Это изобретение относится к новому классу химических соединений и к их использованию в медицине. В частности, изобретение касается новых 4-замещенных-2-дезокси 2,3-дидегидропроизводных α -D-неураминовой кислоты, способов их получения, фармацевтической композиции на их основе и их использованию в качестве антивирусных агентов.
Ферменты, способные отщеплять N-ацетилнеураминовую кислоты (NANA), известную также как сиаловая кислота, от других сахаров, имеются во многих микроорганизмах. К ним относятся бактерии, такие, как Vibrio cholerae, Clostridium perfringens, Streptococcus pneumoniae и Arthrobacter sialophilus и вирусы, такие, как вирус (эпидемического) гриппа, вирус парагриппозной инфекции, вирус эпидемического паротита, вирус болезни Newcastle, птичий чумной (fowe plague) вирус и вирус Sendai. Большая часть из этих вирусов относится к ортомиксовирусной или парамиксовирусной группам и несут неураминидазную активность на поверхности вирусных частиц.
Многие из организмов, содержащих неураминидазу, являются главными возбудителями заболеваний человека и животных, а некоторые, такие, как вирус (эпидемического) гриппа, вирус болезни Newcastle, птичий чумной вирус вызывают заболевания, имеющие огромное экономическое значение.
В течение долгого времени считалось, что ингибиторы неураминидазной активности могут предотвращать инфекцию, вызываемую вирусами, несущими неураминидазу. Большая часть известных неураминидазных ингибиторов являются аналогами неураминовой кислоты, такими как 2-дезокси-2,3-дидегидро-N-ацетилнеураминовая кислота (DANA) и ее производные. См., например, Meindl и др., Virology 1974, 58, 457-63. Наиболее активным из этих соединений является 2-дезокси-2,3-дегидро-N-трифторацетил-неураминовая кислота (FANA), которая подавляет мультициклическое размножение вирусов (эпидемического) гриппа и парагриппозной инфекции in vitro. См. Palese и др., Virology 1974, 59, 490-498.
Ряд производных 2-дезокси-2,3-дидегидро-N-ацетилнеураминовой кислоты является известными в данной области техники. См., например, P.Meindl и др. Virology, 58, 457-463 (1974); P.Meindl и H.Turry, Mh.Chem., 100(4), 1295-1306 (1969); M.Elashner и др., Carbohydrate Research 103, 281-285 (1982); E. Zbiral и др., Liebigs Ann. Chem. 159-165 (1989); T. Ogawa и Y.Ito, Tetrahedron Letters 28 (49), 6221-6224 (1987); T.Goto и др., Tetrahedron Let. 27 (43), 5229-5232 (1986); H.Ogura и др., Chem. Pham.Bull., 36 (12), 4807-4813 (1988); German Offen- legungschrift P. 1439249. Многие из этих соединений являются активными in vitro противонеураминидазы из вирусов заболеваний V. Cholerae или болезни Newcastle, а также и от вируса (эпидемического) гриппа. Отмечено также, что неураминидаза ингибируется in vitro в штаммах по крайней мере вирусов (эпидемического) гриппа или парагриппозной инфекции δ - лактоном 3-аза-2,3,4-тридез окси-4-оксо-D-арабиноктоновой кислоты и 0- α -N-ацетил-D-неураминосил-(2 ---> 3)-2-ацетамидо-2-дезокси- D-глюкозой. См. Zakstel'skaya и др., Vop. Virol 1972, 17, 223-28.
Неураминидаза из Arthrobacter Sialophilus ингибируется in vitro гликолями 2,3-дегидро-4-эпи-N-ацетил-неураминовой кислоты, 2,3-дегидро-2-дезокси-N-ацетилнеураминовой кислоты и 5-ацетамидо-2,6-ангидро-2,3,5-тридезокси-D-манно-нон-2- ен-4-улозонатом и их метиловыми эфирами. См. Kumar и др. Carbohydrate Res. , 1981 94 123-130; Carbohydrate Res., 1982, 103, 281-285. Было отмечено, что тиоаналоги 2- α -азидо-6-тионеураминовая кислота и 2-дезокси-2,3-дидегидро-6-тионеураминовая кислота. Mack & Brossmer, Tetrahedron Letters, 1987, 28, 191-194 и фторированный аналог N-ацетил-2,3-дифтор- α -D-неураминовая кислота, Nakajima и др., Agric. Biol. Chem., 1988, 52, 1209-1215, ингибируют неураминидазу, хотя тип неураминидазы не был идентифицирован.
Schmid и др. , Tetrahedron Letters, 1958, 29, 3843-3846 описали синтез 2-дезокси-N-ацетил- α -D-неураминовой кислоты, но не отметили ее активности или пассивности по отношению к неураминидазе.
Ни один из известных ингибиторов активности неураминидазы in vitro не обнаружил наличия антивирусной активности in vitro, и, действительно, было показано, в частности, что некоторые из них, такие как FANA являются неактивными in vitro. Таким образом, на основании обычных соображений соответственно можно полагать, что соединения, проявляющие in vitro ингибирование в отношении вирусной неураминидазы, не будут оказывать in vivo блокирующего действия на вирусную инфекцию.
Meindl и Tuppy, Hoppe-Seyler's Z.Physiol. Chem., 1969, 350, 1088, описали гидрогенизацию олефиновой двойной связи 2-дезокси-2,3-дегидро-N-ацетилнеураминовой кислоты с образованием β -аномера. Этот β -аномер не ингибирует неураминидазу Vibrio cholerae.
Наиболее вероятные ингибиторы in vitro вирусной неурминидазы, таким образом, могут быть идентифицированы как соединения, которые основываются на структуре неураминовой кислоты, и их можно рассматривать как аналоги переходного состояния. (Miller и др. , Biochem. Biophys. Res. Comm. 1978 83 1479). Но хотя многие из упомянутых выше аналогов неураминовой кислоты являются конкурирующими ингибиторами неураминидаз, на сегодняшний день ни об одном из них не сообщается как проявляющем антивирусную активность in vivo. Например, хотя для ингибирующей активности и считается важным полупланарная система ненасыщенного 6-членного, см. Dernick и др. в Antiviral Chemotherapy (K. K Gauried) Academic Press, 1981, на стр. 327-336, некоторые соединения, характеризующиеся наличием такой системы, особенно FANA, не были отмечены как соединения, обладающие in vivo антивирусной активностью. См. Palese и Schulman и Chemoprophylaxis infection of the Upper respiratory tract, т. 1 (J.S.Oxforded) CRC Press, 1977, на стр. 189-205.
В настоящее время мы нашли новые 4-замещенные 2-дезокси-2,3-дидегидро производные α -D-неураминовой кислоты, которые являются активными in vivo.
Следовательно, объектом настоящего изобретения прежде всего являются производные 2-дезокси-2,3-дидегидро-N-ацетилнеураминовой кислоты общей формулы (1b):
где
R3b представляет азид или группу -NR6bR7b, причем
R6b представляет водород, C1-6 алкил или амидин,
R7b представляет водород, C1-6 алкил или аллил,
R4b представляет NHCOR9b, где R9b представляет C1-4 алкил,
или их фармацевтически приемлемые соли, эфиры и соли эфиров.
Предпочтительны соединения, где R3b представляет NR6bR7b.
Другими предпочтительными соединениями являются соединения, где R3b представляет NH2 или NHC(=NC)•NH2.
Под фармацевтически приемлемым производным подразумевается любой фармацевтически приемлемый эфир или соль такого эфира соединений формулы (1b) или любое другое соединение, которое при его приеме пациентом способно давать (прямо или косвенно) соединение формулы (1b) или антивирусно активный промежуточный продукт обмена или его остаток.
Специалисту в данной отрасли понятно, что соединения формулы (1b) могут быть модифицированы с получением их фармацевтически приемлемых производных при любых функциональных группах в соединениях. Особый интерес в качестве таких производных представляют собой соединения, модифицированные при карбоксильной функции C-1, гидроксильных функциях C-7 или C-9 или при амино группах. Таким образом, соединения, представляющие интерес, включают сложные эфиры C1-4 алкилов (таких, как метил, этил или пропил, например, изопропил) или арилов (например, фенил, бензоил) соединений формулы (1b), C-7 или C-9, сложные эфиры соединений формулы 1b, таких как ацетиловые эфиры, C-7 или C-9 простые эфиры, такие, как фениловые эфиры, бензиловые эфиры, n-толиловые эфиры и ацилированные амино производные, такие, как формил, ацетамидо.
Специалисту в данной области понятно, что фармацевтически приемлемые производные соединений формулы (1b) могут быть производными более чем в одном положении.
Фармацевтически приемлемые соли соединений формулы (1b) включают такие соли, которые получаются из фармацевтически приемлемых неорганических и органических кислот и оснований. Примерами соответствующих кислот являются хлористоводородная, бромистоводородная, серная, азотная, перхлорная, фумаровая, малеиновая, фосфорная, гликолевая, молочная, салициловая, янтарная, толуол-п-сульфоновая, винная, уксусная, лимонная, метансульфоновая, муравьиная, бензойная, малоновая, нафталин-2-сульфоновая и бензолсульфоновая кислоты. Другие кислоты, такие, как щавелевая, хотя и не являются сами по себе фармацевтически приемлемыми, тем не менее, могут быть использованы при получении солей, полезных в качестве промежуточных соединений при получении соединений изобретения и их фармацевтически приемлемых кислотно-аддитивных солей.
Соли, полученные из соответствующих оснований, включают соли щелочных металлов (например, натрия), щелочноземельных металлов (например, магния), аммониевые и N+ 4 (где R представляет собой C1-4 алкил) соли.
Приводимые здесь далее ссылки на соединение изобретения включают соединения формулы (1b) и их фармацевтически приемлемые соли и производные.
Особенно предпочтительными соединениями изобретения являются:
5-ацетамидо-4-амино-2,3,4,5-тетрадезокси-D-глицеро-D-галакто- нон-2-энопиранозоновая кислота (известная также как 5-(ацетиламино)- 4-амино-2,6-ангидро-3,4,5-тридезокси-D-глицеро-D-галакто-нон-2- еноиковая кислота), ее соли, включая ее натриевую соль, и
5-ацетамидо-4-гуанидино-2,3,4,5-тридезокси-D-глицеро-D-галакто- нон-2-енопиранозоновая кислота (известная также как 5-(ацетиламино)-2,6-ангидро-4-гуанидино-3,4,5-тридезокси-D-глицеро- D-галакто-нон-2-еноиковая кислота) и ее соли, включая аммониевую соль.
Соединения формулы (1b) обладают антивирусной активностью. В частности, эти соединения являются ингибиторами вирусной неураминидазы ортоксовирусов и парамиксовирусов, например, вирусной неураминидазы (эпидемического) гриппа A и B, парагриппозной инфекции, эпидемического паротита и болезни Newcastle, эпидемической чумы и вируса Sendai.
Другой аспект изобретения, таким образом, касается соединения формулы (1b) или его фармацевтически приемлемой соли, или его производного для их использования в качестве активного терапевтического агента, в частности, антивирусного агента, например, для лечения ортомиксовирусной или парамиксовирусной инфекций.
Дальнейшим аспектом изобретения является способ лечения вирусной инфекции, например, ортомиксовирусной и парамиксовирусной инфекции у животных, включая человека, включающего этапы приема названным животным эффективного количества соединения формулы (1b) или его фармацевтически приемлемой соли, или производного.
Далее в качестве еще одного аспекта изобретения предлагается использование соединения изобретения - производство лекарственного средства для лечения вирусной инфекции.
Специалисту в данной области понятно, что приведенная здесь ссылка для лечения распространяется также и на профилактику, как и на лечение установленных инфекций или симптомов.
Далее существенным является то, что количество соединения изобретения, требующееся для использования при лечении, будет меняться в зависимости не только от конкретного соединения, выбранного для лечения, но также и от пути приема, природы условий и возраста и условий пациента, и будет в конечном счете рассматриваться обслуживающим врачом или ветеринаром. Однако в основном подходящей дозой является интервал приблизительно от 0,01 до 750 мг/кг веса в день, предпочтительно интервал от 0,1 до 100 мг/кг/день, наиболее предпочтительным интервалом является от 0,5 до 25 мг/кг/день.
В частности, мы установили, что эффективные дозы испытанных соединений связаны с их активностью in vitro. Так, для DANA (которое имеет IC50 снижения чумы 5 мкг/мл) было установлено, что соединение является эффективным при дозах между 1 и 10 мг/кг при лечении. Соответствующий метиловый эфир DANA (IC50 5-100 мкг/мл) является эффективным в пропорционально более высокой дозе.
Лечение предпочтительно начинается до или через некоторое время после инфекции и продолжается до тех пор, пока вирус не будет более присутствовать в дыхательном тракте. Однако соединения являются также эффективными и для данной постинфекции, например, после появления установленных симптомов.
Соответственно лечение осуществляется 1-4 раза в день и продолжается в течение 3-7, например, 5 дней после инфекции - в зависимости от конкретного используемого соединения.
Требуемая доза может быть представлена в виде единичной дозы или в виде отдельных доз, принимаемых через соответствующие интервалы, например, в виде двух, трех, четырех или большего числа субдоз в день.
Соединения обычно принимаются в виде единичной дозированной формы, содержащей, например, от 10 до 1500 мг, обычно от 20 до 1000 мг, наиболее часто от 50 до 700 мг активного ингредиента на единичную дозированную форму.
Хотя и возможно использование соединения изобретения для лечения в виде непосредственного химического продукта, предпочтительным является присутствие активного ингредиента в виде фармацевтической рецептуры.
Таким образом, изобретение дает далее фармацевтическую композицию, включающую соединение формулы (1b) или его фармацевтически приемлемую соль, или его производное, вместе с фармацевтически приемлемым носителем.
Носитель может быть "приемлемым" в том смысле, что он является совместимым с другими ингредиентами композиции и не вредным для того, кто его принимает.
Фармацевтические композиции могут существовать в виде стандартных композиций, предназначенных для определенного способа приема.
Для внутриназального приема в соответствии со способом изобретения ингибиторы неураминидазы могут вводиться любым из способов и рецептур, употребляемых для такого вида приема в данной области.
Таким образом, обычно, соединения могут приниматься в виде раствора для суспензии или эмульсии, или в виде сухого порошка.
Растворы и суспензии в основном являются водными, например, готовятся из одной воды (например, стерильной или очищенной от пирогена) или их воды и физиологически приемлемого сорастворителя (например, этанола, пропиленгликоля и полиэтиленгликолей, такого как ПЭГ 400).
Такие растворы или суспензии могут содержать дополнительно другие наполнители, например, консерванты (такие, как бензалконий хлорид), солюбилизирующие агенты, поверхностно-активные вещества, такие, как полисорбаты (например, Tween 80, Span 80, бензалконий хлорид), буферные агенты, агенты, регулирующие изотоничность (например, хлорид натрия), усилители абсорбции и агенты, повышающие вязкость. Суспензии могут содержать дополнительно суспензирующие агенты (например, микрокристаллическая целлюлоза, натрий карбоксиметилцеллюлоза).
Растворы или суспензии вводятся непосредственно в носовую полость стандартным способом, например, капельницей, пипеткой или распылителем. Композиции могут применяться в виде единичной дозы или множественной дозы. В последнем случае желательно дать способ измерения дозы. В случае капельницы или пипетки это может быть достигнуто пациентом принятием соответствующего, предварительно определенного раствора или суспензии. В случае распыления это может быть достигнуто, например, посредством измерения пульверизации распыляющего насоса.
Интраназальный прием может быть осуществлен также посредством применения аэрозольной композиции, в которой соединение дается в упаковке повышенного давления с соответствующим движущим средством, таким, как хлорфторуглерод (CFC), например, дихлордифторметан, трихлорфторметан или дихлортетрафторэтан, двуокись углерода или другой подходящий газ. Аэрозоль может для удобства содержать также поверхностно-активное вещество, такое, как лицетин. Доза лекарственного средства может контролироваться установлением измерительного клапана.
Или же соединения могут быть даны в виде сухого порошка, например, порошковая смесь соединения в подходящей порошковой основе, такой, как лактоза, крахмал, производные крахмала, такие, как гидроксипропилметилцеллюлоза и поливинилпирролидин (ПЭП). Обычно порошковый носитель образует гель в назальной полости. Порошковая композиция может существовать в виде единичной дозы, например, капсул или гильз, например, в желатиновых или пузырчатых упаковках, из которых порошок может вводиться посредством пульверизатора.
В интраназальных композициях соединение обычно имеет небольшой размер частиц, например, порядка 5 микрон или менее.
Такой размер частиц может быть получен известными в данной области методами, например, микронизацией.
В случае необходимости композиции могут быть приспособлены к тому, чтобы давать поддерживаемое выделение активного ингредиента. Соединения изобретения могут использоваться также в комбинации с другими терапевтическими агентами, например, с другими антиинфекционными агентами. В частности, соединения изобретения могут применяться вместе с другими антивирусными агентами. Таким образом, дальнейшим аспектом изобретения является комбинация, включающая соединение формулы (1b) или его фармацевтически приемлемую соль, или его производное вместе с другим терапевтически активным агентом, в частности, антивирусным агентом.
Комбинации, упомянутые выше, могут быть обычно представлены для использования в виде фармацевтической композиции и, таким образом, эти композиции, включающие комбинацию, определенную выше, вместе с их фармацевтически приемлемым носителем составляют дальнейший аспект изобретения.
Соответствующие терапевтические агенты для использования в таких комбинациях включают другие антиинфекционные агенты, в частности, антибактериальные и антивирусные агенты, такие, как агенты, используемые для лечения респираторных инфекций. Например, в такие комбинации могут быть включены другие соединения, эффективные против вирусов (эпидемического) гриппа, такие, как амантадин, римантадин и рибаверин.
Индивидуальные компоненты таких комбинаций могут приниматься либо последовательно, либо одновременно в отдельных или в комбинированных фармацевтических композициях.
В том случае, когда соединение изобретения используется в сочетании со вторым терапевтическим агентом, активным по отношению к тому же вирусу, доза каждого соединения может либо не меняться, либо будет отличаться от дозы, применяемой в том случае, когда каждое соединение используется в отдельности. Соответствующие дозы могут быть легко установлены специалистом в данной области.
Соединение формулы (1b) и его фармацевтически приемлемые соли и производные могут быть получены любым известным в данной области методом, используемым для получения соединений аналогичного строения.
В соответствии с одним из способов защищенное по карбокси- и гидроксигруппам производное соединение формулы (IIIb):
где
R4b имеет значения, определенные выше, а OL означает удаляемую группу, представляющую собой остаток сульфоновой кислоты, подвергают взаимодействию с нуклеофилом, таким, как азид, полученное при этом соединение общей формулы (1b), где R3b означает азид, в случае необходимости подвергают превращению в другое соединение общей формулы (1b), где R3b означает группу -NR6bR7b, где R6b означает атом водорода, (C1-C6)алкил или амидин, R7b означает атом водорода, (C1-C6) алкил или аллил, и в полученных соединениях удаляют защитные группы и в случае необходимости переводят эти соединения в фармацевтически приемлемые соли, эфиры или соли эфиров.
Соединения формулы (IIIb) могут быть получены из соответствующих соединений формулы (IV):
инверсией 4-OH группы известными в данной области способами, например, взаимодействием с кислотой Льюиса (такой, как этерифицированный BF3), за которым следует гидролиз.
Соединения формулы (IV) являются либо известными в данной области, либо могут быть получены способами, аналогичными тем, которые используются для получения известных соединений.
Как понятно специалисту в данной области, может быть необходимо или желательно на любой стадии описанных выше процессов защитить одну или несколько чувствительных групп в молекуле для того, чтобы предотвратить нежелательные побочные реакции; в последующих реакциях защитная группа может быть удалена любым стандартным способом.
Защитные группы, используемые при получении соединений формулы (1b), могут быть применены в соответствии с существующими способами. См., например, "Защитные группы в органической химии" Изд. J.F.W. McOmie (Plenum Press (1973) или "Защитные группы в органическом синтезе" Teodora W. Green (John Wiley and Sons, 1981).
Гидроксигруппы могут быть защищены, например, аралкильными группами, такими, как бензильные, дифенилметильные или трифенилметильные группы; ацильными группами, такими, как ацетил; силиконовыми защитными группами, такими, как триметилсилильные группы; или такими, как тетрагидрофурановые производные.
Удаление любых присутствующих защитных групп может быть осуществлено стандартными способами. Так, аралкильная группа, такая, как бензильная, может быть расщеплена гидрогенолизом в присутствии катализатора (например, палладия на древесном угле); ацильная группа, такая, как N-бензилоксикарбонильная, может быть удалена гидролизом, например, с бромистым водородом в уксусной кислоте или восстановолением, например, каталитической гидрогенизацией; силиконовые защитные группы могут быть удалены, например, действием иона фтора; тетрагидропирановые группы могут быть расщеплены гидролизом, проводимым в кислотных условиях.
В тех случаях, когда желательно выделить соединение изобретения в виде соли, например, в виде кислотно-аддитивной соли, это может быть достигнуто действием свободного основания общей формулы (1b) с соответствующей кислотой, предпочтительно с эквивалентным количеством, или с соответствующим сульфатом в подходящем растворителе (например, в водном этаноле).
Данное изобретение далее описывается последующими примерами, которые имеют своей целью только проиллюстрировать изобретение и не могут быть истолкованы как ограничивающие изобретение.
Общая методология. Для синтеза соединений изобретения применяются следующие общие методы.
Деацетилирование. Взаимодействие ацетилированного вещества с Amberlite IRA-400 (OH-) при перемешивании в течение периода времени обычно 2 - 3 ч при комнатной температуре приводит в результате к полному де-O-ацетилированию. Образующаяся смола отфильтровывается и фильтрат концентрируется до сухого состояния с образованием нужного де-O-ацетилированного вещества.
Специалисту в данной области понятно, что для полного де-O-ацетилирования того же самого вещества приемлемы и другие стандартные методы, такой, как взаимодействие с метилатом натрия и метаноле.
Деэтерификация. Полностью де-O-ацетилированный материал растворяется в водном растворе гидроокиси натрия и перемешивается при комнатной температуре в течение периода времени в основном 2 - 3 ч. Затем pH смеси доводится до pH 7,0 - 7,5 с помощью смолы Dowex 50X8 (H+). Фильтрование с последующим охлаждением-сушкой фильтрата дает нужный деэтерифицированный материал.
Специалист в данной области может легко идентифицировать несколько альтернативных способов для деэтерификации того же самого вещества, такого, как кислотный гидролиз, альтернативный щелочной гидролиз, например, гидроокисью аммония, гидроокисью калия.
Промежуточные соединения, упомянутые в примерах 1 - 15, идентифицируются следующим образом:
Соединение 2. Метил 5-ацетамидо-7,8,9-три-O-ацетил-2,3,5- тридезокси-D-глицеро-D-тало-нон-2-энопиразонат (4-эпи- Neu 5, 7, 8, 9 Ac4 и 2en 1Me).
Соединение 3. Метил 5-ацетамидо-7,8,9-три-O-ацетил-4-азидо- 2,3,5-тридезокси-D-глицеро-D-галакто-нон-2-энопиранозонат (4-азидо Neu 5,7,8,9 Ac4 2en 1Me).
Соединение 5. Метил 5-ацетамидо-7,8,9-три-O-ацетил-4-амино-2, 3,4,5-тетрадезокси-D-глицеро-D-галакто-нон-2-энопиранозонат(4-амино- Neu 5, 7, 8, 9 Ac4 2en 1Me).
Соединение 8. Метил 5-ацетамидо-7,8,9-три-O-ацетил-4-N,N- диаллиламино-2,3,4,5-тетрадезокси-D-глицеро-D-галакто-нон-2-энопиранозонат (4-N, N-диаллиламино- Neu 5, 7, 8, 9Ac4 2 en 1Me).
Соединение 10. Метил 5-ацетамидо-7,8,9-три-O-ацетил-4-N- аллиламино-2,3,4,5-тетрадезокси-D-глицеро-D-галакто-нон-2-энопиранозонат (4-N-аллиламино- Neu 5, 7, 8, 9 Ac4 2 en 1Me).
Соединение 12. Метил 5-ацетамидо-7,8,9-три-O-ацетил-4-амино- 2,3,4,5-тетрадезоки-D-глицеро-D-тало-нон-2-энопиранозонат(4-эпи-4-амино Neu 5, 7, 8, 9 Ac4 2en 1Me).
Соединение 13. Метил 7,8,9-три-O-ацетил-2,3,5-тридезокси-4',5'-дигидро-2'- метилоксазол[5,4-d]D-глицеро-D-тало-нон-2-энопиразонат (4-эпи- 4,5-оксазало Neu 7,8,9 Ac3 2en 1Me).
Соединение 15. Метил 5-ацетамидо-7,8,9-три-O-ацетил-4-азидо- 2,3,4,5-тетрадезокси-D-глицеро-D-тало-нон-2-энопиранозонат (4-эпи-азидо Neu 5,7,8,9Ac4 2en 1Me).
Соединение 16. Метил 5-ацетамидо-4-азидо-2,3,4,5-тетрадезокси-D- глицеро-D-тало-нон-2-энопиранозонат (4-эпи-азидо Neu 5Ac 2en 1Me).
Соединение 18. Метил 5-ацетамидо-7,8,9-три-O-ацетил-4-N- метиламино-2,3,4,5-тетрадезокси-D-глицеро-D-галакто-нон-2-энопиранозонат (4-N-метиламино Neu 5,7,8,9Ac4 2en 1Me).
Соединение 19. Метил 5-ацетамидо-4-N-метиламино-2,3,4,5- тетрадезокси-D-глицеро-D-галакто-нон-2-энопиранозонат (4-N-метиламино- Neu 5Ac 2en 1Me).
Соединение 21. Метил 5-ацетамидо-7,8,9-три-O-ацетил-4-N,N- диметиламино-2,3,4,5-тетрадезокси-D-глицеро-D-галакто-нон-2- энопиранозонат (4-N,N-диметиламино- Neu 5, 7, 8, 9Ac4 2en 1Me).
Соединение 22. Метил 5-ацетамидо-4-N, N-диметиламино-2,3,4,5-тетрадезокси-D-глицеро-D-галакто-нон- 2-энопиранозонат (4-N,N-диметиламино Neu 5Ac 2en 1Me).
Соединение 24. Метил 5-ацетамидо-7,8,9-три-O-ацетил-4-N-метоксикарбонил-метиламино-2,3,4,5- тетрадезокси-D-глицеро-D-галакто-нон-2-энопиранозонат (4-N-метоксикарбонилметиламино Neu 5,7,8,9 Ac4 2en 1Me).
Соединение 25. Метил 5-ацетамидо-4-N-метоксикарбонилметиламино-2,3,4,5-тетрадезокси-D-глицеро-D- галaкто-нон-2-энопиранозонат (4-N-метоксикарбонилметиламин Neu 5Ac 2en 1Me).
Соединение 27. Метил 5-ацетамидо-7,8,9-три-O-ацетил-4-N-2'-гидроксиэтиламино-2,3,4,5-тетрадезокси-D- глицеро-D-галакто-нон-2-энопиранозонат (4-N-2'-гидрокиэтиламино Neu 5,7,8,9 - Ac4, 2en 1Me).
Соединение 28. Метил 5-ацетамидо-4-N-2'-гидроксиэтиламино-2,3,4,5-тетрадезокси-D-глицеро-D-галакто- нон-2-энопиранозонат (4-N-2'-гидроксиэтиламино Neu 5,7,8,9 Ac4, 2en 1Me).
Соединение 29. Метил 5-ацетамидо-7,8,9-три-O-ацетил-4-N-2'-гидроксиэтиламино-2,3,4,5-тетрадезокси-D- глицеро-D-галакто-нон-2-энопиранозонат (4-N-2'-гидроксиэтиламино Neu 5Ac 2en 1Me).
Соединение 30. 3-дезокси-D-глицеро-D-галакто-2-нонулопиранозоновая кислота (KDN).
Соединение 31. Метил 3-дезокси-D-глицеро-D-галакто-2-нонулопиразонат (KDN 1Me).
Соединение 32. Метил (4,5,7,8,9-пента-O-ацетил-2,3-дидезокси-D-глицеро- β - D-галакто-2-нонулопиранозил хлорид)онат (KDN 4,5,7,8,9 Ac5 2β Cl 1Me).
Соединение 33. Метил 4,5,7,8,9-пента-O-ацетил-2,3-дидезокси-D-глицеро-D-галакто-нон-2-энопиранозонат (KDN 4,5,7,8,9 Ac5 2en 1Me).
Соединение 34. Метил 2,3-дидезокси-D-глицеро-D-галакто-нон-2-энопиранозонат (KDN 2en 1Me).
Соединение 36. Гидразиний 4,5-диамино-2,3,4,5-тетрадезокси-D-глицеро-D-галакто-нон-2-энопиранозонат (гидразиний 4,5-диамино Neu 2en),
Соединение 37. 4,5-диамино-2,3,4,5-тетрадезокси-D-глицеро-D-галакто-нон-2- энопиранозоновая кислота (4,5-диамино Neu 2en).
Пример 1. Получение натрий 5-ацетамидо-4-азидо-2,3,4,5-тетрадезокси-D-глицеро-D-галакто-нон-2- энопиранозоната (4-азидо Neu 5Ac 2en) (4)
Общая схема реакции приведена в конце описания.
Получение соединения (2). К перемешиваемому раствору метил 5-ацетамидо-4,7,8,9-тетра-O-ацетил-2,3,5-тридезокси-D-глицеро-D-галакто- нон-2-энопиранозоната (1) (1500 мг, 3,17 ммоль) в смеси бензола (50 мл) и метанола (300 мг) в течение 30 мин в атмосфере азота при комнатной температуре по каплям добавлялся BF3Et2O (12 мл). Затем перемешиваемую смесь оставляли на 16 ч при комнатной температуре. Раствор разбавлялся этилацетатом (250 мл), интенсивно промывался насыщенным раствором NaHCO3 (30 мл х 3) и водой (20 мл х 3), затем упаривался до небольшого объема (около 10 мл), к которому были добавлены вода (0,5 мл) и уксусная кислота (0,5 мл). Затем общая смесь в течение двух дней перемешивалась при комнатной температуре перед тем, как она подвергалась разбавлению этилацетатом (200 мл). Этилацетатный раствор промывался раствором 5% NaHCO3 (30 мл х 2) и водой (20 мл х 3), затем выпаривался до сухого состояния. Остаток хроматографировался (силикагель, этилацетат в качестве элюирующего растворителя), давая чистое соединение (2) (550 мг, 40%).
1Н-ЯМР (CDCl3) δ (м. д.) 1,95, 2,06, 2,08, 2,10, 2,35 (с, 15н. Ацетил CH3•5), 3,80 (с, 3H, COOCH3), 4,1-4,4 (м, 4H, H4, H5, H6, H9), 4,82 (дд, 1H, J9,8 1,8 Гц, J9,9, 12,3 Гц, H9), 5,27 (м, 1H, H8), 5,45 (дд, 1H, J7,8 3,5 Гц, H7), 6,15 (д, 1H, J3,4 5,4 Гц, H3), 6,47 (д, 1H, JNH,5 8,8 Гц, -CONH).
Получение соединения (3). К перемешиваемому раствору соединения (2) (800 мг, 1,67 ммоль) в безводном дихлорметане (10 мл) и сухом пиридине (316 мг, 4 ммоль) при температуре от -30 до -40oC по каплям добавлялся раствор трифторметансерного ангидрида (Tf2O) (556 мг, 2 ммоль) в дихлорметане (2 мл) в течение 15 мин. Затем реакционная смесь в течение 5 ч при -30oС концентрировалась в вакууме до сухого состояния. После этого остаток промывался в сухом ДМФ (5 мл), содержащем смесь азида натрия (650 мг, 10 ммоль) и тетрабутиламмоний гидросульфат (170 мг, 0,5 ммоль). Реакционная смесь в течение 16 ч перемешивалась при комнатной температуре, после чего выпаривалась до сухого состояния в высоком вакууме. Остаток был разделен между этилацетатом (200 мл) и водой (50 мл). Органический слой отделялся и промывался водой (50 мл x 2), сушился над Na2SO4 и упаривался с образованием остатка (780 мг), который дважды подвергался хроматографированию (силикагель, первый раз система растворителей представляла собой смесь этилацетат/ацетон: 8/1; второй раз - дихлорметан/вода : 10/1), с образованием бесцветного масла (3) (185 мг, 24%).
MC (FAB) 457 (M++1), 414  + 19.1o (Cl, MEOH), ИК (CHCl3) см-1 2100 (N-N3), 1747 (карбонил).
+ 19.1o (Cl, MEOH), ИК (CHCl3) см-1 2100 (N-N3), 1747 (карбонил).
1H-ЯМР (CDCl3) δ (м.д.) 2,04, 2,05, 2,06, 2,12, (c, 12H, Ацетил CH3•4), 3,79 (c, 3H, COOCH3), 3,91 (ддд, 1H, J5,NH 8,4 Гц, J5,4 8,8 Гц, J5,6 9,9 Гц, H5), 4,17 (дд, 1H, J9,8 6,8 Гц, J9,9′ 12,5 Гц, H8, 4,42 (дд, 1H, J4,3 2,9 Гц, J4,5 8,8 Гц, H4), 4,48 (дд, 1H, J6,7 2,3 Гц, J6,5 9,9 Гц, H6), 4,46 (дд, 1H, J9,8 2,7 Гц, J9,9′ 12,5 Гц, H9), 5,31 (м, 1H, J8,7 5,2 Гц, J8,9 2,7 Гц, J8,9′ 6,8 Гц, H8), 5,45 (дд, 1H, J7,6 2,3 Гц, J7,8 5,2 Гц, H7) 5,96 (д, 1H, J3,4 2,9 H, H3), 6,13 (д, 1H, JNH,5 8,4 Гц, -CONH).
13C ЯМР (CDCl3) δ (м.д.) 20,7 (CH3-CO-O-), 23,2 (CH3CO-NH), 48,3 (C5), 52,6 (COOCH3) 57,8 (C4), 62,1 (C9), 67,7, 70,9 (C7, C8), 75,9 (C6), 107,6 (C3), 145,1 (C2), 161,5 (C1), 170,2 170,3, 170,7 (ацетил -C=O • 4).
Получение соединения (4). Соединение (3) (50 мг, 0,11 ммоль) растворялось в безводном метаноле (5 мл), содержащем метилат натрия (8 мг, 0,15 ммоль). Смесь перемешивалась при комнатной температуре в течение 2 ч и концентрировалась в вакууме до сухого состояния. Остаток растворялся в воде (3 мл), перемешивался при комнатной температуре в течение 1,5 ч, величина pH раствора регулировалась до 6-7 с использованием смолы Dowex 50 • 8 (H+), после чего он подвергался лиофилизации, образуя названное в заглавии соединение (4) (35 мг, 94%).
И.К. (KBr) см-1 3400 (уш -OH), 2100 (-N3), 1714 (карбонил).
1H-ЯМР (D2O) δ (м.д.) 2,06 (C, 3H, ацетил CH3), 3,64 (дд, 1H, J9′,8 6,3 Гц J9′,9 11,8 Гц, H9′ 3,65 (дд, 1H, J7,6 3,9 Гц, J7,8 6,8 Гц, H7), 3,88 (дд, 1H, J9,8 2,6 Гц, J9,9′ 11,8 Гц, H9), 3,94 (м, 1H, J8,7 6,8 Гц, J8,9 2,6 Гц, J8,9′ 6,3 Гц, H8), 4,21 (дд, 1H, J5,4 10,4 Гц, J5,6 8,9 Гц, H5, 4,31 (дд, 1H, J4,3 2,2 Гц, J4,5 2,2 Гц, J4,5 10,4 Гц, H4, 4,34 (дд, 1H, J 6,5 8,9 Гц, J6,7 3,9 Гц, H6) 5,82 (д, 1H, J3,4 2,2 Гц, H3).
Пример 2. Получение натрий 5-ацетамидо-4-амино-2,3,4,5-тетрадезокси- D-глицеро-D-галакто-нон-2-энопиранозоната (4-амино-Neu 5Ac 2ен) (6).
Общая схема реакции приведена в конце описания.
Получение соединения (5). Раствор метил 5-ацетамидо-7,8,9-три-О-ацетил-4-азидо. 2,3,4,5-тетрадезокси-D-глицеро-D-галкто-нон-2-энопиранозонана (3), приготовленный в соответствии с примером 1 (95 мг, 0,208 ммоль) в пиридине (6 мл), продувался в течение 16 ч H2S при комнатной температуре. Затем раствор в течение 15 мин сильно продувался азотом и выпаривался в высоком вакууме для удаления пиридина. Остаток подвергался хроматографированию (силикагель, этилацетат/ изопропанол/вода = 5/2/1), образуя бесцветное соединение (5) (50 мг, 56%).
MC (FAB - ионизация 431 (M+ + 1); 414 (M+ - NH2), [α]
1H-ЯМР (CDCl3 + CD3OD) δ (м.д.) 1,96 2,06, 2,07, 2,10 (с, (12H ацетил CH3•4), 3,81 (C, 3H, - COOCH3), 3,92 (уш, 1H, J5,4 & J5,6 10 Гц, H5), 4,17 (дд, 1H, J9′,8/ 7,2 Гц, J9′,9 12,3 Гц, H9′) ), 4,22 (уш, дд, 2H, J4,5 & J6,5 10 Гц, J4,3 & J6,7 2,1 Гц, H4 & H6), 4,71 (дд, 1H, J9,8 2,6 Гц, J9,9′ 12,3 Гц, H9), 5,31 (м, 1H, J8,7 4,9 Гц, J8,9 2,6 Гц, J8,9′ 7,2 Гц, H8), 5,45 (д, 1H, J7,6 2,1 Гц, J7,8 4,9 Гц, H7), 5,97 (д, 1H, J3,4 2,1 Гц, H3).
13C-ЯМР (CDCl3 + CD3OD) δ (м.д.) 20,2, 20,3 (CH3-CO-O-), 22,3 (CH2CO-NH), 48,2 (C2), 50,4 (C4), 52,0 (COOCH3), 52,1 (C9), 67,8 71,2 (C7, C8, 76,5 (C6), 112,5 (C3), 143,5 (C2), 162,0 (C1), 170,2, 170,4 170,8, 172,2 (ацетил - C = 0 • 4).
Получение соединения (6). Соединение (5) (50 мг, 0,116 ммоль) растворялось в безводном метаноле (5 мл), содержащем метилат натрия (12,4 мг, 0,23 ммоль). Смесь перемешивалась при комнатной температуре в течение 1,5 ч и выпаривалась в вакууме до сухого состояния при 30oC. остаток перемешивался в воде (3 мл) при комнатной температуре до тех пор, пока ТЖХ (силикагель, этилацетат/метанол/ 0,1 N HCl = 5/4/1) не показала, что гидролиз прошел полностью. Затем pH раствора (pH около 10,5) постепенно регулировалась до значения около 7,5 с использованием смолы Dowex • 8 (H+). Как только pH раствора достигалa значения 7,5, суспензия быстро фильтровалась с использованием пресс-фильтра. Фильтрат подвергался лиофилизации, давая озаглавленное соединение (6) (30 мг, 83%).
1H-ЯМР (D2O) δ (м.д.) 2,07 (C, 3H, ацетил CH3), 3,59 - 3,70 м, 2H, H7 & H9′) , 3,89 (дд, 1H, J9,8 2,6 Гц, J9,9′ 11,8 Гц, H9), 3,95 (м, 1H, H8 ), 3,99 (ушд, 1H, J4,5 10,6 Гц, H4), 4,21 (уш, т. 1H, J5,4 & J5,6 10,6 Гц, H5), 4,29 (ушд, 1H, J6,5 10,6 Гц, H6), 5,66 (д, 1H, J3,4 1,9 Гц, H3).
Пример 3. Получение аммоний 5-ацетамидо-4-гуанидино-2,3,4,5-тетрадезокси-D-глицеро-D-галакто- нон-2-энопиразоната (7).
Общая схема реакции (5) и (7) приведена в конце описания.
К раствору S-метилизомочевины (546 мг, 3 ммоль) в воде (15 мл) при температуре, поддерживаемой ледяной баней, добавлялся метил 5,7,8,9-три-О-ацетил-4-амино-2,3,4,5-тетрадезокси-D-глицеро-D- галакто-нон-2-энопиранозонат (5), приготовленный в соответствии с примером 2 (40 мг, 0,093 ммоль). Реакционная смесь перемешивалась в течение 7 дней при 5oC и выливалась в колонку со смолой Dowex 50W • (H+) (33 мл). Затем колонка промывалась холодной водой (700 мл) и элюировалась 1,5М раствором NH4OH. Элюант (120 мл) концентрировался в высоком вакууме до сухого состояния. Полученный остаток хроматографировался (силикагель, растворяющая система 1; этилацетат/изопропанол/вода, 1/5/1; растворяющая система 2; 75% изопропанол), образуя озаглавленное соединение (7) (8 мг, 24,5%).
Соединение (7) давало сильную положительную реакцию Sakaguchi, показывающую присутствие группы гуанидина. Данные ЯМР для соединения (7) даются ниже.
1H-ЯМР (D2O + CDOD) δ (м.д.) 2,06 (c, 2H, ацетил CH3), 3,60 (уш, д, 1H, J7,8 9,4 Гц, H7), 3,63 (дд, 1H, J9′,8 6,2 Гц, J9′,9 11,8 Гц H9), 3,76 (уш, д, 1H, J4,5 9,4 Гц, H4), 3,87 (дд, 1H, J9,8 2,6 Гц, J9,9′ 11,8 Гц, H9), 3,93 (ддд, 1H, J8,7 9,4 Гц, J8,9 2,6 Гц, J8,9′ 6,2 Гц, H8), 4,01 (дд, 1H, J5,4 9,4 Гц, J5,6 10,6 Гц, H5), 4,20 (уш, д, 1H, J6,5 10,6 Гц, H6), 5,63 (д, 1H, J3,4 2,1 Гц, H3).
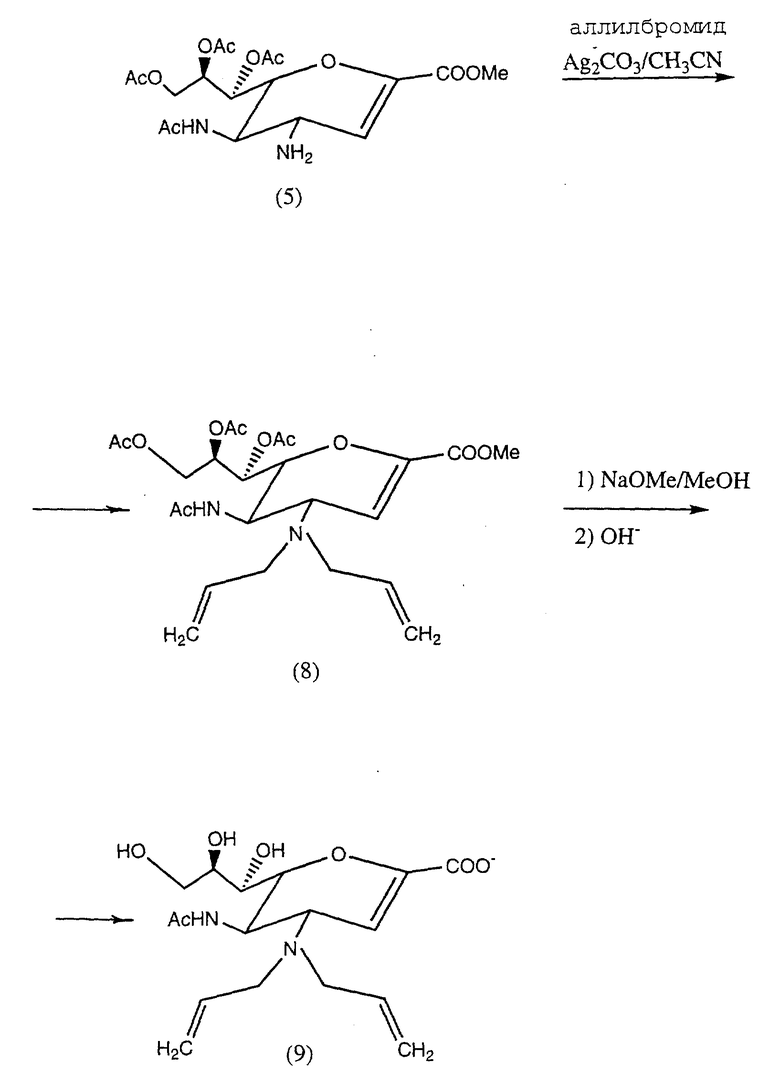
Пример 4. Натрий 5-ацетамидо-4-N,N-аиаллиламино-2,3,4,5-тетрадезокси-D-глицеро- D-галакто-нон-2-энопиранозонат (9).
Общая схема реакции (5), (8), (9) приведена в конце описания.
В раствор алкибромида (60 мг, 0,5 ммоль) и метил 5-ацетамидо-7,8,9-три-О-ацетил-4-амино-2,3,4,5-тетредезокси-D- глицеро-D-галакто-нон-3-энопиранозоната (5) (90 мг, 0,209 ммоль) в ацетонитриле (5 мл) добавлялся карбонат серебра (116 мг, 0,418 ммоль). Смесь перемешивалась в течение 16 ч при комнатной температуре в условиях, защищенных от света. Образующаяся суспензия фильтровалась, и фильтрат выпаривался до сухого состояния. Остаток подвергался флеш-хроматографии с использованием силикагеля и этилацетата, содержащего 10% метанола, образуя метил 5-ацетамидо-7,8,9-три-O- ацетил-4-N, N-диаллиламино-2,3,4,5-тетрадезокси-D-глицеро-D-галакто- нон-2-энопиранозонат (8) (85 мг, 80%).
1H-ЯМР (CDCl3) (м.д.) δ 1,94, 2,05 2,06, 2,11 (с, 12H, ацетил CH • 4), 2,97 (дд, 2H, J10a, 10b & 10'a 10'b 14,3 Гц, J 10a, 11 & 10'a, 11' 7,6 Гц, H 10a & H 10'a), 3,24 (дд, 2H, J 10b, 10a & J 10'b, 10'a 14,3 Гц, J 10b, 11 & J 10'b 11' 4,9 Гц, H10b & H10′b′), 3,58 (дд, 1H, J4,3 2,4 Гц, J4,5 9,3 Гц, H4), 3,79 (с, 3H, COOCH3), 4,12 - 4,26 (м, 3H, H6, H9, H5), 4,70 (дд, 1H, J9,8 2,6 Гц, J9,9′ 12,3 Гц, H9), 5,09 (дд, 2H, J 12 цис, 11 & J 12'цис, 11' 10,6 Гц J 12 гем & 12'гем или ≈ 1,5 Гц, H 12 цис & H12'цис), 5,14 (дд, 2H, J 12 транс. 11 & J 12' транс, 11' 17,7 Гц, J 12 гем & J 12'гем ≈ 1,5 Гц, H 12 транс & H 12'транс), 5,27 - 5,32 (м, 2H, H8 & -CONH), 5,55 (дд, 1H, J7,6 2,1 Гц, J7,8 4,7 Гц, H7), 5,72 (м, 2H, H 11 & H 11'), 6,07 (д, 1H, J3,4 2,4 Гц, H3).
Соединение (8) (80 мг, 0,156 ммоль) растворялось в безводном метаноле (10 мл), содержащем метилат натрия (16,2 мг, 0,30 ммоль).
Раствор перемешивался при комнатной температуре в течение 2 ч, затем выдерживался до сухого состояния. Остаток растворялся в воде (5 мл) и оставлялся на 2 ч при комнатной температуре. Полученный раствор нейтрализовали с использованием Dowex 50 • 80 (H+) и сушили вымораживанием с образованием озаглавленного соединения (9) (49 мг, 80%).
1H-ЯМР (D20) δ (м.д.) 1,94 (с, 3H, Ацетил CH3) 3,24 - 3,44 (м, 4H, H10 • 2 & H10' • 2), 3,48 - 4,33 (м, 7H, H4, H5, H6, H7, H8, H9 & H9′) , 5,24 - 5,29 (м, 4H, H12 • 2 & H 12' • 2), 5,69 (д, 1H, J3,4 ≈ 2 Гц, H3), 5,73 - 5,76 (м, 2H, H11 & H11′). )
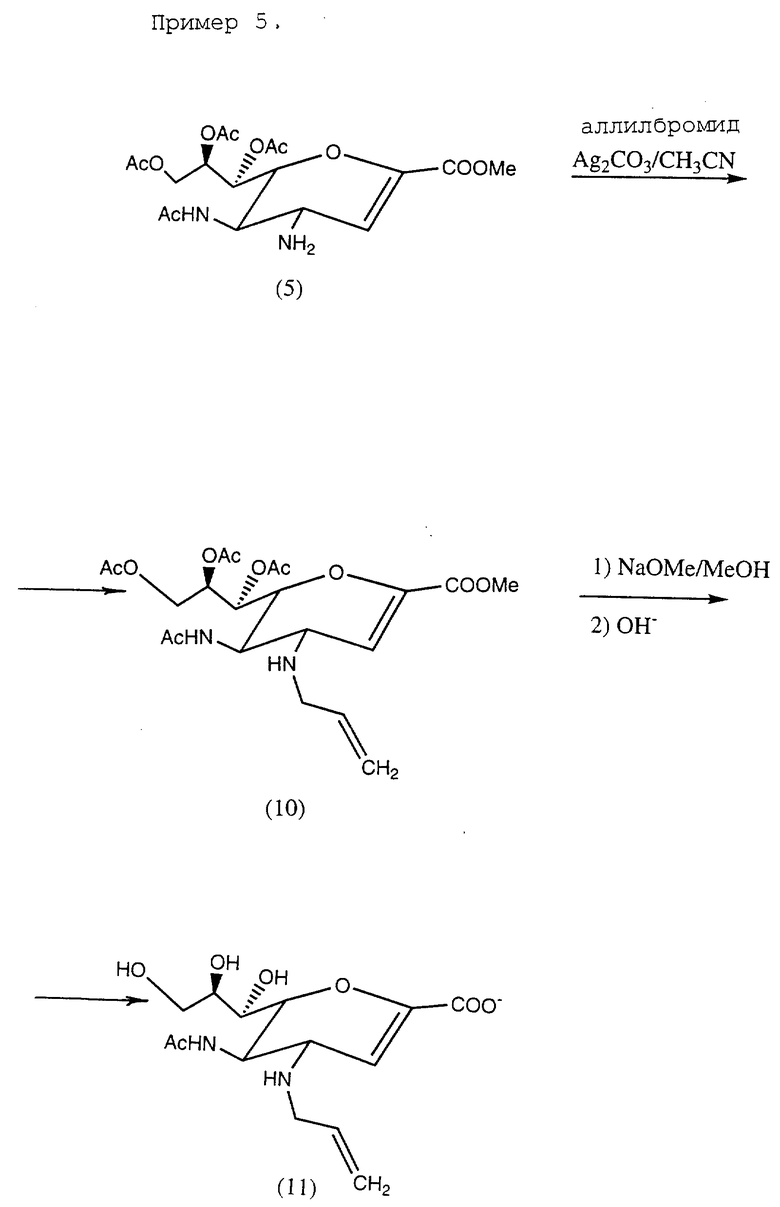
Пример 5. Натрий 5-ацетамидо-4-N-аллиламино-2,3,4,5- тетрадезокси-D-глицеро-D-галакто-нон-2-энопиранозонат (11).
Общая схема реакции приведена в конце описания.
К раствору аллилбромида (48 мг, 0,40 ммоль) и соединения (5) (155 мг, 0,36 ммоль) в ацетонитриле 5 (мл) добавлялся карбонат серебра (107 мг, 0,38 ммоль). Смесь перемешивалась при комнатной температуре в течение 16 ч в условиях, защищенных от света. Образующаяся суспензия отфильтровывалась, а фильтрат упаривался до сухого состояния. Остаток подвергался хроматографированию через силикагелевую колонку (этилацетат/изопропанол/вода = 5:2:1). Фракции со значением Rf 1,5 соединялись и упаривались до сухого состояния, образуя соединение (10) (53 мг, 32%). Исходное вещество (5) со значением Rf 0,9 (20 мг, 11%) были соответственно регенерированы.
1H-ЯМР (CDCl3) соединения (10) показано следующим образом: δ (м.д.) 1,96, 2,05, 2,06, 2,11 (с, 12H, Ацетил CH3 • 4), 3,25 (дд, 1H, J10a,10b - 14,1 Гц, J10a,11 5,8 Гц, H10a), 3,37 (дд, 1H, J10b,10a - 14,1 Гц, J10b,11 5,9 Гц, H10b), 3,43 (дд, 1H, J4,3 3,1 Гц, J4,5 7,5 Гц, H4), 3,79 (с, 3H, COOCH3), 4,09 (ддд, 1H, J5,4 7,5 Гц, J5,NH9 1Гц, J5,6 8,1 Гц, H5), 4,21 (дд, 1H, J9′,8 7,1 Гц, J9′,9 - 12,2 Гц, H9′) ), 4,30 (дд, 1H, J6,5 8,1 Гц, J6,7 4,1 Гц, H6), 4,63 (дд, 1H, J9,8 3,2 Гц, J9,9′ - 12,2 Гц, H9), 5,09 (дд, 1H, J12цис,11 10,2 Гц, J12цис, 12транс - 1,3 Гц, H12цис), 5,18 (дд, 1H, J12транс,11 17,1 Гц, J12транс ,12цис - 1,3 Гц, H12транс), 5,36 (ддд, 1H, J8,7 4,2 Гц, J8,9 3,2 Гц, J8,9′ 7,1 Гц, H8), 5,57 (дд, 1H, J7,6 4,1 Гц, J7,8 4,2 Гц, H7), 5,65 (д, 1H, JNH,5 9,1 Гц, -CONH-), 5,83 (дддд, 1H, J11,12транс 17,1 Гц, J11,12цис 10,2 Гц, J11,10a 5,8 Гц, J11,10b 5,9 Гц, H11), 6,09 (д, 1H, J3,4 3,1 Гц, H3).
Соединение (10) (50 мг, 0,11 ммоль) перемешивалось в безводном метаноле (5 мл), содержащем метилат натрия (12 мг, 0,225 ммоль) в течение 2 ч при комнатной температуре, затем выпаривалось до сухого состояния. Остаток вновь растворяли в воде (5 мл) и оставлялся при комнатной температуре на 2 ч, после чего нейтрализовался с помощью смолы Dowex 50•8 (H+). Водный раствор был выморожен до сухого состояния, образуя соединение (11) (31 мг, 78%).
1H-ЯМР (D 20) δ (м.д.) 2,02 (с, 3H, CH3CO), 3,42 (дд, 1H, J10a,10b - 13,4 Гц, J10a,11 6,6 Гц, H10a), 3,52 (дд, 1H, J10a,10b - 13,4 Гц, J10b,11 6,3 Гц, J10b), 3,51 - 4,27 (м, 7H, H4, H5, H6, H7, H8, H9 & H9′) , 5,30 (дд, 1H, J12цис, 12транс ≈ 1,5 Гц, J12цис,11 10,3 Гц, J12цис), 5,34 Гц, (дд, 1H, J12транс, 12цис ≈ 1,5 Гц, J12транс,11 17,7 Гц, H12транс), 5,72 (д, 1H, J3,4 2,4 Гц, H3), 5,89 (дддд, J11,10a 6,6 Гц, J11,10b 6,3 Гц, J11,12цис 10,3 Гц, J11,12транс 17,7 Гц, H11).
Пример 6. Натрий 5-ацетамидо-4-амино-2,3,4,5-тетрадезокси- D-глицеро-D-тало-нон-2-энопиранозонат (14).
Общая схема реакции (2), (12), (13), (14) приведена в конце описания.
К перемешиваемому раствору соединения (2) (500 мг, 1,04 ммоль) в безводном дихлорметане (8 мл), содержащем пиридин (205 мг, 2,6 ммоль), при -30oС по каплям добавлялся раствор трифторметансульфонового ангидрида (Tf 20) (367 мг, 1,3 ммоль) в дихлорметане (2 мл) в течение 20 мин. После этого реакционная смесь перемешивалась при -30oС в течение 5 ч и, наконец, упаривалась до сухого состояния при пониженном давлении. Образующийся остаток перемешивался в сухом ДМФ, содержащем N,N-диизопропилэтиламин (194 мг, 1,5 ммоль) в течение 16 ч при комнатной температуре. Реакционная смесь концентрировалась в высоком вакууме для удаления ДМФ. После этого остаток перемешивался в двухфазной смеси толуола (5 мл) и воды (5 мл), содержащей тетра-н-бутиламмоний кислый сульфат (950 мг, 2,8 ммоль) и азид натрия (137 мг, 2,1 ммоль). Смесь перемешивалась в течение 16 ч при комнатной температуре и затем упаривалась до сухого состояния. Остаток разделялся между этилацетатом (50 мл) и водой (15 мл), органический слой интенсивно промывался водой (5 мл x 2) и затем упаривался до сухого состояния. Остаток в пиридине (5 мл) продували H2S и затем упаривали до сухого состояния. Остаток подвергали флеш-хроматографированию (силикагель, первая система растворителей была этилацетат, вторая - этилацетат/изопропанол/H2O : 5/2/1).
Этилацетатный элюент содержал соединение (13) (260 мг, 53%). Фракции с положительной нингидринной реакцией, собранные из второй системы растворителей, объединялись и упаривались до сухого состояния, образуя соединение (12) (32 мг, 6,5%).
MC (FAB-ионизация), 431 (M+ + 1), 414 (M+-NH2).
1H-ЯМР (CDCl3 + CD3OD) δ (м.д.) 1,96, 2,06, 2,08, 2,09 (с, 12H, Ацетил, CH3 • 4), 3,52 (дд, 1H, J4,3 5,5 Гц, J4,5 4,5 Гц, H4), 3,80 (с, 3H, COOCH3), 4,16 (дд, 1H, J6,5 10,2 Гц, J6,7 2,3 Гц, H6), 4,17 (дд, 1H, J9′,9 12,4 Гц, J9′,8 7,3 Гц, H9′),, 4,23 (дд, 1H, J5,6 10,2 Гц, J5,4 4,5 Гц, H5), 4,73 (дд, 1H, J9,9′ 12,4 Гц, J9,8 2,7 Гц, H9), 5,34 (ддд, 1H, J8,7 4,7 Гц, J8,9 2,7 Гц, J8,9′ 7,3 Гц, H8), 5,45 (дд, 1H, J7,6 2,3 Гц, 7,8 4,7 Гц, H7), 6,12 (д, 1H, J3,4 5,5 Гц, H3).
13C-ЯМР (CDCl3 + CD3OD) δ (м.д.) 20,7 (CH3C(O)O-), 23,1 (CH3C(O)N-), 43,8 (C3), 46,2 (C4), 52,4 (COOCH3), 62,3 (C9), 68,3, 71,8 (C7, C8), 73,0 (C6), 111,5 (C3), 143,8 (C2), 162,4 (C1), 170,3 & 170,8 (CH3CO • 4).
Соединение (12) перемешивалось в течение 3 ч при комнатной температуре в безводном метаноле (5 мл), содержащем смолу Amberlite JRA400(OH-) (100 мг). Вслед за фильтрованием фильтрат выпаривался до сухого состояния. Остаток растворялся в воде (5 мл) и pH регулировалось до 13 0,1 NaOH. Водный раствор перемешивался в течение 2 ч при комнатной температуре и затем нейтрализовался с помощью смолы Dowex 50•8 (H+). После фильтрования фильтрат подвергали лиофилизации, получая соединение (14) (61 мг, 70%), которое давало положительную нингидринную реакцию.
1H-ЯМР (D2O) δ (м.д.) 2,10 (с, 3H, CH3CO), 3,67 - 3,76 (м, 2H, H4 & H9), 3,92 (дд, 1H, J9,8 2,8 Гц, J9,9′ - 11,9 Гц, H9), 3,90 - 4,02 (м, 2H, H7 & H8), 4,37 - 4,44 (м, 2H, H5 & H6), 5,81 (д, 1H, J3,4 5,14 Гц, H3).
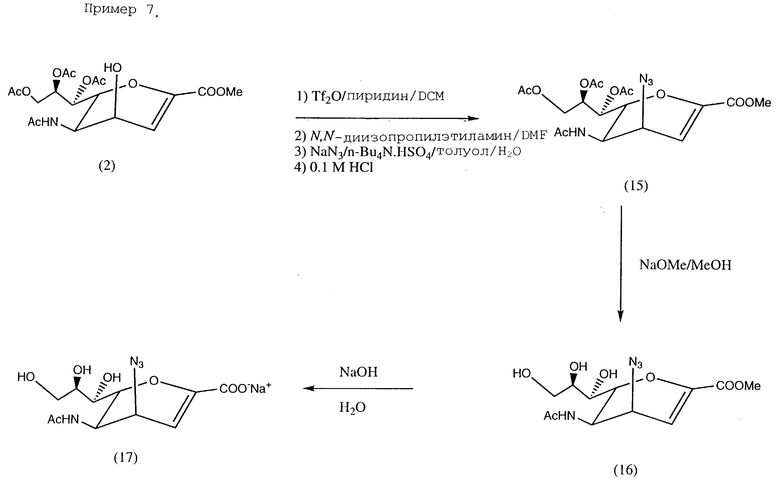
Пример 7. Натрий 5-ацетамидо-4-азидо-2,3,4,5-тетразедокси- D-глицеро-D-тало-нон-2-энопиранозонат (17); см. в конце описания.
К перемешиваемому раствору соединения (2) (500 мг, 1,04 ммоль) в безводном дихлорметане (8 мл), содержащему пиридин (205 мг, 2,6 ммоль) при -30oC добавлялся по каплям раствор трифторметансульфонового ангидрида (Tf2O) (367,8 мг, 1,3 ммоль) в дихлорметане (2 мл) в течение периода времени 20 мин. Затем реакционная смесь перемешивалась в течение 54 ч при 3oC и, наконец, упаривалась до сухого состояния при пониженном давлении. Образующийся остаток перемешивался в течение 16 ч при комнатной температуре в сухом ДМФ, содержащем N,N-диизопропилэтамин (194 мг, 1,5 ммоль). Реакционная смесь концентрировалась в высоком вакууме для удаления ДМФ. Затем остаток перемешивался в двухфазной смеси из толуола (5 мл) и воды (5 мл), содержащей кислый сульфат тетра-н-бутиламмония (950 мг, 2,8 ммоль) и азид натрия (137 мг, 2,1 ммоль). Смесь перемешивалась при комнатной температуре в течение 16 ч и затем разбавлялась 0,2 М раствором HCl (5 мл). Смесь перемешивалась при комнатной температуре в течение 48 ч. К этой реакционной смеси добавлялись этилацетат (50 мл) и 2 М (1 мл). Органический слой отделялся и промывался водой (5 мл х 3), затем упаривался до сухого состояния. Остаток подвергался флеш-хроматографированию (силикагель, этилацетат/гексан = 2/1). Фракции с значением 0,32 (этилацетат/гексан в качестве проявляющего растворителя = 2/1) объединялись и выпаривались до сухого состояния, образуя соединение (15), (40 мг, 8,4%). Колонка затем элюировалась смесью этилацетат/метанол = 10/1 для удаления исходного вещества (2) (280 мг, 56%). Соединение (15) было выделено в виде белого пенящегося вещества.
МС (FAB-ионизация) 457 (M+ + 1), 414 (M+ - N3), ИК (CHCl3) см-1 2108 (-N3), 1748 (карбонил),
1H-ЯМР (CDCl3), δ (м.д.) 1,97, 2,04, 2,06, 2,07 (с, 12H, ацетил, CH3•4), 3,82 (с, 3H, COOCH3), 4,12 ≈4,20 (м, 3H, C6, C4 & C9), 4,51 (ддд, 1H, J5,4 4,4 Гц, J5,6 10,7 Гц, J5, NH, 10,1 Гц, H5), 4,69 (дд, 1H, J9,8 2,6 Гц, J9,9′ 12,4 Гц, H9), 5,31 (м, 1H, J8,7 4,9 Гц, J8,9 2,6 Гц, J8,9′ 7,0 Гц, H8), 5,45 (дд, 1H, J7,6 2,1 Гц, J7,8 4,9 Гц, H7), 5,68 (д, 1H, JNH,5 10,1 Гц, CONH), 6,15 (д, 1H, J3,4 5,7 Гц, H3).
13C-ЯМР (CDCl3), δ (м. д.) 20,7, 20,8 (CH3CO-O•3), 23,1 (O CH3CO-NH), 44,8 (C5), 52,6 (COOCH3), 54,8 (C4), 62,1 (C9), 67,6, 71,3 (C7, C8), 73,5 (C6), 104,5 (C3), 146,3 (C2), 161,5 (C1), 169,9 170,2 170,5 (ацетил, -C= O•4).
Соединение (15) (40 мг, 0,088 ммоль) растворялось в безводном метаноле (4 мл), содержащем метилат натрия (6,4 мг, 0,12 ммоль).
Смесь перемешивалась в течение 2 ч при комнатной температуре и концентрировалась в вакууме до сухого состояния, давая соединение (16), которое растворялось в воде (3 мл), перемешивалось 2 ч при комнатной температуре, pH раствора регулировалась до 6-7 с помощью смолы Dowex 50•8 (H+), затем вещество подвергалось лиофилизации, давая озаглавленное соединение (17) в виде желтоватого порошка (25 мг, 83%).
ИК (KBr) см-1 3400 (уш, -OH), 2108 (-N), 1714 (карбонил).
1H-ЯМР (D2O) δ (м.д.) 1,97 (с, 3H, ацетил), 3,5 ≈ 4,4 (м, 7H, H4, H5, H6, H7, H8, H9 &  , 6,07 (д, J3,4 5,6 Гц, H3).
, 6,07 (д, J3,4 5,6 Гц, H3).
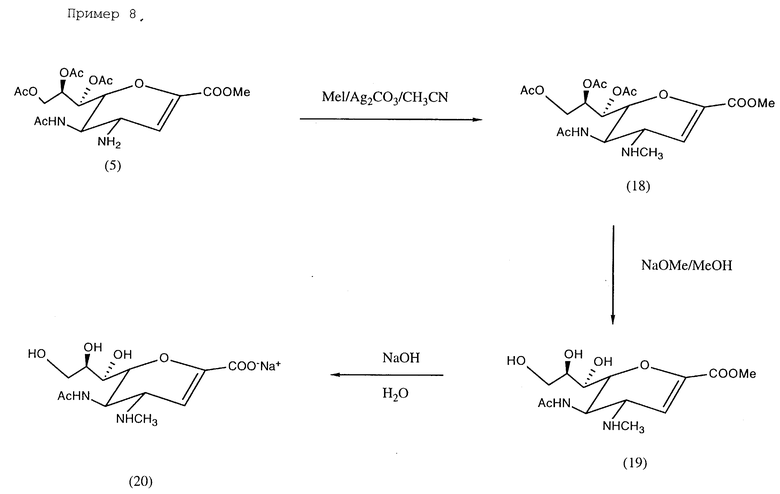
Пример 8. Натрий 5-ацетамидо-4-N-метиламино-2,3,4,5- тетрадезокси-D-глицеро-D-галакто-нон-2-энопиранозонат (20); см. в конце описания.
К раствору метилиодида (15 мг, 0,10 ммоль) и соединения (5) (48 мг, 0,11 ммоль) в ацетонитриле (6 мл) добавлялся карбонат серебра (42 мг, 0,15 ммоль). Смесь перемешивалась при комнатной температуре в течение 16 ч в условиях, защищенных от света. Образующаяся суспензия отфильтровывалась, и фильтрат упаривался до сухого состояния. Остаток подвергался хроматографированию (силикагель, этилацетат/изопропанол/вода = 5/2/1). Фракции с значением Rf 0,36 соединялись и концентрировались в вакууме до сухого состояния, давая соединение (18) (25 мг, 51%).
МС (FAB) 445 (M+ + 1), 414 (M+ - NHCH3).
1H-ЯМР (CDCl3) δ (м.д.) 1,95, 2,05, 2,06 2,12 (с, 12H, ацетил CH3 • 4), 2,45 (с, 3H, N-CH3), 3,72 (дд, 1H, J4,3 2,3 Гц, J4,5, 9,2 Гц, H4), 3,89 (с, 3H, COOCH3), 4,16 (дд, 1H, J9′,8 7,2 Гц, J9′,9 12,3 Гц, H9′), 4,26 (ддд, 1H, J5,4 9,2 Гц, J5,NH 9,1 Гц, J5,6 9,0 Гц, H5), 4,36 (дд, 1H, J6,5 9,0 Гц, J6,7 2,7 Гц, H6), 4,64 (дд, 1H, J9,8 2,9 Гц, J9,9′ 12,3 Гц, H9), 5,34 (м, 1H, J8,7 4,8 Гц, J8,9 2,9 Гц, J8,9′ 7,2 Гц, H8), 5,51 (дд, 1H, J7,6 2,7 Гц, J7,8 4,8 Гц), 6,05 (д, 1H, J3,4 2,3 Гц, H3).
Соединение (18) (25 мг, 0,056 ммоль) перемешивалось при комнатной температуре в течение 2 ч в безводном метаноле (5 мл), содержащем метилат натрия (5,4 мг, 0,1 ммоль), затем выпаривалось до сухого состояния, давая соединение (19), которое растворялось в воде (5 мл) и оставлялось на 2 ч при комнатной температуре, после чего нейтрализовалось с помощью смолы Dowex 50 x 8 (H+). Фильтрат подвергался лиофилизации, давая соединение (20) (15 мг, 82%).
1H-ЯМР (D2O) δ (м.д.) 1,94 (с, 3H, CH3CO), 2,43 (с, 3H, N-CH3), 3,5 ≈4,3 (м, 2H, H4, H5, H6, H7, H8, H9 & H9′), 5,65 (д, 1H, J3,4 2 Гц, H3).
Пример 9. Натрий 5-ацетамидо-4-N,N-диметиламино-2,3,4,5-тетрадезокси- D-глицеро-D-галакто-нон-2-энопиранозонат (23); см. в конце описания.
К раствору метилиодида (65 мг, 0,46 ммоль) и соединению (5) (100 мг, 0,23 ммоль) в ацетонитриле (15 мл) добавлялся карбонат серебра (127 мг, 0,46 ммоль). Смесь перемешивалась и защищалась от света при комнатной температуре в течение 16 ч. Образующаяся суспензия отфильтровывалась, и фильтрат упаривался до сухого состояния. Остаток дважды подвергался хроматографированию (силикагель, этилацетат/изопропанол/вода = 5/2/1), давая соединение (21) (30 мг, 28%) в виде бесцветной пены.
MC (FAB) 459 (M+ + 1), 414 (M+ - N(CH3)2),
1H-ЯМР (CDCl3) δ (м.д.) 1,98, 2,05, 2,06, 2,12 (с, 12H, ацетил, CH3•4), 2,33 (уш. с, 6H, N(CH3)2), 3,42 (дд, 1H, J4,3 2,8 Гц, J4,5 8,6 Гц, H4), 3,79 (с, 3H, COOCH3), 4,17 (дд, 1H, J9′,8 7,4 Гц, J9′,9 12,3 Гц, H9′), 4,18 (ддд, 1H, J5,4 8,5 Гц, J5,NH 8,9 Гц J5,6 9,0 Гц, H5), 4,31 (дд, J6,5 9,0 J6,7 2,9 Гц, H6), 4,68 (дд, 1H), J9,8 3,0 Гц, J9,9′ 12,3 Гц, H9), 5,31 (м, 1H, J8,7 4,4 Гц, J8,9 3,0 Гц, J8,9′ 7,4 Гц, H8), 5,51 (дд, 1H, J7,6 2,9 Гц, J7,8 4,4 Гц, H7), 5,79 (д, 1H, JNH,5 8,9 Гц, CONH), 6,09 (д, 1H, J3,4 2,8 Гц, H3).
Соединение (2) (30 мг, 0,066 ммоль) перемешивалось в безводном метаноле (4 мл), содержащем сухую Amberlite JRA 400 смолу (90 мг) при комнатной температуре в течение 3 ч, после чего смола отфильтровывалась. Фильтрат и промывные фракции собирались и упаривались до сухого состояния, давая соединение (22) (20 мг), которое перемешивалось в воде (5 мл) при pH 12 при комнатной температуре в течение 2 ч, затем перед фильтрованием pH регулировалась до 7,5 с использованием Dowex 50 • 8 (H+). Фильтрат подвергался лиофилизации, давая соединение (23) (15 мг, 66%) в виде белого порошка.
1H-ЯМР (D2O) δ (м.д.) 1,97 (с, 3H, ацетил), 2,33 (с, 6H, N(CH3)2), 3,50 ≈ 4,26 (м, 7H, H4, H5, H6, H7, H8, H9, H9′), 5,71 (д, J3,4 1,8 Гц, H3).
Пример 10. Динатрий 5-ацетамидо-4-N-оксикарбонилметиламино-2,3,4,5- тетрадезокси-D-глицеро-D-галакто-нон-2-энопиразонат (26); см. в конце описания.
К раствору метил α - бромацетата (36 мг, 0,23 ммоль) и соединения (5) (100 мг, 0,23 ммоль) в ацетонитриле (12 мл) добавлялся карбонат серебра (64 мг, 0,23 ммоль). Смесь перешивалась при комнатной температуре в течение 16 ч в условиях, защищенных от света, затем фильтровалась. Фильтрат упаривался до сухого состояния. Остаток хроматографировался на силикагелевой колонке (этилацетат/изопропанол/вода = 5/2/1). Были собраны фракции с значением Rf 0,60 и выпарены до сухого состояния, давая соединение (24) (80 мг, 68,5%).
1H-ЯМР (CDCl3) δ (м.д.) 1,97, 2,044, 2,047, 2,11 (с, 12H, ацетил CH3 • 4), 3,49 (AB, 2H, JAB 17,6 Гц, H10 • 2), 3,50 (дд, 1H, J4,3, 2,9 Гц, J4,5 8,4 Гц, H4), 3,71 (с, 3H, C11 OOMe), 3,79 (с, 3H, C1OOMe), 4,09 (ддд, 1H, J5,4 8,4 Гц, J5,NH 8,8 Гц, J5,6 8,1 Гц, H5), 4,17 (дд, 1H, J9′,8 7,4 Гц, J9′,9 12,3 Гц, H9′), 4,32 (дд, 1H, J6,5 8,1 Гц, J6,7 4,1 Гц, H6), 4,63 (дд, 1H, J9,8 3,1 Гц, J9,9′ 12,3 Гц, H9), 5,37 (м, 1H, J8,7 4,1 Гц, J8,9 3,1 Гц, J8,9′ 7,4 Гц, H8), 5,56 (т, 1H, J7,6 4,1 Гц, J7,8 4,1 Гц, H7), 6,03 (д, 1H, JNH,5 8,8 Гц, CONH), 6,04 (д, 1H, J3,4 2,9 Гц, H3).
Соединение (24) (80 мг, 0,159 ммоль) перемешивалось в безводном метаноле (20 мл), содержащем метилат натрия (18 мг, 0,32 ммоль) в течение 2 ч при комнатной температуре, затем выпаривалось до сухого состояния, давая соединение (26), которое вновь растворялось в воде (15 мл). Раствор оставляли на 2 ч при комнатной температуре, затем регулировали pH до 7 с использованием смолы Dowex 50 • 8 (H+). Фильтрат вымораживался до сухого состояния, образуя соединение (25) в виде белого порошка (59 мг, 94,6%).
1H-ЯРМ (D2O) δ (м.д.), 2,04 (C, 3H, ацетил), 3,58 (AB, 2H, JAB 17,6 Гц, H10• 2), 3,50 ≈ 4,40 (м, 7H, H4, H5, H6, H7, H8, H9 и H9′), 5,68 (д, 1H, J3,4 2,1 Гц, H3).
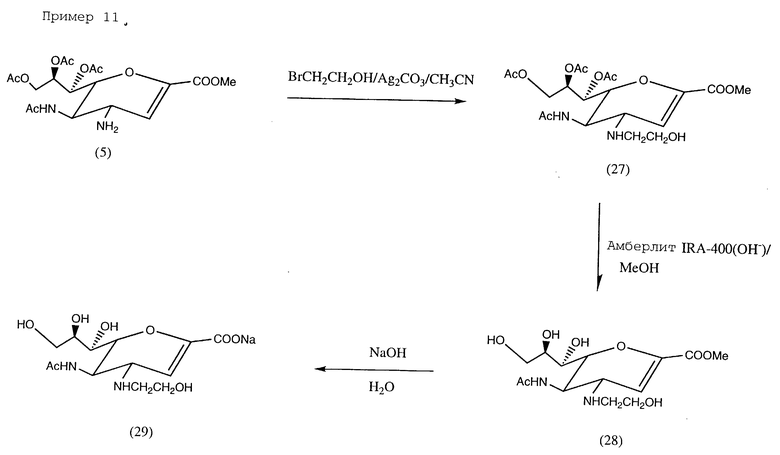
Пример 11. Натрий 5-ацетамидо-4-N-2'-гидроксиэтиламино-2,3,4,5- тетрадезокси-D-глицеро-D-галакто-нон-2-энопиранозонат (29); см. в конце описания.
К раствору бромэтанола (158 мг, 1,26 ммоль) и соединения (5) (84 мг, 0,195 ммоль) в ацетонитриле (10) добавлялся карбонат серебра (100 мг, 0,36 ммоль). Смесь защищалась от света и перемешивалась при комнатной температуре в течение 7 дней. Затем она отфильтровывалась, фильтрат выпаривался до сухого состояния. Остаток хроматографировался с использованием силикалегевой колонки (этилацетат/изопропанол/вода = 5/2/1). Фракции со значением Rf 0,4 соединялись и выпаривались до сухого состояния, образуя соединение (27) (40 мл, 40%).
МС (FAB 475 (M++1), 414 (M+-NHCH2CH2OH).
1H-ЯМР (CDCl3) δ (м.д.) 1,96, 2,05, 2,10, (с, 12H, ацетил CH • 4), 2,29 (уш, с, 2H, NH & OH), 2,76 (ABm, 2H, H10• 2), 3,47 (дд, 1H, J4,3 2,9 Гц, J4,5 7,5 Гц, H4), 3,62 (т, 2H, J11,10 4,9 Гц, H11 • 2), 3,79 (с, 3H, COOCH3), 4,15 (ддд, 1H, J5,4 7,5 Гц), 5,6 8,4 Гц, J5,NH 8,3 Гц, H5), 4,19 (дд, 1H, J9′,8 7,5 Гц, J9′,9, 12,3 Гц, H9′), 4,29 (дд, 1H, J6,5 8,4 Гц, J6,7 3,8 Гц, H6), 4,65 (дд, 1H, J9,8 2,9 Гц, J9,9′ 12,3 Гц, H9), 5,36 (м, 1H, J8,7 4,0 Гц, J8,9 2,9 Гц J8,9′ 7,5 Гц, H8), 5,55 (дд, 1H, J7,6 3,8 Гц, J7,8 4 Гц, H7), 6,08 (д, 1H, J3,4 2,9 Гц, H3), 6,09 (д, 1H, JNH,5 8,3 Гц, CONH).
13C-ЯМР (CDCL3) δ (м.д.) 20,6, 20,8 (CH3-CO-O- • 3), 23,10 (CH3-CO-NH), 46,5 (C5), 47,2 (C10), 52,3 (CH3COOCH), 55,6 (C4), 61,1 (C11), 62,1 (C9), 68,1, 71,1 (C7, C8), 76,7 (C6), 111,6 (C3), 143,7 (C2), 162,1 (C1), 170,1, 170,3, 170,6, 171,0 (ацетил карбонил • 4).
Соединение (27) (4) мг, 0,084 ммоль) перемешивалось в безводном метаноле (10 мл), содержащем сухой Amberlite JRA - 40 (OH-) (120 мг) при комнатной температуре в течение 4 ч, затем фильтровалось. Фильтрат и промывные фракции собирались и упаривались до сухого состояния, давая соединение (28), которое растворялось в воде (10 мл), pH раствора регулировалась до 13 добавлением NaOH. Водный раствор оставляли на 3 ч при комнатной температуре, после чего pH доводили до 6-7 смолой Dowex 50 • 8 (H+). После фильтрации раствор подвергали лиофилизации с образованием соединения (29) в виде белого порошка (20 мг, 66%).
1H-ЯМР (D2O) δ (м.д.) 1,99 (с, 2H, ацетил), 2,91 (AB, 2H, H10 • 2), 3,53 ≈4,25 (м, 9H, H4, H5, H6, H7, H8, H9, H9′, H11 • 2), 5,65 (д, 1H, J3,4 2,24 Гц, H3).
Пример 12. Натрий 2,3-дидезокси-D-глицеро-D-галакто-нон-2- энопиранозонат (35); см. в конце описания.
Соединение (30) (332 мг, 1,24 ммоль) перемешивалось в безводном метаноле (40 мл), содержащем смолу Dowex 50 • 8 (H+), (50 мг) при комнатной температуре в течение 16 ч перед фильтрованием. Фильтрат выпаривался до сухого состояния, образуя соединение (31) (320 мг, 1,13 ммоль, 91,5%), которое перемешивалось в ацетилхлориде (5 мл) при комнатной температуре в течение 3 дней, после чего упаривалось до сухого состояния с образованием соединения (32) (539 мг, 1,057 ммоль, 93,6%). Остаток растворялся в ацетонитриле (20 мл), содержащем нитрат серебра (500 мг, 2,94 ммоль) и карбонат калия (90 мг, 0,65 ммоль) в условиях, защищенных от света, и перемешивался при комнатной температуре в течение 16, после чего фильтровался. Фильтрат упаривался до небольшого объема и разделялся между этилацетатом (75 мл) и водой (15 мл). Органический слой промывался водой (10 мл • 3) и упаривался до сухого состояния. Остаток хроматографировался на силикагелевой колонке (этилацетат/гексан = 2/1), образуя чистое соединение (33) (200 мг, 0,423 ммоль, 40%).
1H-ЯМР (CDCl3) δ (м.д.) 2,062, 2,070, 2,073, 2,094, 2,096 (с, 15H, ацетил CH3 • 5), 3,80 (с, 3H, COOCH3), 4,19 (дд, 1H, J9,8 5,9 Гц, J9′,9 12,3 Гц, H9′), 4,33 (дд, 1H, J6,5 9,4 Гц, J6,7 3,0 Гц, H6), 4,57 (дд, 1H, J9,8 1,9 Гц, J9,9′ 12,3 Гц, H9), 5,20 (дд, 1H, J5,4 7,0 Гц, J5,6 9,4 Гц, H5), 5,38 (м, 1H, J8,7 5,1 Гц, J8,9 1,9 Гц, J8,9′ 5,9 Гц, H8), 5,49 (дд, 1H, J7,6 3,0 Гц, J7,8 5,1 Гц, H7), 5,57 (дд, 1H, J4,3 3,1 Гц, J4,5 7,0 Гц, H4), 5,97 (д, 1H, J3,4 3,1 Гц, H3).
Соединение (33) (100 мг, 0,211 ммоль) перемешивалось в безводном метаноле (10 мл), содержащем метилат натрия (24 мг, 0,423 ммоль) при комнатной температуре в течение 3 ч, затем выпаривалось до сухого состояния, давая соединение (34) (50 мг 90%), которое вновь растворялось в воде (5 мл) и оставлялось на 3 ч стоять при комнатной температуре, после чего pH раствора доводилась до 7 смолой Dowex 50 • 8 (H+). Раствор вымораживался до сухого состояния, образуя соединение (35) (47 мг, 91%).
1H-ЯМР (D2O, δ (м.д.) 3,69 (дд, 1H, J9′,8 5,6 Гц, J9′,9/ 12,0 Гц, H9′), 3,76 (дд, 1H, J5,4 7,8 Гц, J5,6 10,5 Гц, H5), 3,87 ≈ 3,99 (м, 3H, H7, H8, H9), 4,13 (д, 1H, J6,5 10,5 Гц, H6), 4,40 (дд, 1H, J4,3 2,3 Гц, J4,5 7,8 Гц, H4), 6,67 (д, 1H, J3,4 2,3 Гц, H3).
Пример 13. Натрий 4,5-диамино-2,3,4,5-тетрадезокси-D-глицеро-D- галакто-нон-2-энопиранозонат (38); см. в конце описания.
Раствор соединения (6) (125 мг, 0,40 ммоль) в гидразин гидрате (5 мл) в течение 3 дней нагревался в атмосфере азота при 85oC, и образующаяся смесь упаривалась в вакууме до сухого состояния. Остаток растворялся в воде (15 мл) и пропускался через колонку с Amberlite JRA 400 (HCOO-), затем элюировался 1 М раствором HCOOH. Элюат (200 мл) упаривался до сухого состояния. Остаток хроматографировался на силикагеле, деактивированном 10% воды (проявляющий растворитель: изопропанол/вода = 4/1). Фракции со значением Rf 0,1 объединялись и упаривались до сухого состояния, затем сушились вымораживанием. Остаток, представляющий собой соединение (36), растворялся в воде (10 мл), пропускался через небольшую колонку Amberlite IR -4B (OH-) (10 мл). Элюент выпаривался до сухого состояния, образуя соединение (37), МС (FAB) которого была 249 (M+ + 1). Соединение (37) растворяли в воде и доводили pH до 7,5 0,1 М раствором NaOH, затем сушили вымораживанием, получая соединение (38) (20 мг, 20%) в виде белого порошка.
1H-ЯМР (D2O) δ (м.д.) 3,01 (дд, 1H, J5,4, 9,7 Гц, J5,6, 10,2 Гц, H5), 3,58 (м, 2H, H9 & H7), 3,80 - 3,89 (м, 3H, H4, H8 & H9), 4,06 (д, 1H, J6,5 10,2 Гц, H6), 5,54 (д, 1H, J3,4 2,4 Гц, H3).
Пример 14. Метил 5-ацетамидо-2,3,5-тридезокси-9-(п- толуолсульфонил)-D-глицеро-D-галакто-нон-2-энопиранозонат (39).
Раствор, приготовленный из метил 5-ацетамидо-2,3,5- тридезокси-D-глицеро-D-галакто-нон-2-энопиранозоната (1000 мг, 3,16 ммоль) в сухом пиридине (85 мл) охлаждался на ледяной бане. Добавлялся п-толуолсульфонилхлорид (660 мг, 3,46 ммоль) и палево-желтый гомогенный раствор оставлялся на ночь при 4oC при перемешивании.
Добавлялся еще п-толуолсульфонилхлорид (220 мг, 1,15 ммоль) и раствор оставляли перемешиваться еще дополнительно на 4 ч при комнатной температуре.
После первой обработки добавлением воды (1 мл) следовало ротационное испарение с образованием вязкого желтого масла, которое подвергалось флеш-хроматографированию SiO2, EtOAc/i-PrOH / H2O, 6/2/1 об/об/об), образуя основной продукт 1,19 г. Выход 80% соединения (39).
ИК (KBr): νmax (см-1) 2964 (OH), 1730 (CO2CH3), 1656 (NHAc), 1358, 1174 (SO2), 810, 662, 550 (Ar).
МС (FAB) 460 (M + H+).
1H ЯМР (300 МГц, CD3OD/ТМС) δ (м.д.) = 2,03 (с, 3H, NHAc), 2,45 (с, 3H, ArCH3), 3,49 (д, 1H, J6,7 1,70, H6), 3,76 (с, 3H, CO2CH3), 3,91 (дд, 1H, J5,6 10,80, H5), 3,98-4,13 (м, 3H, H8, H9 и H9′). 4,28 (дд, 1H, J7,8 9,55, H7), 4,39 (дд, 1H, J4,5 8,64, H4), 5,92 (д, 1H, J3,4 2,49, H3), 7,74 (д, 2H, ArH), 7,79 (д, 2H, ArH).
Пример 15. Метил 5-ацетамидо-9-азидо-2,3,5,9- тетрадезокси-D-глицеро-D-галакто-нон-2-энопиранозонат (40).
Метил 5-ацетамидо-2,3,5-тридезокси-9-(п-толуолсульфонил)- D-глицеро-D-галакто-нон-2-энопиранозонат (39) (600 мг, 1,27 ммоль) и азид лития (186 мг, 3,80 ммоль) растворялись в сухом ДМФ (20 мл), и желтый гомогенный раствор нагревался до 80oC. Через 2 ч снова добавлялся азид лития (186 мг, 3,80 ммоль) и раствор оставляли при 80oC на ночь. Растворитель удалялся ротационным испарением, а оставшееся темно-коричневое масло растворялось в пиридине (2 мл) и подвергалось флеш-хроматографированию (SiO2), 5/2/1 EtOAc/i-Pr OH/H2O).
Основным продуктом было соединение (40) (370 мг, выход 88%), полученное в виде белой пены.
ИК (KBr): νmax (см-1) 3428 (с, OH), 2104 (с, N3), 1730 (с, CO2CH3), 1656 (с, NHAc).
МС (FAB): 331 (M + H+).
1H-ЯМР (300 МГц, D2O): δ (м.д.) = 1,94 (с, 3H, NHAc), 3,37 (дд, 1H, H9′), 3,48-3,57 (м, 2H, J8,9′ 5,77, H8 и J9,9' 13,16 Hg), 3,66 (с, 3H, CO2CH3), 3,91-3,98 (м, 2H, H5 и H6), 4,15 (д, 1H, J7,8 10,86 H7), 4,38 (дд, 1H, J4,5 8,88, H4), 5,91 (д, 1H, J3,4 2,44 H3).
Пример 16. Метил 3,9-диацетамидо-2,3,5,9-тетрадезокси-D- глицеро-D-галакто-нон-2-энопиранозонат (41).
К метил 5-ацетамидо-9-азидо-2,3,5,9-тетрадезокси-D-глицеро-D-галакто-нон- 2-энопиранозонату (70 мг, 0,21 ммоль) добавлялась тиолуксусная кислота (130 мл, 1,82 ммоль) с образованием желтого целевого раствора, который оставляли при перемешивании на ночь при комнатной температуре.
Избыток тиолуксусной кислоты испаряли при пониженном давлении, а оставшееся твердое вещество повторно обрабатывали водой, после чего проводили выпаривание (3 х 3 мл). Оставшееся твердое вещество растворяли в метаноле (4 мл), фильтровали и фильтрат использовали для приготовления ТЖХ - пластинки (SiO2, 20 см х 20 см х 2 мм с элюированием смесью 5/2/1 EtOAc/i-PrOH/H2O). Был отобран интервал с Rf = 0,47 c получением 51 мг (выход 70%) соединения (41) в виде белого порошка.
ИК (KBr): νmax (см-1) 3400 (с, OH), 1728 (c, CO2CH3), 1656 (c, NHAc).
МС (FAB) 347 (M+H+).
1H ЯМР (300 МГц, D2O): δ (м.д.) = 1,96 (с, 3H, NHAc), 2,00 (с, 3H, NHAc), 3,23 (дд, 1H, H9′), 3,48 (д, 1H, H6), 3,56 (дд, 1H, J9,9′ 14,17, H9), 3,75 (с, 3H, CO2CH3), 3,89 (м, 1H, J8,9,, 2,90, J8,9′ 7,40, H8), 4,02 (дд, 1H, J5,6, 9,10 H5), 4,22 (д, 1H, J7,8 10,85, H7), 4,45 (дд, 1H, J4,5 8,94, H4), 5,61 (д, 1H, J3,4 2,47, H3).
Пример 17. 5,9-диацетамидо-2,3,5,9-тетрадезокси-D-глицеро-D- галакто-нон-2-энопиранозоновая кислота (42)
Получение соединения (42) из соединения (39) приведено в конце описания.
Раствор метил 5,9-диацетамидо-2,3,5,8-тетрадезокси-D-глицеро-D- галакто-нон-2-энопиранозоната (41) (46 мг, 0,13 ммоль), растворенного в 0,1 M водной гидроокиси натрия (5 мл), перемешивался в течение 2,5 ч при комнатной температуре. Затем pH раствора доводилась до 5 с использованием Dewex 5OW•8 (H+), смола отфильтровывалась, а фильтрат подвергался лиофилизации, образуя 40 мг (выход 91%) соединения (42) в виде белого порошка.
ИК (KBr): νmax (см-1) 3376 (с, OH), 1652 (с, NHAc).
МС (FAB): 333 (M + H+).
1H-ЯМР (300 МГц, D2O): δ (м. д.) = 1,89 (с, 3H, NHAc), 1,93 (с, 3H, NHAc), 3,15 (дд, 1H, H9′), 3,40 (д, 1H, H6), 3,48 (дд, 1H, J9,9′ 14,18, H9), 3,82 (м, 1H, J8,9 3,01 J8,9′ 7,43, H8) 3,94 (дд, 1H, J5,6 10,42, H5), 4,13 (д, 1H, J7,8 10,91, H7), 4,36 (дд, 1H, J4,5 8,80, H4), 5,81 (д, 1H, J3,4 2,41, H3).
Пример 18. Метил 5-ацетамидо-9-циано-2,3,5,9-тетрадезокси-D-глицеро-D-галакто-нон-2-энопиранозонат (43).
Раствор метил 5-ацетамидо-2,3,5-тридезокси-9-(п-толуолсульфонил)-D-глицеро-D-галакто- нон-2-энопиранозоната (39) (80 мг, 0,17 ммоль), трет-бутиламмоний цианида (2 мг) и цианида натрия (12 мг, 0,25 ммоль) в сухом ДМСО (1,25 мл) перемешивался в течение 5 дней при комнатной температуре.
Применение препаративной тонкослойной хроматографии (SiO2, 20 см х 20 см х 2 мм, при элюировании смесью EtOAc/i-PrOH/H2O, 5/2/1) дало в качестве основного компонента 30 мг (61% выхода) соединения (43) в виде порошка кремового цвета.
(Rf = 0,74),
ИК (KBr): νmax (см-1) 3440 (с, OH), 2256 (W, CN), 1726 (с, CO2CH3).
1638 (с, NHAc),
МС (FAB) 315 (M + H+,
1H-ЯМР (300 МГц, D2O): δ (м.д.) = 1,92 (с, 3H, NHAc), 2,75 (дд, 1H, H9′), , 2,93 (дд, 1H, J9,9′ 17,22, H9) 3,55 (дд, 1H, J6,7 1,17, H6), 3,67 (с, 3H, CO2CH3) 4,02 (дд, 1H, J5,6 9,05 H5), 4,13 - 4,19 (м, 1H, J8,9 3,91, J8,9′ 6,56, H8), 4,16 (дд, 1H, J7,8 10,90, H7), 4,37 (дд, 1H, J4,5, 8,95, H4), 5,90 (д, 1H, J3,4,) 2,42, H3).
Пример 19. 5-ацетамидо-9-циано-2,3,5,9-тетрадезокси-D-глицеро -D-галакто-нон-2-энопиразоновая кислота (44).
Методология, использованная для получения 5-ацетамидо-9-циано-2,3,5,9-тетрадезокси-D-глицеро-D-галкто-нон-2- энопиразоновой кислоты (44), суммируется ниже и в конце описания.
Метил 5-ацетамидо-9-циано-2,3,5,9-тетрадезокси-D-глицеро-D-галакто-нон-2 -энопиранозонат (43) (80 мг, 0,25 ммолm) растворялся в 0,1 М водном растворе гидроокиси натрия (10 мл) и полученный раствор перемешивали при комнатной температуре в течение 3 ч.
Затем pH регулировалась до 4 с помощью Dowex 50W •8(H+), смола отфильтровывалась, а фильтрат подвергался лиофилизации с образованием 75 мг (98% выход) соединения (43) в виде рыхлого белого порошка.
ИК (KBr): νmax (см-1) 3370 (с, OH), 2254 (W, CN), 1656 (с, NHAc).
MC (FAB) 301 (M + H+).
1H ЯМР (300 МГц, D2O): δ (м.д.) = 1,98 (с, 3H, NHAc), 2,70 (дд, 1H, H9′), 2,88 (дд, 1H, J9,9′ 17,27, H9), 3,48 (д, 1H, H6), 3,97 (дд, 1H, J5,6 9,84 H5), 4,09 - 4,24 (м, 2H, H7 и H8, J8,9 3,90 J8,9′ 6,53), 4,41 (дд, 1H, J4,5 8,87, H4), 5,80 (д, 1H, J3,4 2,42, H3).
Пример 20. Ингибирование неураминидазы вируса (эпидемического) гриппа.
Биологический анализ in vitro описанных выше соединений против неураминидазы вируса (эпидемического) гриппа N 2 проводился в соответствии с работой Warner и O'Brien Biochemistry 1979, 18, 2783-2787. Для сравнения тем же самым анализом было определено Ki для 2-деокси-N-ацетил -α- D-неураминовой кислоты, имеющее значение 3 • 10-4 M.
Значение Ki измерялось с использованием спектрофотометрического способа, который использует фторогенный субстрат 4-метилумбеллиферил N-ацетилнеураминовой кислоты (MUN), как это описано Meyers и др., Anal. Biochem. 1980, 101, 166-174. В случае обоих ферментов анализируемая смесь содержала испытуемое соединение в нескольких концентрациях между 0 и 2 мМ и приблизительно 1 mU фермент в буфере (32,6 мМ MES, 4 мМ CaCl2, pH 6,5 для N2; 32,5 мМ ацетата, 4 мМ CaCl2, pH 5,5 для неураминидазы V.cholerae).
Реакция начиналась с добавления MUN до конечной концентрации 75 или 40 мкМ. Спустя 5 мин при 37oC добавлялось 2,4 мл 0,1 М раствора глицин -NaOH, pH 10,2 и 0,1 мл реакционной смеси для окончания реакции. Флуоресценция наблюдалась при поглощении 365 нм, эмиссии 450 нм и соответствующие контрольные значения (отсутствие фермента) вычитались из отсчета. Ki вычислялось из кривых Dexon (1/флуоресценция в зависимости от концентрации соединения). Результаты суммированы в табл. 1 и, если не оговорено особо, относятся к ингибированию неураминидазы N2.
Пример 21. Ингибирование размножения вируса (эпидемического) гриппа in vitro.
Ингибирование размножения (эпидемического) гриппа A/Singapore/1/5 7 (H2N2) и эпидемического гриппа B/Victoria/102/85 in vitro измерялось снижением вирусных колоний в Madine Darby почечных (MDCK) клетках.
Монослои конфлюентных клеток MDCK, выращенные в шести чашках Петри для яичных тканевых культур, были инокулированы 0,3 мл разбавленного вируса, образуя приблизительно 50-100 колоний/ячейку. Вирус разбавлялся минимальным необходимым количеством освобожденной от сыворотки среды (MEM), содержащей 1 мкг/мл N-толил-1-фенилаланин хлорметилкетона (TPCK), обработанного трипсином (Worthington Enzymes) и испытуемым соединением.
Вирус адсорбировался при комнатной температуре в течение 1 ч и затем клетки покрывали определенной клеточной культурной средой, вариант 1 (DCCM-1)/агаровое покрытие, содержащее испытуемое соединение, 4 мл/ячейку. DCCM-1 представляет собой полностью освобожденную от клеток питательную среду (Biological Industries), к которой добавлялся TPCK, обработанный трипсином, и DEAE декстран до конечной концентрации 2 мкг/мл и 0,001% соответственно. Агар (5%) Indubiose разбавлялся 1:10 перед добавлением в чашку Петри.
Сразу же после нанесения чашки подвергались термостатированию при 37oC, 5% CO2 в течение 3 дней. Затем клетки фиксировались 5%-ным глутаровым альдегидом, окрашивались Carbol fuschin и подсчитывались колонии вирусов. Результаты приведены в табл. 2.
Натрий 5-ацетамидо-4-N-аллил-N-гидорокси-2,3, 4,5-тетрадезокси-D-глицеро-D-галакто-нон-2-энопиранозонат (45) может быть легко получен из соединения (11), описанного в примере 5 с использованием окислительных методов.
Пример 22. In vivo антивирусная активность.
Соединения 2,3 и 6 (4-амино, 4-гуанидин и 4-эпи-амино), а также соединение DANA (2-дезокси-N-ацетил -α- D-неураминовая кислота), которые показали в примере 20 активность in vitro против неураминидазы, были подвергнуты испытаниям на антивирусную активность в стандартных испытаниях in vivo. При интраназальном приеме мышами перед появлением симптомов и в процессе заражения вирусом (эпидемического) гриппа A эти соединения понижали титр вируса в легочной ткани от 1 до 3 дней после инфекции.
Мыши были заражены интраназально с применением 50 мкл вируса эпидемического гриппа A H2N2 103 TCl D50 единиц/мышь (A/Sing/1/57. Испытуемое соединение принималось интраназально при норме дозы или 12,5, или 25 мг/кг веса тела (50 мкл водного раствора/мышь) следующим образом: 24 ч и 3 ч перед инфекцией; спустя 3 часа после инфекции, затем дважды в день каждый день на 1-й, 2-й и 3-й день после инфекции. Для сравнения были использованы также отличные по строению соединения рибаверин и амантадин.
Мыши были умерщвлены на 1-й, 2-й и 3-й дни после инфекции, их легких извлекались и измерялись титры вирусов в легких. Значения титров были изображены графически и выражены как процент площади под кривыми (AUC) при сравнении их с соответствующими данными для мышей, не подвергнутых испытаниям. Полученные результаты суммированы в табл. 3.
Все три испытанных соединения показывают более высокую эффективность, чем DANA.
Пример 23. Фирма "Glaxa Welcome" и исследователи из США показали впервые, что ингибитор вирусной нейроаминидазы GG167 (соединение примера 3) может быть полезен при лечении гриппа у человека.
Нейраминидаза - это основной гликопротеин поверхности вируса, найденный в обоих типах вируса гриппа - A и B, который играет важную роль в репликации вируса. В исследованиях, проведенных в США на добровольцах, была показана эффективность GG167 как для профилактики, так и для лечения гриппа.
Введение через нос.
GG167 вводили в нос 166 добровольцам два или шесть раз в день при дозах от 3,6 до 16 мг, или за 4 ч до искусственного заражения гриппом (профилактика), или через один или два дня после заражения гриппом (раннее или позднее лечение соответственно). Для всех групп суммарных доз GG167 был эффективен для предотвращения жара (p < 0,01 по сравнению с плацебо). Раннее лечение уменьшало пик титра вируса на два порядка (logs), среднее время исчезновения вируса на три дня и частоту возникновения жара на 85% (p < 0,05 для каждого сравнения). Дозировка по два раза в день была так же эффективна, как и по шесть раз в день, и GG167 имел хорошую переносимость.
Пример 24. Следующие рецептуры являются характерными композициями, соответствующими изобретению:
Водный раствор - % по весу
Соединение формулы (1) - 10,0
Бензалконий хлорид - 0,04
Фенилэтиловый спирт - 0,40
Очищенная вода - До 100% по весу
Водный раствор с сорастворителями - % по весу
Соединение формулы (1) - 10,0
Бензалконий хлорид - 0,04
Полиэтиленовый 400 - 10,0
Пропиленгликоль - 30,0
Очищенная вода - До 100% по весу
Аэрозольная рецептура - % по весу
Соединение формулы (1) - 7,5
Лецитин - 0,4
Газ-вытеснитель 11 - 25,6
Газ-вытеснитель 12 - 66,5
Рецептура сухого порошка - % по весу
Соединение формулы (1) - 40,0
Лактоза - 60,0
Эти рецептуры получаются смешением активного ингредиента и наполнителей с использованием стандартных фармацевтических методов.
Несомненно, понятно, что изобретение в своих основных аспектах не ограничивается конкретными, приведенными здесь выше детальными примерами.
| название | год | авторы | номер документа |
|---|---|---|---|
| 5-АЦЕТАМИДО-2,3,4,5-ТЕТРАДЕОКСИ-4-ГУАНИДИНО-D-ГЛИЦЕРО-D-ГАЛАКТО-НОН-2- ЕНОПИРАНОЗОНОВАЯ КИСЛОТА В КРИСТАЛЛИЧЕСКОЙ ФОРМЕ, СПОСОБЫ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1994 |
|
RU2134690C1 |
| ПРОИЗВОДНЫЕ НЕЙРАМИНОВОЙ КИСЛОТЫ, ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ИЛИ ИХ СЛОЖНЫЕ ЭФИРЫ, А ТАКЖЕ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ СИАЛИДАЗУ-ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ | 1997 |
|
RU2124509C1 |
| Фосфониевые соли на основе алантолактона, обладающие противоопухолевой активностью, и способ их получения | 2023 |
|
RU2818095C1 |
| ПРОИЗВОДНЫЕ НЕЙРАМИНОВОЙ КИСЛОТЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ГРИППА | 1997 |
|
RU2169145C2 |
| Противовирусные агенты и их применение | 2015 |
|
RU2730453C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ, КОМПОЗИЦИЯ ДЛЯ ПРИГОТОВЛЕНИЯ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2001 |
|
RU2290409C2 |
| СОЕДИНЕНИЯ ФЛАВОНОИДОВ И ИХ ПРИМЕНЕНИЕ | 2006 |
|
RU2431634C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТАКСАНА | 1992 |
|
RU2128654C1 |
| СПОСОБЫ СЕЛЕКТИВНОГО ВВЕДЕНИЯ ЗАЩИТНЫХ ГРУПП В ПРОИЗВОДНЫЕ РЕЗОРЦИНА | 2019 |
|
RU2738408C1 |
| ПРОИЗВОДНЫЕ ВАРИОЛИНА В, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2293737C2 |

















Предложены производные 2-дезокси-2,3-дидегидро-N-ацетилнеураминовой кислоты формулы I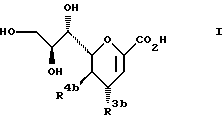
где R3b - азид или группа -NR6bR7b, причем R6b - водород, С1-6 - алкил или аллил; R4b - NHCOR9b, где R9b - С1-6 - алкил, или их фармацевтически приемлемые соли, эфиры и соли эфиров. Способ получения соединений формулы I состоит во взаимодействии защищенного по карбокси- и гидроксигруппам производного формулы III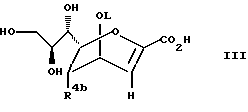
где R4b имеет вышеуказанные значения, OL - удаляемая группа с нуклеофилом и последующем превращении в амин, гуанидин, соль, эфир и соль эфира. Предложена фармацевтическая композиция, обладающая антивирусной активностью в отношении орто-, парамиксовирусов, которая содержит в качестве активного вещества соединении формулы I в эффективном количестве. Предложен способ лечения млекопитающих, включая человека, от вирусных инфекций путем введения соединений формулы I в дозе 0,01 - 750 мг/кг веса тела в день, либо в респираторный тракт, либо внутриназально. 4 с. и 11 з.п. ф-лы, 3 табл.

где R3b представляет азид группу - NR6bR7b, причем
R6b представляет водород, C1-6 алкил или амидин,
R7b представляет водород, C1-6 алкил или аллил,
R4b представляет NHCOR9b, где R9b представляет C1-4 алкил,
или их фармацевтически приемлемые соли, эфиры и соли эфиров. глицеро
глицеро галакто-нон-2-енопиранозонат(4-азидо- Neu 5Ац2ен), натрий 5-ацетамидо-4- N-аллиламино-2,3,4,5-тетрадеокси
галакто-нон-2-енопиранозонат(4-азидо- Neu 5Ац2ен), натрий 5-ацетамидо-4- N-аллиламино-2,3,4,5-тетрадеокси  глицеро
глицеро галакто-нон-2-енопиранозонат, метил 5-ацетамидо-7,8,9-три -0-ацетил-4- N-аллиламино-2,3,4,5-тетрадеокси
галакто-нон-2-енопиранозонат, метил 5-ацетамидо-7,8,9-три -0-ацетил-4- N-аллиламино-2,3,4,5-тетрадеокси глицеро
глицеро галакто- нон-2-енопиранозонат(4- N-аллиламино- Neu 5,7,8,9 Ac42ен IMe), натрий 5-ацетамидо-4- N, N-диметиламино-2,3,4,5-тетра-деокси
галакто- нон-2-енопиранозонат(4- N-аллиламино- Neu 5,7,8,9 Ac42ен IMe), натрий 5-ацетамидо-4- N, N-диметиламино-2,3,4,5-тетра-деокси  глицеро
глицеро  галакто-нон-2-енопиранозонат, 5-ацетамидо-4-амино- 2,3,4,5-тетрадеокси
галакто-нон-2-енопиранозонат, 5-ацетамидо-4-амино- 2,3,4,5-тетрадеокси глицеро
глицеро -галакто-нон-2-енопиранозоновая кислота, натрий 5-ацетамидо-4-амино-2,5,4,5-тетрадеокси
-галакто-нон-2-енопиранозоновая кислота, натрий 5-ацетамидо-4-амино-2,5,4,5-тетрадеокси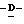 глицеро
глицеро галакто-нон-2-енопиранозонат, аммоний 5-ацетамидо-4-гуанидино-2,3,4,5-тетрадеокси
галакто-нон-2-енопиранозонат, аммоний 5-ацетамидо-4-гуанидино-2,3,4,5-тетрадеокси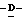 глицеро
глицеро галакто-нон-2-енопиранозонат, и его фармацевтически приемлемые соли, эфиры и соли эфиров.
галакто-нон-2-енопиранозонат, и его фармацевтически приемлемые соли, эфиры и соли эфиров. глицеро-D-галакто-нон-2-енопиранозоновая кислота и ее фармацевтически приемлемые соли, эфиры и соли эфиров.
глицеро-D-галакто-нон-2-енопиранозоновая кислота и ее фармацевтически приемлемые соли, эфиры и соли эфиров.
где R4b имеет значения, определенные выше, а OL означает удаляемую группу, такую как остаток сульфоновой кислоты, подвергают взаимодействию с нуклеофилом, таким как азид, полученное при этом соединение общей формулы Ib, где R3b означает азид, в случае необходимости, подвергают превращению в другое соединение общей формулы Ib, где R3b означает группу -NR6bR7b, где R6b означает атом водорода, (C1 - C6)алкил или амидин, R7b означает атом водорода, (C1 - C6)алкил или аллил, и в полученных соединениях удаляют защитные группы и, в случае необходимости, переводят эти соединения в фармацевтически приемлемые соли, эфиры или соли эфиров.
Приоритет по пунктам и признакам:
24.04.90 по пп. 1 - 3, 6 - 13, за исключением соединений, где R6b-алкил;
24.04.90 по п.4, за исключением соединений: натрий 5-ацетамидо-4-N-аллиламино-2,3,4,5-тетрадеокси-D-глицеро-D-галакто-нон-2-енопиранозата, метил-5-ацетамидо-7,8,9-три-0-ацетил-4-N-аллиламино-2,3,4,5-тетрадеокси-D-глицеро-D-галакто-нон-2-енопиранозоната, натрий 5-ацетамидо-4-N,N -диметиламино-2,3,4,5- тетрадеокси-D-глицеро-D-галакто-нон-2-енопиранозоната;
19.10.90 по п.4 для соединений: натрий 4-N,N-диаллиламино-2,3,4,5-тетрадеокси-D-глицеро-D-галакто-нон-2-енопиранозоната, натрий 5-ацетамидо-4-N-аллиламино-2,3,4,5-тетрадеокси-D-глицеро-D-галакто-нон-2-енопиранозоната;
11.02.91 - для остальных соединений, приведенных в формуле изобретения.
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1987, ч.II, с.380. |

