Изобретение относится к новым соединениям, обладающим двойной активностью, а именно активностью, ингибирующей ангиотензин-конвертирующий фермент, и активностью, ингибирующей нейтральную эндопептидазу, а также к способам получения указанных соединений. Изобретение также относится к фармацевтическим соединениям, содержащим указанные соединения с двойной ингибирующей активностью или их фермацевтически приемлемые соли, и к способу использования этих композиций. Соединения с двойной ингибирующей активностью, рассматриваемые в изобретении, представляют собой соединения формулы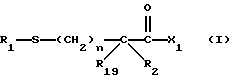
и их фармацевтически приемлемые соли,
где
R1 представляет собой водород,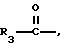
или
R18-S-;
R2 и R19 независимо выбирают из водорода, алкила, циклоалкил-(CH2)m-, замещенного алкила, арил-(CH2)m-, замещенного арил-(CH2)m-, и гетероарил-(CH2)m-;
n= 0 или 1, при условии, что n должно быть равно 0, если R2 и R19 оба не являются водородом;
m=0 или целому числу от 1 до 6;
R3 представляет собой алкил, замещенный алкил, циклоалкил-(CH2)m-, арил-(CH2)m-, замещенный арил-(CH2)m-, или гетероарил-(CH2)m-;
R18 представляет собой алкил, замещенный алкил, циклоалкил-(CH2)m-, арил-(CH2)m-, замещенный арил-(CH2)m-, гетероарил-(CH2)m-, или -S-R18 образует симметричный дисульфид, где R18 имеет формулу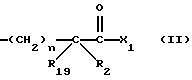
X1 имеет формулу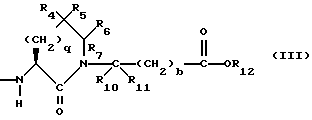
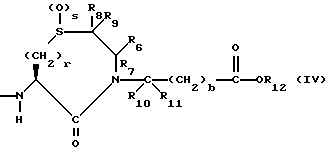
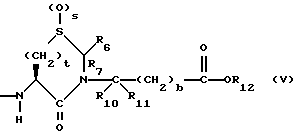


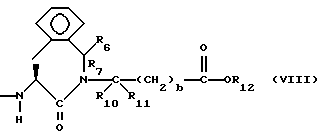
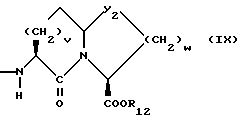
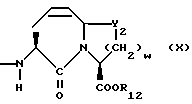
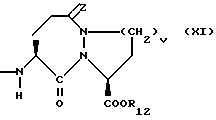
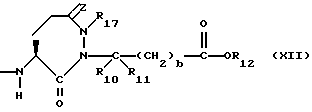
или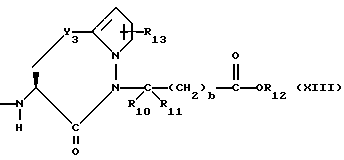
R4 представляет собой водород, алкил, замещенный алкил, алкенил, замещенный алкенил, гидрокси, циклоалкил-(CH2)m-, арил-(CH2)m-, замещенный арил-(CH2)m-, или гетероарил-(CH2)m-;
R5 представляет собой водород, алкил, замещенный алкил, замещенный алкил, алкенил, циклоалкил-(CH2)m-, арил-(CH2)m, замещенный арил-(CH2)m гидрокси, либо R4 и R5, взятые вместе с атомом углерода, с которым они связаны, образуют насыщенное циклоалкильное кольцо, имеющее 3 - 7 атомов углерода, либо R4 и R5, взятые с атомами углерода, с которым они связаны, образуют кето-заместитель, т.е.,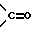
R6, R8 и R10 независимо выбирают из водорода, алкила, замещенного алкила, алкенила, замещенного алкенила, циклоалкил-(CH2)m-, арил-(CH2)m-, замещенного арил-(CH2)m-, и гетероарил-(CH2)m;
R7, R9 и R11 независимо выбирают из водорода, алкила, замещенного алкила, алкенила, замещенного алкенила, циклоалкил-(CH2)m-, арил-(CH2)m-, и замещенного арил-(CH2)m-, либо R6 и R7, взятые вместе с атомом углерода, с которым они связаны, образуют насыщенное циклоалкильное кольцо с 3 - 7 атомами углерода, либо R8 и R9, взятые вместе с атомом углерода, с которым они связаны, образуют насыщенное циклоалкильное кольцо с 3 - 7 атомами углерода;
b = 0 или 1;
q = целому числу от 1 до 4;
r = 1 или 2;
s = 0, 1 или 2;
t = 1, 2 или 3;
v = 1 или 2;
w = 1 или 2;
Y1 представляет собой: -CH2-, -(CH2)2-, -(CH2)3)-, -O-,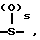
-CH2-O, или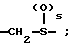
Y2 представляет собой -CH2-,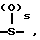
или 0;
Y3 представляет собой -CH2-, или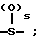
Y4 представляет собой -CH2-, -(CH2)2-, или -(CH2)3, -O- или -CH2-O;
Z представляет собой O или два атома водорода;
R12 представляет собой водород, алкил, замещенный алкил, арил-(CH2)m-, замещенный арил-(CH2)m-, гетероарил-(CH2)m-,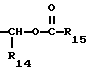
или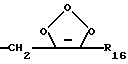
R13 представляет собой водород, низший алкил, или замещенный низший алкил;
R14 представляет собой водород, низший алкил, циклоалкил, или фенил;
R15 представляет собой водород, низший алкил, низший алкокси или фенил;
R16 представляет собой низший алкил, или арил-(CH2)m-; и
R17 представляет собой водород, алкил, замещенный алкил, алкенил, замещенный алкенил, циклоалкил -(CH2)m-, арил-(CH2)m-, замещенный арил-(CH2)m-, или гетероарил-(CH2)m-.
Термин "алкил" относится к радикалам с прямой или разветвленной цепью, имеющим до 7 атомов углерода. Термин "низший" алкил относится к радикалам с прямой или разветвленной цепью, имеющим до 4 атомов углерода и являющимся предпочтительной подгруппой, подпадающей под определение термина "алкил".
Термин "замещенный алкил" относится к таким прямым или разветвленным радикалам с 1 - 7 атомами углерода, в которых один или несколько, а предпочтительно один, два или три атома водорода замещены гидроксида, амино, галогеном, трифторметилом, циано, -NH(низшим алкилом), -N(низшим алкилом)2, низшей алкоксигруппой, низшей алкилтио-группой, или карбокси-группой.
Термин "замещенный низший алкил" относится к таким прямым или разветвленным радикалам с 1 - 4 атомами углерода, в которых один атом водорода замещен гидрокси, амино, галогеном, трифторометилом, циано, -NH(низшим алкилом), -N(низшим алкилом)2, низшей алкокси, низшей алкилтио, или карбокси.
Термин "алкенил" относится к прямым или разветвленным радикалам с 3 - 7 атомами углерода, имеющим одну или две двойные связи. Предпочтительными "алкильными" группами являются радикалы с прямой цепью, имеющие 3 - 5 атомов углерода и одну двойную связь.
Термин "замещенный алкенил" относится к прямым или разветвленным радикалам с 3 - 7 атомами углерода, которые имеют 1 или 2 двойные связи, и в которых водород замещен гидрокси, амино, галогеном, трифторметилом, циано, -NH(низший алкилом), -N-(низшим алкилом)2, низшей алкокси, низшей алкилтио, или карбокси.
Термины "низшая алкокси" и "низшая алкилтио" относятся к низшим алкильным группам, определенным выше, и связанным с атомом кислорода или серы.
Термин "циклоалкил" относится к насыщенным кольцам с 3 - 7 атомами углерода.
Термин "галоген" относится к хлору, брому, фтору или йоду.
Термин "арил" относится к фенилу, 1-нафтилу и 2-нафтилу. Термин "замещенный арил" относится к фенилу, 1-нафтилу, 2-нафтилу, которые имеют заместителя, выбранного из низшего алкила, низшей алкокси, низшей алкилтио, галогена, гидрокси, трифторметила, амино, -NH(низшего алкила), или -N(низшего алкила)2, ди- и три-замещенного фенила, 1-нафтила или 2-нафтила, где указанных заместителей выбирают из метила, метокси, галогена, гидрокси, и амино.
Термин "гетероарил" относится к незамещенным кольцам с 5 - 6 атомами, содержащим один или два атома O и S и/или 1 - 4 атома N, при условии, что полное число гетероатомов в кольце составляет 4 или менее. Гетероарильное кольцо присоединяется посредством соответствующего атома углерода или азота. Предпочтительными гетероарильными группами являются 2-, 3-, или 4-пиридил, 4-имидазолил, 2- и 3-тиенил, и 2- и 3-фурил. Термин гетероарил также включает в себя бициклическое кольцо с 5 и 6 членами, которое содержит атомы O, S и N, как указано выше, и которое является сплавленным с бензольным или пиридильным кольцом. Предпочтительными бициклическими кольцами являются 2- и 3-индолил и 4- и 5-хинолинил. Моно-или бициклическое гетероарильное кольцо может быть также дополнительно замещены в соответствующем атоме углерода низшим алкилом, галогеном, гидрокси, бензилом, или циклогексилметилом. Кроме того, если моно- или бициклическое кольцо имеет соответствующий атом N, то этот атом также может быть замещенным N-защитной группой, такой как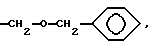
2,4-динитрофенил, низший алкил, бензил, или бензгидрил.
Соединения настоящего изобретения, в которых R1 является водородом или
а R19 является водородом, могут быть получены с помощью реакции взаимодействия ацилмеркапто-содержащей боковой цепи формулы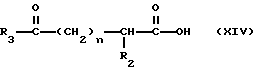
с промежуточным соединением формулы
H-X1 (XV)
в результате чего получают продукт формулы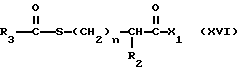
где R12 в определении X1 является предпочтительно легко удаляемой сложноэфирной защитной группой, такой как метил, этил или бензил.
Вышеуказанная реакция может быть осуществлена в органическом растворителе, таком как диметилформамид, и в присутствии сочетающегося реагента, такого как бензотриазол-1-илокситрис диметиламино фосфония гексафторофосфат, дициклогексилкарбодиимид, или карбонилдиимидазо. Альтернативно, ацилмеркаптокарбоновая кислота формулы XIV может быть превращена в активированную форму до проведения реакции сочетания, например, в такую как хлорангидрид, смешанный ангидрид, симметричный ангидрид, активированный сложный эфир и т. п.
Продукт формулы XVI может быть превращен в меркаптановый продукт формулы I, где R1 является водородом, и R12 является водородом, стандартными способами. Напротив, если R3 является метилом, а R12 является метилом или этилом, то после обработки метаноловым гидроксидом натрия получают продукт, в котором R1 и R12 являются водородом, а если R3 является метилом, а R12 является т-бутилом, то после обработки трифторуксусной кислотой, а затем аммиаком, получают продукт, в котором R1 и R12 являются водородом.
Соединения изобретения, в которых R2 и R19 оба не являются водородом, а n = 0, могут быть получены с помощью реакции сочетания боковой цепи, содержащей замещенную меркаптогруппу и имеющей формулу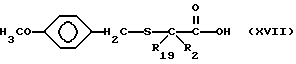
с промежуточным соединением формулы XV, описанной выше, в результате чего получают соединение формулы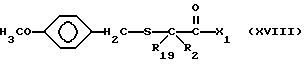
С помощью обработки соединения формулы XVIII сильной кислотой, такой как трифторометансульфоновая кислота, удаляют метоксибензильную защитную группу, и получают соответствующий продукт формулы I, где R1 является водородом.
Соединения, содержащие замещенную меркапто-группу и имеющие формулу XVII, могут быть получены с помощью реакции дизамещенной карбоновой кислоты формулы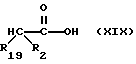
с бис[[(4-метокси)фенил]метил]дисульфидом в присутствии диизопропиламида лития.
Продукты формулы I, где X1 содержит сульфоксид или сульфон, могут быть получены с использованием в качестве меркаптана промежуточного соединения формулы XV, т. е. s = 0, в процессе реакции сочетания. Полученный продукт, имеющий формулу XVI или XVIII, затем окисляют с использованием известного окисляющего реагента, такого как мета-хлоропербензойная кислота, перуксусная кислота, или монопероксифталевая кислота, гексагидрат соли магния, и т.п. Путем регулирования количества окисляющего реагента, и времени протекания реакции получают продукты, где s = 1 или 2.
Продукты формулы I, где R1 является водородом, могут быть ацилированы с использованием ацилгалида или ангидрида формулы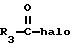
или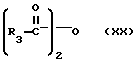
где галогеном является Cl или Br, в результате чего получают другие продукты формулы I, где R1 является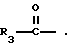
Продукты формулы I, где R1 представляют собой - S - R18, а R18 представляет собой алкил, замещенный алкил, циклоалкил - (CH2)m-, арил-(CH2)m-, замещенный арил (CH2)m - или гетероарил- (CH2)m-, могут быть получены с помощью реакции продуктов формулы I, где R1 является водородом, с сульфониловым соединением формулы.
H3C-SO2-S-R18 (XXI)
в водно-спиртовом растворителе, в результате которой получают целевые продукты. Соединения формулы XXI являются известными соединениями, или они могут быть получены известными способами (Smith и др., Biochemistry, 14, стр. 766-771 (1975)).
Симметричные дисульфидные продукты формулы I могут быть получены путем прямого окисления продукта формулы I, где R1 является водородом, с использованием иода (Ondetti и др., в патенте США 4105776).
Сложноэфирные продукты формулы I, где R12 представляет собой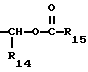
или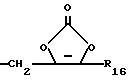
могут быть получены путем обработки продукта формулы I, где R12 является водородом, соединением формулы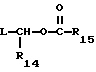
или
где L - является уходящей группой, такой как хлоро, бромо, или толилсульфонилокси.
Известны ацилмеркаптоалкановые кислоты формулы XIV (Ondetti и др. патенты США 4105776 и 4339600, Haslanger и др., патент США 4801609, и др.).
Промежуточные соединения формулы XV также описаны в литературе, либо они могут быть получены с помощью модификаций известных процедур. Например, промежуточные соединения формулы XV, где X1 является таким как он был определен в формуле III раскрывается Thorsett и др., J.Med.Chem. 29, с.251-260 (1988), Harris и др., в патенте США 4587050, 4587238, 4629787 и Yanagisawa и др., в патенте США 4734410. Промежуточные соединения формулы XV, где X1 является таким как он был определен в формуле IV, раскрываются Yanagisawa и др., в J. Med. Chem. , 30, с.1984-1991 (1987) и 31, с.422-428 (1988), Karanewsky в патенте США 4460579, Chevng и др., в патенте США 4594341, и Yanagisawa и др., в патенте США 4699905. Промежуточные соединения формулы XV, где X1 является таким как он был определен в формуле V, раскрываются Karanewsky в патентах США 4460579 и 4711884. Промежуточные соединения формулы XV, где X1 являются таким, как он был определен в формуле VI, а Y1 является -CH2-, -(CH2)2-, или -(CH2)3-, раскрываются Watthey и др., J. Med.Chem., 28, с.1511-1516 (1985) и Watthey в патентах США 4410520, 4470988, 4473575, 4537885 и 4575503, а также Parsons и др., в Biochemical and Biophysical Research Comm., 117 с. 108-113 (1983) и в патенте США 4873235. Промежуточные соединения формулы XV, где X1 является таким, как он определен в формуле VI, а Y1 является S или O, раскрываются Slade и др., в J.Med.Chem., 28, c.1517-1521 (1985) и в патенте США 4477464, и Itoh и др., в Chem.Pharm.Bull., 34, c.1128-1147 (1986) и 34, с. 2078-2089 (1986), а также Sugihara и др., в патенте США 4548932 (Y=0) и Katakami и др., в патенте США 4539150 (Y=S). Промежуточные соединения формулы XV, где X является таким, как он был определен в формуле VII, могут быть получены путем восстановления соответствующих промежуточных соединений, в которых X1 является таким, как он был определен в формуле VI. Промежуточные соединения формулы XV, где X1 является таким, как он был определен в формуле VIII, раскрываются Flynn и др., в патенте США 4973585. Промежуточные соединения формулы XV, где X определен в формуле IX, а Y2 является S, -SO или -SO2, раскрываются Harris и др., и Patchett и др., в патенте США 4415496 и 4617301. Промежуточные соединения формулы XV, где X1 является таким, как он был определен в формуле IX, а Y2 является CH2, раскрываются Thorsett в Actual. Chim. Ther. , 13, с.257-268 (1986). Промежуточные соединения формулы XV, где X1 является таким, как он был определен в формуле XI, раскрываются Attwood и др., в Federation of European Biochemical Studies, 165, с.201-206 (1984) и в патенте США 4512994, и Nataff и др., в Drugs of the Future, 12, с. 475-483 (1987). Промежуточные соединения формулы XV, где X1 является таким, как он был определен в формуле XII, раскрываются Huang и др., в патенте США 4465679. Промежуточные соединения формулы XV, где X1 является таким, как он был определен в формуле XIII, раскрываются Bolos и др., в Tetrahedron 48, с.9567-9576 (1992).
Промежуточные соединения формулы XV, где X1 является таким, как он был определен в формуле III, g = 1 или 2, а R17 является водородом, могут быть получены описанным ниже способом, который также является частью изобретения.
N-фталимидо- α -аминокислоту формулы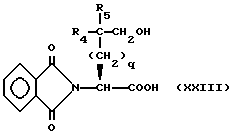
и сложный эфир аминокислоты формулы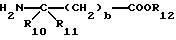
где R12 представляет собой легко удаляемую сложноэфирную защитную группу, такую как метил, этил или бензил, подвергают реакции сочетания и получают соединение формулы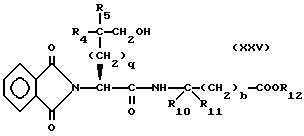
Эту реакцию сочетания предпочтительно осуществляют в присутствии связывающего реагента, такого как бензотриазол-1-ил-окситрис диметиламино фосфония гексафторофосфат или этил-3-(3-диметил-амино)пропилкарбодиимид.
Соединение формулы XXV подвергают окислению, например путем обработки оксалилхлоридом и диметилсульфоксидом или тетра-н-пропиламмония перрутенатом и N-метилморфолина N-оксидом, в результате чего получают соединение формулы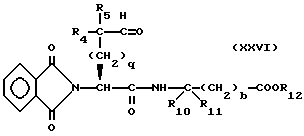
Соединение формулы XXVI подвергают циклизации путем обработки безводной кислотой, такой как трифторуксусная кислота, трифторометансульфоновая кислота, соляная кислота, и т.п., в результате чего получают соединение формулы
Промежуточное соединение формулы XV, где X1 является таким, как он был определен в формуле III, g = 1 или 2, а R6 и R7 оба являются водородом, могут быть получены путем обработки соединения формулы XXVII силилгидридом, таким как триэтилсилан, дифенилметилсилан, и т.п., и катализатором, таким как кислота Льюиса, например, хлористое олово, тетрахлорид титана, бромистое олово, этерат трехфтористого бора, и т. п., с последующей этерификацией карбоксильной группы, а затем обработкой гидратом гидразина для удаления N-фталоильной защитной группы.
Промежуточное соединение формулы XV, где X1 является таким, как он был определен в формуле III, g = 1 или 2, R7 является водородом, а R6 не является водородом, может быть получено из соединения формулы XXVII. Например, если R6 является алкенилом или замещенным алкенилом с 3-7 атомами углерода, то нужное промежуточное соединение получают путем обработки соединения XXVII алкил- или замещенным алкенилсиланом в присутствии кислоты Льюиса в качестве катализатора, примеры которой указаны выше, с последующей этерификацией карбонильной группы, а затем удалением N-фталоильной защитной группы. Алкенильная часть может быть подвергнута гидрогенизации с получением нужного промежуточного соединения, в котором R6 является алкилом или замещенным алкилом с 3-7 атомами углерода. Если R6 не является алкенилом или замещенным алкенилом, то нужное промежуточное соединение получают путем обработки соединения XXVII алюминиевым или титановым соединением, содержащим соответствующий R6 в присутствии кислоты Льюиса с последующей этерификацией карбоксильной группы, а затем удалением N-фталоильной защитной группы.
Промежуточное соединение формулы XV, где X1 является таким, как он был определен в формуле IX или X, Y2 является -CH2-, а v=2, может быть получено описанным ниже способом, который также является частью изобретения.
N-фталимидо α- аминокислоту формулы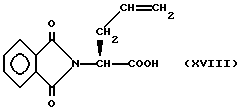
подвергают реакции сочетания со сложным эфиром α- аминокислоты формулы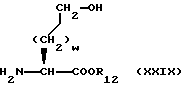
в присутствии катализатора реакции сочетания, описанного выше, в результате чего получают спирт формулы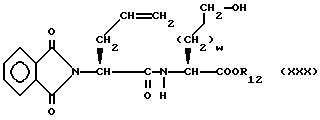
Спирт формулы XXX подвергают окислению, например, путем обработки оксалилхлоридом и получают соответствующий альдегид, который затем обрабатывают кислотой, такой как трифторуксусная кислота или соляная кислота, в результате чего получают сложный эфир карбоновой кислоты формулы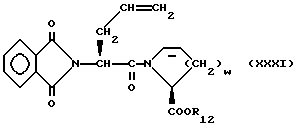
После циклизации соединения формулы XXXI сильной кислотой, такой как трифторметансульфоновая кислота, с последующей перекристаллизацией, осуществляемой традиционным способом, и обработкой иодидом натрия получают смесь соединений формул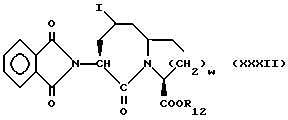

После удаления N-нафталоильной защитной группы из промежуточного соединения формулы XXXIII, как описано выше, получают нужное соединение формулы XV, где X1 является таким, как он был определен в формуле X, а Y2 является -CH2-.
Обработка промежуточного соединения формулы XXXII трис (триметилсилил)силаном или три-н-бутилолова гидридом в присутствии каталитического количества азобисизобутиронитрила способствует иодо-группы. Затем, как описано выше, удаляют N-фталоильную группу, в результате чего получают нужное соединение формулы XV, где X1 является таким, как он был определен в формуле IX, v=2, а Y2 является -CH2-. Аналогичная процедура может быть использована для получения соответствующего соединения формулы XV, где X1 является таким, как он был определен в формуле IX, v=1, а Y2 является -CH2-.
Другие процедуры, используемые для получения промежуточных соединений формулы XV, описаны в примерах.
Соединения формулы I содержат один или несколько асимметричных центров. Таким образом, эти соединения могут существовать в диастереизомерных формах или в виде их смесей, причем все указанные формы входят в объем изобретения. В описанных выше способах, в качестве исходных материалов могут быть использованы рацематы, энантиомеры, или диастереомеры. Если нужно получить диастереизомерные соединения, то они могут быть разделены традиционными методами, например, путем хроматографии или фракционированной кристаллизации.
Соединения формулы I, где R12 является водородом, могут быть получены в виде фармацевтически приемлемой соли. Подходящими для этих целей солями являются соли щелочных металлов, таких как натрий и калий, соли щелочноземельных металлов, таких как кальций или магний, и соли, происходящие от аминокислот, таких как аргинин, лизин и т.п. Эти соли могут быть получены с помощью реакции кислотной формы соединения с эквивалентом основания, отдающего нужный ион в среде, в которой осаждается соль, или в водной среде, с последующей лиофилизацией.
Соединения формулы I представляют собой ингибиторы двойного действия, обладающие способностью ингибировать ангиотензинконвертирующий фермент и нейтральную эндопептидазу. Поэтому соединения формулы I, включая их фармацевтически приемлемые соли, могут быть использованы для лечения физиологических состояний, для которых показано применение либо ингибиторов ангиотензин-трансформирующего фермента, либо ингибиторов нейтральной эндопептидазы. Такими физиологическими состояниями являются сердечно-сосудистые заболевания, например, гипертензин, застойная сердечная недостаточность, почечная недостаточность, цирроз печени, а также аналгезическая активность. Диурез, натрийурез и снижение кровяного давления могут быть стимулированы путем введения млекопитающему, такому как человек, около 1-100 мг на кг веса в день, а предпочтительно около 1-50 мг на кг веса тела в день одного или нескольких ингибиторов двойного действия формулы I, или их фармацевтически приемлемой соли. Ингибирующие соединения с двойным действием формулы I предпочтительно использовать для перорального введения, однако могут быть также использованы и парентеральные способы введения, например, такие как подкожное, внутримышечное и внутривенное введение. Ежедневная доза может быть введена в виде однократной дозы, либо в виде разделенной дозы, рассчитанной на 2-4 приема в день.
Ингибиторы двойного действия формулы I могут быть введены в сочетании с человеческим ANF 99-126. Такая комбинация может содержать ингибитор формулы I в количестве около 1-100 мг веса тела и человеческий ANF 99-126 в количестве около 0,001-0,1 мг на кг веса тела.
Ингибиторы двойного действия формулы I могут быть введены в сочетании с другими классами фармацевтически активных соединений, например, таких как ингибитор кальциевого канала; активатор калиевого канала, средство, снижающее уровень холестерина, и т.п.
Ингибиторы двойного действия формулы I или их фармацевтически приемлемые соли, и другие фармацевтически приемлемые ингредиенты могут быть использованы для составления фармацевтических композиций, предназначенных для вышеуказанных целей. Подходящими композициями для перорального введения могут служить таблетки, капсулы, и эликсиры; а подходящими композициями для парентерального введения могут служить стерильные растворы и суспензии. В соответствии со стандартной фармацевтической практикой, разовая лекарственная форма может содержать около 10-500 мг активного ингредиента в сочетании с физиологически приемлемым наполнением, носителем, связующим, консервантом, стабилизатором, ароматизатором, и т.п.
Предпочтительными соединениями настоящего изобретения в отношении меркапто-содержащей части боковой цепи, имеющей формулу
являются соединения,
где
R1 представляет собой водород,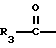
или
R18 -S-,
R3 представляет собой низший алкил с 1-4 атомами углерода, или фенил;
R18 представляет собой низший алкил с 1-4 атомами углерода;
n=0 или 1;
R2 представляет собой арил-CH2-, замещенный арил-CH2, гетероарил-CH2-, или прямой или разветвленный алкил с 1-7 атомами углерода;
R19 представляет собой водород.
Наиболее предпочтительным являются вышеуказанные меркапто-содержащие побочные цепи, в которых R1 представляет собой водород, или
а особенно водород, n = 0, и R2 представляет бензил, 2-тиенил метил, или алкил с прямой или разветвленной цепью, имеющий 1-5 атомов углерода, а более предпочтительно, бензил.
Предпочтительными соединениями настоящего изобретения X1 являются соединения формулы III, в которых g = 1 или 2, R4 представляет собой водород, метил, или фенил, R5 представляет собой водород, R6 и R7 оба являются водородом, либо оба являются метилом, или R6 представляет собой низший алкил с 1-4 атомами углерода, монозамещенный низший алкил с 1-4 атомами углерода, или алкенил с 3-5 атомами углерода, имеющий одну двойную связь, а R7 представляет собой водород, либо R6 и R7, взятые с атомом углерода, с которым они связаны, образуют циклоалкил с 3-5 атомами углерода, R10 и R11 оба являются водородом, либо одним из них является водородом, а другой является низшим алкилом с 1-4 атомами углерода, b=0 или 1, R12 является водородом или низшим алкилом с 1-4 атомами углерода.
Наиболее предпочтительными из вышеуказанных соединений формулы III являются такие соединения, в которых q = 2, R4 и R5 оба являются водородом, R6 и R7 оба являются метилом, либо R6 является пропилом, аллилом, или 2-гидроксиэтилом, а R7 является водородом, b = 0, R10 и R11 оба являются водородом, либо один из них является водородом, а другой является метилом, R12 является водородом.
Предпочтительными соединениями настоящего изобретения в отношении X1 являются соединения формулы IV, в которых v = 1, S = 0, R8 является водородом, фенилом, или низшим алкилом с 1 - 4 атомами углерода, R9 является водородом , R6 и R7 оба являются водородом, или оба являются метилом, либо R6 является низшим алкилом с 1-4 атомами углерода, или фенилом, а R7 является водородом, R10 и R11 оба являются водородом, либо один из них является водородом, а другой низшим алкилом с 1-4 атомами углерода, b = 0, R12 является водородом или низшим алкилом с 1-4 атомами углерода.
Наиболее предпочтительными из вышеуказанных соединений формулы IV являются соединения, в которых R8 является водородом или фенилом, R6 и R7 оба являются водородом, либо R6 является фенилом, а R7 является водородом, R10, R11, и R12 все являются водородом;
Предпочтительными соединениями изобретения в отношении X1 являются соединения формулы V, в которых S = 0, t = 1 или 2, R6 и R7 оба являются водородом, либо оба являются метилом, или R6 является низшим алкилом с 1-4 атомами углерода, или фенилом, а R7 является водородом, R10 и R11 оба являются водородом, либо один из них является водородом, а другой является низшим алкилом с 1-4 атомами углерода, b = 0, R12 является водородом или низшим алкилом с 1-4 атомами углерода.
Наиболее предпочтительными из вышеуказанных соединений формулы V являются соединения, в которых t = 2, R6 является фенилом, а R7 является водородом, R10, R11 и R12, все являются водородом.
Предпочтительными соединениями изобретения в отношении X1 являются соединения формулы VI, в которых Y1 является O, S или CH2, R6 и R7 оба являются водородом, либо R6 является низшим алкилом с 1-4 атомами углерода, а R7 является водородом, R10 и R11 оба являются водородом, либо один из них является водородом, а другой является низшим алкилом с 1-4 атомами углерода, b = 0, R12 является водородом или низшим алкилом с 1-4 атомами углерода.
Наиболее предпочтительными из вышеуказанных соединений формулы VI являются соединения, в которых Y1 является CH2, R6 и R7 оба являются водородом, R10, R11 и R12 все являются водородом.
Предпочтительными соединениями изобретения в отношении X1 являются соединения формулы VII, в которых Y4 является -CH2-, R6 и R7 оба являются водородом, R10 и R11 оба являются водородом, либо один из них является водородом, а другой является низшим алкилом с 1-4 атомами углерода, но особенно предпочтительно, когда оба радикала являются водородом, b = 0, R12 является водородом, или низшим алкилом с 1-4 атомами углерода, но особенно предпочтительно, если R12 является водородом.
Предпочтительными соединениями изобретения в отношении X1 являются соединения формулы VIII, в которых R6 и R7 оба являются водородом, либо один из них является водородом, а другой является низшим алкилом с 1-4 атомами углерода, R10 и R11 оба являются водородом, либо один из них является водородом, а другой является низшим алкилом с 1-4 атомами углерода, а особенно предпочтительно, если оба являются водородом, b = 0, R12 является водородом или низшим алкилом с 1-4 атомами углерода, а особенно предпочтительно, если R12 является водородом.
Предпочтительными соединениями изобретения в отношении X1 являются соединения формулы IX, где v = 1 или 2, а предпочтительно 2, w = 1 или 2, Y2 является S или CH2, R12 является водородом или низшим алкилом с 1-4 атомами углерода, особенно предпочтительно, если R12 является водородом.
Предпочтительными соединениями изобретения в отношении X1 являются соединения формулы X, в которых Y2 является CH2, w = 1 или 2, а предпочтительно 2, R12 является водородом или низшим алкилом с 1-4 атомами углерода, а особенно предпочтительно, если R12 является водородом.
Предпочтительными соединениями изобретения в отношении X1 являются соединения формулы XI, в которых v = 1 или 2, а предпочтительно 2, z является 0, или двумя атомами водорода, а предпочтительно двумя атомами водорода, R12 является водородом или низшим алкилом с 1-4 атомами углерода, а предпочтительно водородом.
Предпочтительными соединениями изобретения в отношении X1 являются соединения формулы XII, в которых z является 0, или двумя атомами водорода, а предпочтительно 0, R17 является низшим алкилом с 1-4 атомами углерода, а предпочтительно метилом, R10 и R11 оба являются водородом, либо один из них является водородом, а другой низшим алкилом с 1-4 атомами углерода, а предпочтительно, если оба являются водородом, b = 0, R12 является водородом или низшим алкилом с 1-4 атомами углерода, а предпочтительно, водородом.
Предпочтительными соединениями изобретения в отношении X1 являются соединения формулы XIII, в которых Y3 является CH2 или S, а предпочтительно CH2, R13 является водородом, R10 и R11 оба являются водородом, либо один из них является водородом, а другой является низшим алкилом с 1-4 атомами углерода, а предпочтительно, если оба являются водородом, b = 0, R12 является водородом или низшим алкилом с 1-4 атомами углерода, а предпочтительно, водородом.
Пример 1.
[3R{S*)]-3,4-Дигидро-3-[[2-(меркаптометил)-1-оксо-3- фенил-пропил]амино] -4-оксо-1,5-бензотиазепин-5-2H-уксусная кислота.
a) (S)-2-[(Ацетилтио)метил]бензолпропановая кислота, эфедриновая соль.
Раствор (1R, 2S)-(-)-эфедрина (17,3 г, 105 мМ) в диэтиловом эфире (175 мл) добавляли одной порцией целиком к раствору 2-[(ацетилтио)метил]бензолпропановой кислоты (50,0 г, 210 мМ) в диэтиловом эфире (175 мл). После выдерживания полученного раствора в течение 16 ч при комнатной температуре, кристаллизованную эфедриновую соль собирали путем фильтрации (19,7 г), т.пл. 114 - 125oC, [α]D = -40,6o (c = 1, метанол). После выдерживания в течение 20 ч при комнатной температуре, из фильтрата выделяли дополнительное количество твердого вещества (8,9 г), т.пл. 121 - 126oC, [α]D = 57,2o (c = 1, метанол). Затем твердые вещества объединяли и перекристаллизовывали из ацетонитрила (1500 мл). После выдерживания в течение 16 ч при комнатной температуре, собирали 20,8 г твердого продукта, т.пл. 125 - 130oC, [α]D = -47,5o (c = 1, метанол). Этот продукт перекристаллизовывали аналогичным способом из ацетонитрила (300 мл), в результате чего получали 18,74 г продукта, т.пл. 128 - 130oC, [α]D = -48,9o (c = 1, метанол). После третьей перекристаллизации из ацетонитрила (225 мл) получали 17,4 г твердой (S)-2-[(ацетилтио)метил]бензопропионовой кислоты эфедриновой соли, т.пл. 128 - 129oC, [α]D = 150,1o (c = 1, метанол).
Элементный анализ для C12H14O3S • C10H15NO:
Вычислено,%: C 65,48; H 7,24; N 3,47; S 7,95;
Найдено,%: C 65,46; H 7,34; N 3,21; S 8,00.
b) [3R(S*)]-3,4-Дигидро-3-[[2-[(ацетилтио)метил]-1- оксо-3-фенилпропил] амино-4-оксо-1,5-бензотиазепин-5(2H)-уксусная кислота, этиловый сложный эфир.
(S)-2-[(Ацетилтио)метил] бензолпропановой кислоты эфедриновую соль (3,34 мМ, 1,01 экв.) суспендировали в этилацетате (66 мл), промывали разбавленной HCl (65 мл воды + 4,7 мл 1,0 н. соляной кислоты, а затем 31 мл воды + 1,6 мл 1,0 н. соляной кислоты), солевым раствором (10 мл), осушали безводным сульфатом магния, фильтровали, и выпаривали досуха. Бесцветный сироп осушали в вакууме в течение 1,0 ч с получением количественного выхода в виде свободной кислоты (836 мг).
Свободную кислоту растворяли в безводном диметилформамиде (19 мл), охлаждали до 0oC (ледяная/солевая баня), обрабатывали 1-гидроксибензотриазолгидратом (470 мг, 3,48 мМ) и 1-этил-3-(3-диметиламинопропил)карбодиимидом (690 мг, 3,60 мМ), а затем размешивали в течение 1 ч в атмосфере аргона. Прозрачный раствор обрабатывали (R)-3,4-дигидро-3-амино-4-оксо-1,5- бензотиазепин-5(2H)-уксусной кислоты, этиловым сложным эфиром, полученным в соответствии с известной процедурой (Slade и др., J. Med. Chem. 28, с. 1517-1521 (1985)) (1,2 г, 3,32 мМ) и 4-метилморфолином (0,37 мл, 3,32 мМ), и продолжали размешивать 1 ч при 0oC и 1 ч при комнатной температуре. Реакционную смесь разводили этилацетатом (80 мл), промывали последовательно водой (12 мл), 5% бисульфатом калия (12 мл), водой (12 мл), насыщенным бикарбонатом натрия (12 мл), и солевым раствором, а затем осушали (безводным сульфатом магния), фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт хроматографировали на колонке с силикагелем (Merck), элюируя смесью этилацетата и гексана (1: 4), в результате чего получали 1,5 г продукта в виде сиропа, Rf = 0,68 (этилацетат:гексан, 1:1)
c) (3R(S*)-3,4-Дигидро-3-[[2-(меркаптометил)-1-оксо-3- фенилпропил] амино]-4-оксо-1,5-бензотиазепин-5(2H)-уксусная кислота
Раствор продукта этилового сложного эфира, полученного в части (a), (1,5 г, 2,996 мМ) в безводном метаноле (15 мл) продували в течение 15 мин аргоном, охлаждали до 0oC (ледяная/солевая баня), а затем по капле обрабатывали предварительно продутым (аргон, 30 мин) раствором 1,0 н. гидроксида натрия (12 мл, 4 экв), причем во время добавления и в течение всей реакции, раствор барботировали аргоном. После этого реакционную смесь размешивали 1 ч при 0oC, подкисляли (при 0oC) 5% бисульфатом калия (50 мл) для доведения pH до 1,0, и экстрагировали этилацетатом (2 • 100 мл). Органический экстракт промывали солевым раствором (25 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали а вакууме. Неочищенный продукт растворяли в смеси метиленхлорида (10 мл) и этилового эфира (5,0 мл), а затем по частям обрабатывали гексаном (30 мл), размешивая полученную смесь до образования твердого вещества. Осадок отфильтровывали, промывали гексаном (50 мл) в течение 48 ч при комнатной температуре в атмосфере азота, фильтровали и осушали в вакууме в течение 5 ч, в результате чего получали 1,01 г продукта в виде аморфного твердого вещества, Rf = 0,62(метиленхлорид: метанол:уксусная кислота, 20:1:1), [α]D = -171,4o (c = 0,74, метанол).
1H-ЯМР(CDCl3): 2,47 - 2,90 (м, 6H); 3,84 (дд, 1H); 4,13 (д, 1H, J = 17 Гц); 4,71 (м, 1H); 4,85 (д, 1H, J = 17 Гц); 6,88 - 7,67 (м, 9H).
Элементный анализ для C21H22N2O4S2:
Вычислено: C 58,59; H 5,15; N 6,51; S 14,89; SH 7,68;
Найдено: C 58,55; H 5,35; N 6,20; S 14,56; SH 7,54.
Пример 2.
[R-(R*, S*)] -3,4-Дигидро-3/-[(2-меркапто-1-оксо-3- фенилпропил)]-амино] -4-оксо-1,5-бензотиазепин-5(2H)-уксусная кислота.
a) (S)-2-(Ацетилтио)бензолпропановая кислота, дициклогексиламиновая соль.
Нитрит натрия (10,3 г, 280 мМ) добавляли в течение 1 ч к раствору D-фенилаланина (30,0 г, 181 мМ) и бромида калия (73,5 г) в серной кислоте (2,5 н. , 365 мл), поддерживая при этом температуру реакционной смеси при 0oC. Затем смесь размешивали при 0oC еще 1 ч, а после этого в течение 1 ч при комнатной температуре. Реакционный раствор экстрагировали эфиром, эфир подвергали обратному экстрагированию водой, а эфирный слой осушали сульфатом натрия. Эфир удаляли в вакууме, маслянистый остаток дистиллировали, и получали 25,7 г (R)-2-бромо-3-бензилпропановой кислоты, т. кип. 141oC (0,55 мм рт. ст. [α]D = +14,5o (с = 2,4, хлороформ).
Смесь тиоуксусной кислоты (7 мл, 97,9 мМ) и гидроксида калия (5,48 г, 97,9 мМ) в ацетонитриле (180,5 мл) размешивали в течение 1,75 ч при комнатной температуре и в присутствии азота. Затем смесь охлаждали в ледяной бане в течение 10 мин. добавляли раствор (R)-2-бромо-3-бензилпропановой кислоты (20,4 г, 89 мМ) в ацетонитриле (20 мл). Реакционную смесь размешивали в течение 5 ч при комнатной температуре в атмосфере аргона, фильтровали и ацетонитрил удаляли в вакууме. Полученный маслянистый остаток снова растворяли в этилацетате и промывали 10% бисульфатом калия и водой. После удаления этилацетата в вакууме, получали 19,6 г неочищенного продукта. Этот продукт очищали в виде его дициклогексиламиновой соли, используя изопропиловый эфир в качестве растворителя для кристаллизации. Аналитический образец (S)-2-(ацетилтио)бензолпропановой кислоты, дициклогексиламиновой соли получали путем перекристаллизации из этилацетата, т. пл. 146 - 147oC, [α]D = -39,6o (с = 1,39, хлороформ).
Анализ для C11H12O3S • C12H23N:
Вычислено: C 68,11; H 8,70; N 3,45; S 7,91;
Найдено: C 67,93; H 8,71; N 3,37; S 7,94.
(b) [R-R*, S*)] -3,4-Дигидро-3-[[2-(ацетилтио)- 1-оксо-3-фенилпропил] амино] -4-оксо-1,5-бензотиазепин-5(2H)-уксусная кислота, этиловый сложный эфир.
Суспензию (S)-2-(ацетилтио)бензолпропановой кислоты, дициклогексиламиновой соли (1,76 г, 4,34 мМ, 1,01 экв.) суспендировали в этилацетате (123 мл), промывали 5% бисульфатом калия (5 • 19 мл) и солевым раствором (25 мл), а затем осушали безводным сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме.
Свободную кислоту растворяли в безводном метиленхлориде (25 мл), охлаждали до 0o в ледяной/солевой бане, обрабатывали 1-гидроксибензтриазолгидратом (611 мг, 4,52 мМ) и 1-этил-3-(3-диметиламинопропил)карбодиимидом (897 мг, 4,68 мМ) и размешивали 1 ч при 0oC. Полученный раствор обрабатывали (R)-3,4-дигидро-3-амино-4-оксо-1,5-бензотиазепин-5(2H)-уксусной кислоты этиловым сложным эфиром, полученным в соответствии с известной процедурой (Slade и др. , J. Med. Chem. vol. 28, с. 1517 - 1521 (1985)) (1,55 г, 4,29 мМ) и 4-метилформолином (0,48 мл, 4,37 мМ), а затем продолжали размешивать 1 ч при 0oC, и 1 ч при комнатной температуре. Реакционную смесь разводили этилацетатом (104 мл), промывали последовательно водой (16 мл), 5% бисульфатом калия (2 • 16 мл), насыщенным бикарбонатом натрия (16 мл) и солевым раствором (16 мл), а затем осушали безводным сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт хроматографировали на колонке с силикагелем (Merck), элюируя смесью этилацетата и гексана (1: 4), в результате чего получали 1,347 г продукта в виде сиропа, Rf = 0,80 (этилацетат:гексан, 1:1).
c) [R-(R*, S*)] -3,4-Дигидро-3-[[(2-меркапто-1- оксо-3-фенилпропил)]-амино]-4-оксо-1,5-бензтиазепин-5(2H) уксусная кислота.
Раствор продукта этилового сложного эфира, полученного в части (b), (1,347 г, 2,768 мМ) в метаноле (14 мл) продували в течение 30 мин аргоном, охлаждали до 0oC в ледяной/солевой бане и по каплям обрабатывали предварительно продутым (аргон, 30 мин) раствором 1,0 н. гидроксида натрия (11 мл, 4 экв. ), причем, во время добавления и в течение всей реакции раствор барботировали аргоном. Затем реакционную смесь размешивали 1 ч при 0oC, подкисляли при 0oC 5% бисульфатом калия (46 мл) для доведения pH до 2,0 и экстрагировали этилацетатом (2 • 100 мл). Органические экстракты промывали солевым раствором (25 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт растворяли в метиленхлориле (10 мл) и порциями обрабатывали гексаном (50 мл), размешивая смесь до образования твердого вещества. Надосадочную жидкость декантировали, а твердый остаток перетирали с дополнительным количеством гексана (50 мл) и пентаном (2 • 100 мл), размешивая 4 ч с первым 100 мл пентана, а затем в течение ночи еще с 100 мл пентана в атмосфере аргона. После этого, полученный продукт осушали в вакууме в течение 12 ч и получали в результате 1,048 г аморфного твердого вещества, Rf = 0,57 (метиленхлорид:метанол:уксусная кислота, 20:1:1), [α]D = -169,9o (с = 0,61, метанол).
1H-ЯМР (CDCl3): 1,99 (д, 1H); 2,76 (т, 1H, J = 11 Гц); 3,10 (м, 2H); 3,54 (м, 1H); 3,76 (м, 1H); 4,10 (д, 1H, J = 17 Гц); 4,65 (м, 1H), 4,86 (д, 1H, J = 17 Гц); 7,12 - 7,68 (м, 9H).
Анализ для C20H22N2O4S • 0,17 C5H12 • 0,52 H2O:
Вычислено: C 57,15; H 5,30; N 6,40; S 14,64; SH 7,55;
Найдено: C 57,15; H 4,99; N 6,14; S 14,72; SH 8,02.
Пример 3.
(S, S)-1,3,4,5-Тетрагидро-4-[[2-(меркаптометил)-1-оксо-3- фенилпропил] амино]-3-оксо-2H-2-бензазепин-2-уксусная кислота.
a) N-(N-фталоил-L-фенилаланил)глицин, этиловый сложный эфир
Гидроксибензтриазолгидрат (1,42 г, 10,5 мМ) и 1-этил-3- (3-диметиламинопропил)карбодиимида гидрохлорид (2,10 г, 11,0 мМ) добавляли к раствору N-фталоил-L-фенилаланинамида (2,95 г, 10,0 мМ) в сухом метиленхлориде (30 мл) при 0oC в присутствии аргона. После 30-минутного размешивания при 0oC полученную смесь обрабатывали глицином, сложным этиловым эфиром, гидрохлоридом (1,675 г, 12,0 мМ) и N-метилморфолином (1,32 мл, 12,0 мМ).
После 1-часового размешивания при 0oC и 3-часового размешивания при комнатной температуре, смесь распределяли между этилацетатом и 5% бисульфатом калия. Органическую фазу промывали последовательно 5% бисульфатом калия, насыщенным бикарбонатом натрия и насыщенным раствором хлорида натрия, после чего осушали сульфатом натрия и выпаривали досуха. Неочищенный продукт растирали с этиловым эфиром и получали 3,542 г белого кристаллического твердого вещества, т. пл. 158 - 159oC, Rf = 0,33 (ацетон:гексан, 2:3), [α]D = -99,7o (с = 0,61, хлороформ).
b) (S)-1,3,4,5-тетрагидро-4-фталимида-3-оксо-2H-2-бензазепин- 2-уксуснаыя кислота, этиловый сложный эфир.
Смесь пентоксида формулы (9,6 г, 68 мМ) и 85% фосфорной кислоты (6,0 мл) нагревали, размешивали, до тех пор, пока не растворится пентоксид фосфора. Полученную вязкую жидкость растворяли в ледяной уксусной кислоте (36 мл), обрабатывали последовательно N-(N-фталоил- L-фенилаланил)глицина этиловым сложным эфиром (2,241 г, 5,90 мМ) и диоксаном (0,75 г, 8,3 мМ), а затем нагревали при 80oC (температура бани) в атмосфере аргона. После 6,5-часового размешивания при 80oC, смесь дополнительно обрабатывали триоксаном (0,75 г, 8,5 мМ). Затем смесь размешивали еще 16 ч при 80oC, после чего ее резко охлаждали смесью льда с водой и экстрагировали этилацетатом. Этилацететный экстракт промывали водой (3х), насыщенным бикарбонатом натрия (пока промывки не будут основными) и насыщенным хлоридом натрия, а затем осушали сульфатом натрия и выпаривали досуха. Неочищенный продукт очищали с помощью флеш-хроматографии на силикагеле (Whatamann LRS-1), элюируя смесью этилацетата и гексана (35: 65), и получали 1,718 г продукта в виде бесцветного пенистого вещества, Rf = 0,38 (этилацетат:толуол, 3:7).
c) (S)-1,3,4,5-тетрагидро-4-[[(фенилметокси)карбонил] амино] -3- оксо-2H-2-бензазепин-2-уксусная кислота, этиловый сложный эфир.
Раствор (S)-1,3,4,5-тетрагидро-4-фталимидо-3-оксо-2H-2- уксусной кислоты, этилового сложного эфира (1,67 г, 4,26 мМ) в 1,0 М гидрата гидразина в этаноле (9,0 мл, 9,0 мМ) размешивали 36 ч при комнатной температуре и в атмосфере аргона. Полученную смесь разводили равным объемом этилацетата, фильтровали, а фильтрат выпаривали досуха. Остаток растворяли в толуоле, а затем снова выпаривали досуха. Бесцветный полутвердый остаток (1,81 г) растворяли в безводном метиленхлориде (20 мл), помещали в ледяную баню и обрабатывали последовательно триэтиламином (0,80 мл, 5,8 мМ) и бензилхлороформатом (0,77 мл, 5,4 мМ). После 2-часового размешивания при 0oC, смесь распределяли между этилацетатом и 5% бисульфатом калия. Органическую фазу промывали последовательно 5% бисульфатом калия, насыщенным бикарбонатом натрия и насыщенным хлоридом натрия, а затем осушали сульфатом натрия и выпаривали досуха. После очистки неочищенного продукта с помощью флеш-хроматографии на силикагеле (Whatmann LRS-1) (элюиент: смесь этилацетата и гексана, 1: 2) получали 1,0747 г продукта в виде бесцветного пенистого вещества, Rf = 0,52 (этилацетат: толуол, 3:7). После простой кристаллизации из гексана получали продукт, т.пл. 80 - 82oC, [α]D = +87,2o (c = 0,53, хлороформ).
d) (S)-1,3,4,5-Тетрагидро-4-амино-3-оксо-2H-2-бензазепин-2-уксусная кислота, этиловый сложный эфир
20% гидроксид палладия/уголь в качестве катализатора добавляли к раствору (S)-1,3,4,5-тетрагидро-4-[[(фенилметокси)карбонил] амино]- 3-оксо-2H-2-бензазепин-2-уксусной кислоты, этилового сложного эфира (1,037 г, 2,62 мМ) в абсолютном этаноле (20 мл) и полученную суспензию размешивали 2 ч в атмосфере водорода (баллонного). Смесь фильтровали через целит и фильтрат выпаривали досуха. Полутвердый остаток растирали с гексаном и получали 0,63 г продукта в виде белого кристаллического твердого вещества, т.пл. 71 - 73oC, Rf = 0,38 (метанол : метиленхлорид, 1:9), [α]D = 77,5o (c = 0,59, хлороформ).
e) (S, S)-1,3,4,5-Тетрагидро-4-[[2-[(ацетилтио)метил]-1-оксо- 3-фенилпропил]амино]-3-оксо-2H-2-бензазепин-2-уксусная кислота, этиловый сложный эфир
(S)-2-[(Ацетилтио)метил] бензолпропановую кислоту [полученную из эфедриновой соли, как описано ранее] подвергали реакции с продуктом из части (b) в соответствии с процедурой примера 1, часть (b) и получали целевой продукт в виде бесцветного пенистого вещества, Rf = 0,30 (этилацетат : гексан, 1:1).
f) (S, S)-1,3,4,5-Тетрагидро-4-[[2-(меркаптометил)-1-оксо-3- фенил-пропил]амино]-3-оксо-2H-2-бензазепин-2-уксусная кислота
Раствор продукта, полученного в части (e), в метаноле обрабатывали согласно процедуре, описанной в примере 1, часть (c) и получали белый твердый продукт, т. пл. 149 - 151oC (разлож.), Rf = 0,47 (метиленхлорид : уксусная кислота, метанол, 20:1:1), [α]D = +112,2o (c = 0,58, метанол).
Анализ для C22H24N2O4S:
Вычислено: C 64,06; H 5,86; N 6,79; S 7,77; SH 8,02;
Найдено: C 64,20; H 6,09; N 6,43; S 7,68; SH 8,33.
Пример 4.
(S, S)-1,3,4,5-Тетрагидро-4-[(2-меркапто-1-оксо-3- фенилпропил)амино]-3-оксо-2H-2-бензазепин-2-уксусная кислота
a) (S,S)-1,3,4,5-тетрагидро-4-[[2-(ацетил)-1-оксо-3- фенилпропил]-амино] -3-оксо-2H-2-бензазепин-2-уксусная кислота, этиловый эфир
(S)-2-(Ацетилтио)-бензолпропановую кислоту [полученную из дициклогексиламиновой соли, как описано ранее] подвергали реакции с продутом примера 3(d) согласно процедуре, описанной в примере 2, часть (b), и получали целевой продукт в виде бесцветного пенистого вещества; Rf = 0,35 (этилацетат : гексан, 1:1).
b) (S,S)-1,3,4,5-тетрагидро-4-[(2-меркапто-1-оксо-3-фенил- пропил)амино] -3-оксо-2H-2-бензазепин-2-уксусная кислота
Раствор продукта, полученного в части (a), в метаноле обрабатывали 1 н. гидроксидом натрия в соответствии с процедурой описанной в примере 2, часть c, в результате чего получали целевой продукт в виде белого твердого вещества, т.пл. 181 - 183oC (с разложением), Rf = 0,49 (метиленхлорид : уксусная кислота : метанол, 20:1:1), [α]D = +103,4o (c = 0,57, метанол).
Анализ для C21H22N2O4S:
Вычисляли: C 63,30; H 5,56; N 7,03; S 8,05; SH 8,30;
Найдено: C 63,35; H 5,65; N 6,87; S 8,07; SH 9,40.
Пример 5.
[S-(R*, R*)] -2,3,4,5-тетрагидро-3-[[(2- (меркаптометил)-1-оксо-3-фенилпропил]амино]-2-оксо-1H-бензазепин-1- уксусная кислота
a) [S-(R*, R*)-2,3,4,5-тетрагидро-3-[[2- [(ацетилтио)метил]-1-оксо-3-фенилпропил] амино]-2-оксо-1H-бензазепин- 1-уксусная кислота, этиловый сложный эфир
(S)-2-[(Ацетилтио)метил]бензолпропановой кислоты эфедриновую соль (1,547 г, 3,83 мМ, 1,01 экв. ) суспендировали в этилацетате (72 мл), промывали разбавленной соляной кислотой (72 мл воды + 5,4 мл 1,0 н. соляной кислоты, а затем 36 мл воды + 1,8 мл 1,0 н. соляной кислоты) и солевым раствором (12 мл), осушали безводным сульфатом магния, фильтровали и выпаривали досуха. Бесцветный сироп осушали в вакууме в течение 1 ч и получали 1,062 г (S)-2-[(ацетилтио)метил]бензолпропановой кислоты.
Свободную кислоту растворяли в безводном диметилформамиде (21,8 мл), охлаждали до 0oC (в ледяной/солевой бане), обрабатывали 1-гидроксибензтриазолгидратом (540 мг, 3,99 мМ) и 1-этил-3-(3-диметиламинопропил)карбодиимидом (792 мг, 413 мМ) и размешивали в течение 1 ч при 0oC в атмосфере аргона. Прозрачный раствор обрабатывали (S)-2,3,4,5-тетрагидро-3-амино-2-оксо-1H-бензазепин-1-уксусной кислоты этиловым сложным эфиром, полученным в соответствии с описанием (Watthey и др., J.Med.Chem., 28, стр. 1511-1516 (1985)) (1,0 г, 3,81 мМ) и продолжали размешивать в течение 1 ч при 0oC, и в течение 1 ч при комнатной температуре. Реакционную смесь разбавляли этилацетатом (100 мл), промывали последовательно водой (15 мл), 5% бисульфатом калия (2•15 мл), насыщенным бикарбонатом натрия 15 мл и солевым раствором (15 мл), а затем осушали безводным сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт хроматографировали на колонке с силикагелем (Merck), элюируя смесями этилацетата и гексана (1: 4, 1:2), в результате чего получали 1,725 г продукта в качестве сиропа, Rf = 0,42 (этилацетат : гексан, 1:1).
b) [S-(R*, R*)] -2,3,4,5-тетрагидро-3-[[2- (меркаптометил)-1-оксо-3-фенилпропил]амино]-2-оксо-1H-бензазепин- 1-уксусная кислота
Раствор продукта этилового сложного эфира, полученного в части (а), (1,725 г, 3,574 мМ), в метаноле (18,0 мл) продували в течение 30 мин аргоном, охлаждали до 0oC в ледяной/солевой бане, а затем по капле обрабатывали предварительно продутым (аргон, 30 мин) раствором 1,0 н. гидроксида натрия (14,3 мл, 4 экв.), при этом во время добавления и в течение всей реакции, раствор барботировали аргоном. После этого реакционную смесь размешивали в течение 1 ч при 0oC, подкисляли при 0oC 5% бисульфатом калия (60 мл) для доведения pH до 1 и затем экстрагировали этилацетатом (2 • 120 мл). Органические экстракты промывали солевым раствором (30 мл), осушали безводным сульфатом натрия, фильтровали и выпаривали досуха. Неочищенный продукт растворяли в метиленхлориде (10 мл) и обрабатывали по частям гексаном (50 мл), размешивая до образования твердого вещества. Надосадочную жидкость декантировали, а твердый остаток растирали с дополнительным количеством гексана (2•50 мл) и пентана (2•50 мл) и пентана (2•100 мл), размешивая 4 ч с первым 100 мл пентана, а затем в течение ночи в атмосфере аргона с последними 100 мл пентана. Полученный продукт осушали в вакууме 12 ч, в результате чего получали 1,438 г аморфного твердого вещества, Rf = 0,53 (метиленхлорид : метанол : уксусная кислота, 20:1:1), [α]D = -140,4o (c = 0,76, метанол).
Анализ для C22H24N2O4S • 0,11 C5H12:
Вычислено: C 64,42; H 6,07; N 6,66; S 7,63; SH 7,87;
Найдено: C 64,07; H 6,13; N 6,34; S 7,25; SH 7,14.
1H-ЯМР CDCl3: 1,42 (т, 1H); 1,95 (м, 1H); 2,44-2,90 (м, 7H); 3,27 (м, 1H); 4,37 (д, 1H, J = 17 Гц); 4,52 (м, 1H); 4,67 (д, 1H, J = 17 Гц); 6,86-8,00 (м, 9H).
Пример 6.
[S-(R*, R*)] -2,3,4,5-тетрагидро-3-[(2-меркапто-1- оксо-3-фенилпропил)амино]-2-оксо-1H-бензазепин-1-уксусная кислота
a) [S-(R*,R*)]-2,3,4,5-тетрагидро-3-[[2-(ацетилтио)-2- оксо-3-фенилпропил]амино]-2-оксо-1H-бензазепин-1-уксусная кислота, этиловый сложный эфир
Суспензию (S)-2-(ацетилтио) бензолпропановой кислоты дициклогексиламиновой соли (1,70 г, 4,19 мМ) суспендировали в этилацетате (120 мл), промывали 5% бисульфатом калия (5•20 мл) и солевым раствором (25 мл), осушали безводным сульфатом магния, фильтровали, выпаривали досуха, осушали в вакууме, в результате чего получали 954 мг свободной кислоты в виде сиропа.
Эту свободную кислоту растворяли в сухом метиленхлориде (25 мл) и охлаждали до 0oC в ледяной/солевой бане, после чего обрабатывали 1-гидроксибензтриазолгидратом (594 мг, 4,40 мМ) и 1-этил-3-3-диметиламинопропил карбодиимидом (870 мг, 4,54 мМ) и размешивали в течение 1 ч при 0oC. Полученный раствор обрабатывали раствором этилового сложного эфира (S)-2,3,4,5-тетрагидро-3-амино-2-оксо-1H-бензазепин-1-уксусной кислоты, полученного в соответствии с известным описанием (Watthey и др., J.Med.Chem., 28, стр. 1511-1516 (1985)), (1,0 г, 3,81 мМ) в сухом метиленхлориде (10 мл) и продолжали размешивать в течение 1 ч при 0oC и в течение 1,5 ч при комнатной температуре. Реакционную смесь разводили этилацетатом (100 мл), промывали последовательно водой (15 мл), 5% бисульфатом калия (2•15 мл), насыщенным бисульфатом натрия (15 мл) и солевым раствором (15 мл), а затем осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт хроматографировали на колонке с силикагелем (Merck), элюируя смесями этилацетата и гексана (1:4, 1:2), и получали 1,632 г продукта в виде сиропа. Кроме того, с колонки также получали дополнительные 127 мл изомерной смеси (в отношении 1:1), Rf = 0,50 (этилацетат : гексан, 1:1).
b) [S-(R*, R*)] -2,3,4,5-тетрагидро-3-[(2-меркапто- 1-оксо-3-фенил-пропил)амино]-2-оксо-1H-бензазепин-1-уксусная кислота
Раствор продукта этилового сложного эфира, полученного в части (a), (1,57 г, 3,35 мМ) в метаноле (17,0 мл) продували аргоном в течение 30 мин, охлаждали до 0oC в ледяной/солевой бане, а затем по капле обрабатывали заранее продутым (аргоном, 30 мин) раствором 1,0 н. гидроксида натрия (13,4 мл, 4,9 экв. ), при этом во время добавления и в течение всей реакции, раствор барботировали аргоном. Затем реакционную смесь размешивали 1 ч при 0oC, подкисляли при 0oC 5% бисульфатом калия (60 мл) для доведения pH до 1 и экстрагировали этилацетатом (2•110 мл). Органические экстракты промывали солевым раствором (30 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт растворяли в метиленхлориде (10 мл) и по частям обрабатывали гексаном (50 мл), размешивая до образования твердого вещества. Затем надосадочную жидкость декантировали, а твердые остатки растирали с дополнительным количеством гексана (2•50 мл) и пентана (2•100 мл), причем, сначала размешивали с первыми 100 мл пентана в течение 4 ч, а затем со следующими 100 мл в течение ночи в атмосфере аргона. Полученный продукт осушали в вакууме в течение 12 часов и получали в результате 1,237 г продукта, Rf = 0,58 (метиленхлорид:метанол:уксусная кислота, 20:1:1), [α]D = -155,6o (c = 0,73, метанол).
Анализ для C21H22N2O4S• 0,1C5H12•0,18H2O:
Вычислено: C 63,14; H 5,81; N 6,85; S 7,84; SH 8,09;
Найдено: C 63,14; H 5,85; N 6,58; S 7,50; SH 7,05.
1H-ЯМР (CDCl3): 1,88 (м, 1H); 1,97 (д, 1H); 2,62 (м, 2H); 3,07 (м, 2H); 3,25 (м, 1H); 3,51 (м, 1H); 4,39 (д, 1H, J=17 Гц); 4,45 (м, 1H); 4,64 (м, 1H, J = 17 Гц); 7,07-7,41 (м, 9H).
Пример 7.
[2S-[ 2α, 5α (R*)] ] -5,6-Дигидро-5-[(2-меркапто-1-оксо- 3-фенилпропил)амино]-4-оксо-2-фенил-2H-1,3-триазин-3(4H)-уксусная кислота
a) (2S-цис)-5,6-Дигидро-5-фталимидо-4-оксо-2-фенил-2H-1,3-тиазин- 3(4H)-уксусная кислота, этиловый сложный эфир
В соответствии с процедурой, описанной в патенте США N 4460579 (пример 1(а) - 1(c) получали 15,6 г смеси диастереомерного продукта. Смесь нагревали с обратным холодильником в течение 4 часов в эфире (500 мл), охлаждали на ледяной бане и фильтровали, в результате чего получали 5,9 г (2R-транс)-5,6-дигидро-5-фталимидо-4-олксо-2-фенил- 2H-1,3-тиазин-3(4H)-уксусной кислоты этилового сложного эфира, т.пл. 166 - 168oC, Rf = 0,40 (гексаны:этилацетат, 1:1), [α]D = -72,9o (c = 1, хлороформ).
Анализ для C22H20N2O5S:
Вычислено: C 62,25; H 4,75; N 6,60; S 7,77;
Найдено: C 62,21; H 4,82; N 6,63; S 7,52.
После растирания оставшейся продуктовой смеси с эфиром (125 мл) при температуре перегонки получали вторую партию (0,9 г) (2R-транс)-изомерного продукта. Остаток снова растирали с эфиром при температуре перегонки и получали в результате 0,75 г нерастворимого вещества, в основном (2R-транс)-изомер, и 7,1 г материала, обогащенного (2S-цис)-изомером. 6,0 г этого обогащенного (2S-цис)-изомером материала хроматографировали на двух соединенных друг с другом колонках Waters Prep LC, элюируя смесью гексанов и этилацетата (3:1). После объединения соответствующих фракций получали 4,8 г (2S-цис)-5,6-дигидро-5-фталимидо-4-оксо-2-фенил-2H-1,3-тиазин- 3(4H)-уксусной кислоты этилового сложного эфира, т.пл. 66 - 68oC, [α]D = -101,2o. ТСХ проводили так же, как для изомера A.
Анализ для C22H20N2O5S•H2O:
Вычислено: C 61,83; H 4,79; N 6,55; S 7,50;
Найдено: C 61,83; H 5,07; N 6,25; S 7,42.
b) (2S-цис)-5,6-Дигидро-5-амино-4-оксо-2-фенил-2H-1,3-тиазин- 3(4H)-уксусная кислота, этиловый сложный эфир
Раствор (2S-цис)-изомерного продукта, полученного в части (а), (прибл. 85% дисатереометрически чистого, 3,03 г, 7,1 мМ) в хлороформе (15 мл) обрабатывали метилгидразином (850 мкл, 736 мг, 16,0 мМ). После 24-часового размешивания при комнатной температуре, смесь разводили этилацетатом и этиловым эфиром, а затем фильтровали. Фильтрат дважды экстрагировали 0,5 г соляной кислотой, а объединенные водные экстракты подщелачивали 1 н. гидроксидом натрия. Водную смесь экстрагировали три раза метиленхлоридом и объединенные метиленхлоридные экстракты осушали сульфатом натрия, фильтровали и упаривали, в результате чего получали 1,648 г продукта в виде бледно-желтого маслянистого вещества, Rf = 0,43 (метиленхлорид:уксусная кислота:метанол, 8: 1:1).
ЯМР-анализ показал, что полученный продукт является приблизительно на 84% диастереометрически чистым.
c) [2S-[ 2α, 5α (R*)]]-5,6-Дигидро-5-[[2-(ацетилтио)-1-оксо- 3-фенилпропил] амино] -4-оксо-2-фенил-2H-1,3-тиазин-3(4H)-уксусная кислота, этиловый сложный эфир
(S)-2-(Ацетилтио)бензолпропановой кислоты дициклогексиламиновую соль (1, 318 г, 3,25 мМ) распределяли между этилацетатом и 5% бисульфатом калия. Этилацетатный слой промывали водой и солевым раствором, а затем осушали сульфатом натрия, фильтровали и упаривали, в результате чего получали свободную кислоту в виде бесцветного маслянистого продукта. Смесь этой свободной кислоты и (2S-цис)-продукта, полученного в части (b), (84% диастереометрической чистоты, 960 мг, 3,26 мМ) растворяли в метиленхлориде (25 мл), обрабатывали 1-гидроксибензтриазолгидратом (622 мг, 4,6 мМ) и охлаждали до -10oC. Затем смесь обрабатывали 1-этил-3-(3-диметил-аминопропил)карбодиимидом (687 мг, 3,58 мМ). После медленного (в течение 1 ч) нагревания до 0oC, эту смесь нагревали до комнатной температуры. Через 4 ч, раствор разбавляли этилацетатом и промывали последовательно водой, 5% бисульфатом калия, снова водой, насыщенным бикарбонатом натрия и солевым раствором, а затем осушали сульфатом натрия, фильтровали и упаривали. Остаток подвергали флеш-хроматографии (Merck-силикагель, этилацетат:гексан = 1: 1) и получали 804 мг продукта в виде бесцветного пенистого вещества, Rf = 0,33 (50% этилацетат в гексане). ЯМР анализ показал, что в отношении ацетилтиоцентра этот продукт имел диастереометрическую чистоту 91%.
d) [2S-[ 2α, 5α (R*)]]-5,6-Дигидро-5-[[2-меркапто-1-оксо- 3-фенилпропил] амино]-4-оксо-2-фенил-2H-1,3-тиазин-3(4H)-уксусная кислота
Раствор этилэфирного продукта, полученного в части (c), (550 мг, 1,0 мМ) в метаноле (5 мл, дезоксигенированном посредством барботирования аргона) при 0oC обрабатывали охлажденной льдом 1 н. гидроокисью натрия (5 мл, дезоксигенированной посредство барботирования аргоном) и полученную смесь размешивали при 0oC в атмосфере азота в течение 30 мин. Раствор подкисляли 10% бисульфитом калия (15 мл), разбавляли водой и экстрагировали этилацетатом. Этилацетатный экстракт промывали водой и солевым раствором, а затем осушали сульфатом натрия, фильтровали и упаривали, в результате чего получали маслянисто-пенистый продукт. Этот продукт подвергали флеш-хроматографии (Merck-силикагель, метиленхлорид: метанол:уксусная кислота = 20:1:1). Фракции, содержащие нужный продукт, объединяли и упаривали, а остаток растворяли в этилацетате и промывали последовательно водой, 0,5 н. соляной кислотой и солевым раствором, а затем осушали сульфатом натрия, фильтровали и упаривали. Полученный маслянистый продукт растворяли в небольшом количестве этилацетате и этиловом эфире и растирали с гексаном. Смесь упаривали досуха, суспендировали в гексане, снова упаривали досуха, осушали в вакууме и получали 392 мг продукта в виде белого пенистого вещества, Rf = 0,24 (метиленхлорид:метанол:уксусная кислота, 20:1:1, [α]D = -13,2o (c =0,5, хлороформ).
ВЭЖХ: колонка УМС S3ODS (6,0х150 мм); элюирование 44% A: 90% воды - 10% метанола - 0,2% фосфорной кислоты; и 56% B: 10% воды - 90% метанола - 0,2% фосфорной кислоты; скорость потока 1,5 мл/мин с детекцией при 220 нм; tR = 26,22 мин и tR = 24,64 мин (9,3% 5α (S*)-изомер).
Анализ для C21H22N2O4S2• 0,17 H2O:
Вычислено: C 58,18; H 5,19; N 6,46; S 14,79; SH 7,63;
Найдено: C 58,20; H 5,51; N 6,44; S 14,46; SH 6,60.
Пример 8.
[2S-[ 2α, 5α (R*)] ] -5,6-Дигидро-5-[[2-(меркаптометил)-1-оксо- 3-фенилпропил]амино-4-оксо-2-фенил-2H-1,3-тиазин-3(4H)-уксусная кислота
a) [2S- 2α, 5α (R*)]]-5,6-Дигидро-5-[[2-[(ацетилтио)метил]-1-оксо- 3-фенилпропил] амино-4-оксо-2-фенил-2H-1,3-тиазин-3(4H)-уксусная кислота, этиловый сложный эфир.
(S)-2-[(Ацетилтио)метил] бензолпропановую кислоту (полученную из эфедриновой соли в соответствии с описанием, приведенным выше) подвергали реакции с продуктом, полученным в примере 7 (часть b) в соответствии с процедурой, описанной в примере 1 (часть b), в результате чего получали целевой продукт в виде белого пенистого продукта, Rf = 0,29 (50% этилацетат в гексане). ЯМР-анализ показал, что полученный продукт является диастереометрически чистым.
b) [2S- 2α, 5α (R*)] ]-5,6-дигидро-5-[[2-(меркаптоэтил)-1-оксо- 3-фенилпропил]амино-4-оксо-2-фенил-2H-1,3-тиазин-3(4H)-уксусная кислота
Раствор продукта, полученного в части (a), в метаноле обрабатывали 1 н. гидроокисью натрия в соответствии с процедурой, описанной в примере 1 (часть (c), в результате чего получали целевой продукт в виде белого, относительно твердого, пенистого вещества, Rf = 0,29 (метиленхлорид:метанол:уксусная кислота, 20:1:1), [α]D = +42,9o (c = 1,0, хлороформ).
ВЭЖХ: колонка УМС S3ODS (6,0•150 мм), элюирование 44% A: 90% воды - 10% метанола - 0,2% фосфорной кислоты, и 56% B: 10% воды - 90% метанола - 0,2% фосфорной кислоты, скорость потока 1,5 мл/мин с детекцией при 220 нм, tR = 33,45 (99,1%).
Анализ для C22H24N2O4S2:
Вычислено: C 59,44; H 5,44; N 6,30; S 14,42; SH 7,44;
Найдено: C 59,63; H 5,84; N 5,99; S 14,54; SH 6,83.
Пример 9.
(S,S)-Гексагидро-3-[[2-(меркаптометил)-1-оксо-3-фенилпропил]амино- 2-оксо-1H-азепин-1-уксусная кислота
a) N2-[1,1-Диметиоэтокси)карбонил]-N6- [(фенилметокси)карбонил]-L-лизин, метиловый сложный эфир
Карбонат цезия (4,28 г, 13,1 мМ) добавляли к смеси N2-[(1,1-диметилэтокси)карбонил] -N6- [(фенилэтокси)карбонил]-L-лизина (10 г, 26,3 мМ) и 20% воды и метанола (60 мл). Через 5 мин раствор становился гомогенным, после чего растворитель выпаривали, а остаточную воду азеотропически удаляли с использованием ацетонитрила (3х). Полученный маслянистый продукт осушали в вакууме, растворяли в безводном диметилформамиде и обрабатывали метилиодидом (3,2 мл, 2,0 экв.). Реакционную смесь размешивали 1,5 ч при комнатной температуре в атмосфере аргона, разводили этилацетатом (200 мл) и промывали последовательно водой (2•26 мл), насыщенным бикарбонатом натрия (25 мл) и солевым раствором (25 мл). Органическую фазу осушали безводным сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме, в результате чего получали 10,11 г продукта в виде светло-желтого сиропа, Rf = 0,30 (этилацетат:гексан, 1:2).
b) (S)-3-[[(1,1-Диметилэтокси)карбонил]амино]-гексагидро-2H-азепин-2-он
Раствор метилэфирного продукта, полученного в части (a), (8,532 г, 21,63 мМ) в сухом метаноле (100 мл) обрабатывали 10% полладированным углем (катализатором, 2,13 г) и гидрогенизировали 2 ч под давлением 50 фунт/кв.дюйм (344,7 кПа). Смесь разбавляли метанолом (100 мл) и фильтровали через слой целита в миллипористом фильтре, тщательно промывая этот слой метанолом (3•100 мл). Прозрачный фильтрат выпаривали досуха, осушали в вакууме и полученный маслянистый продукт растворяли в сухом ксилоле (70 мл). Раствор нагревали с обратным холодильником в присутствии аргона в течение 20 ч, а затем охлаждали до комнатной температуры, разводили этилацетатом (425 мл) и промывали последовательно 5% бисульфатом калия (85 мл), насыщенным бикарбонатом натрия (85 мл) и солевым раствором (85 мл). Органическую фазу осушали сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт хроматографировали на колонке с силикагелем (Merck), элюируя смесью этилацетата и гексана (1:1) и получали 2,76 г белого твердого продукта, т.пл. 146 - 147oC, Rf = 0,58 (этилацетата), [α]D = +4,5o (c = 1,0, метанол).
Анализ для C11H20N2O3:
Вычислено: C 57,87; H 8,83; N 12,27;
Найдено: C 58,12; H 8,63; N 12,26.
c) (S)-3-[[(1,1-диметилэтокси)карбонил] амино] гексагидро-2-оксо- 1H-азепин-1-уксусная кислота, этиловый сложный эфир
Суспензию 2H-азепин-2-он-продукта, полученного в части (b), (2,966 г, 12,99 мМ), в безводном тетрагидрофуране (21,0 мл) обрабатывали 0,1 M т-бутоксида калия/тетрагидрофурана (16,9 мл, 1,3 экв.) и размешивали в атмосфере аргона и при комнатной температуре в течение 5мин. Прозрачный светло-коричневый раствор по капле обрабатывали этилбромацетатом (2,34 мл, 1,7 экв), размешивали 1 ч при комнатной температуре и затем разводили этилацетатом (100 мл). Полученную смесь промывали насыщенным бикарбонатом натрия (20 мл), 5% бисульфатом калия (15 мл) и солевым раствором (15 мл), после чего осушали безводным сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт хроматографировали на колонке с силикагелем (Merck), элюируя гексаном и смесями этилацетата:гексана (1:6, 1:3), в результате чего получали 3,387 г продукта в виде сиропа, Rf = 0,55 (этилацетата: гексан, 1:1).
d) (S)-3-Амино-гексагидро-2-оксо-1H-азепин-1-уксусная кислота, этиловый сложный эфир, гидрохлоридная соль
Раствор продукта этилового сложного эфира, полученного в части (c), (3,278 г, 10,13 мМ) в сухом диоксане (80 мл), обрабатывали 4,0 М соляной кислотой/диоксаном (31 мл, 0,124 М, 12 экв.) и размешивали при комнатной температуре и в атмосфере аргона в течение 20 ч. Реакционную смесь упаривали досуха путем многократного выпаривания из эфира, а затем осушали в вакууме, в результате чего получали 2,541 г продукта в виде белых гигроскопических твердых кристаллов, Rf = 0,32 (метиленхлорид: метанол:уксусная кислота, 8:1: 1).
e) (S, S)-3-[[2-[(Ацетилтио)метил] -1-оксо-3-фенилпропил]амино]-2-оксо- 1H-азепин-1-уксусная кислота, этиловый сложный эфир
(S)-2-[(Ацетилтио)метил]бензолпропановой кислоты эфедриновую соль (1,625 г, 4,028 мМ, 1,01 экв) суспендировали в этилацетате (75 мл), промывали разбавленной соляной кислотой (75 мл воды + 5,7 мл 1,0 н. соляной кислоты, а затем 40 мл воды + 1,9 мл 1,0 н. соляной кислоты) и солевым раствором (15 мл), осушали безводным сульфатом, фильтровали и выпаривали до суха. Бесцветный сироп в течение 1 ч осушали в вакууме и получали 1,05 г свободной кислоты.
Эту свободную кислоту растворяли в сухом диметилформамиде (25 мл), охлаждали до 0oC в ледяной/солевой бане, обрабатывали 1-гидросибензтриазолгидратом (567 мг, 4,20 мМ) и 1-этил-3-(3-диметиламинопропил)карбодиимидом (832 мг, 4,34 мМ), а затем размешивали в течение 1 ч при 0oC и в присутствии аргона. Полученный прозрачный раствор обрабатывали раствором гидролхлоридной соли этилэфирного продукта. Полученный в части (d) (1,0 г, 3,988 мМ) в сухом диметилформамиде) (3,0 мл) и 4-метилморфолином (0,45 мл, 1,0 экв.) и размешивали 1 ч при 0oC и 2 ч при комнатной температуре. Реакционную смесь разводили этилацетатом (100 мл), промывали последовательно водой (15 мл), 5% бисульфатом калия (2х15 мл), насыщенным бикарбонатом натрия (15 мл) и солевым раствором, а затем осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт хроматографировали на колонке с силикагелем (Merck), элюируя смесями этилацетата и гексана (1:4, 1: 2), в результате чего получали 1,618 г продукта в виде сиропа, Rf = 0,030 (этилацетат : гексан, 1:1).
f) (S,S)-Гексагидро-3-[[-меркаптометил)-1-оксо-3-фенилпропил]амино]-2-оксо -1H-азепин-1-уксусная кислота
Раствор продукта этилового эфира, полученного в части (e), (1,537, 3,53 мМ) в метаноле (18 мл) продували в течение 30 мин аргоном, затем охлаждали до 0oC в ледяной/солевой бане и по капле обрабатывали предварительно продутым (аргон, 30 мин) раствором 1,0 н. гидроксида натрия (14,2 мл, 4 экв.), при этом, во время добавления и в течение всей реакции раствор барботировали аргоном. Затем реакционную смесь размешивали 1 ч при 0oC, подкисляя при 0oC 5% бисульфатом калия (60 мл) для доведения pH до 2, а затем экстрагировали этилацетатом (2•100 мл). Органические экстракты промывали солевым раствором (25 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт растворяли в метиленхлориде (5 мл) и частями обрабатывали гексаном (50 мл), размешивая до образования твердого вещества. Надосадочную жидкость декантировали, а твердый остаток растирали с дополнительным количеством гексана (3•50 мл) и полученное твердое вещество выпаривали из пентана (4•40 мл). Полученный продукт осушали в вакууме в течение 12 ч, в результате чего получали 1,261 г аморфного твердого продукта, Rf = 0,43 (метиленхлорид: метанол: уксусная кислота, 20:1:1), [α]D = +20,2o (с = 0,61 метанол).
1 H-ЯМР (400 МГц, CDCl3): 1,46 (т, 3H); 1,81 (м, 6H); 2,60 (м, 2H); 2,87 (м, 3H); 3,21 (м, 1H); 3,71 (м, 1H); 4,17 (с, 2H); 4,68 (м, 1H); 7,13-7,23 (м, 5H).
Анализ для C18H24N2O4S • 0,2 C5H12 • 0,45 H2O:
Вычислено: C 58,96; H 7,11; N 7,24; S 8,28; SH 8,54;
Найдено: C 58,96; H 6,86; N 7,07; S 8,31; SH 8,43.
Пример 10.
(S, S)-Гексагидро-3-[(2-меркапто-1-оксо-3-фенилпропил)амино] -2-оксо-1H- азепин-1-уксусная кислота.
a) (S, S)-3-[[2-(Ацетилтио)-1-оксо-3-фенилпропил]амино]-2-оксо-1H- азепин-1-уксусная кислота, этиловый сложный эфир
Суспензию (S)-2-(ацетилтио) бензолпропановой кислоты дициклогексиламиновой соли (2,81 г, 6,93 мМ или 1,01 экв.) суспендировали в этилацетате (200 мл), промывали 5% бисульфатом калия (5•30 мл) и солевым раствором (40 мл), а затем осушали безводным сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме, в результате чего получали 1,737 г свободной кислоты в виде прозрачного сиропа.
Эту свободную кислоту растворяли в сухом метиленхлориде (30 мл) и охлаждали до 0oC в ледяной/солевой бане, а затем обрабатывали 1-гидроксибензтриазолгидратом (978 мг, 7,23 мМ) и 1-этил-3-3-диметил-аминопропил карбодиимидом (1,435 мг, 7,49 мМ) и размешивали 1 ч при 0oC. Полученный раствор обрабатывали раствором (S)-3-амино-гексагидро-2-оксо-1H-азепин-1-уксусной кислоты, этилового сложного эфира, гидрохлоридной соли (1,58 г, 6,30 мМ) в сухом метиленхлориде (10 мл) и 4-метилморфолина (0,77 мл, 7,01 мМ) и размешивали 1 ч при 0oC и 1,5 ч при комнатной температуре. Реакционную смесь разводили этилацетатом (170 мл) промывали последовательно водой (25 мл), 5% бисульфатом калия (2•25 мл), насыщенным бикарбонатом натрия (25 мл) и солевым раствором (25 мл), а затем осушали безводным сульфатом магния, фильтровали, выпаривали досуха, осушали в вакууме. Неочищенный продукт хроматографировали на колонке с силикагелем (Merck), элюируя смесями этилацетата и гексана (3: 7), в результате чего получали 2,045 г продукта в виде сиропа. Кроме того, с колонки получали еще 261 мг изомерной смеси (в отношении 1:1), Rf = 0,45 (этилацетат: гексан, 1:1).
b) (S,S)-Гексагидро-3-[(2-меркапто-1-оксо-3-фенилпропил)амино]-2-оксо-1H -азепин-1-уксусная кислота
Раствор этилэфирного продукта, полученного в части (a), (2,044 г, 4,86 мМ) в метаноле (25 мл) продували в течение 30 мин аргоном, охлаждали до 0oC в ледяной/солевой бане, а затем по капле обрабатывали предварительно продутым (аргон, 30 мин) раствором 1,0 н. гидроксида натрия (19,55 мл, 4 экв.), при этом во время добавления и в течение всей реакции раствор барботировали аргоном. Реакционную смесь размешивали при 0oC в течение 1 ч, подкисляли при 0oC 5% бисульфатом калия (85 мл) для доведения pH 2,0, а затем экстрагировали этилацетатом (2•140 мл). Органические экстракты промывали солевым раствором (35 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт растворяли в метиленхлориде (10 мл) и по частям обрабатывали гексаном, в результате чего образовывалось твердое вещество. Надосадочную жидкость декантировали, а твердые остатки растирали с дополнительным количеством гексана (2•50 мл) и пентана (2•100 мл), размешивая 4 ч с первыми 100 мл пентана, а затем в течение ночи со следующими 100 мл пентана в присутствии аргона. Полученный продукт осушали в вакууме в течение 12 ч и получали в результате 1,618 г аморфного твердого вещества, Rf = 0,53 (метиленхлорид: метанол: уксусная кислота, 10:1:1); [α]D = +8,04o (с = 0,56, метанол).
1H-ЯМР (400 МГц, CDCl3): 1,71 (м, 6H); 2,02 (д, 1H); 3,16 (м, 3H); 3,61 (м, 1H); 3,70 (м, 1H); 4,17 (с, 2H); 4,65 (м, 1H); 7,18 - 7,72 (м, 5H).
Анализ для C17H22N2O4S:
Вычислено: C 58,27; H 6,33; N 7,99; S 9,15; SH 9,44;
Найдено: C 58,43; H 6,56; N 7,85; S 8,96; SH 10,27.
Пример 11.
[3R- [3α, 6α(S*), 9aβ] - Октагидро-6-[(2-меркапто-1-оксо-3-фенил-пропиламино]-5-оксотиазоло [3,2-а]азепин-3-карбоновая кислота
a) N2фталоил-L-лизин
Смесь N6-[(1,1-диметилэтокси)карбонил] -L-лизина (10,827 г, 43,9 мМ) и карбоната натрия (4,665 г, 44,0 мМ) размешивали в 170 мл этанола и воды (1: 3) в течение 2,5 ч. Затем этанол удаляли с помощью роторного испарителя, а водный раствор обрабатывали N-карботоксифталимидом (9,66 г, 44,0 мМ). После размешивания в течение 1,5 ч при комнатной температуре, раствор фильтровали, обрабатывали метиленхлоридом (200 мл), а водный слой подкисляли 6 н. соляной кислотой до pH 2,8. Слои встряхивали и разделяли. Водный слой экстрагировали снова этилацетатом, а объединенные органические слои осушали сульфатом магния, фильтровали и упаривали, в результате чего получали почти бесцветное маслянистое стеклообразное вещество (19,510 г).
Остаток охлаждали до 0oC, обрабатывали трифторуксусной кислотой (150 мл) и полученную смесь размешивали 3 ч при комнатной температуре. Трифторуксусную кислоту удаляли с помощью роторного испарителя, а остаток азеотропически обезвоживали с использованием толуола (2х). Неочищенный продукт растворяли в воде и пропускали через колонку Dowex AG 50-X2 (100-200 меш, 300 мл, кислотная форма), элюируя последовательно смесью метанола и воды (1:1, 600 мл), водой (500 мл) и наконец смесью пиридина и воды (5:95 (1000 мл). Нужные фракции объединяли, упаривали, растворяли в воде и лиофилизовали, в результате чего получали 7,850 г белого твердого продукта, Rf = 0,57 (н-бутанол:вода, уксусная кислота: этилацетат, 1:1:1:1).
b) [2(S), 4R]-4-(Метоксикарбонил)- α - фталимидо-2-тиазолидипентановая кислота
Суспензию N2-фталоил-L-лизина (7,710 г, 27,9 мМ) в сухом диметилформамиде (135 мл) обрабатывали 4-формил-1-метилпиридиния бензолсульфонатом (7,320 г, 26,2 мМ). Полученная темная смесь постепенно становилась гомогенной. После 1-часового размешивания при комнатной температуре, смесь обрабатывали 1,8-диазобицикло 5.4.0 ундек-7-ен'ом (8,15 мл, 8,30 г, 54,5 мМ), а затем через 5 мин, обрабатывали гидрохлоридом сложного метилового эфира L-цистеина (4,53 г, 26,3 мМ). Темный раствор размешивали 2,5 ч, а затем разводили водной соляной кислотой (pH 1,7, 300 мл) и хлороформом. Водный слой доводили до pH 4,3 - 4,4 с помощью 0,5 н соляной кислоты, а затем 5 раз экстрагировали хлороформом. Объединенные экстракты хлороформа промывали водой (50 мл), осушали сульфатом натрия, фильтровали и упаривали. Остаток растворяли в этилацетате, фильтровали через целит и концентрировали, в результате чего получали 1,84 г неочищенного продукта в виде оранжево-красного маслянистого вещества, Rf = 0,58 большое пятно (уксусная кислота : этилацетат, 15:85). ЯМР показал отношение изомеров 2:1.
c) [3R- (3α, 6α) ]-Октагидро-6-фталимидо-5-оксотиазоло[3,2-а] азепин-3-карбоновая кислота, метиловый сложный эфир
Раствор продукта, полученного в части (b), (1,84 г, 4,7 мМ) в тетрагидрофуране (70 мл) обрабатывали 2-этокси-1-этоксикарбонил-1,2-дигидрохинолином (1,400 г, 5,67 мМ) и полученную смесь размешивали в течение 3 дней при комнатной температуре. Тетрагидрофуран удаляли с помощью роторного испарителя, а остаток растворяли в этилацетате и промывали последовательно 0,5 н. соляной кислотой (2x), 50% насыщенным бикарбонатом натрия, водой и солевым раствором, а затем осушали сульфатом натрия, фильтровали и упаривали. Полученное оранжевое маслянистое вещество подвергали флеш-хроматографии (Merck-силикагель, 40 - 50% этилацетат в гексане), в результате чего получали 1,126 г продукта в виде смеси (1:1) диастереомеров, Rf = 0,22 и 0,20 (40% этилацетат в гексане).
d) [3R- (3α, 6α, 9aβ) ]-Октагидро-6-фталимидо-5-оксотиазоло[3,2-а] азепин-3-карбоновая кислота, метиловый сложный эфир
Смесь продукта метилового сложного эфира, полученного в части (c), (1,110 г) и гидрата п-толуолсульфоновой кислоты (2,33 г) в бензоле (20 мл) нагревали с обратным холодильником в течение 2,5 ч с удалением воды (ловушка Дина-Старка). Охлажденную смесь разводили этилацетатом и промывали 50% насыщенным бисульфатом натрия водой и солевым раствором, а затем осушали сульфатом натрия, фильтровали и упаривали. Остаток подвергали флеш-хроматографии (Merck-силикагель, 10 - 20% этилацетат в метиленхлориде), в результате чего получали почти чистый продукт в виде беловатого пенистого вещества. Это пенистое вещество растворяли в горячей смеси этилацетата и гексана, охлаждали и вносили затравку, в результате чего получали тонкие белые игольчатые кристаллы (845 мг). Еще 50 мг твердого продукта получали из маточного раствора, поэтому в целом количество чистого продукта составляло 895 мг, т.пл. 154 - 155oC, [α]D = -31,8o (c = 0,52, хлороформ), Rf = 0,22 (40% этилацетат в гексане).
e) [3R- (3α, 6α, 9aβ) ]-6-амино-октагидро-5-оксотиазоло[3,2-а]-азепин-3- карбоновая кислота, метиловый сложный эфир
Суспензию продукта метилового сложного эфира, полученного в части (d), (875 мг, 2,34 мМ) в абсолютном этаноле (50 мл) обрабатывали гидратом гидразина (115 мкл, 118 мг, 2,37 мМ) и для стимулирования солюбилизации, раствор периодически подогревали в течение первых 2 ч. После размешивания в течение 3 дней при комнатной температуре, добавляли дополнительно гидрат гидразина (80 мкл, 82,6 мг, 1,65 мМ) и полученную смесь размешивали еще один день. Затем реакционную смесь упаривали досуха, а белый твердый остаток размешивали в холодной (0oC) 0,5 н. соляной кислоте (60 мл) в течение 3,5 ч. Твердое вещество удаляли путем фильтрации и фильтрат подщелачивали до pH 11 - 12 с помощью 1 н. гидроксида натрия, а затем экстрагировали метиленхлоридом (3х) и этилацетатом. Объединенные органические экстракты осушали сульфатом натрия, фильтровали, упаривали и получали почти бесцветный маслянистый продукт (около 600 мг). Этот маслянистый продукт обрабатывали водой (10 мл) и 1,0 н соляной кислотой (3,0 мл), энергично размешивали до тех пор, пока не исчезнет маслянистость продукта, после чего смесь лиофилизовали и получали 674 мг желтоватого твердого продукта, Rf = 0,64 главное пятно (н-бутанол : вода : уксусная кислота : этилацетат 1:1:1:1).
f) [3R- [3α, 6α(S*) 9aβ] ]-Октагидро-6[[2-(ацетилтио)-1-оксо-3-фенилпропил] амино] -5-оксотиазоло 3,2-алазепин-3-карбоновая кислота, метиловый сложный эфир
Гидрохлоридную соль метилэфирного продукта, полученного в части (e), (374 мг, 1,33 мМ) распределяли между насыщенным бикарбонатом натрия и метиленхлоридом. Метиленхлоридный экстракт осушали сульфатом натрия, фильтровали, упаривали и получали свободный амин в виде светло-желтого маслянистого вещества (302 мг, 1,16 мМ). Кроме того, дициклогексиламиновую соль (S)-2-(ацетилтио)бензолпропановой кислоты (648 мг, 1,60 мМ) распределяли между этилацетатом и 5% бисульфатом калия. Этилацетатный слой промывали водой и солевым раствором, а затем осушали сульфатом натрия, фильтровали и упаривали, в результате чего получали свободную кислоту в виде маслянистого вещества. Смесь свободного амина и свободной кислоты в метиленхлориде (9 мл) обрабатывали гидроксибензтриазолгидратом (550 мг, 4,07 мМ), охлаждали до 0oC, а затем обрабатывали 1-этил-3-(3-диметил-аминопропил)карбодиимидом (313 мг, 1,63 мМ). Полученную смесь размешивали 1 ч при 0oC, а затем 3 ч при комнатной температуре. После этого добавляли еще 100 мг 1-этил-3-(3-диметиламинопропил)карбодиимида и продолжали размешивать в течение ночи. Раствор разводили этилацетатом и промывали последовательно водой, 5% бисульфатом калия, водой, 50% насыщенным бикарбонатом натрия и солевым раствором, а затем осушали сульфатом натрия, фильтровали и упаривали. Полученное желтое маслянистое вещество подвергали флеш-хроматографии (Merck-силикагель, 30% ацетона в гексане), в результате чего получали 396 мг продукта в виде смеси (87: 13) диастереомеров боковой цепи. Затем эту смесь снова подвергали флеш-хроматографии (Merck-силикагель, 35 - 40% этилацетат в гексане) и получали 318 мг продукта в виде белого пенистого вещества (94 - 95% диастереометрически чистого), Rf = 0,29, главное пятно и 0,26 (небольшое пятно, [ 6α(R*)- изомер], [α]D = -84,4o (c = 0,36, хлороформ).
g) [3R- [3α, 6α(S*), 9aβ] ]]-Октагидро-6-[(2-меркапто-1-оксо-3-фенилпропил)амино]-5-оксотиазоло 3,2-а азепин-3-карбоновая кислота
Раствор продукта метилового сложного эфира, полученного в части (f), (308 мг, 0,68 мМ) в метаноле (4 мл, дезоксигенированном посредством барботирования аргона) обрабатывали 1 н. гидроксидом натрия (3,5 мл, дезоксигенированном посредством барботирования аргона) и гомогенную смесь размешивали 2 ч в присутствии аргона. Затем смесь подкисляли 10% бисульфатом калия (15 мл), разводили водой и экстрагировали этилацетатом. Этилацетатный экстракт промывали солевым раствором, осушали сульфатом натрия, фильтровали и упаривали, в результате чего получали твердое вещество. Это твердое вещество растворяли в диметилформамиде (1 мл), разбавляли этилацетатом и подвергали флеш-хроматографии (Merck-силикагель, 5% уксусная кислота в этилацетате), в результате чего получали нужный продукт в виде белого твердого вещества, которое затем перетирали с этилацетатом этиловым эфиром, собирали путем фильтрации, осушали и получали 144 мг продукта, т.пл. 217 - 220oC, Rf = 0,52 (5% уксусная кислота в этилацетате), [α]D = -64,3o (c = 0,36, диметилформамид). ЯМР-анализ показал, что полученный продукт имел 93% диастереометрическую чистоту, причем 7% составлял соответствующий 6α(R*)- изомер.
ВЭЖХ: колонка УМС S3 ODS (0,6 • 150 мм); элюирование 44% A: 90% воды - 10% метанола - 0,2% фосфорной кислоты, и 56% B: 10% воды - 90% метанола - 0,2% фосфорной кислоты, скорость потока - 1,5 мл/мин с детектированием при 220 нм, tR = 12,27 мин показало чистоту > 95% (без разделения 6α(S*) и 6α(R*)- изомеров, выявленных с помощью ВЭЖХ).
Анализ для C18H22N2O4S2 • O • 4 H2O:
Вычислено: C 53,82; H 5,72; N 6,97; S 15,96; SH 8,21;
Найдено: C 53,88; H 5,67; N 6,78; S 15,37; SH 4,91.
Пример 12.
[3R- [3α, 6α(S*)-9aβ] ]]-Октагидро-6-[[2-(меркаптометил)-1-оксо-3- фенилпропил]амино]-5-оксо-тиазоло 3,2-а адазепин-3-карбоновая кислота
Повторяя процедуру примера 11, но с использованием в части (f) (S)-2-[(ацетилтио)метил] бензолпропановой кислоты, получали целевой продукт в виде белого твердого вещества, т.пл. 212 - 213oC, Rf = 0,55 (5% уксусная кислота в этилацетате), [α]D = -36,5o (c = 0,36, диметилформамид).
ВЭЖХ: колонка УМС S3 ODS (6,0 • 150 мм): элюирование 44% A: 90% воды - 10% метанола - 0,2 фосфорной кислоты, и 56% B: 10% воды - 90% метанола - 0,2% фосфорной кислоты, скорость потока - 1,5 мл/мин с детекцией при 220 нм, tR = 15,96 мин показало чистоту 99,2%.
Анализ для C19H24N2O4S2:
Вычислено: C 55,86; H 5,92; N 6,86; S 15,70; SH 8,09;
Найдено: C 55,99; H 6,01; N 6,75; S 15,70; SH 7,81.
Пример 13.
[S-(R*, R*)] -3,4-Дигидро-3-[[2-(меркаптометил)-1- оксо-3-фенилпропил] амино]-4-оксо-1,5-бензоксазепин-5(2Н)-уксусная кислота
a) N-[(1,1-Диметилэтокси)карбонил]-O-(2-нитрофенил)-L-серин
Раствор N-[(1,1-диметилэтокси)карбонил] -L-серина (24,3 г, 0,118 М) в сухом диметилформамиде (25 мл) по капле, в течение 1 ч, добавляли к охлажденной (0oC, ледяная/солевая баня) суспензии 60% гидрида натрия (10,1 г, 0,25 М) в сухом диметилформамиде (200 мл) и полученную смесь размешивали при 0oC до тех пор, пока не прекратится пенообразование (около 2 ч). Реакционную смесь по капле в течение 20 мин обрабатывали 1-фторо-2-нитробензолом (14,3 мл, 0,13 М), размешивали при 0oC и в присутствии аргона в течение 4 ч, а затем выливали в ледяную воду (750 мл) и экстрагировали этилацетатом (2 • 100 мл). Водную фазу доводили до pH 1,0 с помощью 6 н. соляной кислоты (70 мл), экстрагировали этилацетатом (3 • 500 мл) и объединенные органические экстракты промывали солевым раствором (100 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенную продуктовую смесь хроматографировали на колонке с силикагелем (Merck), элюируя смесью метиленхлорида, метанола и уксусной кислоты (100:5:0,2), в результате чего получали 27,22 г продукта в виде вязкого желтого сиропа, Rf = 0,27 (метиленхлорид : метанол : уксусная кислота, 100:5:0,5).
b) N-[(1,1-Диметилэтокси)-карбонил]-O-(2-аминофенил)-L-серин
Раствор продукта, полученного в части (a), (27,1 г, 83 мМ) в сухом метаноле (500 мл) обрабатывали 10% палладированным углем (900 мг) и гидрогенизировали при 40 фунт/кв.дюйм (275,76 кПа) в течение 2 ч. Реакционную смесь фильтровали через прокладку из целита в миллипористом фильтре, тщательно промыв эту прокладку метанолом (5 • 100 мл). Темный фильтрат выпаривали досуха и осушали в вакууме, в результате чего получали темный твердый продукт. Этот неочищенный продукт растирали со смесью метиленхлорида и гексана (1:4) и получали 17,69 г светло-рыжевато-коричневатого твердого вещества, Rf = 0,15 (метиленхлорид : метанол : уксусная кислота, 20:1:1).
c) (S)-3-[[(Диметилэтокси)карбонил] амино]-2,3-дигидро-1,5- бензоксазепин-4(5Н)-он
Раствор продукта, полученного в части (b), (16,69 г, 56,3 мМ) в сухом диметилформамиде (121 мл) обрабатывали 1-этил-3-(3-диметиламинопропил) карбодиимидом (10,64 г, 55,5 мМ) и размешивали при комнатной температуре в течение 3 ч. Реакционную смесь распределяли между этилацетатом (2 • 492 мл) и 1,0 н. бикарбонатом натрия (492 мл) и объединенные органические экстракты промывали водой (3 • 492 мл), солевым раствором (492 мл), осушали безводным сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт хроматографировали на колонке с силикагелем (Merck), элюируя смесями этилацетата и гексана (1:4, 1:2, 1:1), в результате чего получали 10,5 г продукта в виде беловатых кристаллов, Rf = 0,40 (этилацетат : гексан, 1:4).
d) (S)-3-[[(Диметилэтокси)карбонил] амино]-3,4-дигидро-4- оксо-1,5-бензоксазепин-5(2Н)-уксусная кислота, этиловый сложный эфир
Раствор продукта, полученного в части (c), (1,0 г, 3,59 мМ) в сухом тетрагидрофуране (10 мл) охлаждали до 0oC (в ледяной/солевой бане) в присутствии аргона, обрабатывали измельченным в порошок гидроксидом калия (259 мг, 4,6 мМ) и бромидом тетрабутиламмония (172 мг, 0,53 мМ), размешивали в течение 5 минут, а затем обрабатывали этил 2-бромацетатом (0,50 мл, 1,2 экв. ). Реакционную смесь размешивали при комнатной температуре и в присутствии в течение 19 ч, распределяли между метиленхлоридом (2 • 25 мл) и водой (13 мл), а объединенные органические экстракты промывали водой (2 • 13 мл), солевым раствором (10 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенную продуктовую смесь хроматографировали на силикагеле (Merck), элюируя смесями этилацетата и гексана (1: 9, 1:4), в результате чего получали 860 мг продукта в виде сиропа, Rf = 0,67 (этилацетат : гексаны, 1:1).
e) (S)-3-Амино-3,4-дигидро-4-оксо-1,5-бензоксазепин-5(2Н)- уксусная кислота, этиловый сложный эфир, гидрохлоридная соль
Раствор продукта, полученного в части (d), (4,0 г, 11 мМ) в сухом диоксане (85 мл) обрабатывали 4,0 М соляной кислоты/диоксаном (33,6 мл, 0,134 М или 12,2 экв.) и размешивали 20 ч при комнатной температуре. Реакционную смесь упаривали досуха, выпаривая полученный сироп из толуола (2х) и этанола (1х), а затем осушали в вакууме, в результате чего получали 3,466 г продукта в виде слегка золотистого сиропа, Rf = 0,48 (метиленхлорид : метанол, 9:1).
f) [S(R*, R*)] -3-[[2-[(Ацетилтио)метил]-1-оксо-3- фенилпропил]амино]-3,4-дигидро-4-оксо-1,5-бензоксазепин-5(2Н)-уксусная кислота, этиловый сложный эфир
(S)-2-[(Ацетилтио)метил] бензолпропановую кислоту [полученную из эфедриновой соли в соответствии с приведенным выше описанием] подвергали реакции с продуктом, полученным в части (e), которую проводили в соответствии с процедурой, описанной в примере 1 (часть (b)), в результате чего получали целевой продукт белого пенистого сиропа, Rf = 0,62 (этилацетат : гексан, 1: 1).
g) [S-(R*,R*)]-3,4-Дигидро-3-[[2-(меркаптометил)-1 -оксо-3-фенилпропил] амино]-4-оксо-1,5-бензазепин-5(2Н)-уксусная кислота
Раствор продукта, полученного в части (f), в метаноле обрабатывали 1,0 н. гидроксилом натрия в соответствии с процедурой, описанной в примере 1 (часть (c)), в результате чего получали целевой продукт в виде аморфного твердого вещества, Rf = 0,62 (метиленхлорид : метанол : уксусная кислота, 20:1:1), [α]D = -140,2o (c = 0,61, метанол).
1H ЯМР (400 МГц, CDCl3): 1,43 (т, 1H); 2,51 - 2,90 (м, 5H); 4,17 (т, 1H, J = 10 Гц); 4,29 (д, 1H, J = 17 Гц); 4,69 (т, 1H, J = 7 Гц); 4,71 (д, 1H, J = 17 Гц); 6,76 (д, 1H); 7,05 - 7,26 (м, 9H).
Анализ для C21H22N2O5S:
Вычислено: C 60,86; H 5,35; N 6,76; S 7,74; SH 7,98;
Найдено: C 60,85; H 5,54; N 6,74; S 7,66; SH 6,29.
Пример 14.
[S, (R*, R*)]-3,4-Дигидро-3-[(2-меркапто-1-оксо-3- фенилпропил)-амино]-4-оксо-1,5-бензоксазепин-5(2Н)-уксусная кислота
a) [S, R*R*)-3-[[2-(Ацетилтио)-1-оксо-3-фенилпропил] амино]-3,4-дигидро-4-оксо-1,5-бензоксазепин-5(2Н)-уксусная кислота, этиловый сложный эфир
(S)-2-(Ацетилтио)бензолпропановой кислоты дициклогексиламиновую соль (1,35 г, 3,81 мМ) суспендировали в этилацетате (120 мл), промывали 5% бисульфатом калия (5 • 20 мл) и солевым раствором (25 мл), осушали безводным сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме, в результате чего получали свободную кислоту.
Эту свободную кислоту растворяли в сухом метиленхлориде (25 мл), охлаждали до 0oC в ледяной/солевой бане, обрабатывали 1-гидроксибензолтриазолгидратом (541 мг, 4,0 мМ) и 1-этил-3-(3-диметиламинопропил)карбодиимидом (792 мг, 4,13 мМ) и размешивали 1 ч при 0oC в атмосфере аргона. Затем раствор обрабатывали (S)-3-амино-3,4-дигидро-4-оксо-1,5-бензоксазепин-5(2Н)- уксусной кислоты этиловым сложным эфиром (1,147 г, 3,81 мМ) и 4-метилморфолином (0,47 мл, 4,2 мМ), размешивали 1 ч при 0oC, 1 ч при комнатной температуре. Реакционную смесь разбавляли этилацетатом (100 мл), промывали последовательно водой (15 мл), 5% бисульфатом калия (2 • 15 мл), водой (15 мл), насыщенным бикарбонатом натрия (15 мл) и солевым раствором (15 мл), а затем осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт хроматографировали на силикагеле (Merck), элюируя смесью этилацетата и гексана (1:4), в результате чего получали 1,21 г продукта в виде сиропа. Было также получено еще 409,6 мг продукта (изомерную смесь, 1:1) Rf = 0,67 (этилацетат : гексан, 1:4, а затем 1:1).
b) (S, R*,R*)-3,4-дигидро-3-[(2-меркапто-1-оксо-1- 3-фенилпропил)амино] -4-оксо-1,5-бензоксазепин-5(2Н)-уксусная кислота
Раствор этилэфирного продукта, полученного в части (a), (1,81 г, 2,51 мМ), в метаноле (13 мл) продували 30 мин аргоном, охлаждали до 0oC в солевой/ледяной бане, а затем по капле обрабатывали предварительно продутым (аргон, 30 мин) раствором 1,0 н. гидроксида натрия (10 мл, 4,0 экв.), при этом во время добавления и в процессе всей реакции, раствор барботировали аргоном. Реакционную смесь размешивали 1 ч при 0oC, подкисляли при 0oC 5% бисульфатом калия (45 мл) до pH 1 и экстрагировали этилацетатом (2 • 80 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт растворяли в метиленхлориде (10 мл) и частями обрабатывали гексаном (100 мл), в результате чего образовывалось твердое вещество. Надосадочную жидкость декантировали, а твердый остаток растирали с гексаном (50 мл) и пентаном (2 • 100 мл), размешивая 4 ч с первыми 100 мл пентана, в течение ночи со следующими 100 мл пентана в атмосфере аргона. Затем полученный продукт осушали в вакууме в течение 6 ч и получали в результате 969,7 мл в виде аморфного твердого вещества, Rf = 0,47 (метиленхлорид : метанол : уксусная кислота, 20:1:1), [α]D = -143,3o (c = 0,54, метанол).
1H ЯМР (400 МГц, CDCl3): 2,02 (д, 1H); 3,07 (м, 2H); 3,59 (м, 1H; 4,09 (т, 1H, J = 10 Гц); 4,24 (д, 1H, J = 17 Гц); 4,63 (т, 1H, J = 8 Гц); 4,73 (д, 1H, J = 17 Гц); 4,93 (м, 1H); 7,04 - 7,26 (м, 9H); 7,42 (д, 1H).
Анализ для C20H20N2O5S • 0,147 C5H12 • 0,43 H2O:
Вычислено: C 59,46; H 5,44; N 6,69; S 7,65; SH 7,89;
Найдено: C 59,46; H 5,25; N 6,65; S 7,88; SH 7,84.
Пример 15.
[3S-[1(R*), 3α (R*)[-3-[(2-Меркапто-1-оксо-3-фенилпропил)амино]- α- метил-2-оксогексагидро-1Н-азепин-1-уксусная кислота
a) [3S-[1(R*), 3α (R*)]-3-[[2-(ацетилтио)-1- оксо-3-фенилпропил]амино]- α- метил-2-оксогексагидро-1Н-азепин-1-уксусная кислота, метиловый сложный эфир
Холодный (0oС) раствор [S-(R*,R*)[-3-аминогексагидро- α- метил-2-оксо-1Н-азепин-1-уксусной кислоты метилового сложного эфира (374 мг, 1,75 мМ), полученного в соответствии с известным описанием (Thotsett и др., "Peptides : Structure and Function", Proceeding 8th American Peptide Sumposium, с. 555 (1983)) и (S)-2-(ацетилтио)бензопропановой кислоты (393 мг, 1,75 мМ) в метиленхлориде (10 мл) обрабатывали триэтиламином (245 мкл, 178 мг, 1,76 мМ), а затем бензотриазол-1-илокси-трис диметиламино фосфора гексафторофосфатом (775 мг, 1,65 мМ). Прозрачный, почти бесцветный раствор размешивали 1 ч при 0oC, а затем 3 ч при комнатной температуре. После этого смесь распределяли между этилацетатом и 0,5 н. соляной кислотой, а этилацетатный экстракт промывали последовательно водой, наполовину насыщенным бикарбонатом натрия и солевым раствором. Раствор осушали сульфатом натрия, фильтровали и упаривали. Остаток подвергали флеш-хроматографии на силикагеле (Merck), элюируя смесью этилацетата и гексана (70:30), в результате чего получали 590 мг чистого продукта в виде бесцветного маслянистого/пенистого вещества, Rf = 0,43 (этилацетат : гексан, 75:25).
b) [3S-[1(R*), 3α (R*)]]-3-[(2-Меркапто-1-оксо-3- фенилпропил)амино]- α- метил-2-оксогексагидро-1Н-азепин-1-уксусная кислота
Охлажденный льдом раствор продукта, полученного в части (a), (570 мг, 1,36 мМ) в метаноле (7 мл, дезоксигенированного посредством барботирования аргоном) обрабатывали 1 н. Гидроксидом натрия (5,5 мл, дезоксигенированного посредством барботирования аргоном). После 1,25-часового размешивания при 0oC, раствор подкисляли 1 н. соляной кислотой (9 мл) и экстрагировали этилацетатом. Этилацетатный экстракт промывали водой и солевым раствором, а затем осушали сульфатом натрия, фильтровали и упаривали, в результате чего получали маслянистое вещество. Это вещество подвергали флеш-хроматографии на силикагеле (Merck), (2% уксусная кислота в этилацетата). Нужные фракции объединяли и упаривали, а остаток растворяли в этилацетате и промывали последовательно водой и солевым раствором, а затем осушали сульфатом натрия, фильтровали и снова упаривали. Полученное маслянистое/пенистое вещество растирали сначала с этиловым эфиром, а затем с гексаном, в результате чего получали белое маслянистое-пенистое вещество. Эту смесь упаривали досуха, суспендировали в гексане, снова упаривали досуха и осушали в вакууме, в результате чего получали 470 мг продукта в виде белой пены; ЯМР-анализ показал, что полученный продукт имел диастереометрическую чистоту свыше 98%, Rf = 0,49 (5% уксусная кислота в этилацетате), [α]D = -37,1o (c = 0,6, хлороформ).
ВЭЖХ: колонка УМС S3 ODS (6,0 • 150 мм), элюирование 44% A: 90 % воды - 10% метанола - 0,2% фосфорной кислоты, и 56% B : 10% воды - 90% метанола - 0,2% фосфорной кислоты, скорость потока 1,5 мл/мин с детекцией при 220 нм, tR = 11,02 (98,0%).
Анализ для C21H22N2O4S2:
Вычислено: C 59,32; H 6,64; N 7,69; S 8,80; SH 9,07;
Найдено: C 59,27; H 6,92; N 7,37; S 8,36; SH 8,75.
Пример 16.
[6R(S*)-2,3,6,7-Тетрагидро-6-[[2-(меркаптометил)-1-оксо-3- фенилпропил] амино]-5-оксо-1,4-тиазепин-4(5Н)-уксусная кислота
a) [2-[(Метилсульфонил)окси] этил]карбаминовая кислота, 1,1-диметилэтиловый сложный эфир
Ди-трет-бутилдикарбонат (21,8 г, 100 мМ) по частям добавляли к размешанному раствору этаноламина (6,1 г, 100 мМ) в дихлорометане (100 мл) при 0 - 5oC. После завершения добавления, охлаждающую баню удаляли и раствор 5 ч размешивали при комнатной температуре. Растворитель удаляли в вакууме, а полученный материал растворяли в дихлорметане (100 мл) и триэтиламине (21 мл, 150 мМ), охлажденном до -10oC и по капле в течение 10 мин обрабатывали метансульфонилхлоридом (9,25 г, 150 мМ). Смесь разбавляли этилацетатом (200 мл) и промывали 1 н. соляной кислотой и водой. Этилацетатный слой осушали сульфатом магния, фильтровали, а растворитель удаляли в вакууме, в результате чего получали продукт в виде вязкого маслянистого вещества, которое затем использовали без очистки.
b) S-[2-[[(1,1-Диметилэтокси)карбонил] амино] этил] -N-[фенил- метокси)карбонил]-L-цистеин, дифенилметиловый сложный эфир
Раствор N-[(фенилметокси)карбонил]-L-цистеина дифенилметилового сложного эфира (12,7 г, 30,1 мМ) и продукта, полученного в части (a), (7,20 г, 30,1 мМ) в диметилформамиде (190 мл) обрабатывали путем распыления осушенного фторида калия (25 г, 430 мМ) и размешивали 20 ч при 70 - 75oC в атмосфере аргона. После охлаждения, смесь распределяли между этилацетатом (800 мл) и водой (400 мл). Органический слой промывали солевым раствором (300 мл), осушали сульфатом магния, фильтровали, раствор удаляли в вакууме и получали в результате маслянистое вещество. Это вещество хроматографировали на силикагеле (Merck, ≈ 1000 мл), элюируя смесями этилацетата и гексана (1:4, 1:3 и 1: 2), в результате чего получали 9,13 г продукта, Rf = 0,35 (этилацетат:гексан, 1:2).
c) (R)-Тетрагидро-6-[[(фенилметокси)карбонил] амино] -1,4- тиазепин-5(4H)он
Раствор продукта, полученного в части (b), (6,13 г, 10,86 мМ) в анизоле (30 мл) и трифторуксусной кислоте (45 мл) и размешивали 2 ч при комнатной температуре, а затем концентрировали в вакууме. Маслянистый остаток разводили изопропиловым эфиром (40 мл), размешивали, а изопропилэфирный слой декантировали. Эту процедуру повторяли еще один раз, после чего остаток осушали в вакууме. Смесь полученного материала и дифенилфосфорилазида (4,4 г, 16 мМ) в сухом диметилфомамиде (75 мл) в атмосфере аргона охлаждали в ледяной бане и, размешивая, по капле добавляли 4-метилморфолин (4,45 г, 44 мМ). После завершения добавления, смесь нагревали до комнатной температуры и оставляли размешиваться в течение 20 ч. Затем эту смесь распределяли между этилацетатом (150 мл) и водой (100 мл). Слои разделяли и водный слой снова экстрагировали этилацетатом (50 мл). Объединенные органические слои промывали насыщенным раствором бисульфата калия (30 мл), насыщенным раствором бикарбоната натрия (50 мл) и солевым раствором (50 мл), осушали сульфатом магния, а растворитель удаляли в вакууме. Остаток растирали с изопропиловым эфиром и получали в результате 2,45 г белого твердого продукта, Rf = 0,27 (этилацетат : гексаны, 1:1).
d) (R)-Тетрагидро-5-оксо-6-[[(фенилметокси)карбонил]амино]-1,4- тиазепин-4(5H)-уксусная кислота, этиловый сложный эфир
Раствор продукта, полученного в части (c), (3,55 г, 12,68 мМ) в дистиллированном тетрагидрофуране (40 мл) в атмосфере аргона охлаждали до 0oC и обрабатывали измельченным в порошок гидроксидом калия (2,16 г, 40 мМ) и бромидом тетрабутиламмония (420 мг, 1,3 мМ). Затем по капле добавляли этилбромоацетат (2,58 г, 15,43 мМ). Холодную смесь размешивали 2 ч, а затем разводили этилацетатом (50 мл) и добавляли безводный сульфат магния. Реакционную смесь фильтровали через слой сульфата магния и этот слой несколько раз промывали этилацетатом. Фильтрат обрабатывали 1 н. раствором соляной кислоты (50 мл) и слои разделяли. Органический слой осушали сульфатом магния, фильтровали, растворитель удаляли в вакууме, в результате чего получали маслянистое вещество, которое очищали с помощью хроматографии на силикагеле, элюируя 10 - 30% этилацетатом в гексане, с получением 3,22 г продукта в виде маслянистого вязкого вещества, Rf = 0,60 (этилацетат : гексан, 1:1).
e) (R)-6-Аминотетрагидро-5-оксо-1,4-тиазепин-4(5H)уксусная кислота, этиловый сложный эфир, гидробромидная соль
Продукт, полученный в части (d), (3,17 г, 8,65 мМ) обрабатывали 30% бромоводородом в уксусной кислоте (14 мл). Смесь размешивали при комнатной температуре в течение 1 ч, а затем добавляли диэтиловый эфир (200 мл). После 1-часового размешивания при комнатной температуре, осажденную соль собирали путем фильтрации и несколько раз промывали простым эфиром. Полученный слегка смолистый материал растворяли в горячем этаноле, а растворитель удаляли в вакууме. Эту процедуру повторяли еще один раз. Затем к остатку добавляли диэтиловый эфир и путем фильтрации собирали гидробромидную соль (2,52 г) в виде желтого твердого продукта.
f) [6R(S*)] -2,3,6,7-Тетрагидро-6-[[2-[(ацетилтио)метил] -1-оксо-3-фенилпропил]амино]-5-оксо-1,4-тиазепин-4(5H)-уксусная кислота, этиловый сложный эфир
(S)-2-[(Ацетилтио)метил] бензопропановую кислоту (полученную из эфедриновой соли, как было описано ранее) подвергали реакции с продуктом, полученным в части (e), в соответствии с процедурой примера 1 (часть (b)), в результате чего получали целевой продукт, Rf = 0,40 (этилацетат : гексан, 1:1).
g) [6R(S*)]-2,3,6,7-Тетрагидро-6-[[2-(меркаптометил)-1- оксо-3- фенилпропил]амино]-5-оксо-1,4-тиазепин-4(5H)-уксусная кислота
Раствор продукта, полученного в части (f), в метаноле обрабатывали 1 н. гидроксидом натрия в соответствии с процедурой описанной в примере 1 (часть (c)), в результате чего получали целевой продукт в виде твердого пенистого вещества, т. пл. 85 - 101oC, Rf = 0,35 (8% метанол в дихлорметане + 2 капли уксусной кислоты/5 мл), [α]D = -9,5o (c = 0,6, метанол).
ВЭЖХ : RT = 13,2 мин, 50% водного метанола, содержащего 0,2% фосфорную кислоту, 1,05 мл/мин, детектирование при 220 нм, YMCS 3 (ODS), 6,0 • 150 мм колонка со сферическим колпачком (3 мкм). H.I => 95%.
Анализ для C17H22N2O4S2 • 0,15 H2O • 0,15 C6H14:
Вычислено: C 60,24; H 5,71; N 6,11; S 13,98;
Найдено: C 60,01; H 5,77; N 5,94; S 13,64.
Пример 17. [6R(S*)-2,3,6,7-Тетрагидро-6-[(2-меркапто-1- оксо-3-фенилпропил)-амино]-5-оксо-1,4-тиазепин-4(5H)-уксусная кислота
a) [6R(S*)-2,3,6,7-Тетрагидро-6-[[2-(ацетилтио)-1-оксо-3- фенилпропил)-амино]-5-оксо-1,4-тиазепин-4(5H)-уксусная кислота, этиловый сложный эфир
(S)-2-(Ацетилтио)бензопропановую кислоту (полученную из дициклогексиламиновой соли, как было описано ранее) подвергали реакции с продуктом, полученным в примере 16 (часть (e)), в соответствии с процедурой, описанной в примере 2 (часть (b)), в результате чего получали целевое соединение, Rf = 0,29 (этилацетат : гексан, 1:1).
b) [6R(S*)-2,3,6,7-Тетрагидро-6-[(2-меркапто-1-оксо-3- фенилпропил)-амино]-5-оксо-1,4-тиазепин-4(5H)-уксусная кислота
Раствор продукта, полученного в части (a), в метаноле обрабатывали 1,0 н. гидроксидом натрия в соответствии с процедурой описанной в примере 2 (часть (c)), в результате чего получали целевой продукт в виде твердого пенистого вещества, т.пл. 69 - 76oC Rf = 0,29 (8% метанол в дихлорметане + 2 капли уксусной кислоты (5 мл).
Анализ для C22H24N2O4S2 • 0,1 C4H8O2:
Вычислено: C 52,21; H 5,56; N 7,43; S 17,00;
Найдено: C 52,27; H 5,54; N 7,35; S 16,98.
Пример 18.
[3R-[ 3α, 6α (S*)] ] -Тетрагидро-6-[(2-меркапто-1-оксо-3- фенилпропил)амино]-5-оксо-фенил-1,4-тиазепин-4(5H)-уксусная кислота
a) (3R-цис)-Тетрагидро-5-оксо-3-фенил-6-[[(фенилметокси) карбонил] -амино]-1,4-тиазепин-4(5H)-уксусная кислота, этиловый сложный эфир
Раствор (3R-цис)-тетрагидро-3-фенил-6-[[(фенилметокси) карбонил]амино] -1,4-тиазепин-5(4H)-он'а (1,16 г, 3,25 мМ), полученного в соответствии с известным описанием (Yanagisawa и др. J. Med. Chem., стр. 1984-1991 (1987)), в дистиллированном тетрагидрофуране (30 мл) и в атмосфере аргона охлаждали до 0oC и обрабатывали измельченным в порошок гидроксидом калия (540 мг, 10 мМ) и бромидом тетрабутиламмония (97 мг, 0,3 мМ). Затем, размешивая, по капле добавляли этилбромоацетат (501 мг, 3 мМ). Эту смесь в охлажденном состоянии размешивали 2 ч, а затем добавляли еще 501 мг (3 мМ) этилбромоацетата. Полученную холодную смесь размешивали еще 2 ч, а затем разводили этилацетатом (50 мл) и добавляли безводный сульфат магния. Реакционную смесь фильтровали через слой сульфата магния, после чего этот слой несколько раз промывали этилацетатом. Фильтрат выливали в холодный 1 н. раствор соляной кислоты (40 мл) и слои разделяли. Органический слой осушали сульфатом магния, фильтровали и, после удаления растворителя в вакууме, получали маслянистое вещество, которое очищали с помощью хроматографии на силикагеле, элюируя 10 - 30% этилацетатом в гексане, в результате чего получали 730 мг продукта, Rf = 0,32 (этилацетат : гексаны, 1:2).
(b) (3R-цис)-6-Аминотетрагидро-5-оксо-3-фенил-1,4-тиазепин- 4(5H)-уксусная кислота, этиловый сложный эфир, гидробромидная соль
Продукт, полученный в части (a), (1,43 г, 3,23 мМ) обрабатывали 30% бромводородом в уксусной кислоте (5,5 мл). Смесь размешивали 2 ч при комнатной температуре, а затем добавляли диэтиловый эфир (100 мл). После 1-часового размешивания при комнатной температуре, осажденную соль собирали путем фильтрации и несколько раз промывали эфиром, в результате чего получали 1,08 гидробромидной соли в виде бежевого твердого продукта.
c) [3R-[ 3α, 6α (S*)]]-6-[[2-(Ацетилтио)-1-оксо-3- фенилпропил]амино]-тетрагидро-5-оксо-3-фенил-1,4-тиазепин-4(5H)- уксусная кислота, этиловый сложный эфир
Суспензию дициклогексиламиновой соли (S)-2-(ацетилтио)бензолпропановой кислоты (736 г, 1,81 мМ) в этилацетате (30 мл) дважды промывали 0,1 н. соляной кислотой (40 мл, 230 мл) и один раз солевым раствором (20 мл), затем осушали сульфатом магния, фильтровали и после удаления растворителя в вакууме получали свободную кислоту в виде маслянистого вещества.
Гидробромидную соль продукта, полученного в части (b), (640 мг 1,65 мМ) растворяли в дихлорометане (30 мл), промывали раствором бикарбоната натрия (2•20 мл), а затем солевым раствором (20 мл), осушали сульфатом магния, фильтровали и после удаления растворителя в вакууме получали свободный амин в виде вязкого маслянистого продукта (495 мг). Этот продукт и свободную кислоту, полученную как описано выше, растворяли в дихлорометане (20 мл) в присутствии аргона и охлаждали до 0oC. После этого добавляли 1-гидроксибензотриазолгидрат (256 мг, 1,90 мМ) и 1-этил-3-(3-диметиламинопропил)карбодиимид (362,5 мг, 1,90 мМ). Полученную холодную смесь размешивали 3 ч, а затем разводили дихлорометаном (30 мл). Раствор промывали водой (15 мл), 5%-ным раствором бисульфата калия (15 мл), раствором бикарбоната натрия (15 мл) и водой (15 мл). Дихлорметановый раствор осушали сульфатом магния, фильтровали, а растворитель удаляли в вакууме. Оставшийся материал хроматографировали на силикагеле (Merck, приблизительно 150 мл), элюируя 25% этилацетатом в гексане и получали 610 мг продукта, Rf = 0,53 (этилацетат : гексан, 1:1).
d) [3R-[ 3α, 6α (S*)] ]-Тетрагидро-6-[(2-меркапто-1-оксо-3- фенилпропил)амино]5-оксо-3-фенил-1,04-тиазепин-4(5H)-уксусная кислота, смесь метилового и этилового сложных эфиров
Раствор продукта, полученного в части (c), (610 мг, 1,185 мМ) в метаноле (12 мл) продували 15 мин аргоном, охлаждали в ледяной/солевой бане и, продолжая продувать аргоном и размешивая, обрабатывали по капле 1 н. раствором гидроксида натрия (4,75 мл), который за 30 мин до его использования продували аргоном. Охлажденную смесь размешивали в течение 75 мин, а затем подкисляли до pH 1 с помощью 5%-ного раствора бисульфата калия. Продукт экстрагировали в этилацетат (2 • 40 мл). Объединенные органические экстракты промывали солевым раствором, осушали сульфатом магния, фильтровали и после удаления растворителя в вакууме получали 533 мг продукта, который медленно кристаллизовался. ЯМР и масс-спектроскопия показали, что этот продукт представляет собой смесь метилового и этилового сложных эфиров, Rf = 0,76 (5% метанол в метиленхлориде + 2 капли уксусной кислоты/5 мл).
e) [3R-[ 3α, 6α (S*)] ]-Тетрагидро-6-[(2-меркапто-1-оксо-3- фенилпропил)амино]-5-фенил-1,04-тиазепин-4(5H)-уксусная кислота
Продукт, полученный в части (d), (480 мг, приблизительно 1,03 мМ) добавляли к метанолу (12 мл). После этого образовывались кристаллы. Затем добавляли дистиллированный тетрагидрофуран (5 мл), и смесь приобретала вид прозрачного раствора, который в течение 15 мин продували аргоном, охлаждали в ледяной/солевой бане и, продолжая при этом продувать аргоном и размешивать, обрабатывали по капле 1 н. раствором гидроксида натрия (4 мл), который за 30 мин до использования продували аргоном. Смесь выдерживали в охлажденном состоянии и в атмосфере аргона в течение 27 ч. Проведенная в это время TCX показала, что гидролиз сложного эфира завершился. Затем смесь подкисляли до pH 1 с использованием 5%-ного раствора бисульфата калия. Продукт экстрагировали в дихлорметан (2•30 мл). Объединенные органические экстракты промывали солевым раствором, осушали сульфатом магния, фильтровали и после удаления растворителя в вакууме получали белое твердое пенистое вещество. Это вещество растворяли в дихлорметане и добавляли гексан до получения мутного раствора. Затем растворитель удаляли в вакууме и получали твердый остаток. Этот остаток растирали с 20% гексаном в эфире. Белое твердое вещество собирали путем фильтрации, промывали холодным 50% гексаном в эфире и получали 350 мг продукта, т.пл. 175 - 182oC, Rf=0,42 (5% метанол в метиленхлориде + 2 капли уксусной кислоты/5 мл), [α]D = +26,7o (с = 0,8, метанол).
ВЭЖХ: RT = 8,0 мин, 66,8% водного метанола, содержащего 0,2% фосфорную кислоту, 1,5 мл/мин, детекция при 220 нм, УМС S-3 (ODS), 6,0 x 150 мм, колонка со сферической крышкой размером 3 микрон. H.1>95%.
Анализ для C22H24N2O4S2:
Вычислено: C 59,44; H 5,44; N 6,30; S 14,42;
Найдено: C 59,52; H 5,54; N 6,08; S 14,14.
Пример 19.
[3R-[ 3α, 6α (S*)] ]-Тетрагидро-6-[[2-(меркаптометил)-1-оксо-3-фенилпропил] амино]-5-оксо-3-фенил-14-тиазепин-4(5H)-уксусная кислота
a) [3R-[ 3α, 6α (S*)]]-тетрагидро-6-[[2-(ацетилтиометил)-1-оксо-3- фенилпропил]амино]-5-оксо-3-фенил-1,4-тиазепин-4(5H)-уксусная кислота, смесь метилового и этилового сложных эфиров
Суспензию эфедриновой соли (S)-2-[(ацетилтио)метил]бензолпропановой кислоты (853 мг, 2,11 мМ) в этилацетате (40 мл) промывали дважды 0,1 н. соляной кислотой (40 мл, 20 мл) и один раз солевым раствором (20 мл), осушали сульфатом магния, фильтровали и после удаления растворителя в вакууме получали свободную кислоту в виде маслянистого вещества.
Смесь метилового и этилового сложных эфиров (3R-цис)-6-аминотетрагидро-5-оксо-3-фенил-1,4-тиазепин-4(5H-уксусной кислоты в виде гидробромидной соли (320 мг этилового сложного эфира, 0,82 мМ и 410 мг метилового сложного эфира, 1,09 мМ) полученную в соответствии с описанием в примере 18 (b) растворяли в дихлорметане (30 мл), промывали раствором бикарбоната натрия (2•20 мл), солевым раствором (20 мл), осушали сульфатом магния, фильтровали, а растворитель удаляли в вакууме, в результате чего получали свободный амин в виде вязкого маслянистого вещества (495 мг, 98%). Это вещество и полученную ранее свободную кислоту растворяли в дихлорметане (20 мл) в атмосфере аргона и охлаждали до 0oC. Затем добавляли 1-гидроксибензотриазолгидрат (285 мг, 2,11 мМ) и 1-этил-3-(3-диметиламинопропил)карбодиимид (403 мг, 2,11 мМ. Эту холодную смесь размешивали 3,5 ч, а затем разводили дихлорметаном (30 мл). Полученный раствор промывали водой (15 мл) 5% бисульфатом калия (15 мл), бикарбонатом натрия (15 мл) и водой (15 мл). Дихлорметановый раствор осушали сульфатом магния, фильтровали, а растворитель удаляли в вакууме. Остаток хроматографировали на силикагеле (Merck, 170 мл), элюируя 25% этилацетатом в гексане, в результате чего получали 535 мг продукта; Rf = 0,68 м 0,63 (этилацетат : гексан, 1:1).
b) [3R-[ 3α, 6α (S*)]]-Тетрагидро-6-[[2-(меркаптометил)-1-оксо-3- фенилпропил]амино]-5-оксо-3-фенил-1,4-тиазепин-4(5H)-уксусная кислота
Продукт, полученный в части (a), (493 мг, около 0,98 мМ) растворяли в метаноле (12 мл) и продували 15 мин аргоном, охлаждали в ледяной/солевой бане и, продолжая продувать аргоном и при этом размешивая, обрабатывали по капле 1 н. раствором гидроксида натрия (4 мл), который за 30 мин до использования предварительно продували аргоном. Эту смесь выдерживали при охлаждении и в атмосфере аргона в течение 48 ч. ТСХ, проведенная в это время, показала, что гидролиз сложного эфира был завершен. Смесь подкисляли до pH 1 5%-ным бисульфатом калия. Полученный продукт экстрагировали в дихлорметан (2 • 30 мл). Объединенные органические экстракты промывали солевым раствором (20 мл), осушали сульфатом магния, фильтровали, а растворитель удаляли в вакууме, в результате чего получали белое стеклообразное пенистое вещество. К этому веществу добавляли 25% гексан в эфире (20 мл), и после размешивания образовывалось белое твердое вещество. Это вещество собирали путем фильтрации и промывали 50% гексаном в эфире, в результате чего получали 342 мг продукта, т. пл. 170 - 174oC, Rf = 0,43, (5% метанол в метиленхлориде + 2 капли уксусной кислоты/5 мл), [α]D = +32,8o (c = 0,9, метанол).
ВЭЖХ: RT = 8,6 мин, 66,8% водного метанола, содержащего 0,2% фосфорной кислоты, 1,5 мл/мин, детекция при 220 мин, УМС S-3 (ODS), 6,0 • 150 мм, колонка со сферической крышкой размером 3 мкм. H.1.>95%.
Анализ для C23H26N2O4S2:
Вычислено: C 60,24; H 5,71; N 6,11; S 13,98;
Найдено: C 60,25; H 5,77; 6,06; S 13,85.
Пример 20.
[2R-[2α, 6β (S*)] ]-Тетрагидро-6-[[2-(меркаптометил)-1-оксо-3-фенилпропил] амино]-5-оксо-2-фенил-1,4-тиазепин-4(5H)-уксусная кислота
a) (2R-транс)-Тетрагидро-2-фенил-6-[[(фенилметокси)карбонил] амино] -1,4-тиазепин-5(4H)-он
Суспензию (2R-транс)-6-аминотетрагидро-1,4-тиазепин-5(4H)-он'а (5,82 г), полученного в соответствии с известным описанием (Yanagisawa и др., J. Med. Shem., vol. 30, с. 1984-1991 (1987)) в смеси дихлорметана (125 мл) и триэтиламина (11 мл) обрабатывали дитретбутилкарбонатом (6 г) и размешивали 4 ч при комнатной температуре. Смесь разводили дихлорметаном (100 мл) и промывали водой. Дихлорметановый раствор осушали сульфатом магния, фильтровали и после удаления растворителя в вакууме получали бледно-желтое твердое вещество. Это вещество хроматографировали на силикагеле, элюируя смесью этилацетата и гексана (1: 1), а затем этилацетатом, в результате чего получали 2,01 г белого твердого продукта.
b) (2R-транс)-Тетрагидро-5-оксо-2-фенил-6-[[(фенилметокси)карбонил] амино]-1,4-тиазепин-4(5H)-уксусная кислота, метиловый сложный эфир
Раствор продукта, полученного в части (a), (1,95 г, 6,05 мМ) в дистиллированном тетрагидрофуране и в атмосфере аргона охлаждали до 0oC и обрабатывали измельченным в порошок гидроксидом калия (1,10 г, 18,15 мМ) и тетрабутиламмония бромидом (195 мг). Затем в течение 30 мин по капле добавляли этилбромоацетат (800 мкл, 7,26 мМ, 1,2 экв.). Смесь в холодном состоянии размешивали в течение 2 ч, а затем разводили дихлорметаном (300 мл) и водой (50 мл). После этого добавляли разбавленный раствор соляной кислоты (75 мл) и слои разделяли. Водный слой снова экстрагировали дихлорметаном (100 мл). Объединенные органические слои осушали сульфатом магния, фильтровали, а после удаления растворителя в вакууме получали белое пенистое вещество. Это вещество растворяли в смеси этилацетата и дихлорметана, а затем обрабатывали избыточным количеством раствора диазометана в эфире. Полученный метиловый сложный эфир очищали с помощью хроматографии на силикагеле, элюируя смесью этилацетата и гексана (1:1), и получали в результате 2,193 г продукта; Rf = 0,49 (этилацетат : гексан, 1:1).
c) (2R-транс)-6-Аминотетрагидро-5-оксо-2-фенил-1,4-тиазепин-5(H)- уксусная кислота, метиловый сложный эфир, гидрохлоридная соль
Продукт, полученный в части (b), (2,19 г, 5,56 мМ) охлаждали в ледяной бане и обрабатывали 4 н. раствором гидрохлоридной соли в диоксане (15 мл). Охлажденную смесь размешивали в течение 1 ч, и в этот период образовывался гель. После этого охлаждающую баню удаляли, а смесь размешивали при комнатной температуре в течение 1 ч. Растворитель удаляли в вакууме, добавляли толуол, а затем его удаляли в вакууме, в результате чего получали 1,84 г продукта в виде твердого вещества.
d) [2R-[ 2α, 6β (S*)]]-Тетрагидро-6-[[2-[(ацетилтио)метил]-1-оксо-3-фенилпропил] амино]-1-оксо-2-фенил-1,4-тиазепин-4(5H)-уксусная кислота, метиловый сложный эфир
(S)-2-[(Ацетилтио)метил] бензоилпропановую кислоту (полученную из эфедриновой соли в соответствии с приведенным выше описанием) подвергали реакции с продуктом, полученным в части (c), согласно процедуре, описанной в примере 1 (часть b), в результате чего получали целевой продукт; Rf = 0,51 (этилацетат : гексан, 1:1).
e) [2R-[ 2α, 6β (S*)] ]-Тетрагидро-6-[[2-(меркаптометил)-1-оксо-3-фенилпропил] амино]-6-оксо-2-фенил-1,4-тиазепин-4(5H)-уксусная кислота
Раствор продукта, полученного в части (d), в метаноле обрабатывали 1 н. гидроксидом натрия согласно процедуре примера 1 (часть (c)) и получали в результате целевой продукт, т.пл. 197 - 199oC, Rf = 0,41 (5% метанол в дихлорметане + 2 капли уксусной кислоты.5 мл), [α]D = +27,1o (c = 0,5, метанол).
Анализ для C23H26N2O4S2:
Вычислено: C 60,24; H 5,71; N 6,11; S 13,98;
Найдено: C 60,01; H 5,77; N 6,94; S 13,64.
Пример 21.
[2R-[ 2α, 6β (S*)]]-Тетрагидро-6-[(2-меркапто-1-оксо-3-фенилпропил)-амино] -5-оксо-2-фенил-1,4-тиазепин-4(5H)-уксусная кислота
a) [2R-[ 2α, 6β (S*)]-Тетрагидро-6-[[2-ацетилтио)-1-оксо-3-фенилпропил] амино]-5-оксо-2-фенил-1,4-тиазепин-4(5H)-уксусная кислота, метиловый сложный эфир
Суспензию дициклогексиламиновой соли (S)-2-(ацетилтио)бензолпропановой кислоты (1,13 г, 2,8 мМ) в этилацетате (50 мл) два раза промывали 0,1 н. соляной кислотой (50 мл, 25 мл) и один раз солевым раствором (25 мл), затем осушали сульфатом магния, фильтровали и после удаления растворителя в вакууме получали свободную кислоту в виде маслянистого вещества. Это вещество растворяли в дихлорметане (15 мл) в атмосфере аргона и охлаждали до 0oC. Затем добавляли 1-гидроксибензотриазолгидрат (405 мг, 3,0 мМ) и 1-этил-3-(3-диметиламинопропил)карбодиимид (592 мг, 3,1 мМ). Эту смесь в охлажденном состоянии размешивали 1 ч, после чего добавляли продукт гидрохлоридной соли амина, полученный в соответствии с описанием в примере 20 (c), (920 мг, 2,79 мМ), а затем добавляли 4-метилморфолин (306 мкл, 2,79 мМ). Смесь размешивали в холодном состоянии в течение 1 ч, а затем в течение 1 ч при комнатной температуре, после чего ее разводили дихлорметаном (70 мл). Раствор промывали водой (15 мл), 5%-ным раствором бисульфата калия (15 мл), раствором бикарбоната натрия (15 мл) и водой (15 мл). Дихлорметановый раствор осушали сульфатом магния, фильтровали, а растворитель удаляли в вакууме. Оставшийся материал хроматографировали на силикагеле (Merck, 160 мл), элюируя смесями этилацетата и гексана (1:3, а затем 1:2), в результате чего получали 880 мг продукта, Rf=0,57 (этилацетат:гексан, 1:1).
b[2R-[ 2α, 6β (SA)] ] -Тетрагидро-6-[2-меркапто-1-оксо-3-фенилпропил)амино]-5-оксо- 2-фенил-1,4-тиазепин-4(5H)-уксусная кислота
Раствор продукта, полученного в части (a), (880 мг, 1,75 мМ) в метаноле (12 мл) продували аргоном в течение 15 мин, охлаждали в ледяной/солевой бане и, продолжая продувать аргоном и размешивая, при этом обрабатывали по капле 1 н. раствором гидроксида натрия (7,0 мл), который за 30 мин до использования продували аргоном. Охлажденную смесь размешивали 70 минут, а затем подкисляли до pH 1 5%-ным раствором бисульфата калия. Полученный продукт экстрагировали в этилацетат (2•40 мл). Объединенные органические экстракты промывали солевым раствором, осушали сульфатом магния, фильтровали и после удаления растворителя в вакууме получали белый частично твердый материал. Затем добавляли эфир и белое твердое вещество собирали путем фильтрации, после чего его промывали добавочным количеством эфира и получали 494 мг продукта; т. пл. 181 - 183oC, Rf=0,43 (5% метанол в дихлорметане + 2 капли уксусной кислоты/5 мл), [α]D = +6,7o (с = 0,7, метанол).
ВЭЖХ: RT= 9,6 мин, 66,8% водного метанола, содержащего 0,2% фосфорной кислоты, 1,5 мл/мин, детекция при 220 нм, УМС S-3 (ODS), (6,0•150 мм), колонка со сферической крышкой размером 3 мкм. H.I.=98%. ЯМР показал присутствие приблизительно 0,1 М этилацетата.
Анализ для C22H24N2O4S2• 0,1 C4H8O2:
Вычислено: C 59,34; H 5,51; N 6,18; S 14,14;
Найдено: C 59,22; H 5,41; N 6,12; S 13,72.
Пример 22.
[R-(R*, S*)] -2,3,4,5-Тетрагидро-3-[(2-меркапто-1-оксо-3-фенилпропил)амино] -2-оксо-1H-бензазепин-1-уксусная кислота
a) (R)-2,3,4,5-Тетрагидро-3-амино-2-оксо-1H-бензазепин-1-уксусная кислота, этиловый спирт сложный эфир
2,3,4,5-Тетрагидро-3-амино-2-2-оксо-1H-беназепин-1-уксусной кислоты этиловый сложный эфир, полученный в соответствии с известным описанием (Watthey и др. , J.Med. Chem., 28, с. 1511 - 1516 (1985)) (14,8 г, 56,4 мМ), растворяли посредством его соли L-винной кислоты описанным выше способом, в результате чего получали S-амин (3,927 г), т.пл. 105 - 107oC; [α]D = -277o (c = 0,99, этанол). Остаток от объединенных маточных растворов (21,5 г, 52,1 мМ) суспендировали в этилацетате (340 мл), два раза промывали 10% NH4OH (41 мл, а затем 30 мл) и солевым раствором (25 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха, осушали в вакууме и получали аминовую смесь (7,01 г), обогащенную R-изомером. Раствор изомерной смеси (2,285 г, 8,71 мМ) в абсолютном этаноле (18 мл) обрабатывали D-винной кислотой (1,31 г, 8,73 мМ) и нагревали на паровой бане до тех пор, пока не образуется раствор. Этот прозрачный раствор охлаждали до комнатной температуры, оставляли на два дня отстаиваться, а затем охлаждали до 0oC (в ледяной/солевой бане) и оставляли в этом состоянии до тех пор, пока не будут образовываться кристаллы. Неочищенную соль (2,585 г) перекристаллизовали из абсолютного этанола (14 мл) и получали нужную соль (2,158 г), [α]D = +154,1o (c = 0,6 метанол). Затем полученную соль суспендировали в этилацетате (35 мл), промывали 10% NH4OH (2•4 мл) и солевым раствором (6 мл), осушали сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме, в результате чего получали 1,31 г продукта, Rf = 0,60 (метиленхлорид: метанол, 9:1), [α]D = +273,1o (c = 0,677, метанол).
b) [R-R*, S*)]-2,3,4,5-Тетрагидро-3-[[2-(ацетилтио)-1-оксо-3-фенилпропил] амино]-2-оксо-1H-бензазепин-1-уксусная кислота, этиловый сложный эфир.
(S)-2-(Ацетилтио)бензопропановую кислоту (полученную из дициклогексиламиновой соли в соответствии с приведенным выше описанием) подвергали реакции с продуктом, полученным в части (a), в соответствии с процедурой, описанной в примере 2 (часть (b)), в результате чего получали целевой продукт в виде сиропов; Rf = 0,57 (этилацетат:гексан, 1:1).
c) [R-(R*, S*)]-2,3,4,5-Тетрагидро-3-[(2-меркапто-1-оксо-3-фенилпропил) амино]-2-оксо-1H-бензазепин-1-уксусная кислота
Раствор продукта, полученного в части (b), в метаноле обрабатывали 1 н. гидроксидом натрия в соответствии с процедурой описанной в примере 2 (часть (c)), в результате чего получали целевой продукт в виде аморфного твердого вещества, Rf = 0,67 (метиленхлорид: метанол: уксусная кислота, 20:1:1), [α]D = -230,9o (c = 0,57, метанол).
Анализ для C21H22N2O4S;
Вычислено: C 63,30; H 5,56; N 7,03; S 8,05; SH 8,30;
Найдено: C 63,23; H 5,80; N 6,76; S 7,99; SH 7,67.
1H-ЯМР (400 МГц, CDCl3) δ : 1,65 (м, 1H), 2,06 (д, 1H), 2,51 (м, 2H), 2,94 (дд, 1H, J= 7,13 Гц), 3,17 (м, 2H), 3,40 (м, 1H), 4,39 (д, 1H), J=17 Гц); 4,45 (м, 1H), 4,67 (д, 1H, J= 17 Гц), 7,05 - 7,31 (м, 9H).
Пример 23.
[R-(R*, S*)] -2,3,4,4,5-Тетрагидро-3-[[2-(меркаптометил)-1-оксо-3- фенилпропил]амино]-2-оксо-1H-бензазапен-1-уксусная кислота
Повторяя процедуру примера 22, но с использованием (S)-2-(ацетилтио) метил бензопропановой кислоты в части (b), получали целевой продукт в виде аморфного твердого вещества; Rf = 0,53 (метиленхлорид:метанол:уксусная кислота, 20:1:1); [α]D = +253,9o (c = 0,38, метанол).
Анализ для C22H24N2O4S:
Вычислено: C 64,06; H 5,86; N 6,79; S 7,77;
Найдено: C 63,83; H 6,08; N 6,40; S 7,75.
1H-ЯМР (400 МГц, CDCl3) δ : 1,51 (м, 2H), 2,32 - 2,52 (м, 4H); 2,77 (м, 3H), 3,17 (м, 1H), 4,36 (д, 1H), J=17 Гц); 4,48 (м, 1H); 4,70 (д, 1H, J= 17 Гц), 6,53 (д, 1H); 7,11 - 7,30 (м, 9H).
Пример 24.
[S-(R*, S*)] -2,3,4,5-Тетрагидро-3-[(2-меркапто-1y-оксо-3-фенилпропил) амино]-2-оксо-1H-бензазепин-1-уксусная кислота
a) (S)- α- Бромобензолпропановая кислота
Раствор L-фенилаланина (30,0 г, 0,175 М) и бромида калия (73,5 г, 0,618 М) в 2,5 н. серной кислоте (365 мл) охлаждали до 0oC (ледяная/солевая баня) и обрабатывали по частям в течение 1 ч нитритом натрия (19,3 г, 0,28 М). После размешивания в течение 1 ч при 0oC и в течение 1 ч при комнатной температуре, реакционную смесь экстрагировали эфиром (3•250 мл). Объединенные органические экстракты промывали водой (100 мл) и солевым раствором (50 мл), осушали безводным сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме, в результате чего получали 34,46 г продукта; Rf = 0,45 (толуол: уксусная кислота, 95:5).
b) (R)- α- (Ацетилтио)бензолпропановая кислота, дициклогексиламиновая соль
Суспензию тиоацетата калия (19,25 г, 0,168 М) в сухом ацетонитриле (300 мл) охлаждали до 0oC в ледяной/солевой бане и обрабатывали по капле в течение 15 мин раствором продукта, полученного в части (a), (34,462 г, 0,15 М) в сухом ацетонитриле (35 мл). Реакционную смесь нагревали до комнатной температуре размешивали 5 ч в атмосфере аргона, а затем фильтровали, тщательно промывая твердый остаток ацетонитрилом (125 мл). Прозрачный фильтрат выпаривали досуха и осушали в вакууме. Свободную кислоту (39,717 г, оранжевый сироп) растворяли в эфире (400 мл), обрабатывали дициклогексиламином (30,2 мл, 1,0 экв.) и размешивали 30 мин при комнатной температуре и в атмосфере аргона. Белый осадок отфильтровывали, тщательно промывали этиловым эфиром (2•100 мл), осушали в вакууме в течение ночи при комнатной температуре, в результате чего получали 41,0 г продукта; [α]D = +31,8o (c = 1,4, метанол).
c) [S-(R*, S*)]-2,3,4,5-Тетрагидро-3-[[(2-ацелтио)-1- оксо-3-фенилпропил]амино]-2-оксо-1H-бензазепин-1-уксусная кислота, этиловый сложный эфир
Суспензию дициклогексиламиновой соли (R) - α- (ацетилтио)бензолпропановой кислоты (850 мг, 2,1 мМ) в этилацетате (60 мл) промывали 5% бисульфатом калия (5•10 мл) и солевым раствором, а затем осушали безводным сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме, в результате чего получали свободную кислоту в виде прозрачного сиропа (497 мг) с количественным выходом.
Эту свободную кислоту растворяли в сухом метиленхлориде (12 мл), охлаждали до 0oC в ледяной/солевой бане и обрабатывали 1-гидроксибензотриазолгидратом (297 мг, 2,20 мМ) и 1-этил-3-(3-диметиламинопропил)карбодиимидом (435 мг, 2,27 мМ). Реакционную смесь размешивали при 0oC в присутствии аргона в течение 1 ч, обрабатывали (S)-2,3,4,5-тетрагидро-3-амино-2-оксо- 1H-бензазепин-1-уксусной кислоты этиловым сложным эфиром, полученным в соответствии с известным Watthey и др. , J.Med. Chem., 28, с. 1511 - 1516 (1985)) (500 мг, 1,91 мМ) и продолжали размешивать 1 ч при 0oC и 1 ч при комнатной температуре. Полученный раствор разбавляли этилацетатом (50 мл), промывали последовательно водой (8,0 мл), 5% бисульфатом калия (2•8 мл), водой (8,0 мл), насыщенным бикарбонатом натрия (8,0 мл) и солевым раствором (8,0 мл), а затем осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт хроматографировали на силикагеле (Merck), элюируя этилацетатом: гексаном (1:4), в результате чего получали 714 мг продукта в виде прозрачного сиропа, Rf = 0,62 (этилацетат: гексаны, 1:1).
d) [S-(R*, S*)] -2,3,4,5-Тетрагидро-3-[(2- меркапто-1-оксо-3-фенилпропил)амино]-2-оксо-1H-1-бензазепин-1- уксусная кислота
Раствор продукта, полученного в части (c), (714 мг, 1,52 мМ) в метаноле (9,0 мл) продували 30 мин аргоном, охлаждали до 0oC в ледяной/солевой бане, а затем по капле обрабатывали предварительно продутым раствором 1,0 н. гидроксида натрия (6,0 мл, 4 экв.), барботируя при этом аргон во время добавления и в течение всей реакции. Реакционную смесь размешивали при 0oC в течение 1 ч, подкисляли до pH 2 5%-ным бисульфатом калия (26 мл), а затем экстрагировали этилацетатом (2 • 50 мл). Объединенные органические экстракты промывали солевым раствором (14 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт растворяли в метиленхлориде (5,0 мл) и по частям обрабатывали гексаном (50 мл), размешивая до образования твердого вещества. Надосадочную жидкость декантировали и твердый остаток растирали с добавочным количеством гексана (50 мл) и пентана (2 • 100 мл), размешивая 4 ч с первыми 100 мл пентана, а затем в течение ночи со следующими 100 мл в атмосфере аргона. Полученный продукт осушали в вакууме и получали аморфное твердое вещество, Rf = 0,47 (метиленхлорид : метанол : уксусная кислота, 20:1:1), [α]D = -228o (c = 0,51, метанол).
Анализ для C21H22N2O4S • 0,26 C5H12 • 0,176 CH2Cl2:
Вычислено: C 62,46; H 5,94; N 6,48; S 7,42;
Найдено: C 62,81, H 5,87; N 6,53; S 7,29.
1H-ЯМР (400 МГц, CDCl3 δ : 1,65 (м, 1H); 2,06 (д, 1H); 2,51 (м, 2H); 2,95 (дд, 1H, J = 7,14 Гц); 3,17 (м, 2H); 4,39 (д, 1H, J = 17 Гц); 4,46 (м, 1H); 4,67 (д, 1H, J = 17 Гц), 7,02 - 7,31 (м, 9H).
Пример 25.
[S-(R*, R*)] -Гексагидро-3-[(2-меркапто-1-оксо-3- фенилпропил)амино]-2-оксо-1(2H)-азоцинуксусная кислота
a) [S-(R*,R*)]-Гексагидро-3-[[2-ацетилтио)-1- оксо-3-фенилпропил]амино] -2-оксо-1(2H)-азоцинуксусная кислота, 1,1-диметилэтиловый сложный эфир
Суспензию дициклогексиламиновой соли (S)-2-(ацетилтио)-бензолпропановой кислоты (2,63 г, 6,6 мМ) в этилацетате (50 мл), промывали два раза 0,1 н. соляной кислотой (50 мл, 25 мл) и один раз солевым раствором (25 мл), а затем осушали сульфатом магния, фильтровали и после удаления растворителя в вакууме получали свободную кислоту в виде маслянистого вещества. Эту свободную кислоту и (S)-3-аминогексагидро-2-оксо-1(2H)-азоцинкусусной кислоты 1,1-диметилэтилэтиловый сложный эфир, полученный в соответствии с известной процедурой (Thotsett и др., в J. Med. Chem., 29, 251-60 (1986)), (1,66 г, 6,5 мМ) растворяли в дихлорметане (30 мл) в атмосфере аргона и охлаждали до 0oC. Затем добавляли 1-гидроксибензотриазолгидрат (891 мг, 6,6 мМ) и 1-этил-2-(3-диметиламино-пропил)карбодиимид (1,26 г, 6,6 мМ). Охлажденную смесь размешивали в течение 4 ч. Затем эту смесь разбавляли дихлорметаном (100 мл). Раствор промывали водой (50 мл), 5%-ным раствором бисульфата калия (50 мл), раствором бикарбоната натрия (50 мл) и солевым раствором (50 мл). Дихлорметановый раствор осушали сульфатом магния, фильтровали, а растворитель удаляли в вакууме. Пенистый остаток хроматографировали на силикагеле (Merck, 500 мл), элюируя 25 - 50% этилацетатом в гексане, в результате чего получали 2,252 г продукта, Rf = 0,35 (этилацетат : гексан, 1:1).
b) [S-(R*,R*]-Гексагидро-3-[[(2-ацетилтио)-1- оксо-3-фенилпропил]-амино] -2-оксо-1-(2H)-азоцинкуксусная кислота
Продукт, полученный в части (a), растворяли в трифторуксусной кислоте (20 мл) и размешивали при комнатной температуре в течение 1,75 ч в атмосфере аргона. Затем трифторуксусную кислоту удаляли в вакууме, дважды добавляли толуол и удаляли в вакууме, в результате чего получали продукт в виде белого пенистого вещества.
c) [S-(R*, R*]-Гексагидро-3-[[(2-меркапто-1- оксо-3-фенилпропил]-амино] -2-оксо-1-(2H)-азоцинуксусная кислота
Раствор продукта, полученного в части (b), (5,45 мМ) в метаноле (30 мл) продували 15 мин аргоном, охлаждали в ледяной/солевой бане и, продолжая продувать аргоном и размешивая, при этом по капле обрабатывали концентрированным раствором гидроксида аммония (2,0 мл). После размешивания в охлажденном состоянии в течение 6,5 ч, добавляли еще 1,0 мл раствора гидроксида аммония, после чего смесь закрывали и оставляли в рефрижераторе на 18 ч. Затем смесь подкисляли до pH 2 5%-ным раствором бисульфата калия. Продукт экстрагировали в дихлорметан (3 • 50 мл). Объединенные органические экстракты осушали сульфатом магния, фильтровали и после удаления в вакууме растворителя получали белое пенистое вещество. К этому веществу добавляли эфир и гексан и полученную суспензию размешивали в атмосфере аргона в течение 1 ч. Белый твердый материал собирали путем фильтрации, промывали еще раз эфиром и гексаном и осушали в вакууме, в результате чего получали 1,531 г продукта в виде белого пенистого вещества, т.пл. 71 - 90oC, Rf = 0,41 (10% метанол в метиленхлориде + 2 капли уксусной кислоты / 5 мл), [α]D = +3,5o (c = 0,75, метанол).
Анализ для C18H24N2O4S:
Вычислено: C 59,32; H 6,64; N 7,69; S 8,80;
Найдено: C 59,02; H 6,77; N 7,67; S 8,70.
ВЭЖХ: RT = 6,1 мин, 62% водного метанола, содержащего 0,2% фосфорной кислоты, 1,5 мл/мин, с детекцией при 220 нм, УМС S-3 (ODS), 6,0 • 150 мм, колонка со сферической крышкой диаметром 3 мкм. H.I > 95%.
Пример 26.
(3S)-2,3,4,5-Тетрагидро-3-[[(2-меркапто-3-(1-нафталенил)-1- оксопропил] амино]-2-оксо-1H-бензазепин-1-уксусная кислота
a) (Ацетиламино)(1-нафталинилметил)пропандионовая кислота, диэтиловый сложный эфир
К раствору этоксида натрия (21% в этаноле, 4,613 г, 67,8 мМ) в этаноле (100 мл) добавляли диэтилацетамидомалонат (14,74 г, 67,8 мМ), а затем 1-(бромометил)нафталин (10,0 г, 45,2 мМ). Полученный раствор размешивали 1 ч при комнатной температуре. Затем реакционную смесь концентрировали до получения маслянистого вещества оранжевого цвета. Это вещество растворяли в этилацетате и промывали 50% насыщенным хлоридом аммония, водой и солевым раствором, а затем осушали сульфатом натрия, фильтровали и концентрировали, в результате чего получали оранжевый твердый остаток. Этот остаток перекристаллизовывали из этилацетата и гексана и получали бежевые кристаллы с примесью малоната. Твердые кристаллы растворяли в 50% этилацетате в гексане и очищали с помощью флеш-хроматографии на силикагеле Merck в 50% этилацетате в гексане. Фракции, содержащие чистый продукт, объединяли и концентрировали, в результате чего получали 10,225 г продукта в виде белого твердого вещества, т.пл. 105 - 108oC, Rf = 0,57 (50% этилацетат в гексане).
b) α- Амино-1-нафталинпропановая кислота
Раствор продукта, полученного в части (a), (16,182 г, 47,5 мМ) суспендировали в 48%-ном бромводороде (100 мл) и нагревали с обратным холодильником в присутствии аргона в течение 14 ч. Бромоводородную соль продукта отфильтровывали из раствора и получали белое твердое вещество, которое затем растворяли в горячей (50oC) воде (500 мл) и полученный раствор нейтрализовали концентрированным гидроксидом аммония. Продукт осаждался из раствора в виде тонкозернистого белого твердого вещества. После фильтрации и осушки в высоком вакууме в течение ночи (18 часов) получали 8,335 г продукта в виде хлопьеобразного белого твердого вещества, т.пл. 264oC.
c) α- Бромо-1-нафталинпропановая кислота
К раствору продукта, полученного в части (b), (4,000 г, 18,6 мМ) и бромида калия (7,63 г, 63,2 мМ) в 2,5 н. серной кислоте (35 мл), который выдерживали при 0oC, добавляли в течение 1 ч нитрит натрия (1,92 г, 27,8 мМ). Полученную смесь размешивали еще 1 ч при 0oC, а затем нагревали до комнатной температуры и размешивали в течение 2,5 ч. Затем реакционную смесь экстрагировали эфиром (3х). Эфирные слои объединяли и промывали водой и солевым раствором, а затем осушали сульфатом натрия, фильтровали и концентрировали, в результате чего получали оранжевый маслянистый продукт. Этот продукт очищали с помощью флеш-хроматографии на силикагеле Merck в 70%-ном этилацетате в гексане с добавлением 1%-ной уксусной кислоты для уменьшения хвостовых фракций. Фракции, содержащие бромид, объединяли и концентрировали, в результате чего получали оранжевый маслянистый продукт с небольшими примесями, который после его выдерживания в течение ночи отверждался. Rf = 0,40 (40% этилацетат в гексане с 1%-ной уксусной кислотой).
d) α- (Ацетилтио)-1-нафталинпропановая кислота
К суспензии тиоацетата калия (0,912 г, 8,00 мМ) в ацетонитриле (300 мл) при 0oC добавляли продукт, полученный в части (c), (2,030 г, 7,27 мМ) в виде раствора в ацетонитриле (3 мл). Полученный раствор размешивали при 0oC в течение 1 ч, а затем нагревали до комнатной температурs и размешивали в течение 15 ч. После этого из реакционной смеси отфильтровывали бромид калия, фильтрат концентрировали и получали оранжевое маслянистое вещество. Это вещество растворяли в этилацетате и промывали 10% бисульфатом калия и солевым раствором, после чего осушали сульфатом натрия, фильтровали, концентрировали и получали оранжевый маслянистый продукт. Этот продукт очищали с помощью флеш-хроматографии на силикагеле (Merck) в 50% этилацетате в гексане с добавлением 1%-ной уксусной кислоты для уменьшения хвостовых фракций. Фракции, содержащие нужный продукт, были все с примесью соединения с Rf = 0,43. Эти фракции объединяли, концентрировали и получали оранжевое маслянистое соединение. Неочищенный продукт очищали с использованием дициклогексиламиновой соли, которую получали путем растворения оранжевого маслянистого продукта в эфире и добавления одного эквивалента дициклогексиламина (18,13 г, 100 мМ) к полученному раствору. Эта дициклогексиламиновая соль была получена в двух сборах в виде коричневых кристаллов (1,450 г) и все еще имела незначительные примеси. Эти кристаллы суспендировали в этилацетате и встряхивали с 10% бисульфатом калия (3x). Органический слой промывали водой и солевым раствором, а затем осушали сульфатом натрия, фильтровали и концентрировали, в результате чего получали 875 мг продукта в виде желтого маслянистого вещества, Rf= 0,40 (40% этилацетат в гексане с 1% уксусной кислотой).
e) (3S)-2,3,4,5-Тетрагидро-3-[(2-ацетилтио)-3-(1-нафталенил)-1- оксопропил]амино]-2-оксо-1H-бензазепин-1-уксусная кислота
Рацемический кислотный продукт, полученный в части (d), (338 мг, 1,23 мМ) и (S)-2,3,4,5-тетрагидро-3-амино-2-оксо-1H-1-1-бензазепин- 1-уксусной кислоты этиловый сложный эфир [полученный в соответствии с описанием Watthey и др., J. Med.Chem., 28, стр. 1511-1516 (1985)] (321 мг, 1,23 мМ) растворяли в метиленхлориде (11 мл) в атмосфере аргона при комнатной температуре. Полученную смесь охлаждали до 0oC и обрабатывали гидроксибензотриазолгидратом (166 мг, 1,23 мМ) и 1-этил-3-(3-диметил-амино-пропил)карбодиимидом (259 мг, 1,35 мМ). После 1-часового размешивания, смесь нагревали до комнатной температуры и размешивали еще 4 ч. Летучие компоненты выпаривали, а остаток растворяли в этилацетата и промывали последовательно 1 н. соляной кислотой, водой, насыщенным бикарбонатом натрия и солевым раствором. Органический слой осушали сульфатом натрия, фильтровали и концентрировали, а остаток подвергали флеш-хроматографии на силикагеле (Merck), элюируя 50% этилацетатом в гексане, в результате чего получали 510 мг маслянистого продукта (смесь диастереомеров, 1:1), Rf =0,40 (5% уксусная кислота в этилацетате).
f) (3S)-2,3,4,5-Тетрагидро-3-[[2-меркапто-3-(1-нафтаденил)-1- оксопропил]амино]-2-оксо-1H-1-бензазепин-1-уксусная кислота
Раствор продукта, полученного в части (e), (508 мг, 0,98 мМ) в метаноле (6 мл, дезоксигенированного посредством барботирования аргона) охлаждали до 0oC и обрабатывали 1 н. гидроксидом натрия (6 мл, дезоксигенорованным посредством барботирования аргона). Полученную смесь размешивали 1 ч в присутствии аргона. Раствор подкисляли 10% бисульфатом калия и экстрагировали этилацетатом. Органический слой промывали солевым раствором, осушали сульфатом натрия, фильтровали, концентрировали и получали прозрачный маслянистый остаток. Этот остаток подвергали флеш-хроматографии на силикагеле (Merck), элюируя 1%-ной уксусной кислотой в смеси гексана и этилацетата (1:1). Фракции, содержащие чистый продукт, объединяли, концентрировали, азеотропически обезвоживали с использованием этилацетата и промывали водой для удаления уксусной кислоты. Органический слой осушали сульфатом натрия, фильтровали и концентрировали. Остаток растворяли в этилацетате и растирали с этиловым эфиром и гексаном. Растворитель удаляли, а остаток суспендировали в гексане, упаривали и осушали в вакууме, в результате чего получали 327 мг продукта в виде белого порошкообразного пенистого вещества, Rf = 0,64 (5% уксусная кислота в этилацетате), [α]D = -187,3o (c = 0,43, хлороформ).
Анализ для C25H23N2O4S • 0,42 H2O:
Вычислено: C 65,98; H 5,28; N 6,16; S 7,05;
Найдено: C 66,29; H 5,29; N 5,85; S 6,65.
ВЭЖХ: tR = 12,3 мин (48,1%) и 14,4 мин (51,8%) (λ = 220 нм) , УМС S-3 ODS(C-18), 6,0•150 мм, 68% (10% воды - 90% метанола - 0,2% фосфорной кислоты) /32% (90% воды - 10% метанола - -1,2% фосфорной кислоты), скорость потока = 1,5 мл/мин, изократич.
Пример 27.
[S-(R*, R*)] -3-[[2-(Ацетилтио)-1-оксо-3- фенилпропил] амино]-2,3,4,5-тетрагидро-2-оксо-1-бензазепин-1-уксусная кислота
Продукт, полученный в соответствии с описанием, приведенным в примере 6 (799 мг, 2,0 мМ) добавляли к дезоксигенированному (путем барботирования аргона) раствору бикарбоната калия (378 мг, 3,8 мМ) в воде (25 мл). После растворения исходного материала, добавляли ангидрид уксусной кислоты (1,5 мл, 1,62 г, 15,9 мМ). В результате получали молочно-белый раствор, который в итоге превращался в смолистый продукт. После его размешивания при комнатной температуре в течение нескольких минут, смесь подкисляли 10% соляной кислотой и экстрагировали этилацетатом. Этилацетатный экстракт промывали водой (3 раза) и солевым раствором, а затем осушали сульфатом натрия, фильтровали и упаривали. Остаток подвергали флеш-хроматографии на силикагеле (Merck) (1% уксусная кислота в этилацетате) и нужные фракции упаривали, азеотропически обезвоживали с использованием этилацетата (3 раза), растворяли в небольшом объеме этилацетата и растирали с гексаном. Растворитель отгоняли, а остаток суспендировали и два раза отгоняли из гексана, в результате чего получали целевое соединение в виде твердого белого пенистого продукта.
ТСХ: (1% уксусная кислота в этилацетате), Rf = 0,41, [α]D = -190,8o (c = 0,68, хлороформ).
Анализ для C23H24N2O5S • 0,13 C4H8O2 • 0,5 H2O:
Вычислено: C 61,28; H 5,69; N 6,08; S 6,96;
Найдено: C 61,18; H 5,64; N 5,60; S 6,70.
Примеры 28 - 30. Повторяя процедуру, описанную в примере 27, но с использованием бензоилхлорида в качестве ацилирующего агента получали [S-(R*, R*)] -3-[[2-(бензилтио)-1-оксо-3- фенилпропил]амино]-2,3,4,5-тетрагидро-2-оксо-1H-бензазепин-1-уксусную кислоту, т.пл. 170 - 171oC, [α]D = -244o (c = 0,30, хлороформ). ТСХ (гексан : этилацетат : уксусная кислота, 40:60:1); Rf = 0,31.
Анализ для C28H26N2O5S • 0,1 C4H8O2 • 0,2 H2O:
Вычислено: C 66,24; H 5,32; N 5,44; S 6,23;
Найдено: C 66,28; H 5,23; N 5,40; S 6,18.
Повторяя процедуру, описанную в примере 27, но с использованием ангидрида пропионовой кислоты в качестве ацилирующего агента получали [S-(R*,R*)] -2,3,4,5, -тетрагидро-[3-[[1- оксо-1-(оксопропил)тио]-3-фенилпропил]амино]-2-оксо-1H- бензазепин-1-уксусную кислоту, т.пл. 156 - 157oC, [α]D = -210o (c = 0,31, хлороформ). ТСХ (гексан : этилацетат : уксусная кислота, 40:60:1); Rf = 0,34.
Анализ для C24H26N2O5S • 0,07 C4H8O2:
Вычислено: C 63,30; H 5,81; N 6,08; S 9,96;
Найдено: C 63,32; H 5,77; N 5,90; S 6,96.
Повторяя процедуру, описанную в примере 27, но с использованием ангидрида триметилуксусной кислоты в качестве ацилирующего агента получали [S-(R*, R*)]-[3-[[2-[(2,2-диметил-1- оксопропилтио]-1-оксо-3-фенилпропил]амино] -2,3,4,5- тетрагидро-2-оксо-1H-бензазепин-1-уксусную кислоту, т. пл. 86 - 90oC, [α]D = -190o (c = 3,06, хлороформ). ТСХ (гексан : этилацетат : уксусная кислота, 30:70:1) Rf = 0,42.
Анализ для C26H30N2O5S • 0,2 C6H14 • 0,25 H2O:
Вычислено: C 64,78; H 6,65; N 5,50; S 6,36;
Найдено: C 64,79; H 6,56; N 5,53; S 6,30.
Пример 31.
[S-(R*, R*)]-3,4-Дигидро-3-[(2-меркапто-1-оксо-3- фенилпропил]амино]-4-оксо-1,5-бензоксазепин-3(2H)пропановая кислота
a) (S)-3-[[(Диметилэтокси)карбонил] амино]-3,4-дигидро-4-оксо- 1,5-бензоксозепин-5(2H)-пропановая кислота, этиловый сложный эфир
Раствор (S)-3-[[(диметилэтокси)-карбонил]амино]2,3-дигидро-1,5- бензоксазепин-4(5H)-он'а, полученного в соответствии с описанием, приведенным в примере 13 (c), (1,49 г, 5,35 мМ) в смеси тетрагидрофурана и т-бутанола (2: 1,21 мл) охлаждали до 0oC и обрабатывали этилацетатом (0,81 мл, 7,50 мМ, 1,4 экв. ), а затем 1,0 н. калиевой солью т-бутанола (535 мкл, 0,1 экв.). Реакционную смесь размешивали 15 мин при 0oC, а затем 2,5 ч при комнатной температуре в присутствии аргона. После этого к реакционной смеси добавляли дополнительное количество 1,0 н. калиевой соли т-бутанола/тетрагидрофурана (270 мкл, 0,05 экв.). Реакционную смесь размешивали при комнатной температуре 45 мин, а затем в течение 2 ч нагревали до 60oC. После этого реакционную смесь гасили 25%-ным хлоридом аммония (50 мл) и экстрагировали этилацетатом (2 • 100 мл). Объединенные органические слои промывали 25% хлоридом аммония (50 мл), водой (50 мл) и солевым раствором (50 мл), а затем осушали сульфатом магния, фильтровали, концентрировали и получали прозрачный сироп. ТСХ остатка показала два пятна.
Реакцию снова инициировали путем обработки раствора смеси (1,42 г 5,10 мМ) в тетрагидрофуране: т-бутаноле (2:1, 15 мл), охлажденной до 0oC, этилакрилатом (0,77 мл, 7,14 мМ, 1,4 экв.), а затем 1,0 н. калиевой соли т-бутанола/тетрагидрофураном (510 мкл, 0,1 экв.). Реакционную смесь размешивали 15 мин при 0oC, а затем 3 ч при комнатной температуре. После этого реакцию гасили 25% хлоридом аммония (50 мл) и экстрагировали этилацетатом (2•100 мл). Объединенные органические слои промывали 25% хлоридом аммония (50 мл), водой (50 мл) и солевым раствором (50 мл), а затем осушали сульфатом магния, фильтровали и концентрировали, в результате чего получали желтый сироп. Остаток очищали с помощью хроматографии на колонке (5 • 15 см) с силикагелем, элюируя смесью 30% этилацетата и гексана (2:1). Нужные фракции объединяли, концентрировали и получали в результате 1,40 г целевого соединения в виде прозрачного сиропа. ТСХ (5% метанол/хлороформ), Rf = 0,63.
b) (S)-3-Аамино-3,4-дигидро-4-оксо-1,5-бензоксазепин-5(2H)-пропановая кислота, этиловый сложный эфир, гидрохлоридная соль
Продукт, полученный в части (a), (1,4 г, 3,7 мМ) обрабатывали смесью 4,0 М соляной кислоты и диоксана (20 мл, 80 мМ) и охлаждали до 0oC. Полученный раствор размешивали при 0oC в течение 30 мин, а затем при комнатной температуре в течение 2 ч. Реакционную смесь концентрировали, дважды упаривали с этиловым эфиром, три раза подвергали азеотропической перегонке с использованием толуола и осушали в вакууме в течение 4 ч, в результате чего получали 1,05 г целевого соединения.
c) [S-(R*, R*)] -3-[[2-(Ацетилтио)-1-оксо-3- фенилпропил]амино]-3,4-дигидро-4-оксо-1,5-бензоксазепин-5(2H)- пропановая кислота, сложный этиловый эфир
Дициклогексиламиновую соль (S)-2-(Ацетилтио)бензопропановой кислоты (1,8 г, 4,40 мМ) суспендировали в этилацетате (100 мл) и промывали 5% бисульфатом калия (5•25 мл) и солевым раствором (30 мл), осушали безводным сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме, в результате чего получали свободную кислоту.
Эту свободную кислоту растворяли в сухом метиленхлориде (10 мл), охлаждали до 0oC в ледяной/солевой бане и обрабатывали раствором продукта, полученного в части (b), (1,05 г, 3,33 мМ) в сухом метиленхлориде (25 мл), а затем триэтиламином (0,62 мл, 4,44 мМ, 1,3 экв.) и гексафторофосфатом бензотриазол-1-илокситрис(диметиламино)фосфония (1,96 г, 4,44 мМ, 1,3 экв.). Реакционную смесь размешивали 1 ч при 0oC, а затем в течение ночи при комнатной температуре. После этого реакционную смесь концентрировали досуха, разводили этилацетатом (150 мл) и промывали 5% бисульфатом калия (2•50 мл), водой (2•50) и солевым раствором (50 мл), а затем осушали безводным сульфатом магния, фильтровали и выпаривали досуха. Неочищенный продукт хроматографировали на колонке (5•15 см) с силикагелем, элюируя смесью 30% этилацетата и гексана. Нужные фракции объединяли и концентрировали, в результате чего получали 740 мг чистого продукта.
ТСХ (10% метанол в хлороформе), Rf = 0,74.
d) [S-(R*,R*)]-3,4-Дигидро-3-[(2-меркапто-1-оксо-3-фенилпропил)- амино] -4-оксо-1,5-бензоксазепин-5(2H)-пропановая кислота
Раствор продукта, полученного в части (c), (740 мг, 1,53 мМ) в метаноле (15 мл) охлаждали до 0oC в ледяной/солевой бане, продували аргоном в течение 30 мин, а затем обрабатывали по капле предварительно продутым (аргоном, 30 мин) раствором 1,0 н. гидроксида натрия (6,1 мл, 4 экв.), барботируя при этом аргон во время добавления и в течение всей реакции. Затем реакционную смесь размешивали 1 ч при 0oC, подкисляли при 0oC 5%-ным бисульфатом калия (150 мл) до pH 1,5 и экстрагировали этилацетатом (3•100 мл). Объединенные органические экстракты промывали солевым раствором (100 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме, в результате чего получали белое пенистое вещество (612 мг). Остаток очищали с помощью колоночной хроматографии (5•15 см) на силикагеле, элюируя 0,5%-ной уксусной кислотой в этилацетате. Остаток растворяли в метиленхлориде (5 мл) и 5 раз упаривали на роторном испарителе с использованием гексана, размешивая смесь до образования белого пенистого твердого вещества. Это вещество осушали в вакууме в течение ночи и получали 590 мг продукта. ТСХ (1% уксусная кислота в этилацетате), Rf = 0,42, [α]D = -127,6o (c = 1,12, метанол).
Анализ для C21H22N2O5S• 0,03 H2O
Вычислено: C 60,86; H 5,36; N 6,75; S 7,73;
Найдено: C 61,08; H 5,68; N 6,45; S 7,51.
Пример 32.
[2R-[ 2α, 3α (S*)]]-3,4-Дигидро-3-[(2-меркапто-1-оксо-3-фенилпропил) амино]-2-метил-4-оксо-1,5-бензоксазепин-5(2H)-уксусная кислота
a) N-[(1,1-Диметилэтокси)карбонил]-O-(2-нитрофенил)-L-треонин
Раствор N-[(1,1-диметилэтокси)карбонил] -L-треонина (5,02 г, 22,9 мМ) в сухом диметилформамиде (10 мл) по капле в течение 30 мин добавляли к охлажденной (0oC) суспензии 60% гидрида натрия (1,93 г, 48,25 мМ, 2,11 экв.) в сухом диметилформамиде (40 мл). Реакционную смесь размешивали при 0oC до тех пор, пока не прекратится пенообразование (около 3,5 ч). Реакционную смесь по капле в течение 20 мин обрабатывали 1-фторо-2-нитробензоллом (2,67 мл, 25,2 мМ, 1,1 экв. ) и размешивали 2 ч при 0oC в присутствии аргона. Затем реакционную смесь помещали в холодную комнату (5oC) и размешивали в течение ночи. Реакционную смесь выливали в ледяную воду (500 мл) и экстрагировали этиловым эфиром (2•200 мл). Водную фазу доводили до pH 1,0 с помощью 6 н. соляной кислоты (200 мл) и экстрагировали этилацетатом (3•300 мл). Объединенные этилацетатные экстракты промывали водой (2•300 мл) и солевым раствором (300 мл), а затем осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный остаток загружали на целит и очищали с помощью хроматографии на колонке (10•20 см) с силикагелем. После элюирования метиленхлоридом (3 л), смесью метиленхлорида и метанола (99:1, 2 л), смесью метиленхлорида и метанола (95:5), смесью метиленхлорида, метанола и уксусной кислоты (100:5:0,5) (5 л), получали 6,07 г целевого соединения. ТСХ (метиленхлорид:метанол:уксусная кислота, 100:5:0,5), Rf = 0,16.
b) N-[(1,1-диметилэтокси)карбонил]-(2-аминофенил)-L-треонин
Раствор продукта, полученного в части (a), (1,0 г, 2,94 мМ) в сухом метаноле обрабатывали 10% палладированным углем в качестве катализатора (35 мг) и гидрировали при 40 фунт/кв.дюйм (275,75 кПа) в течение 5,5 ч. Поскольку реакция не была завершена, то добавляли еще 50 мг 10% палладированного угля и реакционную смесь гидрировали еще 1,5 ч. Затем реакционную смесь фильтровали через слой целита в миллипористом фильтре, промыв этот слой метанолом. Полученный темный фильтрат выпаривали досуха и осушали в вакууме, в результате чего получали 0,856 г целевого продукта в виде темного твердого вещества. ТСХ (метиленхлорид:метанол:уксусная кислота, 20:1:1), Rf = 0,16.
c) (2R-цис)-3-[[(Диметилэтокси)карбонил] амино] -2,3-дигидро-2-метил- 1,5-бензоксазепин-4(5H)-он
Раствор продукта, полученного в части (b), (0,8 г, 2,58 мМ) в сухом диметилформамиде (6 мл) обрабатывали 1-этил-3-(3-диметиламино-пропил)карбодиимидом (495 мг, 2,58 мМ) и размешивали 3 ч при комнатной температуре. Реакционную смесь распределяли между этилацетатом (50 мл) и 50%-ным раствором бикарбоната натрия (50 мл). Водный слой отделяли и экстрагировали этилацетатом (2•50 мл). Объединенные органические экстракты промывали водой (3•50 мл) и солевым раствором (50 мл), а затем осушали безводным сульфатом магния, выпаривали и осушали в вакууме, в результате чего получали неочищенный твердый продукт желтого цвета. Этот продукт хроматографировали на колонке (5•15 см) с силикагелем, элюируя 25% этилацетатом/гексаном и получали 568 мг целевого соединения в виде беловатого твердого продукта, ТСХ (этилацетат:гексан, 1:1), Rf = 0,47.
d) (2R-цис))-3-[[(Диметил)этокси)карбонил]амино]-3,4-дигидро- 2-метил-4-оксо-1,5-бензоксазепин-5(2H)-уксусная кислота, этиловый сложный эфир
Раствор продукта, полученного в части (c), (319 мг, 1,09 мМ) и этилбромоацетата (151,3 мкл, 1,36 мМ, 1,25 экв.) в сухом тетрагидрофуране (3 мл) добавляли к охлажденной (0oC) суспензии 60% гидрида натрия (53 мг, 1,32 мМ, 1,21 экв. ), промытого 3 раза гексаном (в сухом тетрагидрофуране) (2 мл) в течение 5 мин. Реакционную смесь размешивали 1 ч при 0oC, а затем 30 мин при комнатной температуре. Реакцию гасили 25%-ным раствором хлорида аммония (5 мл) и экстрагировали метиленхлоридом (2•20 мл). Объединенные органические экстракты промывали 25% раствором хлорида аммония (10 мл) и солевым раствором (10 мл), а затем осушали безводным сульфатом магния, фильтровали, концентрировали и осушали в вакууме, в результате чего получали 400 мг целевого продукта. ТСХ (этилацетат:гексан, 1:1), Rf = 0,55.
e) (S)-3-Амино-3,4-дигидро-2-метил-4-оксо-1,5-бензоксазепин- 5(2H)-уксусная кислота, этиловый сложный эфир, гидрохлоридная соль
4,0 М соляной кислоты в диоксане (5,5 мл, 22 мМ, 20,8 экв.) добавляли к продукту, полученному в части (d), (400 мг, 1,06 мМ), и охлажденному до 0oC. Полученный раствор размешивали при 0oC в течение 30 мин, а затем при комнатной температуре в течение 3 ч. Реакционную смесь выпаривали на роторном испарителе, концентрировали с этиловым эфиром (3•10 мл) и осушали в вакууме в течение ночи в присутствии гидроксида натрия, в результате чего получали 350 мг целевого продукта. ТСХ (метиленхлорид:метанол, 9:1), Rf = 0,49.
f) [2R-[ 2α, 3α (S*)]]-3-[[(2-(Ацетилтио)-1-оксо-3-фенилпропил]амино]- 2-метил-4-оксо-1,5-бензоксазепин-5(2H)-уксусная кислота
Суспензию (S)-2-(ацетилтио)бензолпропановой кислоты дициклогексиламиновой соли (495 мг, 1,22 мМ, 1,1 экв.) в этилацетате (30 мл) промывали 5%-ным бисульфатом калия (5•5 мл) и солевым раствором (10 мл), а затем осушали безводным сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме, в результате чего получали свободную кислоту в виде прозрачного сиропа (278 мг) с количественным выходом.
Эту свободную кислоту растворяли в сухом метиленхлориде (10 мл), охлаждали до 0oC в ледяной/солевой бане и обрабатывали раствором продукта, полученного в части (e), (350 мг, 1,11 мМ) в сухом метиленхлориде (2 мл), а затем обрабатывали триэтиламином (163 мкл, 1,17 мМ, 1,05 экв.) и бензотриазол-1-илокситрис(диметиламино)фосфония гексафторофосфатом (515,5 мг, 1,17 мМ, 1,05 экв. ). Реакционную смесь размешивали 1 ч при 0oC, а затем 3,5 ч при комнатной температуре. Поскольку реакция не была завершена, как показала ТСХ, то добавляли дополнительное количество триэтиламина (54 мкл, 0,389 мМ) и бензотриазол-1-илокситрис(диметиламино)фосфония гексафторофосфата (172 мг, 0,389 мМ) и размешивали в течение 1 ч. Реакционную смесь концентрировали досуха, промывали 0,5 н. соляной кислотой (2•10 мл), водой (10 мл) и солевым раствором (10 мл), а затем осушали безводным сульфатом магния, фильтровали и выпаривали досуха. Неочищенный продукт хроматографировали на колонке (3•16 см) с силикагелем, элюируя смесью 20% этилацетата и гексана. Нужные фракции объединяли и концентрировали, в результате чего получали 378 мг чистого целевого продукта. ТСХ (этилацетат:гексан, 1:1), Rf = 0,48.
g) [2R-[ 2α, 3α (S*)]]-3,4-Дигидро-3-[(2-меркапто-1-оксо- 3-фенилпропил] амино)-2-метил-4-оксо-1,5-бензоксазепин-5(2H)-уксусная кислота
Раствор продукта, полученного в части (f), (237 мг, 0,61 мМ) в метаноле (4 мл) продували аргоном в течение 30 минут, охлаждали до 0oC в ледяной/солевой бане, а затем по капле обрабатывали предварительно продутым (аргон, 30 мин) раствором 1,0 н. гидроксида натрия (2,45 мл, 4 экв.), барботируя при этом аргон во время добавления и в течение всей реакции. Реакционную смесь размешивали 1 ч при 0oC, подкисляли при 0oC 5%-ным бисульфатом калия (20 мл) для доведения pH до 1,5, а затем экстрагировали этилацетатом (2•50 мл). Объединенные органические экстракты промывали солевым раствором (25 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме, в результате чего получали стеклообразный маслянистый продукт (351 мг). Неочищенный остаток растворяли в метиленхлориде (5 мл) и по частям обрабатывали гексаном (50 мл), размешивая смесь до образования твердого вещества. Надосадочную жидкость декантировали, а твердый остаток растирали с дополнительным количеством гексана (50 мл) и пентана (2•100 мл), размешивая 4 ч с первым 100 мл пентана, а затем в течение ночи со следующими 100 мл пентана в атмосфере аргона. Пентан декантировали, а белый твердый аморфный остаток осушали в вакууме в течение ночи, в результате чего получали 240 мг целевого продукта. ТСХ (2% уксусная кислота в этилацетате), Rf= 0,48; [α]D = -101,1o (с=1, метанол).
Анализ для C21H22N2O5S • 0,40 C4H8O2 • 0,08 H2O:
Вычислено: C 60,36; H 5,65; N 6,23; S 7,13;
Найдено: C 60,17; H 5,57; N 6,16; S 7,45.
Пример 33. Повторяя процедуру, описанную в примере 32, но используя алло-L-треонин, получали [2s-[ 2α, 3β (R*)]] -3,4-дигидро-3-[(2-меркапто-1-оксо-3-фенилпропил)амино] -2-метил-4-оксо-1,5-бензоксазепин-5(2H)-уксусную кислоту, [α]D = -174,3o (c= 0,89, метанол), ТСХ (1% уксусная кислота в этилацетате), Rf = 0,43.
Пример 34.
(2S)-(2S)-3- [(2-Меркапто-1-оксо-3-фенилпропил)амино] -3,4,5,6-тетрагидро-2-оксо-1-бензазоцин-1(2H)уксусная кислота
a) 1-Бензосуберон, оксим
Раствор гидрохлорида гидроксиламина (2,39 г, 34,33 мМ, 1,1 экв.) в воде (16 мл) добавляли к раствору 1-бензосуберона (4,67 мл, 31,21 мМ) в пиридине (9,0 мл) и этаноле (16 мл). Реакционную смесь нагревали с обратным холодильником (температура бани 105oC) в течение 35 мин. Реакционную смесь охлаждали, разбавляли этилацетатом (100 мл) и водой (40 мл) и слои разделяли. Водный слой экстрагировали этилацетатом (2•50 мл). Объединенные органические экстракты промывали 1 н. соляной кислотой (3•100 мл), осушали безводным сульфатом магния, фильтровали и концентрировали, в результате чего получали неочищенный беловатый твердый остаток (5,60 г). Этот остаток очищали с помощью хроматографии на колонке (10•20 см) с силикагелем, элюируя гексаном (2 л), а затем смесью 10% этилацетата и гексана (5 л), в результате чего получали 4,40 г целевого соединения в виде беловатого твердого продукта. ТСХ (10% этилацетат/гексан), Rf=0,35.
b) 3,4,5,6-Тетрагидро-1-бензазоцин-2(1H)-он
Концентрированную серную кислоту (8,8 мл) добавляли целиком (одной порцией) к суспензии продукта, полученного в части (a), (4,33 г, 24,71 мМ), в ледяной уксусной кислоте (4,4 мл). Температура повышалась до 83oC, и реакционную смесь нагревали до 160oC (в масляной бане) в течение 10 мин. Затем реакционную смесь оставляли охлаждаться до комнатной температуры, после чего ее выливали в ледяную воду (100 мл). После этого pH реакционной смеси доводили до значения 11 с помощью 10 н. раствора гидроксида натрия. Смесь разводили этилацетатом (250 мл) и водой (100 мл) и слои разделяли. Водный слой экстрагировали этилацетатом (3•100 мл). Объединенные этилацетатные экстракты промывали водой (150 мл) и солевым раствором (150 мл), затем осушали безводным сульфатом натрия, фильтровали и концентрировали, в результате чего получали 2,8 г целевого продукта. ТСХ (этилацетат), Rf=0,33.
c) 3,4,5,6-Тетрагидро-3-бромо-1-бензазоцин-2(1H)-он
Раствор продукта, полученного в части (b), (8,31 г, 47,42 мМ) в хлороформе (115 мл) охлаждали до 0oC, обрабатывали пентахлоридом фосфора (11,36 г, 54,53 мМ, 1,15 экв.) и иодом (114 мг) и размешивали в течение 30 мин при 0oC, в атмосфере аргона. Реакционную смесь желтого цвета обрабатывали бромом (2,92 мл, 56,0 мМ, 1,2 экв.) нагревали до комнатной температуры, а затем нагревали с обратным холодильником и в атмосфере аргона в течение 3,5 ч. После этого реакционную смесь оставляли охлаждаться до комнатной температуры и выливали в ледяную воду (100 мл). Слои разделяли и слой хлороформа промывали водой (100 мл), осушали сульфатом магния, фильтровали и концентрировали. Неочищенный остаток перекристаллизовывали из горячего этилацетата и получали 8,69 г целевого продукта. ТСХ (ж этилацетат: гексан, 1:1), Rf=0,36.
d) 3,4,5,6-Тетрагидро-3-азидо-1-бензазоцин-2(1H)-он
Раствор продукта, полученного в части (c), (6,87 г, 27,03 мМ) и азида натрия (2,28 г, 35,14 мМ, 1,3 экв.) в диметилсульфоксиде (130 мл) размешивали при 60oC (в масляной бане) и в атмосфере аргона в течение 5 ч. Реакционную смесь охлаждали до комнатной температуры, выливали в холодную воду (400 мл) и размешивали 15 мин. Осадок фильтровали, твердый продукт промывали водой (1 л) и осушали в течение ночи в вакуумной сушилке, в результате чего получали целевой продукт. ТСХ (этилацетат: гексан, 1:1), Rf=0,63.
e) 3-Азидо-3,4,5,6, -тетрагидро-2-оксо-1-бензазоцин-1(2H)-уксусная кислота, сложный этиловый эфир
Раствор продукта, полученного в части (d), (5,454 г, 25,22 мМ) и этилбромоацетата (3,5 мл, 31,53 мМ, 1,25 экв.) в сухом тетрагидрофуране (50 мл) добавляли к охлажденной суспензии (0oC) 60% гидрида натрия [1,23 г 30,77 мМ, 1,22 экв. , промытого гексаном (3•10 мл)] в сухом тетрагидрофуране 15 мл) в течение 15 мин. Затем смесь размешивали 1 ч при 0oC и 30 мин при комнатной температуре. Реакцию гасили 25% раствором хлорида аммония (100 мл) и экстрагировали этилацетатом (2•250 мл). Объединенные этилацетатные экстракты промывали 25%-ным раствором хлорида аммония (100 мл) и солевым раствором (100 мл), а затем осушали безводным сульфатом магния, фильтровали и концентрировали, в результате чего получали целевое соединение в виде желтого маслообразного продукта. ТСХ (35% этилацетат/гексан), Rf=0,35.
f) 3-Амино-3,4,5,6-тетрагидро-2-оксо-1-бензазоцин-1 (2H)-уксусная кислота, сложный этиловый эфир
Раствор продукта, полученного в части (e), (9,0 г, 29,83 мМ), в абсолютном этаноле (50 мл) обрабатывали 10% палладированным углем (900 мг) и гидрировали при давлении 45 фунт/кв.дюйм (310,2 кПа), причем в первые 1,5 ч колбу Парра два раза вентилировали. Смесь фильтровали через миллипористый фильтр, тщательно промывая этанолом. Прозрачный фильтр выпаривали досуха и осушали в вакууме, в результате чего получали 7,59 г целевого продукта в виде густого сиропа желтого цвета. ТСХ (10% метанол/этиленхлорид), Rf=0,29.
g) (2S)-3-[[2-(Ацетилтио)-1-оксо-3-фенилпропил] -амино] -3,4,5,6-тетрагидро-2-оксо-1-бензазоцин-1-(2H)-уксусная кислота этиловый сложный эфир
Суспензию (S)-2-(ацетилтио) бензолпропановой кислоты дициклогексиламиновой соли (2,1 г, 5,18 мМ, 1,1 экв.) в этилацетате (100 мл) промывали 5% бисульфатом калия (5•25 мл) и солевым раствором (30 мл), осушали безводным сульфатом магния, фильтровали, упаривали досуха и осушали в вакууме, в результате чего получали свободную кислоту в виде прозрачного сиропа (1,24 г) с количественным выходом.
Полученную свободную кислоту растворяли в сухом метиленхлориде (45 мл), охлаждали до 0oC (в бане из льда и соли) и обрабатывали раствором продукта, полученного в части (f), (1,3 г, 4,71 мМ) в сухом метиленхлориде (10 мл), с последующей обработкой триэтиламином (723 мкл, 5,18 мМ, 1,1 экв.) и гексафторофосфатом бензотриазол-1-илокситрис(диметиламино)фосфония (2,29 г, 5,18 мМ, 1,1 экв. ). Реакционную смесь размешивали 1 ч при 0oC, а затем в течение ночи при комнатной температуре. После этого смесь концентрировали досуха, разводили этилацетатом (125 мл), промывали 0,5 н. соляной кислотой (2•25 мл), водой (25 мл) и солевым раствором (25 мл), осушали безводным сульфатом магния и выпаривали досуха. Неочищенный продукт хроматографировали на колонке с силикагелем (3•16 см), элюируя 30% этилацетатом/гексаном. Нужные фракции объединяли и концентрировали, в результате чего получали 1,77 г чистого целевого соединения (1:1 - смесь диастереомеров). ТСХ (этилацетат: гексан, 1:1), Rf=0,39.
h) (2S) - 3-[(2-Меркапто-1-оксо-3-фенилпропил)амино]-3,4,5,6-тетрагидро-2-оксо-1 -бензазоцин-1(2H)-уксусная кислота
Раствор продукта, полученного в части (g), (1,75 г, 3,62 мМ) в метаноле (25 мл) продували аргоном в течение 30 мин, охлаждали до 0oC (в бане из льда/соли), а затем по капле обрабатывали предварительно продутым (аргон, 30 мин) раствором 1,0 н. гидроксида натрия (14,5 мл, 4 экв.), барботируя, при этом, аргон во время добавления и в процессе всей реакции. Реакционную смесь размешивали при 0oC в течение 1 ч, подкисляли при 0oC 5%-ным бисульфатом калия (200 мл) до pH 1,5, а затем экстрагировали этилацетатом (2•100 мл). Объединенные органические экстракты промывали солевым раствором (200 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме, в результате чего получали белое пенистое вещество (1,67 г). Неочищенный остаток растворяли в метиленхлориде (10 мл) и порциями обрабатывали гексаном 50 мл), в результате чего образовывалось твердое вещество. Надосадочную жидкость декантировали, а твердый остаток растирали с гексаном (50 мл) и пентаном (2•100 мл), размешивая с первым 100 мл пентана в течение 4 ч и с последующими 100 мл в течение ночи в атмосфере аргона. Пентан декантировали и полученное белое аморфное твердое вещество осушали в вакууме в течение ночи, в результате чего получали 1,388 г целевого соединения (смесь диастереомеров, 1:1), т.пл. 107 - 111oC, [α]D = +28,3o (с = метанол).
ТСХ (метиленхлорид: метанол: уксусная кислота, 20:1:1), Rf=0,44.
Пример 35.
[4S- [4α, 7α(R*), 10aβ] ] -Декагидро-7-[(2-меркапто-1-оксо-3-фенилпропил)амино]-6-оксопиридо [1,2-a]-азепин-4-карбоновая кислота
a) (S)-2-фталамино-4-пентеновая кислота, дициклогексиламиновая соль
Гомогенный раствор (S)-2-амино-4-пентоновой кислоты (2,988 г, 25,9 мМ) и карбоната натрия (2,600 г, 24,5 мМ) в воде (55 мл) обрабатывали твердым N-(карбетокси)фталамидом (5,677 г, 25,9 мМ). После 2,5-часового размешивания при комнатной температуре, смесь подкисляли 10%-ной соляной кислотой и экстрагировали этилацетатом. Этилацетатный экстракт промывали солевым раствором, осушали сульфатом натрия, фильтровали и упаривали. Остаток два раза подвергали флеш-хроматографии (2%-ная уксусная кислота в этилацетате), после чего нужные фракции продукта объединяли, упаривали и подвергали азеотропной отгонке с использованием толуола. Остаток растворяли в этиловом эфире, обрабатывали 5,3 мл дициклогексиламина и вводили затравочный кристалл. Полученный белый осадок собирали путем фильтрации, промывали этиловым эфиром и осушали в вакууме, в результате чего получали 7,620 г чистого целевого продукта, т.пл. 196 - 197oC, [α]D = -13,9o (c = 0,8, метанол). ТСХ (5% уксусная кислота в этилацетате), Rf = 0,57.
b) [S-(R*,R*)]-2-[(2-фталимидо-1-оксо-4-пентил амино]-6-гидрогексановая кислота, метиловый сложный эфир
Суспензию (S)-2-амино-6-гидроксигексановой кислоты (2,42 г, 16,4 мМ) в сухом метаноле (60 мл) обрабатывали газообразным хлороводородом до тех пор, пока не начнется обратный сток смеси. Затем гомогенный раствор размешивали при комнатной температуре в течение 2,5 ч. После этого растворитель упаривали, а остаток подвергали азеотропной обработке (3 раза) с использованием толуола, в результате чего получали неочищенный метиловый эфир (S)-2-амино-6-гидроксигексановой кислоты гидрохлоридную соль в виде маслообразного продукта. Этот продукт растворяли в диметилформамиде (20 мл) и метиленхлориде (50 мл) и обрабатывали 4-метилморфолином (3,20 мл, 2,94 г, 29,1 мМ). Полученную смесь охлаждали до 0oC и обрабатывали (S)-2-фталамидо-4-пентеновой кислотой, полученной из 7,0 г, 16,4 мМ солевого продукта части (a) путем распределения между 10% бисульфатом калия и этилацетатом, в метиленхлориде (10 мл), а затем твердым гидроксибензотриазолом (2,22 г, 16,4 мМ) и гидрохлоридной солью этил-3-(3-диметиламино) пропилкарбодиимида (3,458 г, 18,0 мМ). Затем смесь размешивали 0,5 ч при 0oC и 2 ч при комнатной температуре и распределяли между этилацетатом и 0,5 н. соляной кислотой. Этилацетатный экстракт промывали последовательно водой, 50% насыщенным бикарбонатом натрия и солевым раствором, а затем осушали сульфатом натрия, фильтровали и упаривали. Остаток подвергали флеш-хроматографии (силикагель Merck, этилацетат) и получали чистое целевое соединение в виде маслообразного вещества, которое затем отверждалось после отстаивания. Твердый продукт растирали с этиловым эфиром и гексаном, собирали путем фильтрации и получали в результате 5,149 г аналитически чистого продукта, т.пл. 90 - 92oC, [α]D = +25,6o (c= 1,1, хлороформ). ТСХ (этилацетат) Rf = 0,36.
c)[S-(R*, R*)] -2-[(2-фталамидо-1-оксо-4-пентил) амино]-6-оксогексановая кислота, метиловый сложный эфир
Раствор (-78oC) оксалилхлорида (1,38 мл, 0,95 г, 7,5 мМ) в метиленхлориде (70 мл) по капле обрабатывали раствором сухого диметилсульфоксида (2,20 мл, 2,00 г, 25,6 мМ) в метиленхлориде (2 мл). Через 10 минут смесь обрабатывали раствором продукта части (b) (5,04 г, 13,0 мМ) в метиленхлориде (15 мл). Затем еще через 10 мин добавляли триэтиламин (9,0 мл) и смесь размешивали 5 мин при -78oC, а затем оставляли постепенно нагреваться до 0oC. После этого смесь распределяли между этилацетатом/этиловым эфиром и 0,5 н. соляной кислотой. Органический экстракт промывали 50% насыщенным бикарбонатом натрия и солевым раствором, а затем осушали сульфатом натрия, фильтровали и упаривали. Остаток кристаллизовали из этилацетата/этилового эфира и получали 4,567 г целевого продукта в виде белого твердого вещества. Маточный раствор подвергали флеш-хроматографии (Merck силикагель, 6/4-этилацетат/гексан) и кристаллизовали, в результате чего получали еще 184 мг продукта (всего 4,751 г), т. пл. 74 - 76oC, [α]D = +25,2o (c = 1,3, хлороформ). ТСХ (этилацетат: гексаны, 6:4), Rf = 0,22.
d) [S-(R*, R*)-1,2,3,4-Тетрагидро-(1-оксо-2-фталимидо-4- пентенил)-2-пиридинкарбоновая кислота, метиловый сложный эфир
Раствор продукта, полученного в части (c), (3,657 г, 9,46 мМ) трифторуксусной кислоты (190 мкл) в метиленхлориде (70 мл) нагревали в атмосфере аргона в течение 1,5 ч. Охлажденную смесь промывали разбавленным водным бикарбонатом натрия, осушали сульфатом натрия, фильтровали и упаривали. Остаток подвергали флеш-хроматографии (силикагель Merck, этилацетат: гексан = 1: 1) и получали 3,417 г целевого продукта в виде маслообразного и пенистого продукта. ТСХ (этилацетат:гексан), Rf = 0,50.
e) [4S- (4α, 7α, 10aβ) ]-Декагидро-9-иодо-6-оксо-7-фталимидо-[1,2-a]азепин, 4-карбоновая кислота, метиловый сложный эфир
Раствор продукта, полученного в части (d), (1,427 г, 3,87 мМ) в метиленхлориде (8,8 мл) по капле добавляли к смеси трифторметансульфоновой кислоты (2,2 мл) и ангидрида трифторметансульфоновой кислоты (210 мкл) при комнатной температуре. Раствор ярко-желтого цвета размешивали в течение 5,5 ч, а затем выливали в ледяную воду и экстрагировали этилацетатом. Этилацетатный экстракт промывали солевым раствором, осушали сульфатом натрия, фильтровали и упаривали. Неочищенный остаток (в основном смесь карбоновых кислот) растворяли в метаноле (3 мл) и метиленхлориде (20 мл) и обрабатывали в течение 25 мин избыточным количеством эфирного диазометана. Этот избыточный диазометан удаляли путем барботирования аргона, а растворитель выпаривали. Остаток растворяли в метилэтиленкетоне (40 мл) и обрабатывали иодидом натрия (2,48 г). После 1-часового размешивания при комнатной температуре, смесь распределяли между этилацетатом и водой, которая содержала небольшое количество бисульфата натрия. Органический слой промывали солевым раствором, осушали сульфатом натрия, фильтровали и упаривали. Остаток подвергали флеш-хроматографии (силикагель Merck, этилацетат: гексан, 1:1), а затем этилацетат: гексан) и получали 1,125 г целевого продукта в виде белого пенистого вещества, [α]D = -17,1o (c = 0,7, хлороформ). ТСХ (этилацетат:гексан, 6:4), Rf = 0,43.
Кроме того, получали 206 мг [4S - (4α, 7α, 10aβ) ]- 1,2,3,4,6,7,8,10a-октагидро-6-оксо-7-фталимида-пиридо [1,2-a] азепина 4-карбоновой кислоты, метилового сложного эфира и растирали с эфиром, в результате чего получали белое твердое вещество, т.пл. 162 - 166oC, [α]D = -106,0o (c = 0,8, хлороформ). ТСХ (этилацетат: гексан, 6:4), Rf = 0,36.
f) [4S- (4α, 7α, 10aβ) ] -Декагидро-6-оксо-7-фталимидо-пиридо [1,2-a] азепин, 4-карбоновая кислота, метиловый сложный эфир
Раствор целевого продукта из части (e) (1,068 г, 2,15 мМ) и трис(триметилсилил)-силана (1,0 мл, 806 мг, 3,2 мМ) в сухом бензоле (10 мл) нагревали до 50oC и каждые 30 мин обрабатывали каталитическим количеством (2-3 мг) 2,2'-азобисизобутиронитрила. Через 3,5 ч добавляли еще 400 мкл силана и реакцию продолжали. Через 5 ч слегка мутноватый раствор охлаждали до комнатной температуры и концентрировали. Остаток растирали с этиловым эфиром, полученное твердое вещество собирали путем фильтрации и тщательно промывали этиловым эфиром, в результате чего получали 522 мг в основном чистого целевого продукта. Маточный раствор подвергали флеш-хроматографии (силикагель Merck, этилацетат: гексан, 1:1) и получали еще 261 мг чистого продукта (всего 783 мг), т.пл. 179 - 181oC, [α]D = -10,6o (c = 0,9, хлороформ). ТСХ (этилацетат: гексан, 1:1), Rf = 0,25.
g) [4S- [4α, 7α(R*), 10aβ] ]-Декагидро-7-[[2-(ацетилтио)-1-оксо-3-фенилпропил]амино]-6-оксопиридо- [1,2-a]азепин-4-карбоновая кислота, метиловый сложный эфир
Продукт, полученный в части (f) (786 мг, 2,12 мМ) в метаноле (10 мл) и метиленхлориде (2 мл) обрабатывали моногидратом гидразина (135 мкл, 2,8 мМ) и полученный раствор размешивали 66 ч при комнатной температуре. Затем смесь фильтровали, а твердый остаток промывали метанолом. Фильтрат упаривали, растирали с метиленхлоридом и снова фильтровали. Фильтрат промывали водой, а водный слой подвергали обратному экстрагированию метиленхлоридом. Объединенные метиленхлоридные экстракты осушали сульфатом натрия, фильтровали и упаривали, в результате чего получали 479 мг неочищенного метилового эфира [4S- (4α, 7α, 10aβ) ]-декагидро-7-амино-6-оксопиридо [1,2-a] азапин-4-карбоновой кислоты в виде бесцветного маслообразного продукта. ТСХ (10% метанол в метиленхлориде), Rf = 0,18.
Холодный (0oC) раствор дициклогексиламиновой соли (S)-2-(ацетилтио)бензолпропановой кислоты, полученной из дициклогексиламиновой соли, как описано выше, (524 мг, 2,33 мМ), триэтиламина (295 мкл, 214 мг, 2,11 мМ) и полученный ранее амин в метиленхлориде (15 мл) обрабатывали гексафторофосфатом бензотриазол-1-илокситрис- диметиламино фосфония (940 мг, 2,12 мМ). Полученный раствор размешивали 1 ч при 0oC, а затем 2 ч при комнатной температуре. После этого растворитель выпаривали, а остаток распределяли между этилацетатом и 1 н. соляной кислотой. Органический слой промывали последовательно водой, 50% насыщенным бикарбонатом натрия и солевым раствором, а затем осушали сульфатом натрия, фильтровали и упаривали. Остаток подвергали флеш-хроматографии (силикагель Merck, этилацетат: гексан, 1:1) и получали чистое целевое соединение (770 мг) в виде белого пенистого продукта. ТСХ (этилацетат:гексан), Rf= 0,27.
h) [4S- [4α-, 7α(R*), 10aβ] ] - Декагидро-7-[(2-меркапто-1-оксо-3-фенилпропил)амино]-6-оксопиридо [1,2-a]-алазепин-4-карбоновая кислота
Раствор продукта, полученного в части (g), (755 мг, 1,70 мМ), комнатной температуры в метаноле (8 мл, дезоксигенированного посредством барботирования аргона) обрабатывали 1 н. гидроксидом натрия (10 мл, дезоксигенированного посредством барботирования аргона). После 3-часового размешивания, смесь окисляли 10% соляной кислотой, разводили водой и экстрагировали этилацетатом. Этилацетатный экстракт промывали солевым раствором, осушали сульфатом натрия, фильтровали и упаривали. Остаток подвергали флеш-хроматографии (силикагель Merck, 1%-ная уксусная кислота в этилацетате). Нужные продуктовые фракции упаривали, подвергали азеотропной обработке (3 раза) с использованием этилацетата, растворяли в небольшом количестве этилацетата и растирали с гексаном. Растворители удаляли на роторном испарителе, а остаток снова растирали с гексаном, упаривали и осушали в вакууме, в результате чего получали 654 мг целевого продукта в виде белого аморфного порошка, [α]D = -31,0o (c = 0,8, хлороформ). ТСХ (2% уксусная кислота в этилацетате), Rf = 0,51.
Элементный анализ для C20H26N2O4S • 0,16 C4H8O2 • 0,3 H2O;
Вычислено: C 60,46; H 6,85; N 6,83; S 7,82;
Найдено: C 60,57; H 7,07; N 6,57; S 7,63.
Пример 36.
[3S- [3α(R*), 7β] ] -Гексагидро-3-[(2-меркапто-2-оксо-3- фенилпропил)амино]-2-оксо-7-(2-профенил-1H-азепин-1-уксусная кислота.
a) (S)-N-(2-Фталимидо-1-оксогексил)глицин этиловый сложный эфир
Суспензию глицина, этилового эфира гидрохлоридной соли (2,718 г, 19,5 мМ) в диметилформамиде (36 мл) обрабатывали 4-метилморфолином (2,60 мл, 2,39 г 23,6 мМ) и 5 мин размешивали при комнатной температуре. Затем смесь обрабатывали (S)-2-фталимидо-6-гидроксигексановой кислотой (4,50 г, 16,2 мМ) и гидроксибензотриазолом (2,225 г, 16,5 мМ), охлаждали до 0oC, а затем обрабатывали этил-3-(3-диметиламино)пропилкарбодиимида гидрохлоридной солью (3,438 г, 17,9 мМ). Затем смесь размешивали 1 ч при 0oC и 2 ч при комнатной температуре, после чего ее распределяли между этилацетатом и 0,5 н. гидрохлоридной солью и три раза экстрагировали этилацетатом. Объединенные этилацетатные экстракты промывали последовательно водой, насыщенным бикарбонатом натрия и солевым раствором, а затем осушали сульфатом натрия, фильтровали и упаривали, в результате чего получали 5,77 г целевого продукта в виде бесцветного маслообразного вещества. ТСХ (этилацетат), Rf = 0,34.
b) (S)-N-(2-Фталимидо-1,6-дизоксогексил)глицин, этиловый эфир
Раствор -78oC оксалилхлорида (1,67 мл, 2,43 г, 19,1 мМ) в метиленхлориде (50 мл) по капле обрабатывали раствором сухого диметилсульфоксида (2,70 мл, 2,97 г, 38,0 мМ) в метиленхлориде (2 мл). Через 15 мин добавляли раствор продукта части (a) (5,770 г, 15,9 мМ) в метиленхлориде (25 мл). Еще через 15 мин, смесь обрабатывали триэтиламином 10,0 мл, размешивали 5 мин при -78oC, а затем оставляли нагреваться до 0oC. Полученную смесь промывали 1 н. соляной кислотой и солевым раствором, осушали сульфатом натрия, фильтровали и упаривали. Остаток подвергали флеш-хроматографии (силикагель Merck, Этилацетат: гексан, 80:20) и получали 5,170 г целевого соединения в виде бесцветного маслообразного вещества.
c) (6S-транс)-Тетрагидро-6-фталимидо-оксазоло[3,2-b] -азепин- 2,5-(3H, 6H)-дион
Раствор продукта, полученного в части (b), (5,16 г, 14,3 мМ) в трифторуксусной кислоте (40 мл) и хлороформе (160 мл) нагревали с обратным холодильником в атмосфере аргона в течение 42 ч. Полученную смесь охлаждали до комнатной температуры и нейтрализовали насыщенным водным бикарбонатом натрия. Слои разделяли, а водный слой экстрагировали метиленхлоридом. Объединенные органические слои промывали солевым раствором, осушали сульфатом натрия и фильтровали через небольшой слой силикагеля, промывая смесью (1:1) этилацетата и метиленхлорида. Фильтрат упаривали и получали твердый остаток. Этот твердый остаток суспендировали в метиленхлориде и растирали с этиловым эфиром, в результате чего получали 3,437 г целевого продукта в виде твердого белого вещества, т.пл. 234-240oC. TCX (ацетон: гексан, 1:1), Rf = 0,51.
d) (3S-транс)-Гексагидро-3-фталимидо-2-оксо-7-(2-пропенил)- 1H-азепин-1-уксусная кислота
Раствор продукта, полученного в части (c), (2,6 г, 8,27 мМ) и аллилтриметилсилана (10,0 мл, 7,19 г, 62,9 мМ) в метиленхлориде (75 мл) обрабатывали при комнатной температуре бромидом олова 4 (1,0 М в метиленхлориде, 16,5 мл, 16,5 мМ). Полученную смесь размешивали 9 ч при комнатной температуре, а затем 14 ч при -20oC, после чего реакцию гасили водой и экстрагировали смесью этилацетата и этилового эфира. Экстракт промывали солевым раствором, осушали сульфатом натрия, фильтровали и упаривали, в результате чего получали маслообразное вещество молочно-белой окраски. Это вещество подвергали флеш-хроматографии (силикагель Merck, 2%-ная уксусная кислота в этилацетате) в результате чего получали 2,810 г диастереометрически чистого целевого соединения в виде белой пены. TCX (5%-ная уксусная кислота в этилацетате), Rf = 0,55.
e) (3S-транс)-Гексагидро-3-фталимидо-2-оксо-7-(2-пропенил)- 1H-азепин-1-уксусная кислота, метиловый эфир
Холодный (0oC) раствор продукта части (d) (2,50 г, 7,0 мМ) в метаноле (20 мл) и этиловым эфире (30 мл) обрабатывали избыточным количеством эфирного диазометана в течение 10 мин. Этот избыточный диазометан нейтрализовали путем добавления уксусной кислоты, а растворитель удаляли на роторном испарителе, в результате чего получали желтое маслообразное вещество. Это вещество подвергали флеш-хроматографии (силикагель Merck, 1:1 - этилацетат:гексан) и получали в результате пенистый продукт. Это продукт растворяли в горячем этиловом эфире, содержащем небольшое количество метиленхлорида, а затем вносили затравку, в результате чего получали 2,072 г кристаллического целевого продукта. Маточный раствор дал еще 262 мг продукта (всего 2,334 г), т.пл. 107 - 109oC. ТСХ (этилацетат:гексан, 1:1), Rf = 0,29.
f) [3S- [3α(R*), 7β] -Гексагидро-3-[[2-(ацетилтио)-1-оксо- 3-фенилпропил]амино]-2-оксо-(2-пропенил)-1H-азепин-1-уксусная кислота, метиловый сложный эфир
Суспензию кристаллического продукта, полученного в части (e), (492 мг, 1,33 мМ) в метаноле (10 мл) обрабатывали моногидратом гидразина (142 мкл, 147 мг, 2,93 мМ), после чего (для осуществления солюбилизации исходного материала) раствор быстро нагревали. После 18-часового размешивания при комнатной температуре, смесь разбавляли метиленхлоридом и фильтровали. Фильтрат упаривали, суспендировали в метиленхлориде, фильтровали и снова упаривали, в результате чего получали сырой амин (270 мг) в виде бесцветного маслообразного вещества. Холодный (0oC) раствор амина и (S)-2-(ацетилтио) бензолпропановой кислоты (полученной из дициклогексиламиновой соли как описано выше, 287 мг, 1,28 мМ) в метиленхлориде (12 мл) обрабатывали триэтиламином (172 мкл, 125 мг, 1,23 мМ), а затем гексафторофосфатом трис(диметиламино) фосфония (547 мг, 1,24 мМ). Прозрачный, почти бесцветный раствор размешивали при 0oC в течение 1 ч, а затем при комнатной температуре в течение 2 ч. Смесь упаривали, а затем распределяли между этилацетатом и 1 н. соляной кислотой. Этилацетатный экстракт промывали последовательно водой, 50%-ным насыщенным бикарбонатом натрия и солевым раствором. Полученный раствор осушали сульфатом натрия, фильтровали и упаривали. Остаток подвергали флеш-хроматографии (силикагель Merck, гексан:ацетон:этиловый эфир, 6: 3: 1) и получали 352 мг целевого продукта в виде смеси диастереомеров (84:16). Этот материал объединяли с партией аналогичного примесного продукта и этот объединенный материал очищали с помощью препаративной обращенно-фазовой жидкостной хроматографии высокого разрешения (УМС SH-365-10 S-10 120A ODS, колонка 30 • 400 мм, изократная смесь 66% метанол/34% вода, 50 мл/мин, детектирование при 220 нм, целевой продукт tR = 22,6 мин, примесь: tR = 28,7 мин). Целевой продукт был получен в виде бесцветного маслообразного вещества с чистотой 98,2% (ацетон:гексан, 4:6), Rf = 0,38.
g) [3S- [3α(R*), 7β] -Гексагидро-3-[2-меркапто-1-оксо-3-фенилпропил) амино]-2-оксо-7-(2-пропенил)-1H-азепин-1-уксусная кислота
Раствор комнатной температуры продукта, полученного в части (f), (720 мг, 1,61 мМ) в метаноле (12 мл, дезоксигенированного посредством барботирования аргона) обрабатывали 1 н. гидроксидом натрия (10 мл, дезоксигенированного посредством барботирования аргоном). После 20-минутного размешивания, раствор подкисляли 10%-ный соляной кислотой (5 мл) и экстрагировали этилацетатом. Этилацетатный экстракт промывали водой и солевым раствором, а затем осушали сульфатом натрия, фильтровали и упаривали, в результате чего получали маслообразное вещество. Этот материал подвергали флеш-хроматографии (силикагель Merck, 2-5%-ная уксусная кислота в этилацетате). Продуктовые фракции объединяли и упаривали, а остаток подвергали азеотропной обработке (три раза) с использованием этилацетата. Полученный маслообразный/пенистый продукт растворяли в небольшом количестве этилацетата, а затем растирали с гексаном, в результате чего получали белый маслообразный/пенистый продукт. Смесь упаривали досуха, суспендировали в гексан, снова упаривали досуха и осушали в вакууме, в результате получали 616 мг целевого продукта в виде твердого пенообразного продукта белого цвета, [α]D = -48,5o (c = 0,63, хлороформ). ТСХ (5% уксусная кислота в этилацетате) Rf = 0,56.
Анализ для C20H26N2O4S • 0,2 H2O:
Вычислено: C 60,95; H 6,75; N 7,11; S 8,14;
Найдено: C 60,98; H 6,93; N 6,93; S 7,94
Пример 37.
[3S- [3α(R*), 7β] -Гексагидро-3-[(2-меркапто-1-оксо-3-фенилпропил) амино]-2-оксо-7-пропил-1H-азепин-1-уксусная кислота
a) (3S-транс)-Гексагидро-3-фталимидо-2-оксо-7-пропил-1H- азепин-1-уксусная кислота, метиловый сложный эфир
Раствор продукта примера 36 (e) (767 мг, 2,07 мМ) в метаноле (10 мл) и этилацетате (10 мл) гидрировали (баллонный водород) в присутствии палладированного угля (10% палладия на угле, 62 мг) и при комнатной температуре в течение 1 ч. Полученную смесь фильтровали через целит, упаривали, снова растворяли в этилацетате, фильтровали через слой силикагеля, снова упаривали и получали 780 мг целевого продукта в виде бесцветного маслообразного продукта. ТСХ (этилацетат:гексан = 1:1), Rf = 0,29.
b) [3S- [3α(R*), 7β] -Гексагидро-3-[[2-(ацетилтио)-1-оксо-3-фенилпропил] амино]-2-оксо-7-пропил-1H-азепин-1-уксусная кислота, метиловый сложный эфир.
Продукт части (a) (774 мг, 2,07 мМ) в метаноле (8 мл) обрабатывали моногидратом гидразина (222 мкл, 229 мг, 4,58 мМ) и полученный раствор размешивали 23 ч при комнатной температуре. Смесь разводили 0,5 н. соляной кислотой (20 мл) и размешивали при 0oС в течение 2 ч. Затем раствор фильтровали и фильтрат промывали этилацетатом. Этилацетатный экстракт один раз подвергали обратному экстрагированию водой и объединенные водные слои подщелачивали 1 н. гидроксидом натрия. После экстракции метиленхлоридом (три раза), с последующей осушкой сульфатом натрия и выпариванием растворителя, получали неочищенный амин (354 мг) в виде бесцветного маслообразного продукта.
Холодный раствор (0oC) вышеуказанного сырого амина и (S)- (ацетилтио) бензолпропановой кислоты полученной из дициклогексиламиновой соли, как описано выше, 344 мг, 1,53 мМ) в метиленхлориде (12 мл) обрабатывали триэтиламином (214 мкл, 154 мг, 1,53 мМ), а затем бензотриазол-1-илокситрис (диметиламино)фосфония гексафторофосфатом (679 мг, 1,54 мМ). Прозрачный, почти бесцветный раствор размешивали 1 ч при 0oC, а затем 1 ч при комнатной температуре. После этого смесь упаривали и распределяли между этилацетатом и 0,5 н. соляной кислотой. Этилацетатный экстракт промывали последовательно водой, 50% насыщенным бикарбонатом натрия и солевым раствором. Затем раствор осушали сульфатом натрия, фильтровали и упаривали. Остаток подвергали флеш-хроматографии (силикагель Мerck, гексан:ацетон = 6:4) и получали 510 мг чистого целевого продукта в виде бесцветного маслообразного вещества. ТСХ (ацетон:гексан, 4:6) Rf = 0,31.
c) [3S- [3α(R*), 7β] ] -Гексагидро-3-[(2-меркапто-1-оксо-3- фенилпропил)амино]-2-оксо-7-пропил-1H-азепин-3-уксусная кислота.
Раствор (комнатной температуры) продукта части (b) (502 мг, 1,12 мМ) в метаноле (8 мл, дезоксигенированном посредством барботирования аргона) обрабатывали 1 н. NaOH (6 мл, дезоксигенированным посредством барботирования аргона). После 25-минутного размешивания, раствор подкисляли 10%-ной соляной кислотой и экстрагировали этилацетатом. Этилацетатный экстракт промывали водой и солевым раствором, а затем осушали сульфатом натрия, фильтровали, упаривали и получали маслообразное вещество. Это вещество подвергали флеш-хроматографии (силикагель Merck, 2%-ная уксусная кислота в этилацетате). Продуктовые фракции объединяли и упаривали, а остаток два раза подвергали азеотропной обработке с использованием этилацетата. Полученный маслообразный/пенистый материал растворяли в небольшом количестве метиленхлорида, а затем растирали с гексаном, в результате чего получали белое маслообразное/пенистое вещество. Смесь упаривали досуха, суспендировали в гексане, упаривали снова досуха и осушали в вакууме, в результате чего получали 394 мг целевого продукта в виде твердого белого пенистого продукта; [α]D = -51,2o (с = 0,65, хлороформ). ТСХ (2% уксусная кислота в этилацетате), Rf = 0,47.
Анализ для C20H28N2O4S • 0,2 H2O:
Вычислено: C 60,95; H 6,75; N 7,11; S 8,14;
Найдено: C 60,98; H 6,93; N 6,93; S 7,94.
Пример 38.
[3S- [3α(R*), 7β] ]-7-Циклопропилметил)гексагидро-3-[(2-меркапто-1- оксо-3-фенилпропил)амино]-2-оксо-1H-азепин-1-уксусная кислота.
a) (3S-транс-7-(Циклопропилметил)гексагидро-3-фталимидо-2- оксо-1H-азепин-1-уксусная кислота, метиловый сложный эфир.
К раствору продукта примера 36 (e) (700 мг, 1,879 мМ) в 5 мл метиленхлорида, охлажденному до 0oC, добавляли 75 мл-порцию раствора диазометана/этилового эфира (полученного в соответствии с процедурой Fieser & Fieser т. 1, с. 192), а затем добавляли 7 мг (0,03 мМ) ацетата палладия (II). В реакционной смеси происходило энергичное выделение пузырьков, и она становилась прозрачной. После 10-минутного размешивания, к реакционной смеси добавляли еще 25 мл раствора диазометана/этилового эфира. Затем реакционную смесь 50 мин размешивали при 0oC, фильтровали через целит и концентрировали в вакууме, в результате чего получали сырой маслообразный продукт. Этот продукт подвергали флеш-хроматографии (силикагель Merck, 50 • 150 нм, этилацетат/гексан, 1:2) и получали 717 мг целевого продукта в виде белого пенистого вещества.
b) [3S- [3α(R*), 7β] ]-7-(Циклопропилметил)гексагидро-3-[[2-(ацетилтио)- 1-оксо-3-фенилпропил] амино] -2-оксо-1H-азепин-1-уксусная кислота, метиловый сложный эфир.
К раствору продукта части (a) (697 мг, 1,81 мМ) в 8 мл метанола, размешанному при комнатной температуре, по капле добавляли 92 мкл (1,90 мМ) моногидрата гидразина. Реакционную смесь размешивали 48 часов при комнатной температуре, а затем фильтровали для удаления твердых побочных продуктов. Фильтрат концентрировали в вакууме, растворяли в метиленхлориде, снова фильтровали и снова концентрировали, в результате чего получали сырой маслообразный материал. Этот материал осушали в вакууме и получали 441 мг неочищенного амина в виде белого пенистого вещества. Это пенистое вещество использовали в следующей реакции без дополнительной очистки.
К холодному (0oC) раствору 420 мг (1,64 мМ) полученного неочищенного амина и 384 мг (1,84 мМ (S)-2-(ацетилтио)бензолпропановой кислоты (полученной из дициклогексиламиновой соли, как описано выше) в 10 мл хлороформа добавляли 280 мкл (1,97 мМ) триэтиламина. Реакционную смесь размешивали 30 мин, а затем добавляли 799 мг (1,81 мМ) гексафторофосфата бензотриазол 1 илокситрис(диметиламино)фосфония. Реакционную смесь размешивали 48 ч при 0oC, а затем добавляли еще 399 мг (0,904 мМ) гексафторофосфата бензотриазол-1-илокситрис- (диметиламино)фосфония и 4 капли триэтиламина. Реакционную смесь размешивали еще 24 ч при 0oC, а затем распределяли между 50 мл этилацетата и 50 мл 5%-ного раствора бисульфата калия. Водный слой отделяли и экстрагировали 2 - 25 мл-порциями этилацетата. Объединенные этилацетатные слои промывали 25 мл 5%-ным раствором бисульфата калия, 40 мл насыщенным бикарбонатом натрия и 40 мл солевым раствором, затем осушали сульфатом магния и концентрировали в вакууме, в результате чего получали неочищенное маслообразное вещество. Это вещество подвергали флеш-хроматографии (силикагель Merck, 50 • 200 нм, смесь этилацетата и гексана, 1:3, а затем 1:2) и получали 678 мг целевого продукта в виде белого пенистого продукта.
c) [3S- [3α(R*), 7β] ]-7-(Циклопропилметил)гексагидро-3-[(2-меркапто-1- оксо-3-фенилпропил)амино]-2-оксо-1H-азепин-1-уксусная кислота
Раствор 658 мг (1,43 мМ) продукта части (b) в 10 мл метанола продували аргоном в течение 30 мин и охлаждали до 0oC. К этому раствору по капле добавляли 6 мл 1M гидроксида натрия и снова 30 мин продували аргоном, а затем охлаждали до 0oC. Реакционную смесь размешивали в течение 1 ч при 0oC, продолжая продувать аргоном, а затем подкисляли до pH 1,5%-ным раствором бисульфата калия. Смесь экстрагировали 3-80 мл-порциями этилацетата, а объединенные этилацетатные слои осушали сульфатом магния и концентрировали в вакууме, в результате чего получали неочищенное маслообразное вещество. Это вещество подвергали флеш-хроматографии (силикагель Merck, 50 • 150 мм, 0,2%-ная уксусная кислота/этилацетат) и получали белое пенистое вещество. Это пенистое вещество растирали с метиленхлоридом/гексаном, в результате чего 490 мг целевого продукта в виде белого твердого продукта, [α]D = -78,9o (с = 1,0, CDCl3). ТСХ (5% метанол в метиленхлориде + 3 капли уксусной кислоты), Rf = 0,41.
Анализ для C21H28N2SO4 • 0,50 H2O:
Вычислено:C 61,00; H 7,07; N 6,77; C 7,93;
Найдено: C 61,40; H 6,75; N 6,37; S 7,75.
Пример 39.
[3S- [3α(R*), 7β] ]-7-(Циклопентил)гексагидро-3-[(2-меркапто-1-оксо-3- фенилпропил)амино]-2-оксо-1H-азепин-1-уксусная кислота.
a) (3S-транс)-7-(2-Циклопентил)гексагидро-3-фталимидо-2- оксо-1H-азепин-1-уксусная кислота, метиловый сложный эфир
Тетрахлорид олова (5,1 л, 5,1 мМ, 1 М метиленхлорида) по капле добавляли к раствору продукта примера 36(e) (800 мг, 2,55 мМ) и (2-циклопентенил)триметилсилан (2,85 г, 20,4 мМ) в метиленхлориде (60 мл). Смесь размешивали при комнатной температуре в течение 18 ч, после чего реакцию гасили путем добавления 100 мл воды. Смесь экстрагировали 3 - 100 мл-порциями этилацетата и 2 - 100 мл-порциями этилового эфира, а объединенные органические слои промывали 100 мл солевого раствора, осушали сульфатом натрия и концентрировали в вакууме, в результате чего получали неочищенное маслообразное вещество. Это вещество подвергали флеш-хроматографии (силикагель Merck, 50 • 100 мм, 1: 1 - смесь этилацетата/гексана, а затем 2% уксусной кислоты/этилацетата) и получали 788 мг (3-транс)-7-(2-циклопентил)гексагидро-3-фталимидо-2- оксо-1H-азепин-1-уксусной кислоты в виде белого пенистого вещества.
Раствор этой кислоты (768 мг, 2,01 мМ) в метаноле (7 мл)/этиловом эфире (10 мл), размешанный при комнатной температуре, по капле обрабатывали раствором диазометана/этилового эфира, полученным в соответствии с известной процедурой (Fieser & Fieser т. 1, с. 192). Затем добавляли раствор диазометана/этилового эфира до тех пор, пока реакционная смесь не будет желтого цвета и пока не прекратится выделение пузырьков. После этого смесь размешивали 10 мин, а затем реакцию гасили путем добавления по капле 0,3 мл уксусной кислоты. Бесцветный раствор концентрировали в вакууме и получали неочищенный маслообразный продукт. После флеш-хроматографии этого продукта (силикагель Merck, 50 • 100 мм, 1:2 - смесь этилацетата/гексана) получали 640 мг целевого продукта в виде пенистого белого вещества.
b) (3S-транс)-7-(Циклопентил)гексагидро-3-фталимидо-2-оксо- 1H-азепин-1-уксусная кислота, метиловый сложный эфир.
Раствор продукта, полученного в части (a), (700 мг, 1,89 мМ) в 5 мл метанола продували 15 мин аргоном, а затем добавляли 150 мг (25 мас.%) 10% палладированного угля. Затем реакционный сосуд удаляли и наполняли водородом (3 раза) и 5 ч размешивали в атмосфере водорода (1 атм, баллон). Реакционную смесь фильтровали через целит и фильтрат концентрировали в вакууме, в результате чего получали 594 мг целевого продукта в виде белого пенистого вещества.
c) [3S- [3α(R*), 7β] ]-7-(Циклопентил)гексагидро-3-[[2-(ацетилтио)-1- оксо-3-фенилпропил] амино] -2-оксо-1H-азепин-1-уксусная кислота, метиловый сложный эфир.
Раствор продукта, полученного в части (b), в метаноле обрабатывали моногидратом гидразина в соответствии с процедурой, описанной в примере 38, части (b), в результате чего получали неочищенный метиловый сложный эфир (3S-транс)-7-циклопентил)- гексагидро-3-амино-2-окси-1H-азепин-1-уксусной кислоты в виде белого пенистого продукта.
Этот пенистый продукт обрабатывали (S)-(ацетилтио)бензолпропановой кислотой в соответствии с процедурой, описанной в примере 38, части (b), в результате чего получали целевой продукт в виде прозрачного маслообразного вещества.
d) [3S- [3α(R*), 7β] ]-7-(Циклопентил)гексагидро-3-[(2-меркапто-1-оксо-3- фенилпропил)амино]-2-оксо-1H-азепин-1-уксусная кислота.
Раствор продукта, полученного в части (с), в метаноле обрабатывали 1 М гидроксидом натрия в соответствии с процедурой, описанной в примере 38, часть (c), в результате чего получали целевой продукт в виде белого твердого вещества, [α]D = -78,1o (с = 1, CDCl3). ТСХ (5% метанол в метиленхлориде + 3 капли уксусной кислоты), Rf = 0,39.
Анализ для C22H30N2SO4 • 0,03 H2O:
Вычислено: C 63,06; H 7,23; N 6,69; S 7,65;
Найдено: C 63,25; H 7,25; N 6,50; S 7,55.
Пример 40.
[S-(R*, R*)] -Гексагидро-3-[2-меркапто-2-оксо-3- фенилпропил)амино]-2-оксо-1H-азепин-1-пропановая кислота
a) (S-3-[[(1,1-Диметилотокси)карбонил]амино-гексагидро-2- оксо-1H-азепин-1-пропановая кислота, этиловый сложный эфир
Раствор продукта получают 9(b) (1,75 г, 7,67 мМ) в смеси тетрагидрофурана и т-бутанола (2:1, 30 мл) охлаждали до 0oC и обрабатывали этилацетатом (1,16 мл, 10,71 мМ, 1,4 экв.), а затем 1,0 н. т-бутанола калиевой солью (767 мкл, 0,1 экв. ). Реакционную смесь размешивали 15 мин при 0oC, а затем 1 ч при комнатной температуре. После этого реакцию гасили 25%-ным хлоридом аммония (50 мл) и экстрагировали этилацетатом (2 • 125 мл). Объединенные органические экстракты промывали 25%-ным хлоридом аммония (50 мл), водой (100 мл) и солевым раствором (100 мл), а затем осушали сульфатом магния, фильтровали и концентрировали, в результате чего получали прозрачный сироп. Остаток очищали с помощью хроматографии на колонке (5 • 15 см) с силикагелем, элюируя смесью 30% этилацетата и гексана. Нужные фракции объединяли и концентрировали, в результате чего получали 2,18 г целевого продукта в виде прозрачного сиропа. ТСХ (этилацетат : гексан, 1:1), Rf = 0,38.
b) [S-(R*,R*)]-Гексагидро-3-[[2-(ацетилтио-1-оксо- 3-фенилпропил]амино] -2-оксо-1H-азепин-1-пропановая кислота, этиловый эфир.
Продукт, полученный в части (a), (2,15 г, 6,55 мМ) обрабатывали в течение 30 мин и при 0oC 4 н. смесью 4 н. соляной кислоты и диоксана (30 мл, 120 мМ), а затем размешивали 2 ч при комнатной температуре. Реакционную смесь концентрировали, два раза упаривали с этиловым эфиром, 3 раза подвергали азеотропной отгонке с использованием толуола и в течение 5 ч осушали в вакууме, в результате чего получали гидрохлоридную соль (S)-3-амино-гексагидро-2-оксо-1H-азепин-1-пропановой кислоты сложного этилового эфира.
Полученный амин подвергали реакции с (S)-(ацетилтио)бензолпропановой кислотой в присутствии триэтиламина и бензотриазол-1-илокситрис-(диметиламино)фосфония гексафторосфата, как описано в примере 31, часть (c), в результате чего получали целевой продукт. ТСХ (этилацетат : гексан, 1:1), Rf = 0,19.
c) [S-(R*, R*)-Гексигидро-3-(2-меркапто-1-оксо-3- фенилпропил)-амино]-2-оксо-1H-азепин-1-пропановая кислота.
Продукт, полученный в части (b), в метаноле обрабатывали 1 н. гидроксидом натрия, как описано в примере 31, часть (d), в результате чего получали целевой продукт в виде белого пенистого вещества, [α]D = +4,44o (c = 1,24, метанол). ТСХ (1%-ная уксусная кислота в этилацетате), Rf = 0,27.
Анализ для C18H24N2O4S • 0,17 H2O:
Вычислено: C 58,83; H 6,67; N 7,62; S 8,72;
Найдено: C 58,83; H 6,87; N 7,33; S 8,49.
Пример 41.
[S-(R*, R*)] -Гексагидро-3-[(2-меркапто-1-оксо-3- фенилпропил]амино-2-оксо- α- (фенилметил)-1H-азепин-уксусная кислота
a) (R*)-N-(2-фталимидо-6-гидрокси-1-оксогексил)-L- фенилаланин, этиловый сложный эфир
К раствору гидрохлоридной соли этилового эфира L-фенилаланина (998 мг, 4,3 мМ) в диметилформамиде (10 мл) добавляли 4-метилморфолин (575 мкмл, 529 мг, 5,2 мМ). После 5-минутного размешивания при комнатной температуре, раствор охлаждали до 0oC и обрабатывали последовательно (S)-2-фталамидо-6-гидроксигексановой кислотой (1,002 г, 3,6 мМ), гидроксибензотриазолом (582 мг, 4,3 мМ) и гидрохлоридной солью этил-3-(3-диметиламино)пропилкарбодиимида (770 мг, 4,0 мМ). Полученную смесь размешивали полчаса при 0oC и 1,5 ч при комнатной температуре. Затем раствор распределяли между этилацетатом и водой, а органический экстракт промывали последовательно 0,5 н. соляной кислотой, водой, 50% насыщенным бикарбонатом натрия и солевым раствором, осушали сульфатом натрия, фильтровали и упаривали, в результате чего получали 1,63 г в основном чистого целевого продукта в виде маслообразного/пенистого вещества. ТСХ (ацетон:гексан, 1:1), Rf = 0,30.
b) (R*)-N-(2-фталимидо-1,6-диоксогексид)-L-фенилаланин, этиловый сложный эфир
Раствор - 78oC оксалилхлорида (370 мкл, 538 мг, 4,2 мМ) в метиленхлориде (10 мл) по капле обрабатывали раствором сухого диметилсульфоксида (610 мкл, 672 мг, 8,6 мМ) в метиленхлориде (1,5 мл). Через 10 мин добавляли раствор продукта, полученного в части (a), (1,616 г, 3,6 мМ) в метиленхлориде (10 мл). Еще через 15 мин, смесь обрабатывали триэтиламином (4,0 мл), затем 5 мин размешивали при -78oC и оставляли нагреваться до 0oC. Полученную белую суспензию распределяли между 0,5 н. соляной кислотой и этилацетатом. Органический экстракт промывали солевым раствором, осушали сульфатом натрия, фильтровали и упаривали. Остаток подвергали флеш-хроматографиии (силикагель Merck 40:60 - ацетон:гексан) и получали 1,474 г целевого продукта в виде маслообразного/пенистого вещества. ТСХ (ацетон : гексаны, 12:1) Rf = 0,45.
c) [3S- (3α, 6β, 9aα) ] -Тетрагидро-3-(фенилметил)-6-фталимидооксазол[3,2- a]азепин-2,5(3H,6H)-дион
Смесь продукта, полученного в части (b), (6,11 г, 13,6 мМ) и трифторуксусной кислоты (34 мл) в хлороформе (205 мл) нагревали с обратным холодильником в течение 6 дней. Раствор охлаждали до комнатной температуры и нейтрализовали насыщенным бикарбонатом натрия. Слои разделяли и водный слой экстрагировали метиленхлоридом. Объединенные органические слои промывали водой, осушали сульфатом натрия, фильтровали и упаривали, в результате чего получали маслообразное вещество темно-желто-оранжевого цвета. Остаток подвергали флеш-хроматографии (силикагель Merck, 40-60% этилацетат в гексане) и получали 3,075 г целевого продукта в виде белого пенистого вещества.
d) (S-(R*, R*] -Гексагидро-2-оксо-3-фталимидо- α- (фенилметил)-1H-азепин-1-уксусная кислота, метиловый сложный эфир
Раствор продукта, полученного в части (с), (1,40 г, 3,46 мМ) и триэтилсилана (4,4 мл, 3,20 г, 27,5 мМ) в метиленхлориде (42 мл) при комнатной температуре по капле обрабатывали тетрахлоридом титана (7,0 мл, 7,0 мМ). После добавления тетрахлорида титана прозрачный бесцветный раствор становился ярко-желтым, но осадка при этом не образовывалось. Полученный раствор размешивали в течение 66 ч, после чего реакцию гасили водой и экстрагировали этилацетатом. Этилацетатный экстракт промывали водой и солевым раствором, осушали сульфатом натрия, фильтровали и упаривали, в результате чего получали маслообразное вещество. После флеш-хроматографии (силикагель Merck, этилацетат, а затем 2%-ная уксусная кислота в этилацетате) получали 870 мг [S-(R*,R*)]-гексагидро-2-оксо-3-фталимидо- α- (фенилметил)-1H-азепин-1-уксусной кислоты в виде белого пенистого вещества.
Холодный раствор (0oC) указанной кислоты (898 мг, 2,21 мМ) в метаноле (8 мл) и метиленхлориде (12 мл) обрабатывали 5 мин избыточным количеством эфирного диазометана. Затем избыток этого диазометана нейтрализовали путем добавления уксусной кислоты, а растворитель удаляли на роторном испарителе, в результате чего получали желтое маслообразное вещество. Этот остаток подвергали флеш-хроматографии (силикагель Merck, 50 - 60% этилацетат в гексане) и получали 858 мг целевого продукта в виде белого пенистого вещества. ТСХ (этилацетат : гексан, 1:1), Rf = 0,30.
e) [S-(R*,R*)]-Гексагидро-3-[(2-(ацетилтио)-1- оксо-3-фенилпропил]амино] -2-оксо- α- (фенилметил)-1H-азепин-1- уксусная кислота, метиловый сложный эфир
Раствор продукта, полученного в части (d), в метаноле обрабатывали гидразинмоногидратом в соответствии с процедурой, описанной в примере 36, часть (f), в результате чего получали [S-(R*,R*)]-Гексагидро-3-амино-2-оксо- α- (фенилметил)-1H-азепин-1-уксусной кислоты метиловый эфир в виде бледно-желтого маслообразного вещества. ТСХ (метиленхлорид : уксусная кислота : метанол, 8:1:1), Rf = 0,60.
Холодный (0oC) раствор полученного амина подвергали реакции с (S)-2-(ацетилтио) бензолпропановой кислотой в присутствии триэтиламина и гексафторосфосфата бензотриазол-1-илокситрис диметиламина фосфония в соответствии с процедурой, описанной в Примере 36 (f), в результате чего получали бесцветное маслянистое/пенистое вещество. ТСХ (ацетон : гексан : 1:1), Rf = 0,54.
f) [S-(R*, R*)]-Гексагидро-3-(2-меркапто-1- оксо-3-фенилпропил]амино]-2-оксо- α- (фенилметил)-1H-азепин-1- уксусеная кислота
Раствор продукта, полученного в части (e), в метаноле обрабатывали 1 н. гидроксидом натрия в соответствии с процедурой, описанной в примере 36, часть (g), в результате чего получали целевой продукт в виде относительно твердого пенистого вещества; [α]D = -64,2oC (c = 0,54, хлороформ). ТСХ (5% уксусная кислота в этилацетате), Rf = 0,60.
Анализ для C24H28N2O4S • 0,31 H2O:
Вычислено: C 64,61; H 6,47; N 6,28; S 7,19;
Найдено: C 64,61; H 6,78; N 5,89; S 7,46.
Пример 42.
[6(S)] -Гексагидро-6-[(2-меркапто-1-оксо-3-фенилпропил амино]-2,2-диметил-7-оксо-1H-азепин-1-уксусная кислота
a) 2,2-Диметилциклогексанон, оксим
Раствор 2,2-диметилциклогексанона (8,960 г, 65 мМ), пиридина (12,0 мл, 12,27 г, 155 мМ) и гидрохлорида гидроксиламина (10,30 г, 148 мМ) в абсолютном этаноле (50 мл) нагревали 2 ч при 85oC. Охлажденную смесь распределяли между этиловым эфиром и водой и эфирный слой промывали последовательно 1 н. соляной кислотой и солевым раствором, а затем осушали сульфатом натрия, фильтровали и упаривали. Твердый остаток растворяли в небольшом количестве горячего гексана и охлаждали до 0oC, в результате чего получали 6,801 г целевого продукта в виде белых хлопьев, т.пл. 91 - 93oC. ТСХ (этилацетат: гексан, 4:6), Rf = 0,60.
b) Гексагидро-3,3-дихлоро-7,7-диметил-2-оксо-2H-азепин
Холодную суспензию (0 - 10oC) пентахлорида фосфора (32,50 г, 156,1 мМ) в метиленхлориде (250 мл) обрабатывали раствором продукта, полученного в части (a), (7,353 г, 52,1 мМ) в метиленхлориде (50 мл) в течение 10 мин. Полученную смесь нагревали до комнатной температуре и через раствор барботировали газообразный хлор через полчаса и через полтора часа после добавления оксима. Через 3 ч (в целом) реакцию гасили путем добавления измельченного льда (70 мл), а затем насыщенного бикарбоната натрия (150 мл). При этом требовалось проводить периодическое охлаждение. Двухфазную смесь энергично размешивали 18 ч при комнатной температуре, после чего слои разделяли и метиленхлоридный экстракт промывали насыщенным бикарбонатом натрия и солевым раствором. Органический слой осушали сульфатом натрия, фильтровали, упаривали и подвергали флеш-хроматографии (силикагель Merck, 2:8 - этилацетат:метиленхлорид). Нужные продуктовые фракции упаривали и остаток растворяли в горячем метиленхлориде/этилацетате и охлаждали, в результате чего получали 5,152 г чистого целевого продукта. Маточный раствор снова хроматографировали (силикагель Merck, 1: 9 - этилацетат:метиленхлорид) и получали еще 2,054 г чистого продукта (полный выход 7,206 г), т. пл. 148 - 155oC (шир. TCX (этилацетат:метиленхлорид, 1:9), Rf = 0,36.
c) Гексагидро-3-хлор-7,7-диметил-2H-азепин
Раствор продукта, полученного в части (b), (7,180 г, 34,2 мМ) в ледяной уксусной кислоте (150 мл) гидрировали (баллон.) в присутствии 10% палладированного угля (2,65 г) в течение 1,75 ч. Смесь фильтровали через целит, а растворитель удаляли на роторном испарителе. Остаток подвергали флеш-хроматографии (силикагель, 3/7-этилацетат/метиленхлорид) и получали примесный целевой продукт в виде маслообразного твердого вещества. Остаток обрабатывали этиловым эфиром и растирали с гексаном, в результате чего получали 2,845 г чистого целевого продукта в виде белого твердого вещества. Маточный раствор снова хроматографировали (силикагель Merck, 3/7-этилацетат/метиленхлорид), в результате чего получали еще 1,93 г чистого твердого продукта. Твердые фракции объединяли и получали 4,755 г чистого целевого продукта, т.пл. 115 - 117oC. ТСХ (этилацетат:метиленхлорид, 3:7), Rf = 0,41.
d) Гексагидро-3-азидо-7,7-диметил-2-оксо-2H-азепин
Суспензию продукта, полученного в части (c), (4,670 г, 26,6 мМ) и азида натрия (4,360 г, 67,0 мМ) в сухом диметилсульфоксиде (100 мл) нагревали 6 ч при 80oC. Раствор охлаждали до 0oC и обрабатывали 175 миллилитрами ледяной воды. Полученный белоснежно-белый осадок собирали путем фильтрации, промывали водой и осушали, в результате чего получали 2,37 г чистого целевого продукта. Фильтрат экстрагировали этилацетатом и органический экстракт два раза промывали водой и один раз солевым раствором, а затем осушали сульфатом натрия, фильтровали и упаривали. После растирания остатка с этиловым эфиром и гексаном, получали 1,637 г добавочного продукта. Выделенные твердые фракции объединяли и получали 4,007 г чистого целевого продукта; т.пл. 98 - 100oC ТСХ (этилацетат:гексан, 75:25) Rf = 0,51.
e) Гексагидро-3-[[(1,1-диметилэтокси)карбонил] амино] - 7,7-диметил-2-оксо-2H-азепин
Раствор продукта, полученного в части (d), 3,469 г, 19,0 мМ) в метаноле (80 мл) гидрировали (баллон) 1,5 ч в присутствии 10% паллидированного угля (850 мг). Смесь фильтровали через целит, а растворитель удаляли на роторном испарителе. Остаток выпаривали из метиленхлорида и получали неочищенный амин в виде белого твердого вещества. Это твердое вещество снова растворяли в хлороформе и обрабатывали последовательно триэтиламином (2,65 мл, 1,92 г, 19,0 мМ) и ти-трет-бутилдикарбонатом (5,00 г, 22,9 мМ). После 1-часового размешивания при комнатной температуре, раствор выпаривали и остаток растирали с этилацетатом/этиловым эфиром, в результате чего получали 2,668 г целевого продукта в виде белого твердого вещества. Фильтрат подвергали флеш-хроматографии (силикагель Merck, 2/8-этилацетат/метиленхлорид) и получали добавочный чистый продукт, который объединяли с вышеуказанным твердым веществом и получали в целом 4,082 г чистого целевого продукта, т.пл. 156 - 158,5oC. ТСХ (этилацетат:метиленхлорид, 2:8), Rf = 0,26.
f) Гексагидро-3-[[1,1-диметилэтокси)карбонил] амино-7,7- диметил-2-оксо-1H-азепин-1-уксусная кислота, этиловый сложный эфир
Раствор комнатной температуры продукта, полученного в части (e), (3,512 г, 13,7 мМ) в сухом тетрагидрофуране (110 мл) обрабатывали гексаметилдисилазаном лития (1,0 М в тетрагидрофуране, 21 мл, 21 мМ) а затем этилбромоацетатом (3,0 мл, 4,52 г 27,0 мМ). Через 20 мин добавляли еще 21 мл гексаметилдисилазана лития, а затем еще 1,5 мл этилбромоацетата. Полученный темно-оранжевый раствор размешивали еще 30 мин при комнатной температуре, после чего реакцию гасили путем добавления насыщенного хлорида аммония и воды. Смесь экстрагировали этилацетатом и органический экстракт промывали солевым раствором, осушали сульфатом натрия, фильтровали и упаривали, в результате чего получали темное маслообразное вещество. Этот остаток подвергали флеш-хроматографии (силикагель Merck, 35 - 50% этилацетат в гексане) и получали 1,363 г неочищенного целевого продукта в виде бледно-желтого маслообразного вещества. ТСХ (этилацетат:гексан, 1:1), Rf = 0,48.
g) [6(S)] -Гексагидро-6-[[2-ацетилтио)-1-оксо-3-фенилпропил] амино]-2,2-диметил-7-оксо-1H-азепин-1-уксусная кислота, этиловый сложный эфир.
Раствор продукта, полученного в части (f), (1,349 г, 3,94 мМ) в п-диоксане (3 мл) обрабатывали соляной кислотой (4,0 М в п-диоаксане, 12 мл). Смесь размешивали при комнатной температуре в течение 1,5 ч. Растворитель упаривали, а остаток распределяли между метиленхлоридом и 0,2 н. гидроксидом натрия (26 мл). Слои разделяли и водный слой экстрагировали этиловым эфиром/этилацетатом. Объединенные органические экстракты осушали сульфатом натрия, фильтровали и упаривали, в результате чего получали 982 мг неочищенного свободного амина в виде желтого маслообразного вещества. ТСХ (метанол:метиленхлорид, 1:9), Rf = 0,16.
Раствор (S)-2-(ацетилтио)бензолпропановой кислоты (полученной из дициклогексиламиновой соли как описано ранее (973 мг, 4,3 мМ)] и вышеуказанного сырого амина в метиленхлориде (30 мл) обрабатывали триэтиламином (1,15 мл, 835 мг, 8,2 мМ) и полученную смесь охлаждали до 0oC. Затем добавляли бензотриазол-1-илокси-трис(диметиламино)фосфония гексафторофосфат (1,915 г, 4,3 мМ) в виде твердого вещества. Раствор размешивали 1 час при 0oC, а затем 1,5 ч при комнатной температуре. Растворитель выпаривали, а остаток распределяли между этилацетатом и 0,5 н. соляной кислотой. Органический слой промывали последовательно водой, 50% насыщенным бикаробонатом натрия и солевым раствором, а затем осушали сульфатом натрия, фильтровали и выпаривали. Остаток подвергли флеш-хроматографии (силикагель Merck, ацетон:гексан, 3:7) и получали 1,564 г целевого продукта в виде маслообразного вещества (смесь диастереомеров, 1:1). ТСХ (ацетон:гексан, 3:7), Rf = 0,30.
h) [6(S)] -Гексагидро-6-[(2-меркапто-1-оксо-3-фенилпропил) амино]-2,2-диметил-7-оксо-1H-азепин-1-уксусная кислота
Раствор комнатной температуры продукта, полученного в части (g), (1,548 г, 3,45 мМ) в метаноле (20 мл, дезоксигенированном посредством барботирования аргона) обрабатывали 1 н. гидроксидом натрия (15 мл, дезоксигенированным посредством барботирования аргона). После 30-минутного размешивания добавляли еще 15 мл 1 н. гидроксида натрия и смесь продолжали размешивать в атмосфере аргона. Через 3 ч (в целом), раствор подкисляли 6 н. соляной кислотой и экстрагировали этилацетатом. Этилацетатный экстракт промывали водой и солевым раствором, осушали сульфатом натрия, фильтровали и упаривали, в результате чего получали маслообразное вещество. Это вещество подвергали флеш-хроматографии (силикагель Merck, этилацетат, а затем 1,5%-ная уксусная кислота в этилацетате). Фракции, содержащее, в основном, чистый целевой продукт, объединяли, упаривали и два раза подвергали азеотропной отгонке с использованием этилацетата. Смесь растворяли в небольшом количестве этилацетата и растирали с гексаном, в результате чего получали пенистое вещество. Летучие вещества выпаривали, а остаток суспендировали в гексане, выпаривали досуха и осушали в вакууме, в результате чего получали 863 мг целевого продукта в виде относительно твердой белой пены (смесь диастереомеров, 45:55). ТСХ (2%-ная уксусная кислота в этилацетате), Rf = 0,40.
Анализ для C19H26N2O4S • 0,2 H2O:
Вычислено: C 59,73; H 6,96; N 7,33; S 8,39;
Найдено: C 59,79; H 7,13; N 7,13; S 7,99.
Пример 43.
[S-(R*, S*)] -3,4-Дигидро-3-[(меркапто-1-оксо-3-фенилпропил)амино] -4- оксо-1,5-бензотиазепин-5(2H)-пропановая кислота
a) 2,3-Дигидро-3-амино-4-оксо-1,5-бензотиазепин-5(4H)-пропановая кислота, этиловый сложный эфир
2,3-Дигидро-3-[[(фенилметокси)карбонил] амино] -1,5-бензотиазепин -4(5H)-он, полученный в соответствии с известной процедурой (Slade и др. в J. Med. Chem. 28, с. 1517-1521 (1985)) (2,04 г, 6,21 мМ) подвергали азотропной обработке смесью метиленхлорида и толуола (3x) и осушали в вакууме в течение 1 ч. Суспензию этого соединения в смеси тетрагидрофурана и трет-бутанола (2: 1, 21 мл), охлажденной до 0oC, обрабатывали этилакрилатом (1,08 мл, 9,94 мМ, 1,6 экв.), а затем 1,0 н. калиевой солью т-бутанолом/тетрагидрофураном (621 мкл, 0,1 экв.). Реакционную смесь размешивали 15 мин при 0oC, а затем 20 ч при комнатной температуре в атмосфере аргона. После этого реакцию гасили 50% хлоридом аммония (50 мл) и экстрагировали этилацетатом (2•150 мл). Объединенные органические экстракты промывали насыщенным хлоридом аммония (75 мл), водой (75 мл) и солевым раствором (100 мл), осушали сульфатом магния, фильтровали и концентрировали, в результате чего получали прозрачный желтый сироп. Остаток очищали с помощью хроматографии на колонке (5х-20 см) с силикагелем, элюируя смесью 20% этилацетата и гексана (2л). Нужные фракции объединяли и концентрировали, в результате чего получали 2,33 г смеси сложного этилового эфира 2,3-дигидро-3-[(3-этокси-3- оксопропил)-[(фенилметокси)карбонил] амино] -4-оксо-1,5-бензотиазепин- 5(4H)-пропановой кислоты (25%) и сложного этилового эфира 2,3-дигидро-3-[[(фенилметокси)карбонил] амино] -4-оксо-1,5- бензотиазепин-5(4H)пропановой кислоты (75%) в виде прозрачного сиропа.
Полученную выше смесь (698 мг, 1,32 мМ) первого соединения и 1,698 г, 3,96 мМ второго соединения) и раствора 30% бромоводорода в уксусной кислоте (8,71 мл, 41,98 мМ) размешивали при комнатной температуре и в атмосфере аргона в течение 1,5 ч. Реакционную смесь разбавляли этиловым эфиром (200 мл), размешивали до тех пор, пока не образуются беловатые осадки, после чего смесь фильтровал и полученное оранжевое твердое вещество промывали этиловым эфиром (4•50 мл). Сырой остаток растворяли в 1 н. соляной кислоте (100 мл) и экстрагировали этилацетатом (2•100 мл). Затем водную фазу доводили до pH 9 с помощью 1 н. гидроксида натрия и экстрагировали этилацетатом (3•100 мл) Объединенные органические экстракты промывали солевым раствором (75 мл), осушали сульфатом магния, фильтровали и концентрировали, в результате чего получали 1,50 г неочищенного желтого маслообразного вещества. Остаток очищали с помощью хроматографии на колонке (5•20 см) с силикагелем, элюируя 1% (2 л), 2% (1 л), и наконец 5% (1 л) метанолом в метиленхлориде. Нужные фракции концентрировали и осушали в вакууме, в результате чего получали 1,163 г целевого продукта. ТСХ (метиленхлорид: метанол, 9:1), Rf = 0,40.
b) [S-(R*, S*)]-3,4-Дигидро-3-[[2-(ацетилтио)-1-оксо-3-фенилпропил] амино]-4-оксо-1,5-бензотиазепин-5(2H)-пропановая кислота, этиловый сложный эфир
(S)-2-(ацетилтио)бензолпропановую кислоту (полученную из дициклогексиламиновой соли как описано выше) растворяли в сухом метиленхлориде, охлаждали до 0oC и обрабатывали раствором продукта, полученного в части (a), в сухом метиленхлориде, а затем триэтиламином и бензотриазол-1-илокси(диметиламино)фосфония гексафторофосфатом в соответствии с процедурой, описанной в примере 36, часть (f), в результате чего получали целевой продукт. ТСХ (этилацетат: гексан, 1:1), Rf = 0,39.
c) [S-(R*, R*)]-3,4-Дигидро-3-[(2-меркапто-1-оксо-3-фенилпропил)амино] -1,5-бензотиазепин-5(2H)-пропановая кислота
Раствор продукта, полученного в части (b), в метаноле обрабатывали 2 н. гидроксидом натрия в соответствии с процедурой, описанной в примере 36, часть (g), в результате чего получали целевой продукт в виде белого пенистого вещества; [α]D = -161,6o (c = 1,16, метанол). ТСХ (1% уксусная кислота в этилацетате), Rf = 0,45.
Анализ для C21H22N2O4S • 0,4 H2O:
Вычислено: C 57,63; H 5,25; N 6,40; S 14,65;
Найдено: C 57,65; H 5,04; N 6,38; S 14,44.
Пример 44.
[3(S)] -2,3,4,5-Тетрагидро-3-[(2-меркапто-1-оксо-3-фенилпропил)- амино] -2-оксо-1H-1-бензазепин-1-пропановая кислота
a) 3-Амино-2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-пропановая кислота, этиловый сложный эфир
Раствор 3-азидо-1,3,4,5-тетрагидро-2H-1-бензазепин-2-он'а, полученного в соответствии с описанием Watthey и др., в J. Med. Chem 28, с. 1511-1516 (1985), в тетрагидрофуране/т-бутаноле охлаждали до 0oC и обрабатывали этилацетатом и 1 н. калиевой солью т-бутанола/тетрагидрофураном в соответствии с процедурой, описанной в примере 40, часть (a), в результате чего получали этиловый сложный эфир 3-азидо-2,3,4,5-тетрагидро-2-оксо-1H-1-бензазепин-1-пропановой кислоты в виде сиропа. ТСХ (этиловый эфир: гексан, 1:1), Rf = 0,45.
Раствор этого соединения (2,258 г, 6,94 мМ) в абсолютном этаноле обрабатывали 10% палладированным углем, а затем гидрировали 4 ч при давлении 40 фунт/кв. дюйм (275,76 кПа), причем после первого часа гидрирования Парра вентилировали. После этого смесь разбавляли этанолом (50 мл) и фильтровали через прокладку этанолом (2•50 мл). Прозрачный фильтрат выпаривали досуха и осушали в вакууме, в результате чего получали 2,098 г целевого продукта в виде прозрачного слегка желтоватого сиропа. ТСХ (метиленхлорид: метанол, 9: 1), Rf = 0,32.
b) [3(S)] -2,3,4,5-Тетрагидро-3-[[2-(ацетилтио)-1-оксо-3- фенилпропил] амино]-2-оксо-1H-1-бензазепин-1-пропановая кислота
(S)-2-(Ацетилтио)бензолпропановую кислоту, полученную из дициклогексиламиновой соли как описано ранее, растворяли в метиленхлориде, охлаждали до 0oC, обрабатывали последовательно раствором продукта части (a) в сухом метиленхлориде, триэтиламином и гексафторфосфатом бензотриазол-1-илокситрис диметиламино фосфония в соответствии с процедурой примера 36, часть (f), в результате чего получали целевой продукт в виде сиропа. ТСХ (этилацетат: гексан, 1:1), R1 = 0,56 и 0,52.
c) [3-(S)] -2,3,4,5-Тетрагидро-3-[(2-меркапто-1-оксо-3-фенилпропил] -амино] -2-оксо-1H-1-бензазепин-1-пропановая кислота
Раствор продукта, полученного в части (b), в метаноле охлаждали до 0oC и обрабатывали 1 н. гидроксидом натрия в соответствии с процедурой, описанной в примере 36, часть (g), в результате чего получали целевой продукт в виде твердого пенистого вещества; [α]D = +41,8o (с = 0,608, метанол). ТСХ (метиленхлорид: метанол: уксусная кислота, 20:1:1), Rf = 0,47.
Анализ для C22H24N2O4S • 0,25 C5H12 • 0,413 H2O:
Вычислено: C 63,76; H 6,21; N 6,40; S 7,32;
Найдено: C 63,76; H 6,18; N 6,38; S 7,45.
Пример 45.
[3(S)-2,3,4,5-Тетрагидро-3-[[2-(меркаптометил)-1-оксо-3- фенил-пропил] амино]-2-оксо-1H-1-бензазепин-1-пропановая кислота
a)[3(S)] -2,3,4,5-Тетрагидро-3-[[2-[(ацетилтио)метил] -1- оксо-3-фенилпропил]амино]-2-оксо-1H-1-бензазепин-1-пропановая кислота
(S)-2-[(Ацетилтио)метил] бензопропановую кислоту, полученную из эфедриновой соли в соответствии с описанием, приведенным выше, растворяли в сухом метиленхлориде, охлаждали до 0oC и обрабатывали 1-гидроксибензолгидратом и 1-этил-3-(3-диметиламинопропил)карбодиимидом и раствором продукта примера 44 (a) в сухом метиленхлориде в соответствии с процедурой, описанной в примере 8, часть (a), в результате чего получали целевой продукт в виде сиропа. ТСХ (этилацетат: гексан, 1:1), Rf = 0,58.
b) [3(S)] -2,3,4,5-Тетрагидро-3-[[2-(меркаптометил)-1-оксо- 3-фенилпропил]амино]-2-оксо-1H-1-бензазепин-1-пропановая кислота
Раствор продукта, полученного в части (a), в метаноле охлаждали до 0oC и обрабатывали 1 н. гидроксидом натрия в соответствии с процедурой, описанной в примере 8, части (b), в результате чего получали целевой продукт в виде аморфного твердого вещества, [α]D = +46,9o (c = 0,608, метанол). ТСХ (хлороформ: метанол: уксусная кислота, 20:1:1), Rf = 0,42.
Анализ для C23H26N2O4S • 0,233 C5H12 • 0,61 H2O:
Вычислено: C 63,89; H 6,65; N 6,17; S 7,06;
Найдено: C 63,89; H 6,40; N 6,18; S 6,82.
Пример 46.
[1S-[ 1α,9α (R*)]]-Октагидро-9-[2-меркапто-1-оксо-3-фенил- пропил)амино] -6,10-диоксо-6H-пиридазино [1,2-a][1,2] диазепин-1- карбоновая кислота
a)(1S-цис)-Октагидро-9-фталимидо-6,10-диоксо-6H-пиридазино-[1,2-a] [1,2] диазепин-1-карбоновая кислота, 1,1-диметилэтиловый эфир
5,6-Дигидро-1,3-(2H, 4H)-пиридазинкарбоновой кислоты 3-(1,1-диметилэтил)-1-(фенилметил)эфир, полученный известным способом, (Adams и др. , Synthetic c Communications, 18(18), 2225-2231 (1988)), подвергали реакции с 5-(фенилметил) эфиром (S)-2-фталимидопентандионовой кислоты в соответствии с известной процедурой (Attwood и др. (J. Chem. Soc. Perkins Trans I, 1986, с. 1011-1019), в результате чего получали (S)-2-[2-фталимидо-1,5-диоксо-5-(фенилметокси)пентил]-5,6-дигидро-1,3- (2H,4H)-пиридазиндикорбоновой кислоты 3-(1,1-диметилэтил)-1-(фенилметио)эфир.
Раствор полученного материала (12,11 г, 18,5 мМ) в сухом диметилформамиде (190 мл) обрабатывали 10% палладированным углем (372 мг) и гидрировали при комнатной температуре в течение 24 ч. Затем смесь фильтровали через прокладку из целита и полученный прозрачный темный фильтрат упаривали досуха, а образовавшийся сироп снова растворяли в метаноле (200 мл). Раствор фильтровали через другую прокладку из целита для удаления оставшихся следовых количеств катализатора и полученный прозрачный фильтрат выпаривали досуха и осушали в вакууме. Полученный продукт растворяли в сухом метиленхлориде (185 мл), охлаждали до 0oC в ледяной-солевой бане и обрабатывали тионилхлоридом (1,56 мл, 21,35 мМ, 1,15 экв.). Реакционную смесь размешивали при комнатной температуре и в атмосфере аргона в течение получаса, обрабатывали раствором бикарбоната калия (3,95 г) в воде (34 мл) и размешивали в течение 10 мин. Фазы разделяли, водную фазу снова экстрагировали еще 270 мл метиленхлорида и объединенные органические экстракты осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме.
Неочищенную продуктовую смесь хроматографировали на колонке с силикагелем (Merck), элюируя смесями этилацетата и гексана (1:3, 1:1), в результате чего получали 3,053 г целевого соединения в виде твердого вещества. (ТСХ (этилацетат:гексан, 1:1), Rf = 0,40.
b) (1S-цис)-Октагидро-9-амино-6,10-диоксо-6H-пиридазино[1,2-a] (1,2] диазепин-1-карбоновая кислота, 1,2-диметилэтиловый эфир
Суспензию продукта, полученного в части (a), (1,0 г, 2,41 мМ) в абсолютном этаноле обрабатывали гидратом гидразина (0,26 мл, 2,2 экв.) и размешивали 1 ч при комнатной температуре в атмосфере аргона. Затем смесь упаривали досуха, выпаривали из толуола (25 мл), а полученный остаток размешивали с 2,0 М уксусной кислоты (10 мл) в атмосфере аргона в течение 24 ч. Твердые вещества отфильтровывали, промывали 2,0 М уксусной кислоты (5,0 мл) и водой (2•2,0 мл) и прозрачный фильтрат доводили до pH 10,0 с использованием твердого бикарбоната натрия (1,8 г). Полученную смесь экстрагировали метиленхлоридом (2•25 мл) и объединенные органические экстракты промывали солевым раствором (6,0 мл), осушали безводным сульфатом натрия и фильтровали. Прозрачный раствор выпаривали досуха и осушали в вакууме, в результате чего получали 755 мг целевого продукта в виде сиропа. ТСХ (метиленхлорид: метанол, 9:1), Rf = 0,20.
c) [1S-[1 1α,9α (R*)]]-Октагидро-9-[[2-(ацетилтио)-1-оксо- 3-фенилпропил]амино]-6,10-диоксо-6Н-пиридазино[1,2-а][1,2]диазепин-1- карбоновая кислота, 1,1-диметилэтиловый эфир
Дициклогексиламиновую соль (S)-2-(ацетилтио)бензолпропановой кислоты (1,07 г, 2,64 мМ) суспендировали в этилацетате (80 мл), промывали 5% бисульфатом калия (5 • 12 мл) и солевым раствором (14 мл), осушали безводным сульфатом магния, фильтровали, выпаривали досух и осушали в вакууме.
Свободную кислоту растворяли в сухом метиленхлориде (20 мл), охлаждали до 0oC (в ледяной/солевой бане), обрабатывали последовательно раствором продукта части (b) (755 мг, 2,41 мМ) в сухом метиленхлориде (5,0 мл), триэтиламином (0,33 мл, 2,37 мМ) и гексафторофосфатом бензотриазол-1-илокситри(диметиламино) фосфония (1,08 г, 2,44 мМ). Реакционную смесь размешивали 1 ч при 0oC и 2 ч при комнатной температуре в атмосфере аргона. После этого смесь упаривали досуха, а полученный сироп снова растворяли в этилацетате (80 мл), промывали 0,5 н. соляной кислотой (2 • 14 мл), водой (14 мл) и солевым раствором (14 мл), затем осушали безводным сульфатом магния, фильтровали и выпаривали досуха. Сырой продукт хроматографировали на колонке с силикагелем (Merck), элюируя смесями этилацетата и гексана (1:3, 1:1), в результате чего получали 747 мг целевого продукта в виде прозрачного сиропа. ТСХ (этилацетат : гексан, 1:1), Rf = 0,23.
d) [1S-[ 1α,9α (R*)]]-Октагидро-9-[[2-(ацетилтио)-1-оксо-3- фенилпропил] амино]-6,10-диоксо-6Н-пиридазино[1,2-а][1,2]диазепин-1- карбоновая кислота
Раствор продукта, полученного в части (c), (747 мг, 1,48 мМ) в сухом метиленхлориде (10 мл) обрабатывали анизолом (0,69 мл, 6,35 мМ), а затем трифторуксусной кислотой (1,04 мл, 13,5 мМ) и полученную реакционную смесь размешивали при комнатной температуре и в атмосфере аргона в течение 5,5 дней. Прозрачный раствор упаривали досуха, выпаривали из этилацетата (2 • 100 мл) и сырой продукт хроматографировали на колонке с силикагелем Merck, элюируя смесью этилацетата и гексана (4: 1) а затем смесью этилацетата, гексана и уксусной кислоты (85:10:5). Нужные фракции объединяли, упаривали досуха и выпаривали из толуола (2 • 100 мл). Полученный сироп снова растворяли в этилацетат (59 мл), промывали водой (3 • 10 мл) и солевым раствором (5,0 мл), а затем осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме, в результате чего получали 556 мг целевого продукта в виде сиропа. ТСХ (этилацетат : уксусная кислота, 95:5), Rf = 0,30.
e) [1S-[ 1α,9α (R*)] ] -Октагидро-9-[(2-меркапто-1-оксо-3-фенилпропил)амино]-6,10- диоксо-6Н-пиридазоно[1,2-а][1,2] диазепин-1-карбоновая кислота
Раствор продукта, полученного в части (d), (556 мг, 1,24 мМ) в метаноле (7,0 мл) охлаждали до 0oC (в ледяной/солевой бане), продували 30 мин аргоном и обрабатывали предварительно продутым (аргон, 30 мин) раствором 1,0 н. гидроксида натрия (2,5 мл, 2,5 мМ), продолжая барботировать аргон во время добавления и в течение всей реакции. Затем реакционную смесь размешивали при 0oC в течение 30 мин, доводили до pH 2,0 с помощью 5% бисульфата калия (11,0 мл) при 0oC, нагревали до комнатной температуры и экстрагировали этилацетатом (2 • 40 мл). Объединенные органические экстракты промывали солевым раствором (11,0 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Сырой продукт растворяли в метиленхлориде (5,0 мл) и порциями обрабатывали гексаном (50 мл) до тех пор, пока не получали твердый материал. Этот материал растирали с еще 50 мл гексана и 2 • 50 мл пентана, причем указанный твердый материал 2 ч размешивали с первым 50 мл и в течение ночи - со следующими 50 мл при комнатной температуре и в атмосфере аргона. Полученный продукт осушали в вакууме и получали 468,5 мг целевого продукта в виде твердого пенистого вещества; [α]D = -83,5o (c = 0,492, метанол). ТСХ (этилацетат : уксусная кислота, 95:5), Rf = 0,33.
Анализ для C19H23N3O5S • 0,34 H2O:
Вычислено: C 55,45; H 5,80; N 10,21; S 7,79;
Найдено: C 55,45; H 5,95; N 9,84; S 7,49.
Пример 47.
[1S-[ 1α,9α (R*)]]-Октагидро-9-[(2-меркапто-1-оксо-3- фенилпропил)амино] -10-оксо-6Н-пиридазино[1,2-а][1,2]диазепин-1-карбоновая кислота
a) (1S-цис)-Октагидро-9-фталимидо-10-оксо-6Н-пиридазино[1,2-а] [1,2] диазепин-1-карбоновая кислота, 1,1-диметилэтиловый эфир
Раствор продукта примера 46 а (1,0 г, 2,35 мМ) в сухом тетрагидрофуране (8,0 мл) охлаждали до около 5oC (в ледяной/солевой бане), обрабатывали в течение 30 мин 1,0 М диборана/тетрагидрофурана (2,88 мл, 2,88 мМ), нагревали до комнатной температуры и размешивали в течение ночи в атмосфере аргона. Реакционную смесь разводили метиленхлоридом (14,0 мл), обрабатывали 2,0 н. соляной кислотой (6,8 мл) и размешивали 15 минут. Затем pH смеси доводили до значения 10,0 с помощью безводного бикарбоната натрия (1,92 г), фазы разделяли, водную фазу снова экстрагировали еще 30 мл метиленхлоррида. Объединенные органические экстракты промывали солевым раствором (5,0 мл), осушали безводным сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт хроматографировали на колонке с силикагелем (Merck), элюируя смесями этилацетата и гексана (1:3, 1:1), и получали 877 мг целевого продукта в виде сиропа. ТСХ (этилацетат : гексан, 1:1), Rf = 0,63.
b) (1S-цис)-Октагидро-9-амино-10-6Н-пиридазино[1,2-а][1,2] диазепин-1-карбоновая кислота, 1,1-диметилэтиловый эфир
Раствор продукта, полученного в части (a), (1,257 г, 3,0 мМ) в абсолютном этаноле (13,0 мл) обрабатывали гидратом гидразина (0,33 мл, 2,2 экв.) в соответствии с процедурой, описанной в примере 46, часть (b), в результате чего получали 704 мг целевого продукта в виде сиропа. ТСХ (метилен : метанол, 9:1), Rf = 0,35.
c) [1S-[ 1α,9α (R*)]]-Октагидро-9-[[2-(ацетилтио)-1-оксо-3- фенилпропил] амино] -10-оксо-6Н-пиридазино[1,2-а] [1,2] диазепин-1- карбоновая кислоты, 1,1-диметилэтиловый эфир
Дициклогексиламиновую соль (S)-2-(ацетилтио)бензолпропановой кислоты (1,10 г, 2,71 мМ) суспендировали в этилацетате (85 мл), промывали 5% бисульфатом калия (5 • 12,4 мл) и солевым раствором (14,4 мл), осушали безводным сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме.
Свободную кислоту растворяли в сухом метиленхлориде (21 мл), охлаждали до 0oC (в ледяной/солевой бане), обрабатывали последовательно раствором продукта, полученного в части (b), (704 мг, 2,48 мМ) в сухом метиленхлориде (5,0 мл), триэтиламином (0,34 мл, 2,44 мМ) и гексафторофосфатом бензотриазол-1-илокситрис диметиламино фосфония (1,113 г, 2,55 мМ). Реакционную смесь 1 ч размешивали при 0oC и 2,5 ч при комнатной температуре в атмосфере аргона. Затем смесь упаривали досуха и полученный сироп снова растворяли в этилацетате (80 мл), промывали 0,5 н. соляной кислотой (2 • 14,0 мл), водой (14,0 мл) и солевым раствором (14,0 мл), осушали безводным сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт хроматографировали на колонке с силикагелем (Merck), элюируя смесью этилацетата : гексана (1: 3) и получали 1,085 г целевого продукта в виде сиропа. ТСХ (этилацетат : гексан, 1:1), Rf = 0,53.
d) [1S-[[ 1α,9α (R*)] ] -Октагидро-9-[(2-меркапто-1-оксо-3- фенилпропил)амино]-10-оксо-6Н-пиридазино[1,2-а][1,2]диазепин-1- карбоновая кислота
Раствор продукта, полученного в части (c), (1,085 г, 2,216 мМ) в сухом метиленхлориде (15 мл) обрабатывали анизолом (1,03 мл, 9,5 мМ), а затем трифторуксусной кислотой (1,56 мл, 20,2 мМ) и полученную реакционную смесь размешивали 4 дня при комнатной температуре в атмосфере аргона. Прозрачный раствор упаривали досуха, выпаривали из этилацетата (2 • 150 мл) и неочищенный продукт суспендировали в смеси этилацетата (10 мл) и гексана (50 мл). Твердые вещества отфильтровывали, тщательно промывали гексаном (50 мл), выпаривали из толуола (2 • 100 мл) и осушали в вакууме, в результате чего получали 865 мг [1S-[ 1α,9α (R*)]]-Октагидро-9-[[2-(ацетилтио)-1-оксо-3-фенилпропил] амино] - 10-оксо-6Н-пиридазино[1,2-а][1,2]диазепин-1-карбоновой кислоты в виде твердого вещества.
Полученный материал суспендировали в метаноле (15 мл), охлаждали до 0oC (в ледяной/солевой бане), продували в течение 30 мин аргоном, а затем по капле обрабатывали предварительно продутым раствором 1,0 н. гидроксида натрия (6,03 мл, 3 экв.), продолжая при этом барботирование аргона во время добавления и в процессе всей реакции. Реакционную смесь размешивали 30 мин при 0oC твердые вещества уходят в раствор в первые 5 мин доводили до pH 2 с помощью 5% бисульфата калия (26,6 мл), нагревали до комнатной температуры и экстрагировали этилацетатом (2 • 65 мл). Объединенные органические экстракты промывали солевым раствором (18 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Продукт растворяли в метилехлориде (7,0 мл) частями обрабатывали гексаном (70 мл) до тех пор, пока не получали твердое вещество. Это твердое вещество растирали с гексаном (100 мл) и пентаном (2 • 100 мл), размешивая 2 ч с первыми 100 мл и в течение ночи со следующими 100 мл при комнатной температуре в атмосфере аргона. Продукт осушали в вакууме и получали 790,6 мг целевого соединения в виде твердого пенистого вещества; [α]D = -44,4o (c = 0,63, метанол). ТСХ (этилацетат : уксусная кислота, 95:5), Rf = 0,67.
Анализ для C19H25N3O4S • 0,20 C5H12 • 0,80 H2O:
Вычислено: C 57,11; H 6,95; N 9,99; S 7,62;
Найдено: C 57,11; H 6,72; N 9,83; S 7,44.
Пример 48.
[S-(R*,R*)]-Гексагидро-6-[(2-меркапто-1-оксо-3- фенилпропил)амино]-2-метил-3,7-диоксо-1Н-1,2-диазепин-1-уксусная кислота
a) [2-[(1,1-Диметилэтокси карбонил]гидразино]уксусная кислота, этиловый сложный эфир
Триэтиламин (13,94 мл, 0,1 М) добавляли к раствору 1,1-диметилэтилового эфира гидразинкарбоновой кислоты (13,216 г, 0,1 М) в бензоле (100 мл), а затем добавляли этилбромоацетат (11,09 мл, 0,1 М). Реакционную смесь нагревали с обратным холодильником (масляная баня, 95 - 100oC) в течение 14 ч, после чего добавляли триэтиламин (1,4 мл, 0,01 М), а затем бромоацетат (1,1 мл, 0,01 М). После нагревания с обратным холодильником еще 8 часов, добавляли триэтиламин (2 мл, 0,015 М) и этилбромоацетат (1,4 мл, 0,013 М). Смесь нагревали с обратным холодильником еще 14 ч (всего 36 ч). После охлаждения до комнатной температуры, реакционную смесь фильтровали и полученную гидрохлоридную соль триэтиламина промывали смесью этилацетата/гексана (1:1). Объединенный фильтрат разбавляли этилацетатом, промывали насыщенным бикарбонатом натрия, водой, солевым раствором, осушали сульфатом натрия и фильтровали. Фильтрат концентрировали в вакууме и получали 19,38 г неочищенного продукта в виде желтого сиропа, который использовали в следующей реакции без очистки.
b) [2-[(1,1-Диметилэтокси)карбонил]-1-[(фенилметокси)карбонил] гидразино]уксусная кислота, сложный этиловый эфир
Триэтиламин (12,4 мл, 89 мМ) добавляли к раствору неочищенного продукта, полученного в части (a), (19,38 г) в бензоле (120 мл), а затем по капле добавляли [(фенилметокси)карбонил]-хлорид (13,4 мл, 89 мМ) в бензоле (30 мл) в течение 30 мин. Полученную суспензию размешивали 20 ч при комнатной температуре в атмосфере аргона, а затем распределяли между этилацетатом (500 мл) и 1М раствором бисульфата калия. Органическую фазу отделяли, промывали насыщенным бикарбонатом натрия, водой, солевым раствором, осушали сульфатом натрия и фильтровали. Фильтрат концентрировали в вакууме и остаток кристаллизовали из этилацетата/гексана, в результате чего (после вакуумной осушки над пентоксидом фосфора) получали 15,736 г целевого продукта в виде белого кристаллического соединения с т.пл. 117 - 119oC.
c) [2-[[(1,1-Диметилэтокси)карбонил]-1-[(карбонил)-1-фенилметокси карбонил]-2-метил]гидразино]уксусная кислота, этиловый эфир
Карбонат калия (измельченный в порошок и осушенный, 11,8 г, 85,15 мМ) добавляли к раствору продукта, полученного в части b, (6,0 г, 17,03 мМ) в безводном диметилформамиде (30 мл), а затем добавляли метилиодид (5,3 мл, 85,15 мл). После 17-часового размешивания при 40oC, добавляли еще 64 мМ метилиодида и реакционную смесь размешивали при 40oC еще 28 ч. Затем суспензию фильтровали и к фильтрату добавляли 6,0 г свежего карбоната калия (измельченного в порошок и осушенного), а затем 5,0 мл метилиодида. Суспензию размешивали при 40oC в течение 21 ч, после чего добавляли 1,2 мл метилиодида и реакционную смесь размешивали еще 17 ч до тех пор, пока исходный материал не будет обнаруживаться ТСХ. Затем реакционную смесь фильтровали и собранное твердое вещество промывали этилацетатом. Фильтрат концентрировали в вакууме для удаления наибольшего количества диметилформамида, а затем разбавляли 500 мл этилацетата, промывали водой, солевым раствором и осушали сульфатом магния. Фильтрат концентрировали и остаток осушали в вакууме, в результате чего получали 6,285 г неочищенного продукта в виде бесцветного маслообразного вещества.
d) [2-[[(1,1-Диметилэтокси)карбонил]-2-метил]гидразино]уксусная кислота, этиловый эфир
Суспензию продукта, полученного в части (c), (6,285 г, 17,03 мМ) и гидроксида палладия (800 мг) в этаноле (75 мл) энергично размешивали 4 ч в присутствии водорода (баллонного). Затем суспензию фильтровали с использованием миллипористого фильтра. Фильтрат концентрировали, а остаток осушали в вакууме, в результате чего получали 4,3 г целевого продукта в виде слегка желтоватого маслообразного вещества.
e) (S)-2-Фталимидо-1,5-пентадионовая кислота, 5-(фенилметил)эфир)
N-(Карбетокси)фталимид (9,7 г, 44,24 мМ, 1,05 экв) добавляли к суспензии L-глутаминовой кислоты 5-(фенилметил) эфира (10,0 г, 42,14 мМ) и бикарбоната натрия (4,47 г, 42,14 мМ) в воде (80 мл) и диоксану (40 мл). После 2- часового размешивания добавляли еще 4,4 г (0,1 экв.) N-карбетокси фталимида. Полученную смесь размешивали еще 2 ч. После этого pH реакционной смеси доводили до 1-2 с помощью твердого бисульфата калия и полученную смесь экстрагировали этилацетатом (3•150 мл). Объединенные этилацетатные экстракты промывали 1М бисульфатом калия, солевым раствором, осушали сульфатом натрия и фильтровали. Фильтрат концентрировали в вакууме. Маслообразный остаток разводили этилацетатом и обрабатывали дициклогексиламином (8,40 мл, 42,14 мМ). После кристаллизации неочищенной соли из этилацетата/гексана получили (после вакуумной осушки) 13 г целевого продукта в виде дициклогексиламиновой соли. Маточный раствор промывали 1 М дисульфата калия (2х), водой, солевым раствором и осушали сульфатом натрия. Фильтрат концентрировали в вакууме. Маслянистый остаток хроматографировали с использованием 0,5% уксусной кислоты в смеси гептана и этилацетата (3:7) в качестве подвижной фазы. Нужные фракции собирали и концентрировлаи. Маслянистый остаток осушали в вакууме и получали 9,644 г целевого продукта.
f) (S)-4-Фталимидо-5-оксо-5-[2-[(1,1-диметилэтокси)карбонил] -1-(2-этокси-2- оксоэтил)-2-метилгидразино] пентановая кислота, фенилметиловый сложный эфир
К раствору продукта, полученного в части (e), (около 17,10 мМ) (охлажденного до 0oC) в простом эфире (80 мл) добавляли пентахлорид фосфора (5,10 г, 24,5 мМ). После 30-минутного размешивания при 0oC и 30-минутного размешивания при комнатной температуре, реакционную смесь концентрировали в вакууме. Остаток упаривали с толуолом (3х) и осушали в вакууме в течение 2 ч, в результате чего получали маслянистое соединение, которое использовали непосредственно в следующей реакции.
Раствор бикарбоната натрия (4 г, в 46 мл воды) добавляли к холодному (0oC) раствору продукта, полученного в части (d), в толуоле (40 мл). Размешивая, полученное выше соединение в толуоле (30 мл) по капле добавляли через канюлю. После добавления, реакционную смесь размешивали при комнатной температуре и в атмосфере аргона в течение 19 ч. Затем реакционную смесь разбавляли этилацетатом (300 мл), промывали водой, 5% бисульфатом калия, водой, солевым раствором, а затем осушали сульфатом натрия. Фильтрат концентрировали, а остаток осушали в вакууме, в результате чего получали целевое соединение в виде маслообразного вещества.
g) (S)-4-Фталимидо-5-оксо-5-[1-(2-этокси-2-оксоэтил)-2-метилгидразино] пентановая кислота, фенилметиловый эфир
К охлажденному (-10oC) раствору неочищенного продукта, полученного в части (f), в метиленхлориде (55 мл) добавляли по капле анизол (7,4 мл) а затем тетрафторуксусную кислоту (13,10 мл, 170 мМ). После завершения добавления, реакционную смесь размешивали при -10oC в течение 1 ч. Затем реакционную смесь нагревали до комнатной температуры и продолжали размешивать в течение 14 ч. После удаления летучих веществ в вакууме, остаток разводили 200 мл этилацетата, промывали насыщенным бикарбонатом натрия осторожно, затем водой, солевым раствором и осушали сульфатом натрия. Фильтрат концентрировали в вакууме, а остаток хроматографировали, используя в качестве подвижной фазы 20-40% этилацетат/гексан. Нужные фракции собирали и концентрировали, а остаток осушали в вакууме, в результате чего получали 7,254 г целевого продукта в виде бледно-желтого маслообразного вещества.
h) (S)-4-Фталимидо-5-оксо-5-[-(2-этокси-3-оксоэтил)-2-метилгидразино] пентановая кислота
Гидроксид палладия (1,0 г) добавляли к раствору продукта, полученного в части (g), (6,436 г, 13,38 мМ) в теплом этаноле (100 мл). Суспензию энергично размешивали в присутствии водорода (баллонного) в течение 4 ч. Затем суспензию фильтровали, а катализатор промывали этанолом. Объединенный фильтрат концентрировали. Остаток упаривали с толуолом (2х) и осушали в вакууме в течение ночи, в результате чего получали 5,86 г продукта в виде бесцветного маслообразного вещества, которое использовали в следующей реакции без очистки.
i) (S)-Гексагидро-2-метил-3,7-диоксо-6-фталимидо-1H-1,2-диазацин-1- уксусная кислота, этиловый сложный эфир.
Тионилхлорид (1,1 мл) добавляли по капле к охлажденному раствору (0oC) продукта, полученного в части (h), (5,86 г, 13,38 мМ) в метиленхлориде (120 мл). Реакционную смесь размешивали 15 мин при 0oC, а затем 3 ч при комнатной температуре. Затем добавляли еще 0,3 мл тионилхлорида (всего 1,4 мл) и реакционную смесь размешивали еще 3 ч, после чего реакцию гасили раствором бикарбонта калия (2,7 г бикарбоната калия в 27 мл в воде). Выделенную органическую фазу промывали насыщенным бикарбонатом натрия. Водную фазу экстрагировали метиленхлоридом (2х). Объединенную метиленхлоридную фазу промывали водой, солевым раствором и осушали сульфатом магния. Фильтрат концентрировали, а остаток осушали в вакууме, в результате чего получали 4,70 г нужного продукта.
j) (S)-Гексагидро-2-метил-3,7-диоксо-6-амино-1H-1,2-диазецин-1-уксусная кислота, этиловый сложный эфир
Моногидрат гидразина (255 мкл, 5,25 мМ) добавляли к суспензии продукта, полученного в части (i), (1,867 г, 5,0 мМ) в этаноле (30 мл) и метиленхлориде (10 мл). После размешивания смеси в течение 2 ч, летучие вещества удаляли в вакууме, а остаток упаривали с толуолом (2х), а затем осушали в вакууме. Остаток разводили 2М водной уксусной кислотой (20 мл), а полученный раствор размешивали 14 ч при комнатной температуре. Суспензию фильтровали. Фильтрат подщелачивали твердым бикарбонатом натрия до pH 10 и экстрагировали метиленхлоридом (3х). Объединенную метиленхлоридную фазу промывали 50% солевым раствором, осушали сульфатом натрия, фильтровали и выпаривали. Остаток хроматографировали с использованием в качестве подвижной фазы 2,5% метанол/метиленхлорид и получали 568 мг целевого соединения в виде бледно-желтого маслообразного продукта.
k) [S-(R*,R*)-Гексагидро-6-[[2-(ацетилтио)-1-оксо-3- фенилпропил]амино] -2-метил-3,7-диоксо-1H-1,2-диазепин-1-уксусная кислота, этиловый сложный эфир
Триэтиламин (302 мкл, 2,171 мМ) добавляли к охлажденному (0oC) раствору (S)-2-(ацетилтио)бензопропановой кислоты (полученной из дициклогексиламиновой соли, как описано ранее, 486 мг 2,17 мМ), а затем добавляли продукт, полученный в части (j), (480 мг, 1,973 мМ) в метиленхлориде (3 мл) и гексафторофосфат бензотриазол-1-илокситрис диметиламино фосфенил (960 мг, 2,171 мМ). Полученную смесь размешивали 1 ч при 0oC, а затем 2 ч при комнатной температуре. Летучие вещества удаляли в вакууме. Остаток растворяли в этилацетате (100 мл), промывали 5% бисульфатом калия, 50% насыщенным бикарбонатом натрия, солевым раствором, осушали сульфатом магния, фильтровали и выпаривали досуха. Сырой продукт подвергали флеш-хроматографии, используя в качестве подвижной фазы 50% - 70% этилацетат/гексан и получали 824 мг целевого продукта в виде белого пенящегося вещества.
l) [S-(R*, R*)]-Гексагидро-6-[(2-меркапто-1-оксо-3- фенилпропил)-амино] -2-метил-3,7-диоксо-1H-1,2-диазепин-1-уксусная кислота
Раствор продукта, полученного в части (k), (800 мг, 1,78 мМ) в метаноле (9 мл), и охлажденного до 0oC, продували 30 мин аргоном, а затем по капле обрабатывали предварительно продутым 1М раствором гидроксида натрия (7,12 мг, 4,0 экв). Реакционную смесь размешивали при 0oC, барботируя аргон в течение 1,5 ч, после чего смесь подкисляли 1М бисульфатом калия для доведения pH до 1-2 и экстрагировали этилацетатом (3х). Объединенные этилацетатные экстракты промывали солевым раствором (2х), осушали сульфатом магния, фильтровали и выпаривали досуха. Остаток подвергали флеш-хроматографии, используя в качестве подвижной фазы 2% уксусную кислоту/этилацет, и получали 668 мг целевого продукта в виде белого пенящегося вещества. При объединении продуктовых колоночных фракций использовали хлороформ, [α]D = -54,8o (c=0,8, метанол). ТСХ (3% уксусная кислота/этилацетат), Rf = 0,12.
Анализ для C17H21N3O5S • 0,57 CHCl3:
Вычислено: C 47,16; H 4,86; N 9,39; S 7,16;
Найдено: C 47,48; N 4,56; N 9,01; S 7,17.
Пример 49.
[S-(R*, R*)] -Гексагидро-6-[(2-меркапто-1-оксо-3-фенилпропил) амино]-2-метил-7-оксо-1H-1,2-диазепин-1-уксусная кислота
a) (S)-Гексагидро-2-метил-7-оксо-6-фталимидо-1H-1,2-диазепин-1- уксусная кислота, этиловый сложный эфир
Раствор продукта, полученного в примере 48, части (i), (375 мг, 1,0 мМ) в сухом тетрагидрофуране (2,4 мл) охлаждали до 0oC и обрабатывали до капли 1М-раствором диборана в тетрагидрофуране. После завершения добавления, реакционную смесь размешивали 30 мин при 0oC (за это время смесь становилась гелеобразной). Затем охлажденную смесь размешивали еще 1 ч при комнатной температуре, после чего смесь разводили 2 мл метиленхлорида и 2 мл 2 М водной соляной кислоты. После 15-минутного размешивания, смесь разбавляли 20 мл метиленхлорида и осторожно промывали насыщенным бикарбонатом натрия. Выделенную водную фазу экстрагировали метиленхлоридом (3х). Объединенный метиленхлоридный экстракт промывали 5-%-ным солевым раствором, солевым раствором и осушали сульфатом натрия. Фильтрат выпаривали в вакууме досуха и получали 344 мг сырого продукта в виде маслообразного вещества, которое использовали в следующей реакции без очистки.
b) (S)-Гексагидро-2-метил-7-оксо-6-амино-1H-1,2-диазепин-1-уксусная кислота, этиловый сложный эфир
Раствор продукта, полученного в части (a), (344 мг, 0,958 мМ) в этаноле (3 мл) обрабатывали моногидратом гидразина (54,8 мкл, 1,13 мМ) в соответствии с процедурой, описанной в примере 48, части (j), в результате чего получали 169 мг целевого соединения в виде бледно-желтого маслообразного продукта.
c) [S-(R*, R*)-Гексагидро-6-[(2-ацетилтио)-1-оксо-3-фениламино]-2-метил -7-оксо-1H-1,2-диазепин-1-уксусная кислота, этиловый сложный эфир
Холодный (0oC) раствор (S)-2-(ацетилтио)бензолпропановой кислоты (полученной из дициклогексиламиновой соли как описано ранее, 198 мг, 0,885 мМ) в метиленхлориде (1 мл) обрабатывали триэтиламино (123 мкл, 0,88 мМ), продуктом, полученным в части (b) (169 мг, 0,738 мМ) в метиленхлориде ( 2 мл) и гексафторофосфатом бензотриазол-1-илокситрис диметиламино фосфония (392 мг 0,885 мМ). Полученную смесь размешивали 1 ч при 0oC, а затем 2 ч при комнатной температуре. Летучие вещества удаляли в вакууме. Остаток растворяли в этилацетате (50 мл), промывали 5% бисульфатом калия, насыщенным бикарбонатом натрия, 50%-ным солевым раствором, солевым раствором и осушали сульфатом магния. Фильтрат выпаривали досуха и остаток подвергали флеш-хроматографии, используя 40 - 50% этилацетат/гексан в качестве подвижной фазы, и получали 260,5 мг целевого продукта в виде маслообразного бесцветного вещества.
d) [S-(R*, R*)]-Гексагидро-6-[(2-меркапто-1-оксо-3-фенилпропил)-амино] -2-метил-7-оксо-1H-1,2-диазепин-1-уксусная кислота
Раствор продукта, полученного в части (c), (235 мг, 0,54 мМ) в метаноле (3 мл) и охлажденного до 0oC, продували 30 мин аргоном, а затем по капле обрабатывали предварительно продутым 1М раствором гидроксида натрия (2,20 мл, 4,0 экв). Реакционную смесь размешивали при 0oC, барботируя при этом аргон, в течение 2 ч, а затем подкисляли 1М бисульфата калия до pH 1 - 2 и экстрагировали этилацетатом (3х). Объединенные этилацетатные экстракты промывали солевым раствором (2х), осушали сульфатом натрия, фильтровали и выпаривали досуха. Остаток подвергали флеш-хроматографии, используя 2% уксусную кислоту/этилацетат в качестве подвижной фазы, в результате чего получали 182,2 мг целевого продукта в виде белого пенящегося соединения; [α]D = -9,6o (c = 0,3, метанол). ТСХ (3% уксусная кислота в этилацетате), Rf = 0,26.
Анализ для C17H23N3O4S • 0,2 H2O:
Вычислено: C 55,32; H 6,39; N 11,39; S 8,69;
Найдено: C 55,73; H 6,44; N 10,98; S 8,56.
Пример 50.
[S-(R*, R*)]-2,3,4,5-Тетрагидро-3-[(2-меркапто-1-оксогексил)-амино]-2- оксо-1H-бензазепин-1-уксусная кислота
a) (S)-2-Бромогексановая кислота
Бромид калия (15,9 г, 133 мМ) добавляли в размешанному раствору D-норлейцина (5,0 г, 38 мМ) в 2,5 н. серной кислоте (77 мл) при комнатной температуре. Реакционную смесь охлаждали до -10oC и частями добавляли твердый нитрит натрия (3,94 г, 57 мМ), поддерживая при этом температуру (-10) - (-5)oC. После завершения добавления, пенистую реакционную смесь размешивали в течение 1 ч, а затем нагревали до комнатной температуры и размешивали еще 1 ч. После этого реакционную смесь два раза экстрагировали эфиром, эфирные экстракты один раз промывали водой, осушали сульфатом магния, фильтровали и выпаривали, в результате чего получали 3,3 г сырого целевого продукта.
b) (S)-2-Ацетилтио)гексановая кислота, дициклогексиламиновая соль
К размешанной суспензии тиоацетата калия (2,11 г, 18,5 мМ) в 50 мл сухого ацетонитрила при комнатной температуре и в атмосфере аргона добавляли раствор продукта, полученного в части (a), (3,27 г, 16,8 мМ) в 26 мл ацетонитрила. Реакционную смесь размешивали в течение 5 часов. Полученную суспензию фильтровали и выпаривали. Остаток снова растворяли в этиловом эфире, один раз промывали 5% раствором бисульфата калия и один раз солевым раствором, после чего осушали сульфатом магния и выпаривали. Остаток растворяли в эфире (64 мл) и обрабатывали дициклогексиламином (3,4 мл, 16,8 мМ). Эфирный раствор концентрировали в вакууме и растирали с гексаном, в результате чего получали белое твердое вещество, которое перекристаллизовывали из смеси этилового эфира и гексана и получали целевой продукт. Маточный раствор концентрировали и два раза перекристаллизовывали, в результате чего в целом получали 2,2 г целевого продукта; т.пл. 145 - 147oC; [α]D = -33,8o (c = 1,08, хлороформ).
c) [S-(R*, R*)]-2,3,4,5-Тетрагидро-3-[(2-меркапто-1-оксогексил)-амино] -2- 1H-бензазепин-1-уксусная кислота, этиловый эфир
Размещенную суспензию продукта, полученного в части (b), (295 мг, 0,795 мМ) в 5 мл этилацетата дважды промывали 5 мл-порциями 5%-ного раствора бисульфата калия. Органический экстракт осушали сульфатом натрия, фильтровали, концентрировали и осушали в вакууме в течение 20 мин. Полученное маслообразное вещество растворяли в 3 мл метиленхлорида и размешивали в атмосфере аргона при 0oC. К этому раствору добавляли раствор (S)-2,3,4,5-тетрагидро-3-амино-2-оксо-1H- бензазепин-1-уксусной кислоты этилового эфира, полученного известным способом (Watthey и др., в J. Med. Chem, 28, с. 1511 - 1516 (1985)) (149 мг, 0,57 мМ) в метиленхлориде (3 мл), после чего добавляли триэтиламин (83 мкл, 0,60 мМ) и наконец бензотриазол-1-илокситрис (диметиламино)фосфония гексафторофосфат (264 мг, 0,60 мМ). Через 1 ч реакцию нагревали до комнатной температуры и размешивали в течение 90 мин. Полученный раствор выпаривали при менее чем 30oC и остаток снова растворяли в этилацетате. Раствор промывали один раз 1М соляной кислоты, один раз водой, один раз насыщенным раствором бикарбоната натрия и один раз солевым раствором. Органический слой осушали сульфатом магния, фильтровали и выпаривали. После очистки с помощью флеш-хроматографии (с использованием смеси (1:3) этилацетата: гексана) получали 193 мг целевого продукта; [α]D = -284,3o (c = 0,21, хлороформ).
d) [S-(R*, R*)]-2,3,4,5-тетрагидро-3-[(2-меркапто- 1-оксо-гексил-3-фенилпропил)амино]-2-оксо-1H-бензазепин-1-уксусная кислота
Раствор продукта, полученного в части (c), (183 мг, 0,42 мМ) в 3,8 мл метанола продували азотом в течение 5 мин, а затем охлаждали до 0oC. К этому раствору по капле добавляли 3,8 мл продутого азотом 1М гидроксида натрия. В процессе реакции, раствор медленно барботировали азотом. Через 1 ч реакционную смесь нагревали до комнатной температуры и размешивали в течение 1 ч. Затем смесь подкисляли 5%-ным раствором бисульфата натрия, экстрагировали этилацетатом, а экстракты промывали водой и насыщенным раствором хлорида натрия, осушали сульфатом магния и выпаривали. После повторного выпаривания из гексана получали белое твердое вещество. Это твердое вещество слегка нагревали в этиловом эфире и кристаллизовали с гексаном, в результате чего получали 117 мг целевого соединения в виде белого твердого продукта; т.пл. 151 - 153oC; [α]D = -214,1o (c = 0,46, хлороформ).
ТСХ (этилацетат/гексан/уксусная кислота, 4:4:0,1),
Rf = 0,23.
Анализ для C18H24N2O4S:
Вычислено: C 59,32; H 6,64; N 7,69; S 8,80;
Найдено: C 59,10; H 6,62; N 7,72; S 8,63.
Пример 51.
[3S- [3α(R*), 7β] ] - Гексагидро-7-(2-гидроксиэтил)-3-[(2-меркапто-1-оксо-3-фенилпропил) амино]-2-оксо-1H-азепин-1-уксусная кислота
a) (3S-транс)-Гексагидро-3-[[(1,1-диметилэтокси)карбонил] амино] -2-оксо-7-(2-пропенил)-1H-азепин-1-уксусная кислота, метиловый сложный эфир
Моногидрат гидразина 133 мкл, 2,70 мМ по капле добавляли к раствору (3S-транс)гексагидро-3-фталимидо-2-оксо-7-(2-пропенил)- 1H--уксусной кислоты метилового эфира, полученного в соответствии с процедурой, описанной в примере 36, части (e), 609 мг, 2,45 мМ. Реакционную смесь размешивали при комнатной температуре в течение 64 ч, а затем фильтровали для удаления побочных продуктов. Фильтрат концентрировали в вакууме, растворяли в метиленхлориде, снова фильтровали и снова концентрировали, в результате чего получали неочищенное маслообразное вещество. Это вещество осушали в вакууме и получали 656 мг сырого метилового эфира (3S-транс)-гексагидро-3-амино-2-оксо-7-(2-пропенил)-1H- азепин-1-уксусной кислоты в виде желтого маслообразного продукта.
К раствору этого сырого амина (589 г, 2,455 мл) в метиленхлориде (10 мл), продутому аргоном, добавляли 510 мкл (3,68 мМ) триэтиламина. Реакционную смесь размешивали 10 мин, после чего добавляли 642 мг (2,94 мМ) ди-т-бутилдикарбоната. Реакционную смесь размешивали при комнатной температуре в течение 16 ч, а затем добавляли вторую порцию 170 мкг (1,23 мМ) триэтиламина и 160 мг (0,74 мМ)ди-т-бутилдикарбоната. Реакционную смесь размешивали еще 16 ч при комнатной температуре и разводили 20 мл метиленхлорида. Органический слой промывали 2-10 мл-порциями воды, осушали сульфатом магния и концентрировали в вакууме, в результате чего получали сырой маслообразный продукт. Этот продукт подвергали флеш-хломатографии (двуокись кремния Merck, 25 • 120 мм, 1:6 - этилацетат:гексан, а затем 1:4 - этилацетат:гексан) и получали 651 мг целевого продукта в виде прозрачного маслообразного вещества.
Моногидрат гидразина 133 мкг, 2,70 мМ по капле добавляли к раствору (3S-транс)гексагидро-3-фталимидо-2-оксо-7-(2-пропенил)-1H- уксусной кислоты метилового эфира, полученного в соответствии с процедурой, описанной в примере 36, части (e), 609 мг, 2,45 мМ. Реакционную смесь размешивали при комнатной температуре в течение 64 ч, а затем фильтровали для удаления побочных продуктов. Фильтрат концентрировали в вакууме, растворяли в метиленхлориде, снова фильтровали и снова концентрировали, в результате чего получали неочищенноое маслообразное вещество. Это вещество осушали в вакууме и получали 656 мг сырого метилового эфира (3S-транс)-гексанидро-3-амино-2-оксо-7-(2-пропенил)-1H-азепин-1- уксусной кислоты в виде желтого маслообразного продуккта.
К раствору этого сырого амина (589 г, 2,455 мл) в метиленхлориде (10 мл), продутому аргоном, добавляли 510 мкл (3,68 мМ) триэтиламина. Реакционную смесь размешивали 10 мин, после чего добавляли 642 мг (2,94 мМ) ди-т-бутилкарбоната. Реакционную смесь размешивали при комнатной температуре в течение 16 ч, а затем добавляли вторую порцию 170 мкг (1,23 мМ) триэтиламина и 160 мг (0,74 мМ) ди-т-бутиодикарбоната. Реакционную смесь размешивали еще 16 ч при комнатной температуре и разводили 20 мл метиленхлорида. Органический слой промывали 2 - 10 мл-порциями воды, осушали сульфатом магния и концентрировали в вакууме, в результате чего получали сырой маслообразный продукт. Этот продукт подвергали флеш-хроматографии (двуокись кремния Merck, 25х120 мм, 1:6 - этилацетат:гексан, а затем 1:4 - этилацетат:гексан) и получали 651 мг целевого продукат в виде прозрачного маслообразного вещества.
b) (3S-транс)-Гексагидро-3-[[(1,1-диметилэтокси)карбонил] амино] -2-оксо-7-(гидроксиэтил)-1H-азепин-1-уксусная кислота, метиловый сложный эфир.
К раствору продукта, полученного в части (a), (554 мг, 1,63 мМ) в 3 мл воды и 3 мл диоксана добавляли 731 мг (3,42 мМ) периодата натрия, а затем по капле в течение 10 мин добавляли 400 мкл (0,98 мМ) тетраксида осмия. Реакционную смесь размешивали при комнатной температуре 16 ч фильтровали и промывали этилацетатом. Полученный фильтрат концентрировали в вакууме без нагревания и подвергали флеш-хроматографии (двуокись кремния Merck, 25• 120 мм, 1: 4 - этилацетат/гексан - без давления), в результате чего получали 309 мг 3(S)-транс)-гексагидро-3-[[(1,1-диметилэтокси)карбонил]амино]-2-оксо-7- ацетальдегид-1H-азепин-1-уксусной кислоты метилового сложного эфира в виде прозрачного маслообразного вещества.
К раствору этого альдегида (309 мг, 0,90 мМ) в метаноле (5 мл), охлажденному до 0oC, добавляли порциями 68 мг (1,80 мМ) борогидрида натрия. Реакционную смесь размешивали 1,5 ч при 0oC, после чего реакцию гасили путем добавления по капле 0,5 мл воды и нагревали до комнатной температуры. Затем смесь распределяли между 25 мл этилацетат и 25 мл 1М соляной кислоты, а водный слой экстрагировали 2-20 мл порциями этилацетата. Объединенные этилацетатные слои промывали 10 мл насыщенного бикарбоната натрия и 10 мл солевого раствора, осушали сульфатом магния и концентрировали в вакууме, в результате чего получали сырое маслообразное вещество. Это вещество подвергали флеш-хроматографии (двуокись кремния Merck, 25 • 90 мм, 2:1 - этилацетет/гексан, а затем этилацетат) и получали 292 мг целевого продукта в виде прозрачного маслообразного вещества.
c) [3S- [3α(R*), 7β] ] - Гексагидро-3-[(2-(ацетилтио)-1-оксо-3- фенилпропиламино]-7-(2-гидроксиэтил)-2-оксо-1H-азепин-1-уксуснная кислота, метиловый сложный эфир
К раствору продукта, полученного в части (b), (292 мг, 0,85 мМ) в диоксане (1 мл), охлажденному до 0oC, добавляли порциями (2,5 мл 9,35 мМ, 4 М) соляной кислоты/диоксана. Реакционную смесь нагревали до комнатной температуры и размешивали 16 ч, а затем концентрировали в вакууме и подвергали азеотропной обработке с использованием толуола, в результате чего получали 248 мг (3S-транс)-гексагидро-3-амино-2-оксо-7-(2-гидроксиэтил)-1H-азепин-1- уксусной кислоты метилового эфира гидрохлоридной соли в виде неочищенного маслообразного вещества.
К раствору этого амина гидрохлоридной соли (189 мг, 0,67 мМ) и (S)-2-(ацетилтио)бензопропановой кислоты (полученной из дициклогексиламиновой соли, как описано ранее, 157 мг, 0,74 мМ) в метиленхлориде (10 мл), охлажденному до 0oC, добавляли триэтиламин (230 мкл, 1,68 мМ). Эту смесь размешивали в течение 20 мин, а затем добавляли гексафторофосфат бензоатриазол-1-илокситрис-диметиламино фосфония (327 мг, 0,74 мМ). Реакционную смесь размешивали 1 ч при 0oC, а затем 3 ч при комнатной температуре. Затем реакционную смесь распределяли между 20 мл этилацетата и 20 мл 5%-ного раствора бисульфата калия. Водный слой отделяли и экстрагировали 2-20 мл-порциями этилацетат, а объединенные этилацетатные слои промывали 20 мл 5%-ного раствора бисульфата калия, 20 мл насыщенного бикарбоната натрия, 2-20 мл-порциями солевого раствора, осушали сульфатом магния и концентрировали в вакууме, в результате чего получали неочищенное маслообразного вещества. Это вещество подвергали флеш-хроматографии (двуокись кремния Merck, 25 •80 мм, 1:4 - этилацетат/гексан) и получали 84 мг целевого продукта в виде белого пенистого вещества.
d) [3S- [3α(R*), 7β] ]- Гексагидро-7-(2-гидроксиэтил)-3-[(2-меркапто-1-оксо-3-фенилпропил) амино]-2-оксо-1H-азепин-1-уксусная кислота
Раствор продукта, полученного в части (c), (843 мг, 0,19 мМ) в метаноле (2 мл) продували аргоном в течение 30 мин и охлаждали до 0oC. К этому раствору по капле добавляли 2 мл 1М гидроксида натрия, также продутого аргоном в течение 30 мин, и охлаждали до 0oC. Реакционную смесь размешивали при 0oC в течение 1 ч, продолжая продувать аргоном, а затем подкисляли 5% раствором бисульфата калия для доведения pH до 2. Полученную смесь экстрагировали 3-20 мл-порциями этилацетат, а объединенные этилацетатные слои осушали сульфатом магния и концентрировали в вакууме, в результате чего получали неочищенное пенистое вещество. Это вещество подвергали флеш-хроматографии (двуокись кремния Merck, 15• 60 мм, 0,5:5:95 - уксусная кислота: метанол: метиленхлорид) и получали белое пенистое вещество, которое подвергали азеотропной обработке с использованием толуола и осушали в вакууме, в результате чего получали 48 мг целевого продукта в виде белого пенистого вещества; [α]D = -42,9o (c = 1,0, CDCl3). ТСХ (уксусная кислота: метанол: метиленхлорид, 1:10:90), Rf = 0,25.
Анализ для C19H26N2O5S • 0,5 H2O:
Вычислено: C 56,55; H 6,75; N 6,94; S 7,94;
Найдено: C 56,55; H 6,64; N 6,77; S 7,51.
Пример 52.
[1S- [ 1α, 9α ]] -Октагидро-9-[[2-меркапто-1-оксо-3-(2-тиенил) пропил] амино]-10-оксо-6H-пиридазино[1,2-a][1,2]диазепин-1-карбоновая кислота
a) (S)- α- -(ацетилтио)-2-тиофенпропановая кислота
Хлорид калия (3,0 г, 40,1 мМ) добавляли к раствору β- -(2-тиенил)-D-аланина (1,37 г, 8,03 мМ) в 2,5 н соляной кислоты (25 мл) при комнатной температуре и в атмосфере аргона. После 10-минутного размешивания, полученную смесь охлаждали до 0oC и обрабатывали нитридом натрия (720 мг, 10,44 мМ). Через 2,5 ч реакционную смесь нагревали до комнатной температуры и размешивали в течение 1 ч. После этого смесь распределяли между водой и этилацетатом и органический слой осушали сульфатом натрия, хроматографировали и концентрировали. Остаток подвергали флеш-хроматографии (силикагель Merck), элюируя 1% уксусной кислотой в смеси (3:1) гексана и этилацетата, в результате чего получали 760 мг (R) - α- -хлоро-2-тиофенкарбоновой кислоты в виде желтого маслообразного продукта.
К раствору, содержащему вышеуказанный хлорид (750 мг, 4,73 мМ) в диметилформамиде (15 мл) при комнатной температуре и в атмосфере аргона добавляли тиоацетат цезия (2,95 г, 14,19 мМ). После 2-часового размешивания, реакционную смесь распределяли между 10% бисульфатом калия и этилацетатом. Органический слой промывали солевым раствором, осушали сульфатом натрия, фильтровали и концентрировали, а остаток подвергали флеш-хроматографии (силикагель Merck), элюируя 1%-ной уксусной кислотой в смеси (4:1) гексана и этилацетата, в результате чего получали 500 мг целевого продукта в виде маслообразного вещества. ТСХ (2% уксусная кислота в смеси (3:1) этилацетат и гексана), Rf = 0,73.
b) [1S-[ 1α, 9α (R*)]]-Октагидро-9-[[2-(ацетилтио)-1-оксо-3-(2-тиенил) пропил] амино]-10-оксо-6H-пиридазино[1,2-a][1?2]-диазепин-1- карбоновая кислота
Продукт, полученный в части (a), (246 мг, 1,07 мМ) и (1S-цис)-октагидро-9-амино-10-оксо-6H-пиридазино [1,2-a][1,2]диазепин-1-карбоновой кислоты, 1,1-диметилэтиловый эфир, полученный, как описано в примере 47 (b), (370 мг, 1,17 мМ) растворяли в метиленхлориде (10 мл) при комнатной температуре в атмосфере аргона. Полученную смесь охлаждали до 0oC и добавляли триэтиламин (0,15 мл, 1,07 мМ). Полученную смесь размешивали 5 мин, а затем добавляли гексафторофосфат бензотриазол-1-илоксистрис диметиламино фосфония (517 мг, 1,17 мМ). После 1-часового размешивания при 0oC, реакционную смесь нагревали до комнатной температуры и размешивали в течение 16 ч. Летучие вещества выпаривали, а остаток растворяли в этилацетат и промывали последовательно 1 н. соляной кислотой, водой, 50% насыщенным бикарбонатом натрия и солевым раствором. Органический слой осушали сульфатом натрия, фильтровали и концентрировали, а остаток подвергали флеш-хроматографии (силикагель Merck), элюируя смесью (3:2) гексана и этилацетата. Затем продукт снова подвергали флеш-хроматографии, элюируя смесью (3:1) гексана и этилацетата, и получали 395 мг [1S- [ 1α, 9α (R*)]]-окстагидро-9- [[2-(ацетилтио)-1-оксо-3-(2-тиенил)пропил] амино]-10-оксо-6H-пиридазино [1,2-a] [1,2] диазепин-1-карбоновой кислоты, 1,1-диметилэтилового сложного эфира в виде белого пенистого вещества. ТСХ (этилацетат: гексан, 1:1), Rf = 0,43.
К раствору полученного выше продукта (390 мг, 0,82 мМ) в метиленхлориде (6 мл) при комнатной температуре и в атмосфере аргона добавляли анизол (0,38 мл, 3,52 мМ). Полученную смесь обрабатывали трифторуксусной кислотой (0,58 мл, 7,5 мМ) и размешивали в течение 36 ч. Летучие вещества удаляли, а остаток отделяли с использованием толуола и удаляли, в результате чего получали 380 мг желтого маслообразного вещества. Это вещество очищали с помощью колоночной хроматографии (силикагель Merck), элюируя 1%-ной уксусной кислотой в смеси (1:1) этилацетата и гексана, а затем 1%-ной уксусной кислотой в смеси этилацетата и гексана, в результате чего получали 360 мг целевого продукта.
ТСХ (1% уксусная кислота в смеси (1:1) этилацетата и гексана), Rf = 0,28.
c) [1S-[ 1α, 9α (R*)] ] -Оксагидро-9-[[(2-меркапто-1-оксо-3- (2-тиенил)пропил] амино] -10-оксо-6H-пиридазино [1,2-a][1,2]диазепин- 1-карбоновая кислота
Раствор продукта, полученного в части (b), (328 мг, 0,75 мМ) в метаноле (5 мл, дезоксагенированном посредством барботирования аргона) охлаждали до 0oC и обрабатывали 1 н. гидроксидом натрия (4 мл, дезоксигенированным посредством барботирования аргона. Полученную смесь размешивали 1 ч в атмосфере аргона. Смесь подкисляли 10% бисульфатом калия и экстрагировали этилацетатом. Органический слой промывали последовательно водой и солевым раствором, осушали сульфатом натрия, фильтровали и концентрировали, в результате получали прозрачное маслообразное вещество. Остаток подвергали флеш-хроматографии (силикагель Merck), элюируя 1%-ной уксусной кислотой в смеси (1:2) гексана и этилацетата. Фракции, содержащие чистый целевой продукт, объединяли, упаривали, подвергали азеотропной обработке с использованием этилацетата и промывали водой для удаления уксусной кислоты. Органический слой осушали сульфатом магния, фильтровали и концентрировали. Остаток растворяли в этилацетате и растирали с гексаном. Растворитель удаляли, а остаток суспендировали в гексане, упаривали и осушали в вакууме, в результате чего получали 165 мг целевого продукта в виде белого порошкообразного пенистого вещества. [α]D = -131,6o (c = 0,43, метиленхлорид), Rf = 0,36.
Анализ для C17H23N4O4S2•0,21 H2O•0,09 C6H14•0,13 C4H8O2:
Вычислено: C 51,58; H 6,17; N 9,99; S 15,25;
Найдено: C 51,24; H 6,08; N 9,69; S 15,22
Пример 53.
[IS-[1α, 8α (R*)] ] -Гексагидро-8-[[(2-меркапто-1-оксо-3-фенилпропил] амино]-9- оксо-IH,5H-пиразоло[1,2-a][1,2]диазепин-1-карбоновая кислота
a) I-[(Фенилметокси) карбонил)-3-пиразолидин-карбоновая кислота, 1,1-диметилэтиловый сложный эфир
Раствор 3-пиразолидинкарбоновой кислоты 1,1-диметилэтилового сложного эфира (10,935 г, 63,5 мМ) в сухом ацетонитриле (90 мл) охлаждали до 0oC (ледяная-солевая баня) и обрабатывали сухим пиридином (11,0 мл), а затем раствором бензилхлороформатом (12,54 г, 10,5 мл, 69,9 мМ) в сухом ацетонитриле (25 мл) реакционную смесь размешивали 3 ч при 0oC, выпаривали досуха и полученный сироп снова растворяли в этилацетате (250 мл). Раствор промывали 5% бикорбонатом натрия (2•25 мл) и солевым раствором (25 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенную продуктовую смесь хроматографировали на колонке с силикагелем (Merck), элюируя смесями этилацетата и гексана (1:4, 1:2) в результате чего получали 11,144 г целевого продукта в виде сиропа. ТСХ (этилацетат: гексан, 1:1), Rf = 0,25.
b) (S-2-[2-Фталимидо-1,5-диоксо-5-(фенилметокси)-пентил] -1,3- пиразолидинкарбоновая кислота, 3-(1-диметилэтил)-1-фенилметиловый эфир
Дициклогесиламиновую соль (S)-2-фталимидопентадионовой кислоты 5-(фенилметил)эфир (11,548 г, 21 мМ) суспендировали в этилацетате (700 мл) промывали 5% бисульфатом калия (5•100 мл) и солевым раствором (100 мл), осушали сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме.
Полученную свободную кислоту растворяли в сухом эфире (100 мл), охлаждали до 0oC (в ледяной-солевой бане), обрабатывали пентахлоридом фосфора (4,45 г, 21 мМ) и размешивали 30 мин при 0oC, а затем 15 мин при комнатной температуре. Реакционную смесь упаривали досуха, выпаривали из толуола (2•230 мл) и осушали в вакууме. Неочищенный хлорангидрид растворяли в толуоле (46 мл), добавляли к охлажденному раствору (10oC, ледяная-солевая баня) продукта, полученного в части (a), (5,52 г, 18 мМ) в толуоле (35 мл) и бикорбанате натрия (4,77 г, в 46 мл) воды, нагревали до комнатной температуры и размешивали в течение 20 ч в атмосфере аргона. Реакционную смесь разбавляли этилацетатом (650 мл), промывали 5% бисульфатом калия (2•140 мл) и солевым раствором (140 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме, в результате чего получали 11,587 г целевого продукта.
c) (IS-цис)-Гексагидро-8-фталимидо-5,9-диоксо-IH, 5H-пиразоло-[1,2-a] [1,2]диазепин-1-карбоновая кислота, 1,1-диметилэтиловый эфир
Раствор продукта, полученного в части (b), (12,653 г, 18 мМ) в сухом диметилформамиде (196 мл) обрабатывали 10% палладированным углем (2,19 г) и гидрировали 24 ч при комнатной температуре. Реакционную смесь разводили метанолом (200 мл) и фильтровали через прокладку из целита, тщательно промывая эту прокладку метанолом (2•200 мл). Прозрачный фильтрат выпаривали досуха и осушали в вакууме, в результате чего получали сироп слегка золотистого цвета. Неочищенный продукт растворяли в сухом метиленхлориде (185 мл), охлаждали до 0oC (ледяная-солевая баня), обрабатывали тионилхлоридом (1,62 мл, 22,2 мМ) и размешивали 15 минут при 0oC, а затем 6,0 ч при комнатной температуре. Реакционную смесь гасили раствором бикорбаната калия (3,93 г) в воде (34 мл), размешивали 10 - 15 мин, а затем экстрагировали метиленхлоридом (2•290 мл). Объединенные органические экстракты осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Полученную неочищенную продуктовую смесь хроматографировали на колонке с силикагелем (Merck), элюируя смесями этилацетата и гексана (1:3, 1:2), в результате чего получили 2,427 г целевого соединения. ТСХ (этилацетат: гексан, 1:1), Rf = 0,37.
d) (IS-цис)-Гексагидро-8-фталимидо-9-оксо-IH, 5H-пиразоло [1,2-a] [1,2] диазепин-1-карборновая кислота, 1,1-диметилэтиловый эфир
Раствор продукта, полученного в части (c), (607 мг, 1,43 мМ) в сухом тетрагидрофуране (12 мл) охлаждали до 0oC (ледяная-солевая баня) и обрабатывали 1,0 М диборана/тетрагидрофурана в соответствии с процедурой, описанной в примере 47, часть (a), в результате чего получали 528 г целевого продукта в виде сиропа. ТСХ (этилацетат: гексан, 1:1), Rf = 0,38.
e) (IS-цис)-Гексагидро-8-амино-9-оксо-IH, 5H-пиразоло[1,2-a] [1,2] диазепин-1-карбоновая кислота, 1,1-диметилэтиловый эфир
Раствор продукта, полученного в части (d), (505 мг, 1,25 мМ) в абсолютном этаноле (5,5 мл) обрабатывали гидратом гидразина (0,14 мл, 2,2 экв.) в соответствии с процедурой, описанной в примере 46, части (b), в результате чего получали 312 мг целевого соединения в виде сиропа. ТСХ (метиленхлорид: метанол, 9:1), Rf = 0,63.
f) [IS-[ 1α, 8α (R*)]]-Гексагидро-8-[[2-(ацетилтио)-1-оксо-3-фенилпропил]амино]-9- оксо-1H, 5H-пиразоло [1,2-a] [1,2]диазепин-1-карбоновая кислота
(S)-2-(Ацетилтио)бензолпропановую кислоту, полученную из 554 мг дициклогексиламиновой соли, как описано ранее, растворяли в метиленхлориде (11 мл), охлаждали до 0oC (ледяная/солевая баня) и обрабатывали последовательно раствором продукта, полученного в части (e), (350 мг, 1,25 мМ) в сухом метиленхлориде (2,5 мл), триэтиламином (0,17 мл, 1,22 мМ) и гексафторофосфатом бензотриазол-1-илокситрис диметиламино фосфония (561 мг, 1,29 мМ) в соответрствии с процедурой в примере 47, части (c), в результате чего получали 400 мг [IS - [ 1α, 8α (R*)]]-гексагидро-8-[[2-(ацетилтио)-1-оксо-3-фенилпропил] амино] -9-оксо-IH, 5H-пиразоло [1,2-a] [1,2] диазепин-1-карбоновой кислоты 1,1-диметилэтилового эфира в виде сиропа. ТСХ (этилацетат: гексан, 1:1), Rf = 2,28.
Раствор полученного материала в сухом метиленхлориде обрабатывали анизолом и трифторуксусной кислотой в соответствии с процедурой, описанной в примере 47, части (d), в результате чего получали целевой продукт в виде твердого вещества; [α]D = -111,5o (с = 0,48, метанол). ТСХ (этилацетат: уксусная кислота, 95:5), Rf = 0,38.
q) [IS-[ 1α, 8α (R*)] ] -Гексагидро-8-[(2-меркапто-1-оксо-3-фенилпропил)амино]-9-оксо- IH,5H-пиразоло[1,2-a][1,2]диазепин-1-карбоновая кислота
Раствор продукта, полученного в части (f), в сухом метаноле обрабатывали 1,0 н. гидроксидом натрия в соответствии с процедурой, описанной в примере 47, части (d), в результате чего получали целевой продукт в виде белого твердого аморфного пенистого вещества: [α]D = -71o (с = 0,50, метанол). ТСХ (этилацетат: уксусная кислота, 95:5), Rf = 0,25.
Анализ для C18H23N3O4S• 0,13 C5H12•0,20 H2O:
Вычислено: C 57,36; H 6,44; N 10,76; S 8,21
Найдено: C 57,36; H 6,50; N 10,47; S 8,10.
Пример 54.
[IS-[ 1α, 8α (R*)] ] -Гексагидро-8-[(2-меркапто-1-оксо-3-фенилпропил)амино] -5,9- диоксо-1H, 5H-пиразоло[1,2-a] [1,2] диазепин-1-карбоновая кислота
a) (IS-цис)-Гексагидро-8-амино-5,9-диоксо-1H, 5H-пиразоло [1,2-a][1,2] диазепин-1-карбоновая кислота, 1,1-диметилэтиловый эфир
Раствор продукта, полученного в примере 53, части (c), (700 мг 1,6 мМ) в абсолютном этаноле (7,4 мл) обрабатывали гидратом гидразина (0,19 мл, 2,2 экв. ) в соответствии с процедурой, описанной в примере 53, части (e), в результате чего получали 436 мг целевого продукта в виде белого пенистого вещества. ТСХ (метиленхлорид: метанол, 9:1), Rf = 0,30.
b) [IS-[ 1α, 8α (R*)] ] -Гексагидро-8-[(2-меркапто-1-оксо-3-фенилпропил)амино] -5,9-диоксо-IH, 5H-пиразоло[1,2-a] [1,2] диазепин-1-карбоновая кислота
Продукт, полученный в части (a), подвергали реакции с (S)- 2-ацетилтио)бензолпропановой кислотой в присутствии триэтиламина и гексафторофосфата бензотриазол-1-илокситрис(диметиламино)-фосфин в соответствии с процедурой, описанной в примере 53, части (f), в результате чего получали [1S-[1α, 8α (R*)]]-гексагидроу-8-[[2-ацетилтио)-1-оксо-3-фенилпропил]амино]-5,9- диоксо-1H,5H-пиразоло[1,2-а][1,2]диазепин-1-карбоновой кислоты 1,1-диметилэтиловый сложный эфир в виде сиропа. ТСХ (этилацетат: гексан, 1:1), Rf = 0,22.
Этот материал обрабатывали анизолом, а затем трифторуксусной кислотой в соответствии с процедурой, описанной в примере 53, часть (f). Полученный кислотный продукт растворяли в метаноле и обрабатывали 1,0 н. гидроксидом натрия в соответствии с процедурой примера 53 (g), в результате чего получали целевой продукт в виде белого твердого аморфного пенистого вещества; [α]D = -95,8o (с = 0,72: метанол). ТСХ (метиленхлорид: метанол:уксусная кислота, 20:1:1), Rf = 0,13.
Анализ для C18H21N3O5S • 0,07 C6H14 • 1,14 H2O:
Вычислено: 52,89; H 58,83; N 10,06; S 7,67;
Найдено: 52,89; H 5,80; N 9,18; S 7,01.
Пример 55.
[3S-[1(R*), 3α(R*), 7β ] ] -Гексагидро - α- метил-3-[(2-меркапто-1-оксо-3-фенилпропил]амино]-2-оксо-7-пропил-1H-азепин -1-уксусная кислота
a) [3S-[1(R*), 3α, 7β ]]-Гексагидро- α- метил-3-фталимидо-2-оксо-7-(2-пропенил)-1H-азепин-уксусная кислота, метиловый эфир
Аллилтриметилсилан (1,53 мл, 9,6 мМ) добавляли к раствору [S-(R*,R*)]- гексагидро- α- - метил-3-фталимидо-2-оксо-1H-азепин-1-уксусной кислоты, метиловому сложному эфиру (485 мг, 1,48 мМ) в метиленхлориде (20 мл) при комнатной температуре и в атмосфере аргона. После добавления бромида олова (1,0 М) в метиленхлориде (2,52 мл, 2,52 мМ), полученную смесь размешивали в течение 4 ч. Затем добавляли другую порцию бромида олова (1,48) мл и смесь размешивали в течение 48 ч. Реакционную смесь гасили водным раствором хлорида аммония и экстрагировали этилацетатом. Органический слой промывали солевым раствором, осушали сульфатом натрия, фильтровали и концентрировали, в результате чего получали 1,2 г желтого остатка. Неочищенную кислоту растворяли в метаноле, охлаждали до 0oC и обрабатывали диазометаном для получения метилового сложного эфира. Летучие вещества удаляли, а остаток подвергали флеш-хроматографии (силикагель Merck), элюируя смесью этилацетата и гексана (1: 3), а затем смесью 1:1 этилацетата и гексана, в результате чего получали 283 мг целевого продукта в виде белого пенообразного вещества; [α]D = -13,7o (с = 0,54, метиленхлорид).
ТСХ (этилацетат: гексан, 1:1), Rf = 0,50.
b) [3S-[1(R*), 3α, 7β ]]-Гексагидро- α- -метил-3-фталимидо-2-оксо-7-пропил-1H-азепин-1-уксусная кислота, метиловый эфир
Раствор продукта, полученного в части (a), (463 мг, 1,24 мМ) в смеси метанола/метилацетата (1: 1, 14 мл) гидрировали 3 ч в присутствии 10% палладированного угля (41 мг). Полученную смесь фильтровали через целит, летучие вещества удаляли и получали 483 мг целевого продукта в виде желтого маслообразного продукта.
c) [3S-[(R*), 3α(R*), 7β ]]-Гексагидро- α- - метил-3-[(2-ацетилтио)-1-оксо-3-фенилпропил] амино] -2-оксо-7-пропил-1H- азепин-1-уксусуная кислота, метиловый эфир
Продукт, полученный в части (b), (483 мг) суспендировали в метаноле (7 мл) при комнатной температуре и в атмосфере аргона. Полученную смесь обрабатывали моногидратом гидразина (0,07 мл, 1,45 мМ), в результате чего эта смесь становилась гомогенной и размешивали в течение 18 ч. Смесь фильтровали для удаления белого осадка, а фильтрат упаривали, обрабатывали метиленхлоридом, фильтровали и снова упаривали, в результате чего получали 282 мг [3S-[1(R*), 3α, 7β ]]-гексагидро- α- - метил-3-амино-2-оксо-7-пропил-1H-азепин-1-уксусной кислоты, метилового эфира в виде желтого маслообразного вещества.
(S)-2-(Ацетилтио)бензолпропановую кислоту (полученную из 491 мг дициклогексиламиновой соли, как описано ранее) растворяли в метиленхлориде (15 мл) при комнатной температуре в присутствии аргона. После добавления вышеуказанного амина (282 мг, 1,10 мМ) смесь охлаждали до 0oC и обрабатывали триэтиламином 0,17 мл, 1,21 мМ и гексафторофосфатом бензотриазол-1-илокситрис диметиламино фосфония (511 мг, 1,16 мМ) в соответствии с процедурой, описанной в примере 36 части (f), в результате чего получали 385 мг целевого продукта в виде белого пенообразного вещества.
ТСХ (этилацетат: гексан, 1:1), Rf = 0,43.
d) [3S-[1(R*), 3α(R*), 7β ]]-Гексагидро - α- метил-3-[(2-меркапто-1-оксо-3-фенилпропил)амино]-2-оксо-7-пропил-1H-азепин- 1-уксусная кислота.
Раствор продукта, полученного в части (с), (385 мг, 0,83 Мм) в метаноле (10 мл) обрабатывали 1 н. гидроксидом натрия (8 мл) в соответствии с процедурой, описанной в примере 36, части (g), в результате чего получали 142 мг целевого продукта в виде белого порошкоообразного пенистого вещества; [α]D = -51,6o (с = 0,48, метиленхлорид ). ТСХ (1% уксусная кислота в смеси (2:1) этилацетата/гексана), Rf = 0,60.
Анализ для C21H30N2O4S, 0,14 H2O:
Вычислено: C 61,67; H 7,46; N 6,85; S 7,84;
Найдено: C 61,97; H 7,50; N 6,55; S 7,62.
Пример 56
[S-(R*, R*)] 2,3,4,5-Тетрагидро-3-[(2-меркапто-1-оксо-3-фенилпропил)амино] -2-оксо-1H-пирроло[1,2-b][1,2]диазепин-1-уксусная кислота
a) [S-(R*,R*)]-2,3,4,5-Тетрагидро-3-[[2-(ацетилтио)-1-оксо -3-фенилпропил] амино]-2-оксо-1H-пирроло[1,2-b][1,2]диазепин-1-уксусная кислота, этиловый сложный эфир
(S)-3-Амино-2-оксо-1H-пирроло[1,2-b] [1,2] диазепин-1-укусусной кислоты этиловый сложный эфир, полученный известным способом ((Bolos и др. J.Org. Chem. 57, с. 3535-3339, 1992), подвергали реакции с (S)-2-(ацетилтио) бензолпропановой кислотой в присутствии триэтиламина и гексафторфосфата бензотриазол-1-илокситрис(диметиламино)фосфония в соответствии с процедурой, описанной выше в примере 36, части (f), в результате чего получали целевой продукт в виде бесцветного маслообразного вещества.
b) [S-(R*, R*)] -2,3,4,5-Тетрагидро-3-[(2-меркапто-1-оксо-3- фенилпропил)амино]-2-оксо-1H-пирроло[1,2-b][1,2]диазепин-1-уксусная кислота
Раствор продукта, полученного в части (a), (135 мг, 0,30 мМ) в метаноле (2 мл) тетрагидрофуране (1 мл) и воде (1 мл) барботировали в течение 30 мин аргоном. К этому раствору при комнатной температуре добавляли моногидрид гидроокиси лития (50 мг, 1,2 мМ). Реакционную смесь размешивали 2 ч, продолжая барботировать аргон, а затем подкисляли путем добавления 1M водного раствора соляной кислоты (1,4 мл), после чего смесь добавляли к 20 мл воды, а затем экстрагировали этилацетатом (2•15 мл). Органические экстракты объединяли, осушали сульфатом натрия и концентрировали в вакууме, в результате чего получали маслообразное вещество. Это вещество очищали с помощью флеш-хроматографии (двуокись кремния Merck, 1:99 уксусная кислота:этилацетат) и получали 115 мг целевого соединения в виде белого твердого пенообразного продукта; [α]D = -67o (с = 0,26, метанол), ТСХ (уксусная кислота: метанол: метиленхлорид, 1:10:90), Rf = 0,43.
Анализ для C19H21N3O4S:
Вычислено: C 58,90; H 5,46; N 10,85; S 8,27;
Найдено: C 58,55; H 5,50; N 10,74; S 8,17.
Пример 57.
[1S-[1α, 9α (R*)] ] -Октагидро-9-[[2-метилдитио)-1-оксо-3-фенилпропил] амино]-10- оксо-6-H-пиридазино [1,2-а][1,2]диазепин-1-карбоновая кислота
Раствор продукта примера 47 (100 мг, 0,247 мМ) в этаноле (1,5 л) и воде (0,15 мл) охлаждали до 0oC и обрабатывали метилметантиосульфонатом (30,5 мкл, 37,4 мг, 0,296 мМ). После размешивания в течение 2 ч при 0oC и 6 ч при комнатной температуре, реакционную смесь разбавляли водой и экстрагировали смесью этилацетата и этилового эфира. Органический экстракт промывали водой и солевым раствором, осушали сульфатом магния и концентрировали в вакууме, в результате чего получали 157 мг неочищенного продукта. После флеш-хроматографии на 12 г силикагеля (Merck) с элюированием сначала гексаном, затем смесью гексана: этилацетата (1: 1) и наконец смесью гексана: этилацетата: уксусной кислоты (4:4:0,1), получали очищенный продукт. Этот продукт растирали со смесью этилацетата и гексана и получали 85,4 мг целевого соединения в виде белого твердого пенообразного продукта; [α]D = -104,2o (с = 0,18, хлороформ). ТСХ (гексан: этилацетат: уксусная кислота, 4:4:0,1), Rf = 0,16.
Анализ для C20H27N3O4S • 0,05 C4H8O2 • 0,01 C6H14:
Вычислено: C 54,95; H 6,27; N 9,49; S 14,48;
Найдено: C 55,03; H 6,22; N 9,49; S 14,40.
Пример 58.
[S-((R*, R*)]-2,3,4,5-Тетрагидро-3-[[2-(метилтио)-1-оксо-3- фенилпропил] -амино]-2-оксо-1H-бензазепин-1-уксусная кислота
Раствор продукта примера 6 (100 мг, 0,235 мМ) в этаноле (1,2 мл) и в воде (0,112 мл) охлаждали до 0oC и обрабатывали метилметансульфонатом (29 мкл, 35,6 мг, 0,282 мМ). После 1-часового выдерживания при 0oC, реакционную смесь выдерживали 8 ч при комнатной температуре, после чего смесь помещали на ночь в холодильник. Затем смесь разбавляли водой и экстрагировали смесью этилацетата и этилового эфира. Органический экстракт промывали водой и солевым раствором. Осушали сульфатом магния и концентрировали в вакууме, в результате чего получали 123 мг неочищенного продукта. После флеш-хроматографии на 9 г силикагеля (Merck) с элюированием сначала гексаном, затем смесью 1:1 гексана: этилацетата и наконец смесью 4:4:0,1 гексана: этилацетата: уксусной кислоты, получали 108 мг продукта. Этот продукт растирали со смесью этилацетата и гексана и получали 97 мг целевого продукта в виде белого твердого вещества; т. пл. 83 - 95oC; [α]D = -185,17o (с = 0,3, хлороформ). ТСХ (гексан: этилацетат: уксусная кислота, 4:4:0,1), Rf = 0,16.
Анализ для C22H24N2O4S2 • 0,84 H2O • 0,07 C4H8O2 • 0,07 C6H14:
Вычислено: C 57,78: H 5,81: N 5,94: S 13,59;
Найдено: C 57,70: H 6,13: N 6,27: S 13,20.
Пример 59.
[S-(R*, R*)] -2,3,4,5-Тетрагидро-3-[(2-меркапто-1-оксо-4- метилпентил)амино]-2-оксо-1H-бензазепин-1-уксусная кислота
a) (S)-2-Бромо-4-метилпентановая кислота
Бромид калия (9,5 г, 80 мМ) добавляли к размешанному раствору D-лейцина (3,0 г, 23 мМ) в 2,5 н. серной кислоте (47 мл) при комнатной температуре. Реакционную смесь охлаждали до -10oC и порциями добавляли твердый нитрит натрия (2,4 г, 34 мМ), поддерживая при этом температуру (-10)-(-5)oC. После завершения добавления, реакционную смесь размешивали в течение 1 ч затем нагревали до комнатной температуры и размешивали еще 1 ч. После этого, реакционную смесь два раза экстрагировали эфиром и эфирные экстракты один раз промывали водой, осушали сульфатом магния, фильтровали и выпаривали, в результате чего получали 2,7 г неочищенного целевого продукта.
b) (S)-2-(Ацетилтио)-4-метилпентановая кислота, дициклогексиламиновая соль
К размешанной суспензии тиоцетата калия (1,7 г, 15,0 мМ) в 50 мл сухого ацетонитрила при комнатной температуре м в атмосфере аргона добавляли раствор продукта, полученного в части ( ), (2,6 г, 13 мМ) в 17 мл ацетонитрила. Реакционную смесь размешивали в течение 4 ч. Полученную суспензию фильтровали и выпаривали. Остаток снова растворяли в этиловом эфире, один раз промывали 5% бисульфатом калия и один раз солевым раствором, а затем осушали сульфатом магния и выпаривали. Остаток растворяли в эфире (64 мл) и обрабатывали дициклогексиламином (2,7 мл, 14 мМ). Сразу после обработки из раствора начинал выпадать белый твердый осадок. Этот раствор фильтровали, белый твердый осадок собирали и получали в результате 2,0 г целевого продукта; т.пл. 153 - 158oC; [α]D = -54,5o , (с = 0,61, хлороформ).
c) [S-(R*, R*)] -2,3,4,5-Тетрагидро-3-[[2-(ацетилтио)-1-оксо- -4-метилпентил]амино]-2-оксо-1H-бензазепин-1-уксусная кислота, этиловый сложный эфир
Размешанную суспензию продукта, полученного в части (b), (312 мг, 0,840 мМ) в 5 мл этилацетата два раза промывали 5 мл порциями 5%-ного раствора бисульфата калия. Органический экстракт осушали сульфатом натрия, фильтровали, концентрировали и осушали в вакууме в течение 20 мин. Полученный маслообразный остаток растворяли в 2,5 мл метиленхлорида и размешивали при 0oC в атмосфере аргона. К этому раствору добавляли раствор (S)-2,3,4,5-тетрагидро-3-амино-2-оксо-1H-бензазепин-1-уксусной кислоты этилового сложного эфира, полученного известным способом (Watthey) и др. J.Med. Chem, 28, с. 1511-1516) (142 мг, 0,54 мМ) в метиленхлориде (3 мл), а затем добавляли триэтиламин (80 мкл, 0,58 мМ) и наконец гексафторофосфат бензотриазол-1-илокситрис (диметиламино)фосфония (251 мг, 0,57 мМ). Через 1 ч реакционную смесь нагревали до комнатной температуры и размешивали в течение 90 мин. Полученный бесцветный раствор выпаривали при менее чем 30oC и маслянистый остаток снова растворяли в этилацетате. Полученный раствор один раз промывали 1М соляной кислоты, один раз водой, один раз насыщенным раствором бикарбоната натрия и один раз солевым раствором. Органический слой осушали сульфатом магния, фильтровали и выпаривали. После очистки с помощью флеш-хроматографии (с элюированием смесью (2:5) этилацетата/гексана) получали 223 мг целевого продукта; [α]D = -243,9o (с = 0,62, хлороформ).
d) [S-(R*, R*)] -2,3,4,5-Тетрагидро-3- [(2-меркапто-1-оксо-4-метилпентил)амино] -2-оксо-1H-бензазепин-1-уксусная кислота
Раствор продукта, полученного в части (c), (190 мл, 0,44 мМ) в 4 мл метанола продували 5 мин азотом и охлаждали до 0oC. К этому раствору по капле добавляли 4 мл продутого азотом 1М гидроксида натрия. В процессе реакции, азот медленно барботировали через раствор. Через 1 ч реакцию нагревали до комнатной температуры и размешивали в течение 1 ч. Затем реакцию подкисляли 5%-ным раствором бисульфата калия и экстрагировали этилацетатом, а экстракты промывали водой и насыщенным раствором хлорида натрия, осушали сульфатом натрия и выпаривали. После повторного выпаривания из гексана получали белый твердый остаток. Этот твердый остаток слегка нагревали в этиловом эфире, фильтровали для удаления нерастворившегося маслообразного вещества и кристаллизовали с гексаном, в результате чего получали 117 мг целевого продукта в виде белого маслообразного твердого вещества; т.пл. 143 - 144oC; [α]D = -224,3o (с= 0,46, хлороформ). ТСХ (этилацетат/гексан/уксусная кислота, 4:4:0,1), Rf= 0,19.
Анализ для C18H24N2O4S • 0,02 C4H10O:
Вычислено: C 59,34; H 6,67; N 7,66; S 8,76;
Найдено: C 58,97; H 6,72; N 7,52; S 8,67.
Пример 60.
[R - (R*, S*)] -2,3,4,5-Тетрагидро-3- [(2-меркапто-2-метил-1-оксо-фенилпропил)амино]-2-оксо-1H-бензазепин -уксусная кислота
a) S,S'-Бис(4-метокси- α- толуолтиол)
Раствор 4-метокси- α- толуолтиола (25 г, 0,16 М) в бензоле (200 мл) добавляли к раствору карбоната калия (44,8 г, 0,32 М) в воде (200 мл) и полученную смесь обрабатывали порциями, быстро размешивая при этом, иода (23,6 г) до тех пор, пока не установится стойкая окраска иода. Реакционную смесь размешивали 15 мин, после чего избыточное количество иода нейтрализовали путем добавления твердого сульфита натрия (10 г). Реакционную смесь разбавляли бензолом (200 мл) и распределяли, снова экстрагируя водную фазу еще 100 мл бензола. Объединенные органические экстракты промывали водой (100 мл), 5% сульфитом натрия (100 мл) и солевым раствором (50 мл), затем осушали безводным сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт перекристаллизовывали из этилацетата (200 мл) и фильтровали, промывая осадки кремового цвета этилацетатом (25 мл), в результате чего получали 16,596 г целевого продукта; т.пл. 99 - 100oC, ТСХ (метиленхлорид: метанол, 9:1), Rf=0,57.
b) α- Метил-гидрокоричная кислота
Раствор α- метилкоричной кислоты (10,0 г, 61,7 мМ) в сухом метаноле (250 мл) обрабатывали 10% палладированным углем и гидрировали (с использованием баллона) при комнатной температуре в течение 16 ч. Реакционную смесь разбавляли метанолом (250 мл), фильтровали через прокладку из целита в миллипористом фильтре, тщательно промыв указанную прокладку метанолом (2•100 мл).
прозрачный фильтр выпаривали досуха и получали 10,225 г целевого продукта в виде густого сиропа.
c) α- Метил- α- [[(4-метоксифенил)метил)тио] -гидрокоричная кислота
Раствор диизопропиламина (1,69 мл, 12,2 мМ) в сухом тетрагидрофуране (9,0 мл) охлаждали до -30oC (в бане из ацетонитрила и сухого льда), обрабатывали 2,5 М бутиллития (4,84 мл, 12,1 мМ) и размешивали 20 мин при -30oC. Полученный раствор диизопропиламида лития обрабатывали раствором α- метил-гидрокоричной кислотой (1,0 г, 6,1 мМ) в сухом тетрагидрофуране (1,0 мл), нагревали до комнатной температуры и размешивали в атмосфере аргона 1,5 ч. Затем реакционную смесь охлаждали до 0oC (в ледяной-солевой бане), обрабатывали раствором S,S'-бис(4-метокси- α- толуолтиола) (1,869 г, 6,1 мМ) в сухом тетрагидрофуране (6,0 мл) и размешивали 45 мин при 0oC в присутствии аргона. После этого смесь медленно добавляли к 1,0 н. соляной кислоте (19,0 мл) и полученную водную смесь экстрагировали этилацетатом (2•250 мл). Объединенные органические экстракты промывали водой (15 мл) и солевым раствором (15 мл), осушали безводным сульфатом магния, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенную продуктовую смесь хроматографировали на силикагеле (Merck), элюируя смесями этилацетата и гексана (1:4, 1:2), а затем смесью этилацетата и уксусной кислоты (100:2), в результате чего получали 1,45 г целевого продукта.
d) [R-(R*, S*)] -2,3,4,5-Тетрагидро-3-[[2-[[(4-метоксифенил) метил]-тио] -2-метил-1-оксо-3-фенилпропил] амино] -2-оксо-1H-бензапин-1-уксусная кислота, 1,1-диметилэтиловый сложный эфир и [S- (R*, R*)]-2,3,4,5,тетрагидро-3-[[2-[[(4-метоксифенил)метил] тио] -2-метил-1-оксо-3-фенилпропил]амино]-2-оксо-1H-бензазепин-1- уксусная кислота, 1,1-диметилэтиловый сложный эфир
Раствор продукта, полученного в части (c), (497 мг, 1,57 мМ, 1,09 экв.) охлаждали до 0oC (в ледяной/солевой бане), обрабатывали раствором (S)-2,3,4,5-тетрагидро-3-амино-2-оксо-1H- бензазепин-1-уксусной кислоты, 1,1-диметилэтилового сложного эфира, полученного в соответствии с известной процедурой (Watthey и др., J. Med. Chem., 28, c. 1511-1516), (417 мг, 1,44 мМ) в сухом метиленхлориде (2,9 мл), а затем обрабатывали триэтиламином (0,2 мл, 1,45 мл) и гексафторофосфатом бензотриазол-1-илокситрис диметиламино фосфония (642 мг, 1,45 мМ). Реакционную смесь размешивали 1 ч при 0oC, а затем 2,5 ч при комнатной температуре, после чего смесь выпаривали досуха. Полученный сироп снова растворяли в этилацетате (50 мл) и раствор промывали 0,5 н. соляной кислотой (2•8,3 мл), водой (8,3 мл) и солевым раствором (8,3 мл), осушали безводным сульфатом натрия, фильтровали, выпаривали досуха и осушали в вакууме. Неочищенный продукт хроматографировали на колонке с силикагелем (Merck), элюируя смесью этилацетата и гексана (1:4), в результате чего получали 709 мг смеси изомерных продуктов.
После повторной хроматографии смеси изомерных продуктов на колонке с силикагелем с использованием в качестве элюента смеси этилацетата и метиленхлорида (1: 99, 2: 98) получали 340 мг целевого [R-(R*, S*)]-продукта; [α]D = -72o (с = 0,66, метанол); ТСХ (этилацетат:гексан, 1:1), Rf= 0,70 и 316 мг целевого [S-(R*, R*)]-продукта; [α]D = -142o (с = 0,5, метазол); ТСХ (этилацетат: гексан, 1:1) Rf=0,87.
e) [R- (R*, S*)] -2,3,4,5-Тетрагидро-3- [(2-меркапто-2-метил-1-оксо-3-фенилпропил)амино] -2-оксо-1H-бензазепин-1-уксусная кислота
Раствор (R-, R*, S*)-продукта из части (d) (300 мг, 0,51 мМ) в трифтороуксусной кислоте (2,5 мл) охлаждали до 0oC (в ледяной-солевой бане), обрабатывали анизолом (0,25 мл, 2,3 мМ) и трифторометансульфоновой кислотой (0,13 мл, 1,47 мМ) и размешивали при 0oC в присутствии аргона в течение 2,5 ч. Реакционную смесь упаривали досуха, выпаривая остаточный сироп из этилацетата (2•15 мл) и осушали в вакууме. Неочищенную продуктовую смесь хроматографировали на колонке с силикагелем (Merck), элюируя этилацетатом и смесью этилацетата и уксусной кислоты (100:2). Первую партию нужных фракций объединяли и распределяли между этилацетатом и 1,0 н. соляной кислотой, в результате чего получали 91,0 мг неочищенного продукта. Этот продукт объединяли со второй партией нужных фракций, которые также были отдельно хроматографированы на другой колонке с силикагелем, в результате чего получали всего 301 мг продукта, который все еще содержал примеси. После повторной хроматографии на третьей колонке с силикагелем с элюированием смесью толуола: уксусной кислоты, 6:1 получали 156 мг чистого целевого продукта в виде твердого пенообразного вещества; [α]D = -248,3o (c = 0,73, метанол). ТСХ (толуол: уксусная кислота: 5:1), Rf = 0,23.
Анализ для C22H24N2O4S• 0,21 C6H14•0,54 H2O:
Вычислено: C 63,82; H 5,85; N 6,40; S 7,32;
Найдено: C 63,82; H 6,14; N 6,21; S 7,16.
Пример 61.
[S-(R*, R*)]-2,3,4,5-Тетрагидро-3-[(2-меркапто-2-метил-1-оксо-3- фенилпропил)амино]-2-оксо-1H-бензазепин-1-уксусная кислота
Раствор [S-(R*, R*)]-продукта примера 60, часть (d) (316 мг, 0,54 мМ) в трифторуксусной кислоте (2,63 мл) охлаждали до 0oC и обрабатывали анизолом (0,26 мл, 2,30 мМ) и трифторметансульфоновой кислотой (0,14 мл, 1,58 мМ) в соответствии с процедурой, описанной в примере 60, части (e), в результате чего получали 209,8 мг целевого продукта; [α]D = -123o (c = 0,48, метанол). ТСХ (толуол: уксусная кислота, 5:1), Rf = 0,35.
Анализ для C22H24N2O4S• 0,4 C6H14:
Вычислено: C 64,22; H 5,95; N 6,73; S 7,70;
Найдено: C 64,38; H 6,19; N 6,27; S 7,05.
Пример 62.
[3R- [3α, 6α(S*), 9αβ] ]-Октагидро-6-[(2-меркапто-1-оксо-3-фенилпропил] амино]- 5-оксотиазоло[3,2-a]азепин-3-карбоновой кислоты 1-оксид
a) [3R- [3α, 6α(S*), 9αβ] ]-Октагидро-6-[[2-(ацетилтио)-1-оксо-3- фенилпропил] амино] -5-оксотиазоло[3,2-a] азепин-3-карбоновой кислоты 1-оксид, метиловый сложный эфир
Раствор продукта примера 11, части (f), (301 мг, 0,67 мМ) в хлороформе (9,5 мл) охлаждали до 0 - 5oC. К этому раствору добавляли раствор мета-хлорпероксибензойной кислоты (137 мг, 0,79 мМ) в хлороформе (7 мл). Реакционную смесь размешивали при 0oC в присутствии азота. Через 1 ч ТСХ показала отсутствие исходного материала. Реакционную смесь разбавляли хлороформом (66 мл), промывали разбавленным бикарбонатом натрия (2•13 мл) (pH 10), водой (27 мл) и осушали сульфатом натрия. Затем смесь фильтровали и концентрировали в вакууме, в результате чего получали пенообразное твердое вещество. Это твердое вещество подвергали флеш-хроматографии на силикагеле [Merck (30 мл), используя смесь этилацетата и ацетонитрила (95:5) (400 мл)] и получали 263 мг целевого продукта в виде 55: 45 - смеси диастереомеров (R,S - смесь сульфоксида). ТСХ (этилацетат:ацетонитрил, 95:5), Rf = 0,23.
b) [3R- [3α, 6α(S*), 9αβ] ]-Октагидро-6-[(2-меркапто-1-оксо-3-фенилпропил)амино]- 5-оксотиазоло[3,2-a]азепин-3-карбоновой кислоты 1-оксид
Раствор продукта, полученного в части (a), (263 мг, 0,56 мМ) в метаноле (3,4 мл), дезоксигенированном посредством барботирования аргона, обрабатывали 1 н. гидроксидом натрия (2,8 мл, 2,8 мМ), дезоксигенированным посредством барботирования аргона, и прозрачный раствор размешивали при комнатной температуре и в присутствии аргона. Через 2 ч ТСХ показала отсутствие исходного материала. Раствор подкисляли 10% бисульфатом калия (12,5 мл), разбавляли водой (17 мл) и экстрагировали этилацетатом (3•60 мл). Этилацетатный экстракт промывали водой (25 мл), солевым раствором (50 мл) и осушали сульфатом натрия. После фильтрации и концентрирования получали маслянистый остаток. Этот остаток обрабатывали гексаном и этиловым эфиром и получали твердое вещество (230 мг). Полученное твердое вещество подвергали флеш-хроматографии на силикагеле (Merck, 50 мл), используя смесь этилацетата/ацетонитрила/уксусной кислоты (50:50:6-7, 600 мл) и получали 142 мг твердого вещества. Это вещество (142 мг) растворяли в метаноле (2 мл) и разбавляли этилацетатом (25 мл). Затем раствор промывали 10% бисульфатом калия (3 мл, pH 2) и разводили водой (8 мл). Органический слой отделяли. Водный слой экстрагировали этилацетатом (3•10 мл). Объединенный этилацетатный экстракт промывали водой (5 мл), солевым раствором (5 мл), осушали сульфатом натрия, фильтровали и концентрировали, в результате чего получали маслянистый остаток. После растирания этого остатка с этилацетатом/этиловым эфиром, получали 102 мг целевого продукта в виде одного изомера; т.пл. 129 - 132oC (пенист.). [α]D = -89,3o (C = 0,3, диметилформамид). ТСХ (этилацетат/ацетонитрил/уксусная кислота), Rf = 0,26.
Анализ для C18H22N2O5S2• 0,12 C4H8O2•0,56 H2O:
Вычислено: C 51,48; H 6,62; N 6,49; S 14,87;
Найдено: C 51,32; H 5,34; N 6,74; S 14,50.
Активность in vitro соединений примеров определяли согласно следующим методикам.
Ингибирующую активность по отношению к ангиотензинпревращающему ферменту (АПФ) определяли в соответствии с известным способом (Cushman et al., Biochem. Pharmacol., т. 20, с. 1637-1648 (1971)). В качестве источника АПФ использовали экстракт легких кролика и фиксировали скорость образования гиппуровой кислоты из субстрата гиппурил-L-гистидин-L-лейцина (ГГЛ). После 30 мин инкубирования тестируемого соединения с ферментом и субстратом реакцию прекращали добавлением HCl. Добавляли этилацетат. Слой этилацетата удаляли и выпаривали до получения сухого продукта, образовавшуюся гиппуровую кислоту вновь растворяли в воде и спектрофотометрически определяли ее концентрацию. Определяли концентрацию тестируемого соединения, которая ингибировала реакцию на 50% (ИК50).
Активность в отношении нейтральной эндопептидазы почек (НЭПП) определяли, сравнивая ее с очищенной нейтральной эндопептидазой почек крысы с помощью флюорометрического анализа. Этот анализ основан на расщеплении флюоресцентного дансилата трипептида Дис-Гли-Фен-Арг до Дис-Гли, последний экстрагируется из смеси, подкисленной этилацетатом и является сильным флюоресцентом. Тестируемые соединения предварительно инкубировали в течение 10 мин с ферментным препаратом до того, как начинали реакцию путем добавления субстрата. Через 30 мин инкубирования реакцию прекращали добавлением HCl. Дис-Гли экстрагировали в этилацетат. Относительную флюоресцентную интенсивность экстракта измеряли с помощью спектрометра. Определяли концентрацию тест-соединения, которая ингибировала реакцию на 50 % (ИК50) ( см. таблицу).
Определяли также ингибирующую активность in vivo по отношению к АПФ в соответствии с модификацией известного способа (Rubin et al., J. Pharmacol. Exsper., т. 204, с. 271-280 (1978)). Самцам крыс Sprague-Dawley устанавливали катетеры в брюшную аорту и полую вену. Спустя две недели или более у ненаркотизированных крыс производили фиксацию давления крови в аорте прямым способом. Прессорные реакции на в/в инъекции ангиотензина 1 (A1) в дозе 310 мг/кг получали до (контроль) и с 2-30 мин интервалами, вплоть до 6 ч, после в/в введения тестируемых соединений. Для каждого интервала определяли ингибирование прессорной реакции на A1 (в % изменения по сравнению с контролем). Кривую ответа на дозу строили, используя точку времени после каждой дозы, в которой отмечалось максимальное ингибирование. Среднюю эффективную дозу (ЭД50) определяли путем интерполяции.
Тест-соединение - Прессор A1, в/в ЭД50
1 - 3,96
2 - 0,143
5 - 0,517
6 - 0,020
9 - Более 5
10 - 0,838
11 - 0,071
12 - 0,799
13 - 5,84
14 - 0,262
15 - 1,22
17 - 4,4
18 - 0,49
20 - Более 1,5
21 - Менее 1,5
26 - 0,27
31 - 1,2
32 - 0,015
33 - 2,14
35 - 0,12
36 - 0,04
37 - 0,06
42 - 0,27
43 - 0,33
44 - 1,34
45 - 13,3
48 - 0,13 (короткая длительность)
50 - 0,06
51 - 0,08
52 - 0,025 (хорошая длительность)
55 - 0,12
59 - 0,28 (хорошая длительность)
62 - 0,11
Пример 63. 1000 таблеток, каждая из которых содержала следующие ингредиенты:
[S-(R*, R*)-2,3,4,5-Тетрагидро-3-[(2-меркапто-1-оксо-3- фенилпропил)амино]-2-оксо-1H-бензазепин-2-уксусная кислота - 200 мг
Кукурузный крахмал - 100 мг
Желатин - 20 мг
Авицел (микрокристаллическая целлюлоза) - 50 мг
Стеарат магния - 5 кг - 375 мг
получили из достаточного объемного количества путем смешивания продукта примера 6 и кукурузного крахмала с водным раствором желатина. Полученную смесь осушали и измельчали до получения тонкодисперсного порошка. Затем к смеси подмешивали авицел, а после этого стеарат магния и смесь гранулировали. После этого смесь прессовали в таблетки (всего 1000 таблеток), каждая из которых содержала 200 активного ингредиента.
Аналогичным образом могут быть получены таблетки, содержащие 200 мг продукта, полученного в соответствии с процедурой, описанной в любом из примеров 1-5 и 7-62.
Аналогичные процедуры могут быть использованы для получения таблеток или капсул, содержащих 50 - 500 мг активного ингредиента.
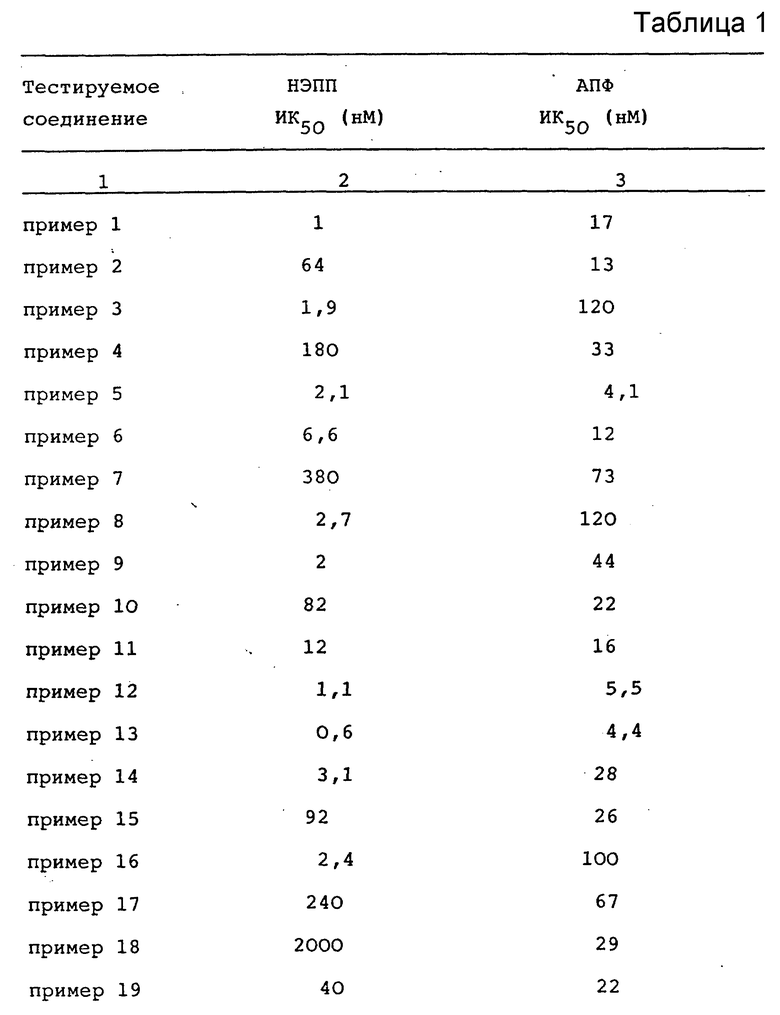


Предложены гетероциклические азотсодержащие производные карбоновой кислоты или их соль формулы I, где R1 - атом водорода, группа R3(C=O)- и группа R18S-; R2 и R19 - атом водорода, C1-C7-алкил, группа арил-(CH2)m-, где арил - фенил или нафталенил; группа гетероарил-(CH2)n-, где гетероарил - тиенил; n = 0 или 1; m = 0 или целое число от 1 до 6; R3 - C1-C7-алкил, арил-(CH2)m-, где арил - фенил; R18 - C1-C7-алкил; X1 - группа формулы III, где R4 и R5 - атом водорода, R6, R7 - атом водорода и др. или X1 - группа формулы IV, где R6, R7, R8 и R9 - атом водорода или фенил-(CH2)m-, где m имеет указанные значение, S = 0, r = 1 или X1 - группа формулы V или группа формулы VI, где Y1 - атом кислорода, серы или группа -CH2- и -CH2-CH2- или X1 - группа формулы VIII или IX, где Y2 - атом серы или группа -CH2- и 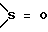 v = 1 или 2; w = 1 или 2; или X1 - группа формулы XI, где Z - атом кислорода, v = 1 или 2 или X1 - группа формулы XII, где Z - атом кислорода или два атома водорода, или X1 - группа формулы XIII, где Y3 - -CH2-, R13 - атом водорода. Соединения формулы I получают взаимодействием ацилмеркаптокарбоновой кислоты формулы XIV с промежуточным соединением формулы XV : H - X1, с получением соединения формулы XVI и затем удаляют ацильную группу R3 - (C= O)- и сложноэфирную защитную группу R12. Предложена фармацевтическая композиция, обладающая активностью, ингибирующей антиотензин-конвертирующий фермент и ингибирующей нейральную эндопептидазу, которая содержит соединения формулы I и фармацевтически приемлемый носитель. 3 с. и 5 з.п.ф-лы.
v = 1 или 2; w = 1 или 2; или X1 - группа формулы XI, где Z - атом кислорода, v = 1 или 2 или X1 - группа формулы XII, где Z - атом кислорода или два атома водорода, или X1 - группа формулы XIII, где Y3 - -CH2-, R13 - атом водорода. Соединения формулы I получают взаимодействием ацилмеркаптокарбоновой кислоты формулы XIV с промежуточным соединением формулы XV : H - X1, с получением соединения формулы XVI и затем удаляют ацильную группу R3 - (C= O)- и сложноэфирную защитную группу R12. Предложена фармацевтическая композиция, обладающая активностью, ингибирующей антиотензин-конвертирующий фермент и ингибирующей нейральную эндопептидазу, которая содержит соединения формулы I и фармацевтически приемлемый носитель. 3 с. и 5 з.п.ф-лы. 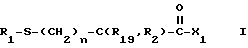
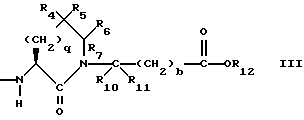
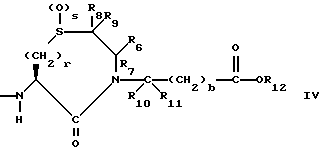
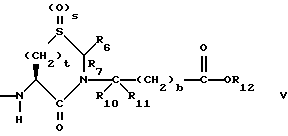
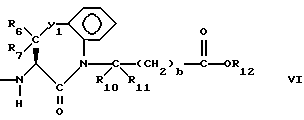
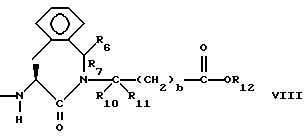
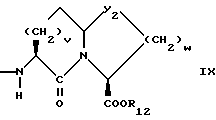
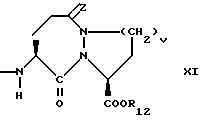
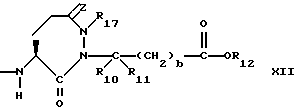
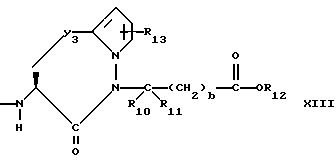
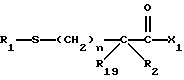
и их фармацевтически приемлемые соли,
где R1 представляет собой атом водорода, группу 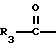 и группу R18-S-;
и группу R18-S-;
R2 и R19 независимо друг от друга выбирают из атома водорода, С1-С7-алкила, группы арил -(CH2)m -, где арил обозначает фенил или нафталенил, и группы гетероарил -(CH2)m-, где гетероарил означает тиенил;
n равно 0 или 1;
m равно 0 или целому числу от 1 до 6;
R3 представляет собой С1-С7-алкил, арил -(CH2)m-, где арил обозначает фенил;
R18 представляет собой С1-С7-алкил;
X1 выбирают из ряда, содержащего группу общей формулы III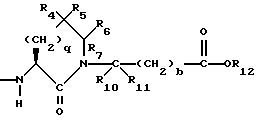
где R4 и R5 обозначают атом водорода; R6 и R7 независимо друг от друга обозначают атом водорода, С1-С7-алкил, возможно замещенный гидроксигруппой, С3-С7-алкенил, С3-С7-циклоалкил-(CH2)m -, где m имеет указанные значения;
q равно 2 или 3;
группу общей формулы IV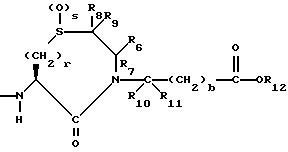
где R6 и R7, R8 и R9 независимо друг от друга обозначают атом водорода или фенил -(CH2)m-, где m имеет указанные значения;
s = 0;
r = 1;
группу общей формулы V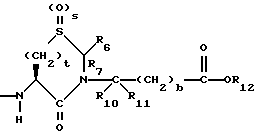
где R6 и R7 независимо друг от друга обозначают атом водорода или фенил -(CH2)m-, где m имеет указанные значения;
s = 0;
t = 1;
группу общей формулы VI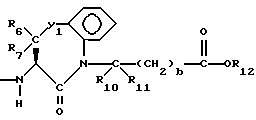
где R6 и R7 независимо друг от друга обозначают атом водорода и С1-С7-алкил;
Y1 обозначает атом кислорода, серы или группы -CH2- и -CH2-CH2-;
группу общей формулы VIII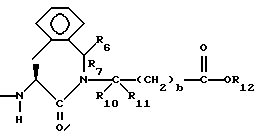
где R6 и R7 - водород;
группу общей формулы IX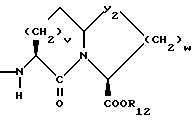
где Y2 обозначает атом серы или группы -CH2- и 
v равно 1 или 2;
w равно 1 или 2;
группу общей формулы XI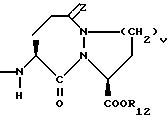
где Z обозначает атом кислорода или два атома водорода;
v равно 1 или 2;
группу общей формулы XII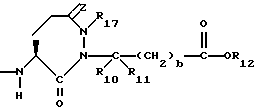
где Z обозначает атом кислорода или два атома водорода;
R17 обозначает С1-С7-алкил;
группу общей формулы XIII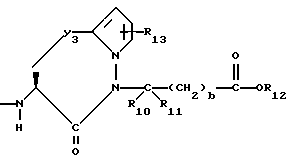
где Y3 обозначает группу -CH2-;
R13 обозначает атом водорода;
где R10 и R11 независимо друг от друга обозначают атом водорода С1-С7-алкил или фенил -(CH2)m-, где m имеет указанные значения;
R12 обозначает атом водорода;
b равно 0 или 1.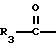 и группу R18-S-; R3 представляет собой C1-С4-алкил или фенил; R18 представляет собой С1-С4-низший алкил; n = 0 или 1; R2 представляет собой арил -CH2-, где арил обозначает фенил или нафталенил, гетероарил -CH2-, где гетероарил обозначает тиенил С1-С7-алкил; R19 представляет собой водород.
и группу R18-S-; R3 представляет собой C1-С4-алкил или фенил; R18 представляет собой С1-С4-низший алкил; n = 0 или 1; R2 представляет собой арил -CH2-, где арил обозначает фенил или нафталенил, гетероарил -CH2-, где гетероарил обозначает тиенил С1-С7-алкил; R19 представляет собой водород.
[S-(R*, R*)]-2,3,4,5-тетрагидро-3-[(2-меркапто-1-оксогексил)-амино]-2-оксо-1H-бензазепин-1-уксусная кислота;
[S-(R*, R*)] -2,3,4,5-тетрагидро-3-[(2-меркапто-1-оксо-4-метилпентил)амино] -2-оксо-1H-бензазепин-1-уксусная кислота; [S-(R*, R*)]-2,3,4,5-тетрагидро-3-[(2-меркапто-1-оксо-3-фенилпропил)амино] -2-оксо-1H-бензазепин-1-уксусная кислота; [1S-[1α, 9α(R*)]]-октагидро-9-[-(2-меркапто-1-оксо-3-(2-тиенил)пропил)амино] -10-оксо-6H-пиридазино[1,2-a] [1,2]диазепин-1-карбоновая кислота; [1S-[1α, 9α(R*)]]-октагидро-9-[(2-меркапто-1-оксо-3-фенилпропил)амино] -10-оксо-6H-пиридазино[1,2-a] [1,2]-диазепин-1-карбоновая кислота; [1S-[1α, 9α(R*)] ] -октагидро-9-[(2-меркапто-1-оксо-3-фенилпропил)амино] -6,10-диоксо-6H-пиридазино[1,2-a][1,2]-диазепин-1-карбоновая кислота; [4S-[4α, 7α(R*), 10aβ] ] -декагидро-7-[(2-меркапто-1-оксо-3-фенилпропил)амино] -6-оксопиридо[1,2-a]азепин-4-карбоновая кислота; [3R-[3α, 6α(S*), 9aβ] ] -октагидро-6-[(2-меркапто-1-оксо-3-фенилпропил)амино] -5-оксотиазол[3,2-a] азепин-3-карбоновая кислота; [3S-[1(R*), 3α(R*), 7β]]-гексагидро-α-метил-3-[(2-меркапто-1-оксо-3-фенилпропил)амино] -2-оксо-7-пропил-1H-азепин-1-уксусная кислота; [6(S)] -гексагидро-6-[(2-меркапто-1-оксо-3-фенилпропиламино] -2,2-диметил-7-оксо-1Н-азепин-1-уксусная кислота; [3S-[3α( R*), 7β] ]-гексагидро-3-[(2-меркапто-1-оксо-3-фенилпропил)амино]-2-оксо-7-пропил-1Н-азепин-1-уксусная кислота; и [3S-[3α(R*), 7β]]-гексагидро-3-[(2-меркапто-1-оксо-3-фенилпропил)амино] -2-оксо-7-(2-пропенил)-1Н-азепин-1-уксусная кислота.
где R1, R2, R19, X1 и n определены в п.1,
отличающийся тем, что если R1 является водородом или 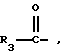 а R19 является водородом, то ацилмеркаптокарбоновую кислоту формулы
а R19 является водородом, то ацилмеркаптокарбоновую кислоту формулы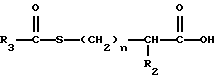
или активированную форму кислоты формулы XIV подвергают реакции сочетания с промежуточным соединением формулы XV
H - X1,
в результате чего получают продукт формулы XVI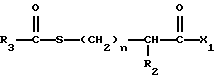
где R12 в определении X1 является легко удаляемой сложноэфирной защитной группой,
а затем удаляют ацильную группу  и сложноэфирную защитную группу R12.
и сложноэфирную защитную группу R12.
| УСТРОЙСТВО ДЛЯ МЕХАНИЗИРОВАННОГО ВСКРЫТИЯ И ЗАКРЫТИЯ ЛЕТКИ | 0 |
|
SU254032A1 |
| Способ обработки волокон при прядении льна, джута и др. лубовых волокон | 1925 |
|
SU2624A1 |
| СПОСОБ ПОЛУЧЕНИЯ ХЛОРПИКРИНА | 1949 |
|
SU80012A1 |
| US 5118806 A, 02.01.92 | |||
| DE 3823919 A1, 1989 | |||
| DE 3825368 A1, 1989 | |||
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1988, ч | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ получения твердых неплавких и нерастворимых продуктов уплотнения формальдегида с фонолами | 1925 |
|
SU435A1 |

