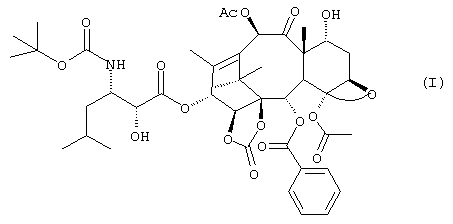
Настоящее изобретение относится к новым таксанам, полезным в качестве химиотерапевтического агента, фармацевтическим композициям, содержащим их, и способам получения производных 14-β -гидрокси-1,14-карбонат-баккатина III и V, замещенных в положении 13 изосериновым остатком.
Таксаны - один из наиболее важных классов противораковых лекарственных средств, разработанных в последнее время. Значительная эффективность паклитакселя и его аналога доцетакселя в лечении различных опухолей сосредоточила исследования на веществах с антимикротубулярной активностью. Однако таксаны характеризуются специфическим механизмом действия, при котором они активизируют сборку микротрубочек и ингибируют деполимеризацию тубулина.
Главными недостатками применяемых в настоящее время таксанов являются: (а) нерастворимость в воде, делающая обязательным применение специфических носителей, которые могут вызывать реакции гиперсенсибилизации; (b) токсичность, которая ограничивает дозировки; (с) развитие механизмов резистентности. Клеточная резистентность к таксанам связана с MDR-фенотипом ("множественная лекарственная устойчивость (резистентность)"),обусловленным переносчиком Р-гликопротеина, путем альтераций тубулина и путем изменений в экспрессии апоптотических регуляторных белков.
Для поиска новых активных молекул, имеющих более высокую растворимость и лучшую толерантность, синтезировали производные таксана 14β -гидрокси-10-деацетилбаккатина III и V.
Некоторые производные 14-гидроксибаккатина III, замещенные в положении-13 изосериновыми остатками, раскрыты в патенте США 5705508, вместе со способом их получения.
В настоящее время установлено, что соединение формулы (I), производное 14-β -гидрокси-1,14-карбонатбаккатина V
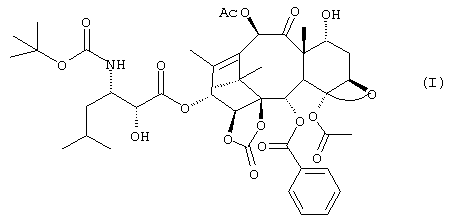
имеет значительную цитотоксичную и антираковую активности и способно к преодолению резистентности клеточных линий, экспрессирующих MDR-фенотип.
Указанное соединение отличается от производных, описанных в вышеупомянутом американском патенте, гидроксилом в положении 7, который в данном случае находится в альфа-конфигурации. 13-(N-Boc-β -изобутилизосеринил)-14β -гидрокси-баккатин III - 1,14 карбонат, соответствующий производному, упомянутому в патенте США №5705508 как SB-T-101131, может применяться как исходный продукт для получения соединения (I).
В этом случае указанное баккатин III производное или подвергают обработке с DBU (диазобицикло[5,4,0]-7-ундецен) в метаноле или ТГФ (THF), или просто оставляют в растворе метиленхлорида или в хлорированных растворителях в присутствии алифатических спиртов типа метанола, этанола или пропанола с основным оксидом алюминия на время, в пределах от одного часа до 14 дней. Соединение, имеющее С-7 бета-конфигурацию, преобразовывают (переводят) при нейтральном или слегка основном рН в более устойчивый альфа-изомер (производное баккатина V).
Альтернативно, соединение (I) можно получить способом, который также позволяет получать соответствующий С-7 бета-эпимер.
Указанный способ (А) включает следующие стадии:
a) преобразование 14β -гидрокси-10-деацетилбаккатин III или V в триэтилсилилированное в положении 7 производное;
b) получение 1,14-карбонатного производного из соединения со стадии (а);
с) селективное ацетилирование 10-гидроксила;
d) реакция продукта со стадии (с) с (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислотой;
е) отщепление защитных групп триэтилсилила и диметоксибензилидена от продукта со стадии (d).
Согласно предпочтительному воплощению способа (А), триэтилхлорсилан применяют как силилирующий агент на стадии (а), тогда как 1,14-карбонатное производное на стадии (b) получено с применением фосгена в толуоле в растворе 3:1 метиленхлорид/пиридин в атмосфере азота. На следующей стадии (с) 14-β -гидрокси-10-деацетилбаккатин III или V 7-Tes-1,14-карбонат образует соль с литийгексаметилдисилилазидом (LiHMDS) в безводном ТГФ и таким образом получают литиевую соль 10-гидрокси производного, которую затем ацетилируют с ацетилхлоридом. Реакцию конденсации между 14-β -гидрокси-7-Теs-1,14-карбонат-баккатином III или V и (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислотой (стадия (d)) осуществляют в безводном неполярном органическом растворителе, в присутствии основания и конденсирующего агента типа дициклогексилкарбодиимида (DCC).
В результате, на стадии (е) триэтилсилил удаляют с пиридиний фторидом в растворе ацетонитрил/пиридин в атмосфере азота, тогда как диметоксибензилиденовую группу удаляют в метиленхлоридном растворителе при добавлении метанола - НСl, а затем NaHCO3.
Последовательность стадий описанного способа можно поменять местами, получая конечный продукт с практически сопоставимым выходом. Указанный альтернативный процесс (В) включает следующие стадии:
а') селективное ацетилирование гидроксила в С-10 14β -гидрокси-10-деацетилбаккатин III или V;
b') получение 1,14 карбонатного производного продукта со стадии (а');
с') силилирование гидроксила в С-7 положении;
d') реакция продукта со стадии (с') с (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислотой;
е') отщепление защитных групп триэтилсилила и диметоксибензилидена от продукта со стадии (d).
В последнем способе заключен целый ряд преимуществ, таких как возможность получить требуемый синтон (1,14-карбонат-7-Теs-баккатина III или V) без хроматографических очисток, по существу только кристаллизацией.
Согласно предпочтительному воплощению, селективное ацетилирование стадии (а') выполняют с уксусным ангидридом в присутствии солей церия, скандия, иттербия, предпочтительно СеСl3·7 Н2O, тогда как остальные стадии выполняют, как обозначено выше.
Настоящее изобретение также включает в себя, в качестве промежуточных продуктов способа получения 14-β -гидрокси-1,14-карбонатбаккатина III или V, следующие соединения: 14β -гидроксибаккатин III или V, 14-β -гидроксибаккатина III или V 1,14 карбонат, 14β -гидрокси-7-Теs-10-деацетилбаккатин III или V, 14β -гидрокси-7-Теs-баккатин III или V, 1,14 карбонат 14β -гидрокси-7-Теs-баккатина III или V.
Следующий аспект изобретения касается способа получения (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислоты по следующей схеме:
СХЕМА

Указанный способ включает следующие стадии:
a) защита аминогруппы лейцинола Нос-группой;
b) переведение N-Boc-L-лейцинола в N-Boc-L-лейциналь;
c) получение циангидринного производного продукта со стадии (b);
d) преобразование циангидриннитрила в соответствующую карбоновую кислоту;
e) образование сложного метилового эфира карбоновой кислоты;
f) очистка сложного метилового эфира (2R,3S)-3-(N-Вос)амино-2-гидрокси-5-метилгексановой кислоты;
g) конденсация продукта со стадии (f) с 2,4-диметоксибензальдегид-диметил-ацеталь;
h) преобразование сложного метилового эфира (4S,5R)-N-Вос-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислоты в соответствующую карбоновую кислоту.
Согласно предпочтительному воплощению, на стадии (а) проводят реакцию лейцинола с Вос-ангидридом, и затем окисляют до альдегида в растворителе ДМСО/СН2Сl12, применяя оксалилхлорид при температуре ниже -60° С, нейтрализуя образовавшуюся кислоту триэтиламином, либо окисляют его гипохлоритом натрия при температуре от -2 до -5° С, Циангидрин на стадии (с) получают, заменяя сульфо-группу промежуточного 1-гидрокси-2-(N-Boc)амино-4-метилпентансульфоната цианид-ионом, Циангидрин затем гидролизуют до соответствующей карбоновой кислоты на стадии (d) кипячением с обратным холодильником в концентрированной хлористоводородной кислоте.
На стадии (е), (2R/S,3S)-3-(N-Вос)амино-2-гидрокси-5-метилгексановую кислоту превращают в соответствующий сложный метиловый эфир взаимодействием с диазометаном в эфирном растворе. На стадии (f) диастереомер(2R,3S) очищают кристаллизацией из циклогексана или смеси гексан/толуол. Стадию (g) выполняют в ТГФ в присутствии пиридиний-п-толуолсульфоната, удаляя выделяющийся метанол; после завершения реакции пиридиний-п-толуолсульфонат нейтрализуют бикарбонатом. На стадии (h) сложный эфир гидролизуют в смеси метанол/вода с карбонатом калия. Реакционную смесь затем подкисляют и готовый продукт экстрагируют метиленхлоридом.
Изобретение также включает (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновую кислоту, как промежуточное соединение для синтеза баккатин III и V производных, замещенных в 13-положении N-Boc-β -изобутилсеринильным остатком.
Новый таксан настоящего изобретения показал сильную антираковую активность против злокачественных клеток груди, легкого, яичника, толстой кишки, простаты, почек, поджелудочной железы, и также против клеток, стойких к известным антираковым лекарствам, таким как адриамицин, винбластин и платиновых производных.
Таким образом, изобретение относится к фармацевтическим составам, содержащим эффективное количество соединения данного изобретения, вместе с фармакологически приемлемыми носителями и наполнителями. Более подробно, состав можно сформировать в форме таблеток, порошков, гранулятов, капсул, инъекций, растворов, свечей, эмульсий, дисперсий, и т.п. Для внутривенного введения главным образом применяются смеси Chremophor L и этанола, полисорбата и этанола, или липосомный состав, полученный с натуральным или синтетическим фосфатидилхолином, или смеси натуральных фосфолипидов в присутствии холестерина; для орального введения лучше приготовить мягкие желатиновые капсулы, в которых продукт растворяют в полисорбатах, ПЭГ (PEG) или в смесях из него, необязательно в присутствии фосфолипидов. Соединение (I) можно вводить людям при концентрациях от 50 до 500 мг/м2.
Следующие примеры иллюстрируют изобретение в больших деталях.
Пример 1: Синтез 1,14 карбоната 13-(N-Boc-β -изобутил-серинил)-14β -гидроксибаккатина III
43,26 г 14β -гидрокси-деацетилбаккатина III вместе с 22,3 мл N-метилимидазола растворяли в 230 мл ДМФ (DMF) в 500 мл стеклянной круглодонной колбе; в раствор добавляли 14 мл триэтилхлорсилана при сильном перемешивании при комнатной температуре в течение 1 часа. После окончания реакции реакционную смесь выливали в 2 л воды при сильном перемешивании. Образовался обильный осадок, который оставляли при 4° С с вечера (на ночь). Затем осадок фильтровали, тщательно промывая водой, а потом н-гексаном. После сушки в вакууме получали 48,1 г 7-Теs-10-деацетилбаккатин III (XII), содержащий небольшой процент 7,10-производного, и имеющий следующие химико-физические характеристики:

1Н ЯМР (СDСl3, 200 МГц): δ (ppm)=0,55 (6H, t, J=7,8 Гц, 7-OTES CH2), 0,94 (9H, q, J=7,8 Гц, 7-OTES СН3), 1,18 (3H, s, С16Н3), 1,20 (3Н, s, С17Н3), 1,77 (3Н, s, С19Н3), 1,90 (1Н, ddd, J=2,4, 10,8, 13,2 Гц, С6Нβ ), 2,12 (3Н, d, J=1,6 Гц, С18Н3), 2,31 (3Н, s, 4-ОСОСН3), 2,48 (3Н, ddd, J=14,3, 9,8, 6,5 Гц, C6Hα ), 2,73 (1Н, d, J=5,5 Гц, ОН), 3,79 (1Н, d, J=7,1 Гц, С3Н), 4,20 (1Н, dd, J=1,0, 8,3 Гц, С20Нβ ), 4,31 (1Н, d, J=8,6 Гц, С20Нα ), 4,39 (1Н, dd, J=6,4, 10,7 Гц, С7Н), 4,77 (1Н, d, J=5,8 Гц, С14Н), 4,94 (1Н, dd, J=2,1, 9,7 Гц, (С5Н), 5,05 (1Н, m, C13H), 5,13 (1Н, d, J=1,9 Гц, С10Н), 6,05 (1Н, d, J=7,3 Гц, С2Н), 7,41-8,09 (5Н, m, Ph),
Масс-спектр (NH3, DEP/CI, положительные ионы): (m/z) 718 [(M+NH4)+, 100%], 701 [М+Н)+, 39%].
Полученное соединение растворяли в 300 мл 3:1 смеси метиленхлорид/пиридин в атмосфере азота; данный раствор добавляли при перемешивании к раствору фосгена (214 мл 1,9 М раствора в толуоле), предварительно охлажденному при -10° С, сохраняя температуру от -5 до -10° С в ходе добавления.
Реакционную смесь перемешивали 30’, затем встряхивали с 700 мл насыщенного раствора NаНСО3, сохраняя температуру 2° С или ниже. Фазы разделяли и органическую фазу промывали, чтобы удалить пиридин. Органическую фазу сушили над МgSО4 и упаривали досуха. Получали 46,6 г 7-Tes-l,14-карбоната 10-деацетилбаккатина III, который можно непосредственно применять для следующих реакций.
31 г соединения растворяли в 250 мл абсолютно безводного ТГФ; раствор охлаждали до -50° С, добавляли 48 мл раствора 1М LiHMDS в течение 2 минут и перемешивали в течение 20 минут при той же самой температуре. Добавляли 3,7 г ацетилхлорида в течение 40 минут при перемешивании. Температуру реакции оставляли подниматься до 0° С, продолжая перемешивание в течение 2 час. После завершения взаимодействия, смесь обрабатывали насыщенным раствором NН4Сl и разбавляли этилацетатом. Фазы разделяли и водный раствор разбавляли этилацетатом до извлечения продукта. Объединенные органические фазы промывали водой, затем высушивали над MgSO4 и упаривали досуха. Получали 33 г 14-β -гидрокси-7-Теs-1,14-карбонат-баккатина III, загрязненного соединениями предшествующих реакций. Соединение хроматографировали на силикагеле, элюируя чистый продукт смесью 9:1 этилацетат/СН2Сl2, Получали 30 г требуемого соединения (XIII), имеющего следующие характеристики:

1Н ЯМР (CDCl3, 200 МГц): δ (ppm) = 0,55 (6Н, t, J=7,8 Гц, 7-OTES СН2), 0,95 (9Н, q, J=7,8 Гц, 7-OTES СН3), 1,16 (3Н, s, С16Н3), 1,32 (3Н, s, С17Н3), 1,77 (3Н, s, С19Н3), 1,88 (1Н, ddd, J=2,4, 10,8, 13,2 Гц, С6Нβ ), 2,21 (3Н, d, J=1,6 Гц, С18Н3), 2,19 (3Н, s, 10-ОСОСН3), 2,31 (3Н, s, 4-ОСОСН3), 2,48 (3Н, ddd, J=14,3, 9,8, 6,5 Гц, C6Hα ), 2,73 (1Н, d, J=5,5 Гц, ОН), 3,72 (1Н, d, J=7,1 Гц, С3Н), 4,20 (1Н, d, J=8,3 Гц, С20Нβ ), 4,31 (1Н, d, J=8,6 Гц, C20Hα ), 4,46 (1H, dd, J=6,4, 10,7 Гц, С7Н), 4,79 (1Н, d, J=5,8 Гц, С14Н), 4,94 (1Н, dd, J=2,1, 9,7 Гц (С5Н), 5,02 (1Н, m, С10Н), 5,05 (1Н, m, С13Н), 6,09 (1Н, d, J=7,3 Гц, С2Н), 7,41-8,09 (5Н, m, Ph).
Масс-спектр (NH3, DEP/CI, положительные ионы): (m/z) 759 [(M+NH4)+, 19%], 743 [М+Н)+, 100%].
20 г 14β -гидрокси-7-Теs-1,14-карбонат-баккатина III вместе с 300 мл строго безводного толуола помещали в круглодонную колбу емкостью 1 л, добавляли 10 г (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислоты, 2 г N,N-диметиламинопиридина (DMAP) и 9,5 г дициклогексилкарбодиимида (DCC), растворенного в СН3Сl2. Реакционную смесь кипятили с обратным холодильником в течение 3 час, затем охлаждали, продукт мочевины осаждали, и маточный раствор промывали насыщенным раствором NaHCO3 для удаления непрореагировавшей кислоты и затем разбавленной хлористоводородной кислотой для удаления (DMAP) и, наконец, снова NаНСО3 для нейтрализации. Органическую фазу упаривали досуха с получением 41,5 г продукта, который можно непосредственно применять на следующей стадии.
40 г этого соединения подвергли снятию защиты в две стадии, удаляя первым Теs и затем 2,4-диметоксибензальдегид, 40 г соединения растворяли в 100 мл смеси (80:100) ацетонитрил/пиридин в атмосфере азота и дали возможность охладиться при 0° С; добавляли 13 мл пиридиний фторида и перемешивали в течение 24 час. Раствор выливали в 2 л воды, отфильтровывали продукт и высушивали в вакууме.
Остаток растворяли в 60 мл метиленхлорида и этот раствор добавляли в 40 мл 0,6 н. НСl в метаноле при сильном перемешивании и при 0° С. Реакционную смесь оставляли на 2 час при перемешивании, затем разбавляли в 150 мл метиленхлорида и встряхивали с раствором МаНСО3, доводя рН до 6-7. Органическую фазу упаривали досуха и кристаллизовали остаток из ацетона гексана. После высыхания получали 16 г 13-(N-Boc-β -изобутилизосеринил)-14β -гидроксибаккатин-1,14-карбоната, имеющего следующие химико-физические и спектральные характеристики:
Формула: C44H57NO17.
Внешний вид: белый порошок.
Температура плавления: 245° С.

2
Химические сдвиги (ppm) 13С ЯМР в растворе СDСl3 (50,308 Мгц) ОСН3
ОСН3 ОСН3
ОСН3 ОPh
ОPh =O (BOC)
=O (BOC) =O (карбонат)
=O (карбонат) H3)3C Boc
H3)3C Boc (Вос)
(Вос) Н3
Н3 Н3
Н3
Масс-спектр: (NН3, DEP/CI, положительные ионы): (m/z) 889 [(MNH4)+], 832 [(MNH4-(CH3)3С)+], 772 [(MNH4-BocNH2)+]
(NН3, DEP/CI, отрицательные ионы): (m/z) 871 (М-), 260 (боковая цепь)
Инфракрасный спектр (КВr диск): 3521, 3321, 2971, 2953, 1826, 1762, 1706, 1526, 1366, 1238, 1165, 1072, 723 см-1
УФ спектр (МеОН): 231, 276 и 284 нм;
-Е1% при 231 нм = 180,99
-E1% при 276 нм = 14,094
-E1% при 284 нм = 12,182
Пример 2: Синтез 13-(N-Boc-β -изобутилсеринил)-14β -гидроксибаккатин V, 1,14 карбонат
5 г 13-(N-Boc-β -изобутилсеринил)-14β -гидроксибаккатин III, 1,14 карбоната растворяли в 500 мл толуола в атмосфере аргона, полностью дезоксигенируя раствор; добавляли 80 мг DBU (диазобицикло[5,4,0]-7-ундецен), и реакционную смесь кипятили с обратным холодильником в течение 1 часа в атмосфере аргона. Раствор разбавляли 100 мл этилацетата и промывали водой. Упаривали органическую фазу досуха, чтобы получить 4,5 г 1,14 карбоната 13-(N-Вос-β -изобутилсеринил)-14β -гидроксибаккатина V, со следующими химико-физическими и спектральными характеристиками:
Формула: C44H57NO17
Внешний вид: белый порошок
Температура плавления: 245° С
Химические сдвиги (ppm) 1H ЯМР в растворе СDС13 (200 МГц)

Химические сдвиги (ppm) 13С ЯМР в растворе СDСl3 (50,308 МГц) ОСН3
ОСН3 ОСН3
ОСН3 OPh
OPh =O (Воc)
=O (Воc) =O (Карбонат)
=O (Карбонат) Н3)3C
Н3)3C (Вос)§
(Вос)§  Н3
Н3 Н3
Н3
Химические сдвиги (ppm) 13С ЯМР в растворе СDСl3 (50,308 МГц)
Масс-спектр (TSP+): (m/z) 872 (МН+); 816 (МН+-(СН3)2С=СН2); 772 (816-СO2); 756 (816-АсОН); 712 (772-АсОН)
Инфракрасный спектр (KBr диск): 3450, 2963, 1813, 1740, 1702, 1247, 1091, 710 см-1
УФ спектр (МеОН): 200 е 230 нм
-E1% при 200 нм = 370,9
-E1% при 230 нм = 193,2
Пример 3: Получение (4S,5R)-N-Boc-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислоты.
Получение N-Boc-L-лейцинола (III):
46,8 г L-лейцинола II (400 ммоль) растворяли в 300 мл CH2Cl12 в 2 л трехгорлой круглодонной колбе, оборудованной механической мешалкой, термометром и капельной воронкой. Затем к перемешиваемому раствору добавляли по капле при комнатной температуре раствор Вос-ангидрида (87,2 г, 400 ммоль) в CH2Cll2 (100 мл) в течение 90 мин. При добавлении первых 25% Вос-ангидрида, взаимодействие было экзотермическим, достигало 20-30° С, образуя суспензию, которая становилась прозрачной после перемешивания при комнатной температуре в течение следующих трех часов. Все оставляли при комнатной температуре на ночь. Растворитель испаряли в высоком вакууме, чтобы получить нужный продукт в виде густого масла при количественном выходе (87 г). Продукт затем обрабатывали без дальнейших очисток.
Получение N-Boc-L-лейцинола (IV)
В раствор оксалилхлорида (26,274 мл, 300 ммоль) в 130 мл метиленхлорида, предварительно охлажденный при -60/-65° С, медленно добавляли ДМСО (28,4 мл, 400 ммоль).
Раствор становится прозрачным после завершения добавления ДМСО. После 20 мин перемешивания при той же самой температуре реакционную смесь обрабатывали раствором спирта III (43,7 г, 200 ммоль) в CH2Cl2 (200 мл) в течение 25 мин, сохраняя температуру ниже -60° С. Во время добавления спирта реакционная смесь помутнела и образовался белый осадок. После 20-25 мин перемешивания при той же самой температуре добавляли по капле раствор триэтиламина (112 мл, 800 ммоль) в CH2Cl2 (100 мл) в течение 40 мин, сохраняя температуру между -68 и -62° С. Реакционную смесь затем перемешивали при температуре между -60 и -65° С в течение следующих 50 мин. ТСХ реакционной смеси осуществляли, применяя смесь 8% метанола в CH2Cl2, поскольку элюент не обнаруживал исходного продукта.
Холодный раствор затем переливали в 800 мл ледяного раствора, содержащего 68 г (0,5 моль) KHSO4. Органический слой отделяли и водную фазу экстрагировали CH2Cl2 (100 мл). Объединенные органические фазы промывали водным KHSO4 (5%, 1× 200 мл), насыщенным раствором соли (100 мл, 50 мл) и выпаривали до половины объема (≈ 250 мл). Указанное вещество применяли непосредственно на следующей стадии.
Альдегид (V) бисульфит производное
Метиленхлоридный раствор альдегида (IV) в 2 л трехгорлой круглодонной колбе, оборудованной механической мешалкой, термометром и капельной воронкой, обрабатывали в течение 10 мин, при -5° С раствором бисульфита натрия (41,7 г, 400 ммоль) в воде (200 мл) и затем H-Bu4NKSO4 (678 мг, 2 ммоль). Раствор охлаждали до -5° С. Реакционную смесь перемешивали при температуре от -5 до 0° С в течение 5-6 часов и затем в течение ночи при комнатной температуре. Водную фазу, содержащую состав V, отделяли и промывали с CH2Cl2 (2× 20 мл).
(2-циано-3-(N-Вос)-амино-S-метилгексанол (VI)
В вышеупомянутый водный раствор (≈ 250 мл) добавляли СН2Сl2 (120 мл) и реакционную смесь охлаждали до 0-5° С на бане со льдом. Затем добавляли твердый KCN (15 г, 230 ммоль) к реакционной смеси, и раствор перемешивали при комнатной температуре в течение ночи. Органическую фазу отделяли, а водную фазу экстрагировали CH2Cl2. Объединенные органические фазы промывали соляным раствором (1× 50 мл), высушивали над МgSO4 и выпаривали до получения продукта в виде бесцветной вязкой жидкости (43 г). Продукт имеет [α ]D 51,11 (с=2, МеОН) и является смесью VI 2(R),3(S) и 2(S),3(S) производных в соотношении около 2:1. Выход составляет 89% по сравнению с исходным L-лейцинолом.
(2RS,3S)-3-амино-2-гидрокси-5-метилгексановая кислота (VII)
Смесь вышеупомянутого неочищенного нитрила VI (43 г) обрабатывали 150 мл концентрированной НСl (37%) (150 мл) и кипятили с обратным холодильником в течение ночи, чтобы получить неочищенную кислоту VII*. Избыток хлористоводородной кислоты удаляли на роторном испарителе и остаток упаривали с водой (100 мл) для удаления НСl. Затем остаток растворяли в 150 мл воды и добавляли 100 мл ацетона, обрабатывали 33 мл 6,25 М раствора NaOH, доводя рН до 5. Дополнительное количество ацетона (500 мл) добавляли к раствору, который был оставлен, с вечера при 4° С. Осадок затем отфильтровывали, промывали твердую лепешку на фильтре ацетоном и высушивали в вакууме, чтобы получить неочищенную кислоту VII (6,5 г), содержащую смесь 2(R), 3(S) и 2(S), 3(S) производных соединения VI в соотношении около 3:1.
Фильтрат упарили и добавили воду, чтобы довести объем раствора до 75 мл.
Затем ацетон (1 л) добавляли к раствору, который оставили на ночь при 4° С в холодильнике. Осадок отфильтровывали и твердую лепешку на фильтре промывали ацетоном и высушивали в вакууме, чтобы получить дополнительное количество продукта (18 г), содержащего твердый NaCl и смесь 2(R), 3(S) и 2(S), 3(S) производных VII в соотношении около 1:1.
Извлеченный первый продукт VII (22,5 г) нагревали в воде (120 мл), не получая полного растворения, затем охлаждали на льду и отфильтровывали, чтобы получить 12,5 г кислоты VII, все еще загрязненной примерно до 10% нежелательным 2(R), 3(S) производным VII. Продукт высушивали и смешивали с вышеупомянутой 1:1 смесью кристаллов второго сбора (общее количество 27 г).
(2RS,3S)-3-(N-Boc)амино-2-гидрокси-5-метилгексановая кислота (VIII)
(A) Неочищенную кислоту VI 2(R), 3(S), приблизительно 90% чистоты (2,5 г; 77,6 ммоль), растворяли в смеси 1:1 вода-ТГФ (80 мл), затем триэтиламин (13,5 мл) и затем Вос-ангидрид (18,5 г; 85 ммоль) добавляли к реакционной смеси, весь раствор перемешивали в течение 40 часов при комнатной температуре. Растворитель испаряли на роторном испарителе, добавляли 60 мл воды и 60 мл этилацетата, продолжая перемешивание. Водную фазу отделяли и экстрагировали с этилацетатом (30 мл). Объединенные органические фазы экстрагировали 10% водным раствором карбоната натрия (30 мл, 20 мл). Основной экстракт затем объединяли с водной фазой, подкисленной 2 М хлористоводородной кислоты (≈ 55 мл), для доведения рН раствора до 2. Кислоту VIII затем экстрагировали из водной фазы этилацетатом (3× 40 мл) и гетероуксусные экстракты промывали водой (20 мл), высушивали (MgSO4) и выпаривали, чтобы получить неочищенное VIII Вос-производное в виде сиропа (20 г, 99%).
(B) Неочищенную кислоту VII 2R, 3S, с чистотой около 50%, загрязненную NaCl (27 г), растворяли в смеси 1:1 вода-диоксан (120 мл). Затем добавляли триэтиламин (20 мл) к реакционной смеси, потом - Воc ангидрид (26,15 г, 120 ммоль). Раствор перемешивали в течение 40 часов при комнатной температуре. Растворитель испаряли в роторном испарителе, воду и этилацетат (100 мл) добавляли к остатку, продолжая перемешивание еще в течение нескольких минут. Органическую фазу отделяли и экстрагировали с 10% водным раствором карбоната натрия (45 мл, 30 мл). Экстракты карбоната натрия затем объединяли с водной фазой, подкисленной 1 М хлористоводородной кислотой (≈ 165 мл) и экстрагированной с этилацетатом (3× 60 мл), а затем промытой водой (30 мл), высушенной (МgSO4) и выпаренной, чтобы получить неочищенный VII Воc в виде сиропа (16 г), состоящего из смеси 2R, 3S и 2S, 3S изомеров в соотношении 1:1.
Сложный метиловый эфир (2R,3S)-3-(N-Вос)амино-2-гидрокси-5-метилгексановой кислоты (IX)
Диазометан получали из диазальдегида, следуя способу, описанному Т.Н. Black [Aldrichimica Acta, 16, 3 (1983)].
(A) Раствор неочищенной кислоты VIII (20 г, 56,6 ммоль) в CH2Cl2 (75 мл) медленно добавляли к холодному эфирному раствору диазометана (77 ммоль) и смесь оставляли в течение двух часов на ледяной бане. Цвет раствора на этой стадии становился белым, таким образом указывая, что большая часть диазометана адсорбирована. Затем раствор выпаривали и остаток кристаллизовали из смеси толуола (20 мл) и гексана (70 мл). После охлаждения в течение ночи в холодильнике при 4° С, кристаллы чистого IXA 2R, 3S производного собирали фильтрацией. Выход составлял 15 г. Маточные растворы давали приблизительно 5 г смеси изомеров в соотношении 1:1.
(B) Применяя тот же самый способ, 1:1 смесь кислоты VIII (16 г) преобразовывали в 1:1 смесь IXA и IXB эфиров.
Добавляли материал от маточных растворов (5 г со стадии А), материал объединяли и разделяли на хроматографической колонке, применяя гексан-этилацетат в качестве элюента (от 9:1 до 7:3). Нингидрин применяли как проявитель для ТСХ пластин, Неполярное соединение Rf 0,75 (гексанэтилацетат: 7:3) идентифицировали как требуемый эфир IXA (2R,3S), который перекристаллизовывали из циклогексана, чтобы получить IXA в виде бесцветных игл (8 г) t° пл. 95-96° С, [α ]D 72,4° (с=l, МеОН).
Полярное соединение Rf 0,5 (гексан-этилацетат 7:3) идентифицировали как IXB (2S,3S) и перекристаллизовывали из циклогексана, чтобы получить 10 г IXB в виде бесцветных игл.
2,4-диметоксибензальдегиддиметилацеталь
Смесь 2,4-диметоксибензальдегида (41,25 г, 0,25 моль), обезвоженного триметилортоформиата (50 мл) и нитрата аммония (2 г растворенного в 20 мл метанола) кипятили с обратным холодильником в течение 6 часов (1Н ЯМР реакционной смеси показал 65-70% конверсии). Сначала горячая реакционная смесь была прозрачным раствором, но поскольку взаимодействие продолжалось, выпадал твердый осадок. Добавляли вторую часть безводного триметилортоформиата (20 мл), и часть метанола отгоняли.
Когда температура реакционной смеси достигала 95-100° С, весь осадок в колбе растворялся. Раствор охлаждали до комнатной температуры и добавляли обезвоженный Nа2СО3 (5 г), перемешивая 30 мин. Впоследствии раствор отфильтровывали и остаток перегоняли фракционной дистилляцией в вакууме при 0,25 мм рт.ст. Первая фракция при низкой температуре состояла, главным образом, из избытка триметилортоформиата, а вторая фракция, которая перегонялась в виде бесцветного масла при 175-180° С, представляла собой требуемый ацеталь. Выход: 37 г (70%).
Метиловый эфир (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислоты (X)
В раствор сложного метилового эфира (2R,3S)-3-(N-Вос)амино-2-гидрокси-5-метилгексановой кислоты (IXA) (34,375 г, 125 ммоль) в безводном ТГФ (150 мл) добавляли дистиллированный 2,4-диметоксибензальдегид диметилацеталь (30 г, 142 ммоль) и далее пиридиний-п-толуолсульфонат (Ру. Тоs; 400 мг).
Раствор нагревали при умеренном кипении в 500 мл трехгорлой колбе с обратным холодильником, оборудованной насадкой Дина-Старка. После приблизительно 6 час кипения с обратным холодильником, удаляли около 60 мл ТГФ, содержащего метанол, полученный в ходе реакции. Образец подвергали 1H ЯМР анализу (в CDCL3).
Пик при δ =1,41 ppm исчезал (1), а появлялся новый пик при δ =1,24 ppm для защищенного метилового эфира (2). После 6 часов кипения с обратным холодильником превращение составляло около 70-75%.
Добавляли новую порцию безводного ТГФ (50 мл), затем 2,4-диметоксибензальдегидацеталь (5,0 г; 24 ммоль). Реакционную смесь нагревали с обратным холодильником в течение дальнейших 2,5 часов, во время которых удаляли около 50 мл ТГФ, используя насадку Дина-Старка. Последующий 1Н ЯМР анализ показал полную конверсию исходного материала.
В реакционную смесь добавляли насыщенный водный раствор NaHCO3 (15 мл) и перемешивали смесь в течение 15 минут, чтобы нейтрализовать Py. Tos. Затем добавляли трет-бутилметиловый эфир (85 мл) и воду (15 мл) и отделяли органическую фазу. Водную фазу экстрагировали с трет-бутилметиловым эфиром (20 мл) и объединенные органические фазы промывали водой (30 мл) и выпаривали с получением остатка (66 г) неочищенного продукта X.
Гидролиз сложного эфира Х для получения кислоты XI
Неочищенный сложный эфир Х (22 г, 42 ммоль) растворяли в 100 мл метанола и добавляли воду (50 мл), содержащую 8,7 г карбоната калия. После перемешивания в течение ночи при комнатной температуре считалось, что реакция завершена при контроле ТСХ (толуол-этилацетат: 4,5:1). ТСХ анализ подтвердили 1H ЯМР исследованиями, проверяя исчезновение пика метилового эфира.
Метанол испаряли при температуре не выше 40° С в вакууме (около 60 г остатка) и к остатку добавляли воду (150 мл). Водную суспензию экстрагировали этилацетатом (5× 50 мл), чтобы удалить избыток бензальдегида и бензальдегиддиметилацеталя. К водной фазе добавляли 90 мл метиленхлорида, смесь охлаждали на ледяной бане, и двухфазную систему обрабатывали приблизительно 125 мл 1 М NaHSО4 (pH 3) при сильном перемешивании. Фазы разделяли и водную фазу экстрагировали метилен хлоридом (75 мл). Объединенные метиленхлоридные экстракты промывали водой (30 мл), солевым раствором (30 мл) и высушивали над МgSO4. Затем раствор хранили при -60° С. Выход конечного продукта в виде бесцветного твердого вещества составлял 16 г, около 93%, относительно исходного продукта.
Пример 4: Получение 14β -гидрокси-7-Теs баккатин III 1,4 карбонат
К раствору 11,2 г 10-деацетил-14-гидроксибаккатин III в 50 мл сухого тетрагидрофурана добавляли 0,72 г СеСl3·7 Н2О и 7,3 мл уксусного ангидрида. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов; в течение этого времени смесь становилась однородной. Добавляли 10 г льда, и все перемешивали в течение 1 часа. Тетрагидрофуран выпаривали в вакууме и остаток растворяли в 200 мл H2O. Осадок отфильтровывали и высушивали в вакууме в присутствии P2O5: продукт кристаллизовали из этилацетата, чтобы получить 10 г 14-гидроксибаккатина III, обладающего следующими характеристиками:
Т.пл.: 236-8° С; ИК (КВr): 3474, 1739, 1400, 1240, 1090, 1049 см-1.
1H ЯМР (CDCl3, 200 МГц); 8,07 (d, J=8 Гц, Bz), 7,55 (d, J=8 Гц, Bz), 7,44 (t, J=8 Гц, Bz), 6,31 (s, H-10), 5,80 (d, J=7 Гц, Н-2), 4,97 (br d, J=8 Гц, Н-5), 4,73 (br, d, J=4 Гц, Н-13), 4,41 (m, H-7), 4,24 (d, J=4 Гц, Н-14), 4,20 (d, J=7 Гц, Н-20a), 4,06 (d, J=7 Гц, Н-20b), 3,89 (J=0 Гц, Н-3), 2,29 (s, Oаc), 2,22 (s, Oac), 2,04 (s, H-18), 1,66 (s, H-19), 1,25, 1,11 (S, Н-16 и Н-17).
В четырехгорлую колбу, оборудованную мешалкой, капельной воронкой, термометром и парциальным конденсатором горячего орошения, охлажденным до -12° С, помещали 52,8 мл 1,9 М раствора фосгена в толуоле. В этот раствор по капле добавляли 11,6 г 14-гидроксибаккатина III, растворенного в 53 мл метиленхлорида и 17,5 мл пиридина при перемешивании в течение 30 минут. Температуру поддерживали между -6 и -10° С. Через 30 минут добавляли 50 мл насыщенного раствора NаНСО3 при перемешивании и точном контроле температуры. После нагревания до комнатной температуры фазы разделяли. Водную фазу экстрагировали метиленхлоридом и органические фазы промывали 45 мл 2 н. НСl, доводя рН до около 1. Органическую фазу промывали 0,1 н. НСl и затем NаНСО3, высушивали над Na2SO4 и выпаривали досуха, чтобы количественно получить 11,5 г 1,14 карбоната 14-гидроксибаккатина.
11,5 г 1,14 карбоната 14-гидроксибаккатина растворяли в 50 мл ДМФ (DMF), добавляли 1,1 эквивалента хлортриэтилсилана и 3 эквивалента N-метилимидазола при комнатной температуре. После завершения взаимодействия смесь выливали в 500 мл Н2O и осадок отфильтровывали и основательно промывали H2O, затем сушили, чтобы получить 12,8 г 1,14 карбоната 14β -гидрокси-7-Tes-баккатина III с теми же самыми характеристиками, что приведены в примере 1.
Пример 5: Синтез 13-(N-Boc-β -изобутилсеринил)-14β -гидроксибаккатин III, 1,14 карбонат
Исходя из 14β -гидрокси-7-Теs-баккатин III-1,14 карбоната, полученного, как описано в вышеупомянутом примере, методика была следующей.
В 1 л круглодонную колбу помещали 20 г 14β -гидрокси-7-Теs-1,14-карбонат-баккатин III вместе с 300 мл полностью обезвоженного толуола; добавляли 10 г (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислоты, растворенной в CH2Cl2 и 2 г N,N-диметиламинопиридина (DMAP), и добавляли 9,5 г дициклогексилкарбодиимида (DCC). Реакционную смесь кипятили с обратным холодильником в течение 3 час, затем охлаждали, чтобы ускорить выпадение осадка продукта производного мочевины, а маточные растворы промывали насыщенным раствором NаНСО3 для удаления непрореагировавшей кислоты, затем разбавленной хлористоводородной кислотой, чтобы удалить DMAP и, наконец, снова NaHCO3 для нейтрализации. Органическую фазу выпаривали досуха, чтобы получить 41,5 г продукта, который можно непосредственно использовать на следующей стадии.
40 г этого состава подвергали снятию защиты на двух стадиях, расщепляя сначала Теs и затем 2,4-диметоксибензальдегид. 40 г состава растворяли в 100 мл смеси (80:100) ацетонитрил/пиридин в атмосфере азота, и смесь охлаждали до 0° С; добавляли 13 мл пиридиний фторида, и оставляли при перемешивании на 24 час. Раствор выливали в 2 л воды, продукт фильтровали и сушили в вакууме. Остаток растворяли в 60 мл метиленхлорида, и в этот раствор добавляли 40 мл метанольного 0,6 н. НСl при сильном перемешивании и при 0° С. Реакционную смесь оставляли на 2 час при перемешивании, затем разбавляли с 150 мл метиленхлорида и встряхивали с раствором NaHCO3, доводя рН до 6-7. Органическую фазу выпаривали досуха, и остаток кристаллизовали из ацетонгексана, затем сушили с получением 16,5 г 1,14-карбоната 13-(N-Boc-β -изобутилизосеринил)-14β -гидроксибаккатина III.
Пример 6: Синтез 1,4-карбоната 13-(N-Boc-β -изобутилсеринил)-14β -гидроксибаккатина-V.
Растворяли 3 г 1,4-карбоната 13-(N-Boc-β -изобутилсеринил)-14β -гидроксибаккатина-III в 500 мл смеси дихлорметан:метанол, 9:1. Затем добавляли 10 г основного оксида алюминия и суспензию оставляли при перемешивании на ночь. Затем суспензию фильтровали и растворитель упаривали досуха. Остаток очищали хроматографией на колонке (гексанацтон 1:1) с получением 1,4-карбоната 13-(N-Boc-β -изобутилсеринил)-14β -гидроксибаккатина-V, имеющего следующие физико-химические и спектроскопические характеристики.
Формула: C44H57NO17
Внешний вид: белый порошок
Температура плавления: 245° С.
Пример фармацевтической композиции:
Соединение I 300 мг
Этиловый спирт (дегидратированный спирт) 5 мл
Полисорбат 80 10 мл
ДАННЫЕ БИОЛОГИЧЕСКИХ ИСПЫТАНИЙ
1. Фармакологическая активность
Анализ на цитотоксичность
Соединение формулы I по пункту 1 (Соединение I) и известное ссылочное соединение паклитаксел (таксол, одно из наиболее активных известных химиотерапевтических средств) исследовали для сравнения их цитотоксической активности в отношении различных опухолевых клеточных линий.
Использовали следующие клеточные линии карциномы человека: клетки карциномы молочной железы MCF-7(34), устойчивые к лекарственным средствам клетки MCF-7 (MCF7/R), устойчивые к лекарственным средствам клетки карциномы яичников А2789 (38) и устойчивую к лекарственным средствам линию клеток карциномы человека НСТ-15.
Ингибирующее действие испытываемых соединений на рост клеток оценивали с использованием сульфородамина В (SRB) (Sigma Chemical Co.) в анализе, основанном на окрашивании, который косвенно определяет число клеток посредством измерения белков, ассоциированных с мембранами. Кратко, 1× 105 экспоненциально пролиферирующих клеток высевали в 96-луночные микротитровальные планшеты в полной среде роста и инкубировали при 37° С в течение 15-18 часов, давая возможность клеткам прикрепиться к субстрату перед добавлением таксанов. Для каждого испытываемого таксана повергали испытаниям параллельно пять 96-луночных микротитровальных планшетов. Все клеточные линии подвергали воздействию 10-12 различных концентраций каждого лекарства, включающие концентрации 5-6-log диапазона, в течение 72 часов при 37° С, 5% СO2. Клетки фиксировали in situ в течение 1 часа при 4° С с помощью охлажденной льдом 50% трихлоруксусной кислоты. Планшеты затем промывали 6 раз водой, и в каждую лунку добавляли 150 мл 0,4% SRB. После 5 минут инкубации при комнатной температуре планшеты промывали (ополаскивали) 0,1% уксусной кислотой и сушили на воздухе. Связанный SRB солюбилизировали добавлением 100 мкл 10 мМ Трис-основания (рН 10,5) на лунку и давали выдерживали при комнатной температуре в течение 5 минут. Оптическую плотность (OD) каждой лунки измеряли при 570 нм. При этих условиях число клеток является пропорциональным OD. Определяли концентрацию каждого лекарства, которая ингибирует рост клеток на 50% (IC50), используя эмпирически полученные концентрации лекарств в интервале 10 пМ - 30 мМ; IС50 получали путем построения графика зависимости концнтрация-эффект. Индекс устойчивости RI, который является мерой клеточной устойчивости к конкретному лекарственному средству, вычисляли путем деления значения IC50, полученного для устойчивой к лекарственному средству клеточной линии на IС50 соответствующей клеточной линии, чувствительной к лекарственному средству (IC50MCF-7R/IC50 MCF7).
Цитотоксическая активность представлена в таблице 5.
Результаты
Силу цитотоксичности Соединения I сравнивали с таковой паклитаксела для опухолевых клеток человека, указанных выше (таблица 5). Результаты показывают, что Соединение I демонстрирует широкий спектр цитотоксичности и является более сильным, чем паклитаксел на всех испытанных клеточных линиях. Соединение I было особенно эффективно против устойчивых к лекарственным средствам клеточным линиям, значительно снижая RI МСF7 клеток.
2. Фармакокинетика
Фармакокинетический профиль Соединения I является более благоприятным в терминах абсорбции и биодоступности - как при внутривенном, так и пероральном введении, чем β -эпимера (US 3705508), как показано данными, представленными в таблице 6.
мин.
мкг/мл· ч
5
3,3
86,4
98,7
60, в.в
5
3,5
90,3
3. Токсичность
Исследование токсичности Соединения I проводили на крысах согласно известным процедурам. Результаты предствлены в таблице 7.
Вышеуказанные данные по активности и токсичности демонстрируют, что Соединение I является значительно более активным и менее токсичным, чем паклитаксел.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТАКСАНА | 2001 |
|
RU2275365C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ТАКСАНА | 2007 |
|
RU2434014C2 |
| С-2'-МЕТИЛИРОВАННЫЕ ПРОИЗВОДНЫЕ ПАКЛИТАКСЕЛА ДЛЯ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ АГЕНТОВ | 2002 |
|
RU2287528C2 |
| СПОСОБ ПОЛУЧЕНИЯ 14БЕТА-ГИДРОКСИБАККАТИН III-1,14-КАРБОНАТА | 2002 |
|
RU2285000C2 |
| ПОЛУСИНТЕТИЧЕСКИЕ ТАКСАНЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 2001 |
|
RU2259363C2 |
| СПОСОБ ПОЛУЧЕНИЯ 14БЕТА-ГИДРОКСИБАККАТИН III-1,14-КАРБОНАТА | 2002 |
|
RU2291866C2 |
| ПРОИЗВОДНЫЕ 10-ДЕАЦЕТИЛБАККАТИНА III И 10-ДЕАЦЕТИЛ-14β-ГИДРОКСИБАККАТИНА III, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2152936C1 |
| ПРОИЗВОДНЫЕ 10-ДЕАЦЕТИЛ-14-БЕТА-ГИДРОКСИБАККАТИНА III, СПОСОБ ИХ ПОЛУЧЕНИЯ И КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ | 1996 |
|
RU2161615C2 |
| ПРОИЗВОДНЫЕ ТАКСАНА, ФУНКЦИОНАЛИЗИРОВАННЫЕ ПО 14-ПОЛОЖЕНИЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2320652C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ БАККАТИНА III | 2001 |
|
RU2264394C2 |
Изобретение относится к новому производному таксана формулы I: 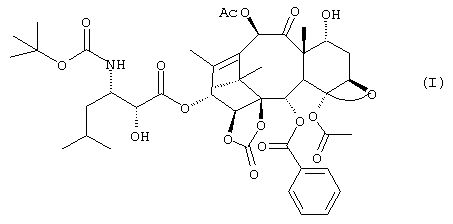 которое обладает сильным противоопухолевым действием. Изобретение также относится к промежуточным продуктам, способу его получения соединения формулы I, способу получения 1,14-β-гидрокси-1,14-карбонат-баккатин III производных, в положении 3 замещенных изосериновым остатком, и фармацевтической композиции на основе соединений формулы 1. Технический результат - получение нового производного таксана, которое более активно и менее токсично, чем паклитаксел. 7 н. и 3 з.п. ф-лы, 7 табл.
которое обладает сильным противоопухолевым действием. Изобретение также относится к промежуточным продуктам, способу его получения соединения формулы I, способу получения 1,14-β-гидрокси-1,14-карбонат-баккатин III производных, в положении 3 замещенных изосериновым остатком, и фармацевтической композиции на основе соединений формулы 1. Технический результат - получение нового производного таксана, которое более активно и менее токсично, чем паклитаксел. 7 н. и 3 з.п. ф-лы, 7 табл.
a) преобразование 14β -гидрокси-10-деацетилбаккатина III в триэтилсилилированное в 7-м положении производное;
b) получение 1,14-карбонатного производного продукта стадии (а);
с) селективное ацетилирование гидроксила в положении 10;
d) взаимодействие продукта стадии (с) с (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоковой кислотой;
е) отщепление защитных групп триэтилсилила и диметскси-бензилидена от продукта со стадии(d).
а') селективное ацетилирование гидроксила в положении С-10 14β -гидрокси-10-деацетилбаккатина III;
b') получение 1,14-карбонатного производного продукта со стадии (а');
с') силилирование гидроксила в положении С-7;
d') взаимодействие продукта со стадии (с) с (4S,5R)-N-Вос-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислотой;
е') отщепление триэтилсилильных и диметоксибензилиденовых защитных групп от продукта со стадии (d').
a) защита аминогруппы лейцинола Вос-группой;
b) преобразование N-Boc-L-лейцинола в N-Boc-L-лейциналь;
c) получение циангидринного производного продукта со стадии (b);
d) преобразование циангидриннитрила в соответствующую карбоновую кислоту;
e) получение метилового эфира карбоновой кислоты;
f) очистка метилового эфира (2R,3S)-3-(N-Boc)амино-2-гидрокси-5-метилгексановой кислоты;
g) конденсация продукта стадии (f) с 2,4-диметоксибензальдегиддиметилацеталем;
h) преобразование метилового эфира (4S,5R)-N-Boc-2-(2,4-диметоксифенил)-4-изобутил-1-оксазолидин-5-карбоновой кислоты в соответствующую карбоновую кислоту.
| US 5705508 А 06.01.1998 | |||
| US 5750562 А 12.05.1998 | |||
| RU 95110687 A1 27.12.1996 | |||
| RU 95113421 A1 20.03.1997. |

