Настоящее изобретение относится к способу получения соединения 13-(N-Boc-β-изобутилизосеринил)-14β-гидроксибаккатин III 1,14-карбоната формулы (I):

Соединение (I), описанное в РСТ WO 01/02407, является особенно активным по отношению к опухолям молочной железы, легкого, яичников, ободочной кишки, простаты, почки и поджелудочной железы, а также по отношению к клеткам, устойчивым к известным противоопухолевым препаратам, таким как адриамицин, винбластин и некоторые производные Pt. Производные 14β-гидрокси-1,14-карбонатдеацетилбаккатина III обычно готовят, используя в качестве исходного вещества 14β-гидроксидеацетилбаккатин III, природное соединение, получаемое в небольших количествах экстракцией листьев Taxus wallichiana, как описано в ЕР 559019. Существует необходимость в альтернативных легких и эффективных способах получения производных 14β-гидрокси-1,14-карбонатдеацетилбаккатина III, в частности соединений (I).
Согласно способу настоящего изобретения в качестве исходного материала используют 10-деацетилбаккатин III, который в противоположность 14β-гидроксибаккатину III, можно легко выделить в значительных количествах из листьев Taxus baccata.
Таким образом изобретение относится к способу получения соединений формулы (I), который включает следующие стадии:
а) защиты гидроксилов 10-диацетилбаккатина III в 7- и 10-положениях:

где R и R1, которые могут быть одинаковыми или различными, выбраны из C1-С10 алкила или арила, C1-С10 алкил- или арилкарбонила, трихлорацетила, C1-C4 триалкилсилила;
предпочтительно, когда R и R1 являются одинаковыми, они представляют собой трихлорацетил, тогда как если они различные, предпочтительно, R представляет собой трихлорацетил, а R1 является ацетилом, или R представляет собой триэтил- или триметилсилил или ВОС, а R1 является ацетилом;
b) двухстадийного окисления, чтобы получить производное, окисленное до карбонила в положении 13 и гидроксилированное в положении 14:
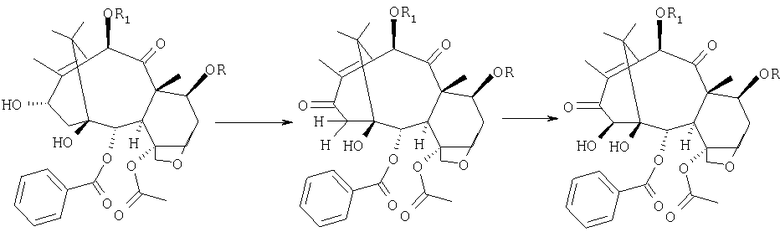
c) карбоксилирования вицинальных гидроксилов в 1- и 14-положениях, чтобы получить 1,14-карбонатное производное:

d) восстановления карбонила в положении 13:

e) снятия защитных групп в 7- и 10-положениях:

f) селективного ацетилирования гидроксила в положении 10:
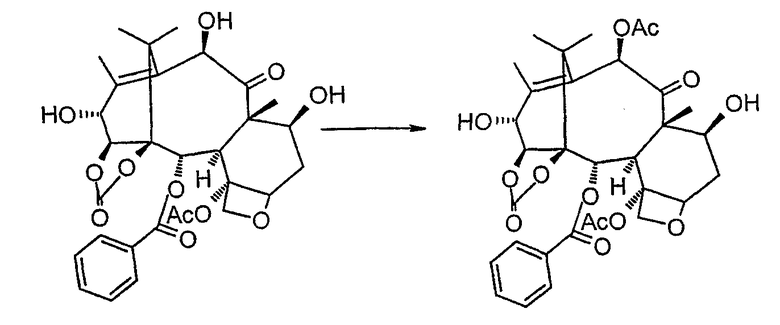
g) превращения 14β-гидроксибаккатин-1,14-карбоната III в производное, триэтилсилилированное в положении 7:

h) реакции соединения, полученного на стадии (g), с (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислотой:

i) удаления триэтилсилильной и диметоксибензилиденовой защитных групп из соединения, полученного на стадии (h):

Процедуры селективной защиты гидроксилов в 7- и 10-положениях описываются в работе Holton et al., Tetrahedron Letters 39 (1998), 2883-2886. Селективная защита гидроксилов исходного соединения деацетилбаккатина III является возможной благодаря их различной реакционной способности. В частности, реакционная способность по отношению к ацилирующим, алкилирующим или силилирующим агентам, как было обнаружено, изменяется в порядке С(7)-ОН>C(10)-OH>C(13)-OH>C(1)-OH, поэтому группы в 7- и 10-положении можно селективно защитить, в то же время сохраняя гидроксилы в 1- и 13-положениях свободными. Более того, изменяя условия реакции, можно повернуть в противоположном направлении порядок реакционной способности гидроксилов в 7- и 10-положениях, таким образом давая возможность их дифференциального замещения. Примеры реагентов и реакционных условий, которые удобны для защиты гидроксилов в 10- и 7-положении, сообщаются в цитируемой выше публикации. Аналогичные селективности получают, используя в качестве исходного материала 14β-гидроксибаккатин-1,14-карбонат.
В соответствии с предпочтительным вариантом осуществления изобретения на практике деацетилбаккатин III реагирует с трихлорацетилхлоридом в метиленхлориде в присутствии триэтиламина и с использованием диметиламинопиридина (ДМАП) в каталитических количествах. Использование защитных трихлорацетатных групп, как оказалось, является очень благоприятным на последующих стадиях окисления, карбоксилирования и восстановления (соответственно, (b), (c) и (d)). В частности, 7,10-бис-трихлорацетатное производное, которое с количественными выходами получают из исходного вещества, окисляют и карбоксилируют, затем легко восстанавливают в положении 13 с одновременным снятием защитных трихлорацетатных групп, получая 14-гидрокси-1,14-карбонат-деацетилбаккатин III. Использование ДМАП в каталитических количествах предоставляет очевидные преимущества с промышленной и экологической точки зрения, поскольку ацилирование данного вещества до настоящего времени проводили в пиридине, что приводит к проблеме удаления остаточного растворителя.
Стадию окисления (b) гидроксила в положении 13 проводят диоксидом марганца или диоксидом висмута или озоном в растворителе, выбранном из ацетонитрила, ацетона или смесей этилацетат/метиленхлорид 9:1, при энергичном перемешивании, предпочтительно озоном или диоксидом марганца в ацетонитриле или ацетоне. В реакции с озоном происходит быстрое образование соединения, окисленного в положении 13, тогда как в случае MnO2 реакция протекает быстро, давая соединение, окисленное в положении 13, которое можно выделить из реакционной среды, поскольку более длительная реакция дает производное, окисленное в положении 13 и гидроксилированное в положении 14.
Последующую стадию карбоксилирования (с) гидроксилов в 1- и 14-положениях обычно осуществляют фосгеном или в смеси метиленхлорид/толуол в присутствии пиридина. Затем полученное в результате 1,14-карбонатное производное можно легко восстановить в положении 13, получая соответствующее 13-гидрокси производное (стадия (d)). Указанное восстановление происходит региоселективно на карбониле в 13 положении, в то время как карбонил в положении 9 остается неизменным. Данную реакцию обычно осуществляют боргидридом натрия в метаноле или боргидридом тетрабутиламмония, и она идет с высокими выходами. Последующая стадия (е) заключается в снятии защиты гидроксилов в положениях 7 и 10, давая 14β-гидрокси-1,14-карбонатдеацетилбаккатин III. Условия и реагенты, которые можно использовать при селективном снятии защиты гидроксилов в положениях 7 и 10, описывается в работах Zheng et al., Tetrahedron Lett., 1995, 36, 2001 и Datta et al., J. Org. Chem., 1995, 60, 761.
Селективное ацетилирование в положении 10 (стадия (f)) проводят уксусным ангидридом в присутствии солей церия, скандия или иттербия, предпочтительно CeCl3·7H2O. Затем гидроксил в положении 7 защищают силилированием (стадия (g)). Последующая стадия (h) включает реакцию конденсации 14β-гидрокси-7-Tes-1,14-карбонат-баккатина III с (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислотой. Последнюю готовят способом, описанным в РСТ WO 01/02407. Реакцию проводят в сухих неполярных органических растворителях в присутствии основания и конденсирующего агента, такого как дициклогексилкарбодиимид (ДЦК).
Наконец, на стадии (i) триэтилсилильную группу можно удалить фторидом пиридиния в растворе ацетонитрил/пиридин под атмосферой азота, тогда как диметоксибензилиденовую группу можно удалить в метиленхлориде добавлением HCl в метаноле и затем NaHCO3.
Следующие далее примеры иллюстрируют изобретение более детально.
Пример I Получение 7,10-бис-трихлорацетил-10-деацетилбаккатина III
1я Альтернатива:
В раствор 10 г 10-деацетилбаккатина III (18,4 ммоль) в 125 мл сухого метиленхлорида и 42 мл пиридина по каплям добавляют 4,77 мл трихлоруксусного ангидрида (42,32 ммоль). Реакционную смесь перемешивают в течение 3 часов или иным способом, пока реакция не закончится, что контролируют тонкослойной хроматографией (ТСХ) на силикагеле, используя смесь н-гексан/этилацетат 1:1 в качестве элюента. После завершения реакции добавляют 5 мл метанола, чтобы ликвидировать избыток трихлоруксусного ангидрида, и затем добавляют воду. Органическую фазу тщательно промывают водой, подкисленной HCl, чтобы удалить пиридин, в то время как оставшуюся органическую фазу сушат над MgSO4 и концентрируют до сухого состояния под вакуумом, получая бледно-желтое твердое вещество (17 г), которое после кристаллизации из хлороформа имеет: [α]D -34° (CH2Cl2 C5,8) ИК (KBr) 3517, 1771, 1728, 1240, 981, 819, 787, 675 см-1;
1Н-ЯМР (200 МГц): δ 8,11 (Bz С), 7,46 (Bz, ВВ'), 6,50 (с, Н-10), 5,72 (м, Н-7 Н-29), 5,02 (д, J=8 Гц, Н-5), 4,95 (8 м, Н-13), 4,37 (д, J=8 Гц, Н-20a), 4,18 (д, J=8 Гц, Н-20b), 4,02 (д, J=6 Гц, Н-3), 2,32 (с, 4-Ас), 2,22 (с, Н-18), 1,91 (с, Н-19), 1,25 и 1,11 (с, Н-16, Н-17), 1,94 (м, Н14α), 1,89 (м, Н14β).
2я Альтернатива
10-ДАБ III (10 г, 18,38 ммоль) суспендируют в CH2Cl2 (120 мл), добавляют ДМАП (220 мг, 1,4 ммоль, 0,1 экв.) и смесь охлаждают до 0°С на ледяной бане. Затем в потоке азота в течение 5 мин добавляют Et3N (10,26 мл, 73,6 ммоль, 4 экв.) и сразу после этого Cl3COCl (4,12 мл, 36,8 ммоль, 2 экв.), поддерживая температуру ниже 10°С. После завершения добавления смесь продолжают перемешивать на ледяной бане в течение 15 мин, затем баню удаляют и смесь перемешивают при комнатной температуре в течение 1 ч. После 1 ч реакцию контролируют ТСХ (AcOEt/н-гексан 2:3, Rf 10-ДАБ III = 0,05, Rf 7,10-бис-трихлорацетил-10-ДАБ III = 0,26) и добавляют Cl3CCOCl (1 мл, 0,5 экв.). Продолжают перемешивание при комнатной температуре в течение 10 мин, затем смесь выливают в химический стакан, содержащий 160 г измельченного льда, перемешивают при комнатной температуре до равновесного состояния (примерно 1 час). После этого отделяют водную фазу и экстрагируют CH2Cl2 (3 х 40 мл). Объединенные органические фазы промывают 1 н. HCl (20 мл), затем насыщенным раствором NaHCO3 (20 мл), сушат над Na2SO4 и растворитель выпаривают. Масса неочищенного вещества: 16,5 г. После кристаллизации из хлороформа, ИК, 1Н-ЯМР и [α]D спектры согласуются со спектрами соединения, полученного с использованием пиридина и трихлоруксусного ангидрида.
Пример II
Окисление 10-деацетилбаккатин III 7,10-бис-трихлорацетата в положении 13 и гидроксилирование в положении 14.
К раствору 10-деацетилбаккатин III 7,10-бис-трихлорацетата (3 г) в ацетонитриле (40 мл) добавляют 30 г активированного MnO2, суспензию перемешивают магнитной мешалкой при комнатной температуре и реакцию контролируют ТСХ (петролейный эфир-этилацетат 5:5; Rf исходного материала примерно 0,31). Примерно через час образование 13-дегидропроизводного завершается (анализ ТСХ, Rf 13-дегидропроизводного примерно 0,50). Затем перемешивание продолжают примерно в течение 72 часов, в течение которых 13-дегидропроизводное медленно окисляется до своего 14β-гидроксипроизводного (Rf примерно 0,36). Реакционную смесь фильтруют через Celite и осадок несколько раз промывают этилацетатом. Растворитель выпаривают и остаток очищают колоночной хроматографией на силикагеле (100 мл, элюент:петролейный эфир-этилацетат 7:3), получая 170 мг 13-дегидропроизводного и 1,05 г 14β-гидрокси-13-дегидропроизводного.
13-Дегидро-14β-гидрокси-10-деацетилбаккатин III 7,10-бис-трихлорацетат: белый порошок, Тпл=97°С; ИК (KBr диск): 3440, 1780, 1767, 1736, 1686, 1267, 1232, 1103, 1010, 854 см-1;
1Н-ЯМР (200 МГц, CDCl3): δ 8,07 (Bz AA'), 7,60 (Bz, C), 7,49 (Bz, BB'), 6,52 (с, Н-10), 5,92 (д, J=6,7 Гц, Н-2), 5,70 (шир. т, J=8,0 Гц, Н-7), 4,95 (шир. д, J=8,2 Гц, Н-5), 4,37 (д, J=8,2 Гц, Н-20а), 4,31 (д, J=8,2 Гц, Н-20b), 4,17 (с, Н14), 4,02 (д, J=6,7 Гц, Н-3), 2,71 (м, Н-6), 2,29 (с, OAc), 2,17 (с, OAc), 1,96 (с, Н-18), 1,27, 1,01 (с, Н-16, Н-17 и Н-19).
Пример III
Окисление 7-триэтилсилилбаккатина III в положении 13 и гидроксилирование в положении 14.
К раствору 7-триэтилсилилбаккатина III (1,0 г) в ацетонитриле (10 мл) добавляют 10 г активированного MnO2, суспензию перемешивают магнитной мешалкой при комнатной температуре и реакцию контролируют ТСХ (петролейный эфир-этилацетат 6:4, Rf исходного материала около 0,25). Примерно через 2 часа образование 13-дегидропроизводного завершается (анализ ТСХ, Rf 13-дегидропроизводного около 0,45). Перемешивание продолжают в течение примерно 188 часов, в течение которых добавляют дополнительное количество MnO2 (10 г). 13-дегидропроизводное медленно окисляют до его 14β-гидроксипроизводного (Rf примерно 0,38). Реакционную смесь фильтруют через Celite и осадок промывают этилацетатом. Растворитель выпаривают и остаток очищают колоночной хроматографией на силикагеле (40 мл, элюент:петролейный эфир-этилацетат 7:3, получая 126 мг 13-дегидропроизводного, 479 мг (46%) 14β-гидрокси-13-дегидропроизводного и 189 мг их смеси.
13-Дегидро-7-триэтилсилилбаккатин III: белый порошок, Тпл=168°С [α]D 25 -35 (CH2Cl2, C 0,67) ИК (KBr): 3488, 1726, 1711, 1676, 1373, 1269, 1244, 1230, 1105 см-1; 1Н-ЯМР (200 МГц, CDCl3): δ 8,07 (Bz AA'), 7,60 (Bz, C), 7,49 (Bz, BB'), 6,59 (с, Н-10), 5,69 (д, J=6,9 Гц, Н-2), 4,92 (д, J=8,2 Гц, Н-5), 4,48 (дд, J=10,6 Гц, Н-7), 4,33 (д, J=8,0 Гц, Н-20а), 4,12 (д, J=8,0 Гц, Н-20b), 3,91 (д, J=6,9 Гц, Н-3), 2,96 (д, J=20 Гц, Н-14а), 2,65 (д, J=20 Гц, Н-20b), 2,50 (м, Н-6α), 2,23 (с, OAc), 2,19 (с, ОАс + Н-18), 1,67, 1,28, 1,19 (с, Н-16, Н-17 и Н-19), 0,19 (м, TES).
13-Дегидро-14β-гидрокси-10-деацетилбаккатин III 7,10-бис-трихлорацетат: белый порошок, Тпл.=153 °С [α]D 25 +20(CH2Cl2, C 0,75) ИК (KBr): 3431, 1723, 1692, 1371, 1269, 1242, 1223, 1096 см-1; 1Н-ЯМР (200 МГц, CDCl3): δ 8,06 (Bz AA'), 7,60 (Bz, C), 7,48 (Bz, BB'), 6,51 (с, Н-10), 5,88 (д, J=6,9 Гц, Н-2), 4,90 (д, J=8,2 Гц, Н-5), 4,47 (дд, J=10,6 7 Гц, Н-7), 4,30 (д, J=8,0 Гц, Н-20а), 4,28 (д, J=8,2 Гц, Н-20b), 4,13 (шир. д, J=2 Гц, Н-14), 3,84 (д, J=6,9 Гц, Н-3), 3,69 (шир. д, J=2 Гц, 14-ОН), 3,62 (с, 1-ОН) 2,52 (м, Н-6α), 2,24 (с, ОАс), 2,21 (с, ОАс), 2,11 (с, Н-18), 1,92 (м, Н-6β), 1,74, 1,56, 1,28 (c, H-16, H-17 и Н-19), 0,94 (м, TES), 0,59 (м, TES). HRNS: 714,3092 (рассчитано для С37H50O12Si 714,3092).
Пример IV
Получение 1,14-карбонат-13-дегидро-7-TES-14β-гидрокси-баккатина III.
К раствору фосгена (1,8 мл 20% раствора в толуоле, 3,4 ммоль, 20 мол. экв.) и пиридина (0,56 мл, 6,8 ммоль, 20 мол. экв.) в CH2Cl2 (2 мл) по каплям в течение 5 мин добавляют раствор 13-дегидро-14β-гидрокси-7-триэтилсилилбаккатина III (124 мг, 1,17 ммоль) в CH2Cl2 (1 мл). Смесь перемешивают при комнатной температуре в течение 1 часа, затем гасят избыток фосгена, добавляя насыщенный раствор NaHCO3, и экстрагируют CH2Cl2. Органическую фазу промывают насыщенным раствором NaHCO3, насыщенным солевым раствором и сушат (Na2SO4). Растворитель выпаривают, получая красноватый остаток, который очищают на короткой силикагелевой колонке (примерно 5 мл, элюент гексан/этилацетат 8:2), получая 118 мл (92%) карбоната. Если реакцию проводят, используя в качестве основания триэтиламин и без обратного добавления, то получают смесь 1:15 1,17-карбоната и 2-дебензоил-1,2-карбонат-14-бензоата.
13-Дегидро-14β-гидрокси-7-триэтилсилилбаккатин III 1,14-карбонат. Белый порошок, Тпл = 153°С [α]D 25 +23 (CH2Cl2, C 0,75) ИК (KBr) ОН полоса отсутствует 1834, 1734, 1709, 1373, 1242, 1225, 1088, 1057 см-1; 1Н-ЯМР (200 МГц CDCl3): δ 7,99 (Bz AA'), 7,60 (Bz, C), 7,48 (Bz, BB'), 6,51 (с, Н-10), 6,12 (д, J=6,9 Гц, Н-2), 4,90 (д, J=8,2 Гц, Н-5), 4,78 (с, Н-14), 4,44 (дд, J=10,7 Гц, Н-7), 4,34 (д, J=8 Гц, Н-20а), 4,19 (д, J=8,2 Гц, Н-20b), 3,80 (д, J=6,9 Гц, Н-3), 2,50 (м, Н-6α), 2,23 (с, ОАс), 2,22 (с, ОАс), 2,19 (с, Н-18), 1,92 (м, Н-6β), 1,72, 1,39, 1,26 (c, H-16, H-17 и Н-19), 0,90 (м, TES), 0,56 (м, TES). HRNS: 740,2851 (рассчитано для С38H48O13Si 740,2864).
13-Дегидро-14β-гидроксибаккатин III 1,14-карбонат. Белый порошок, 240°С [α]D 25 - 2,5 (CH2Cl2, C 0,4) ИК (KBr) 3539, 1831, 1736, 1240, 1088, 1068, 1057, 1024 см-1; 1Н-ЯМР (200 МГц CDCl3): δ 7,98 (Bz AA'), 7,61 (Bz, C), 7,50 (Bz, BB'), 6,39 (с, Н-10), 6,14 (д, J=6,9 Гц, Н-2), 4,98 (д, J=8,2 Гц, Н-5), 4,80 (с, Н-14), 4,43 (дд, J=10,7 Гц, Н-7), 4,35 (д, J=8 Гц, Н-20а), 4,24 (д, J=8,2 Гц, Н-20b), 3,80 (д, J=6,9 Гц, Н-3), 2,50 (м, Н-6α), 2,30 (с, ОАс), 2,20 (с, ОАс), 2,15 (с, Н-18), 1,90 (м, Н-6β), 1,74, 1,34, 1,25 (c, H-16, H-17 и Н-19). HRNS: 626,2005 (рассчитано для С33H34O1 626, 1999).
Пример V
Получение 1,14-карбонат-7-О-триэтилсилилбаккатина III.
К раствору 13-дегидро-14β-гидрокси-7-триэтилсилилбаккатин III 1,14-карбоната (50 мг) в метаноле (5 мл) небольшими частями добавляют избыточное количество NaBH4 (примерно 20 мг). Через 30 мин в реакционную смесь добавляют насыщенный раствор NH4Cl, экстрагируют этилацетатом, промывают соляным раствором и сушат над Na2SO4. Растворитель выпаривают и остаток очищают колоночной хроматографией на силикагеле (примерно 5 мл, элюирование смесью гексан/этилацетат 8:2), получая 35 мг 13α-гидроксипроизводного и 9 мг 13β-гидроксипроизводного.
14β-гидрокси-7-триэтилсилилбаккатин III 1,14-карбонат [α]D 25 - 35 (CH2Cl2, C 0,60) ИК (KBr) 3054, 1819, 1736, 1603, 1371, 1261, 1238, 1090, 1069 см-1; 1Н-ЯМР (200 МГц CDCl3): δ 8,06 (Bz AA'), 7,65 (Bz, C), 7,50 (Bz, BB'), 6,47 (с, Н-10), 6,12 (д, J=6,9 Гц, Н-2), 5,05 (шир. д, J=5,5 Гц, Н-13), 4,98 (шир. д, J=9 Гц, Н-5), 4,83 (д, J= 5 Гц, Н-14), 4,50 (дд, J=10,7 Гц, Н-7), 4,34 (д, J=8,0 Гц, Н-20а), 4,23 (д, J=8 Гц, Н-20b), 3,75 (д, J=6,9 Гц, Н-3), 2,56 (м, Н-6α), 2,34 (с, ОАс), 2,22 (с, ОАс), 1,78 (м, Н-6β), 1,35 (с, Н-18), 1,75, 1,18, 0,95 (c, H-16, H-17 и Н-19), 0,90 (м, TES), 0,62 (м, TES).
14β-гидрокси-7-триэтилсилил-13-эпибаккатин III 1,14-карбонат.
Аморфный, [α]D 25 - 13 (CH2Cl2, C 0,60) ИК (KBr) 3630, 1825, 1734, 1603, 1375, 1262, 1091, 1071, 1049 см-1; 1Н-ЯМР (200 МГц CDCl3): δ 8,01 (Bz AA'), 7,63 (Bz, C), 7,48 (Bz, BB'), 6,44 (с, Н-10), 6,12 (д, J=7,2 Гц, Н-2), 4,90 (шир. д, J=9 Гц, Н-5), 4,81 (д, J= 8 Гц, Н-14), 4,48 (шир. д, J=8, Н-13), 4,50 (дд, J=10,7 Гц, Н-7), 4,41 (д, J=8 Гц, Н-20а), 4,31 (д, J=8 Гц, Н-20b), 3,68 (д, J=7,2 Гц, Н-3), 2,60 (м, Н-6α), 2,32 (с, ОАс), 2,26 (с, Н-18), 2,21 (с, ОАс), 1,80 (м, Н-6β), 1,72, 1,43, 1,27 (c, H-16, H-17 и Н-19), 0,93 (м, TES), 0,61 (м, TES).
Пример VI
Получение 13-дегидро-14β-гидрокси-7,10-бистрихлорацетил-баккатин III 1,14-карбоната.
Раствор 13-дегидро-14β-гидрокси-7,10-бистрихлорацетил-баккатина III (200 мг) в CH2Cl2 (2 мл) добавляют в течение 5 мин к раствору фосгена (20% в толуоле, 3,6 мл, 20 экв.) и пиридина (1,12 мл, 20 экв.) в CH2Cl2 (2 мл). Смесь перемешивают при комнатной температуре в течение 1 часа, затем избыток фосгена гасят насыщенным раствором NaHCO3 (3 мл). Смесь экстрагируют CH2Cl2, органическую часть промывают насыщенным раствором NaHCO3, затем насыщенным раствором NaCl и сушат над Na2SO4. Растворитель выпаривают и остаток очищают колоночной хроматографией на силикагеле (элюент:гексан/AcOEt 9:1), получая 175 мг (89%) карбоната.
13-дегидро-14β-гидрокси-7,10-бис-трихлорацетилбаккатин III 1,14-карбонат. Белое аморфное твердое вещество. ИК (KBr) 1834, 1771, 1735, 1709, 1232, 1103, 1010, 854 см-1.
1Н-ЯМР (200 МГц, CDCl3): δ 8,03 (Bz AA'), 7,60 (Bz, C), 7,50 (Bz, BB'), 6,52 (с, Н-10), 5,92 (д, J=6,7 Гц, Н-2), 5,70 (шир. т, J=8,0 Гц, Н-7), 4,95 (шир. д, J=8,2 Гц, Н-20b), 4,77 (c, Н-14), 4,02 (д, J=6,7 Гц, Н-3), 2,71 (м, Н-6), 2,29 (с, OAc), 1,96 (с, Н-18), 1,27- 1,01 (м, Н-16, Н-17 и Н-19).
Пример VII
Получение 14β-гидрокси-10-деацетилбаккатин III 1,14-карбоната.
Раствор 13-дегидро-14β-гидрокси-7,10-бис-трихлорацетил-баккатин III 1,14-карбоната (500 мг) в MeOH (8 мл) охлаждают до 0°С на ледяной бане и к нему в течение 5 мин добавляют твердый NaBH4 (44 мг). Смесь перемешивают при комнатной температуре в течение 1 часа, затем охлаждают до 0°С, в течение 5 мин добавляют ацетон (2 мл) и концентрируют при умеренном вакууме, затем добавляют AcOEt (10 мл) и фильтруют через Celite. Прозрачный раствор промывают насыщенным раствором NaCl и сушат над Na2SO4. Растворитель выпаривают, остаток (смесь С13 эпимеров 4,5:1) очищают хроматографией на силикагелевой колонке (элюент:гексан/AcOEt), получая 251 мг указанного в заголовке соединения и 55 мг 13-эпимера (88% от общего) карбоната со снятыми защитными группами.
14β-гидрокси-10-деацетилбаккатин III 1,14-карбонат. Белое аморфное твердое вещество. ИК (KBr): 3520 (ОН), 1834, 1709, 1232, 1103, 1010, 854 см-1.
1Н-ЯМР (200 МГц, CDCl3): δ 8,03 (Bz AA'), 7,60 (Bz, C), 7,50 (Bz, BB'), 6,27 (с, Н-10), 5,92 (д, J=6,7 Гц, Н-2), 4,95 (шир. д, J=8,2 Гц, Н-20b), 4,85 (м, Н-13), 4,77 (c, Н-14), 4,42 (шир. т, J=8,0 Гц, Н-7), 4,02 (д, J=6,7 Гц, Н-3), 2,71 (м, Н-6), 2,29 (с, OAc), 1,96 (с, Н-18), 1,27- 1,01 (м, Н-16, Н-17 и Н-19).
Пример VIII
Получение 13-(N-Boc-β-изобутилизосеринил)-14β-гидроксибаккатин III 1,14-карбоната
К раствору 14β-гидрокси-10-деацетилбаккатин III 1,14-карбоната (126 мг) в 3 мл сухого тетрагидрофурана добавляют 7,5 мг CeCl3·7H2O и 0,078 мл уксусного ангидрида. Реакционную смесь перемешивают при комнатной температуре в течение 5 часов, в течение которых реакционная смесь становится гомогенной. Добавляют 1,5 г льда, продолжая перемешивание в течение 1 часа. Органический растворитель выпаривают под вакуумом и остаток разбавляют 5 мл Н2О. Образовавшийся осадок отфильтровывают и сушат отсосом под разрежением в течение 18 часов. Полученный в результате продукт (белый порошок, 135 мг) имеет следующие характеристики:1Н-ЯМР (400 МГц, CDCl3): δм.д = 1,25, 1,11 (с, Н-16 и Н-17), 1,66 (с, Н-19), 2,04 (С,Н-18), 2,22 (с, ОАс), 2,29 (с, ОАс), 3,89 (д, J=0,9 Гц, Н-3), 4,06 (д, J=7 Гц, С20b), 4,20 (д, J=7 Гц, Н-20a), 4,41 (м, Н-7), 4,77 (д, J=4 Гц, Н-14), 4,85 (шир. д, J=4 Гц, Н-13), 4,97 (шир. д, J=8 Гц, Н-5), 5,8 (д, J=7 Гц, Н-2), 6,31 (с, Н-10), 7,44 (т, J (Гц, Bz), 7,55 (д, J=8 Гц, Bz), 8,07 (д, J=8 Гц, Bz).
14β-гидроксибаккатин III 1,14-карбонат (130 мг) растворяют в диметилформамиде (4 мл) и добавляют N-метилимидазол (0,07 мл). Через 1 час к данному раствору, интенсивно перемешиваемому при комнатной температуре, добавляют триэтилхлорсилан (0,042 мл). Затем смесь выливают в 10 мл Н2О при интенсивном перемешивании. Суспензию оставляют при 4°С на 18 часов, образовавшийся белый осадок отфильтровывают и промывают Н2О (5 мл), затем гексаном (2х3 мл). Полученное в результате белое твердое вещество (150 мг) имеет такие же спектроскопические характеристики, как соединение, полученное в примере V.
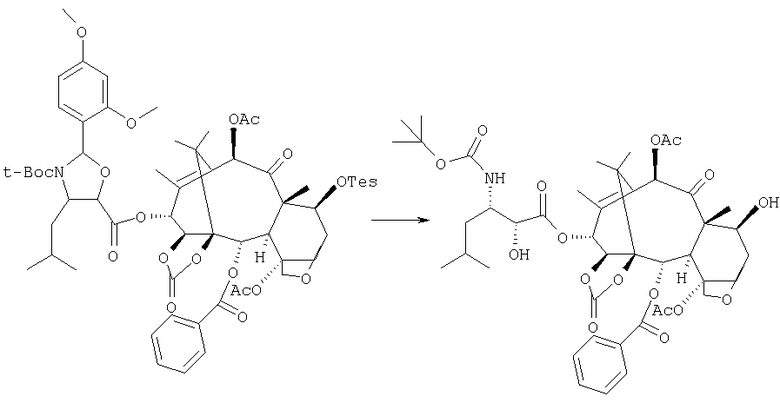
В 1 л круглодонную колбу помещают 20 г 14β-гидрокси-7-Tes-1,14-карбонатбаккатина III вместе с 300 мл тщательно осушенного толуола; затем добавляют 10 г (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислоты, 2 г N,N-диметиламинопиридина (ДМАП) и 9,5 г дициклогексилкарбодиимида (ДЦК), растворенного в СН2Cl2. Реакционную смесь кипятят с обратным холодильником в течение 3 часов, затем охлаждают и удаляют осажденный продукт ацетилмочевины. Маточные растворы промывают насыщенным раствором NaHCO3 для удаления непрореагировавшей кислоты, затем разбавленной хлористоводородной кислотой для удаления ДМАП, затем снова NaHCO3 до нейтральности. Органическую фазу концентрируют до сухого состояния, получая 41,5 г продукта, который можно использовать в последующей стадии без очистки.
40 г данного соединения подвергают реакции снятия защиты двумя стадиями, сначала удаляя Tes и затем 2,4-диметоксибензальдегид, 40 г данного соединения растворяют в 100 мл смеси ацетонитрил/пиридин (80:100) под атмосферой азота и смесь охлаждают до 0°С, затем добавляют 13 мл фторида пиридиния и продолжают перемешивание в течение 24 часов. Раствор выливают в 2 л воды, продукт отфильтровывают и сушат под вакуумом.
Остаток растворяют в 60 мл метиленхлорида, данный раствор энергично перемешивают при 0°С и к нему добавляют 40 мл 0,6 н. раствора HCl в метаноле. Реакционную смесь перемешивают в течение 2 часов, затем разбавляют 150 мл метиленхлорида и взбалтывают с раствором NaHCO3, регулируя рН до 6-7. Органическую фазу концентрируют до сухого состояния, остаток кристаллизуют из смеси ацетон/гексан и сушат, получая 16 г 13-(N-Boc-β-изобутилизосеринил)-14β-гидроксибаккатин-1,14-карбоната, имеющего следующие физико-химические и спектроскопические характеристики:
Формула:C44H57NO17
Внешний вид: белый порошок
Температура плавления: 245°С
Химические сдвиги (м.д) 1Н-ЯМР в растворе CDCl3 (200 МГц)
Химические сдвиги (м.д 13С ЯМР в растворе CDCl3 (50.308 МГц)
Масс-спектр: (NH3, быстрый нагрев на проволоке/хим. иониз., положительные ионы): (m/z) 889 [(MNH4)+], 832 [(MNH4-(CH3)3C)+, 772 [(MNH4-BocNH2)+];
(NH3, быстрый нагрев на проволоке/хим. иониз., отрицательные ионы): (m/z) 871 (М-), 260 (боковая цепь).
Инфракрасный спектр (KBr таблетка): 3521, 3321, 2971, 2953, 1826, 1762, 1706, 1526, 1366, 1238, 1165, 1072, 723 см-1.
УФ-спектр (МеОН): 231, 276 и 284 нм.
-Е1% при 231 нм = 180,99.
-Е1% при 276 нм = 14,094.
-Е1% при 284 нм = 12,182.
Изобретение относится к способу получения производных таксана, а именно 13-(N-Вос-β-изобутилизосеринил)-14β-гидроксибаккатин III 1,14-карбоната формулы (I) 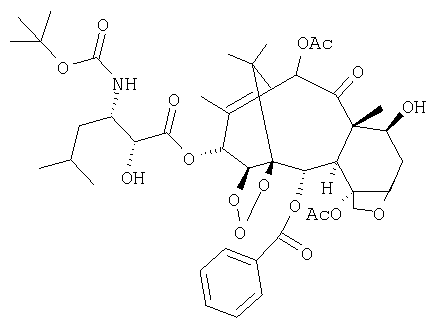 из 10-деацетилбаккатина III в девять стадий. Технический результат - эффективный способ получения ценного биологически активного соединения из более доступного исходного соединения. 10 з.п. ф-лы, 2 табл.
из 10-деацетилбаккатина III в девять стадий. Технический результат - эффективный способ получения ценного биологически активного соединения из более доступного исходного соединения. 10 з.п. ф-лы, 2 табл.

который включает:
а) защиту гидроксилов 10-деацетилбаккатина III в 7- и 10-м положениях:

где R и R1, которые могут быть одинаковыми или различными, выбраны из C1-С10 алкила или арила, C1-С10 алкил- или арилкарбонила, трихлорацетила, C1-C4 триалкилсилила;
b) двухстадийное окисление, чтобы получить производное, окисленное до карбонила в положении 13 и гидроксилированное в положении 14:

с) карбоксилирование вицинальных гидроксилов в 1- и 14 -м положениях, чтобы получить 1,14-карбонатное производное:

d) восстановление карбонила в положении 13:

е) снятие защитных групп в 7- и 10-м положениях:

f) селективное ацетилирование гидроксила в положении 10:
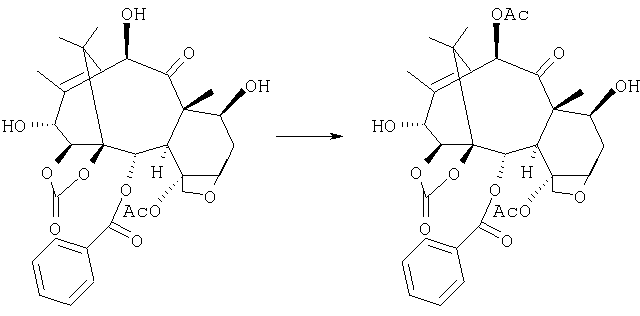
g) превращение 14β-гидроксибаккатин-1,14-карбоната III в производное, содержащее триэтилсилильную группу в положении 7:

h) реакцию соединения, полученного на стадии (g), с (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислотой:

i) удаление триэтилсилильной и диметоксибензилиденовой защитных групп из соединения, полученного на стадии (h):
| US 5917056 А, 29.06.1999 | |||
| US 5705508 А, 06.01.1998 | |||
| Телескопический буровой став | 1975 |
|
SU559019A1 |
| СПОСОБЫ ПОЛУЧЕНИЯ 10-ДЕЗАЦЕТИЛ-БАККАТИНА III | 1993 |
|
RU2108330C1 |
| RU 97112894 А, 27.05.1999. | |||

