Область техники, к которой относится изобретение
Настоящее изобретение относится к новому способу селективного введения защитных групп в производные 5,7-дигидроксикумарина, к применению агентов для такой селективной защиты, а также к промежуточным соединениям.
Уровень техники
Моно ОН-замещенные производные 5,7-дигидроксикумаринов обладают высокой нейропротекторной и противораковой активностью (например, алкалоиды минутин A и B). В то же время, химическая эквивалентность гидроксильных групп в положениях 5 и 7 делает получение таких производных трудоемкой задачей, поскольку селективная защита OH групп в производных кумаринов является нерешенной проблемой органической химии. Не существует опубликованных общих и селективных способов защиты одного из гидроксилов в 5,7-дигидроксикумаринах [см., например, D1 и цитированную в нем литературу]. Описанные способы (при условии, что в положении 4 кумарина нет карбонильной или имино группы, см. D2 и D3) дают, как правило, смеси 5-OH и 7-OH защищенных изомеров, при этом зачастую с низким выходом, как, например, описано в D4 и D5. Единственным общим способом селективного введения защитных групп является исчерпывающая защита всех гидроксилов с последующим селективным снятием защиты. Иллюстративным примером этого подхода является одна из стадий получения соединений с нейропротекторной активностью, описанная в D5 (Схема 1).
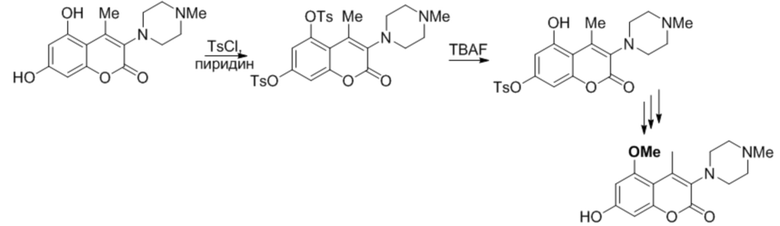
Схема 1
Еще одним примером использования подхода с исчерпывающим тозилированием является ключевая стадия получения аналога анти-ВИЧ-препарата Каланолид А (Схема 2), описанный в D6. Синтез включает исчерпывающее тозилирование дигидроксипроизводного 1 с последующим удалением одной из защитных групп с получением дитозильного производного 2. Такой способ требует дополнительной стадии снятия защиты и, как результат, увеличение количества побочных продуктов, числа стадий и снижение выхода. С другой стороны, воспроизводя процедуру, описанную в D6, мы обнаружили в реакционной смеси существенное количество непрореагировавшего 2, дигидроксипроизводного 1 и побочного изомера 3b.
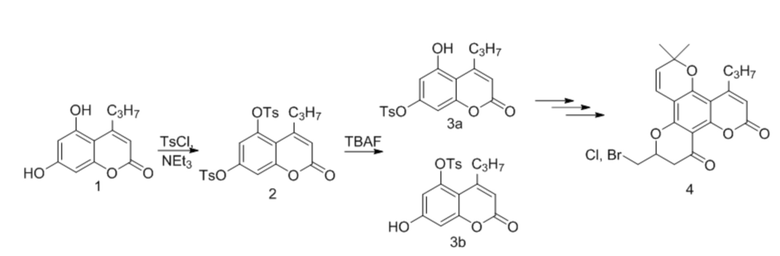
Схема 2
В результате, полученную смесь соединений 1, 2, 3а и 3b удается эффективно очистить только при помощи хроматографии.
Таким образом, существует потребность в новых методах селективной защиты гидроксилов в производных 5,7-дигидроксикумаринов.
Цитированные документы уровня техники
D1 Wuts, P. G. M., Greene T. W. Greene's protective groups in organic synthesis. Wiley, 2008.
D2 Pandey, G.; Muralikrishna, C.; Bhalerao, U. T. Tetrahedron 1989, 45, 6867–6874.
D3 Chang, C.-F. et al. Tetrahedron 2008, 64, 3661–3666.
D4 Flavin, M. T. et al. J. Med. Chem. 1996, 39, 1303–1313.
D5 Sun, M. et al. Eur. J. Med. Chem. 2013, 67, 39–53.
D6 Ma, T. et al. J. Med. Chem. 2008, 51, 1432–1446.
Раскрытие изобретения
В первом аспекте настоящее изобретение относится к способу селективного введения защитной группы в соединение формулы (I), включающему
взаимодействие соединения формулы (I) с соединением формулы (II) с получением соединения формулы (III):
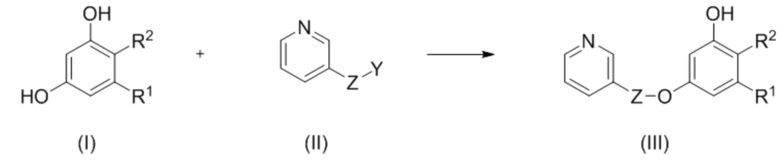 ,
,
где R1 и R2 независимо представляют собой водород или галоген;
или R1 и R2 образуют вместе с несущими их атомами углерода 6-членный цикл, в частности, пироновый цикл, который замещен R3 и R4, где R3 и R4 независимо выбраны из водорода, С1-С6 алкила, С1-С6 галогеналкила, С6-С10 арил С1-С6 алкила, NHCOC1-C6 алкила; или R3 и R4 соединены вместе с образованием 5 или 6 членного карбоциклического фрагмента, необязательно содержащего одну или несколько двойных связей;
Z представляет собой CO или SO2; и
Y представляет собой гидроксил или хорошую уходящую группу, такую как галоген, –N3 или бензотриазолил.
Реакция необязательно может проводится в присутствии агентов сочетания, известных в данной области техники, таких как 1,3-дициклогексилкарбодиимид (DDC), гексафторфосфат азабензотриазол тетраметил-урония (HATU), 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC), 1,3-диизопропилкарбодиимид и т.п.
Взаимодействие можно проводить в инертном апротонном растворителе, в частности, в тетрагидрофуране, ацетоне, ацетонитриле, ДМФА, этилацетате или диоксане.
Взаимодействие можно проводить в присутствии основания, в частности, органического амина; например, триэтиламина, N-метилморфолина, N-этилдиизопропиламина, 1,4-диазабицикло[2.2.2]октана или этилендиамина.
Полноту протекания реакции можно установить при помощи тонкослойной хроматографии (ТСХ), высокоэффективной жидкостной хроматографии (ВЭЖХ), спектроскопии ядерного магнитного резонанса (ЯМР) или других известных методов. Как правило, реакция протекает полностью или по существу полностью в течение от 1 часа до 24 часов.
Температура проведения реакции не ограничивается специальным образом, однако, с точки зрения технологии, предпочтительная температура составляет от –20 до 100 °С, более предпочтительно от 0 до 60 °С и наиболее предпочтительно около 25°С.
Давление, при котором проводится реакция также не ограничивается специально, однако наиболее предпочтительным является использование атмосферного давления.
Предпочтительные варианты осуществления
В одном из вариантов осуществления заявленного способа R1 и R2 представляют собой независимо водород или галоген, в частности, R1 и R2 представляют собой водород; или R1 представляет собой водород, и R2 представляет собой хлор.
В другом варианте осуществления R1 и R2 образуют пироновый цикл, так что соединение формулы (I) описывается соединением формулы (Ia)
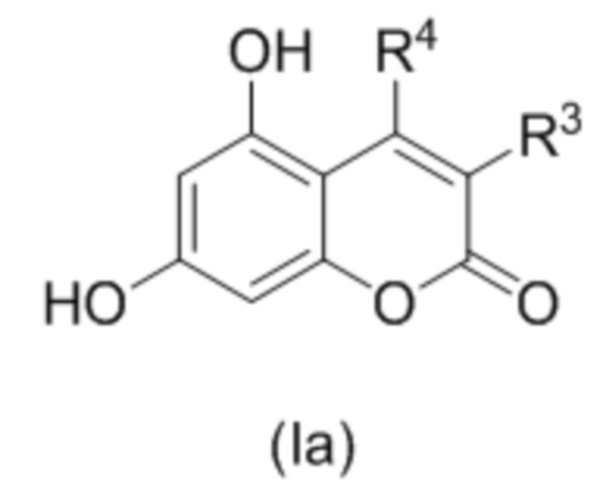 ,
,
где R3 и R4 независимо выбраны из водорода, С1-С6 алкила, С1-С6 галогеналкила, С6-С10 арил С1-С6 алкила, NHCOC1-C6 алкила; или R3 и R4 соединены вместе с образованием 5 или 6 членного карбоциклического фрагмента, необязательно содержащего одну или несколько двойных связей.
В одном из вариантов осуществления Z представляет собой CO.
В одном из вариантов осуществления Y представляет собой бензотриазолил, в частности, 1-бензотриазолил.
В следующем аспекте настоящее изобретение относится к соединению формулы (III)
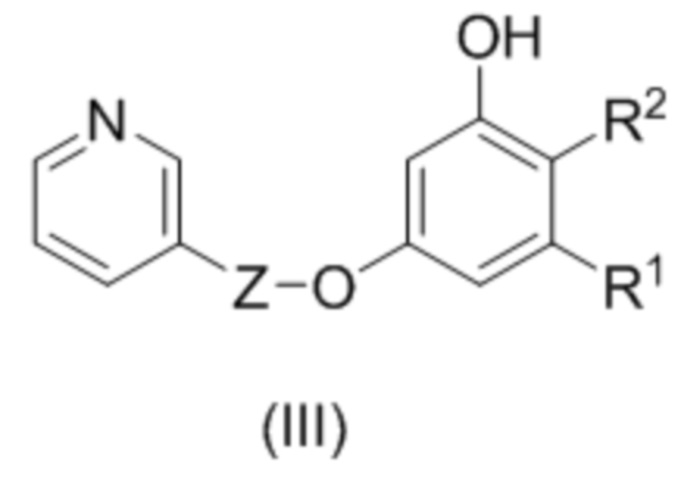 , где
, где
где R1 и R2 и Z такие как описано выше;
при условии, что соединение формулы (III) не является
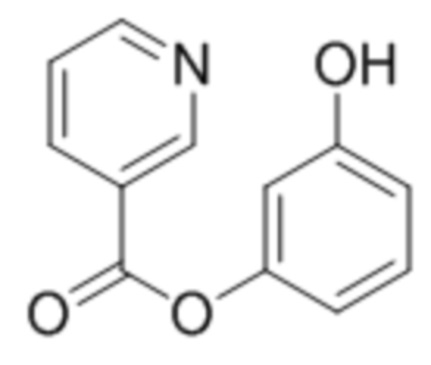 .
.
В одном из вариантов в соединении (III) R1 и R2 представляют собой независимо водород или галоген, в частности R1 представляет собой водород, и R2 представляет собой хлор.
В другом варианте в соединении (III) R1 и R2 образуют пироновый цикл, так что соединение формулы (III) описывается соединением формулы (IIIa)
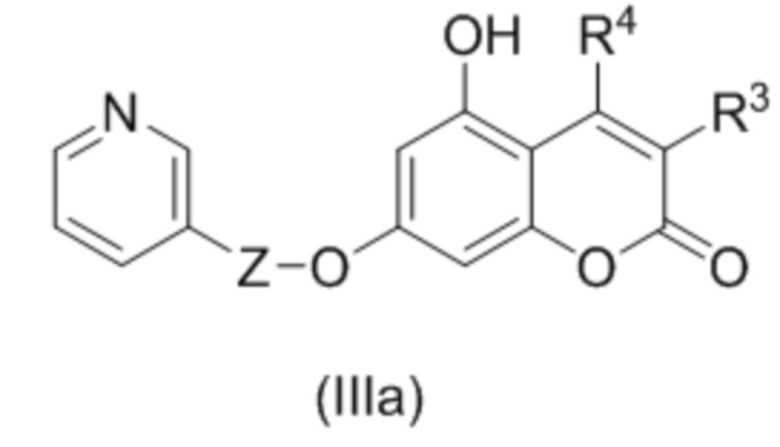 ,
,
где R3 и R4 независимо выбраны из водорода, С1-С6 алкила, С1-С6 галогеналкила, С6-С10 арил С1-С6 алкила, NHCOC1-C6 алкила; или R3 и R4 соединены вместе с образованием 5 или 6 членного карбоциклического фрагмента, необязательно содержащего одну или несколько двойных связей.
В одном из вариантов в соединении (III) Z представляет собой CO;
В конкретных вариантах осуществления соединение (III) выбрано из группы, состоящей из:
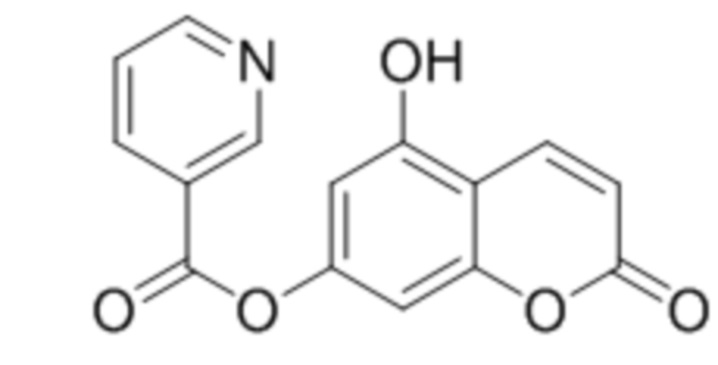 ,
,  ,
,  ,
,  ,
,  ,
, 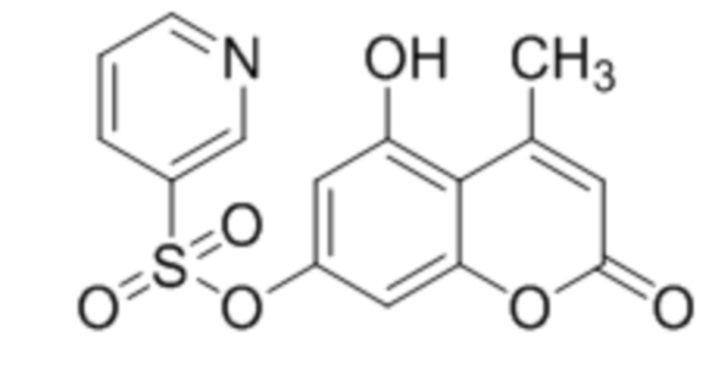 ,
, 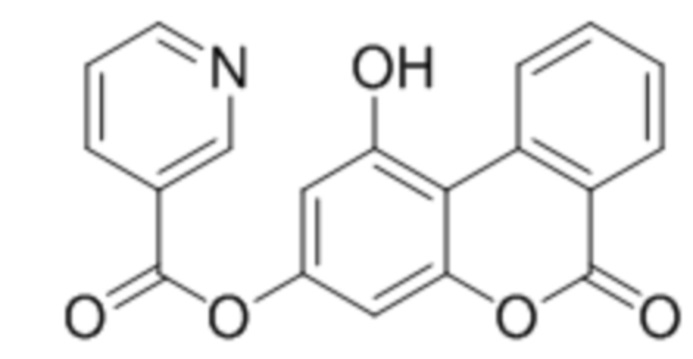 ,
,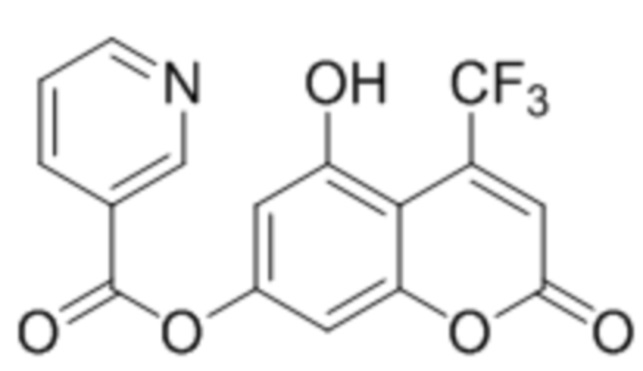 и
и 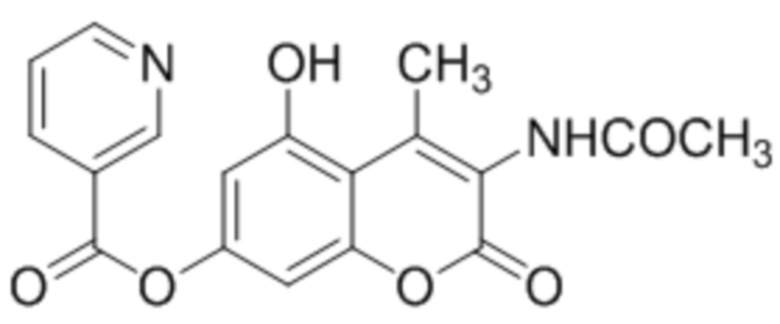 .
.
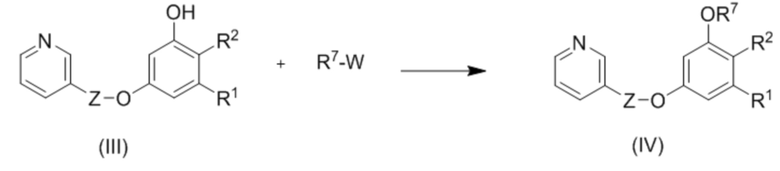
Соединения формулы (III) содержат одну свободную гидроксильную группу, и могут быть использованы, например, для получения соединений (IV).
Соответственно, в еще одном аспекте изобретение относится к способу получения соединений (IV), включающему взаимодействие
 ,
,
где Z, R1 и R2 такие как описано выше;
R7 представляет собой С1-С6 алкил(диС6-С10 арил)силил, триС1-С6 алкилсилил, С6-С10 арилсульфонил, необязательно замещенный С1-С6 алкилом, С1-С6 алкоксикарбонил или С1-С6 алкил-CO; и
W представляет собой хорошо уходящую группу.
В одном из вариантов осуществления хорошо уходящая группа W представляет собой атом галогена или ацетоксигруппу.
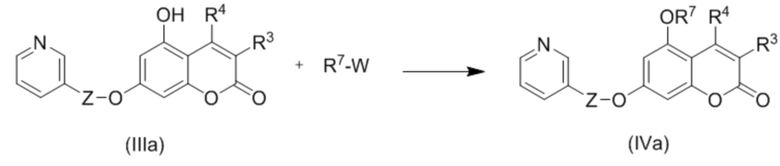
В частности, изобретение относится к способу получения соединений (IVa), включающему взаимодействие соединений IIIa с соединениями R7-W:
 ,
,
где Z, R3 и R4 такие как описано выше;
R7 представляет собой С1-С6 алкил(диС6-С10 арил)силил, триС1-С6 алкилсилил, С6-С10 арилсульфонил, необязательно замещенный С1-С6 алкилом, С1-С6 алкоксикарбонил или С1-С6 алкил-CO; и
W представляет собой хорошо уходящую группу, в частности, атом галогена или ацетоксигруппу.
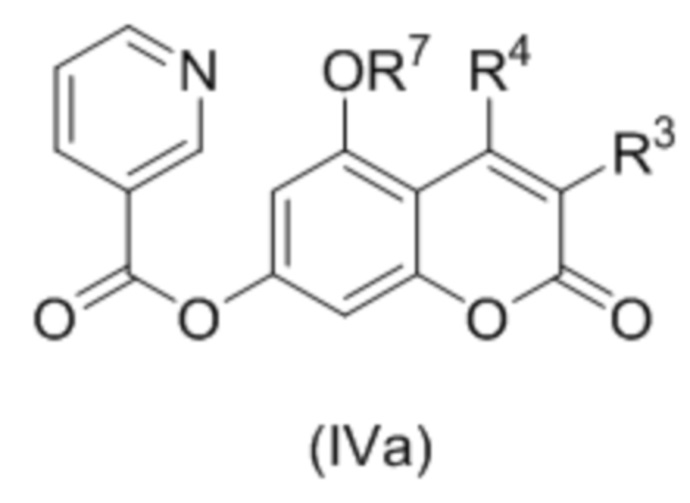
В следующем аспекте изобретение относится к соединениям формулы (IVа)
 ,
,
где R3 и R4 независимо выбраны из водорода, С1-С6 алкила, С1-С6 галогеналкила, С6-С10 арил С1-С6 алкила, NHCOC1-C6 алкила; или R3 и R4 соединены вместе с образованием 5 или 6 членного карбоциклического фрагмента, необязательно содержащего одну или несколько двойных связей;
R7 представляет собой С1-С6 алкил(диС6-С10 арил)силил, триС1-С6 алкилсилил, С6-С10 арилсульфонил, необязательно замещенный С1-С6 алкилом, С1-С6 алкоксикарбонил или С1-С6 алкил-CO.
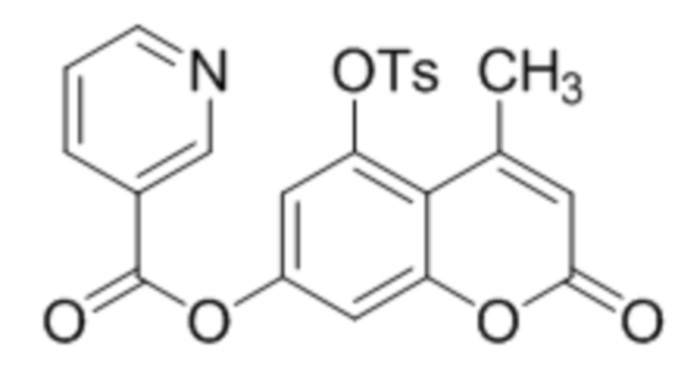
В конкретных вариантах осуществления соединение (IVa) выбрано из группы, состоящей из:
 ,
, 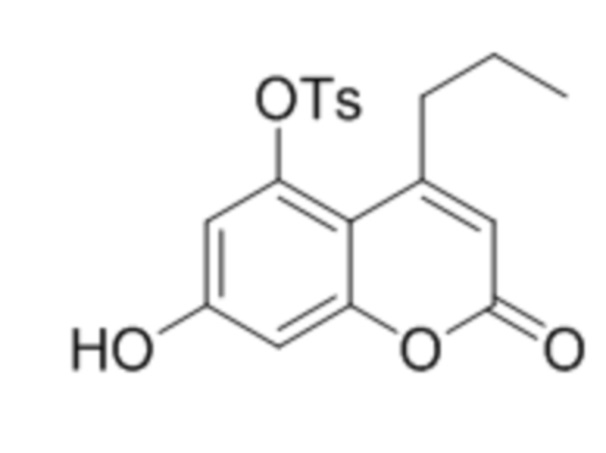 ,
, 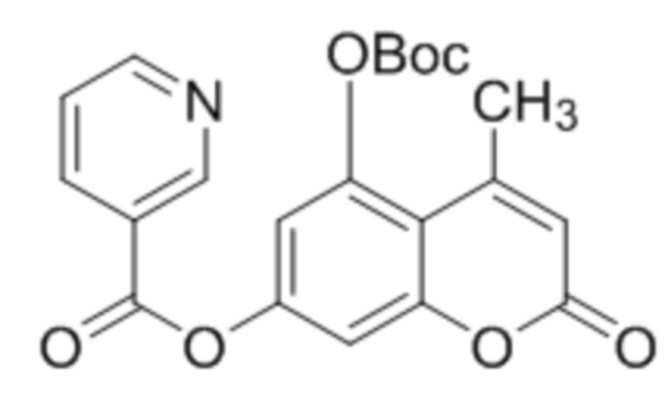 и
и 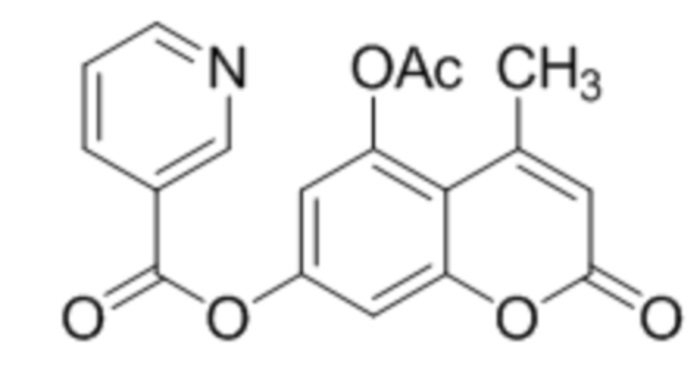 .
.
В еще одном аспекте изобретение относится к способу получения соединений формулы (Vа) из соединений (IVa):
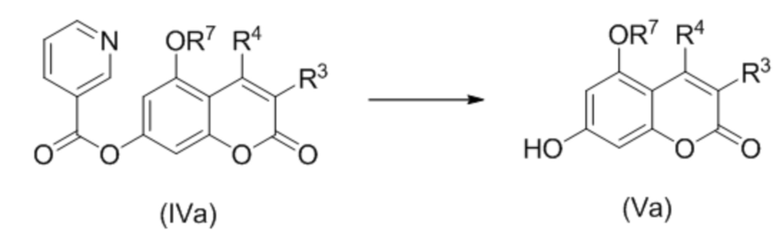 ,
,
где R3 и R4 независимо выбраны из водорода, С1-С6 алкила, С1-С6 галогеналкила, С6-С10 арил С1-С6 алкила, NHCOC1-C6 алкила; или R3 и R4 соединены вместе с образованием 5 или 6 членного карбоциклического фрагмента, необязательно содержащего одну или несколько двойных связей;
R7 представляет собой С1-С6 алкил(диС6-С10 арил)силил, триС1-С6 алкилсилил, С6-С10 арилсульфонил, необязательно замещенный С1-С6 алкилом, С1-С6 алкоксикарбонил или С1-С6 алкил-CO.
В следующем аспекте изобретение относится к соединениям формулы (Vа)
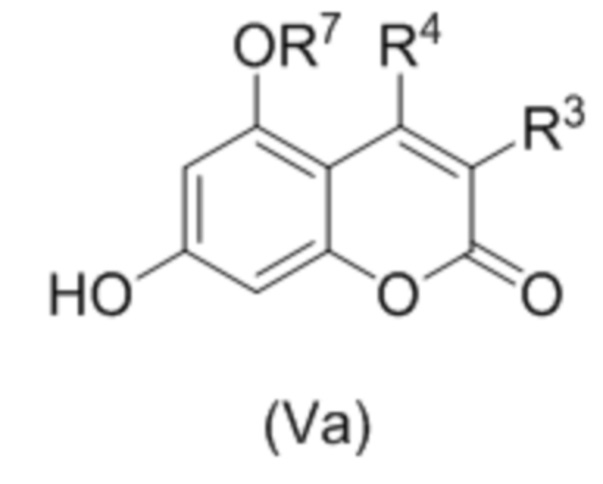 ,
,
где R3 и R4 независимо выбраны из водорода, С1-С6 алкила, С1-С6 галогеналкила, С6-С10 арил С1-С6 алкила, NHCOC1-C6 алкила; или R3 и R4 соединены вместе с образованием 5 или 6 членного карбоциклического фрагмента, необязательно содержащего одну или несколько двойных связей;
R7 представляет собой С1-С6 алкил(диС6-С10 арил)силил, триС1-С6 алкилсилил, С6-С10 арилсульфонил, необязательно замещенный С1-С6 алкилом, С1-С6 алкоксикарбонил или С1-С6 алкил-CO.
В конкретных вариантах осуществления соединение (Va) выбрано из группы, состоящей из:
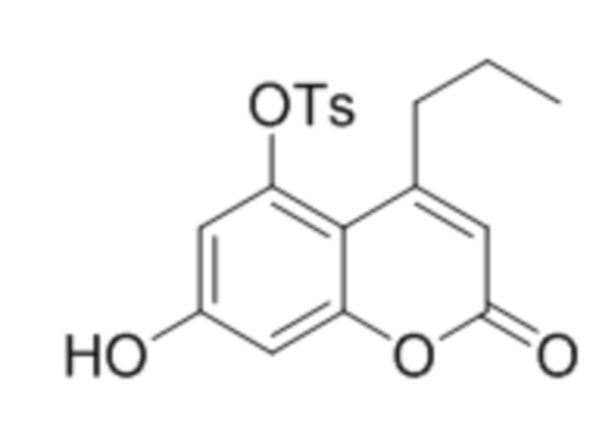 ,
, 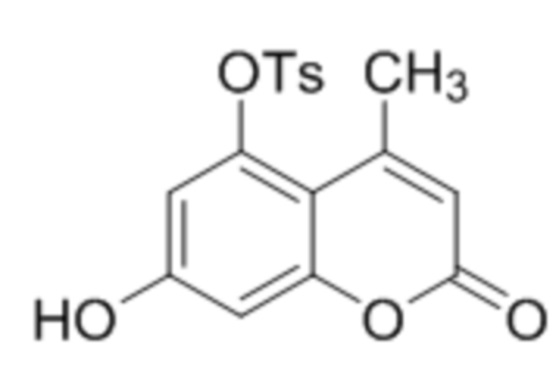 и
и 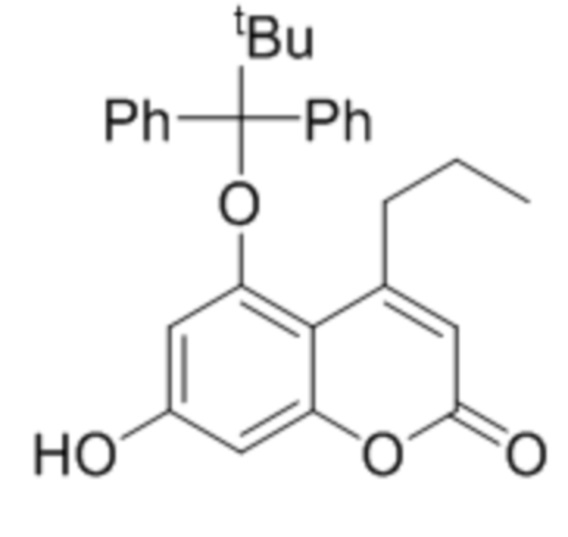 .
.
Таким образом, технический результат настоящего изобретения заключается в том, что предложен способ введения защиты в производные резорцина, в том числе в производные 5,7-дигидроксикумарина при помощи производных никотиновой кислоты, причем предложенный способ позволяет ввести защитную группу в одну простую стадию; при этом, полученное соединение представляет собой твердое вещество, которое может быть легко выделено; кроме того, получаемое твердое вещество предпочтительно является кристаллическим и не требует очистки.
Примеры
Представленные ниже примеры иллюстрируют некоторые предпочтительные варианты осуществления настоящего изобретения, но не ограничивают его.
Пример 1
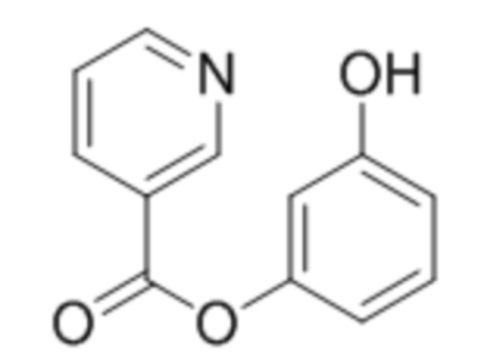
3-Гидроксифенил никотинат
К раствору резорцина (1,0 ммоль, 110 мг) в этилацетате при перемешивании последовательно добавили триэтиламин (1,1 ммоль, 111 мг) и 1-никотиноилбензотриазол (1,0 ммоль, 224 мг). Через сутки отфильтровали выпавший осадок 3-гидроксифенил никотината. Выход 66%.
1Н ЯМР: (ДМСО-d6+CCl4) 9.49 (с, 1H, OH), 9.24 (дд, J=0.7 Гц, J=1.9 Гц, 1H, H2’), 8.83 (дд, J=1.9 Гц, J=4.8 Гц, 1H, H6’), 8.43 (дт, Jd=8.0 Гц, Jt=1.9 Гц, 1H, H4’), 7.57 (ддд, J=0.7 Гц, J=4.8 Гц, J=8.0 Гц, 1H, H5’), 7.19 (м, 1H, H2), 6.69 (м, 1H, H6), 6.61 (м, 2H, H4+H5)
ИК: 1143, 1238, 1427, 1484, 1597, 1735.
Пример 2
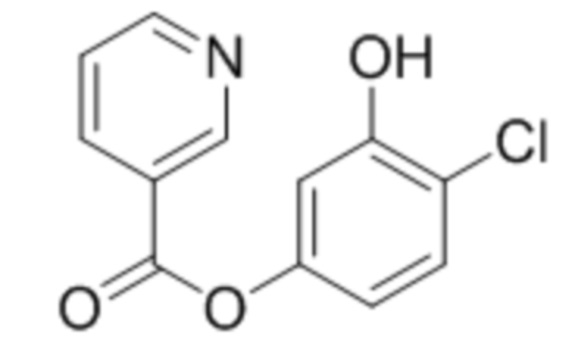
4-Хлор-3-гидроксифенил никотинат
К раствору 4-хлоррезорцина (1,0 ммоль, 146 мг) в этилацетате при перемешивании последовательно добавили триэтиламин (1,1 ммоль, 111 мг) и 1-никотиноилбензотриазол (1,0 ммоль, 224 мг). Через сутки упаривали растворитель и остаток перекристаллизовывали из ацетонитрила с получением указанного в заголовке соединения. Выход 150 мг (60%).
1Н ЯМР: (ДМСО-d6+CCl4) 10.28 (с, 1H, OH), 9.28–9.19 (уш с, 1H, H-2’), 8.88–8.79 (м, 1H, H-6’), 8.47–8.37 (м, 1H, H-4’), 7.62–7.53 (м, 1H, H-5’), 7.31 (д, J=8.6 Гц, 1H, H-5), 6.86 (д, J=1.8 Гц, 1H, H-2), 6.67 (дд, J=8.6 Гц, J=1.8 Гц, 1H, H-6)
ИК: 1741, 1612, 1391, 1221, 1094, 727.
Пример 3
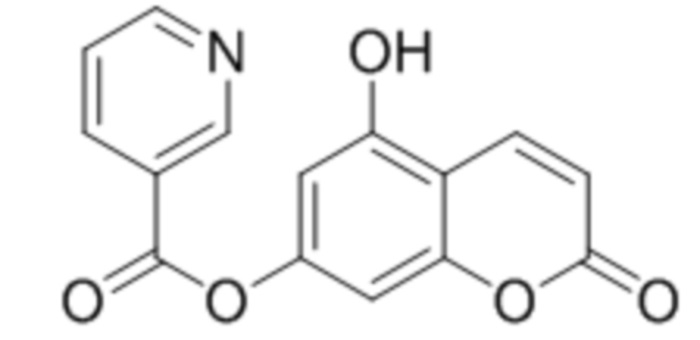
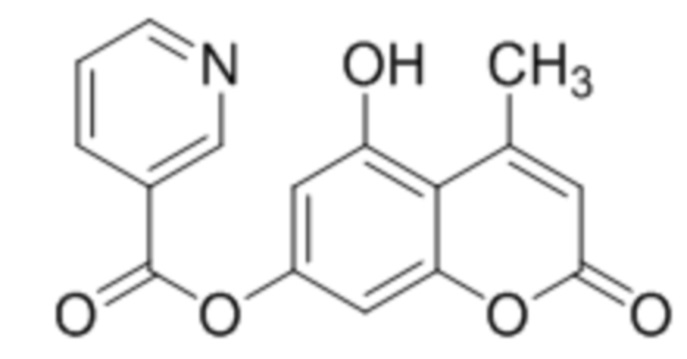
5-Гидроксикумарин-7-ил никотинат
К раствору 5,7-дигидроксикумарина (1 ммоль, 178 мг) и триэтиламина (1,1 ммоль, 111 мг) в 10 мл ацетона при перемешивании добавили 1-никотиноилбензотриазол (1,0 ммоль, 224 мг). Через сутки отфильтровали выпавший осадок 5-гидроксикумарин-7-ил никотината. Выход 72%. Тпл. 246–248 °С.
1Н ЯМР: (ДМСО-d6+CCl4) 11.16 (с, 1H, OH-5), 9.25 (д, J=1.3 Гц, 1H, H2’), 8.88 (дд, J=1.3 Гц, J=4.8 Гц, 1H, H6’), 8.45 (дт, Jd=8.0 Гц, Jt=1.7 Гц, 1H, H4’), 8.10 (д, J=9.7 Гц, 1H, H4), 7.64 (дд, J=4.8 Гц, J=8.0 Гц, 1H, H5’), 6.85 (д, J=1.7 Гц, 1H, H8), 6.72 (д, J=1.7 Гц, 1H, H6), 6.32 (д, J=9.7 Гц, 1H, H3);
ИК: 1075, 1109, 1287, 1444, 1614, 1737.
Пример 4a
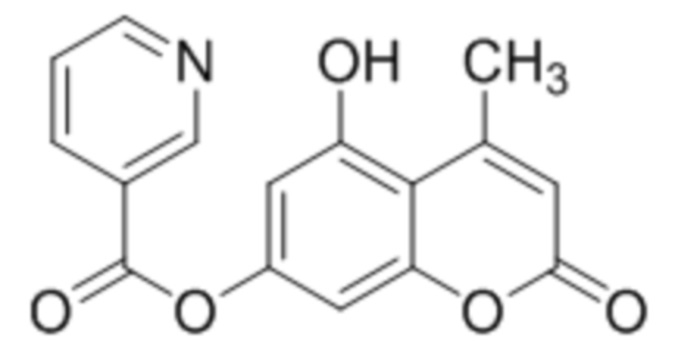
5-Гидрокси-4-метилкумарин-7-ил никотинат
К раствору 5,7-дигидрокси-4-метилкумарина (1 ммоль, 192 мг) и триэтиламина (1,1 ммоль, 111 мг) в 10 мл ацетона при перемешивании добавили 1-никотиноилбензотриазол (1,0 ммоль, 224 мг). Через сутки отфильтровали выпавший осадок 5-гидрокси-4-метилкумарин-7-ил никотината. Выход 87%. Тпл. 269-271 °С.
1Н ЯМР: (ДМСО-d6+CCl4) 10.58-11.00 (уш с, 1H, OH-5), 9.24 (м, 1H, H2’), 8.87 (м, 1H, H6’), 8.43 (м, 1H, H4’), 7.61 (м, 1H, H5’), 6.73 (с, 2H, H6, H8), 6.01 (с, 1H, H3), 2.61 (с, 3H, CH3);
ИК: 1085, 1292, 1428, 1615, 1737.
Пример 4b
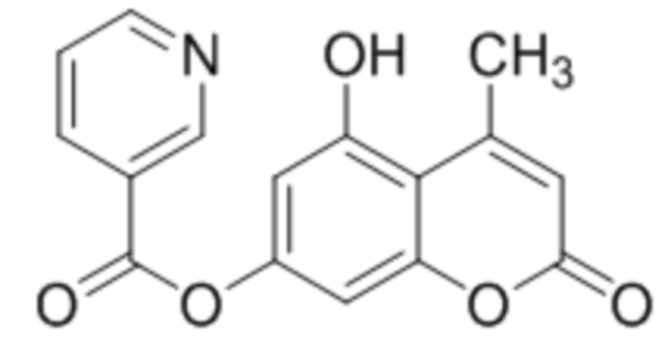
5-Гидрокси-4-метилкумарин-7-ил никотинат
(альтернативная процедура)
К раствору 5,7-дигидрокси-4-метилкумарина (1 ммоль, 192 мг) и триэтиламина (1,1 ммоль, 111 мг) в 10 мл ацетона при перемешивании добавили азид никотиноивой кислоты (1,0 ммоль, 148 мг). Через сутки отфильтровали выпавший осадок 5-гидрокси-4-метилкумарин-7-ил никотината. Выход 71%.
Пример 4c
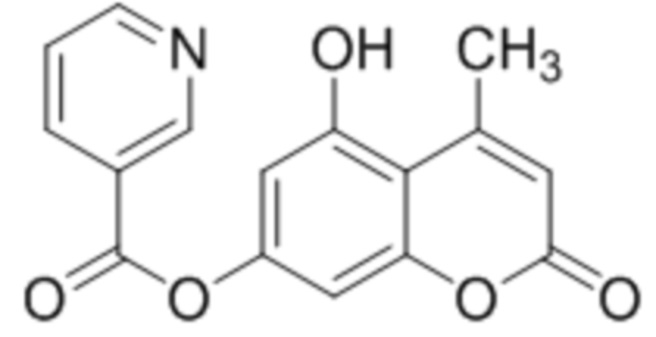
5-Гидрокси-4-метилкумарин-7-ил никотинат
(альтернативная процедура)
К раствору 5,7-дигидрокси-4-метилкумарина (1 ммоль, 192 мг) и триэтиламина (1,1 ммоль, 111 мг) в 10 мл ацетона при перемешивании добавили никотиновую кислоту (1,0 ммоль, 148 мг), DCC (1,5 ммоль, 309 мг). Через неделю отфильтровали выпавший осадок 5-гидрокси-4-метилкумарин-7-ил никотината и перекристаллизовали из спирта. Выход 60%.
Пример 5
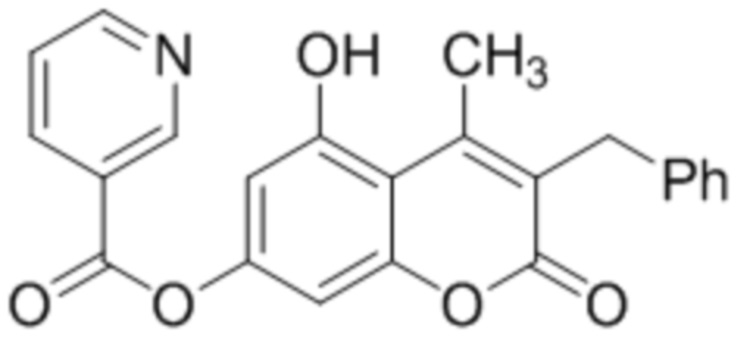
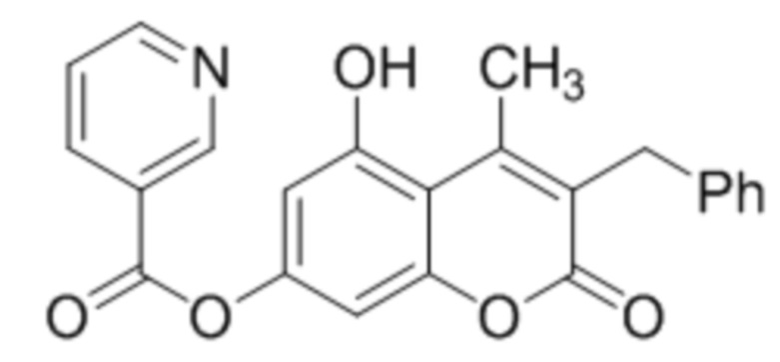
3-Бензил-5-гидрокси-4-метилкумарин-7-ил никотинат
К раствору 3-бензил-5,7-дигидрокси-4-метилкумарина (1 ммоль, 282 мг) и триэтиламина (1,1 ммоль, 111 мг) в 10 мл ацетона при перемешивании добавили 1-никотиноилбензотриазол (1,0 ммоль, 224 мг). Через сутки отфильтровали выпавший осадок 3-бензил-5-гидрокси-4-метилкумарин-7-ил никотината. Выход 69%. Тпл. 223-225 °С.
1Н ЯМР: (ДМСО-d6+CCl4) 10.80 (с, 1H, OH-7), 9.28 (уш с, 1H, H2’), 8.85 (м, 1H, H6’), 8.44 (м, 1H, H4’), 7.60 (м, 1H, H5’), 7.13–7.26 (м, 5H, Ph), 6.71 (с, 2H, H6, H8), 3.97 (с, 2H, CH2), 2.65 (с, 3H, CH3);
ИК: 1157, 1285, 1428, 1604, 1700, 1743.
Пример 6
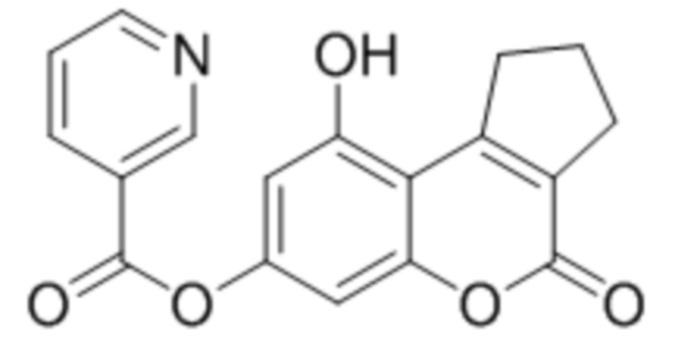
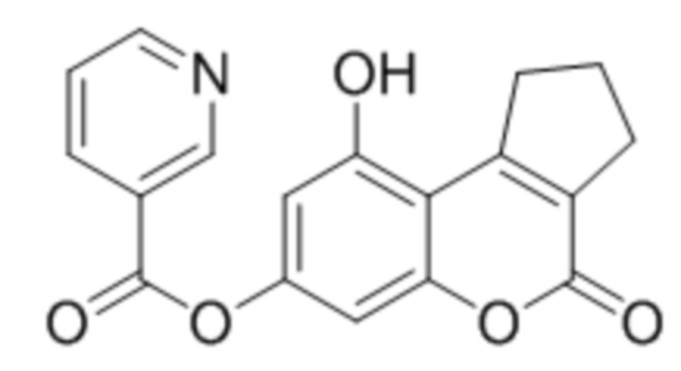
9-Гидрокси-4-оксо-2,3-дигидроциклопента[c]хромен-4(1H)-он-7-ил никотинат
К раствору 7,9-дигидрокси-2,3-дигидроциклопента[c]хромен-4(1H)-она (1 ммоль, 218 мг) и триэтиламина (1,1 ммоль, 111 мг) в 10 мл ацетона при перемешивании добавили 1-никотиноилбензотриазол (1,0 ммоль, 224 мг). Через сутки отфильтровали выпавший осадок 9-гидрокси-4-оксо-2,3-дигидроциклопента[c]хромен-4(1H)-он-7-ил никотината. Выход 84%. Тпл. 255–257 °С.
1Н ЯМР: (ДМСО-d6+CCl4) 10.71 (с, 1H, OH-5); 9.24 (уш с, 1H, H2’); 8.84 (м, 1H, H6’), 8.44 (м, 1H, H4’); 7.60 (м, 1H, H5’); 6.74 (д, J=1.8 Гц, 1H, H8); 6.66 (д, J=1.8 Гц, 1H, H6); 3.35 (т, J=7.5 Гц, 2H, CH2); 2.72 (т, J=7.5 Гц, 2H, CH2); 2.1 (p, J=7.5 Гц, 2H, CH2);
ИК: 1006, 1292, 1372, 1576, 1599, 1662, 1695.
Пример 7
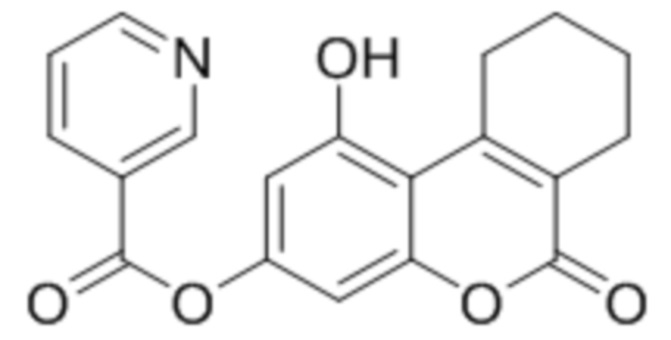
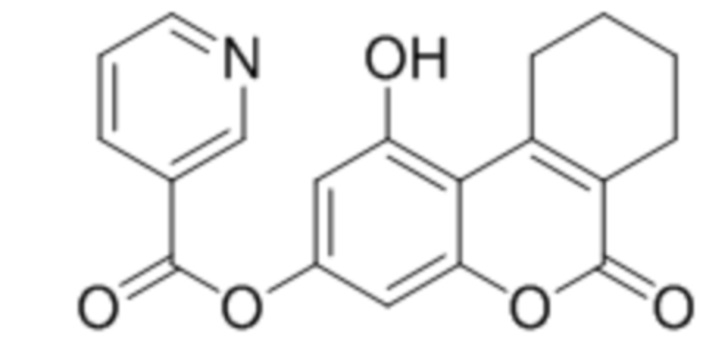
1-Гидрокси-6-оксо-7,8,9,10-тетрагидро-6H-бензо[c]хромен-3-ил никотинат
К раствору 1,3-дигидрокси-6-оксо-7,8,9,10-тетрагидро-6H-бензо[c]хромен-6-он (1 ммоль, 232 мг) и триэтиламина (1,1 ммоль, 111 мг) в 10 мл ацетона при перемешивании добавили 1-никотиноилбензотриазол (1,0 ммоль, 224 мг). Через сутки отфильтровали выпавший осадок 1-гидрокси-6-оксо-7,8,9,10-тетрагидро-6H-бензо[c]хромен-3-ил никотината. Выход 68%. Тпл. 223–225 °С.
1Н ЯМР: (ДМСО-d6+CCl4) 10.93 (уш с, 1H, OH-5); 9.23 (уш с, 1H, H2’); 8.89 (м, 1H, H6’), 8.43 (м, 1H, H4’); 7.63 (м, 1H, H5’); 6.82 (д, J=1.8 Гц, 1H, H8); 6.71 (д, J=1.8 Гц, 1H, H6); 3.09 (м, 2H, CH2); 2.39 (м, 2H, CH2); 1.67 (м, 2H, CH2+CH2);
ИК: 1080, 1277, 1292, 1614, 1703, 1733, 1742.
Пример 8
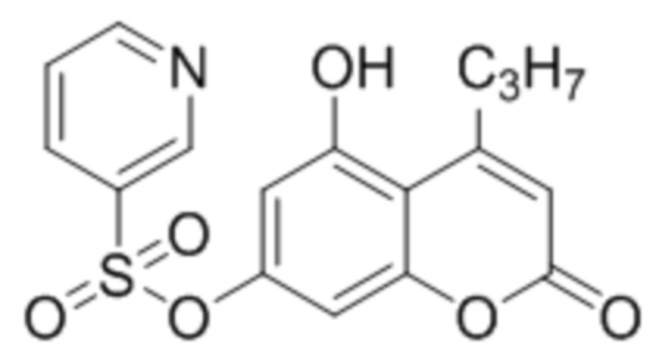
5-Гидрокси-4-пропилкумарин-7-ил пиридин-3-сульфонат
К раствору 5,7-дигидрокси-4-пропилкумарина (1 ммоль, 220 мг) и триэтиламина (1,1 ммоль, 111 мг) в 10 мл ацетона при перемешивании добавили 1-(3-пиридилсульфонил)-бензотриазол (1,0 ммоль, 260 мг). Через сутки ацетон упарили и остаток перекристаллизовали из этилацетата с получением 5-гидрокси-4-пропилкумарин-7-ил пиридин-3-сульфоната. Выход 58%.
1Н ЯМР: (400 МГц, ДМСО-d6) δ 10,99 (уш с., 1Н), 9,00 (дд, 1Н, J=2,4, 0,6 Гц), 8.94 (дд, 1Н, J=1,6, 4,9 Гц), 8.30 (ддд, 1Н, J=1,7, 2,3, 8,1 Гц), 7.71 (ддд, 1Н, 0,7, 4,9, 8,1 Гц), 6.56, (д, 1Н, J=2,4 Гц), 6.45 (д, 1Н, J=2,4 Гц), 5.97 (с, 1Н), 2.89 (т, 1Н, J=7,4 Гц), 1.63 (м, 2Н), 1.00 (т, 3Н, J=7,3 Гц).
Пример 9
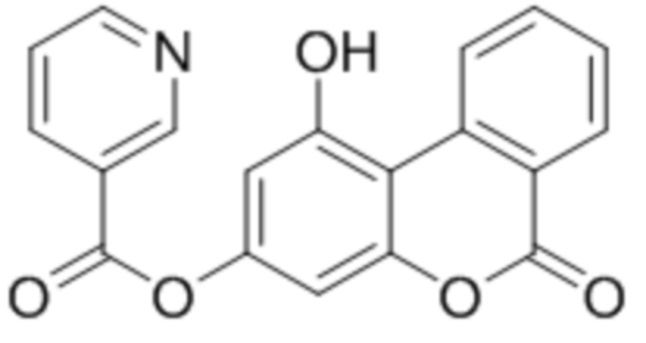
1-Гидрокси-6-оксо-6H-бензо[c]хромен-3-ил никотинат
К раствору 1,3-дигидрокси-6-оксо-6H-бензо[c]хромен-6-она (1 ммоль, 228 мг) и триэтиламина (1,1 ммоль, 111 мг) в 10 мл ацетона при перемешивании добавили 1-никотиноилбензотриазол (1,0 ммоль, 224 мг). Через сутки отфильтровали выпавший осадок 1-гидрокси-6-оксо-6H-бензо[c]хромен-3-ил никотината. Выход 87%. Тпл. >300 °С.
1Н ЯМР: (ДМСО-d6+CCl4) 11.54 (с, 1H, OH-5), 9.31–9.25 (уш с, 1H, H-2’), 9.15-9.11 (м, 1H, H-10), 8.94–8.89 (м, 1H, H-6’), 8.50–8.46 (м, 1H, H-4’), 8.30–8.26 (м, 1H, H-7), 7.96–7.92 (м, 1H, H-9), 7.69–7.62 (м, 2H, H-3’ + H-8), 6.98 (д, J=2.4 Гц, 1H, H-4), 6.90 (d, J=2.4 Гц, 1H, H-2);
ИК: 1744, 1735, 1608, 1416, 1289, 1087.
Пример 10
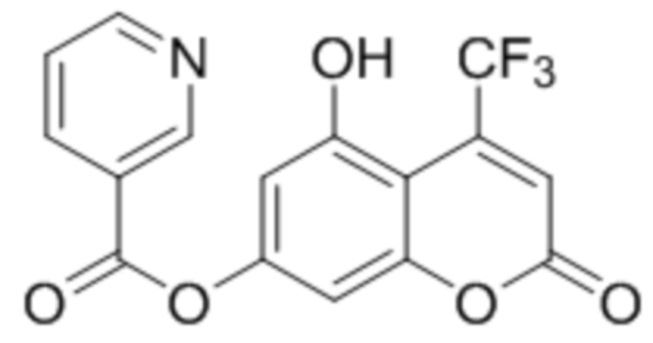
5-Гидрокси-4-трифторметилкумарин-7-ил никотинат
К раствору 5,7-дигидрокси-4-трифторметилкумарина (1 ммоль, 351 мг) и триэтиламина (1,1 ммоль, 111 мг) в 10 мл этилацетата при перемешивании добавили 1-никотиноилбензотриазол (1,0 ммоль, 224 мг). Через сутки отфильтровали выпавший осадок 5-гидрокси-4-трифторметилкумарин-7-ил никотината. Выход 65%. Тпл. 257–259 °С.
1Н ЯМР: (ДМСО-d6+CCl4) 11.61–11.44 (уш с, 1H, OH-5), 9.26 (д, J=1.8 Гц, 1H, H-2’), 8.91 (дд, J=1.8 Гц, J=4.8 Гц, 1H, H-6’), 8.47 (дт, Jd=1.8 Гц, Jt=8.0 Гц, 1H, H-4’), 7.66 (дд, J=4.8 Гц, J=8.0 Гц, 1H, H-5’), 7.02 (д, J=2.3 Гц, 1H, H-8), 6.87 (с, 1H, H-3), 6.83 (д, J=2.3 Гц, 1H, H-8);
ИК: 1738, 1614, 1402, 1200, 1285, 1084.
Пример 11
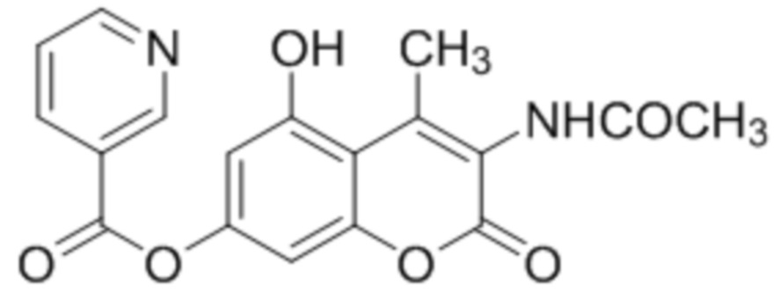
3-Ацетиламидо-5-гидрокси-4-метилкумарин-7-ил никотинат
К раствору 3-ацетиламидо-5,7-дигидрокси-4-метилкумарина (1 ммоль, 249 мг) и триэтиламина (1,1 ммоль, 111 мг) в 10 мл ацетона при перемешивании добавили 1-никотиноилбензотриазол (1,0 ммоль, 224 мг). Через сутки отфильтровали выпавший осадок 3-ацетиламидо-5-гидрокси-4-метилкумарин-7-ил никотината. Выход 68%. Тпл. > 300 °С.
1Н ЯМР: (ДМСО-d6+CCl4) 11.30–11.01 (уш с, 1H, OH-5), 9.40 (с, 1H, NH), 9.31–9.21 (уш с, 1H, H-2’), 8.91–8.86 (м, 1H, H-6’), 8.51–8.42 (м, 1H, H-4’), 7.70–7.63 (м, 1H, H-5’), 6.91 (д, J=2.1 Гц, 1H, H-8), 6.79 (д, J=2.1 Гц, 1H, H-8), 2.44 (с, 3H, CH3), 2.05 (с, 3H, CH3 (Ac));
ИК: 1741, 1732, 1716, 1670, 1614, 1292, 1088.
Пример 12
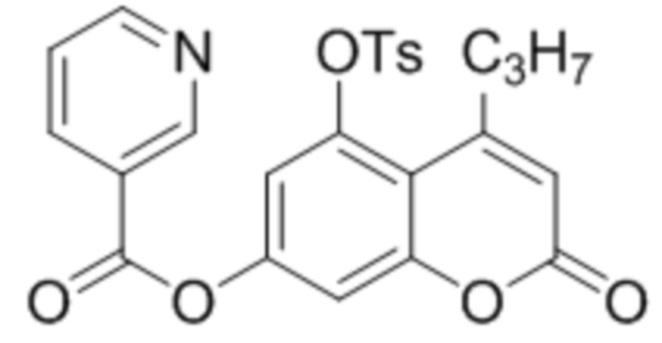
5-Тозилокси-4-пропилкумарин-7-ил никотинат
К суспензии 5-гидрокси-4-пропилкумарин-7-ил никотината (1,0 ммоль, 297 мг) в 10 дихлорметане при перемешивании при комнатной температуре добавили триэтиламин (1 ммоль, 101 мг), п-(N,N-диметиламино)пиридин (0.2 ммоль, 24 мг) и тозилхлорид (1.5 ммоль, 285 мг). Через 2 часа раствор промыли водой, высушили сульфатом натрия и упарили дихлорметан. Сырой 5-тозилокси-4-пропилкумарин-7-ил никотинат перекристаллизовали из этанола. Выход 90%.
1Н ЯМР: (ДМСО-d6+CCl4) 9.39 (дд, J=0.7 Гц, J=1.6 Гц, 1H, H2’), 8.92 (дд, J=1.6 Гц, J=4.8 Гц, 1H, H6’), 8.46 (дт, Jd=8.0 Гц, Jt=2.0 Гц, 1H, H4’), 7.85 (д, J=8.3 Гц, 2H, H2’’+H6’’ (Ts)), 7.54 (ддд, J=0.7 Гц, J=4.8 Гц, J=8.0 Гц, 1H, H5’), 7.41 (д, J=8.3 Гц, 2H, H3’’+H5’’ (Ts)), 7.23 (д, J=2.4 Гц, 1H, H6), 7.16 (д, J=2.4 Гц, 1H, H8), 6.24 (с, 1H, H3), 2.84 (м, 2H, CH2), 2.48 (с, 3H, CH3 (Ts)), 1.61 (м, 2H, CH2), 0.95 (м, 3H, CH2-CH3).
Пример 13
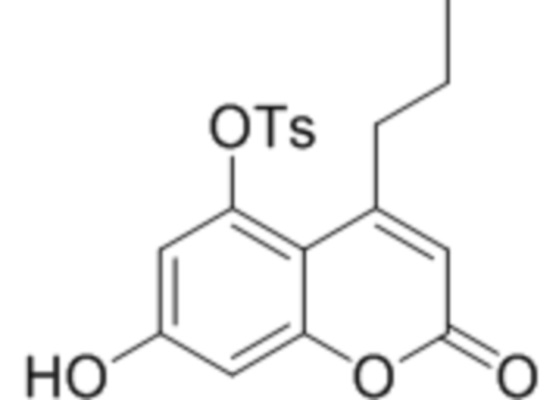
7-Гидрокси-5-тозилокси-4-метилкумарин
Кипятили 5-тозилокси-4-пропилкумарин-7-ил никотинат в 10 мл раствора соляной кислоты (20%) 5 часов. Выпавший осадок отфильтровали, промыли водой и метанолом. Выход 90%.
1Н ЯМР: (ДМСО-d6+CCl4): 10.97 (с, 1H, OH-7), 7.85 (д, J=8.2 Гц, 2H, H’o), 7.54 (д, J=8.2 Гц, 2H, H’m), 6.68 (д, J=2.4 Гц, 1H, H8), 6.52 (д, J=2.4 Гц, 1H, H6), 6.08 (с, 1H, H3), 2.69 (м, 2H, CH2), 2.44 (с, 3H, CH3’) 1.45 (м, 2H, CH2), 0.80 (м, 3H, CH3);
Пример 14
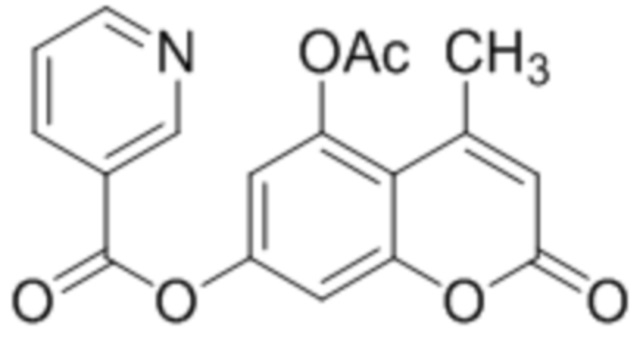
5-Ацетокси-4-метилкумарин-7-ил никотинат
К пиридину (5 мл) добавляли 5-гидрокси-4-метилкумарин-7-ил никотинат (1,0 ммоль, 297 мг), п-(N,N-диметиламино)пиридин (0.2 ммоль, 24 мг) и уксусный ангидрид (4,0 ммоль, 408 мг) и перемешиваем при комнатной температуре. Через 2 часа раствор вылили на лед. Выпавший 5-ацетилокси-4-пропилкумарин-7-ил никотинат отфильтровали и промыли водой. Выход 85%.
1Н ЯМР: (ДМСО-d6+CCl4) 9.40 (д, J=1.6 Гц, 1H, H2’), 8.91 (дд, J=1.6 Гц, J=4.8 Гц, 1H, H6’), 8.46 (дт, Jd=8.0 Гц, Jt=2.0 Гц, 1H, H4’), 7.54 (дд, J=4.8 Гц, J=8.0 Гц, 1H, H5’), 7.24 (д, J=2.4 Гц, 1H, H6), 7.04 (д, J=2.4 Гц, 1H, H8), 6.26 (д, J=1.1 Гц, H3), 2.55 (д, J=1.1 Гц, 3H, CH3), 2.41 (с, 3H, CH3).
Пример 15
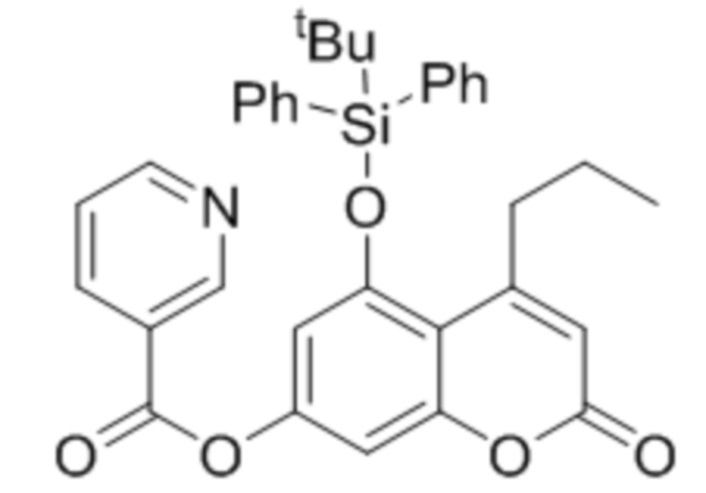
5-((трет-Бутилдифенилсилил)окси)-2-оксо-4-пропил-2Н-хромен-7-ил никотинат
К суспензии 5-гидрокси-4-пропилкумарин-7-ил никотината (195 мг, 0,6 ммоль) в CH2Cl2 (5 мл) добавляли триэтиламин (125 мкл, 0,9 ммоль) и TBDPSCl (247 мг, 0,9 ммоль). Смесь перемешивали при комнатной температуре до завершения, что контролировали с помощью ТСХ (около 5 часов). Затем смесь промывают водой, сушат над Na2SO4 и растворитель упаривали в вакууме. Остаток кристаллизовали из метанола с получением 210 мг указанного в заголовке соединения в виде бесцветных кристаллов. Выход 62%.
ИК: 1716, 1607, 1139, 1098, 1076, 812 см–1.
1H ЯМР (400 МГц, CDCl3): 9.09 (уш с, 1H, H-2′), 8.79–8.78 (м, 1H, H-6′), 8.21–8.18 (м, 1H, H-4′), 7.73–7.71 (м, 4H, Ph), 7.48–7.36 (м, 7H, H-5′ + Ph), 6.83 (д, J=2.3 Гц, 1H, H-8), 6.24 (с, 1H, H-3), 6.23 (д, J=2.3 Гц, 1H, H-6), 3.33 (т, J=7.4 Гц, 2H, C-4-CH2), 1.79 (секстет, J=7.4 Гц, 2 H, CH2CH3), 1.14 (с, 9H, tBu), 1.04 (т, J=7.4 Гц, 3H, CH2CH3).
Пример 16
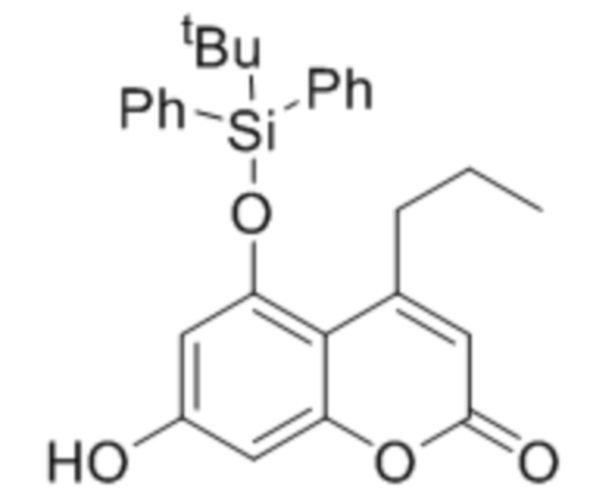
5-((трет-Бутилдифенилсилил)окси)-7-гидрокси-4-пропил-2Н-хромен-2-он
5-((трет-Бутилдифенилсилил)окси)-2-оксо-4-пропил-2Н-хромен-7-ил никотинат
(165 мг, 0,293 ммоль) растворяли в МеОН, добавляли смолу Amberlyst 16, (4,8 экв / кг) (165 мг) и реакционную смесь кипятили с обратным холодильником при перемешивании до завершения, что контролировали с помощью ТСХ (около 5 часов). Затем реакционную смесь охлаждали до комнатной температуры и отфильтровывали смолу. Фильтрат охлаждали до -15 ° C, и образовавшийся осадок отфильтровывали и сушили на воздухе с получением 76 мг указанного в заголовке соединения в виде бесцветных кристаллов. Выход 57%.
ИК: 1694, 1582, 1449, 1355, 1168, 813 см–1.
1H ЯМР (400 МГц, CDCl3): 7.73–7.71 (м, 4 H, Ph), 7.46–7.37 (м, 6 H, Ph), 6.56 (д, J=2.3 Гц, 1H, H-8), 6.48 (с, 1H, OH), 6.05 (с, 1H, H-3), 5.90 (d, J=2.3 Гц, 1H, H-6), 3.29 (т, J=7.3 Гц, 2 H, C-4-CH2), 1.76 (секстет, J=7.3 Гц, 2 H, CH2CH3), 1.11 (с, 9 H, tBu), 1.03 (т, J=7.3 Гц, 3 H, CH2CH3).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ ДЛЯ СИНТЕЗА КАЛАНОЛИДОВ И ИХ АНАЛОГОВ | 2019 |
|
RU2733731C1 |
| МЕХАНОХИМИЧЕСКИЙ СПОСОБ СИНТЕЗА КУМАРИНОВ | 2022 |
|
RU2799566C1 |
| НОВЫЕ ЗАМЕЩЕННЫЕ 8-ГЕТЕРОАРИЛКСАНТИНЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2004 |
|
RU2357969C2 |
| ПРОИЗВОДНЫЕ АРИЛСУЛЬФОНИЛАМИНОГИДРОКСАМОВОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ МАТРИЧНЫХ МЕТАЛЛОПРОТЕИНАЗ И СПОСОБ ЛЕЧЕНИЯ | 1996 |
|
RU2145597C1 |
| ПРОИЗВОДНЫЕ 3-[2-(3-АЦИЛАМИНО-2-ОКСО-2Н-ПИРИДИН-1-ИЛ)-АЦЕТИЛАМИНО]-4-ОКСО-ПЕНТАНОВОЙ КИСЛОТЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КАСПАЗЫ | 2005 |
|
RU2412936C2 |
| ПРОИЗВОДНЫЕ 6-АМИНО-МОРФИНАНА И ИХ ПРИМЕНЕНИЕ | 2002 |
|
RU2306314C2 |
| 2-ТИОЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА И ИХ ПРИМЕНЕНИЕ В ФАРМАЦЕВТИКЕ | 2003 |
|
RU2331638C2 |
| ПРОИЗВОДНЫЕ ИЗОХИНОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2006 |
|
RU2443688C2 |
| ИНГИБИТОРЫ КАСПАЗ И ИХ ПРИМЕНЕНИЕ | 2004 |
|
RU2372335C2 |
| ИНДАЗОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ FGFR, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2719428C2 |
Изобретение относится к области органического синтеза, в частности к способу селективного введения защитной группы в соединение формулы (I). Способ включает взаимодействие соединения формулы (I) с соединением формулы (II) в инертном апротонном растворителе в присутствии основания с получением соединения формулы (III). В формулах (I)-(III) R1 и R2 независимо представляют собой водород или галоген; или R1 и R2 образуют вместе с несущими их атомами углерода пироновый цикл, который замещен R3 и R4, где R3 и R4 независимо выбраны из водорода, С1-С6 алкила, С1-С6 галогеналкила, С6-С10 арил С1-С6 алкила, NHCOC1-C6 алкила; или R3 и R4 соединены вместе с образованием 5- или 6-членного карбоциклического фрагмента, необязательно содержащего одну или несколько двойных связей; Z представляет собой CO и Y представляет собой OH или –N3. Предлагаемый способ позволяет селективно вводить защитную группу в производные резорцина и 5,7-дигидроксикумарина в одну простую стадию. 8 з.п. ф-лы, 16 пр.
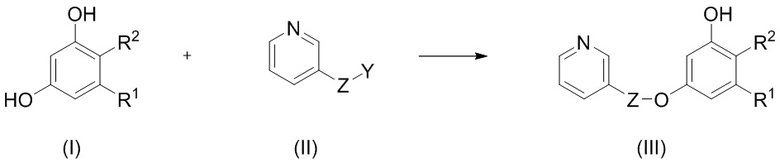
1. Способ селективного введения защитной группы в соединение формулы (I), включающий:
взаимодействие в инертном апротонном растворителе в присутствии основания соединения формулы (I) с соединением формулы (II) с получением соединения формулы (III):
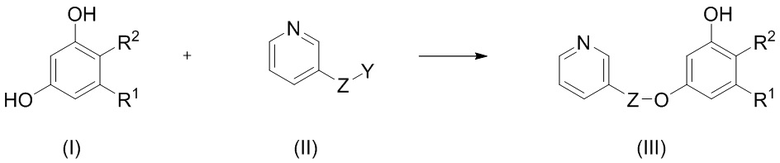 ,
,
где R1 и R2 независимо представляют собой водород или галоген;
или R1 и R2 образуют вместе с несущими их атомами углерода пироновый цикл, который замещен R3 и R4, где R3 и R4 независимо выбраны из водорода, С1-С6 алкила, С1-С6 галогеналкила, С6-С10 арил С1-С6 алкила, NHCOC1-C6 алкила; или R3 и R4 соединены вместе с образованием 5- или 6-членного карбоциклического фрагмента, необязательно содержащего одну или несколько двойных связей;
Z представляет собой CO и
Y представляет собой OH или –N3.
2. Способ по п. 1, где R1 и R2 представляют собой водород.
3. Способ по п. 1, где R1 представляет собой водород и R2 представляет собой хлор.
4. Способ по п. 1, где R1 и R2 образуют пироновый цикл, так что соединение формулы (I) описывается соединением формулы (Ia)
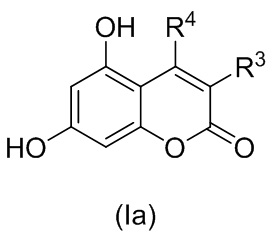 ,
,
где R3 и R4 независимо выбраны из водорода, С1-С6 алкила, С1-С6 галогеналкила, С6-С10 арил С1-С6 алкила, NHCOC1-C6 алкила; или R3 и R4 соединены вместе с образованием 5- или 6-членного карбоциклического фрагмента, необязательно содержащего одну или несколько двойных связей.
5. Способ по любому из пп. 1-4, где Y представляет собой –N3.
6. Способ по любому из пп. 1-4, где Y представляет собой OH и реакцию проводят в присутствии агента сочетания, выбранного из 1,3-дициклогексилкарбодиимида (DDC), гексафторфосфата азабензотриазол тетраметил-урония (HATU), 1-этил-3-(3-диметиламинопропил)карбодиимида (EDC) и 1,3-диизопропилкарбодиимида.
7. Способ по любому из пп. 1-6, где растворитель выбран из тетрагидрофурана, ацетона, ацетонитрила, ДМФА, этилацетата и диоксана.
8. Способ по любому из пп. 1-7, где основание выбрано из органического амина, такого как триэтиламин, N-метилморфолин, N-этилдиизопропиламин или этилендиамин.
9. Способ по любому из пп. 1-8, где взаимодействие проводят в течение от 1 до 24 ч при температуре 0-60°С, наиболее предпочтительно при температуре около 25°С.
| А.К | |||
| ИНЮТИНА И ДР | |||
| Паровоз для отопления неспекающейся каменноугольной мелочью | 1916 |
|
SU14A1 |
| Новый подход к синтезу биологически активных производных пиранокумаринов, 2-я Конференция "Современные синтетические методологии для создания лекарственных препаратов и функциональных материалов" (MOSM2018), ТЕЗИСЫ ДОКЛАДОВ, Екатеринбург, Россия, 15-17 ноября 2018, стр | |||
| Способ получения суррогата олифы | 1922 |
|
SU164A1 |
| M | |||
| SUN et al., Coumarin derivatives protect | |||

