в воде (55,0 л), и образовавшуюся двухфазную смесь перемешивали в течение 1 часа, затем разбавляли водой. Разделяли фазы, водную фракцию экстрагировали дихлорметаном (23,8 л). Органические фракции объединяли, промывали 20% соляной кислотой (40 л), затем водой (30,0 л) и насыщенным раствором хлорида натрия (40 л). Отгоняли часть растворителя под вакуумом, и раствор целевого соединения (VI) непосредственно использовали на следующей стадии.
1H ЯМР (300 МГц, CDCl3): 1,24 (с, 3Н), 1,28 (с, 3 Н), 1,56 (ОН, с), 1,75 (с, 3Н), 1,92 (ддд, 1Н), 2,13 (д, 3Н), 2,60 (ддд, 1Н), 2,28 (с, 3Н), 2,34 (с, 3Н), 2,82 (ОН, 1Н), 3,76 (д, 1Н), 4,25 (д, 1Н), 4,34 (д, 1Н), 4,46 (дд, 1Н), 4,83 (д, 1Н), 5,01 (дд, 1Н), 5,09 (д, 1Н), 6,12 (д, 1Н), 6,34 (с, 1Н), 7,29 (т, 1Н), 7,52 (т, 2Н), 8,06 (д, 2Н).
Пример 3
7-Трихлорацетил-14β-гидроксибаккатин III-1,14-карбонат (VII) (стадия в)
К раствору из предыдущей стадии добавляли пиридин (2 л), и смесь охлаждали до -10°С. Добавляли трихлорацетил хлорид (1,6 кг) в течение 15 минут, поддерживая температуру в диапазоне от -10 до 0°С. Реакционную смесь перемешивали при той же температуре в течение 2 часов. Реакционную смесь разбавляли добавлением раствора NaHSO4 (2 кг) в воде (20 л). Разделяли фазы и водную фракцию экстрагировали дихлорметаном (2 л). Объединенные органические фракции упаривали до небольшого объема и добавляли толуол (20 л). Растворитель удаляли дистилляцией при атмосферном давлении до тех пор, пока температура кипения дистиллята не достигла 110°С. После охлаждения целевое вещество кристаллизовалось в виде белого осадка, который отфильтровывали и сушили в вакууме. (4,96 кг, выход на две стадии 85%).
1H ЯМР (300 МГц, CDCl3): 1,20 (с, 3Н), 1,28 (с, 3 Н), 1,93 (с, 3Н), 2,03 (ддд, 1Н), 2,17 (д, 3Н), 2,20 (с, 3Н), 2,38 (с, 3Н), 2,71 (ддд, 1Н), 3,02 (д, ОН), 3,91 (д, 1Н), 4,24 (д, 1Н), 4,37 (д, 1Н), 4,46 (дд, 1Н), 4,83 (д, 1Н), 5,00 (дд, 1Н), 5,04 (м, 1Н), 5,71 (дд, 1Н), 6,17 (д, 1Н), 6,44 (с, 1Н), 7,52 (т, 2Н), 7,66 (т, 1Н), 8,04 (д, 2Н).
Пример 4
(7-Трихлорацетил)-13-(N-Boc-2-(2,4-диметоксифенил)-4-изобутил-5-оксозалидинил)-14β-гидроксибаккатин-1,14-карбонат (VIII) (стадия г)
К раствору (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислоты (4,0 кг) в дихлорметане (60 л) добавляли 7-трихлорацетил-14β-гидроксибаккатин III-1,14-карбонат (VII) (4,96 кг) и диметиламинопиридин (ДМАП) (100 г). Реакционную смесь охлаждали до 5°С и добавляли раствор дициклогексилкарбодиимида (2,5 кг) в дихлорметане (18 л) в течение 30 минут, получая белую суспензию, которую перемешивали в течение 3 часов. Дициклогексилмочевину (ДГМ) отфильтровывали, промывали дихлорметаном (4 л). Полученный раствор промывали фосфатным буфером, имеющим значение рН 3,5, (100 л) и насыщенным раствором хлорида натрия (50 л), добавляли метанол, который приводил к кристаллизации целевого соединения (VIII), которое высушивали в вакууме при 60°С (выход 6,9 кг, 92%).
1H ЯМР (300 МГц, CDCl3): 1,10 (д, 6Н), 1,33 (с, 2Н), 1,37 (с, 2Н), 1,37 (с, 9Н), 1,60 (м, 1Н), 1,95 (с, 3Н), 1,97 (м, 2Н), 2,04 (ддд, 1Н), 2,16 (д, 3Н), 2,20 (с, 3Н), 2,34 (с, 3Н), 2,68 (ддд, 1Н), 3,85 (с, 3Н), 3,95 (с, 3Н), 4,26 (д, 1Н), 4,36 (д, 3Н), 4,63 (м, 1Н), 4,88 (д, 1Н), 4,97 (дд, 1Н), 5,76 (дд, 1Н), 6,19 (д, 1Н), 6,46 (с, 3Н), 6,50 (т, 1Н), 6,50 (д, 2Н), 6,53 (дд, 1Н), 7,27 (д, 1Н), 7,49 (т, 1Н), 7,64 (т, 2Н), 8,03 (д, 2Н).
Пример 5
13-(N-Boc-2-(2,4-диметоксифенил)-4-изобутил-5-оксазалидинил)-14β-гидроксибаккатин-1,14-карбонат (IX) (стадия д)
(7-Трихлороацетил)-13-(N-Boc-2-(2,4-диметоксифенил)-4-изобутил-5-оксозалидинил)-14β-гидроксибаккатин-1,14-карбонат (VIII) (6,9 кг) растворяли в N-метилпирролидине (11 л). К реакционной смеси добавляли 2М раствор аммиака в метаноле (293 мл) в течение 10 минут, перемешивали при комнатной температуре в течение 45 минут. Реакционную смесь добавляли в воду (110 л) в течение 1 часа и перемешивали в течение 30 минут. Продукт отфильтровывали, промывали водой (50 л). Целевое соединение (IX) высушивали при 60°С в вакууме (6,14 кг, 99%).
1H ЯМР (300 МГц, CDCl3): 1,09 (д, 6Н), 1,30 (с, 3Н), 1,37 (с, 12Н), 1,72 (c, 3Н), 1,79 (м, 3Н), 1,85 (м, 1Н), 2,04 (д, 3Н), 2,26 (с, 3Н), 2,31 (с, 3Н), 2,55 (м, 1Н), 3,76 (д, 1Н), 3,83 (с, 3Н), 3,88 (с, 3Н), 4,23 (д, 1Н), 4,30 (д, 1Н), 4,45 (дд, 1Н), 4,85 (д, 1Н), 4,95 (дд, 1Н), 6,14 (д, 1Н), 6,33 (с, 1Н), 6,48 (м, 1Н), 6,52 (м, 2Н), 7,25 (м, 1Н), 7,47 (т, 1Н), 7,61 (т, 2Н), 8,01 (д, 2Н).
Пример 6
13-(N-Boc-4-изобутил-5-оксазолидинил)-14β-гидроксибаккатин-1,14-карбонат (I) (стадия е)
13-(N-Boc-2-(2,4-диметоксифенил)-4-изобутил-5-оксозалидинил)-14β-гидроксибаккатин-1,14-карбонат (IX) (6,1 кг) растворяли в дихлорметане (20 л). Раствор охлаждали до 0°С и при температуре 0°С добавляли по каплям 0,5М раствор HCl в метаноле (12 л), полученную смесь перемешивали при комнатной температуре в течение 4 часов.
Реакционную смесь выливали в интенсивно перемешиваемую двухфазную смесь дихлорметана (27 л) и водного раствора NaHCO3 (0,6 кг в 21 л воды), поддерживая значение рН во время прибавления в диапазоне от 6 до 7. Отделяли органическую фракцию, водную фракцию дважды экстрагировали дихлорметаном (2×2 л). Органическую фракцию упаривали до объема 18 л, добавляли этилацетат (18 л), упаривали полученный раствор до объема 18 л. Раствор оставляли кристаллизоваться в течение ночи. Осадок отфильтровывали, промывали этилацетатом (7 л), высушивали в течение ночи в вакууме при 45°С (4,53 кг). Сухой белой порошок растворяли при 40°С в ацетоне (20 л) и осаждали добавлением н-гексана (40 л). Смесь оставляли кристаллизоваться при комнатной температуре в течение ночи. Продукт отфильтровывали, промывали н-гексаном и высушивали в вакууме, получая 3,75 кг целевого вещества с чистотой 99,9%.
1H ЯМР (300 МГц, CDCl3): 0,95 (д, 3Н), 0,96 (д, 3Н), 1,21 (м, 1Н), 1,25 (с, 3Н), 1,32 (с, 3Н), 1,35 (с, 9Н), 1,43 (м, 1Н), 1,65 (м, 1Н), 1,69 (с, 3Н), 1,86 (м, 1Н), 1,87 (д, 3Н), 2,22 (с, 3Н), 2,40 (с, 3Н), 2,52 (ддд, 1Н), 3,68 (д, 1Н), 4,08 (м, 1Н), 4,20 (д, 1Н), 4,27 (д, 1Н), 4,30 (дд, 1Н), 4,37 (м, 1Н), 4,72 (NH, д), 4,84 (д, 1Н), 4,91 (дд, 1Н), 6,09(д, 1Н), 6,25 (с, 1Н), 6,44 (д, 1Н), 7,46 (м, 2Н), 7,58 (м, 1Н), 8,01 (м, 2Н).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ТАКСАНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2000 |
|
RU2245882C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТАКСАНА | 2001 |
|
RU2275365C2 |
| С-2'-МЕТИЛИРОВАННЫЕ ПРОИЗВОДНЫЕ ПАКЛИТАКСЕЛА ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ АГЕНТОВ | 2002 |
|
RU2287528C2 |
| СПОСОБ ПОЛУЧЕНИЯ 14БЕТА-ГИДРОКСИБАККАТИН III-1,14-КАРБОНАТА | 2002 |
|
RU2291866C2 |
| ПОЛУСИНТЕТИЧЕСКИЕ ТАКСАНЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2001 |
|
RU2259363C2 |
| ПРОИЗВОДНЫЕ 10-ДЕАЦЕТИЛ-14-БЕТА-ГИДРОКСИБАККАТИНА III, СПОСОБ ИХ ПОЛУЧЕНИЯ И КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ | 1996 |
|
RU2161615C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПАКЛИТАКСЕЛА | 2001 |
|
RU2276147C2 |
| ПРОИЗВОДНЫЕ 10-ДЕАЦЕТИЛБАККАТИНА III И 10-ДЕАЦЕТИЛ-14β-ГИДРОКСИБАККАТИНА III, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2152936C1 |
| СПОСОБ ПОЛУЧЕНИЯ 14БЕТА-ГИДРОКСИБАККАТИН III-1,14-КАРБОНАТА | 2002 |
|
RU2285000C2 |
| ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ПОЛУСИНТЕЗА ТАКСАНОВ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2159237C2 |
Изобретение относится к улучшенному способу синтеза 13-(N-Boc-β-изобутилсеринил)-14β-гидроксибаккатин III-1,14-карбоната (I),
в котором карбонилирование 1,14-гидроксигрупп остова баккатина проводят бис(трихлорметилкарбонатом) и 7-гидроксигруппа защищена трихлороацетильной группой. Изобретение также относится к новым промежуточным соединениям формулы VII и VIII:
 и
и 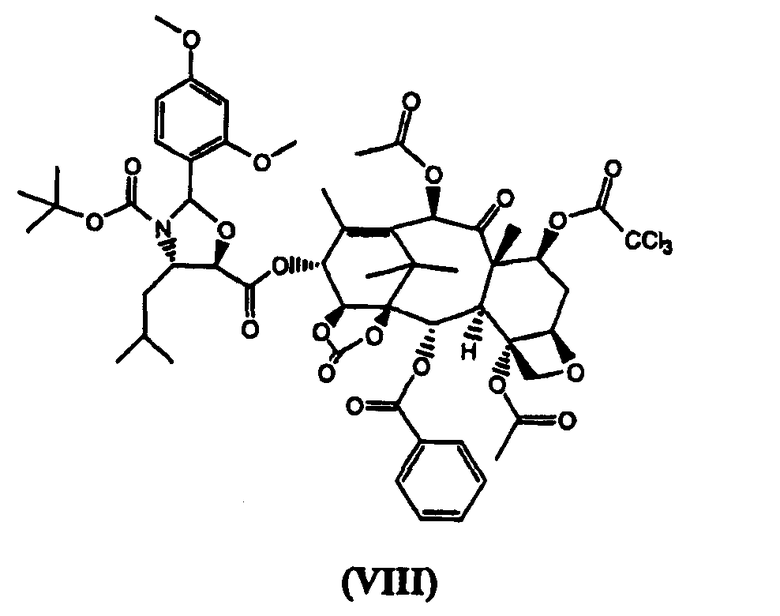
Цель изобретения упрощения процесса. 3 н.п. ф-лы.
1. Способ получения 13-(N-Вос-β-изобутилсеринил)-14β-гидроксибаккатин III-1,14-карбоната (I):
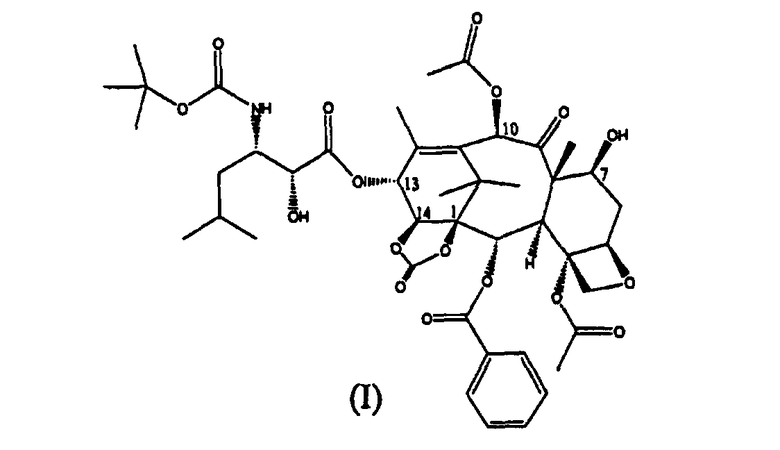
который включает следующие стадии:
а) ацетилирование 10-гидроксигруппы 14β-гидрокси-10-дезацетилбаккатина III (IV)
б) реакцию (V) с бис(трихлорметил)карбонатом с получением 1,14-карбонатного производного (VI)
в) реакцию (VI) с трихлорацетилхлоридом с получением (VII)
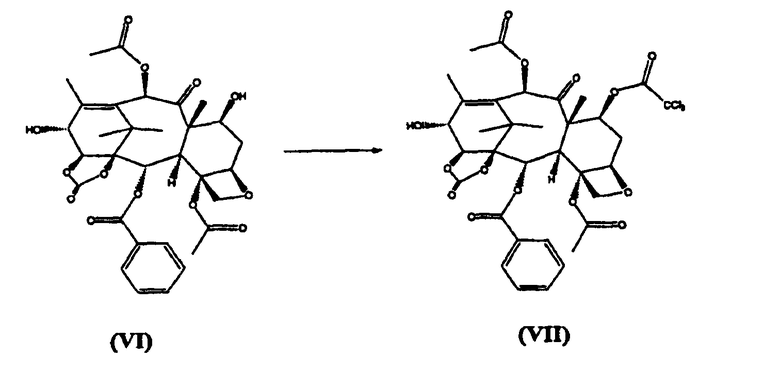
реакцию (VII) с (4S,5R)-N-Вос-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислотой с получением (VIII)
г) удаление щелочью защитной трихлорацетильной группы из соединения (VIII)
д) удаление диметоксибензилиденовой защитной группы из соединения (IX)
2. Соединение формулы (VII):
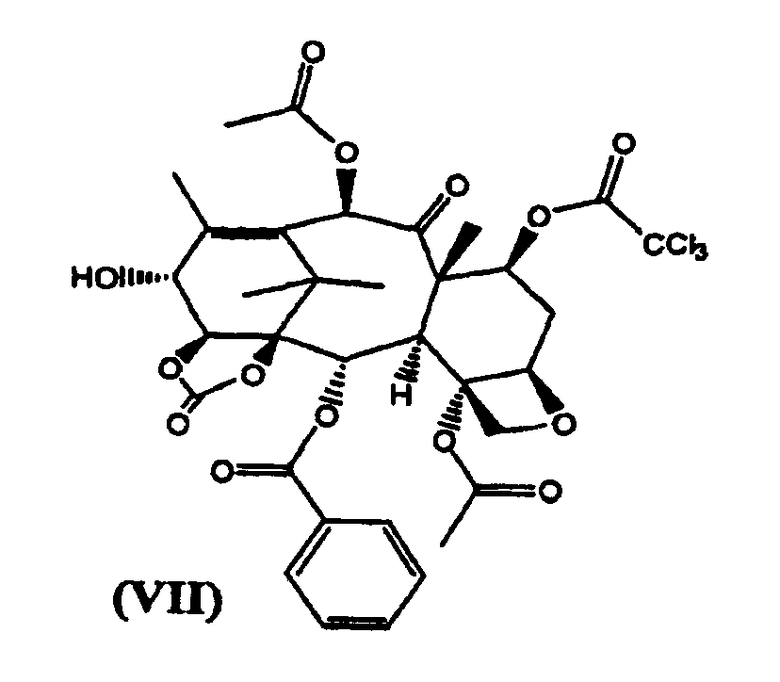
3. Соединение формулы (VIII):
| Способ изготовления бесконечных лент из полос шлифовальной шкурки | 1955 |
|
SU102407A1 |
| US 20040030164 A1, 12.02.2004 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТАКСАНА | 2001 |
|
RU2275365C2 |

