Изобретение было разработано при поддержке Национального института здравоохранения, Национального института рака (разрешение N 5 P37 CA22215). В соответствии с этим правительство США имеет определенные права на это изобретение.
Данная заявка является частичным продолжением заявки на патент США рег. N 07/849769, поданной 12 марта 1992 г.
Изобретение относится к галихондринам, и к родственным им соединениям, таким, как синтетические промежуточные соединения галихондринов, а также к производным указанных промежуточных соединений.
Галихондрины представляют собой класс полиэфирных макролидов, первоначально выделенных из морских губок Halichondria okadai Kadota. Примерами галихондринов являются галихондрин B, норгалихондрин B и гомогалихондрин B, структуры которых были полностью выявлены. Галихондрины обладают исключительно высокой in vitro и in vivo - противоопухолевой активностью. Однако очень ограниченные запасы галихондринов в природе не позволяют полностью оценить их возможное клиническое применение.
В одном из своих вариантов изобретение относится к новым соединениям, которые могут быть использованы для синтеза галихондрина B и норгалихондрина B, а также к производным этих новых соединений.
Один класс соединений по изобретению имеет следующую формулу:
где каждый из R1 и R2 представляет собой H- или C1-C10-алкил, алкенил или алкинил (предпочтительно, C1-C6-алкил, например, метил); а каждый из A и B представляет собой H-, HO- со спиртозащитной группой или без таковой; незамещенный углеводород, или замещенный углеводород со спиртозащитной группой или без таковой; где полное число атомов углерода в A и B составляет 0 - 18 (без учета атомов углерода в любых спиртозащитных группах, если они присутствуют); и каждый из m и n является 0 - 3 (предпочтительно 1). Если оба A и B являются замещенными или незамещенными углеводородами, то они могут быть соединены вместе в одном или нескольких положениях.
Следует отметить, что линия между двумя атомами в структурных формулах, приведенных в описании, означает простую связь, если это не оговорено особо.
Термин "замещенный углеводород" или "замещенный алкил" означает углеводород или алкил, который содержит функциональную группу, такую, как галоген, кето, сложноэфирная группа, аминогруппа, или спиртовая группа (-OH). Если функциональной группой является -OH, то эта группа может быть защищена спиртозащитной группой. Примерами спиртозащитных групп служат, но не ограничиваются ими, следующие группы: R5-O-, R5-CO-O-,R5-O-CO-O-, или 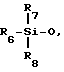 , где R5 представляет собой C1-C10-алкил, C2-C10-алкенил, C2-C10-алкинил, C7-C20-аралкил, C7-C20-алкарил, фенил, или тетрагидропиранил, а каждый из R6, R7 и R8 представляет собой C1-C6-алкил, C2-C6-алкенил или фенил. При этом следует отметить, что кислород, присутствующий в правом конце перечисленных спиртозащитных групп, является тем же самым кислородом, который присутствует в защищаемой -OH-группе.
, где R5 представляет собой C1-C10-алкил, C2-C10-алкенил, C2-C10-алкинил, C7-C20-аралкил, C7-C20-алкарил, фенил, или тетрагидропиранил, а каждый из R6, R7 и R8 представляет собой C1-C6-алкил, C2-C6-алкенил или фенил. При этом следует отметить, что кислород, присутствующий в правом конце перечисленных спиртозащитных групп, является тем же самым кислородом, который присутствует в защищаемой -OH-группе.
Ниже приводятся примеры четырех спиртозащитных групп: p-метоксифенилметил (MePhCH2O- или "MPM"), 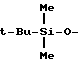 (т-бутилдиметилсилил или "TBS"), пивалоил ("Py") и CH2-CH-CH2-O-CO-O-.
(т-бутилдиметилсилил или "TBS"), пивалоил ("Py") и CH2-CH-CH2-O-CO-O-.
Предпочтительно, чтобы каждый из A и B в указанной формуле представлял собой HO-; HO-группу, связанную со спиртозащитной группой; или замещенный углеводород, выбранный из группы, состоящей из R3-CO-R4-; R3-CH(OH)-R4; R3-CH(OH)-R4-, связанной со спиртозащитной группой; R3-O-CO-; R3-O-CO-R4; HO-R4-; и HO-R4-, связанной со спиртозащитной группой, где каждый из R3 (одновалентный) и R4 (двухвалентный) представляет собой алкил, алкенил или алкинил. Предпочтительно также, чтобы полное число атомов углерода в A и B составляло 0 - 15 или 0 - 12 (без учета углерода, присутствующего в спиртозащитных группах, если они присутствуют), а каждый из m и n составлял 0 - 2.
В некоторых предпочтительных соединениях рассматриваемого класса каждый из A и B представляет собой HO-; HO-, связанную со спиртозащитной группой; HO-R4-; или HO-R4-, связанную со спиртозащитной группой. Особенно предпочтительно, чтобы A представлял собой HO-R4- или HO-R4-связанную со спиртозащитной группой.
В соединении, заявленном в п. I формулы изобретения, стереохимия C.25, C.31, C.35 и C.36 представляет собой либо R, S, R и R, либо S, R, S и S, соответственно.
Изобретение также относится к способу введения терапевтически эффективного количества одного или нескольких описанных соединений в целях ингибирования роста опухоли (например, меланомы, фибросаркомы, моноцитарного лейкоза, карциномы толстой кишки, карциномы яичника, карциномы молочной железы, остеосаркомы, или ras-трансформирующей фибробластомы) у млекопитающих.
Другим классом соединений по изобретению являются соединения, имеющие формулу: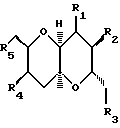
где R1 представляет собой H или C1-C10-алкил (например, метил), каждый из R2 и R4 представляет собой -OH-группу, или защищенную -OH-группу; R3 представляет собой 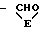 -CH2-B, -CO-O-D, или -CH-O; а R5 представляет собой -CHO, -CH2-B, или -CO-O-D, B является -OH-группой или защищенной -OH-группой, D является -H или C1-C10-алкилом, а E является замещенным или незамещенным C1-C30 или C1-C10-алкилом. Например, R2 может представлять собой -OMPM, R4 может представлять собой -OTBS, а R5 может представлять собой -CH2-OTBS (т. е., -CH2-B, где B является -OTBS). Предпочтительно, R4 является TBSO-, а R5 является TBSO-CH2-.
-CH2-B, -CO-O-D, или -CH-O; а R5 представляет собой -CHO, -CH2-B, или -CO-O-D, B является -OH-группой или защищенной -OH-группой, D является -H или C1-C10-алкилом, а E является замещенным или незамещенным C1-C30 или C1-C10-алкилом. Например, R2 может представлять собой -OMPM, R4 может представлять собой -OTBS, а R5 может представлять собой -CH2-OTBS (т. е., -CH2-B, где B является -OTBS). Предпочтительно, R4 является TBSO-, а R5 является TBSO-CH2-.
В одном примере соединений этого класса, R3 имеет следующую формулу: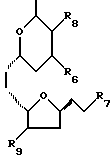
где R6 представляет собой -H или C1-C10-алкил (например, метил); R7 представляет собой -COH, -CH2-B, или -CO-O-D; а каждый из R8 и R9 представляет собой -H, метилиден (т.е., =CH2), C1-C5-алкил (например, метил) или C2-C5-алкенил. Предпочтительно, чтобы R1 представлял собой метил, а R5 представлял собой CH2-B. Линия между R8 (или R9) и атомом на кольце, с которым он связан, означает либо простую связь (если R8/R9 является H), либо двойную связь (если R8/R9 является =CH2).
Другим примером соединений этого класса являются соединения формулы: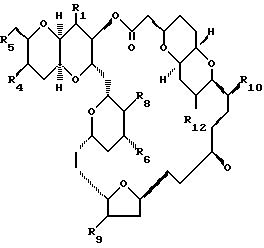
где каждый из R10, R11 и R12 является -OH или защищенной -OH. Предпочтительно, чтобы каждый из R1 и R6 представлял собой метил, R4 представлял собой защищенную -OH-группу, R5 представлял собой -CH2-B, а каждый из R8 и R9 представлял собой метил или метилиден. Особенно предпочтительно, чтобы R4 представлял собой TBSO-, а R5 представлял собой TBSO-CH2-.
Еще одним примером соединений этого класса являются соединения формулы: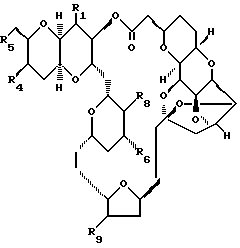
В этих соединениях предпочтительно, если каждый из R1 и R6 являются метилом, R4 является защищенной -OH-группой (например, TBSO-), R5 является -CH2-B (например, TBSO-CH2-), а каждый из R8 и R9 является метилом или метилиденом.
Еще одним классом соединений изобретения являются соединения следующей структуры.
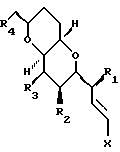
где X представляет собой галоген (например, -I или -Br) или активированный спирт (например, -OSO2CF3); каждый из R1, R2 и R3 представляет собой -OH или защищенную OH; а R4 представляет собой -CHO, -CH2-B, или -CO-O-D; где B является -OH или защищенной -OH, и D является -H или C1-C10-алкилом. Предпочтительно, если каждый из R1, R2 и R3 представляет собой -OTBS, а R4 представляет собой -CO-OCH3.
Еще одним классом соединений изобретения являются соединения следующей структуры: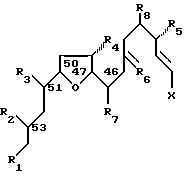 где каждый из R1, R2, R3, R4 и R5 представляет собой -OH или защищенную -OH; R6 представляет собой O или защищенный кетон; каждый из R7 и R8 представляет собой -H, C1-C5-алкил, или C2-C5-алкенил; а X представляет собой галоген (-I или -Br) или активированный спирт (например, -OSO2CF3). Примерами подходящих защищенных кетонов по изобретению являются, но не ограничиваются ими,
где каждый из R1, R2, R3, R4 и R5 представляет собой -OH или защищенную -OH; R6 представляет собой O или защищенный кетон; каждый из R7 и R8 представляет собой -H, C1-C5-алкил, или C2-C5-алкенил; а X представляет собой галоген (-I или -Br) или активированный спирт (например, -OSO2CF3). Примерами подходящих защищенных кетонов по изобретению являются, но не ограничиваются ими, 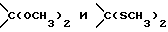 . Углерод у левого конца является углеродом кетона (т.е.,
. Углерод у левого конца является углеродом кетона (т.е., 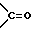 , который требует защиты. В предпочтительном соединении, каждый из R1, R2, R3 и R4 является TBSO-; R5 является -OMPM; R6 является O; каждый из R7 и R8 является метилом, а X является йодом. Стереохимия C.46, C.47, C.50, C.51, и C.53 представляет собой, предпочтительно, S, S, S, S и R, соответственно.
, который требует защиты. В предпочтительном соединении, каждый из R1, R2, R3 и R4 является TBSO-; R5 является -OMPM; R6 является O; каждый из R7 и R8 является метилом, а X является йодом. Стереохимия C.46, C.47, C.50, C.51, и C.53 представляет собой, предпочтительно, S, S, S, S и R, соответственно.
Другим классом соединений изобретения являются соединения следующей структуры: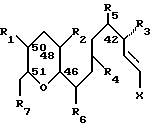
где каждый из R1, R2 и R3 представляет собой -OH или защищенную -OH; R4 представляет собой O или защищенный кетон; каждый из R5 и R6 представляет собой -H или C1-C5-алкил; R7 представляет собой -CH2-B, -CHO или -CO-O, X представляет собой галоген или активированный спирт, где B является -OH или защищенной -OH, а D является -H или C1-C10-алкилом. Стереохимия C.42, C.48 и C. 50 предпочтительно представляет собой S, S и R соответственно. В наиболее предпочтительном соединении, каждый из R1 и R2 является TMSO-; R3 является -OMPM; R4 является O; каждый из R5 и R6 является метилом; R7 является -CO-OCH3; X является йодом; а стереохимия C.46 и C.51 представляет собой S и R соответственно.
Еще одним классом соединений изобретения являются соединения следующей структуры
где каждый из R1, R2, R3, R4, R5 и R6 представляет собой -OH или замещенную -OH; каждый из R8, R9, R10 и R11 представляет собой -H, C1-C5-алкил, или C2-C5-алкенил; каждый из R12 и R13 представляет собой метил или метилиден; каждый из R14 и R15 представляет собой O или защищенный кетон. Стереохимия C.42, C.46, C.47, C.48, C.50, C.51 и C.53 предпочтительно представляет собой S, S, S, S, S, S и R соответственно. В наиболее предпочтительном соединении, каждый из R1, R2, R3, R4 и R5 является TBSO-; R5 является -OMPM; R7 является O или защищенным кетоном; каждый R8, R9, R10 и R11 является -H или C1-C5-алкилом; а каждый из R12 и R13 является метилом или метилиденом.
В объем изобретения также входят соединения формулы:
где каждый из R1, R2, R3 и R4 представляет собой -OH или защищенную -OH; каждый из R5, R6, R7 и R8 представляет собой -H, C1-C5-алкил, или C2-C5-алкенил; каждый из R9 и R10 представляет собой метил или метилиден; R11 представляет собой - CHO, -CH2-B или -CO-O-D; каждый R12 и R13 представляет собой O или защищенный кетон; где B представляет собой -OH или защищенную -OH, а D представляет собой -H или C1-C10-алкил. Стереохимия C.42, C. 48 и C.50 предпочтительно представляет собой S, S и R соответственно. В наиболее предпочтительном соединении, каждый из R1, R2 и R4 представляет собой TBSO-; R3 представляет собой -OMPM; каждый из R5, R6, R7 и R8 представляет собой метил; каждый из R9 и R10 представляет собой метилиден; R11 представляет собой -CO-OCH3.
Изобретение также относится к способу синтеза новых соединений и использования этих соединений в химическом синтезе. Один из наиболее предпочтительных способов предусматривает осуществление Ni(II)/Cr(II)-опосредованной реакции взаимодействия альдегида с винилгалидом. Эта реакция приводит к непосредственному образованию углерод-углеродной связи между химически нестабильными группами. Как будет показано ниже, для синтеза галихондрина B и норгалихондрина B с хорошим выходом может быть использована Ni(II)/Cr(II)-опосредованная реакция с образованием углерод-углеродных связей между C11 и C12, 13 и C14, 26 и C27, C29 и C30, и C38 и C39.
Таким образом, соединения и методы по изобретению дают возможность синтезировать галихондрины с относительно хорошим выходом. Кроме того, изобретение позволяет получить конечные материалы в количествах, достаточных для проведения исследований полного спектра их биологических активностей. Указанный метод позволяет также получать новые соединения в чистом виде.
Другие отличительные особенности и преимущества изобретения будут понятны из нижеследующего подробного описания предпочтительных вариантов его осуществления, иллюстрируемых фиг.1-3, а также из формулы изобретения.
На фиг.1 приведены структуры галихондрина B, норгалихондрина B, и гомогалихондрина B, на фиг.2 - структуры енона галихондрина B и енона норгалихондрина; на фиг. 3 - структуры четырех испытуемых соединений, родственных галихондрину.
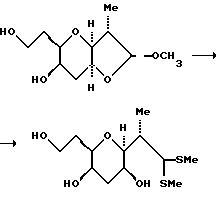
Соединения по изобретению могут быть использованы для синтеза галихондрина B1 и норгалихондрина B2 (структуры которых показаны на фиг.1).
Схему 1 и 2, приведенные в конце текста описания, в совокупности иллюстрируют общий метод синтеза двух указанных соединений. Далее будет показано, что предпочтительные соединения могут быть модифицированы, например, с использованием различных стандартных спиртозащитных групп и той же самой общей схемы для синтеза соединений 1 и 2. Будет также показано, что для синтеза аналогов соединений 1 и 2 исходные соединения могут быть слегка модифицированы (например, путем замещения метильной группы этилом).
Схема 1 иллюстрирует синтез правой половины ряда соединений галихондрина B (т.е., галихондрина B, норгалихондрина B и гомогалихондрина B). Связь C.21 - C.22 (т.е., связь между атомами углерода в положениях 21 и 22) может быть образована в результате реакции окисления первичного спирта 3 (реакция Десс-Мартина) с получением альдегида; реакции Хорнера-Эммонса в условиях строгого контроля; и сопряженной реакции восстановления полученного енона с использованием реагента Страйкера без изомеризации двойной связи. Восстановлением водородом полученного насыщенного кетона дает смесь двух возможных диастереоизомеров в соотношении приблизительно 1:1. Без установления стереохимии диастереомерных спиртов оба диастереомера затем трансформируют отдельно в соответствующие мезилаты, которые используют в последующей реакции сочетания. Однако важно отметить, что указанные два диастереоизомерных спирта легко превращаются друг в друга с помощью реакции Mitsunomu.
В результате Ni(II)/Cr(II)-опосредованной реакции сочетания соединения 5 с соединением 6 получают смесь двух возможных аллиловых спиртов в соотношении приблизительно 6:1, которую затем подвергают реакции циклизации, индуцированной с помощью основания, и получают нужный тетрагидропиран с полным выходом 50 - 60%, и небольшое количество нежелательного диастереомера. Стереохимия в положениях C.23 и C.27 может быть установлена с помощью NOE-экспериментов. Соединение 5 (мезилат) является абсолютно лабильным, очевидно благодаря участию C.20-эфирного кислорода вместе с мезилатной группой. При этом было обнаружено, что соединение 5 является достаточно устойчивым в условиях Ni(II)/Cr(II)-реакции сочетания.
Ni(II)/Cr(II)-опосредованная реакция сочетания C.14-альдегида, полученного из соединения 7, с соединением 8, и последующая реакция окисления Dess-Martin дают транс-енон с полным выходом 77%. После удаления р-метоксифенилметильной ("МРМ") группы в положении С.30 и гидролиза метилового сложного эфира в положении С.1, указанный енон подвергают лактонизации в условиях Yamaguchi, в результате чего получают лактоненон 9 с полным выходом 63%.
Полициклическая кольцевая система вокруг С.8 - С.14 - части четко и эффективно образуется после обработки соединения 9 соединением (n-Bu)4 NF ("TBAF"), а затем p-Ts OH•Py ("PPTS"), с выходом 64%. 1H-ЯМР-спектр показал, что продукт TBAF-стадии является главным образом насыщенным кетоном. Региоселективность реакции Михаэля является исключительно высокой в отношении образования нужного пятичленного кольца, тогда как стереоселективность в отношении нужного диастереомера составляет приблизительно (5-6):1. Нежелательный аддукт реакции Михаэля, выделенный из нужного продукта после PPTS - обработки, может быть затем рециркулирован в TBAF-условиях. И, наконец, после корректировки защитных групп полициклического продукта получают правую половину 10 соединений ряда галихондринов B. Альтернативный синтез полициклической кольцевой системы из соединений 7 и 8 показан ниже (см. пример 2, часть, озаглавленную "Соединение 10").
Теперь рассмотрим схему 2. Реакцию сочетания правой половины 10 галихондрина B с левой половиной II осуществляют с использованием Ni(II)/Cr(II), после реакции окисления Dess-Martin, в результате чего получают ожидаемый транс-енон с полным выходом 60% (схема 2). Затем, в трех стадиях без выделения продукта в каждой стадии, полученный енон превращают в галихондрин B. 1Р-ЯМР-спектр показал, что продукт TBAF-стадии имел частично структуру A. Этот способ предусматривает разблокирование т-бутилдиметилсилильной ("TBS") группы в положении С.48, образование гемикеталя С.48-гидроксильной группой и С. 44-кетоном и присоединение (реакция Михаэля) гидроксильной группы гемикеталя к ненасыщенному α,β -кетону. Затем завершают образование 5,5-спирокеталя путем разблокирования С.41 МРМ-группы с последующей обработкой кислотой. Гидроксильная группа у С.41 должна быть защищена отдельно от других, для того, чтобы избежать образование спирокеталя между гидроксильными группами у С. 41 и С. 48 и С.44-кетоном. Хотя эта трехстадийная трансформация вводит три новых хиральных центра, однако ее стереоселективность является очень высокой. Полный выход описанной трехстадийной трансформации составляет 50 - 50%, а полученный в результате этой трансформации синтетический галихондрин B идентичен натуральному галихондрину B1, что было подтверждено спектроскопическими (1Н-ЯМР, МС, ИК, [α]
Синтез норгалихондрина B2 осуществляют практически тем же способом, что и галихондрин B за исключением того, что гидролиз С.53-метилового сложного эфира необходимо проводить в самой последней стадии синтеза. Полный выход синтеза норгалихондрина B составляет величину, сравнимую с полным выходом галихондрина B. Сравнение данных спектроскопии (1Н-ЯМР, МС, ИК, [α]
Относительно структуры галихондрина B, норгалихондрина B и гомогалихондрина B ранее были сделаны предварительные заключения, которые опирались главным образом на три очевидных факта, а именно: 1) сравнение данных спектроскопии с данными, полученными для структуры норгалихондрина A, которая была совершенно точно установлена с помощью рентгеновского анализа; 2) биогенетические соображения относительно стереохимии галихондрина Bs в положении С. 50 и далее и 3) предположение о том, что абсолютная стереохимия галихондрина Bs идентична стереохимии норгалихондрина A, которая была установлена исходя из экситонной хиральности его С.12, С.13-бис-n-бромбензоатов. Благодаря описанному синтезу была точно установлена относительная и абсолютная стереохимия галихондрина B и норгалихондрина B.
Процедуры полного химического синтеза галихондрина B1 и норгалихондрина B2 более подробно описаны ниже и включают в себя получение различных соединений, входящих в объем изобретения. Кроме того, ниже описаны процедуры для получения других соединения по изобретению, которые являются родственными галихондринам. В описании изобретения представлены также результаты биологических тестов, проиллюстрировавших противоопухолевую активность некоторых синтетических соединений, которые являются родственными галихондринам. Эти результаты показали, что противоопухолевая активность галихондрина Bs может быть в значительной степени отнесена на счет правой половины его молекулы.
При этом следует отметить, что в следующих примерах во избежание повторений не использовали тех же самых или сходных реакционных условий. В любом случае любой специалист исходя из подробных описаний приведенных примеров, может получить соединения по изобретению.
Синтез галихондрина B и норгалихондрина B.
Соединение 1.
Соединение 1, галихондрин B получали из соединений 10 и 11 с помощью следующей процедуры:
(1) Получение C38-альдегида.
К перемешанному раствору C38-спирта (т.е. соединения 10, имеющего 38-углеродный скелет) (8,1 мг, 9,0 мкМ) в метиленхлориде (1,0 мл), при комнатной температуре добавляли твердый NaHCO3 (50 мг), а затем периодинановый реагент Dess-Martin (15 мг, 36 мкМ). Через 30 мин добавляли еще 15 мг (36 мкМ) реагента Dess-Martin. Через 90 мин (всего), ТСХ (этилацетат/гексан, 2:1) указывала на отсутствие эфира (59 мл) и водного раствора насыщенного гидрокарбоната натрия (5 мл), содержащего Na2S2O3 (10 мас.%). Затем полученную двухфазовую смесь перемешивали при комнатной температуре в течение 20 мин. Выделенную органическую фазу еще раз промывали смесью NaHCO3/Na2S2O3 в течение 10 мин, а затем водой (5 мл) и солевым раствором (5 мл). После этого органическую фазу осушали сульфатом натрия, фильтровали через стекловату и концентрировали. Полученный таким образом C38-альдегид (≈ 8 мг, 8,9 мкМ) использовали в последующей стадии без дополнительной очистки.
(2) Получение енона галихондрина B (C38-кетона).
К перемешанному раствору C38-альдегида (8 мг, 8,9 мкМ) и соединения 11 (24 мг, 22 мкМ) в диметилформамиде (≈ 1 мл), в атмосфере азота добавляли порошкообразный CrCl2, содержащий NiCl2 (0,1 мас.%) (всего прибл. 30 мг). После перемешивания при комнатной температуре в течение 11,5 ч, ТСХ (элюент: гексан/этилацетат/CHCl3, 1: 2:1) указывала на отсутствие остаточного альдегида. После этого смесь разбавляли насыщенным водным NH4Cl (10 мл) и H2O (2 мл), а затем экстрагировали этилацетатом (4 • 5 мл). Объединенные этилацетатные экстракты промывали водой (2 • 10 мл), солевым раствором (10 мл), осушали сульфатом натрия, фильтровали через слой стекловаты и концентрировали. Остаток очищали с помощью препаративной ТСХ ("ПТСХ"), элюируя гексаном/этилацетатом/-CHCl3 (1: 2:1), и получали C38-аллиловые спирты (11 мг, 6,0 мкМ, выход 67%) в виде прозрачного бесцветного маслообразного вещества при соотношении диастереомеров приблизительно 1:1.
К перемешанному раствору C38-спиртов (8,0 мг, 4,4 мкМ) в метиленхлориде (2,00 мл) при комнатной температуре добавляли твердый гидрокарбонат натрия (50 мг), а затем периодинановый реагент Dess-Martin (14,8 мг, 35 мкм). После перемешивания при комнатной температуре в течение 1 ч добавляли еще 14,8 мг реагента Dess-Martin. Через 90 мин (всего) реакционную смесь разводили диэтиловым эфиром (6 мл) и водным раствором (10 мл), насыщенным гидрокарбонатом натрия и содержащим Na2S2O3 (10 мас.%). Полученную двухфазовую смесь перемешивали при комнатной температуре в течение 20 мин. Отделенную органическую фазу промывали в течение 10 мин водным NaHCO3/Na2S2O3, водой и солевым раствором (прибл. 10 мл). Органическую фазу осушали сульфатом натрия, фильтровали через стекловату и концентрировали. Остаток очищали с помощью препаративной ТСХ (элюент: гексан/этилацетат, 1:1) и получали енон галихондрина B (7,4 мг, 4,1 мкМ, выход 93%) в виде прозрачного бесцветного маслообразного вещества. Структура этого енонового продукта показана на фиг.2 (верхняя часть).
(3) Получение галихондрина B (соединение 1).
К перемешанному раствору енона (3,7 мг, 2,0 мкМ) в ДМФ (750 мкл) при комнатной температуре добавляли безводный метилацетат (50 мкл), а затем приблизительно 1 моль раствора фторида тетрабутиламмония (TBAF) в ТГФ (25 мкл, pH прибл. 7,6). Полученный раствор перемешивали при комнатной температуре в течение 34 ч и на этот период времени высокоэффективная ТСХ (ВЭТСХ) ([5642-пластины (E. Merck Art)] покрывали реакционным раствором, осушали с помощью высоковакуумного трубопровода в течение 20-30 мин, а затем элюировали этилацетатом/метанолом, 10: 1) показывала мажорное пятно при Rf - 0,53. Затем реакционный раствор фильтровали через 2 см слой силикагеля 60 (230 - 400 меш), добавляя этилацетат для удаления фторида тетрабутиламмония. Фильтрат концентрировали в вакууме и получали желтое маслообразное вещество, которое использовали без дополнительной очистки. 1Н-ЯМР-анализ этого неочищенного продукта не обнаруживал протонного резонанса α,β- -ненасыщенного кетона, но обнаруживал два мажорных продукта в отношении, приблизительно равном 2: 1. Это соединение соответствует соединению, имеющему неполную структуру A (см. схему 2).
Смесь описанного продукта растворяли в смеси CH2Cl2 (1,00 мл), водного фосфатного буфера (Na2HPO4/KH2PO4H2O, pH 7, 100 мкл) и т-бутанола (20 мкл). К полученной быстро перемешанной смеси добавляли 2,3-дихлоро-5,6-дицианобензохинон (1,8 мг, 8 мкМ). После этого смесь обрабатывали ультразвуком в водяной бане в течение 30 с, перемешивали при комнатной температуре (не обрабатывая ультразвуком) в течение 3 мин, а затем обрабатывали ультразвуком еще 30 с и перемешивали без обработки ультразвуком в течение 16 мин. ВЭТСХ (этилацетат/метанол, 10: 1) не обнаруживала остатков исходного материала. Реакционную смесь промывали насыщенным водным гидрокарбонатом натрия (2 • 1 мл), водой и солевым раствором (прибл. 1 мл). Затем объединенные водные фракции экстрагировали метиленхлоридом (2 • 0,5 мл), а объединенные органические фракции осушали сульфатом натрия, фильтровали и концентрировали с получением оранжевого маслообразного вещества. Неочищенный продукт кроме того осушали с использованием трубопровода высокого вакуума в течение 1 ч, после чего этот продукт непосредственно использовали в последующей реакции.
Смесь описанного продукта растворяли в безводном метиленхлориде (1,00 мл) и к перемешанному при комнатной температуре раствору добавляли раствор (+/-)-камфорсульфоновой кислоты (CSA) в метиленхлориде (10 мкл, 1,00 мг CSA/1,00 мл CH2Cl2-раствора). После перемешивания при комнатной температуре в течение 2 ч ВЭТСХ (элюент:этилацетат/метанол, 10:1) указывала на почти полную конверсию в мажорное пятно. После этого реакционный раствор промывали насыщенным водным гидрокарбонатом натрия (2 • 0,5 мл), водой, солевым раствором (прибл. 0,5 мл). Объединенные водные фракции экстрагировали метиленхлоридом (2 • 0,5 мл), а объединенные органические фракции осушали сульфатом натрия, фильтровали и концентрировали. Полученный остаток хроматографировали на ВЭТСХ-пластинах (элюент:этилацетат/метанол, 10:1), в результате чего получали синтетический галихондрин B, т.е. соединение 1 (1,27 мг, 1,1 мкМ, полный выход за все три стадии составлял 57%) в виде бесцветного маслообразного вещества.
Этот совместно элюированный синтетический продукт не отличался от образца натурального продукта на ВЭТСХ-пластинах в следующей системе пяти растворителей и многократного элюирования: (1) этилацетат/метанол, 10:1; (2) этилацетат/CH2Cl2, 10: 1; этилацетат/CHCl3/метанол, 10:5:1; (4) этилацетат/CH2Cl2/метанол, 10: 5: 1; (5) этилацетат/т-бутилметиловый эфир/метанол, 10:5:1.
ИК (см-1: 1017 см-1, 1073, 1187, 1737, 2852, 2953, 3438, (шир.).
1Н-ЯМР (C6D6, 500 МГц):
BPMC (FAB) для C60H86O19: Вычислено: 1111.5841. Найдено: 1111.5878 (М+).
[α]D : -51,2o (с = 0,127, MeOH).
Соединение 2.
Соединение 2, норгалихондрин B, получали из C38-альдегида (происходящего от соединения 10, описанного выше) и соединения 12.
(1) Получение енона норгалихондрина B (C38-кетона).
К перемешанному раствору C38-альдегида (5,4 мг, 6,1 мкМ) и соединения 12 (13 мг, 12,2 мкМ) в ДМФ (прибл. 750 мкл), в атмосфере азота добавляли порошкообразный CrCl2, содержащий 0,1% NiCl2 (мас.%) (всего прибл. 20 мг). Полученную зеленую смесь перемешивали при комнатной температуре в течение 16 ч и в этот период времени ТСХ (элюент:этилацетат/гексан/CHCl3) не обнаруживала остаточного количества альдегида. После этого добавляли насыщенный водный NH4Cl (1 мл), H2O (0,5 мл) и этилацетат (1 мл) и полученную смесь перемешивали в течение 20 мин. Верхний органический слой отделяли, а нижнюю фазу кроме того экстрагировали этилацетатом (3 • 0,5 мл). После этого объединенные этилацетатные фракции промывали водой (2 • 1 мл) и солевым раствором (1 мл), а затем осушали сульфатом натрия, фильтровали через стекловату и концентрировали. Полученный остаток очищали с помощью ПТСХ (элюент:гексан/этилацетат/CHCl3 1:1:1), в результате чего получали аллиловые C38-спирты (8 мг, 5 мкМ, выход 83%) в виде прозрачного бесцветного маслообразного вещества (смеси диастереомеров = прибл. 1:1).
К перемешанному раствору C38-спирта (8 мг, 5 мкМ) в CH2Cl2 (1,00 мл), при комнатной температуре, добавляли твердый гидрокарбонат натрия (50 мг), а затем периодинановый реагент Dess-Martin (17 мг, 40 мкМ). После перемешивания при комнатной температуре в течение 90 мин, ТСХ (элюент:гексан/этилацетат/CHCl3, 1: 1: 1) указывала на отсутствие исходного материала. Полученную реакционную смесь разводили диэтиловым эфиром (3 мл) и перемешивали 20 мин с водным раствором (5 мл) насыщенным гидрокарбонатом натрия и содержащим Na2S2O3 (10 мас.%). Отделенную органическую фазу промывали еще в течение 10 мин смесью водного NaHCO3/Na2S2O3, а затем промывали водой и солевым раствором (прибл. 5 мл). Эту органическую фазу осушали безводным сульфатом натрия, фильтровали через стекловату и концентрировали. Остаток очищали с помощью ПТСХ (элюент:гексан/этилацетат/CHCl3, 1:1:1) и получали енон норгалихондрина B (6,3 мг, 3,9 мкМ, выход 79%) в виде прозрачного бесцветного маслообразного продукта. Структура этого енона показана в нижней части фиг.2.
ИК (см-1): 835 см-1, 1088, 1252, 1463, 1737, 2929, 2953,
1Н-ЯМР (C6D6, 500 МГц): BPMC (FAB): m/z (M+Na+) Вычислено: [C86H136O21Si3+Na]+: 1611.8779, Найдено: 1611.8811.
[α]D : - 32o (с = 0,56, MeOH).
(2) Получение метилового сложного эфира норгалихондрина B.
К перемешанному раствору енона (3,15 мг, 2,4 мкМ) в ТГФ (400 мкл) и безводного метилацетата (200 мкл) добавляли приблизительно 1М раствор фторида тетрабутиламмония (TBAF) в ТГФ (20 мкл, pH прибл. 7,6). Полученный раствор перемешивали при комнатной температуре в течение 14,5 ч и в этот период времени ВЭТСХ ([5642-пластины E.Merck Art.] покрывали реакционным раствором, осушали в течение 20 - 30 мин с использованием трубопровода высокого вакуума, а затем элюировали этилацетатом] указывала на одно мажорное пятно при Rf = 0,45. Затем реакционный раствор фильтровали через 2 см-слой силикагеля 60 (230 - 400 меш.) этилацетатом для удаления TBAF. Фильтрат концентрировали в вакууме и получали желтое маслообразное вещество, которое использовали без дополнительной очистки.
Смесь описанного продукта растворяли в метиленхлориде (1 мл). После этого добавляли водный фосфатный буфер (pH = 7,00, 100 мкл) и т-бутанол (20 мкл), а к полученной быстро перемешанной смеси добавляли DDQ (1,8 мг, 8 мкМ). Затем смесь обрабатывали ультразвуком в водяной бане в течение 3 • 30 с, причем в интервалах между обработками ультразвуком, составляющими 3-5 мин, смесь просто размешивали при комнатной температуре. В этот период времени ТСХ (элюент:этилацетат) указывала на отсутствие остаточного исходного материала. После этого реакционную смесь промывали насыщенным водным гидрокарбонатом натрия (2 • 1 мл), водой (1 мл) и солевым раствором (1 мл). Объединенные водные фракции экстрагировали метиленхлоридом (2 • 0,5 мл), а объединенные органические фракции осушали сульфатом натрия, фильтровали и концентрировали с получением оранжевого маслообразного продукта. Этот неочищенный продукт осушали с помощью высоковакуумного трубопровода в течение 30 мин, после чего этот продукт использовали непосредственно в последующей стадии.
Описанный продукт растворяли в безводном метиленхлориде (1,00 мл) и к перемешанному при комнатной температуре раствору добавляли раствор (+/-)-камфорсульфоновой кислоты ("CSA") в CH2Cl2 (10 мкл, 1,00 мг CSA/1,00 мл раствора метиленхлорида). Через час, ВЭТСХ (этилацетат) указывала на почти полное завершение реакции. Реакционный раствор промывали насыщенным водным гидрокарбонатом натрия (2 • 0,5 мл), водой (0,5 мл) и солевым раствором (0,5 мл). Объединенные водные фракции экстрагировали метиленхлоридом (2 • 0,5 мл), а объединенные органические фракции осушали безводным сульфатом натрия, фильтровали и концентрировали. Остаток хроматографировали на ВЭТСХ-пластинах (этилацетат), в результате чего получали синтетический метиловый сложный эфир норгалихондрина B (1,2 мг, 1,1 мкМ, выход за все три стадии составлял 45%) в виде бесцветного маслообразного вещества.
Совместно элюированный синтетический продукт не отличался от аутентичного образца (полученного путем обработки натурального норгалихондрина B диазометаном в метаноле) после многократного элюирования на ВЭТСХ-пластинах с использованием системы двух растворителей: (1) этилацетата и (2) CH2Cl2/метанола, 25:1.
ИК (см-1): 1021, 1073, 1189, 1435, 1739, 2924, 3609.
BPMC (FAB): m/z (M+Nz+) для C60H84O19 + Na+: Вычислено: 11315504. Найдено: 11315502.
[α]D : -46,4o (с = 0,22, MeOH).
(3) Получение норгалихондрина B.
К перемешанному раствору метилового сложного эфира (2,2 мг, 2,0 мкМ) в ТГФ (300 мкл), при комнатной температуре добавляли 100 мкл водного 1М раствора LiOH. После перемешивания в течение 90 мин при комнатной температуре ВЭТСХ (этилацетат) указывала на завершение реакции. Затем тетрагидрофуран удаляли в потоке азота и полученный раствор разводили водой (200 мкл) и охлаждали до 0oC. К быстро перемешанному раствору добавляли водную 1М соляную кислоту (100 мкл). Полученную смесь экстрагировали этилацетатом (4 • 0,5 мл), а объединенные экстракты осушали сульфатом натрия, фильтровали через стекловату и концентрировали. Остаток очищали на полистироловой колонке TSKG300S (2 см-колонку уравновешивали водой, карбоновую кислоту загружали в 50%-ный водный этанол и элюировали H2O → этанолом), в результате чего получали карбоновую кислоту (1,3 мг, 1,2 мкМ, выход 60%) в виде бесцветного маслообразного вещества. Полученным соединением является норгалихондрин B (соединение 2).
Процедуры, которые были использованы для получения промежуточных соединений (соединения 3 - 12, схемы 1 и 2), необходимых для описанного синтеза галихондрина B1 и норгалихондрина B2, описаны ниже.
Соединение 3.
Соединение 3 синтезировали в соответствии со следующей процедурой.

К моноацетониду (50,4 г, 0,179 М) добавляли 250 мл смеси HOAc/H2O (4:1) и раствор перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали при пониженном давлении и очищали с помощью флеш-хроматографии (элюент: 100% EtOAc), в результате чего получали триол (40,6 г, выход 94%).
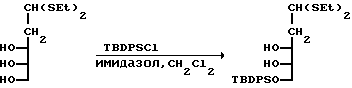
К триолу (40,6 г, 0,169 М) в метиленхлориде при 0oC, добавляли имидазол (50,0 г, 0,734 М), т-бутилдифенилсилилхлорид ("TBDPSCl"; 51,0 г, 0,186 М), и полученную смесь перемешивали при 0oC в течение 1 ч и при комнатной температуре в течение 1 ч. Реакционную смесь разводили метиленхлоридом и дважды промывали водным раствором гидрокарбоната натрия. Водный слой подвергали обратному экстрагированию (4 раза) метиленхлоридом, а объединенные органические слои осушали сульфатом натрия. В результате концентрирования при пониженном давлении получали маслообразный продукт, который очищали с помощью флеш-хроматографии (элюент:гексан/EtOAc, 4:1). Таким образом был получен диол (75,7 г, выход 94%) в виде бесцветного маслообразного вещества.
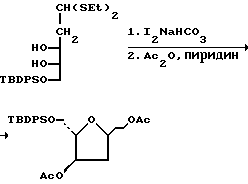
При 0oC к перемешанному раствору тиоацеталя (75,7 г, 0,158 М) и NaHCO3 (79,7 г, 0,949 М) в ацетоне (550 мл) и H2O (90 мл), добавляли 120,4 г йода (0,474 М). Через полчаса, ТСХ (элюент:гексан/EtOAc, 1:1) указывала на полное отсутствие исходного тиоацеталя. После этого реакционную смесь гасили путем добавления водного Na2S2O3, а ацетон удаляли при пониженном давлении. Смесь экстрагировали 4 раза этилацетатом, а объединенные органические слои один раз промывали солевым раствором, один раз водой, а затем осушали сульфатом натрия и концентрировали при пониженном давлении с получением фуранозы в виде желтого маслообразного вещества. К неочищенной фуранозе добавляли пиридин (75 мл), Ac2O, (80,5 г, 0,789 М, 74 мл) и диметиламинопиридин ("DMAP"; 1,9 г, 0,0157 М) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение ночи, концентрировали при пониженном давлении, а затем очищали с помощью флеш-хроматографии (элюент: гексан/EtOAc), в результате чего получали диацетат (67,7 г, выход 94%) и маслообразное вещество.
ИК (пленка): 606 см-1, 702, 997, 1113, 1230, 1742, 2932, 2958, 3072.
1Н-ЯМР (CDCl3: δ 1,91 (OAc, с); 2,06 (OAc, с), 2,08 (OAc, с); 2,09 (OAc, с); 2,18 (0,5H, м); 2,27 (0,5H, м); 2,49 (0,5H, м); 2,56 (0,5H, м); 3,78 (1,5H, м); 3,86 (0,5H, дд); 4,18 (0,5H, м); 4,31 (0,5H, м); 5,37 (0,5H, дд); 5,42 (0,5H, м); 6,38 (0,5H, дд); 6,42 (0,5H, д); 7,39 (6H, м); 7,69 (4H, м).
BPMC (FAB) для C25H32O6Si + Na: Вычислено: 4791866, Найдено: 4791891.
[α]D -18,6o (с = 1,81, MeOH).

К охлажденному льдом раствору диацетата (67,7 г, 0,148 М) и аллилтриметилсилана (50,8 г, 0,444 М, 70,6 мл) в CH3CN по капле в течение 10 мин добавляли BF3 • OEt2 (21,0 г, 0,148 М, 18,2 мл). Реакционную смесь перемешивали еще 15 мин, а затем гасили разбавленным водным гидрокарбонатом натрия. Полученную смесь три раза экстрагировали этилацетатом, а объединенные органические слои осушали сульфатом натрия. После концентрирования при пониженном давлении и очистки с помощью флеш-хроматографии (элюент: гексан/EtOAc, 5: 1) получали маслообразное вещество (58,9 г, выход 90%).
ИК (пленка) 702 см-1, 1113, 1247, 1759, 2867, 3079.
1H-ЯМР (CDCl3, 500 МГц): δ 1,05 (9H, с, т-Бут.); 1,74 (1H, м); 2,06 (3H, с, O-CH3); 2,31 (1H, м); (2,44 (1H, м); 2,49 (1H, м); 3,71 (1H, дд, J = 4,5, 10,9 Гц); 3,76 (1H, дд, J = 3,7, 10,9 Гц); 4,09 (1H, м); 4,24 (1H, р, J = 6,6 Гц); 5,09 (2H, м); 5,36 (1H, м, CH-OAc); 5,81 (1H, м); 7,39 (6H, м); 7,67 (4H, м).
BPMC (FAB) для C26H34O4Si (M+Na)+: Вычислено: 4612124; Найдено: 4612138.
[α]D : -16,9o (с = 1,25, MeOH).
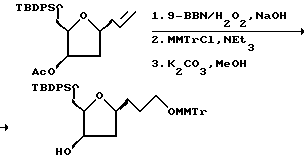
К раствору олефина (24,1 г, 55 мМ) в ТГФ (400 мл) при 0oC добавляли 0,5 М 9-BBN в ТГФ (142 мл, 71,5 мМ). Полученную реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Этот раствор охлаждали до 0oC и добавляли 10% NaOH (90 мл), а затем 30% H2O2 (90 мл). После перемешивания в течение 2 ч, реакцию завершали путем добавления водного NH4Cl и экстрагировали этилацетатом (3 • 600 мл). Объединенные органические слои дважды промывали водным карбонатом калия, осушали сульфатом натрия и концентрировали при пониженном давлении. Следует отметить, что наблюдался частичный гидролиз ацетата. Таким образом, неочищенную смесь использовали для селективной функционализации первичного спирта без очистки.
К раствору неочищенного спирта в CH2Cl2 (300 мл) при 0oC добавляли Et3N (54 мл, 385 мМ), а затем п-анизилхлородифенилметан (18,7 г, 60,5 мМ). Полученную реакционную смесь перемешивали в течение 8 ч, а затем гасили путем добавления водного гидрокарбоната натрия и экстрагировали метиленхлоридом (2 • 100 мл). Объединенные органические слои осушали сульфатом натрия и концентрировали при пониженном давлении. К раствору неочищенного ацетата в ТГФ (10 мл) и MeOH (300 мл) порциями добавляли порошкообразный карбонат калия (4 г). Реакционную смесь перемешивали в течение 2,5 ч при комнатной температуре, а затем фильтровали целит и очищали с помощью флеш-хроматографии (элюент: 20% EtOAc/гексан) в результате чего получали нужный продукт (26,7 г, выход за описанные три стадии составлял 70,8%).
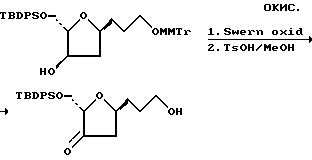
К раствору оксалилхлорида (1,33 мл, 15,3 мМ) в метиленхлориде (150 мл) по капле в течение 3 мин и при -78oC, добавляли ДМСО (2,16 мл, 30,2 мМ). После перемешивания в течение 10 мин к раствору на протяжении 5 мин добавляли раствор спирта в CH2Cl2 (10 мл). Пустую колбу еще раз промывали метиленхлоридом (3 мл) и указанный раствор добавляли к реакционной смеси. После перемешивания в течение 1 ч, к реакционной смеси добавляли Et3N (8,55 мл, 60,4 мМ). Полученную реакционную смесь перемешивали еще 15 мин, а затем в течение 45 мин нагревали до комнатной температуры. После этого реакционную смесь гасили путем добавления насыщенного NH4Cl, а органический слой отделяли. Этот органический слой промывали водой, солевым раствором, осушали сульфатом натрия и концентрировали. Остаток очищали с помощью колоночной хроматографии (элюент: 20% EtOAc в гексане), в результате чего получали 2,37 г (выход 97%) кетона.
К раствору MMTr-кетона (9,3 г, 13,5 мМ) в CH2Cl2 (200 мл) и MeOH (50 мл), добавляли TsOH (1 г) (при комнатной температуре). После перемешивания в течение 2 ч к реакционной смеси для нейтрализации TsOH добавляли твердый гидрокарбонат натрия. После часового перемешивания реакционную смесь фильтровали, концентрировали и очищали с помощью колоночной хроматографии (элюент: 33% EtOAc в гексане), в результате чего получали кето-спирт (5,4 г, 96,6% выход).
ИК (пленка): 703 см-1, 743, 823, 1077, 1113, 1428, 1759, 2893, 2930, 3071, 3438.
1Н-ЯМР (CDCl3): δ 1,01 (9H, с); 1,57 (1H, шир. с); 1,75 (2H, м); 1,80 (2H, м); 2,28 (1H, дд, J = 8,3, 17,9 Гц); 2,66 (1H, дд, J = 6,5, 17,9 Гц); 3,73 (2H, м); 3,87 (1H, дд, J = 2,2, 11,1 Гц); 3,97 (1H, дд, J = 2,4, 11,1 Гц); 4,03 (1H, шир. с); 4,73 (1H, м); 7,43 (6H, м); 7,68 (2H, м), 7,70 (2H, м).
BPMC (FAB для C24H32O4Si + Na: Вычислено: 4351968; Найдено: 4351954.
[α]D = 19,2o (с = 1,2, MeOH).
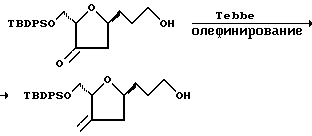
К тщательно перемешанному раствору кето-спирта (747 мг, 1,81 мМ) в смеси толуола/ТГФ/пиридина (3:1:1), в течение 15 мин по капле и при 0oC добавляли свежеприготовленный раствор реагента Tebbe (полученного in situ в соответствии с процедурой, описанной Grubbs, R.M.; Cannazzo, L.F. J.Org. Chem., 50, 2386 (1985) (8,3 мл, около 3 экв.). ТСХ (элюент: 50% этилацетат/гексан) через полчаса указывала на полное отсутствие исходного материала. Реакцию завершали путем осторожного добавления 0,1 н. NaOH (10 мл). Полученную смесь разводили эфиром и раствор энергично перемешивали до тех пор, пока органический слой не становился светло-желтым. После этого слои отделяли и водную фазу экстрагировали эфиром. Объединенные органические фракции исключительно тщательно промывали водой для удаления пиридина, а затем промывали солевым раствором. Органические слои осушали сульфатом натрия и удаляли в вакууме. Полученный остаток очищали с помощью флеш-хроматографии (элюент: 40% этилацетат/гексан) и получали экзоциклоолефин (647 мг, выход 87%).

К перемешанному раствору спирта (5,25 г, 12,8 мМ) в метиленхлориде (90 мл) при комнатной температуре добавляли пиридин (62 мл, 76,6 мМ), DMAP (50 мг) и пивалоилхлорид (8,3 мл, 67,7 мМ). После перемешивания в течение часа реакционную смесь гасили насыщенным NH4Cl, разводили метиленхлоридом и экстрагировали. Объединенные органические слои промывали 10%-ной соляной кислотой, водой, насыщенным гидрокарбонатом натрия, солевым раствором, а затем осушали сульфатом натрия. Растворители концентрировали при пониженном давлении.
К раствору неочищенного пивалоата в ТГФ (140 мл) добавляли по капле при комнатной температуре 1 М TBAF в ТГФ (20 мл, 20 мМ). После перемешивания в течение 1,1 ч реакционную смесь концентрировали при пониженном давлении, а остаток очищали с помощью колоночной хроматографии (элюент: 15% EtOAc в гексане), в результате чего получали нужный продукт (2,98 г, выход за указанные две стадии составлял 91%).
ИК (пленка): 1157 см-1, 1284, 1480, 1727, 2872, 2959, 3078, 3453.
1Н-ЯМР (CDCl3): δ 1,19 (9H, с); 1,55 (1H, м); 1,67 (2H, м); 1,77 (1H, м); 1,93 (1H, м); 2,29 (1H, дд); 2,70 (1H, дд); 3,61 (2H, м); 4,09 (3H, т, J = 6,3 Гц); 4,50 (1H, шир. с.); 4,92 (1H, м); 5,08 (1H, м).
BPMC (Cl) для C14H24O4 + H (M+H)+: Вычислено: 2571753; Найдено: (M+H)+: 2571744.
[α]D -27,2o (с = 1,1, MeOH).
Соединение 4.
Соединение 4 синтезировали в соответствии со следующей процедурой.
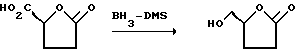
К раствору карбоновой кислоты (42 г, 0,32 М) в безводной ТГФ (300 мл) при комнатной температуре, добавляли BH3-ДМС (39 мл, чистый, 0,39 М) таким образом, чтобы обеспечивалось мягкое нагревание с обратным холодильником. Еще через 3 ч смесь охлаждали до 0oC, а затем осторожно гасили путем добавления избыточного количества метанола (500 мл). Растворители удаляли путем дистилляции при атмосферном давлении, а остаток очищали путем дистилляции в вакууме, в результате чего получали 34,3 г чистого спирта.
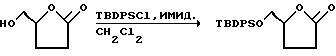
К перемешанному раствору лактона 1 (12,5 г, 108 мМ) и имидазола (15,6 г, 229 мМ) в CH2Cl2 (300 мл) при 0oC добавляли т-бутилдифенилсилилхлорид (29,0 мл, 112 мМ). Полученную смесь оставляли для перемешивания при комнатной температуре в течение ночи. После этого смесь разводили метиленхлоридом и промывали насыщенным гидрокарбонатом натрия, водой, солевым раствором, а затем осушали сульфатом натрия и концентрировали при пониженном давлении. Затем эту смесь очищали путем кристаллизации из гексана и получали силиловый эфир 11 (30,05 г в первом сборе и еще 5,40 г во втором сборе; объединенный выход составлял 93%, т.пл. 84oC).
ИК (пленка): 3072 см-1, 3049, 2998, 2931, 2893, 2858, 1777, 1590, 1473, 1461, 1427, 1391, 1346, 1174, 1113, 1084, 1032, 995, 941, 858, 822, 741, 703.
1Н-ЯМР (CDCl3): δ 1,07 (9H, с); 2,19 - 2,34 (2H, м); 2,52 (1H, м); 2,69 (1H, м); 3,70 (1H, дд, J = 3,3, 11,3 Гц); 3,90 (1H, дд, J = 3,2, 11,3 Гц); 4,61 (1H, м); 7,30 - 7,50 (6H, м); 7,60 - 7,80 (4H, м).
13С-ЯМР (CDCl3): δ 19,22, 23,65, 26,78, 28,55, 65,45, 79,89, 127,70, 129,77, 135,38, 135,47, 177,37. \\\ [α]D : + 24,9o (с = 5,91, CHCl3).
Вычислено,%: C21H26O3Si. C 71,15; H 7,39. Найдено,%: C 70,91: H 7,42.

К перемешанному раствору диизопропиламина (3,88 мл, 27,69 мМ) в ТГФ (10 мл) при -78oC добавляли 2,35 М раствора н-бутиллития в гексане (11,8 мл, 27,69 мМ). После перемешивания полученной смеси в течение 20 мин медленно с использованием канюли добавляли раствор лактона 11 (9,347 г, 26,4 мМ) в ТГФ (20 мл). После перемешивания смеси в течение 25 мин при -78oC добавляли метилиодид (4,85 мл, 77,9 мМ). Затем через 35 мин реакцию завершали путем осторожного добавления насыщенного NH4Cl. Смесь оставляли для нагревания до 0oC, а затем разводили эфиром. Слои отделяли и водный слой снова экстрагировали эфиром. Объединенные органические слои промывали водой, солевым раствором, осушали сульфатом натрия и концентрировали при пониженном давлении. После кристаллизации из гексана получали мажорный метилированный продукт III (т. пл. 84oC, 7,3 г, выход 75%). Маточный раствор содержал 1,4 г смеси двух метилированных соединений.
ИК (пленка): 3072 см-1, 3051, 2958, 2932, 2859, 1774, 1473, 1462, 1428, 1362, 1349, 1202, 1173, 1113, 1067, 1022, 998, 954, 935, 822, 742, 727, 702, 623.
1Н-ЯМР (CDCl3): δ 1,06 (9H с); 1,27 (3H, д, J = 7,1 Гц); 1,98 (1H, м); 2,47 (1H, м); 2,86 (1H, м); 3,67 (1H, дд, J = 3,3 , 11,3 Гц); 3,88 (1H, дд, J = 3,5, 11,3 Гц); 4,56 (1H, м); 7,30 - 7,50 (6H, м); 7,70 - 7,80 (4H, м).
13С-ЯМР (CDCl3): δ 16,47, 19,25, 26,84, 32,27, 34,24, 65,57, 77,49, 127,74, 129,81, 132,49, 132,87, 135,42, 135,51, 179,82.
MC (FAB): 369 AEM (M++H, интенсивность 2%), 313(6), 312(18), 311(74), 293(7), 292(24), 291(100), 233(17), 199(29), 187(36), 163(27), 135(86).
[α]D : +1,3o (с = 1,43, CHCl3).
C22H28O3Si • 1/4H2O. Вычислено,%: C 71,70; H 7,66. Найдено,%: C 70,84; H 7,54.
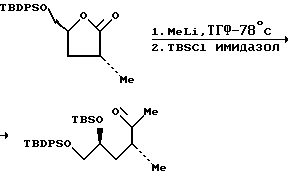
К раствору лактона (46,2 г, 0,125 М) в ТГФ (400 мл), в течение 10 мин и при -78oC, добавляли 1,4 М MeLi в эфире (89,5 мл, 0,125 М). После перемешивания в течение 10 мин реакционную смесь выливали в насыщенный раствор NH4Cl (300 мл) и экстрагировали этилацетатом (3 • 300 мл). Объединенные органические слои промывали солевым раствором (200 мл), осушали сульфатом натрия и концентрировали при пониженном давлении.
К раствору неочищенного гемикеталя в метиленхлориде (600 мл) добавляли имидазол (22,2 г, 0,150 М), а затем т-бутилдиметилсилилхлорид (22,2 г, 0,325 М). Полученную реакционную смесь перемешивали 36 ч при комнатной температуре. После этого реакционную смесь промывали насыщенным гидрокарбонатом натрия, водой и солевым раствором. Органический слой осушали сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (10% EtOAc в гексане), в результате чего получали 51,3 г дисилилового эфира (выход 82,4%) в виде бесцветного маслообразного вещества.
ИК (пленка): 1112 см-1, 1254, 1462, 1473, 1716, 2886, 2930, 2957.
1Н-ЯМР (CDCl3): δ -1,20 (3H, с); -0,50 (3H, с); 0,81 (9H, с); 1,06 (9H, с); 1,11 (3H, д, J = 7,1 Гц); 1,47 (1H, м); 2,12 (3H, с); 2,15 (1H, м), 2,71 (1H, м); 3,46 (1H, дд, J = 7,2, 10,1 Гц); 3,57 (1H, дд, J = 4,6, 10,1 Гц); 3,70 (1H, м); 7,42 (6H, м); 7,66 (4H, м).
BPMC (FAB) для C29H46O3Si2 + Na:
Вычислено: 5212853; Найдено: 5212885.
[α]D -13,0o (с = 1,15, MeOH).

К раствору кетона (7,1 г, 14,26 мМ) в ТГФ (16 мл) добавляли Трис-NHNH2 (5,1 г, 1,2 экв.), а затем 1 каплю концентрированной соляной кислоты. После 4-часового перемешивания реакционную смесь непосредственно концентрировали, осушали путем азеотропного удаления воды с использованием бензола (процедуру повторяли дважды), а затем эту смесь помещали в высокий вакуум. Неочищенный гидразон растворяли в смеси TMEDA/пентана (30/120 мл) и охлаждали до -78oC, а затем реакционную смесь обрабатывали 2,06 молями н-бутиллития (27,5 мл, 4 экв. ) в течение 30 мин. После этого реакционную смесь нагревали до 0oC и поддерживали в течение 10 мин в ледяной бане (красный цвет менялся на желтый). Виниллитиевый раствор охлаждали до -78oC и очень медленно добавляли н-Bu3SnCl (15 мл, 3,9 экв.). Если перемешивание было затруднено, то температуру доводили до -15oC и реакционную смесь перемешивали 1 ч при той же температуре и 1 ч при 0oC (почти бесцветный раствор). После этого реакционную смесь разводили эфиром (100 мл) и промывали насыщенным NH4Cl, водой и солевым раствором. Органический слой осушали сульфатом натрия, концентрировали и очищали с помощью колоночной хроматографии (элюент: гексан/10% толуол в гексане), в результате чего получали винилолово.
Полученное винилолово растворяли в метиленхлориде (100 мл) и подвергали титрованию раствором иода до тех пор, пока не появлялся багряный цвет (0oC). После этого реакционную смесь промывали раствором NaHSO3, водой и солевым раствором. Органический слой концентрировали, а остаток очищали с помощью колоночной хроматографии (элюент: 10% толуол в гексане), в результате чего получали 6,76 г винилиодида (выход 78%).

К раствору силилового эфира в MeCN (90 мл) и ТГФ (10 мл) добавляли 48% HF (1,2 мл, 3 экв.). После перемешивания в течение 10 ч добавляли твердый гидрокарбонат натрия (5 г) и этилацетат (300 мл), а затем перемешивали до тех пор, пока не прекращалось выделение пузырьков. Затем эту смесь фильтровали, концентрировали и очищали с помощью колоночной хроматографии (элюент: градиент 10% EtOAc в CHCl3/EtOAc), в результате чего получали 1,79 г диола (выход 70%). Вследствие летучести диола выход был низким. Чтобы получить лучший выход, необязательно полностью осушать диол, поскольку следующую стадию проводили в водной среде.
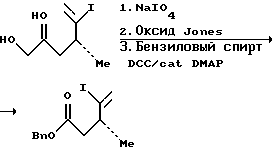
К раствору диола (1,79 г, 7,02 мМ) в ТГФ (15 мл) и H2O (7 мл) при комнатной температуре добавляли Na104. После перемешивания в течение 1 ч реакционную смесь разводили водой до образования прозрачного раствора, а затем экстрагировали эфиром (2 • 20 мл). Органический слой промывали раствором NaHSO3 для удаления избытка оксиданта. Затем этот органический слой концентрировали при пониженном давлении без осушки. Неочищенный альдегид разводили 50 мл ацетона. Полученный раствор охлаждали до 0oC и обрабатывали реагентом Джонса. После завершения реакции избыток реагента Джонса гасили путем добавления изопропанола. Затем реакционную смесь фильтровали через целит, концентрировали, разбавляли эфиром и промывали водой и солевым раствором. Органический слой осушали сульфатом магния, концентрировали и дважды осушали путем азеотропного удаления воды с использованием бензола.
К раствору неочищенной кислоты в CH2Cl2 (20 мл) при комнатной температуре, добавляли бензиловый спирт (1 мл, 1,4 экв.), а затем диизопропилкарбодиимид ("DCC"; 1,74 г, 1,2 экв.) с каталитическим количеством DMAP (5%). После перемешивания в течение 12 ч к реакционной смеси добавляли еще 0,5 г (0,34 экв.) диизопропилкарбодиимида вместе с бензиловым спиртом (0,5 мл, 0,7 экв. ). Через 6 ч реакционную смесь концентрировали, разводили эфиром (5 мл) и фильтровали через целит. Полученный фильтрат концентрировали и очищали с помощью колоночной хроматографии (элюент: 9% EtOAc в гексане), в результате чего получали бензиловый сложный эфир (2,16 г, выход 93,5%).

К раствору фосфоната (3,4 г, 27,2 мМ) в ТГФ (16 мл) (при -78oC) добавляли 2,13 молей н-бутиллития. После часового перемешивания к раствору по капле добавляли бензоат (2,5 г, 7,58 мМ) в ТГФ (4 мл). Через 20 мин реакционную смесь гасили насыщенным NH4Cl и экстрагировали этилацетатом (3 • 20 мл). После осушки сульфатом натрия и концентрирования остаток очищали с помощью колоночной хроматографии (элюент: EtOAc в гексане, 50% - 80%) и получали 1,90 г кетофосфоната (выход = 78,4%) вместе с выделенным бензоатом (249 мг, выход 10%). Выход по отношению к выделенному исходному материалу составлял 87%.
ИК (пленка): 1028 см-1, 1262, 1613, 1715, 2959.
1Н-ЯМР (CDCl3): δ 1,03 (3H, д, J = 6,1 Гц); 2,57 (2H, м); 2,81 (1H, м); 3,08 (2H, м); 3,78 (3H, с, -OMe); 3,80 (3H, с, -OMe); 5,72 (1H, д, J = 1,4 Гц); 6,19 (1H,с).
BPMC (FAB) для C9H16O4PI + Na:
Вычислено: 3689729; Найдено: 3689744.
[α]D -2,4o (с = 4,9, MeOH).
Соединение 5.
Соединение 5 синтезировали в соответствии со следующей процедурой: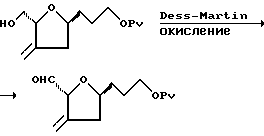
К раствору спирта (180 мг, 0,706 мМ) в дихлорметане (12 мл) при комнатной температуре добавляли реагент Dess-Martin (449 мг, 1,5 экв.) и 2 г молекулярных сит  . После 25-минутного перемешивания, реакционную смесь разводили эфиром (60 мл) и фильтровали через целит. Фильтрат промывали 6 эквивалентами раствора дитионата натрия в насыщенном растворе гидрокарбоната натрия (20 мл), а затем насыщенным гидрокарбонатом натрия, водой и солевым раствором. Органический слой осушали, концентрировали и снова осушали путем азеотропной дистилляции. Неочищенный альдегид использовали в последующей стадии без дополнительной очистки.
. После 25-минутного перемешивания, реакционную смесь разводили эфиром (60 мл) и фильтровали через целит. Фильтрат промывали 6 эквивалентами раствора дитионата натрия в насыщенном растворе гидрокарбоната натрия (20 мл), а затем насыщенным гидрокарбонатом натрия, водой и солевым раствором. Органический слой осушали, концентрировали и снова осушали путем азеотропной дистилляции. Неочищенный альдегид использовали в последующей стадии без дополнительной очистки.

К раствору кетофосфоната (333 мг, 1,06 мМ) в безводном ТГФ (4 мл) при 0oC добавляли NaH (38 мг, 0,95 мМ). После получасового перемешивания по капле и в течение 5 мин добавляли альдегид в ТГФ (2 мл). После дополнительного перемешивания в течение получаса, реакцию завершали путем добавления насыщенного хлорида аммония и смесь экстрагировали этилацетатом. Органические слои осушали сульфатом натрия и концентрировали в вакууме. После очистки с помощью флеш-хроматографии получали (элюент: 20% этилацетат/гексан) получали чистый енон (295 мг, выход 88%).
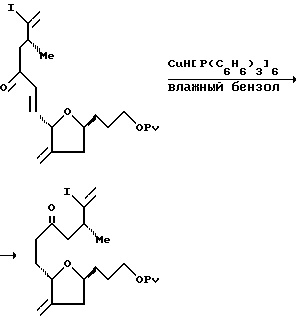
К раствору енона (650 мг, 1,37 мМ) в 65 мл дегазированного бензола и 0,4 мл (16 экв.) дегазированной воды, добавляли [CuH(Ph3P)]6 (806 мг, 1,8 экв.). Полученную красную реакционную смесь перемешивали 3 ч в атмосфере аргона, а затем реакционный сосуд открывали и осушали воздухом для разложения избыточного количества реагента. Через час черную реакционную смесь фильтровали через целит. Фильтрат концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии (элюент: 6% этилацетат в гексане), в результате чего получали кетон (605 мг, выход 93%).
ИК (пленка): 898 см-1, 1174, 1336, 1726, 2934, 2962.
1Н-ЯМР (CDCl3) δ 1,02 (3H, д, J = 6,5 Гц, CH3); 1,58 (9H, с, -OPv); 1,50 (1H, м); 1,61 (1H, м); 1,72 (2H, м); 1,87 (1H, м); 2,25 (1H, м); 2,33 (1H, дд, J = 6,9, 16,3 Гц); 2,54 (3H, м); 2,67 (2H, м); 3,99 (1H, м); 4,06 (2H, м); 4,35 (1H, м); 4,87 (1H, д, J = 2,0 Гц); 5,00 (1H, д, J = 2,1 Гц); 5,70 (1H, д, J = 1,7 Гц); 6,17 (1H, д, J = 1,3 Гц).
BPMC (FAB) для C21H33O4I + Na:
Вычислено: 4991323; Найдено: 4991334.
[α]D -37,0o (с = 1,01, MeOH).

К раствору кетона (1,26 г, 2,65 мМ) в MeOH (20 мл) при 0oC добавляли NaBH4 (130 мг, 3,38 мМ). После 20-минутного перемешивания при той же температуре реакционную смесь гасили насыщенным хлоридом аммония и экстрагировали этилацетатом (3 • 20 мл). Объединенные органические слои осушали сульфатом натрия, концентрировали и очищали с помощью колоночной хроматографии (гексанEtOAc/CHCl3, 8/2/1), в результате чего получали нужный спирт с высоким Rf (877 мг, выход 69,6%), побочный нежелательный спирт с низким Rf (372 мг, выход 29,5%) и смешанные фракции (56 мг, выход 4,4%).
ИК (пленка): 1157 см-1, 1285, 1728, 2930, 2959, 3440.
1Н-ЯМР (CDCl3): δ 0,98 (3H, д, J = 6,5 Гц); 1,19 (9H, с, -OPv); 1,25 - 1,70 (11H, м); 2,08 (1H, м); 2,27 (1H, м); 2,47 (1H, шир. с); 2,70 (1H, м); 3,58 (1H, м); 4,07 (2H, м); 4,39 (1H, м); 4,86 (1H, м); 5,00 (1H, м); 5,75 (1H, д, J = 1,3 Гц); 6,24 (1H, шир. с.).
BPMC (FAB) для C21H35O4I + Na:
Вычислено: 5011480; Найдено: 5011485.
[α]D -82,2o (с = 0,9, MeOH).

К раствору спирта (364 мг, 0,76 мМ) в эфире (16 мл) добавляли Ph3P (478 мг, 2,4 экв.), п-нитробензойную кислоту (280 мг, 2,4 экв.), а затем диэтилазодикарбоксилат (250 мкл, 2,4 экв.). После этого реакционную смесь перемешивали в течение 1 ч и добавляли 20 мл гексана. Для удаления избыточного количества реагента реакционную смесь фильтровали через слой SiO2 (элюент: 30% этилацетат в гексане). Фильтрат концентрировали, а остаток растворяли в бензоле. Затем суспензию помещали на колонку с силикагелем и фильтровали через слой стекловаты для удаления нерастворимых веществ. После очистки с помощью колоночной хроматографии (элюент: 10% этилацетат в гексане) получали бензоат (422 мг, выход 88,9%).
Полученный бензоат растворяли в MeOH (20 мл) с использованием карбоната калия (2 мг). После этого реакционную смесь перемешивали до полного израсходования исходного материала. Для нейтрализации или подкисления реакционной смеси добавляли HOAc (1 или 2 капли). После перемешивания в течение 10 мин реакционную смесь концентрировали и очищали с помощью колоночной хроматографии (элюент: 13% EtOAc в гексане) в результате чего получали инвертированный спирт (319 мг, выход 99%).
ИК (пленка): 1157 см-1, 1285, 1728, 2930, 2959, 3440.
1Н-ЯМР (CDCl3) δ 0,98 (3H, д, J = 6,5 Гц); 1,19 (9H, с, -OPv); 1,25 - 1,70 (11H, м); 2,08 (1H, м); 2,27 (1H, м); 2,47 (1H, шир. с); 2,70 (1H, м); 3,58 (1H, м); 4,07 (2H, м); 4,39 (1H, м); 4,86 (1H, м); 5,00 (1H, м); 5,75 (1H, д, J = 1,3 Гц); 6,24 (1H, шир. с).
BPMC (FAB) для C21H35O4I + Na:
Вычислено: 5011480; Найдено: 5011485.
[α]D -82,2o (с = 0,9, MeOH).
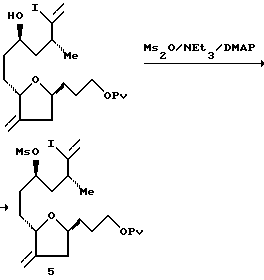
К раствору спирта (14,6 мг, 0,03 мМ) в метиленхлориде (1 мл) добавляли каталитическое количество DMAP, NEt3 (7,7 мкл, 1,8 экв.) и Ms2O (7,5 мг, 1,5 экв. ). Через 30 мин реакцию завершали путем добавления насыщенного раствора гидрокарбоната натрия, а затем экстрагировали этилацетатом (2 • 5 мл). Объединенные органические слои промывали солевым раствором, осушали и концентрировали. Неочищенный остаток быстро фильтровали через прокладку из двуокиси кремния (элюент: 25% этилацетат в гексане) и получали 16,3 мг мезилата (соединение 5) с выходом 96%.
ИК (пленка): 898 см-1, 1173, 1337, 1355, 1725, 2871, 2934, 2962.
1Н-ЯМР (CDCl3): δ 1,01 (3H, д, J = 6,5 Гц); 1,19 (9H, с); 1,45 - 1,80 (9H, м); 1,84 (1H, м); 1,90 (1H, м); 2,27 (1H, дд, J = 5,6, 15,3 Гц); 2,68 (1H, дд, J = 6,4, 15,3 Гц); 3,02 (1H, с); 4,05 (1H, м); 4,07 (1H, м); 4,37 (1H, м); 4,69 (1H, м); 4,88 (1H, с); 5,01 (1H, с); 5,85 (1H, с); 6,36 (1H, с).
Соединение 6
Соединение 6 синтезировали в соответствии со следующей процедурой.
(1) Получение 3,5-ди-O-трет-бутилдиметилсилил-D-галактала (Kinzy, W. et. al. Tetrahedron Letters 28: 1981-1984, 1987; и Horton, D. et al. O. Carbohydrate Research 144:325-330, 1990).
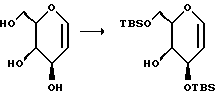
К перемешанному раствору D-галактала (Kozikowski, A. P.; Lee. J.J. Org. Chem. 55:863-870, 1990) (18,188 г, 125 мМ) в безводном N,N-диметилформамиде (62 мл) при комнатной температуре добавляли имидазол (42,35 г, 622, 9 мМ), а затем трет-бутилдиметилсилилхлорид (41,36 г, 274 мМ). Полученную смесь перемешивали при комнатной температуре в течение 2,2 ч. В это время ТСХ (элюент: гексан/этилацетат/хлороформ, 2:1:1) указывала на полное дисилилирование. Реакционную смесь выливали в H2O (1 л) и полученную смесь экстрагировали гексаном (3 • 500 мл). Затем объединенные экстракты промывали водой и солевым раствором (прибл. 500 мл). Органическую фазу осушали сульфатом магния, фильтровали и концентрировали, в результате чего получали неочищенный дисилиловый эфир (46,93 г) в виде прозрачного маслообразного вещества. Этот материал бензилировали без дополнительной очистки.
(2) Получение 4-O-бензил-3,5-ди-O-трет-бутилдиметилсилил-D-галактала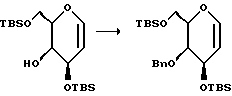
К перемешанному при 0oC раствору неочищенного спирта (46,93 г, прибл. 125 мМ) в смеси ТГФ и ДМФ (4:1 по объему, всего 500 мл) в атмосфере аргона добавляли бензилбромид (29,7 мл, 249 мМ). Затем к смеси порциями в течение 1 ч добавляли 50%-ную суспензию NaH в минеральном масле (15 г, 312,5 мМ). Полученную смесь оставляли для нагревания до комнатной температуры и перемешивали при этом. Через 2 ч после завершения добавления NaH ТСХ указывала на отсутствие исходного материала. Затем смесь охлаждали до 0oC и в течение 30 мин осторожно добавляли 20 мл безводного метанола. Полученную смесь перемешивали еще 30 мин, нагревая при этом до комнатной температуры. После этого добавляли H2O (100 мл) и смесь выливали в дополнительное количество воды (900 мл), а затем экстрагировали диэтиловым эфиром (3 • 500 мл). Объединенные эфирные экстракты промывали водой и солевым раствором, а затем осушали сульфатом магния, фильтровали и концентрировали, в результате чего получали желтое маслообразное вещество (прибл. 80 г). Полученный материал использовали без дополнительной очистки.
(3) Получение 4-O-бензил-3,5-дипропионил-D-галактала.

К раствору неочищенного 4-O-бензил-3,5-ди-O-трет-бутилдиметилсилил-D-галактала прибл. 80 г) в ТГФ (200 мл) медленно добавляли 1,0 М раствора фторида тетрабутиламмония в ТГФ (275 мл, 275 мМ). Полученный прозрачный оранжевый раствор перемешивали при комнатной температуре в течение 2 ч и в этот период времени ТСХ не обнаруживала остаточного силилового эфира. Смесь концентрировали на роторном испарителе, а остаток непосредственно подвергали реакции ацилирования.
Неочищенный диол растворяли в метиленхлориде (500 мл) и полученную перемешанную смесь охлаждали до 0oC в атмосфере аргона. Затем приблизительно через 15 мин добавляли триэтиламин (104,5 мл, 750 мМ), N,N-диметиламинопиридин (1,00 г) и пропионовый ангидрид (48,1 мл). Полученный раствор перемешивали при 0oC в течение 30 мин, а затем оставляли для нагревания до комнатной температуры. После этого через 1 ч добавляли еще триэтиламин (33 мл) и пропионовый ангидрид, а затем еще через 4,5 ч реакционную смесь промывали насыщенным водным гидрокарбонатом натрия (500 мл) и концентрировали. Остаток суспендировали в диэтиловом эфире (500 мл) и промывали насыщенным водным гидрокарбонатом натрия (3 • 500 мл), водой (2 • 500 мл) и солевым раствором (500 мл). Органическую фазу осушали сульфатом магния, фильтровали и концентрировали. Полученный таким образом остаток хроматографировали (элюент: гексан/этилацетат, 10:1 - 1:1) и получали дипропиониловое соединение (прибл. 60 г) и монопропиониловый побочный продукт (3,4 г).
(4) Получение карбоновой кислоты (Ireland R.E. et al. J. Am. Chem. Soc. 110:854-860, 1988).

При -78oC и в атмосфере азота к перемешанному раствору гексаметилдисилазана (35,9 мл, 172,2 мМ) в ТГФ (320 мл) приблизительно в течение 10 мин добавляли раствор н-бутиллития в гексане (63,4 мл, 2,5 М, 158,5 мМ). Полученный раствор перемешивали в течение 30 мин при -78oC а затем приблизительно в течение 10 мин добавляли раствор т-бутилдиметилсилилхлорида (25,88 г, 172,5 мМ) в гексаметилфосфорамиде (85 мл). Полученный раствор перемешивали 5 мин, а затем по капле в течение 25 мин добавляли раствор дипропионата (20,00 г, 57,4 мМ) в ТГФ (80 мл). После этого полученный раствор перемешивали еще 5 мин при -78oC, медленно нагревали до около 0oC и при этой температуре выливали в делительную воронку, содержащую смесь (0oC) ледяной воды (1 л) и петролейного эфира (1,5 л, т.пл. 40 - 60oC). Затем смесь энергично перемешивали и органическую фазу отделяли и промывали при 0oC насыщенным водным раствором NaCl (500 мл), осушали сульфатом натрия, фильтровали и концентрировали на роторном испарителе при 25 - 30oC. Полученное желтое маслообразное вещество растворяли в бензоле (1 л), а раствор нагревали при температуре с обратным холодильником в течение 6 ч. После частичного охлаждения раствор концентрировали на роторном испарителе и остаток растворяли в смеси тетрагидрофурана и воды (прибл. 250 мл). Полученную таким образом смесь перемешивали при комнатной температуре в течение 24 ч (эта процедура может быть пропущена). Тетрагидрофуран удаляли на роторном испарителе и к смеси добавляли 1 М водный раствор NaOH, а полученную суспензию перемешивали 3 ч при комнатной температуре. Затем смесь экстрагировали диэтиловым эфиром (2 • 250 мл), а водную фазу подкисляли до значения pH, примерно равного 2,5, путем добавления 1 М водной HCl (200 мл). Полученную суспензию экстрагировали диэтиловым эфиром (3 • 250 мл), а затем этилацетатом (2 • 250 мл). Объединенные органические экстракты осушали сульфатом натрия, фильтровали и концентрировали, в результате чего получали неочищенную карбоновую кислоту в виде прозрачного маслообразного вещества (18,74 г).
(5) Получение йодолактона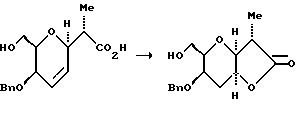
К механически перемешанному раствору неочищенного карбоксилата (30,12 г, прибл. 86 мМ) в насыщенном водном гидрокарбонате натрия (1,00 л) при комнатной температуре добавляли раствор I2 (59,25 г, 234 мМ) и KI (220,5 г, 1,329 М) в H2O (375 мл). Полученную смесь перемешивали при комнатной температуре в течение 14 ч, и в этот период времени ТСХ (элюент: гексан/этилацетат, 1:1) указывала на отсутствие исходного материала. После этого добавляли насыщенный водный раствор Na2S2O3 (400 мл) и полученную смесь экстрагировали этилацетатом (4 • 500 мл). Объединенные органические экстракты осушали сульфатом натрия, фильтровали и концентрировали с получением желтого маслообразного вещества. Этот неочищенный йодолактон использовали без дополнительной очистки. В отдельном эксперименте для проведения реакции было достаточно 6 ч. Также отмечалось, что диастереометрические метиловые эпимеры могут быть хроматографически разделены с помощью хроматографии на силикагеле (гексан/этилацетат, 70:30).
(6) Получение лактона.
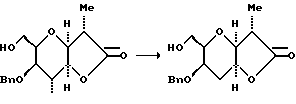
В атмосфере азота перемешанный раствор йодолактона три-н-гидрида бутилолова (28,0 мл, 104 мМ) и 2,2'-азобисизобутиронитрила (AIBN) (100 мг) в бензоле (500 мл) помещали в масляную баню (80oC) и нагревали с обратным холодильником в течение 1 ч. ТСХ (элюент: гексан/этилацетат, 1:1) указывала на отсутствие остаточного исходного материала. Раствор охлаждали до комнатной температуры и концентрировали на роторном испарителе. Остаток хроматографировали (толуол/ацетат) и получали лактон (23,30 г, 79,7 мМ, выход по отношению и дипропионату составлял 92,2%) в виде бесцветного кристаллического твердого вещества.
(7) Получение метилацеталей.
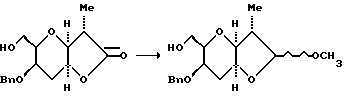
При -78oC и в атмосфере азота к перемешанному раствору лактона (23,25 г, 79 мМ) в ТГФ (500 мл) с использованием капельной воронки в течение примерно 30 мин добавляли 1 М раствор гидрида диизобутилалюминия в гексане (258 мл, 258 мМ). После выдерживания смеси в течение 1 ч при -78oC осторожно в течение 15 мин добавляли безводный метанол (90 мл), а затем насыщенный водный хлорид аммония (90 мл). После этого охлаждающую баню удаляли, добавляли диэтиловый эфир (500 мл) и перемешанную смесь оставляли для нагревания до комнатной температуры. Белую гелеобразную суспензию фильтровали через слой целита, а остаток промывали водой, эфиром (4 • 250 мл) и этилацетатом (2 • 250 мл). Объединенный фильтрат и промывки концентрировали с получением желтого маслообразного продукта После этого добавляли сухой толуол (500 мл) и раствор снова концентрировали на роторном испарителе, в результате чего получали неочищенный гемиацеталь (18,46 г, прибл. 0,8 мМ, выход 79%) в виде прозрачного желтого маслообразного вещества.
Этот полученный гемиацеталь растворяли в безводном метаноле (1 л) и добавляли моногидрат п-толуолсульфоновой кислоты (200 мг). Полученный раствор перемешивали при комнатной температуре в течение 14 ч. ТСХ (гексан/этилацетат/хлороформ, 1: 1: 1) указывала на наличие трех продуктов. После добавления 2 г твердого гидрокарбоната натрия смесь концентрировали на роторном испарителе. Остаток непосредственно помещали на колонку с силикагелем и элюировали гексаном/этилацетатом (1:1 - 0:1), в результате чего получали два наименее полярных продукта (13,98 г, 45,39 мМ, выход 72%) и отдельные наиболее полярные продукты (4,65 г, 15,1 мМ, выход 24%). Эти оба полярных продукта (А и С) имели нужную метильную конфигурацию в то время, как промежуточный Rf - продукт (В) имел нежелательную метильную конфигурацию.
Получение нитрила
К перемешанному раствору спирта (2,70 г, 8,77 мМ) в метиленхлориде (200 мл) в атмосфере аргона и при -42oC добавляли пиридин (1,56 мл, 19,3 мМ), а затем в течение 5 мин по капле добавляли ангидрид трифторометансульфоновой кислоты (2,22 мл, 13,15 мМ). Полученную смесь перемешивали при -42oC в течение 40 мин, и ТСХ указывала на отсутствие исходного материала. Затем добавляли насыщенный водный гидрокарбонат натрия (250 мл) и диэтиловый эфир (400 мл). Отделенный органический слой промывали водой (2 • 400 мл) и солевым раствором (200 мл). Полученный раствор осушали сульфатом натрия, фильтровали и выпаривали на роторном испарителе при температуре, близкой к комнатной, в результате чего получали неочищенный трифторацетат в виде прозрачного желтого маслообразного вещества. Это вещество кроме того концентрировали с использованием всасывающего трубопровода в течение 10 мин, а затем использовали непосредственно в последующей стадии.
Неочищенный трифторацетат растворяли в N,N-диметилформамиде (40 мл) при 0oC и в атмосфере аргона. Затем к перемешанному прозрачному бледно-желтому раствору добавляли NaCN (1,718 г, 35,05 мМ). Полученную смесь нагревали до комнатной температуры и перемешивали в течение 40 мин до тех пор, пока смесь не приобретала темный оттенок. В этот период времени ТСХ указывала на отсутствие остаточного исходного материала. После этого добавляли насыщенный водный гидрокарбонат натрия (200 мл) и диэтиловый эфир (250 мл). Органическую фазу отделяли и промывали водой (2 • 250 мл), а объединенные органические фракции промывали водой (2 • 250 мл) и солевым раствором (200 мл). После осушки сульфатом натрия, фильтрации, концентрирования и хроматографии на силикагеле получали нитрил (1,211 г, 3,82 мМ, выход в двух указанных стадиях составлял 44%) в виде прозрачного маслообразного вещества.
(9) Получение C38 - первичного спирта.
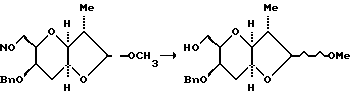
К перемешанному раствору нитрила (5,29 г, 16,7 мМ) в метиленхлориде (250 мл) при -78oC и в атмосфере аргона в течение 15 мин добавляли 1 М раствор гидрида диизобутилалюминия в гексане (25,0 мл, 15,0 мМ). Полученный раствор перемешивали при -78oC в течение 45 мин, после чего добавляли 1 М водной соляной кислоты (50 мл). Охлаждающую баню удаляли и полученную смесь нагревали до 0oC в течение 30 мин. После этого добавляли диэтиловый эфир (600 мл), а смесь промывали еще 1 М соляной кислотой, а затем солевым раствором (прибл. 50 мл). Объединенные водные фазы экстрагировали эфиром (2 • 50 мл), а объединенные органические фракции осушали сульфатом натрия, фильтровали и концентрировали с получением желтого маслообразного продукта. Этот продукт использовали непосредственно без дополнительной очистки.
Неочищенный альдегид растворяли в метаноле (100 мл) и перемешанный раствор охлаждали до 0oC, а затем добавляли NaBH4 (1,00 г, 26,7 мМ). Охлаждающую баню удаляли и через 10 мин растворитель удаляли на роторном испарителе. Остаток суспендировали в H2O (100 мл) и экстрагировали этилацетатом (4 • 100 мл). Объединенные экстракты промывали солевым раствором (100 мл), осушали сульфатом натрия, фильтровали, концентрировали и хроматографировали, в результате чего получали первичный спирт (4,384 г, 13,6 мМ, выход по отношению к двум стадиям составлял 81%) в виде прозрачного бесцветного маслообразного вещества.
(10) Получение диола.
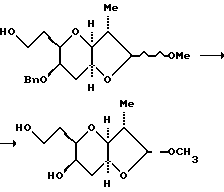
К перемешанному раствору полученного таким образом первичного спирта (5,27 г, 16,3 мМ) в метаноле (200 мл) добавляли 10% Pd (OH)2 на угле (1 г). Быстро перемешанную смесь откачивали и снова наполняли H2 (4 раза), а затем в течение 13 ч перемешивали при давлении H2 в 1 атмосферу. В этот период времени ТСХ указывала на отсутствие остаточного бензилового эфира. Смесь фильтровали через целит, промывали метанолом, а объединенный фильтрат и промывки концентрировали и получали прозрачное маслообразное вещество (3,685 г, 15,9 мМ, выход 97%).
(11) Получение диметилацеталя.
При -78oC и в атмосфере аргона к перемешанному раствору метилацеталя (3,571 г, 15,37 мМ) в CH2Cl2 (60 мл) добавляли конденсированный метилмеркаптан (прибл. 20 мл). Полученный раствор нагревали до 0oC и добавляли BF3•OEt3 (2 мл). После перемешивания в течение 30 мин при 0oC, ТСХ (элюент; этилацетат) указывала на полную конверсию в одно пятно с высоким Rf. После этого по капле осторожно добавляли насыщенный водный гидрокарбонат натрия (50 мл) и H2O (50 мл), а затем водную фазу отделяли и экстрагировали метиленхлоридом (4 • 150 мл). Объединенные органические фракции осушали безводным K2CO3, фильтровали и концентрировали, в результате чего получали дитиоацеталь (4,206 г, 14,19 мМ, выход 92,3%) в виде прозрачного бесцветного пенистого вещества.
(12) Получение диметилацеталя трисилилэфира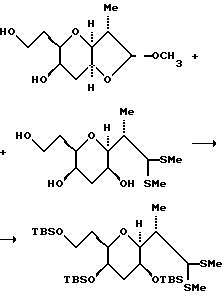
При 0oC к раствору триола (1,26 г, 4,29 мМ) в метиленхлориде (30 мл) добавляли триэтиламин (4,5 мл, 32,2 мМ), а затем т-бутилдиметилсилилтрифтороацетат (3,7 мл, 16,2 мМ). Через 1 ч ТСХ-анализ (гексан/EtOAc, 20:1) указывал на присутствие исходного материала в дополнение к моно-, ди- и трисилированным продуктам присоединения. В это время к реакционной смеси добавляли еще триэтиламин (4,5 мл, 32,2 мМ) и т-бутилдиметилсилилтрифтороацетат (3,7 мл, 16,2 мМ), после чего реакционный раствор перемешивали 2 ч. Затем реакцию завершали путем добавления насыщенного водного бикарбоната натрия (50 мл). Полученную таким образом смесь тщательно экстрагировали этилацетатом и объединенные органические экстракты промывали солевым раствором, осушали сульфатом натрия и концентрировали в вакууме. Остаточное маслообразное вещество очищали с помощью флеш-хроматографии (элюент: гексан/этилацетат, 50:1) и получали трисилиловый эфир (2,2 г, выход 80%) в виде светло-желтого маслообразного вещества.
(13) Получение альдегида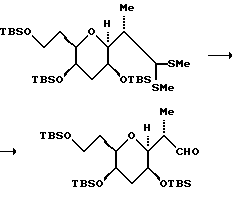
К перемешанному раствору дитиоацеталя (640 мг, 1,00 мМ) в ацетоне /H2O (9:1, об/об, 50 мл), при комнатной температуре добавляли твердый NaHCO3 (252 мг, 1 мМ), а затем I2 (254 мг, 1 мМ). Через 30 мин красную реакционную смесь охлаждали до 0oC и добавляли еще NaHCO3 (252 мг) и I2 (254 мг). После выдерживания смеси в течение 30 мин при 0oC добавляли еще NaHCO3 (252 мг и I2 (254 мг) и смесь оставляли для нагревания до комнатной температуры. Через 190 мин (всего) ТСХ указывала на отсутствие остаточного исходного материала. После этого реакционную смесь выливали в делительную воронку, содержащую этилацетат (50 мл) и 10%-ный водный Na2S2O3 (50 мл). После встряхивания и удаления водной фазы прозрачную бесцветную органическую фазу промывали водой и солевым раствором (прибл. 50 мл). Объединенные водные фракции экстрагировали этилацетатом (2 • 50 мл), а объединенные органические фракции осушали сульфатом натрия, фильтровали и концентрировали. Остаток хроматографировали на силикагеле (элюент: гексан/этилацетат, 10:1) и получали альдегид (509 мг, 907 мкМ, выход 91%) в виде прозрачного бесцветного маслообразного продукта.
(14) Получение метилакрилатов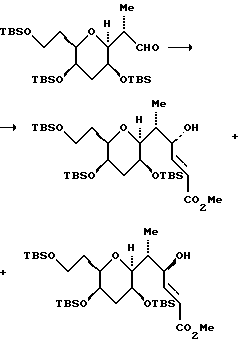
В атмосфере азота, смесь альдегида (989 мг, 1,75 мМ) и транс- β -йодо-метилакрилата (1,85 г, 8,74 мМ) растворяли в ТГФ (10 мл). К перемешанному при комнатной температуре раствору добавляли порошкообразный CrCl2, содержащий приблизительно 750 мг NiCl2 (1 мас.%). Через 50 мин добавляли еще 1% NiCl2/CrCl2 (прибл. 500 мг) к бледно-зеленой суспензии и полученную смесь перемешивали в течение 22 ч при комнатной температуре. После этого реакционную смесь разводили насыщенным водным хлоридом аммония (20 мл) и экстрагировали этилацетатом, а затем диэтиловым эфиром (4 • 10 мл). Объединенные экстракты концентрировали на роторном испарителе, а остаток суспендировали в этилацетате (20 мл) и промывали водой (2 • 20 мл) и солевым раствором (10 мл). Этилацетатный раствор осушали сульфатом натрия, фильтровали и концентрировали. Остаток 2 раза хроматографировали на силикагеле (прибл. 150 г SiO2, гексан/трет-бутилметиловый эфир, 6:1, гексан/этилацетат/CHCl3, 5:1:1) и получали два диастереомерных продукта (912 мг, 1,47 мМ, объединенный выход 84%).
(15) С30-инверсия.
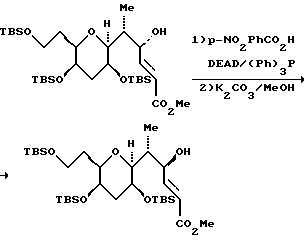
К перемешанному раствору трифенилфосфина (373 мг, 1,42 мМ) и п-нитробензойной кислоты (238 мг, 1,42 мМ) в диэтиловом эфире (20 мл) и толуоле (10 мл) при комнатной температуре добавляли раствор спирта (441 мг, 712 мкМ) в диэтиловом эфире/толуоле (2:1, об/об, 10 мл). Затем к полученному прозрачному раствору по капле добавляли диэтилазидодикарбоксилат (224 мкл, 1,42 мМ). Образовавшийся прозрачный желтый раствор перемешивали при комнатной температуре в течение 4 ч. ТСХ (элюент: гексан/этилацетат/хлороформ, 5:1:1) указывала на отсутствие остаточного исходного материала. После этого добавляли насыщенный водный хлорид аммония (30 мл) и выделенную органическую фазу промывали насыщенным водным гидрокарбонатом натрия, водой и солевым раствором (прибл. 10 мл). Органическую фазу осушали сульфатом натрия, фильтровали, концентрировали и хроматографировали, в результате чего получали п-нитробензоат в виде прозрачного желтого маслообразного вещества (522 мг).
К перемешанному при 0oC раствору п-нитробензоата (522 мг) в метаноле (10 мл) добавляли K2CO3 (5 мг). После перемешивания в течение 30 мин ТСХ не обнаруживала остаточного исходного материала. К смеси добавляли 5 мкл уксусной кислоты и полученную смесь концентрировали, а остаток хроматографировали, в результате чего получали спирт (404 мг, выход 91%) в виде бесцветного маслообразного вещества.
(16) Образование метоксифенилметилового эфира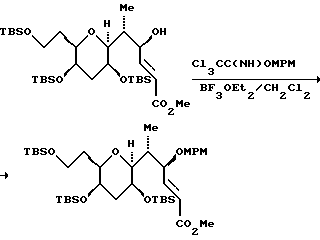
При 0oC к перемешанному раствору спирта (755 мг, 1,22 мМ) и п-метоксибензилтрихлороацетамида (3,446 г, 12,2 мМ) в метиленхлориде (120 мл) добавляли 0,1 М раствор BF3•OEt2 в CH2Cl2 (50 мкл.). Полученный оранжевый раствор перемешивали в течение 10 мин и в этот период времени ТСХ указывала на отсутствие исходного материала. После этого добавляли насыщенный водный NaHCO3 (40 мл) и смесь энергично перемешивали, а органическую фазу отделяли, осушали сульфатом натрия, фильтровали и концентрировали. Остаток хроматографировали на силикагеле (элюент: гексан/этилацетат, 5:1) и получали п-метоксибензиловый эфир (866 мг, 1,17 мМ, выход 96%) в виде прозрачного маслообразного вещества.
(17) Получение триола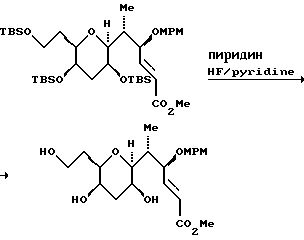
При 0oC к перемешанному раствору п-метоксибензилового эфира (986 мг, 1,33 мМ) в ацетонитриле (25 мл) добавляли пиридин (500 мкл), а затем в течение 1 мин добавляли реагент HF•пиридин (5,0 мл, Aldrich). Через 75 мин ТСХ (этилацетат) указывала на почти полное завершение реакции. Затем к раствору порциями осторожно добавляли насыщенный водный гидрокарбонат натрия (200 мл). Полученную смесь экстрагировали этилацетатом (3 • 200 мл), а объединенные экстракты осушали сульфатом натрия, фильтровали и концентрировали, в результате чего получали неочищенный триол (590 мг) в виде прозрачного оранжевого маслообразного вещества.
[α]
(18) Получение ацетонида.
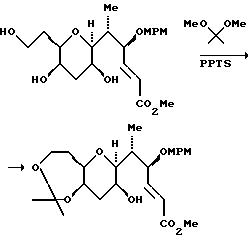
К раствору неочищенного триола (560 мг, прибл. 1,3 мМ) в метиленхлориде (10 мл) при комнатной температуре добавляли 2,2-диметоксипропан (327 мкл, 2,66 мМ), а затем п-толуолсульфонат пиридиния (5 мг). Через 1 ч добавляли еще 327 мкл (2,66 мМ) 2,2-диметоксиропана. Реакционный раствор оставляли для перемешивания на 24 ч и в этот период времени ТСХ указывала на полную конверсию в одно пятно с высоким Rf. После этого реакционную смесь промывали насыщенным водным гидрокарбонатом натрия (50 мл), водой (50 мл) и солевым раствором (50 мл), а полученную органическую фазу осушали сульфатом натрия, фильтровали, концентрировали и полученный остаток хроматографировали (элюент: этилацетат/гексан/триэтиламин, 1:1:0,001 - 1:0:0,001), в результате чего получали 7-позиционный ацетонид (516 мг) в виде бесцветной пены.
(19) Добавление Michael-типа.
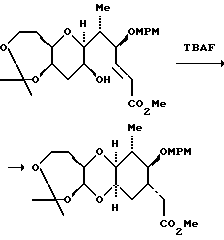
К перемешанному (0oC) раствору гидроксиакрилата (420 мг, 904 мкМ) в ТГФ (36 мл) и безводного метилацетата (4 мл) добавляли 1 М раствор фторида тетрабутиламмония (TBAF) в ТГФ (9 мл, 9 мМ, Aldrich). Через 1 ч добавляли еще метилацетат (4 мл) и раствор TBAF (4,5 мл). После выдерживания раствора в течение 6 ч при 0oC этот раствор разводили насыщенным водным гидрокарбонатом натрия (175 мл) и солевым раствором (175 мл), а затем экстрагировали этилацетатом (3 • 100 мл). Объединенные экстракты промывали водой (250 мл) и солевым раствором (250 мл), после чего осушали сульфатом натрия, фильтровали и концентрировали. Остаток хроматографировали на силикагеле и в процессе элюирования получали исходный материал (57 мг), нужный циклизованный продукт (336 мг, 723 мкМ, выход 80%) и нежелательный С29-эпимерный циклизованный продукт (16 мг), причем все эти продукты имели вид прозрачных маслообразных веществ.
(20) Получение диола.
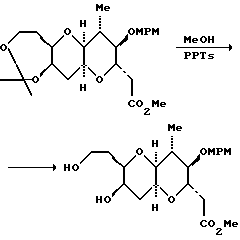
К перемешанному раствору ацетонида (268 мг, 577 мкМ) в метаноле (10 мл) при комнатной температуре добавляли паратолуолсульфонат пиридиния (5 мг). После 20-минутного перемешивания при комнатной температуре ТСХ указывала на отсутствие остаточного исходного материала. После этого к раствору добавляли твердый гидрокарбонат натрия (50 мг) и смесь концентрировали на роторном испарителе. Остаток фильтровали через узкую прокладку из силикагеля с использованием этилацетата и фильтрат концентрировали, в результате чего получали неочищенный диол (232 мг, 547 мкМ, выход 95%) в виде бесцветного маслообразного вещества. Этот материал использовали без дополнительной очистки.
(21) Получение дисилилового эфира.
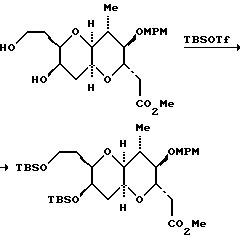
При 0oC и в атмосфере аргона к перемешанному раствору диола (230 мг, 542 мкМ) добавляли триэтиламин (605 мкл, 4,33 мМ), а затем т-бутилдиметилсилилтрифторометансульфонат (498 мкл, 2,17 мМ). Через 1 ч к раствору добавляли еще триэтиламин (303 мкл) и трет-бутилдиметилсилилтрифторометансульфонат (249 мкл). Через 2 ч (всего) реакционную смесь промывали насыщенным водным NaHCO3, осушали (Na2SO4), фильтровали и концентрировали. Остаток хроматографировали на силикагеле (гексан/этилацетат, 5:1) и получали дисилиловый эфир (308,8 мг, 473 мкМ, выход 87%) в виде прозрачного бесцветного маслообразного вещества.
(22) Получение дисилилового спирта.
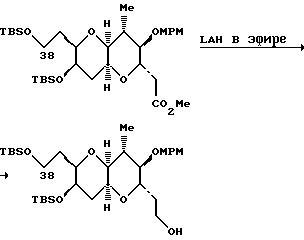
При 0oC к раствору сложного эфира (306 мг, 0,480 мМ) в эфире (10 мл) добавляли 1,6 М раствор алюмогидрида лития (0,60 мл, 2 экв.) в диэтиловом эфире. Через 5 мин добавляли насыщенный водный раствор сегнетовой соли и хлорида аммония и полученную смесь перемешивали до тех пор, пока не образовывались две прозрачные фазы. После этого смесь экстрагировали этилацетатом (2 • 10 мл), а объединенные экстракты осушали сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюент:гексан/этилацетат/хлороформ, 5:1:1) и получали спирт (278 мг, выход 95,2%).
ИК (пленка): 775 см-1, 385, 1251, 1472, 1514, 2930, 2956, 3453.
1Н-ЯМР (CDCl3): δ 0,47 (6H, с); 0,53 (3H, с); 0,80 (3H, с); 0,89 (9H, с, т-бут. ); 0,90 (9H, с, т-бут); 1,15 (3H, д, J = 7,1 Гц); 1,60 (1H, м); 1,76 (2H, м); 1,82 (1H, м); 1,93 (1H, м); 2,08 (2H, м); 2,98 (1H, шир. с.); 3,06 (1H, м); 3,37 (1H, т, J = 4,2 Гц); 3,50 (1H, м); 3,71 (2H, м); 3,79 (1H, м); 3,80 (3H, с, -OMe); 4,13 (1H, м); 4,45 (2H, д, J = 10,9 Гц); 4,56 (2H, д, J = 10,9 Гц); 6,87 (2H, д, J = 8,5 Гц); 7,24 (2H, д, J = 8,5 Гц).
BPMC (FAB) для C33H60O7Si2 + Na:
Вычислено: 6473775; Найдено: 6473763.
[α]D -20,2o (с = 0,99, MeOH).
(23) Получение дисилилового альдегида (соединение 6)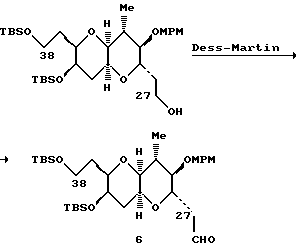
К перемешанному раствору спирта (100 мг, 0,164 мМ) в дихлорметане (5 мл) при комнатной температуре добавляли периодинановый реагент Dess-Martin (150 мг, 2,5 экв. ). Примерно через 1 ч ТСХ указывала на отсутствие остаточного исходного материала. Реакционную смесь разводили диэтиловым эфиром (25 мл) и добавляли водный раствор тиосульфата натрия и насыщенного гидрокарбоната натрия. Полученную смесь перемешивали до образования двух прозрачных фаз, а затем экстрагировали диэтиловым эфиром (2 • 15 мл). Объединенные экстракты осушали безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: 12% этилацетат в гексане) и получали альдегид (92,5 мг, выход 92,8%).
Соединение 7.
Соединение 7 синтезировали в соответствии со следующей процедурой.

К раствору альдегида (102 мг, 0,168 мМ) и мезилата (141,6 мг, 0,255 мМ) в 17% (об. /об.) ДМФ в ТГФ (2,08 мл) добавляли примерно 20 мг 0,1% NiCl2 в LiCl2 и примерно 10 мг 1% NiCl2 в CrCl2. Затем еще 4 раза в течение 26 ч в тех же пропорциях добавляли металлические реагенты. Реакционную смесь разводили насыщенным водным хлоридом аммония (8 мл) и экстрагировали этилацетатом (3 • 10 мл). Объединенные органические слои осушали безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный остаток осушали путем азеотропного удаления воды с использованием бензола, а затем осушали в высоком вакууме. Этот остаток растворяли в 1,2-диметоксиэтане (25 мл) и 35% дисперсию KH (прибл. 10 мг) в минеральном масле добавляли к полученному раствору. Затем реакционную колбу помещали в масляную баню (80oC) в течение 2,5 мин, а затем охлаждали до 0oC в ледяной/водяной бане. Реакционную смесь разводили безводным эфиром (30 мл), фильтровали через слой силикагеля вместе с дополнительными промывками безводного эфира (30 мл). Фильтрат концентрировали и остаток очищали с помощью колоночной хроматографии на силикагеле (элюент: 12% этилацетат в гексане), в результате чего получали 86 мг продукта (выход 53%).
ИК (пленка): 722 см-1, 835, 1040, 1250, 1463, 1514, 1755, 2854, 2926.
BPMC (FAB) для C54H92O10Si2 + Na: Вычислено: 9796124; Найдено: 9796149.
[α]D :-20,0oC (с = 3,3, MeOH).
1Н-ЯМР (C6D6): δ 0,04 (3H, с); 0,10 (3H, с); 0,11 (3H, с); 0,14 (3H, с); 0,97 (3H, д, J = 6,5 Гц); 1,00 (9H, с); 1,02 (9H, с); 1,11 (1H, м); 1,16 (9H, с); 1,26 (4H, м); 1,51 - 1,90 (11H, м); 1,98 - 2,20 (5H, м); 2,27 (2H, м); 2,39 (1H, м); 2,45 (1H, м); 3,23 (2H, т, J = 9,0 Гц); 3,31 (3H, с); 3,53 (1H, м); 3,68 (2H, м); 3,75 (1H, м); 3,86 (1H, м); 3,93 (1H, м); 4,02 (2H, м); 4,19 (1H, м); 4,31 (1H, м); 4,44 (1H, м); 4,54 (2H, кв., J = 7,9 Гц); 4,83 (1H, с); 4,86 (1H, шир. д.,); 4,91 (1H, шир. д.); 5,14 (1H, с); 6,80 (2H, д, J = 8,4 Гц); 7,28 (2H, д, J = 8,4 Гц).
При 0oC к раствору C14-пивалоата (27,8 мг, 0,024 мМ) в безводном эфире (2 мл) добавляли 1,6 М раствор алюмогидрида лития в диэтиловом эфире (38 мкл, 61 мкМ). Реакционную смесь перемешивали в течение 5 мин, разводили этилацетатом (4 мл) и добавляли насыщенный водный раствор сегнетовой соли и хлорида аммония. После этого реакционную смесь перемешивали до образования прозрачного водного слоя. Органический слой отделяли, а водный слой экстрагировали этилацетатом (2 • 4 мл). Объединенные органические слои осушали сульфатом натрия и концентрировали при пониженном давлении. Маслообразный остаток очищали с помощью колоночной хроматографии (элюент: 50% гексан в этилацетате) и получали C14-спирт (25 мг, выход 98,5%).
Соединение 8.
Соединение 8 синтезировали из диацетон глюкозы в соответствии с двумя нижеследующими процедурами.
Пример 1.
К перемешанному раствору ДМСО (10,65 мл, 150,1 мМ) в метиленхлориде (300 мл) при -78oC и в атмосфере аргона по капле добавляли ангидрид трифтороуксусной кислоты (16,00 мл, 113,3 мМ). Полученную белую суспензию перемешивали при -78oC в течение 10 мин, а затем медленно через канюлю добавляли раствор 1,2 : 5,6-ди-O-изопропилиден- α -D-глюкофуранозы. После этого полученную смесь перемешивали в течение 1 ч при -78oC, медленно добавляли триэтиламин (30,4 мл, 218 мМ) и реакционную смесь оставляли для нагревания до комнатной температуры. Затем добавляли насыщенный водный хлорид аммония (100 мл), а органическую фазу отделяли и промывали холодным водным 1 М раствором HCl (2 • 100 мл), водой (100 мл), насыщенным водным раствором NaHCO3 (100 мл), снова водой (100 мл) и после этого насыщенным водным раствором NaCl (100 мл). Органическую фазу осушали, фильтровали и концентрировали с получением неочищенного кетона (1,2:2,5-ди-O-изопропилиден- α -D-рибо-гексофуранос-3-улозы, 22,3 г) в виде бледно-желтого маслообразного вещества. Это вещество использовали непосредственно без дополнительной очистки.
При 0oC к перемешанному раствору неочищенного кетона (22,3 г, прибл. 75 мМ) в 95%-ном этаноле (200 мл) порциями в течение 20 мин добавляли NaBH4 (5,8 г, 154 мМ). После дополнительного перемешивания в течение 10 мин ТСХ указывала на отсутствие остаточного исходного материала. Растворитель удаляли на роторном испарителе и остаток суспендировали в этилацетате (200 мл), а затем промывали водой (2•100 мл) и насыщенным водным раствором NaCl (100 мл). Органическую фазу осушали сульфатом натрия, фильтровали и концентрировали, в результате чего получали неочищенную 1,2:5,6-ди-O-изопропилиден- α -D-аллофураносеаллозу в виде белого твердого продукта (19,64 г, прибл. 75 мМ, выход в описанных двух стадиях составлял 98%).

При 0oC и в атмосфере аргона к перемешанному раствору диацетонида аллозы (19,6 г, прибл. 75 мМ) в ТГФ/ДМФ (500 мл, 4:1 по объему) порциями в течение 30 мин добавляли 60%-ную дисперсию гидрида натрия в минеральном масле (6,0 г, 150 мМ). Полученную смесь нагревали до комнатной температуры в течение 1 ч, а затем снова охлаждали до 0oC, после чего добавляли бензилбромид (25,7 г, 17,9 мл, 150 мМ) и иодид т-н-бутиламмония (1,0 г). Эту смесь оставляли для нагревания до комнатной температуры и перемешивали в течение 16 ч. После охлаждения до 0oC к реакционной смеси осторожно в течение 30 мин добавляли безводный метанол (20 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1 ч, после чего ТГФ удаляли с помощью роторного испарителя. Остаток суспендировали в воде (250 мл) и экстрагировали этилацетатом (4 • 200 мл). Объединенные органические фракции промывали водой (500 мл) и солевым раствором (200 мл), а затем осушали сульфатом натрия, фильтровали и концентрировали с получением неочищенной 3-O-бензил- 1,2:5,6-ди-O-изопропилиден- α -D-аллофуранозы в виде прозрачного желтого маслообразного вещества (46,8 г). Этот материал использовали без дополнительной очистки.
При 0oC к перемешанной смеси неочищенного бензилового эфира и диацетонида (46,8 г, примерно 75 мМ) в метаноле/H2O (500 мл, 4:1 по объему) добавляли водный 1 М раствор соляной кислоты (50 мл). Полученную смесь нагревали до комнатной температуры и перемешивали в течение 7,5 ч, после чего ее нейтрализовали, добавляя порциями твердый гидрокарбонат натрия (20 г). Метанол удаляли с помощью роторного испарителя, а полученную водную суспензию экстрагировали этилацетатом (6 • 100 мл). Объединенные органические фракции осушали сульфатом натрия, фильтровали и концентрировали, в результате чего получали неочищенную 3-O-бензил-1,2-O-изопропилиден- α -D-аллофуранозу в виде прозрачного желтого маслообразного вещества. Полученный материал использовали без дополнительной очистки.
ИК (пленка): 3420 см-1, 3070, 3065, 3032, 2986, 2934, 2901, 1498, 1454, 1434, 1423, 1409, 1382, 1373, 1316, 1250, 1214, 1167, 1137, 1122, 1095, 1025, 697.
1Н-ЯМР (CDCl3): δ 1,35 (3H, с); 1,58 (3H, с); 2,71 (1H, шир. т., J = 5,7 Гц); 2,82 (1H, д, J = 3,5 Гц); 3,67 (2H, м); 3,93 (1H, дд, J = 4,3, 8,9 Гц); 4,00 (1H, м); 4,09 (1H, дд, J = 3,2, 8,9 Гц); 4,55 (1H, д, J = 11,4 Гц); 4,61 (1H, м); 4,77 (1H, д, J = 11,4 Гц); 5,75 (1H, д, = 3,8 Гц); 7,25 - 7,50 (5H, м).
13С-ЯМР (CDCl3): 26,56, 26,74, 63,05, 71,00, 72,13, 77,00, 77,33, 79,06, 104,21, 113,15, 128,20, 128,53, 136,85.
[α]D :+ 107,3oC (с = 6,65, CHCl3).

При 0oC к раствору неочищенного диола (прибл. 75 мМ) в метиленхлориде (300 мл) добавляли пиридин (90 мл, 1,1 М), а затем метансульфонилхлорид (30 мл, 388 мМ). Полученный раствор перемешивали при 0oC в течение 1 ч, нагревали до комнатной температуры и перемешивали еще 3 ч. После этого к раствору осторожно порциями в течение 30 мин добавляли насыщенный водный раствор гидрокарбоната натрия (100 мл). Органическую фазу отделяли и промывали насыщенным водным раствором гидрокарбоната натрия (100 мл), водным 1 М раствором HCl (2 • 100 мл), водой (100 мл) и солевым раствором (100 мл). Органическую фазу осушали сульфатом натрия, фильтровали и концентрировали с получением оранжевого маслообразного вещества. Этот неочищенный димезилат, т.е., 3-O-бензил-1,2-O-изопропилиден-5,6-ди-O-метансульфонил- α -D-аллофуранозу использовали без дополнительной очистки. (См.: Brimacombe, J.S.; Mofti, A.M.; Tucker, L. C.N. J. Chem. Soc. (C) 1971, 2911-2915, где они использовали 3-O-метиловый аналог).
ИК (пленка): 3032 см-1, 2970, 2939, 1455, 1361, 1247, 1241, 1219, 1176, 1135, 1122, 1104, 1093, 1027, 1010, 972, 924, 872, 831, 797, 746, 701, 665.
1Н-ЯМР (CDCl3): δ 1,36 (3H, с); 1,58 (3H, с); 3,00 (3H, с); 3,03 (3H, с); 3,95 (1H, дд, J = 4,3, 8,8 Гц); 4,20 (1H, дд, J = 3,2, 8,8 Гц); 4,39 (2H, д, J = 3,5 Гц); 4,56 (1H, д, J = 11,2 Гц); 4,59 (1H, м); 4,75 (1H, д, J = 11,2 Гц); 5,10 (1H, м); 5,75 (1H, д, J = 3,6 Гц); 7,25 - 7,50 (5H, м).
13С-ЯМР (CDCl3): 26,53, 26,83, 37,78, 38,74, 66,67, 72,29, 76,46, 77,11, 77,65, 104,34, 113,70, 128,41, 128,61.
[α]D : +70,0oC (с = 2,86, CHCl3).
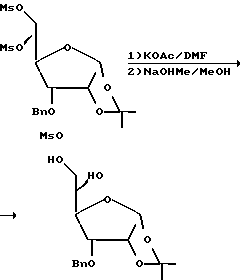
К раствору неочищенного димезилата (прибл. 75 мМ) в ДМФ (350 мл) добавляли порошкообразный ацетат калия (20 г, 204 мМ). Полученную смесь перемешивали в течение 1 ч при комнатной температуре, а затем нагревали в течение 18 ч до 145 - 151oC. ТСХ указывала на отсутствие исходного материала, но также свидетельствовала о наличии трех продуктов. После этого смесь охлаждали до температуры, близкой к комнатной, разводили водой (1,4 л) и экстрагировали этилацетатом (4 • 500 мл). Объединенные экстракты промывали водой и солевым раствором, осушали сульфатом натрия, фильтровали и концентрировали с получением темно-коричневого маслообразного вещества. Это вещество растворяли в безводном метаноле (250 мл) и полученный раствор охлаждали (в атмосфере аргона) до 0oC, перемешивая при этом. Затем медленно с использованием канюли к раствору добавляли свежеприготовленный раствор натрия (5 г) в метаноле (200 мл) и полученный раствор перемешивали при 0oC в течение 1 ч, после чего нагревали до комнатной температуры (при перемешивании) в течение 3 ч. К этому раствору добавляли 10 г твердого хлорида аммония, а растворитель удаляли с помощью роторного испарителя. Остаток суспендировали в этилацетате (100 мл) и фильтровали через воронку из спеченного стекла. Твердый остаток промывали этилацетатом (50 мл), а объединенный фильтрат и промывки концентрировали и получали коричневое маслообразное вещество. После хроматографии на силикагеле (элюент: этилацетат/гексан, 4:1 - 4:0) получали диол(3-O-бензил-1,2-O-изопропилиден- α -D-талофураноза (6)6, в виде бесцветного прозрачного маслообразного вещества (14,6 г, 47,2 мМ; при этом, выход за пять стадий составлял 63%).
ИК (пленка): 3431 см-1, 3064, 3032, 2987, 2935, 1653, 1496, 1454, 1382, 1373, 1309, 1216, 1168, 1129, 1102, 1075, 1025, 921, 872, 739, 699, 666.
1Н-ЯМР (CDCl3): δ 1,38 (3H, с); 1,59 (3H, с); 2,51 (2H, шир. с); 3,64 - 3,80 (3H, м); 3,93 (1H, дд, J = 4,3, 8,9 Гц); 4,05 (1H, дд, J = 2,2, 9,0 Гц); 4,53 (1H, м); 4,58 (1H, д, J = 11,7 Гц); 4,76 (1H, д, J = 11,7 Гц); 5,73 (1H, д, J = 3,6 Гц); 7,25 - 7,50 (5H, м).
13С-ЯМР (CDCl3): δ 26,55, 26,84, 64,84, 69,85, 72,33, 77,18, 77,68, 79,75, 104,41, 113,13, 127,94, 128,36, 137,25.
[α]D : + 93,1oC (с = 9,99, CHCl3).
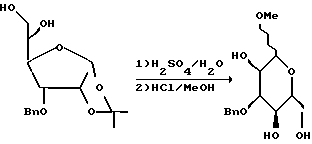
Перемешанный раствор ацетонида (14,6 г, 47,2 мМ) в водном 0,25 М растворе H2SO4 (500 мл) нагревали в течение 1 ч при 60oC, а затем в течение 3 ч охлаждали до комнатной температуры, перемешивая при этом. Затем раствор осторожно нейтрализовали путем добавления (порциями) твердого BaCO3 (12,33 г, 62,5 мМ). Суспензию фильтровали через воронку из спеченного стекла вместе с H2O-промывками. После этого объединенные промывки и фильтрат концентрировали на роторном испарителе с получением сиропа, который кроме того осушали в течение ночи на всасывающем трубопроводе и получали 12,7 г (прибл. 47,2 мМ) неочищенной L-3-O-бензилталозы.
К раствору L-3-O-бензилталозы (11,7 г, 43,5 мМ) в безводном метаноле (250 мл) осторожно по капле добавляли ацетилхлорид (3,0 мл, 34,6 мМ). Полученный раствор нагревали с обратным холодильником в течение 72 ч, а затем охлаждали до комнатной температуры. После добавления твердого гидрокарбоната натрия (5 г) растворитель удаляли на роторном испарителе. Остаток суспендировали в этилацетате (250 мл) и фильтровали. Образовавшиеся твердые продукты еще раз промывали этилацетатом и объединенные промывки и фильтрат концентрировали с получением α,β- -L-талопиранозидов в виде оранжевых маслообразных веществ (10,87 г, 38,4 мМ, выход 81%).
Лит. : α D-метил-талопиранозид: [α]D +98o (с = 1,3, H2O). β -D-метил-талопиранозид: [α]D :24 +28,5o (с = 1,5, H2O). См.: Angyal, S.J.; Bodkin, C. L.; Parrish, F.W. Aust. J.Chem. 28:1541-1549, 1975.
Лит. : α -D-метил-талопиранозид: [α]D : +105o (H2O). См. Gorin, P.A. J. Can. J. Chem. 38:641-651, 1960.
Лит. : α -D-метил-талопиранозид: [α]D : +106,5o (с = 0,97, H2O). См.: Evans, M.E. и др., Carb. Res. 54:105-114, 1977.
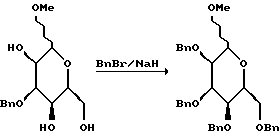
В атмосфере аргона (при 0oC) к перемешанному раствору неочищенного метилталозида (23,8 г, 84 мМ) в ДМФ (250 мл), порциями в течение 30 мин добавляли гидрид натрия (15,2 г 60%-ной дисперсии в минеральном масле; 380 мМ). Полученную смесь нагревали до комнатной температуры и перемешивали в течение 1 ч, после чего снова охлаждали до 0oC. Затем к смеси по капле в течение 20 мин добавляли бензилбромид (38 мл, 318 мМ), после чего еще через 20 мин добавляли 2 г иодида тетра-н-бутиламмония (TBAI). Эту смесь перемешивали при 0oC в течение 30 мин, а затем нагревали до комнатной температуры и перемешивали в течение 18 ч. В этот период времени ТСХ указывала на наличие остаточного исходного материала. После повторного охлаждения смеси до 0oC добавляли еще гидрид натрия (5,1 г 60%-ной дисперсии, 128 мМ), бензилбромид (12,7 мл, 106 мМ) и TBAI (2 г). Указанную смесь снова нагревали до комнатной температуры и перемешивали в течение 6 ч. Через 24 ч реакционную смесь охлаждали до 0oC, осторожно добавляли 20 мл безводного метанола, и смесь перемешивали в течение 1 ч. После этого реакционную смесь разводили водой (1 л) и экстрагировали диэтиловым эфиром (4 • 500 мл). Объединенные органические экстракты промывали водой (500 мл), солевым раствором (500 мл), осушали сульфатом натрия, фильтровали и концентрировали. Остаток подвергали хроматографии на силикагеле и получали α,β- -L-метил-2,3,4,6-тетра-O-бензилталопиранозид в виде прозрачного маслообразного вещества (44,31 г, 80 мМ, выход 95%).
ИК (CCl4): 3064 см-1, 3030, 2897, 1497, 1454, 1360, 1134, 1101, 1028, 735, 697.
1Н-ЯМР (CDCl3): δ 3,34 (3H, с); 3,71 - 3,76 (4H, м); 3,91 (1H, шир. с); 3,93 (1H, м); 4,49 (1H, J = 11,8 Гц); 4,52 (2H, с); 4,56 (1H, д, J = 11,8 Гц); 4,75 (2H, д, J = 11,7 Гц); 4,87 (1H, д, J = 1,5 Гц); 4,88 (1H, д, J = 11,7 Гц); 4,97 (1H, д, J = 11,7 Гц); 7,20 - 7,50 (20H, м).
13С-ЯМР (CDCl3): 55,94 (м.д.), 69,57, 70,61, 71,04, 73,07, 73,28, 73,52, 73,70, 74,18, 100,23, 127,14, 127,24, 127,36, 127,45, 127,58, 127,75, 128,03, 128,14, 128,19, 128,32, 138,24, 138,36, 138,70, 139,06.
[α]D : - 24,8oC (с = 0,81, CHCl3).
Лит. : α -D-аналог) 1Н-ЯМР (CDCl3): 4,79 (J 1,2 1,8 Гц, H1); 5,08 (H2); 5,25 (H4); 4,22 (H5); 4,22 (H6); 3,42 (OCH3), 2,00, 2,08, 2,16. См.: Banaszek, A. и др., Pol. J. Chem. 53:2029-2039, 1979.
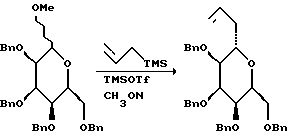
При 0oC и в атмосфере аргона к перемешанному раствору метилталозида (20,00 г, 36,1 мМ) в ацетонитриле (220 мл) добавляли аллилтриметилсилан (23 мл, 145 мМ), а затем по капле добавляли триметилсилилтрифторометансульфонат (7,1 мл, (37 мМ). Полученный прозрачный желтый раствор перемешивали в течение 10 мин при 0oC, а затем нагревали до комнатной температуры. Через 1 ч ТСХ указывала на отсутствие остаточного исходного материала. После этого реакционную смесь разводили диэтиловым эфиром (400 мл) и промывали насыщенным водным раствором NaHCO3 (2 • 200 мл), водой (200 мл) и солевым раствором (200 мл). Органическую фазу осушали сульфатом натрия, фильтровали концентрировали. Остаток хроматографировали (SiO2; элюент: гексан/этилацетат, 7:1) и получали мажорный аллилированный продукт в виде прозрачного бесцветного маслообразного вещества (16,207 г, 28,74 мМ, выход 80%).
ИК (пленка): 3064 см-1, 3030, 2898, 1453, 1093, 1074, 913, 734, 696.
1Н-ЯМР (CDCl3): δ 2,23 (1H, дт, J = 7,7, 14,3 Гц); 2,59 (1H, м); 3,13 (1H, дд, J = 2,6, 9,4 Гц); 3,60 (1H, дд, J = 2,3, 6,3 Гц); 3,81 (1H, дд, J = 1,8, 12,0 Гц); 3,92 (1H, м); 4,12 (1H, т, J = 2,3 Гц); 4,17 (1H, дд, J = 8,8, 12,0 Гц); 4,31 (1H, м); 4,37 (1H, д, J = 11,5 Гц); 4,51 (1H, д, J = 11,5 Гц); 4,53 - 4,61 (4H, м); 4,69 - 4,77 (2H, м); 5,06 (1H, дд, J = 0,8, 10,1 Гц); 5,10 (1H, дд, J = 0,8, 17,2 Гц); 5,89 (1H, м); 7,20 - 7,42 (20H, м).
13С-ЯМР (CDCl3): 35,72 (млн. д. ), 66,31, 67,33, 71,00, 71,28, 73,11, 73,91, 75,06, 76,87, 78,01, 116,90, 127,32, 127,40 127,71, 127,75, 127,78, 127,84, 128,15, 128,28, 128,39, 128,42, 134,83, 137,82, 138,01, 138,72, 138,81.
[α]D : -24,8oC (с = 0,81, CHCl3).
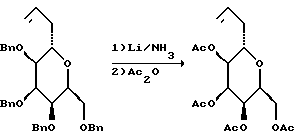
При -78oC и в атмосфере азота к жидкому NH3 (1 л) порциями добавляли 4,5 г (652 мМ) лития. Полученный синий раствор перемешивали в течение 20 мин, а затем через канюлю добавляли раствор аллилированного продукта (32,07 г, 56,86 мМ) в ТГФ (200 мл). Этот синий раствор перемешивали при -78oC в течение 30 мин, после чего по капле добавляли безводный метанол до исчезновения синего цвета. Охлаждающую баню удаляли, а растворители выпаривали в потоке азота. Остаток суспендировали в CH2Cl2 (500 мл) и охлаждали до 0oC в атмосфере азота. К перемешанной суспензии добавляли триэтиламин (178,4 мл, 1,28 мМ), уксусный ангидрид (60 мл, 640 мМ) и N,N-диметиламинопиридин (1,2 г). Полученную смесь нагревали до комнатной температуры и перемешивали в течение 12 ч. Затем эту смесь промывали водой (2 • 500 мл) и солевым раствором (500 мл), после чего ее осушали сульфатом натрия, фильтровали и концентрировали. Остаток хроматографировали на силикагеле и получали алкентетраацетат в виде прозрачного маслообразного вещества (19,34 г, 51,9 мМ, при этом выход за две стадии составлял 91,4%).
ИК (пленка): 3077 см-1, 2941, 1749, 1643, 1497, 1434, 1372, 1226, 1116, 1074, 1043, 915.
1Н-ЯМР (CDCl3): δ 2,03 (3H, с); 2,06 (3H, с); 2,07 (3H, с); 2,10 (3H, с); 2,27 - 2,38 (2H, м); 4,02 (1H, ддд, J = 4,5, 7,5, 7,5 Гц); 4,10 (1H, дд, J = 3,6, 12,2 Гц); 4,20 (1H, ддд, J = 3,6, 5,1, 8,9 Гц); 4,63 (1H, дд, J = 8,9, 12,2 Гц); 4,84 (1H, дд, J = 3,2, 7,3 Гц); 5,08 (1H, т, J = 1,3 Гц); 5,11 (1H, м); 5,19 (1H, дд, J = 3,2, 5,1 Гц); 5,45 (1H, т, J = 3,2 Гц); 5,78 (1H, м).
13С-ЯМР (CDCl3): 20,60 млн.д, 20,78, 34,75, 60,47, 66,54, 67,27, 68,82, 69,32, 70,61, 117,79, 132,96, 169,47, 169,58, 169,73, 170,77.
MC (FAB): 374 AEM (отн. интенсивность 12%); 373 (M+ +H, 91), 331(9), 313(38), 271(7), 154(76), 136(78), 91(98), 77(100), 63(70, 51(97).
[α]D 2: - 31,6o (с = 0,93, CHCl3).

При 0oC и в атмосфере аргона к перемешанному раствору алкентетраацетата (20,30 г, 54,55 М) в ТГФ (200 мл) добавляли 0,5 М раствор 9-борабицикло[3.3.1] нонана ("9-BBN") в ТГФ (185 мл, 92,5 мМ). Полученный раствор нагревали до комнатной температуры и перемешивали в течение 3 ч. В этот период времени ТСХ не обнаруживала исходного материала. Реакционную смесь охлаждали до 0oC, а затем по капле добавляли 10%-ный водный раствор NaOH (25 мл) и 30%-ный водный раствор H2O2 (25 мл). Полученную смесь перемешивали в течение 1,5 ч при 0oC, добавляли насыщенный водный раствор хлорида аммония 91 л), и эту смесь экстрагировали этилацетатом (500 мл). Этилацетатную фазу промывали насыщенным водным раствором NaCl (500 мл), а объединенные водный фазы экстрагировали этилацетатом (2 • 500 мл). После этого объединенные этилацетатные экстракты осушали сульфатом натрия, фильтровали, концентрировали и хроматографировали (SiO2, гексан/этилацетат, 1:2-0:1), в результате чего получали первичный спирт 11 (16,102 г, 41,25 мМ, выход 75,6%) в виде прозрачного маслообразного вещества.
ИК (пленка): 3510 см-1, 3020, 3012, 2940, 2877, 1745, 1451, 1432, 1402, 1371, 1227, 1118, 1091, 1046, 991, 965.
1Н-ЯМР (CDCl3): δ 1,53 - 1,72 (4H м); 2,05 (3H, с); 2,08 (6H, с); 2,10 (3H, с); 3,63 (2H, шир. т.); 3,96 (1H, м); 4,08 (1H, дд, J = 3,6, 12,2 Гц); 4,19 (1H, ддд, J = 3,6, 5,0, 8,9 Гц); 4,63 (1H, дд, J = 8,9, 12,2 Гц); 4,80 (1H, дд, J = 3,2, 7,1 Гц); 5,19 (1H, дд, J = 3,2, 5,0 Гц); 5,43 (1H, т, J = 3,2 Гц).
13С-ЯМР (CDCl3): 20,53 млн.д, 20,67, 20,71, 26,87, 28,40, 60,53, 62,26, 66,64, 67,33, 69,57, 69,99, 70,59, 169,41, 169,65, 170,74.
[α]D : -39,6o (с = 9,59, CHCl3).
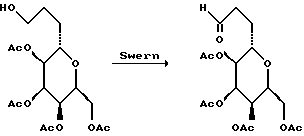
При -78oC и в атмосфере аргона к раствору оксалилхлорида (483 мкл, 5,55 мМ) в метиленхлориде (20 мл) по капле добавляли ДМСО (872 мкл, 12,3 мМ). Полученный раствор перемешивали в течение 15 мин при -78oC, после чего медленно добавляли раствор первичного спирта (1,015 г, 2,60 мМ) в метиленхлориде (10 мл). После выдерживания раствора при -78oC в течение 1 ч, к нему добавляли триэтиламин (2,00 мл) и образовавшийся прозрачный раствор перемешивали еще 20 мин при -78oC, а затем оставляли для нагревания до комнатной температуры. После этого реакционную смесь промывали насыщенным водным раствором хлорида аммония (30 мл), разводили диэтиловым эфиром (50 мл) и промывали 50 мл воды, а затем 50 мл насыщенного водного раствора NaCl. Объединенные водные промывки снова экстрагировали диэтиловым эфиром (50 мл), а объединенные органические фазы осушали сульфатом натрия, фильтровали и концентрировали с получением неочищенного альдегида. Полученный продукт использовали непосредственно без дополнительной очистки.
ИК (пленка): 2939 см-1, 2851, 2731, 1748, 1653, 1617, 1576, 1559, 1539, 1521, 1507, 1436, 1372, 1227, 1119, 1074, 1044, 964, 908.
1Н-ЯМР (CDCl3): δ 1,68 (1H, м); 1,87 (1H, м); 1,95 (3H, с); 1,97 (3H, с); 2,00 (3H, с); 2,03 (3H, с); 2,45 (2H, м); 3,86 (1H, м); 3,93 (1H, дд, J = 3,1, 13,1 Гц); 4,09 (1H, м); 4,68 (1H, дд, J = 9,7, 13,1 Гц); 4,69 (1H, м); 5,08 (1H, дд, J = 3,1, 5,8 Гц); 5,41 (1H, дд, J = 3,0, 3,1 Гц); 9,67 (1H, м).
13С-ЯМР (CDCl3): 20,52 млн.д, 20,60, 20,75, 23,32, 39,37, 59,96, 66,76, 67,52, 67,92, 69,53, 71,31, 169,24, 169,50, 169,61, 170,75, 201,16.
[α]D : - 45,9oC (с = 8,37, CHCl3).
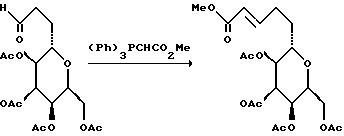
К перемешанному раствору альдегида (16,23 г, 42 мМ) в метиленхлориде (400 мл) при 0oC добавляли карбометоксиметилентрифенилфосфоран (27,8 г, 83,2 мМ). Полученный раствор перемешивали при 0oC в течение примерно 1 ч, а затем нагревали до комнатной температуры и перемешивали в течение 18,5 ч. После этого раствор промывали насыщенным водным хлоридом аммония (200 мл), водой (200 мл) и насыщенным водным раствором NaCl (200 мл). Органическую фазу осушали сульфатом натрия, фильтровали и концентрировали. Остаток хроматографировали (SiO2, гексан/этилацетат, 2:1) и получали метиловый сложный эфир (16,524 г, 37,138 мМ, выход 88%) в виде прозрачного маслообразного вещества.
ИК (пленка): 2953 см-1, 1750, 1658, 1437, 1373, 1321, 1225, 1170, 1117, 1073, 1042, 908.
1Н-ЯМР (CDCl3): δ 1,59 - 1,73 (2H, м); 2,03 (3H, с); 2,06 (3H, с); 2,11 (3H, с); 2,08 (3H, с); 2,23 - 2,40 (2H, м); 3,72 (3H, с); 3,91 (1H, ддд, J = 3,3, 7,8, 9,5 Гц); 4,11 (1H, дд, J = 3,1, 12,3 Гц); 4,18 (1H, ддд, J = 3,1, 5,4, 9,2 Гц); 4,63 (1H, дд, J = 9,2, 12,3 Гц); 4,77 (1H, дд, J = 3,1, 7,8 Гц); 5,18 (1H, дд, J = 3,1, 5,4 Гц); 5,46 (1H, т, J = 3,1 Гц); 5,85 (1H, д, J = 15,6 Гц); 6,94 (1H, м).
13С-ЯМР (CDCl3): δ 20,57 млн. д, 20,65, 20,78, 27,58, 28,93, 51,39, 60,31, 66,68, 67,43, 68,26, 69,47, 70,94, 121,79, 147,87, 166,85, 169,35, 169,54, 169,67, 170,71.
[α]D : -23,7oC (с = 1,41, CHCl3).
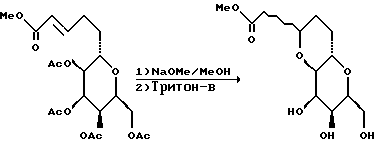
К раствору метоксида натрия (полученному из 8 г Na в 500 мл MeOH) добавляли безводный метилацетат (25 мл) и полученный раствор посредством каннюлирования добавляли к перемешанному раствору метилового сложного эфира (16,2 г, 36,4 мМ) в метаноле (250 мл) и метилацетате (25 мл) при 0oC. Полученный раствор перемешивали в течение 30 мин при 0oC, а затем добавляли раствор тритона В (10 мл 40%-ного раствора в метаноле). Образовавшийся желтый раствор перемешивали при 0oC в течение 1 ч, после чего нагревали до комнатной температуры, перемешивая при этом. После добавления твердого хлорида аммония (20 г) растворители удаляли в вакууме. Остаток суспендировали в метилацетате (200 мл) и фильтровали через воронку из спеченного стекла. Твердые вещества дополнительно промывали метилацетатом (2 • 100 мл) и объединенные фильтрат и промывки концентрировали с получением желтого маслообразного вещества. Остаток подвергали колоночной хроматографии на SiO2 (элюент: метилацетат/этилацетат/метанол, 10:1), в результате чего получали триол в виде прозрачного бледно-желтого маслообразного вещества и смеси (прибл. 2,5:1) C3-диастереомеров (7,26 г, 26,3 мМ, выход 72%).
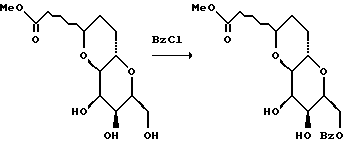
При 0oC и в атмосфере азота, к перемешанному раствору триола (17 мг, 61 мкМ) в CH2Cl2 (1,0 мл) добавляли пиридин (100 мкл, 1,24 мМ), а затем бензоилхлорид (50 мкл, 431 мкМ). Через 10 мин ТСХ (этилацетат) указывала на отсутствие остаточного исходного материала. Реакционный раствор разводили диэтиловым эфиром (2 мл) и промывали водой и солевым раствором. Органическую фазу осушали сульфатом натрия, фильтровали и концентрировали. Остаток хроматографировали на SiO2 (элюент: этилацетат) и получали первичный бензоат в виде прозрачного маслообразного вещества (21 мг, 55 мкМ, выход 89%).

К перемешанному раствору бензоата (436 мг, 1,13 мкМ) в бензоле (20 мл) при комнатной температуре добавляли диметилацетальанизальдегид (412 мл, 2,4 мМ), а затем п-толуолсульфонат пиридиния (10 мг). Полученный раствор перемешивали при комнатной температуре. Через 30 мин к раствору добавляли 0,5 г измельченных молекулярных сит  , после чего продолжали перемешивание. Через 6 ч добавляли твердый NaHCO3 (1 г) и полученную смесь фильтровали через целит вместе с диэтилэфирными промывками. Фильтрат концентрировали, а остаток хроматографировали на SiO2-колонках (элюент: гексан/этилацетат, 1:1 и этилацетат/метанол, 10: 1), в результате чего получали анизилиден (356 мг, 714 мкМ, выход 62%) в виде прозрачного маслообразного бесцветного продукта и 157 мг диола (415 мкМ).
, после чего продолжали перемешивание. Через 6 ч добавляли твердый NaHCO3 (1 г) и полученную смесь фильтровали через целит вместе с диэтилэфирными промывками. Фильтрат концентрировали, а остаток хроматографировали на SiO2-колонках (элюент: гексан/этилацетат, 1:1 и этилацетат/метанол, 10: 1), в результате чего получали анизилиден (356 мг, 714 мкМ, выход 62%) в виде прозрачного маслообразного бесцветного продукта и 157 мг диола (415 мкМ).
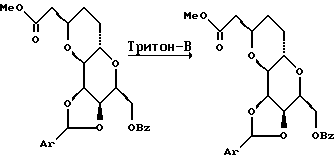
К перемешанному раствору C3-эпимеров анизилидена (356 мг, 714 мкМ) в бензоле (30 мл) при комнатной температуре добавляли метилацетат (100 мкл), а затем раствор тритона В (30 мкл 40%-ного раствора в метаноле). Полученный бледно-желтый раствор перемешивали при комнатной температуре в течение 10 мин, после чего добавляли насыщенный водный раствор хлорида аммония 920 мл) и диэтиловый эфир (30 мл). Отделенную органическую фазу промывали 50 мл воды и 30 мл солевого раствора. После осушки сульфатом натрия фильтрования и концентрирования получали маслообразно вещество (1Н-ЯМР-анализ неочищенного продукта указывал на наличие только одного изомера). Полученное вещество использовали без дополнительной очистки.
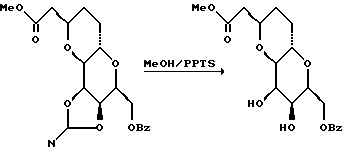
К перемешанному раствору анизилидена (3,37 г, 6,74 мМ) в метаноле (150 мл) при комнатной температуре добавляли п-толуолсульфонат пиридиния (50 мг). После перемешивания при комнатной температуре в течение 13 ч к раствору добавляли еще 50 мг п-толуолсульфоната пиридиния (PPTS). Через 15 ч (всего) ТСХ (элюент: гексан/этилацетат/CHCl3, 1:1:1) указывала на отсутствие остаточного исходного материала. Затем добавляли 250 мг твердого гидрокарбоната натрия и 250 мкл пиридина и смесь концентрировали. Остаток суспендировали в этилацетате, фильтровали, концентрировали и хроматографировали на двуокиси кремния (элюент: этилацетат), в результате чего получали 2,53 г (6,65 мМ, выход 99%) диола в виде бесцветного маслообразного вещества.
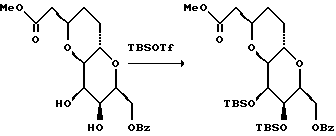
При 0oC и в атмосфере аргона к перемешанному раствору диола (2,53 г, 6,65 мМ) в метиленхлориде (200 мл) добавляли триэтиламин (7,81 мл, 56 мМ), а затем т-бутилдиметилсилилтрифторометансульфонат (6,43 мл, 28 мМ). Полученный раствор медленно нагревали до комнатной температуры и перемешивали в течение 14 ч. ТСХ (гексан/этилацетат/CHCl3, 3:1:1) обнаруживала наличие одного пятна (Rf прибл. 0,85). После этого реакционную смесь разбавляли диэтиловым эфиром (300 мл) и промывали насыщенным раствором хлорида аммония (200 мл), водой (200 мл) и солевым раствором (200 мл). Органическую фазу осушали сульфатом натрия, фильтровали и концентрировали. Остаток подвергали хроматографии на колонках с силикагелем (элюент: гексан/этилацетат, 8:1) и концентрировали в течение ночи на всасывающем трубопроводе, в результате чего получали дисилиловый эфир в виде прозрачного бесцветного маслообразного вещества (3,897 г, 6,41 мМ, выход 96%).
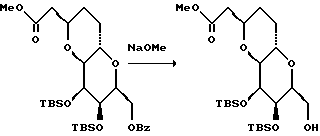
При 0oC к перемешанному раствору дисилилового эфира (3,89 г, 6,40 мМ) в метаноле (150 мл) и метилацетате (6 мл) добавляли раствор метоксида натрия в метаноле (полученный из 0,5 г Na в 50 мл метанола). После перемешивания в течение 30 мин при 0oC охлаждающую баню удаляли, и через 280 мин (всего) ТСХ (гексан/этилацетат/CHCl3, 3: 1: 1) указывала на отсутствие остаточного исходного материала. Затем к этой смеси добавляли твердый NH4Cl (2 г) и полученную белую суспензию концентрировали в вакууме. Остаток суспендировали в диэтиловом эфире (200 мл) и промывали водой (2 • 100 мл) и солевым раствором (100 мл). Органическую фазу осушали сульфатом натрия, фильтровали и концентрировали. После хроматографии на колонках с силикагелем (гексан/этилацетат, 4: 1) получали первичный спирт в виде кристаллического твердого вещества (2,906 г, 5,74 мМ, выход 90%).
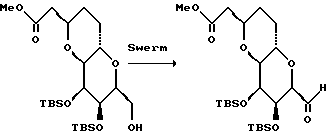
При -78oC и в атмосфере аргона к перемешанному раствору оксалилхлорида (533 мкл, 6,126 мМ) в метиленхлориде (20 мл) по капле добавляли диметилсульфоксид (963 мкл, 13,58 мМ). После 20-минутного перемешивания при -78oC к смеси медленно добавляли раствор спирта (636 мг, 1,26 мМ) в метиленхлориде (10,0 мл). После часового перемешивания при -78oC добавляли триэтиламин (2,21 мл), охлаждающую баню удаляли, а смесь перемешивали еще 30 мин. Затем добавляли насыщенный водный раствор хлорида аммония (50 мл) и диэтиловый эфир (50 мл), а отделенную органическую фазу промывали водой (2 • 50 мл) и солевым раствором (50 мл). После осушки сульфатом натрия концентрирования и хроматографирования на колонках с силикагелем получали альдегид в виде прозрачного маслообразного вещества (551 мг, 1,10 мМ, выход 87%).

К перемешанному раствору альдегида (551 мг, 1,10 мМ) и йодотриметилсилилацетилена (относящегося к 1-йодо-2-триметилсилилэтину; Commercon, A. и др. Tetrahedron. 36:1215, 1980) (1,25 г, 5,6 мМ) в ТГФ (в атмосфере N2 и при комнатной температуре) добавляли приблизительно 200 мг 0,01% NiCl2/CrCl2. Через 10,5 ч добавляли еще примерно 100 мг указанной смеси, а еще через 16 ч добавляли примерно 250 мг той же смеси. Через 31 ч (всего) реакционную смесь разводили насыщенным раствором хлорида аммония (5 мл) и экстрагировали этилацетатом (4 • 4 мл). Объединенные этилацетатные экстракты промывали водой (2 • 10 мл) и солевым раствором (5 мл), осушали сульфатом натрия, фильтровали и концентрировали. После хроматографии на колонке с силикагелем (8: 1 гексан/этилацетат) получали мажорный C11-диастереомер с более высоким Rf (529 мг, 882 мкМ, 80%) и смесь (109 мг) нежелательного С11-диастереомера с более низким Rf и исходный альдегид. 1Н-ЯМР неочищенной продуктовой смеси (до хроматографии) показал соотношение диастереомерных аддуктов приблизительно 10:1.
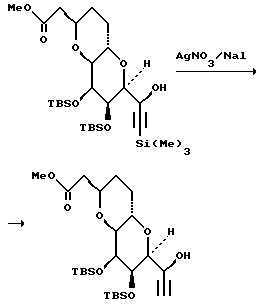
К размешанному раствору мажорного C11-диастереомера (241 мг, 402 мкМ) в этаноле-H2O (5 мл, 4:1 об./об.) при 0oC добавляли раствор AgNO3 (138 мг, 804 мкМ) в этаноле-H2O (0,5 мл, 3:1 об./об.). Полученную суспензию размешивали 20 мин при 0oC (ТСХ не обнаруживала исходного материала). Затем по капле добавляли раствор NaI (240 мг, 1,608 мМ) в воде (0,5 мл), после чего продолжали размешивать еще 30 мин при 0oC. Желтую суспензию разводили диэтиловым эфиром (10 мл) и фильтровали через целит вместе с дополнительными эфирными промывками (4 • 10 мл). Фильтрат, объединенный с промывками, концентрировали с помощью ротационного выпаривания, в результате чего получали водную суспензию, которую затем экстрагировали диэтиловым эфиром (4 • 5 мл). Объединенный органический экстракт осушали сульфатом натрия, фильтровали и концентрировали. После хроматографии на колонке с силикагелем получали де-триметилсилилированный продукт (206 мг, 390 мкМ, выход 97%) в виде кристаллического твердого вещества( см., Tetrahedron Lett. 28:3923-3926, 1987).
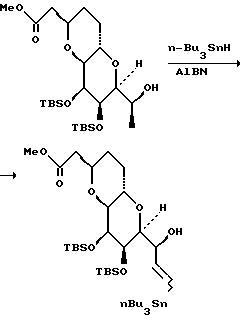
Размешанный раствор де-триметилсилилированного продукта (52 мг, 98,5 мкМ), три-н-бутилстаннана (134 мкл, 493 мкМ), и AIBN (2 мг) в дегазированном толуоле в атмосфере аргона помещали в предварительно нагретую масляную баню при 80oC. Через 30 мин ТСХ (гексан/этилацетат/CHCl3 = 5:1:1) показала полную конверсию в два пятна с более высоким Rf. Затем реакционную смесь охлаждали до комнатной температуры, концентрировали в вакууме, а остаток хроматографировали (SiO2, гексаны __→ гексаны - этилацетат; 5:1), в результате чего получали E-винилстаннан с более высоким Rf (58 мг, 71 мкМ, 72% - выход) и Z-винилстаннан с более низким Rf (18 мг, 22 мкМ, 22% - выход) в виде прозрачного бесцветного маслообразного продукта.

К размешанному раствору E-винилстаннана (58 мг, 70 мкМ) в CH2Cl2 (1 мл) при 0oC добавляли раствор I2 в CH2Cl2 (приблизительно 360 мкл, 50 мг I2 /мл CH2Cl2-раствора около 71 мкМ) до тех пор, пока реакционная смесь оставалась слегка розоватой. После выдерживания еще 30 мин при 0oC, ТСХ (гексан/этилацетат/CHCl3 = 5:1:1) показала полную конверсию в пятно с более высоким Rf. Раствор разводили диэтиловым эфиром (2 мл) и промывали водным 10%-ным Na2S2O3 (2 • 1 мл), водой (1 мл) и солевым раствором (1 мл). Органическую фазу осушали сульфатом натрия, фильтровали и концентрировали. После очистки остатка с помощью колоночной хроматографии на силикагеле (гексан-этилацетат, 4:1) получали E-винилиодид в виде прозрачного бесцветного маслообразного вещества (46 мг, 70 мкМ, выход приблизительно 100%).
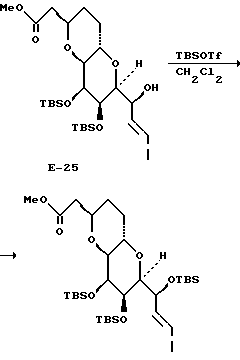
К размешанному раствору E-винилиодида (42 мг, 64 мкМ) в CH2Cl2 (2 мл) при 0oC и в атмосфере аргона добавляли триэтиламин (179 мкл, 1,28 мМ), а затем добавляли т-бутилдиметилсилилтрифторометансульфонат (149 мкл, 640 мМ). После этого охлаждающую баню удаляли, а реакционную смесь оставляли нагреваться до комнатной температуры и размешивали в течение 3 ч. ТСХ (гексан/этилацетат/CHCl3 = 5: 1:1), проведенная на этой стадии, не обнаруживала исходного материала. Затем реакционный раствор разводили диэтиловым эфиром (5 мл) и промывали насыщенным водным раствором NaHCO3 (5 мл), H2O (5 мл) и солевым раствором (2 мл). После осушки сульфатом натрия, фильтрации, концентрирования и колоночной хроматографии на двуокиси кремния (гексан: этилацетат, 5:1) получали соединение 8 в виде прозрачного бесцветного маслообразного продукта (49 мг, 63 мМ, выход 99%).
Пример 2. Был разработан более короткий способ синтеза соединения 8, который изображен на схеме 3.
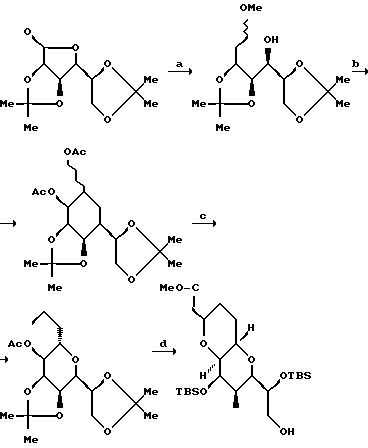
Реагенты и реакционные условия. (a) 1. DIBAL /CH2Cl2 / -78oC. 2. MeOCH2PPh3Cl /t-BuOK/t-BuOH-ТГФ/50oC. (b). 1. OSO4/N,N'-бис-(мезитилметил)-1,2-диаминоэтан/-78oC. 2. Ac2O/ру/комн. тем-ра. (c) CH2-CHCH2TMS/TMSOTf/- MeCN /0oC. (d) Контроль за реакционными условиями показан на с. 73 - 81.
Соединение 9.
Соединение 9 получали из соединений 7 и 8 следующим образом.
(1) Превращение C14-спирта в C14-альдегид.
К размешанному раствору C14-спирта, полученного из соединения 7 (46,9 мг, 53,8 мкМ) в CH2Cl2 (4 мл), добавляли твердый NaHCO3 (50 мг), а затем периодинан (реагент реакции Десс-Мартина) (45,6 мг, 108 мкМ). ТСХ (гексан/этилацетат/CHCl3), проведенная через 1 ч, не обнаруживала исходного материала. Реакционную смесь разводили диэтиловым эфиром (16 мл) и промывали 20 мин водным раствором насыщенного NaHCO3, содержащего 10% Na2S2O3 (по массе). Выделенную органическую фазу промывали 10 мин еще 10 мл водного раствора NaHCO3/Na2S2O3, 10 мл H2O и 10 мл солевого раствора. Органическую фазу осушали сульфатом натрия, фильтровали, и концентрировали. Остаток фильтровали через тонкую прокладку из SiO2 (гексан/этилацетат = 1:1) и полученный фильтрат концентрировали с получением C27-эпимерных C14-альдегидов (44 мг, 51 мкМ, выход 94%) в виде прозрачного бесцветного маслообразного продукта.
(2) Реакция С.13 - С.14-сочетания.
К размешанному раствору C14-альдегида (62 мг, 71 мкМ) и винилиодида (соединение 8) (179 мг, 214 мкМ) в ДМФ (2 мл) в атмосфере азота добавляли 0,1% NiCl2/CrCl2 (прибл. 200 мг). Полученную смесь зеленого цвета размешивали при комнатной температуре. ТСХ, проведенная через 14 ч (гексаны/этилацетат/CHCl3), обнаруживала приблизительно 50%-ную конверсию. Затем добавляли 0,1% NiCl2/CrCl2 (около 200 мг). Через 39 ч ТСХ не обнаруживала остаточного альдегида. После этого реакционную смесь разводили насыщенным водным раствором NH4Cl (12 мл) и H2O (2 мл), экстрагировали этилацетатом (4 • 10 мл), осушали сульфатом натрия, фильтровали, и концентрировали. Остаток очищали с помощью колоночной хроматографии на SiO2 и получали объединенный аллиловый спирт (79,1 мг, 56,7 мкМ, выход 80%) в виде бесцветного пенистого вещества.
(3) Получение пентазилиленонова (С.14-кетон).
К размешанному раствору объединенных аллиловых спиртов (59 мг, 39 мкМ) в CH2Cl2 (дихлорметан) (2 мл) добавляли NaHCO3 (50 мг), а затем периодинановый реагент (реакции Десс-Мартина) (66 мг, 156 мкМ). Через 1 ч добавляли еще 33 мг (78 мкМ) указанного реагента и 50 мг NaHCO3. По истечении 90 мин реакционную смесь разводили диэтиловым эфиром (16 мл) и промывали водным насыщенным раствором NaHCO3, содержащим 10 мас.% Na2S2O3, в течение 20 мин. Отделенную органическую фазу промывали 10 мин еще 10 мл водного раствора NaHCO3/Na2S2O3, 10 мл воды и 10 мл солевого раствора. Органическую фазу осушали сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на SiO2, а затем с помощью препаративной ТСХ (гексан/этилацетат/ CHCl3 = 5:1:1), в результате чего получали енон (50,1 мг, 33 мкМ, выход 85%) в виде прозрачного бесцветного маслообразного продукта.
(4) Удаление С.41-р-метоксифенилметила (МРМ) для получения С.30-спирта
К размешанной смеси С30-МРМ-эфира (смесь С27-эпимеров) (50 мг, 33 мкМ) в дихлорметане (CH2Cl2) (2,00 мл), водного фосфатного буфера (200 мкл, pH 7,00) и трет-бутанола (20 мкл) добавляли DDQ (15,0 мг, 66 мкМ). Реакционную колбу, содержащую полученную смесь, погружали в баню для обработки ультразвуком (H2O, комнатная температура) и обрабатывали 30 мин ультразвуком, затем смесь вынимали из бани и размешивали примерно 3-5 мин без обработки ультразвуком, после чего смесь снова обрабатывали ультразвуком 30 с. ТСХ высокого разрешения (гексан/этилацетат/трет-бутилметиловый эфир = 3:1:1), проведенная на этой стадии, показала приблизительно 50%-ную конверсию. Затем добавляли еще 15,0 DDQ и смесь обрабатывали ультразвуком еще 2 • 30 с, после чего эту смесь размешивали в течение 20 мин. ТСХ не обнаруживала исходного материала. После этого реакционную смесь промывали насыщенным водным раствором NaHCO3 (2 • 2 мл) и H2O (2 мл). Объединенные водные фазы экстрагировали дихлорметаном (2 • 1 мл) и объединенные органические фракции промывали солевым раствором (2 мл), осушали сульфатом натрия, фильтровали, а затем концентрировали. Остаток очищали с помощью препаративной ТСХ (2 элюирования гексаном/этилацетатом/трет-бутилметиловым эфиром = 3:1:1), в результате чего получали нужный C27-эпимер с более высоким Rf (31,4 мг, 22,5 мкМ, выход 66%), и нежелательный C27-эпимер с более низким Rf (6,4 мг, 14%-выход) в виде бесцветных маслообразных продуктов.
(5) Получение C30-С.1-карбоновой кислоты.
К размешанному раствору сложного метилового эфира (C27-эпимера с более высоким Rf, 31,4 мг, 22,5 мкМ) в ТГФ (2,00 мл) при комнатной температуре добавляли 1 М водный раствор LiOH (666 мкл). Полученную смесь размешивали при комнатной температуре в течение 36 ч. ТСХ, проведенная на этой стадии (этилацетат), показала лишь следовые количества исходного материала. ТГФ удаляли путем ротационного выпаривания при комнатной температуре, а полученную водную суспензию разводили водой (2 мл), охлаждали до 0oC и быстро перемешивая осторожно подкисляли до pH 3 с помощью 0,5 М водной HCl. Водную смесь экстрагировали диэтиловым эфиром (5 • 2 мл) и объединенные экстракты промывали 2 мл воды и 2 мл солевого раствора. Эфирную фазу осушали сульфатом натрия, фильтровали и концентрировали. Остаток хроматографировали на короткой колонке (около 2 см) с двуокисью кремния (этилацетат), в результате чего получали карбоновую кислоту (29,7 мг, 21,5 мкМ, выход 96%) в виде бесцветного пенистого продукта.
(6) Получение лактона (соединение 9).
К размешанному раствору C30-гидрокси-C1-карбоновой кислоты (29,7 мг, 21,5 мкМ) в ТГФ (225 мкл) при комнатной температуре в атмосфере аргона добавляли триэтиламин (7,5 мкл, 54 мкМ), а затем 2,4,6-трихлоробензоилхлорид (4,2 мкл, 27 мкМ). Полученную смесь размешивали при комнатной температуре в течение 2 ч, а затем фильтровали через фильтр из спеченного стекла вместе с сухими толуоловыми промывками в атмосфере аргона. Фильтрат, объединенный с промывками, разводили до объема 11,25 мл сухим толуолом и полученный прозрачный бесцветный раствор в течение 14 ч с помощью шприца добавляли к размешанному (70oC) раствору N,N-диметиламинопиридина (16,5 мг, 135 мкМ) в толуоле (10 мл) в атмосфере аргона. Затем шприц промывали сухим толуолом (2 • 0,5 мл) и промывки добавляли к реакционному раствору. Еще через 2 ч (всего 16 ч) реакционный раствор охлаждали до комнатной температуры, разводили диэтиловым эфиром (20 мл) и промывали 0,5 М водным раствором HCl (2 • 5 мл), водой (5 мл) и солевым раствором (5 мл). Объединенные водный фракции экстрагировали диэтиловым эфиром (2 • 5 мл), и объединенные органические фазы осушали сульфатом натрия, фильтровали, а затем концентрировали. Остаток очищали с помощью препаративной ТСХ (гексан/этилацетат = 5:1), в результате чего получали лактон (23,3 мг, выход 81%) в виде прозрачного бесцветного маслообразного продукта.
Соединение 10.
Соединение 10 синтезировали из соединения 9 с использованием одной из описанных двух процедур.
Пример 1. К размешанному раствору соединения 9 (35,7 мг, 26,2 мкМ) в ТГФ (2,62 мл) и безводному метилацетату (262 мкл) добавляли приблизительно 1 М раствор фторида тетрабутиламмония (TBAF) в ТГФ (79 мкл, pH около 6,8). После размешивания при комнатной температуре в течение 36 ч ТСХ (этилацетат/метанол = 10:1) показала полное десилирование. Реакционный раствор фильтровали через прокладку (2 см) из двуокиси кремния (силикагель 60, 230 - 400 меш, этилацетат) для удаления TBAF. Фильтрат концентрировали, а остаток осушали прибл. 1 ч (до использования в следующей стадии, которую проводили без дополнительной очистки) с помощью высоковакуумного трубопровода. 1Н-ЯМР (C6D6) показал присутствие диастереомерных аддуктов типа Михаэля в соотношении приблизительно 5:1.
Указанную продуктовую смесь растворяли в дихлорметане (CH2Cl2) (2,0 мл) при комнатной температуре и к размешанному раствору добавляли п-толуолсульфонат пиридиния (2 мг). После 18-часового размешивания при комнатной температуре ТСХ (этилацетат/метанол = 10:1) показала полную конверсию в нужный полициклический кеталь с более высоким Rf и минорный побочный продукт с более низким Rf. Затем реакционный продукт фильтровали через 1 см прокладку из двуокиси кремния вместе с этилацетатом/метанолом (20:1). Объединенный фильтрат концентрировали, а остаток использовали в следующей стадии без дополнительной очистки.
К размешанному раствору сырого диола в дихлорметане (2,0 мл) при комнатной температуре добавляли пиридин (17 мкл, 210 мкМ), а затем р-нитробензоилхлорид (19,3 мг, 104 мкМ). После размешивания в течение 16 ч при комнатной температуре ТСХ (этилацетат) не обнаруживала исходного материала. Раствор концентрировали путем ротационного выпаривания, а остаток суспендировали в диэтиловом эфире (5 мл), и фильтровали через тонкую прокладку из целита вместе с дополнительными эфирными промывками. Фильтрат концентрировали, а продукты выделяли с помощью препаративной ТСХ (этилацетат), в результате чего получали C38-п-нитробензоат/C35-спирт/C14-C18-полициклический кеталь (15,5 мг, 16,7 мкМ, выход 64% за все 3 стадии ) с более высоким Rf и примесь в виде C14-кетонового побочного продукта с более низким Rf.
К размешанному раствору полициклического кеталя с более высоким Rf (15,5 мг, 16,7 мкМ) в ДМФ (675 мкл) добавляли пиридин (45 мкл, 556 мкМ), а затем добавляли т-бутилдиметилсилилхлорид (16,5 мг, 109 мкМ) и AgNO3 (18,8 мг, 111 мкМ). Полученную белую суспензию размешивали в темноте при комнатной температуре в течение 18 ч, ТСХ, проведенная на этой стадии, не обнаруживала исходного материала. Смесь разводили диэтиловым эфиром (5 мл) и фильтровали через целит вместе с дополнительными эфирными промывками. Объединенный фильтрат промывали насыщенным водным NH4Cl, H2O и солевым раствором (5 мл каждого), а затем осушали сульфатом натрия, фильтровали через тонкую прокладку из двуокиси кремния (этилацетат) и концентрировали. Остаток очищали с помощью препаративной ТСХ (гексан/этилацетат 1:1), в результате чего получали вторичный силиловый эфир 16,1 мг, 15,5 мкМ, выход 93%) в виде прозрачного бесцветного маслообразного продукта.
К размешанному раствору вторичного силилового эфира (16 мг, 15 мкМ) в метаноле (1 мл) при комнатной температуре добавляли твердый K2CO3 (около 0,2 мг). Полученную смесь размешивали 2,5 ч при комнатной температуре и проведенная на этой стадии ТСХ не показала присутствие исходного материала. Затем добавляли толуол (1 мл) и уксусную кислоту (5 мкл), а метанол удаляли путем ротационного выпаривания. Полученную суспензию фильтровали через короткую прокладку из SiO2 вместе с этилацетатом. Объединенный фильтрат концентрировали, а остаток очищали с помощью препаративной ТСХ (этилацетат), в результате чего получали первичный спирт (12,8 мг, 14,4 мкМ, выход 93%) в виде прозрачного бесцветного маслообразного продукта. Этот продукт представлял собой соединение 10.
Пример 2. Соединение 10 также было синтезировано альтернативным способом, показанным ниже.
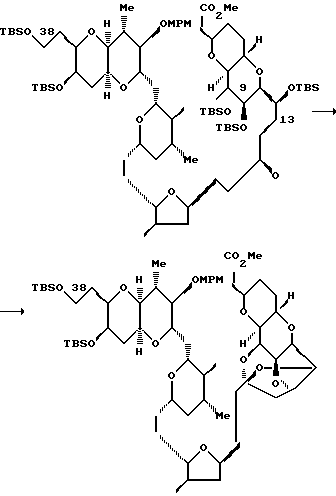
Получение циклического кеталя.
К размешанному раствору пентазилиленона (полученного из альдегида, происходящего от соединений 7 и 8, в соответствии с процедурой, описанной на с. 78 - 79; 35,0 мг 24 мМ) в ТГФ (5,4 мл) и безводном метилацетате (1,8 мл) добавляли 1 М раствор TBAF в ТГФ (1,8 мл, около pH 7,4). После 21-часового размешивания при комнатной температуре реакционную смесь концентрировали и хроматографировали на SiO2 (AcOEt 100% - 5% MeOH/CHCl3), в результате чего получали смесь, содержащую нужный кетон, нежелательный кетон и десилированный енон (35,5 мг).
Полученную продуктовую смесь растворяли в CH2Cl2 (2,3 мл) при комнатной температуре и к размешанному раствору добавляли р-толуолсульфонат пиридиния (PPTS) (2 мг). Затем через 11,5 ч добавляли еще 1 мг PPTS. Через полные 17 ч к реакционной смеси добавляли насыщенный водный NaHCO3, и экстрагировали CH2Cl2 (10 мл • 3). Объединенные органические экстракты промывали солевым раствором, осушали сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на SiO2 (AcOEt 100% - 2% MeOH/AcOEt - 5% MeOH/CHCl3), в результате чего получали циклический кеталь (КТ1-287-1) (15,3 мг, выход 72% на 2 стадии).
Получение ди-ТВ-эфира.
К размешанному раствору диола (КТ1-287-1) (15,3 мг, 16,5 мМ) в CH2Cl2 (1 мл) при комнатной температуре добавляли имидазо (10 мг, 147 мМ), а затем трет-бутилдиметилсилилхлорид (9 мг, 60 мМ). После 2-часового размешивания при комнатной температуре к реакционной смеси добавляли воду и экстрагировали CH2Cl2 (10 мл • 2). Объединенные органические экстракты промывали солевым раствором, осушали сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на колонке с двуокисью кремния (10% AcOEt/гексан - 30% AcOEt/гексан-50% AcOEt/гексан), в результате чего получали первичный TBS-эфир (КТ2-8-1) (13,3 мг, 12,8 мМ, 78%).
Полученный первичный TBS-эфир растворяли в CH2Cl2 (1 мл) и к размешанному раствору при 0oC добавляли триэтиламин (14 мл, 102, 4 мМ), а затем т-бутилдиметилсилилтрифторометансульфонат (TBSOTf) (12 мл, 51,2 мМ). После 30-минутного размешивания при 0oC к реакционной смеси добавляли насыщенный водный раствор NaHCO3 (2 мл), а затем солевой раствор (5 мл) и экстрагировали дихлорметаном (CH2Cl2) (10 мл • 2). Объединенные экстракты осушали сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на колонке с SiO2 (10% AcOEt/гексан-30% AcOEt/гексан), в результате чего получали ди-TBS-эфир (14,4 мг, 12,4 мМ, 98%).
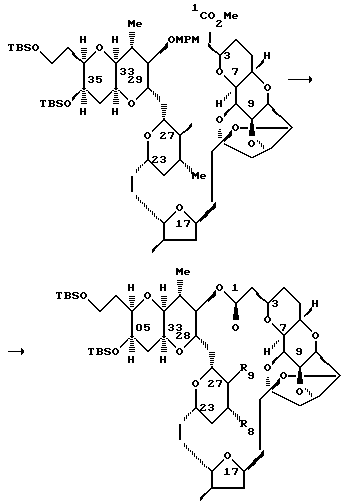
Получение C30-спирта
К размешанной смеси МРМ-эфира (КТ-2-8-2) (14,4 мг, 12,4 мМ) в CH2Cl2 (560 мл) водного фосфатного буфера (56 мл), т-бутанола (5,6 мл) добавляли DDQ (5,6 мг, 24,8 мМ). Полученную смесь обрабатывали 30 с ультразвуком и размешивали в течение 3 мин, после чего снова обрабатывали 30 с ультразвуком и снова размешивали в течение 3 мин. Затем к реакционной смеси добавляли насыщенный водный NaHCO3 (2 мл) и экстрагировали дихлорметаном (10 мл • 3). Объединенные органические экстракты промывали солевым раствором, осушали сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью препаративной ТСХ (гексан: AcOEt: бензол = 1,5:1:1, 2 элюции), и получали нужный C27-диастереомер с более высоким Rf (8,5 мг, 8,2 мМ, выход 66%) (КТ2-9-1) и нежелательный C27-диастереомер с более низким Rf (2,6 мг, 2,5 мМ, выход 20%) (КТ2-9-2) в виде бесцветного маслообразного продукта.
Получение карбоновой кислоты.
К размешанному раствору метилового сложного эфира (КТ2-9-1) (8,5 мг, 8,2 мМ) в ТГФ (550 мл) при комнатной температуре добавляли 1 М водный LiOH (250 мл). После 9-часового размешивания при комнатной температуре ТГФ удаляли при пониженном давлении. К полученному остатку добавляли CH2Cl2 (10 мл) и солевой раствор (10 мл) и смесь подкисляли до pH около 3 с использованием 0,1 М HCl. Полученную смесь встряхивали и органическую фазу отделяли. Водную фазу экстрагировали дихлорометаном (10 мл • 2), а объединенные органические экстракты промывали солевым раствором, осушали сульфатом натрия, фильтровали и концентрировали. Остаток хроматографировали на короткой колонке с двуокисью кремния (CHCl3 - 5% MeOH/CHCl3 - 10% MeOH/CHCl3) и получали карбоновую кислоту (КТ2-9-1) (8,3 мг, 8,2 мМ, 100%).
Получение лактона.
К размешанному раствору карбоновой кислоты (КТ-2-9-1) (8,3 мг, 8,2 мМ) в ТГФ (250 мл) при комнатной температуре и в атмосфере аргона добавляли триэтиламин (4,6 мл, 33,2 мМ), а затем смесь размешивали при комнатной температуре в течение 2 ч, а затем разводили толуолом (6 мл). Этот раствор в течение 8,5 ч с помощью шприца добавляли к размешанному (80oC) раствору N,N-диметиламинопиридина (DMAP) (6,3 мг, 51,4 мМ) в толуоле (2 мл) в присутствии аргона. Затем шприц промывали сухим толуолом (1 мл • 2) и промывки добавляли к реакционному раствору. Через 11 ч добавляли еще 6,3 мг (51,4 мМ) DMAP и 4,6 мл (33,2 мМ) триэтиламина в толуоле (1 мл). Через 32 ч (всего) реакционную смесь охлаждали до комнатной температуры и добавляли солевой раствор (10 мл). Полученную смесь подкисляли приблизительно до pH = 3 путем добавления 0,1 М HCl, и экстрагировали эфиром (10 мл), а затем дихлорметаном (10 мл • 2). Объединенные органические экстракты осушали сульфатом натрия, фильтровали, и концентрировали. Остаток очищали с помощью препаративной ТСХ (40% AcOEt/гексан) и получали в результате лактон 9 (6,6 мг, 6,6 мМ, выход 80%).
При этом следует отметить, что указанные пентазилиленон был также получен с помощью реакции Хорнера - Эммонса, как показано ниже.

Получение C14-карбоновой кислоты.
К размешанному раствору C14-альдегида (77,7 мг, 0,125 мМ) в трет-бутаноле (2,5 мл) и 2-метил-2-бутене (0,6 мл) при комнатной температуре по капле в течение 10 мин добавляли раствор NaClO2 (100 мг, 1,1 мМ) и NaH2PO4 (100 мг, 0,72 мМ) в воде (1 мл). После 30-минутного размешивания при комнатной температуре к реакционной смеси добавляли воду (5 мл) и экстрагировали этилацетатом (10 мл • 3). Объединенные экстракты промывали солевым раствором, осушали сульфатом натрия, фильтровали и концентрировали, в результате чего получали карбоновую кислоту (82,2 мг) в виде прозрачного маслообразного продукта. Этот неочищенный продукт использовали в следующей стадии без дополнительной очистки.
Получение метилового сложного эфира.
К размешанному раствору карбоновой кислоты (82,2 мг) в диэтиловом эфире (5 мл) при 0oC добавляли раствор диазометана в диэтиловом эфире (избыточное кол-во). После 10-минутного размешивания при 0oC смесь концентрировали и остаток очищали с помощью хроматографии на колонке с диоксидом кремния (5% AcOEt/гексан-10% AcOEt/гексан), в результате чего получали C14 метиловый сложный эфир (72,4 мг, 0,11 мМ, выход за две стадии 89%) в виде прозрачного маслообразного продукта.
Получение кетофосфоната.
К размешанному раствору диметилметилфосфоната (120 мл, 1,1 мМ) в ТГФ (2 мл) при -78oC по капле добавляли раствор н-бутиллития в гексане (780 мл, 1,31 М, 1,1 мМ). Полученную смесь размешивали 1 ч при -78oC, после чего по капле при -78oC добавляли раствор метилового сложного эфира (72,4 мг, 0,11 мМ) в ТГФ (3,5 мл). После 50-минутного размешивания при -78oC к реакционной смеси добавляли насыщенный раствор NH4Cl, и экстрагировали этилацетатом (10 мл • 3). Объединенные экстракты промывали солевым раствором, осушали сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью хроматографии на колонке с двуокисью кремния (50% AcOEt/гексан - 80% AcOEt/гексан), в результате чего получали кетофосфат (73,5 мг, 0,099 мМ, 89%) в виде прозрачного маслообразного продукта.
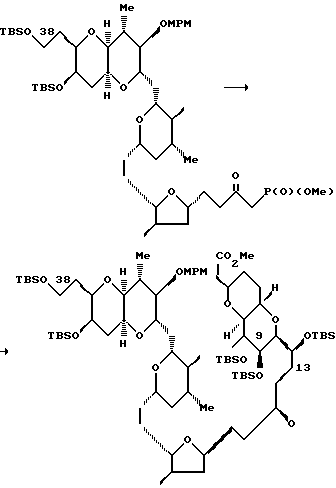
Получение енона.
К размешанному раствору кетофосфоната (56,0 мг, 0,075 мМ) в ТГФ (2 мл) при 0oC добавляли 5% суспензию NaH в минеральном масле. Полученную смесь размешивали при 0oC в течение 1 ч, после чего добавляли при 0oC раствор C12-альдегида (42 мг, 0,065 мМ) в ТГФ (3 мл). Затем реакционную смесь медленно нагревали (в течение 30 мин) до комнатной температуры. После этого реакционную смесь снова охлаждали до 0oC и добавляли насыщенный водный раствор NH4Cl (5 мл). Смесь экстрагировали этилацетатом (10 мл • 3) и объединенные экстракты промывали солевым раствором, осушали сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии на двуокиси кремния (10% AcOEt/гексан-20% AcOEt/гексан-50% AcOEt/гексан-80% AcOEt/гексан), в результате чего получали енон (35,0 мг, 0,023 мМ, 85% превращения исходя из кетофосфоната) и восстановленный кетофосфонат (30 мг, 0,048 мМ, 89%) в виде прозрачного маслообразного продукта.
Соединение 11.
Соединение 11 синтезировали в соответствии с нижеописанной процедурой.
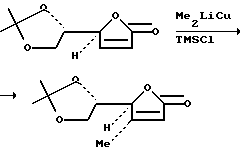
В 250-миллилитровую охлажденную азотом трехгорлую колбу, снабженную магнитной мешалкой и отверстием для впуска азота, вводили свежеприготовленный CuBr - DMS-комплекс (5,57 г, 27,2 мМ). Затем из колбы откачивали газ и заполняли азотом. После этого шприцем вводили ТГФ (80 мл) и полученную суспензию охлаждали до -78oC. Затем в течение 15 мин шприцем добавляли метиллитий (Aldrich, 1,45 М в эфире, 38 мл). В результате желтая суспензия становилась прозрачной и бесцветной. Затем по капле с помощью шприца вводили свежедистиллированный TMSCI (6,9 мл, 54,4 мМ). Полученный бесцветный раствор размешивали 5-10 мин. Бутенолид (2,55 г, 13,9 мМ) растворяли в 5 мл сухого ТГФ и добавляли по капле в течение 1-15 мин. Полученную реакционную смесь размешивали 1 ч при -78oC, причем в этот период желтая окраска смеси менялась на оранжевую. Затем охлаждающую баню удаляли и смесь размешивали 8 ч при комнатной температуре. Темно-зеленую реакционную смесь гасили путем осторожного (по капле) добавления 35 мл насыщенного 10%-ного NH4OH/NH4Cl. Затем колбу открывали (для впуска воздуха) и размешивали 12 ч. За этот период водный слой становился ярко-синим. Органический слой декантировали, а водный слой экстрагировали этилацетатом (3 • 50 мл). Объединенные органические слои промывали насыщенным хлоридом аммония, солевым раствором и осушали сульфатом натрия. Растворители выпаривали и получали почти чистый продукт (2,64 г, 13,2 мМ, выход 95%) в виде маслянистого вещества. Образец этого вещества подвергали хроматографии на силикагеле (65% эфир/гексан), в результате чего получали аналитически чистый продукт.
ИК (пленка): 2985 см-1, 2936, 2887, 1779, 1456, 1419, 1380, 1372, 1257, 1211, 1158, 1128, 1093, 1062, 1025, 987, 966, 948, 944, 870, 827.
1Н-ЯМР (CDCl3): δ 1,17 (2H, д, J = 6,9 Гц); 1,36 (3H, с); 1,38 (3H, с); 2,11 (1H, дд, J = 5,8, 17,5 Гц); 2,59 (1H, ддтд, J = 4,7; 5,8, 6,9, 8,9 Гц); 2,83 (1H, дд, J = 8,9, 17,5 Гц); 3,92 (1H, дд, J = 7,5, 8,2 Гц); 4,04 (1H, дд, J = 2,3, 4,7 Гц); 4,07 (1H, дд, J = 6,9, 8,2 Гц); 4,21 (1H, ддд, J = 2,3, 6,9, 7,5 Гц).
13С-ЯМР (CDCl3): δ 19,49, 25,70, 25,89, 32,20, 36,58, 65,48, 76,26, 84,40, 109,99, 176,33.
MC (FAB): 201 AEM (M++H, относит. интенсивность 25%), 185(21), 147(15), 73(100).
[α]D : +21,5o (с 2,00 CH2Cl2).
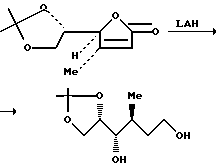
К суспензии LAH (190 мг, 5,0 мМ) в 10 мл сухого эфира, охлажденного до 0oC, по капле добавляли раствор лактона (500 мг, 2,5 мМ) в 1 мл эфира. ТСХ (50% этилацетат/гексан) указывала на полное завершение реакции через 0,5 ч. Затем к реакционной смеси добавляли 200 мкл H2O, 200 мкл 15%-ного водного раствора NaOH и 600 мкл H2O. Полученную смесь размешивали до тех пор, пока не появлялся белый осадок. Затем раствор фильтровали через прокладку из целита и промывали эфиром. растворитель выпаривали и получали слегка желтоватый продукт почти с количественным выходом. Этот продукт использовали в последующей реакции без дополнительной очистки.
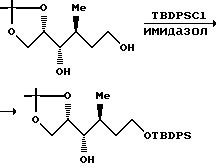
К раствору диола (747 мг, 3,66 мМ) в дихлорметане (10 мл) добавляли имидазол (500 мг, 7,32 мМ). Полученную смесь охлаждали до 0oC и по капле добавляли т-бутилдифенилсилилхлорид. После этого реакционную смесь оставляли нагреваться до комнатной температуры в течение 1 ч, а затем проводили ТСХ (15% этилацетат/гексан), которая указывала на полное завершение реакции. Реакционную смесь гасили водой и экстрагировали дихлорметаном. Органические слои промывали солевым раствором и осушали сульфатом натрия. Растворители удаляли в вакууме, а полученное маслообразное вещество подвергали колоночной хроматографии на силикагеле (15% этилацетат/гексан), в результате чего получали чистый продукт (1,50 г, выход 94%).

Раствор спирта (126 мг, 0,285 мМ) и свежеприготовленный MPMBr (86 мг, 0,428 мМ) растворяли в сухом ТГФ (15 мл) и охлаждали до 0oC. К этой смеси добавляли порциями обезмасленный гидрид калия (200 мг, 5,09 мМ). Реакционную смесь нагревали до комнатной температуры в течение 1,5 ч, а затем разводили эфиром и гасили насыщенным хлоридом аммония. Органический слой удаляли, водный слой экстрагировали эфиром (3 • 10 мл). Объединенные органические слои промывали солевым раствором, осушали сульфатом натрия и фильтровали. Растворители удаляли, а полученное густое маслообразное вещество очищали с помощью колоночной хроматографии на силикагеле (10% этилацетат/гексан), в результате чего получали 148 мг продукта с выходом 92%.
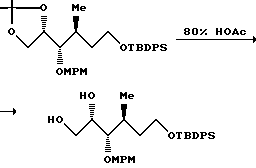
К раствору ацетонида (7,50 г, хх мМ) в ТГФ (10 мл) добавляли 80% HOAc (40 мл) при комнатной температуре. Ход реакции тщательно контролировали с помощью ТСХ (50% этилацетат/гексан). Через 4 ч реакция в основном полностью завершалась. Затем смесь разводили этилацетатом и гасили водой. Для поглощения избыточной уксусной кислоты добавляли твердый бикарбонат натрия. После этого водный слой экстрагировали этилацетатом. Объединенные органические слои осушали сульфатом магния, а растворители удаляли в вакууме, в результате чего получали диол (5,6 г, 80% выход) в виде вязкого маслообразного продукта.
ИК (пленка): 1034 см-1, 1159, 1249, 1514, 1612, 1724, 2961, 3435.
1Н-ЯМР (CDCl3, 500 МГц): δ 1,06 (3H, д, J = 6,9 Гц); 1,19 (9H, с); 1,53 (1H, м); 1,85 (1H, м), 1,96 (1H, м); 2,04 (1H, м); 2,51 (1H, д, J = 6,5 Гц); 3,31 (1H, м); 3,59 (2H, м); 3,72 (1H, м); 3,81 (3H, с, -OCH3); 4,10 (1H, м); 4,14 (1H, м); 4,46 (1H, д, J = 10,9 Гц); 4,63 (1H, д, J = 10,9 Гц); 6,89 (2H, д, J = 8,5 Гц); 7,25 (2H, д, J = 8,5 Гц).
MCBP (FAB): для C20H32O6: вычислено: (M++Na) 3912097, найдено: (M+Na) 3912119.
 -9,2o (с 1,2, MeOH).
-9,2o (с 1,2, MeOH).

К суспензии NaH (60% в масле, 2,33 г, 58 мМ) в ТГФ (300 мл) при комнатной температуре добавляли раствор диола (3,18 г, 6 мМ) в ТГФ (20 мл). После 40-минутного размешивания, смесь охлаждали до 0oC и порциями добавляли TsIm (1,49 г, 6,7 мМ). Полученную смесь размешивали в течение ночи при 4oC. Затем реакцию гасили путем добавления воды и органический слой отделяли. Водный слой экстрагировали эфиром. Органический слой осушали сульфатом натрия, а растворитель удаляли путем ротационного выпаривания. Остаток хроматографировали на колонке (гексан: EtOAc = 8,1), в результате чего получали эпоксид (2,53 г, выход 83%). Для получения лучшего результата, по всей вероятности, необходимо более медленное добавление TsIm в виде раствора.
ИК (пленка): 821 см-1, 1159, 1285, 1514, 1613, 1726, 2970.
1Н-ЯМР (CDCl3): δ 1,07 (9H, с, т-Bu); 1,51 (1H, м); 1,89 (1H, м); 1,95 (1H, м); 2,52 (1H, м); 2,81 (2H, м); 3,05 (1H, м); 3,80 (3H, с); 4,09 (2H, м); 4,49 (1H, д, J = 11,5 Гц); 4,78 (1H, д, J = 11,5 Гц); 6,87 (2H, д, J = 8,6 Гц); 7,29 (2H, д, J = 8,6 Гц).
BPMC (FAB): для C20H30O5: вычислено: + Na 373,1991, найдено: 373,2007.
[α]d -12,2o (с 0,98, MeOH).
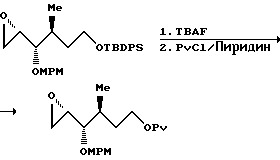
К раствору TBDPS-эфира (780 мг, 1,56 мМ) в ТГФ (30 мл) при комнатной температуре добавляли TBAF (5 мл, 5 мМ). После 2-часового размешивания реакционную смесь концентрировали, разводили этилацетатом и насыщенным хлоридом аммония. Органический слой отделяли и промывали водой, а затем солевым раствором. Органический слой осушали сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии (25% EtOAc в гексане), в результате чего получали 406 мг спирта в виде бесцветного маслообразного вещества.
Очищенный спирт растворяли в CH2Cl2 (10 мл) и обрабатывали пиридином (0,90 мл, 10,9 мМ) и PvCl (1,20 мл, 9,39 мМ), а затем каталитическим количеством DMAP. Реакционную смесь очищали с помощью колоночной хроматографии (33% EtOAc в гексане) и получали пивалоат (505 мг, выход 92% за две стадии) в виде маслообразного продукта.
ИК (пленка): 821 см-1, 1159, 1285, 1514, 1613, 1726, 2970.
1Н-ЯМР (CDCl3): δ 1,07 (9H, с, т-Bu); 1,51 (1H, м); 1,89 (1H, м); 1,95 (1H, м); 2,52 (1H, м); 2,81 (2H, м); 3,05 (1H, м); 3,80 (3H, с); 4,09 (2H, м); 4,49 (1H, д, J = 11,5 Гц); 4,78 (1H, д, J = 11,5 Гц); 6,87 (2H, д, J = 8,6 Гц); 7,29 (2H, д, J = 8,6 Гц).
BPMC (FAB) для C20H30O5: вычислено: + Na 373,1991, найдено: 373,2007.
[α]D -12,2o (с 0,98, MeOH).
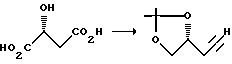
К раствору BH3SMe2 (87,5 мл, 0,923 М) и триметилбората (91 мл, 0,80 М) в 200 мл ТН при 0oC по капле добавляли раствор D-(+)-яблочной кислоты (35 г, 0,261 М) в ТГФ (150 мл). Полученный раствор нагревали до комнатной температуры, и размешивали в течение ночи. Затем по капле добавляли метанол (230 мл), а растворители выпаривали. После еще трех выпариваний совместно с метанолом (100 мл) и концентрирования, получали неочищенный продукт.
Неочищенную продуктовую смесь разделяли на две части. Каждую часть растворяли в 950 мл ацетона и добавляли TsOH•H2O (1,0 г). Каждый раствор размешивали в течение ночи, затем добавляли триэтиламин (1 мл) и полученную смесь размешивали еще некоторое время. Растворитель удаляли путем ротационного выпаривания. Объединенный остаток хроматографировали на колонке (гексан: ацетон = 7:3), в результате чего получали 33 г (выход за две стадии - 86%) продуктовой смеси.
Полученную смесь продуктов (33 г) разделяли на 5 различных партий. Две партии окисляли методом Swern, а остальные три партии окисляли РСС-методом. Для данного субстрата, лучше использовать РСС-метод. Всего было получено 18,0 г (55%) альдегида.
Смесь альдегида (18 г) разделяли на 5-граммовую (34,7 мМ) и 13-граммовую (90,2 мМ) партии. Ниже описывается процедура для реакции в масштабе 13 г. К суспензии Ko+Bu (13,3 г, 119 мМ) в 350 мл ТГФ при -78oC добавляли по капле DAMP (16,2, 108 мМ). После 15-минутного размешивания раствор альдегида в 40 мл ГТФ добавляли по капле в течение прибл. 10 мин. После 12-часового размешивания при этой же температуре добавляли воду (100 мл). Органический слой отделяли и разводили метиленхлоридом (350 мл). Этот органический слой промывали водой (4 • 70 мл) и солевым раствором (50 мл). Объединенный водный слой экстрагировали метиленхлоридом (150 мл и 50 мл). Эти экстракты промывали солевым раствором (30 мл). Все объединенные органические слои осушали (NaSO4). После объединения экстрактов 5 г реакционной смеси растворители отгоняли. Остаток подвергали дистилляции и получали в результате 9,24 г (52%) нужного продукта (т.кип. 62 - 64 oC / прибл. 25 мм рт. ст.).
ИК (пленка): 732 см-1, 910, 1069, 2270, 2901, 2963, 3021, 3309.
1Н-ЯМР (CDCl3): δ 1,36 (3H, с); 1,43 (3H, с); 2,00 (1H, т, J = 2,6 Гц); 2,42 (1H, ддд, J = 2,6, 7,3, 16,5 Гц); 2,53 (1H, ддд, J = 2,6, 5,2, 14,00 Гц); 3,77 (1H, дд, J = 6,2, 8,5 Гц); 4,11 (1H, дд, J = 6,0, 8,5 Гц); 4,24 (1H, м).
BPMC (Cl) для C8H12O2: вычислено: = H 141.0915; найдено (M + H) + 141.0923.
[α]D -38,7o (с 1,68, MeOH).

К раствору ацетилена (257 мг, 1,82 мМ) в ТГФ (13 мл) добавляли н-бутиллитий (1,67 мМ) при -78oC. После 1-часового размешивания, а анионному раствору добавляли BF3•OEt2 (0,20 мл, 1,60 мМ), и реакционную смесь размешивали 20 мин. Затем в течение 20 мин к реакционной смеси добавляли раствор эпоксида (257 мг, 0,733 мМ) в ТГФ ( 4мл). После часового размешивания размешивали еще 20 мин при -45oC, после чего реакцию гасили насыщенным хлоридом аммония. После обычной обработки остаток очищали с помощью колоночной хроматографии (20% этилацетат в гексане) и получали алкин (317 мг, выход 88,8%).
К полученному раствору алкина в гексане (26 мл) добавляли хинолин (0,69 мл), а затем Pd/CaCO3 (173 мг). К сосуду с реакционной смесью подсоединяли баллон с водородом и смесь размешивали в течение 50 мин. После этого реакционную смесь фильтровали через целит, а фильтрат промывали 10% HCl (2 • 10 мл), водой, насыщенным водным гидрокарбонатом натрия и солевым раствором. После осушки и концентрирования получали гомоаллиловый спирт (310 мг, выход 97,8%).
ИК (пленка): 1514 см-1, 1612, 1725, 2978, 3502.
1Н-ЯМР (CDCl3): δ 1,03 (3H, д, J = 6,9 Гц); 1,19 (9H, с); 1,34 (3H, с); 1,41 (3H, с); 1,51 (1H, м); 1,88 (1H, м); 1,96 (1H, м); 2,20 - 2,42 (4H, м); 2,44 (1H, д); 3,14 (1H, м); 3,54 (1H, т, J = 7,6 Гц); 3,69 (1H, м); 3,80 (3H, с); 4,02 (1H, дд); 4,07 ≈ 4,17 (2Н, м); 4,57 (2H, дд); 5,56 (2H, м); 6,88 (2H, д); 7,26 (2H, д).
BPMC для C28H44O7: вычислено: + Na (M+ + Na) 515,2985, найдено: 515,2972.
[α]D -7,3o с 1,1, MeOH).
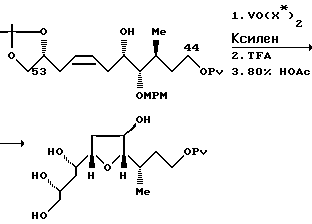
К раствору алкена (1,01 г, 2,23 мМ) в ксилене (18 мл) добавляли VO(X*)2 (10 мг), а затем 3 М т-BuOOH (743 мкл). Полученную реакционную смесь размешивали 30 мин при комнатной температуре. Затем добавляли еще 3 мг катализатора и 185 мкл т-BuOOH. Через 12 ч снова добавляли 3 мг катализатора и 75 мкл BuOOH и реакционную смесь размешивали еще 12 ч. (Реакцию прекращали до того, как на ТСХ-пластинке появлялось более высокое пятно, чем исходный материал. Реакционное время не должно меняться). После восстановительной обработки раствором трисульфата натрия неочищенное соединение растворяли в метиленхлориде (35 мл). Полученный раствор обрабатывали ТФК (1 экв.) в течение 10 мин, в результате чего получали смесь циклизованных продуктов. К этой реакционной смеси добавляли 80% HOAc (40 мл). Реакционную смесь размешивали в течение 3 ч, концентрировали при пониженном давлении и очищали с помощью препаративной ТСХ (9% MeOH в EtOAc), в результате чего получали нужный тетраол (420 мг, выход 59,6%) вместе с нежелательным тетраолом (57 мг, выход 8,2%).
(*X = 2,2,6,6-тетраметил-3,5-гептандион).
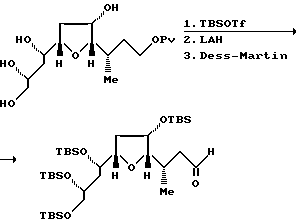
К раствору тетраола (42,1 мг, 0,120 мМ) в CH2Cl2 (2 мл) при 0oC добавляли TBSOTf (0,194 мл, 0,845 мМ) и NEt3 (0,202 мл, 1,45 мМ). Реакционную смесь размешивали в течение 1,6 ч, а затем гасили путем добавления насыщенного водного NaHCO3. Полученную смесь экстрагировали простым эфиром (3 • 5 мл) и объединенные органические слои промывали водой и солевым раствором. Растворитель осушали сульфатом натрия, фильтровали и концентрировали. Остаток очищали с помощью колоночной хроматографии (60% EtOAc в гексане).
Очищенное соединение (89,3 мг) растворяли в эфире (10 мл) и обрабатывали 1,6 М LAH в эфире (2 экв.) при 0oC. После 5-минутного размешивания реакционную смесь гасили насыщенным водным раствором сегнетовой соли. Реакционную смесь размешивали до тех пор, пока не образовывался прозрачный раствор. После обычной экстракции и очистки с помощью колоночной хроматографии (12% EtOAc в гексане) получали спирт (67 мг, выход 83,9%).
ИК (пленка): 775 см-1, 836, 1078, 1254, 1473, 2857, 2929, 2955, 3450.
1Н-ЯМР (C6D5) δ 0,00 (3H, с); 0,04 (3H, с); 0,14 (3H, с); 0,25 (3H, с); 0,27 (3H, с); 0,28 (3H, с); 0,29 (3H, с); 0,39 (3H, с); 0,85 (3H, д, J = 6,8 Гц); 0,97 (9H, с, т-Bu); 1,02 (9H, с, т-Bu); 1,06 (9H, с, т-Bu); 1,07 (9H, с, т-Bu); 1,11 (1H, т, J = 7,1 Гц); 1,55 (1H, м); 1,64 (1H, м); 1,76 (1H, м); 1,89 (1H, м); 1,99 (1H, м); 2,04 (1H, м); 2,18 (1H, м); 3,03 (1H, дд, J = 3,4, 9,4 Гц); 3,25 (1H,. кв., J = 7,1 Гц); 3,64 (1H, м); 3,70 (1H, м); 3,81 (1H, дд, J = 3,9, 10,4 Гц); 3,87 (1H, м); 3,89 (1H, м); 4,04 (1H, м); 4,25 (1H, м).
BPMC (FAB) для C36H80O6Si4: вычислено: = Na 743,4929, найдено: 743,4937.
[α]D -1,5o (с 2,3, MeOH)
К раствору спирта (полученного как описано выше) (35 мг, 0,048 мМ) в CH2Cl2 (2,5 мл) добавляли реагент Десс-Мартина (42 мг, 2 экв.) вместе с NaHCO3 (2 экв. ) для забуферивания этой реакционной системы. Реакционную смесь размешивали в течение 1,5 ч и обрабатывали раствором тиосульфата натрия (6 экв.) в насыщенном водном NaHCO3. После экстракции осушки и концентрации остаток очищали с помощью колоночной хроматографии (20% EtOAc в гексане) и получали 34 мг альдегида с количественным выходом.
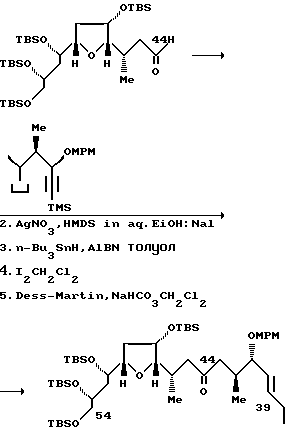
К раствору бромида (46 мг, 0,122 мМ) в эфире (1,5 мл) добавляли 1,7 М т-бутиллития в эфире (0,139 мл, 0,105 мМ) при -78oC. Через 20 мин к анионному раствору добавляли раствор альдегида (16 мг, 0,022 мМ). Реакционную смесь размешивали 30 мин и гасили насыщенным водным NH4Cl. После обычной обработки смесь подвергали хроматографическому разделению (6% EtOAc в гексане), в результате чего получали два эпимерных спирта, т.е., спирт с более высоким Rf (11,4 мг, выход 51%) и спирт с более низким Rf (5,8 мг, выход 26%), вместе со смешанной фракцией (2,9 мг, 13%).
К раствору спирта (11,4 мг, 0,011 мМ) в этаноле (2 мл) добавляли гексаметилдисиламид("HMDS", 16,7 мкл, 0,077 мМ), а затем раствор AgNO3 (11,5 мг, 0,067 мМ) в 65%-ном водном этаноле (1,06 мл). Реакционную смесь размешивали в течение 1 ч до тех пор, пока не образовывался коричневый осадок и прозрачный раствор, указывающие на завершение реакции. Реакционную смесь разбавляли эфиром (2 мл) и обрабатывали раствором NaI (20 мг, 0,132 мМ) в 0,4 мл воды. Затем смесь размешивали в течение 20 мин и фильтровали через целит. Фильтрат экстрагировали этилацетатом (3 • 7 мл). Объединенные органические слои концентрировали, а остаток фильтровали через короткую SiO2-пробку, в результате чего получали ≈ 11 мг ацетилена. Этот ацетилен осушали путем азеотропного удаления воды и растворяли в толуоле (1 мл).
К полученному раствору добавляли n-Bu3SnH (0,1 мл, 0,15 мМ), а затем каталитическое количество AIBN. Реакционную смесь нагревали до 80oC в течение 1 ч, концентрировали и очищали с помощью колоночной хроматографии (6% этилацетат в гексане), в результате чего получали винилолово (10,7 мг, выход 80%).
К очищенному раствору винилолова в дихлорметане (1 мл) добавляли раствор йода (2,6 мг, 0,013 мМ) при 0oC до тех пор, пока не установится стойкая йодная окраска. Реакционную смесь обрабатывали как обычно раствором NaHSO3, а неочищенный остаток очищали с помощью колоночной хроматографии (6% этилацетат в гексане), в результате чего получали винилиодид. Винилиодид в 1 мл CH2Cl2 обрабатывали реагентом Десс-Мартина (2 экв.) в течение 1 ч. После восстановительной обработки остаток очищали с помощью колоночной хроматографии (4% EtOAc в гексане), в результате чего получали 9,8 мг кетона с количественным выходом.
Обычно в результате 3-стадийной реакции сочетания получали соединение 11 в большом масштабе с выходом ≈ 82%.
ИК (пленка): 791 см-1, 863, 1084, 1271, 1622, 1727, 2856, 2929.
1Н-ЯМР (C6D6): δ 0,01 (3Н, с); 0,02 (3Н, с); 0,14 (6Н, с); 0,25 (3Н, с); 0,26 (3H, с); 0,27 (3, с); 0,28 (3H, с); 0,87 (3H, д, J = 6,8 Гц); 0,96 (9H, с); 0,98 (3H, д, J = 6,8 Гц); 1,02 (9H, с); 1,07 (9H, с); 1,08 (9H, с); 1,35 (1H, шир. с); 1,58 (1H, м); 1,76 (1H, м); 1,91 (1H, м); 1,99 (1H, м); 2,20 (1H, дд, J = 8,7, 16,7 Гц); 2,29 (1H, дд, J = 10,2, 16,7 Гц); 2,39 (1H, м); 2,53 (1H, дд, J = 4,2, 16,7 Гц); 2,67 (1H, м); 2,97 (1H, дд, J = 2,2, 16,7 Гц); 3,10 (1H, м); 3,30 (1H, м); 3,32 (3H, с); 3,69 (1H, дд, J = 6,0, 10,3 Гц); 3,76 (1H, м); 3,80 (1H, дд, J = 3,5, 10,3 Гц); 3,92 (1H, м); 4,00 (1H, м); 4,05 (1H, д, J = 11,5 Гц); 4,26 (1H, м); 4,35 (1H, д, J = 11,5 Гц); 6,01 (1HJ, д, J = 14,5 Гц); 6,33 (1H, дд, J = 7,8, 14,5 Гц); 6,79 (2H, д, J = 8,6 Гц); 7,13 (2H, д, J = 8,6 Гц).
BPMC (FAB) для C50H95O8ISi4: вычислено: + Na 1085, 5048; найдено: 1085, 5022.
[α]D -16,8o (с 1,4, CHCl3).
Соединение 12.
Соединение 12 синтезировали в соответствии с процедурой, проиллюстрированной на схеме 4.

Указанная γ -лактоза 001 может быть легко получена из D-галактозогликаля с помощью перегруппировки Ireland-Claisen и иодолактонизации.
Стадия А.
(1) Получение нитрила.
В соответствии со стандартными процедурами, лактозу обрабатывали DEBAL /CH2Cl2 /-78oC _→ 0oC; p-Ts OH /MeOH при комнатной температуре; и Tf2O/Py/CH2Cl2/-42oC, а затем обрабатывали NaCN/DMF при комнатной температуре, в результате чего получали нитрил 002.
(2) Восстановление нитрила 002.
К раствору нитрила 001 (80,0 мг, 0,252 мМ) в метиленхлориде 3,0 мл), охлажденном до -78oC, добавляли диизобутилалюмогидрид ("Dibal-H", 0,60 мл, 0,60 мМ). Реакционную смесь размешивали 2 ч при -78oC, а затем реакцию гасили путем добавления метанола (1 мл) с последующим добавлением насыщенного водного хлорида аммония (1 мл). Смесь разводили эфиром (6,0 мл), а полученный раствор нагревали до комнатной температуры. После 2-часового выдерживания при комнатной температуре получали гетерогенный раствор, а белый осадок удаляли путем фильтрации через слой целита. Фильтрат промывали водной 1 н. HCl (10 мл), насыщенным NaHCO3 (20 мл), и солевым раствором (30 мл). Органический экстракт осушали твердым сульфатом натрия, а растворитель удаляли в вакууме, в результате чего получали альдегид 002 в виде желтого маслообразного продукта (88 мг), который непосредственно использовали в следующей стадии без дополнительной очистки.
(3) Восстановление альдегида 003.
Раствор альдегида 003 (80 мг, 0,25 мМ) в MeOH/CH2Cl2 (3:1, 4,0 мл) охлаждали до 0oC. Затем к этому раствору порциями добавляли борогидрид натрия ("NaBH4", 30 мг, 0,79 мМ), и полученную смесь размешивали при 0oC в течение 1 ч. После этого смесь концентрировали при пониженном давлении, а полученный маслообразный продукт распределяли между этилацетатом (20 мл) и водой (10 мл). Органический слой удаляли, промывали солевым раствором, осушали сульфатом натрия и концентрировали в вакууме. Остаточный маслообразный продукт очищали с помощью флеш-хроматографии (гексан/этилацетат, 3:2) и получали бензиловый эфир 004 (65 мг, 81% - выход) в виде бесцветного маслообразного продукта.
ИК (пленка): 697, 1011, 1097, 1454, 1496, 2877, 2919, 3466 шир. см-1.
1Н-ЯМР (500 МГц, CDCl3): δ 1,12 (3H, д, J = 7,2 Гц); 1,56 (1H, м); 1,63 (1H, ддд, J = 4,1, 4,1, 15,8 Гц); 2,17 - 2,21 (2H, м); 2,41 (1H, шир. с.); 2,57 (1H, ддд, J = 2,0, 2,0, 15,8 Гц); 3,29 (1H, м); 3,41 (3H, с); 3,50 (1H, м); 3,72 - 3,85 (2H, м); 3,83 - 4,09 (1H, м); 4,10 (1H, м); 4,41 (1H, д, J = 12,2 Гц); 4,78 (1H, д, J = 12,2 Гц); 4,85 (1H, д, J = 5,8 Гц).
BPMC для C18H27O5 вычислено: [M + H+] 323,1858; найдено: 327,1837.
[α]D :-49,3o (с 0,41, MeOH).
Стадия b.
(1) Гидрогенолиз бензилэфира 004.
Суспензию гидроксида палладия (катализатор Perleman) в абсолютном этаноле (0,5 мл) добавляли к раствору бензилового эфира 004 (61 мг, 0,196 мМ) в абсолютном этаноле (2,5 мл), охлажденном до 0oC. Атмосферу газообразного водорода создавали с помощью баллона, наполненного газом, а полученную смесь размешивали при комнатной температуре в течение 17 ч. Суспензию фильтровали через целит и фильтрат концентрировали в вакууме, в результате чего получали спирт (45 мг, выход 99%) в виде бесцветного маслообразного продукта. Как показал ЯМР-анализ этого продукта, дополнительной очистки не требовалось.
(2) Получение трисилированного триола 005 из метилгликозида 004.
Этантиол (3 мл) добавляли к раствору метилгликозида 003 (43 мг, 0,186 мМ) в метиленхлориде (3 мл), охлажденном до 0oC. Затем добавляли этерат трифторида бора (0,1 мл, 0,80 мМ) и полученную смесь размешивали в течение 4 ч при 0oC. Реакционную смесь выливали в делительную воронку, содержащую охлажденный льдом этилацетат и насыщенный раствор бикарбоната натрия. Водный слой тщательно экстрагировали этилацетатом, а затем метиленхлоридом и объединенные органические экстракты осушали путем обработки сульфатом натрия. Органический раствор концентрировали в вакууме и маслянистый остаток очищали с использованием короткой пробки из двуокиси кремния (этилацетат), в результате чего получали триол 004А (66 мг) в виде желтого маслообразного вещества.
К раствору триола 004А (66 мг) в метиленхлориде (2мл), охлажденном до 0oC, добавляли триэтиламин (0,15 мл, 1,1 мМ), а затем TBSOTf (0,128 мл, 0,55 мМ). Реакционную смесь размешивали 1 ч при 0oC, после чего добавляли еще 0,15 мл (1,1 мМ) триэтиламина и 0,128 мл (0,55 мМ) TBSOTf. Смесь нагревали до комнатной температуры и размешивали еще 2 ч. Реакционную смесь гасили добавлением насыщенного раствора бикарбоната натрия, после чего смесь тщательно экстрагировали эфиром. Объединенные органические экстракты промывали солевым раствором, осушали сульфатом натрия и концентрировали в вакууме. Остаточное маслообразное вещество очищали с помощью флеш-хроматографии (гексан/этилацетат, 15:1), в результате чего получали трисилированный триол 005 (96 мг, выход 70% исходя из диолметилгликозида) в виде светло-желтого маслообразного продукта.
(3) Превращение диэтилтиоацеталя 005 в спирт 006.
Бикарбонат натрия (83 мг, 0,327 мМ), а затем йод (83,0 мг, 0,327 мМ) добавляли к раствору диэтилтиоацеталя 005 (138 мг, 0,218 мМ) в ацетоне/воде (9: 1) при 0oC. через 2,5 ч добавляли дополнительное количество бикарбоната натрия (4,5 экв.) и йода (1,5 экв.) и смесь размешивали еще 1 ч. Затем реакционную смесь разводили этилацетатом (20 мл) и полученный раствор промывали насыщенным водным тиосульфатом натрия, насыщенным водным NaHCO3, и солевым раствором. Органический раствор осушали сульфатом натрия и концентрировали при пониженном давлении, в результате чего получали 137 мг неочищенного альдегида 004А, который затем использовали непосредственно в следующей стадии.
К раствору альдегида (137 мг) в метаноле/метиленхлориде (15 мл, 2:1) при 0oC добавляли порциями борогидрид натрия 950 мг). Полученный раствор размешивали 30 мин при 0oC, а затем концентрировали при пониженном давлении. Маслообразный остаток распределяли между этилацетатом (50 мл) и водой (20 мл), а органический слой промывали солевым раствором, осушали твердым сульфатом натрия, и концентрировали в вакууме. Неочищенный маслообразный остаток очищали с помощью флеш-хроматографии (гексан:этилацетат, 3:1), в результате чего получали спирт (106 мг, выход 93%) в виде бесцветного маслообразного продукта.
(4) Синтез нитрила 008.
Метансульфонилхлорид (60 мл, 0,774 мМ) добавляли к раствору спирта 006 и триэтиламина (125 мл, 0,838 мМ) в метиленхлориде (5,0 мл) и реакционную смесь размешивали при комнатной температуре в течение 10 мин. Смесь разводили метиленхлоридом (20 мл) и гасили путем добавления насыщенного водного хлорида аммония (20 мл). Органический экстракт промывали водным бикарбонатом натрия, а затем солевым раствором, осушали твердым сульфатом натрия и концентрировали в вакууме. Неочищенный маслообразный продукт использовали непосредственно в следующей стадии без дополнительной очистки.
Цианид натрия (350 мг) добавляли к раствору мезилата (400 мг) в ДМСО (4 мл) и смесь размешивали в течение 3 ч при 60oC. Реакционную смесь охлаждали до комнатной температуры и разводили солевым раствором, охлажденным льдом. Смесь исчерпывающе экстрагировали этилацетатом и объединенные органические слои промывали солевым раствором, осушали сульфатом натрия и концентрировали в вакууме. Маслообразный остаток очищали с помощью флеш-хроматографии (гексан/этилацетат, 9:1), в результате чего получали нитрил 008 (338 мг, выход 92%).
ИК (пленка): 772, 837, 1006, 1019, 1030, 1100, 1255, 1386, 1463, 1472, 2125, 2857, 2884, 2929, 2955 см-1.
1Н-ЯМР (500 МГц, CDCl3): δ 0,02 (9H, с); 0,04 (3H, с); 0,05 (3H, с); 0,09 (3H, с); 0,86 (9H, с); 0,88 (9H, с); 0,89 (9H, с); 1,04 (3H, д, J = 6,7 Гц); 1,49 (1H, м); 1,84 (1H, ддд, J = 4,2, 4,4, 10,6 Гц); 1,93 (1H, м); 2,00 (1H, д, J = 14,9 Гц); 2,30 (1H, м); 2,46 (1H, дд, J = 7,5, 16,7 Гц); 2,60 (1H, дд, J = 3,2, 16,6 Гц); 2,94 (1H, д, J = 8,9 Гц); 3,42 (1H, д, J = 10,2 Гц); 3,60 (1H, м); 3,69 - 3,73 (2H, м); 3,81 (1H, м).
BPMC для C29H62O4Si3N: вычислено: [M + H]+ 572,3986; найдено 572,3997.
[α]D : -0,87o (с 1,95, MeOH).
(5) Получение альдегида 009 из нитрила 008.
Раствор гидрида диизобутилалюминия в гексане (1,0 М, 2,5 мл, 2,5 мМ) по капле добавляли к размешанному раствору нитрила 008 (310 мг, 0,543 мМ) в метиленхлориде (5 мл), охлажденном до -78oC. Реакционную смесь размешивали еще 1,5 ч а затем гасили путем добавления метанола (6 мл), насыщенного раствора хлорида аммония (6 мл) и эфира (6 мл). Смесь нагревали до комнатной температуры и размешивали еще 1 ч. После этого смесь фильтровали через целит и эту прокладку из целита промывали эфиром. Полученный фильтрат промывали насыщенным раствором хлорида аммония (20 мл) и солевым раствором, а затем осушали сульфатом натрия и концентрировали в вакууме. Маслообразный остаток очищали с помощью флеш-хроматографии (гексан/этилацетат, 9:1), в результате чего получали альдегид 009 (296 мг, выход 96%) в виде бесцветного маслообразного продукта.
Стадия с.
Соединение 105 (бромид) синтезировали в соответствии со следующей процедурой. В итоге в результате реакции бромида с альдегидом 009 будет получено соединение 12. Бромид получали из(S)-(+)-метил 3-гидрокси-2-метил-пропионата 100 (Aldrich) в несколько стадий с полным выходом приблизительно 40%.
(1) Превращение сложного β -гидроксиэфира 100 в спирт.
р-Толуолсульфоновую кислоту (80 мг, 0,42 мМ) добавляли к раствору спирта 100 (21,0 г, 0,177 М) и дигидрофурана (26 мл, 0,284 М) в этиловом эфире (180 мл) и полученную смесь размешивали при комнатной температуре в течение 20 ч. Реакционную смесь гасили путем добавления насыщенного раствора бикарбоната натрия, а затем полностью экстрагировали эфиром. Объединенные органические слои промывали солевым раствором, осушали твердым сульфатом натрия и концентрировали в вакууме, в результате чего получали 40 г ТГФ-эфира 100А. Этот неочищенный продукт непосредственно использовали в следующей стадии без дополнительной очистки.
К раствору эфира, охлажденному льдом медленно в течение 1 ч добавляли алюмогидрид лития (10,0 г, 0,263 М). К полученной суспензии по капле добавляли сложный эфир 100А (40 мг) в простом эфире 9200 мл), после чего смесь размешивали 12 ч при 0oC. Затем реакцию гасили путем последовательного добавления этилацетата (40 мл), воды (40 мл), 1 н. раствора гидроксида натрия (40 мл) и воды (40 мл). Полученную суспензию фильтровали через целит, после чего прокладку из целита промывали эфиром. Фильтрат промывали солевым раствором, а органический экстракт осушали твердым сульфатом натрия и концентрировали в вакууме. Маслообразный остаток очищали путем вакуумной дистилляции, в результате чего получали спирт 101 (29,0 г, выход 94%), т. кип. 105oC (0,1 торр.).
(2) Окисление спирта 101 методом Swern.
Оксалилхлорид (12,0 мл, 0,137 М) добавляли к метиленхлориду (60 мл), охлажденному до -78oC. Затем в течение 15 мин по капле добавляли раствор диметилсульфоксида (13 мл, 0,183 М) в метиленхлориде (30 мл). После этого в течение 20 мин по капле добавляли раствор спирта 101 (16,0 г, 0,091 М) в метиленхлориде (30 мл) и полученную смесь размешивали 30 мин при -78oC. Затем добавляли раствор триэтиламина (64,1 мл, 0,459 М) в метиленхлориде (30 мл) и смесь размешивали еще 30 мин при -78oC. После этого реакционную смесь удаляли из охлаждающей бани и нагревали до 0oC в течение 15 мин. Затем реакционную суспензию распределяли между смесью бензола/эфира (600 мл, 4:1) и водой (800 мл). Органический слой удаляли и промывали водой (2 • 500 мл) и солевым раствором. После этого органический экстракт осушали твердым сульфатом натрия и концентрировали в вакууме, в результате чего получали 15,8 г альдегида 102 в виде светло-желтого маслообразного вещества. Этот неочищенный альдегид использовали непосредственно в следующей стадии без дополнительной очистки.
(3) Реакция 1,2-присоединения с альдегидом 102.
Раствор бутиллития в гексане (50, мл, 0,105 М) добавляли к раствору триметилсилилацетилена в эфире (400 мл) при -78oC. Полученную смесь нагревали до 30oC и размешивали в течение 30 мин. После этого раствор охлаждали до -78oC, и в течение 20 мин по капле добавляли раствор альдегида 101 (14,0 г, 0,091 М) в эфире (30 мл). Реакционную смесь выдерживали в течение 1 ч при -78oC, а затем гасили путем добавления насыщенного раствора хлорида аммония. Полученную смесь нагревали до 0oC и полностью экстрагировали этиловым эфиром. Объединенные органические экстракты промывали солевым раствором, осушали твердым сульфатом натрия и концентрировали в вакууме. Маслообразный остаток (22 г) очищали с помощью флеш-хроматографии (гексан/хлороформ/этилацетат, 10: 4: 1), в результате чего получали 5,5 г нужного спирта, 7,1 г смешанных фракций и 6,1 г нежелательного спирта (полный выход исходя из спирта 101 составлял 85%).
(4) МРМ-защита пропаргилового спирта 103.
Раствор пропаргилового спирта (2,0 г, 7,40 мМ) в метиленхлориде (10 мл) добавляли к раствору п-метоксибензилтрихлороимидата в метиленхлориде (500 мл), охлажденном до 0oC. Затем по капле добавляли раствор трифторэтерата бора в метиленхлориде (0,2 н, 0,7 мл, 0,14 мМ), после чего раствор, который был сначала желтого цвета, становился оранжевым. ТСХ, проведенная через 10 мин, указывала на полное завершение реакции. Реакционную смесь гасили путем добавления насыщенного раствора бикарбоната натрия. Органический слой удаляли, а оставшийся водный слой полностью экстрагировали метиленхлоридом. Объединенные органические экстракты промывали солевым раствором, осушали твердым сульфатом натрия и концентрировали в вакууме. Твердый остаток растворяли в равных количествах бензола и гексана и вносили в колонку на двуокиси кремния. После градиентного элюирования гексаном/этилацетатом (50:1,50:5) получали защищенный спирт 104 (2,49 г, выход 83%) в виде светло-желтого маслообразного продукта.
(5) Сольволиз ТНР-защищенного спирта 104.
Камфорсульфоновую кислоту (100 мг, 0,43 мМ) добавляли к раствору ТНР-эфира 104 (2,0 г, 4,9 мМ) в метаноле (50 мл). Реакционную смесь размешивали при комнатной температуре в течение 9,5 ч, после чего реакцию гасили путем добавления твердого бикарбоната натрия (36 мг, 0,43 мМ). Смесь размешивали в течение 20 мин, а затем концентрировали в вакууме. Маслообразный остаток распределяли между эфиром (40 мл) и водой (20 мл), а органический экстракт промывали солевым раствором, осушали сульфатом натрия и концентрировали в вакууме. Маслообразный остаток очищали с помощью флеш-хроматографии (гексан/этилацетат, 9: 1) и получали спирт 104 (1,3 г, выход 87%) в виде бесцветного маслообразного продукта.
(6) Превращение спирта 104 в бромид 105.
Триэтиламин (0,228 мл, 1,52 мМ) добавляли к раствору спирта 104 (380 мг, 1,17 мМ) в метиленхлориде (5 мл), охлажденном до 0oC. Затем добавляли метансульфонилхлорид и полученный раствор размешивали 30 мин при 0oC. После этого реакционную смесь разводили метиленхлоридом (20 мл) и гасили путем добавления насыщенного раствора хлорида аммония (30 мл). Органический слой последовательно промывали 0,3 н. раствором HCl (20 мл), насыщенным раствором бикарбоната натрия и солевым раствором. Затем органический раствор осушали твердым сульфатом натрия и концентрировали в вакууме. Полученное неочищенное маслообразное вещество (470 мг) непосредственно использовали в следующей стадии без дополнительной очистки.
Бромид лития (1,02 г, 11,7 мМ) добавляли к раствору мезилата 104А (470 мг) в ТГФ (10 мл) при комнатной температуре. Реакционную смесь размешивали 2 ч при температуре перегонки, а затем охлаждали до комнатной температуры и переносили в делительную воронку, содержащую простой эфир (40 мл) и воду (30 мл). Органический слой удаляли, промывали солевым раствором, осушали твердым сульфатом натрия и концентрировали в вакууме. Маслообразный остаток очищали с помощью флеш-хроматографии, в результате чего получали бромид 105 (418 мг, выход 93%) в виде светло-желтого маслообразного продукта.
ИК (пленка): 760, 843, 1023, 1037, 1073, 1251, 1514, 1612, 2168, 2835, 2936, 2961 см-1.
1Н-ЯМР (500 МГц, CDCl3): δ 0,18 (3H, с); 1,11 (3H, д, J = 6,7 Гц); 2,11 (1H, м); 3,46 - 3,52 (2H, м); 3,78 (3H, с); 4,04 (1H, д, J = 7,1 Гц); 4,41 (1H, д, J = 11,1 Гц); 4,70 (1H, д, J = 11,1 Гц).
BPMC для C17H25O2Br: вычислено: [M]+ 368,0807; найдено: 368,0810.
[α]D : -70,6o (с 1,33, MeOH).
Стадия d.
(1) Реакция сочетания бромида 105 и альдегида 009.
Раствор т-бутиллития (0,71 мл, 1,21 мМ) в гексане добавляли к раствору бромида 105 (249 мг, 0,675 мМ) в эфире при -78oC. Раствор размешивали в течение 20 мин при -78oC, после чего в течение 1 мин с помощью каннюли добавляли раствор альдегида 009 (73 мг, 0,127 мМ) в эфире (3 мл). Реакционную смесь размешивали в течение 30 мин при -78oC, а затем гасили путем добавления насыщенного хлорида аммония (20 мл). Полученную смесь нагревали до комнатной температуры, а затем полностью экстрагировали простым эфиром. Органические объединенные экстракты промывали солевым раствором, осушали твердым сульфатом натрия и концентрировали в вакууме. Маслообразный остаток очищали с помощью флеш-хроматографии (гексан/этилацетат, 9:1), в результате чего получали спирт 011 (94 мг, выход 86%) в виде смеси диастереомеров.
(2) Разблокирование TMS-защищенного алкина 011.
Раствор нитрата серебра (256 мг, 1,51 мМ) в абсолютном этаноле (10 мл) и воде (6,0 мл) в течение 15 мин по капле добавляли к раствору защищенного алкина 011 (227 мг, 0,262 мМ) в абсолютном этаноле (41 мл) и HMDS (0,387 мл, 1,83 мМ). Реакционную смесь размешивали 2 ч при комнатной температуре, а затем гасили путем добавления иодида натрия (472 мг, 3,14 мМ) в эфире (20 мл). Смесь фильтровали через прокладку из целита, после чего эту прокладку промывали водой. Фильтрат концентрировали и остаток распределяли между эфиром (30 мл) и водой. Органический слой промывали солевым раствором, осушали твердым сульфатом натрия и концентрировали в вакууме. Маслообразный остаток очищали с помощью флеш-хроматографии (гексан/этилацетат, 4:1) и получали конечный алкин 012 (206 мг, выход 99%) в виде маслообразного бесцветного продукта.
(3) Получение винилстанана 012А.
Раствор алкина 012 (45 мг, 0,057 мМ) и AIBH (5 мг) в толуоле (3 мл) и гидрид трибутилолова (0,3 мл) размешивали при 80oC. Через 1 ч добавляли гидрид трибутилолова (0,4 мл) и размешивали еще 1 ч. Полученную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Маслообразный остаток очищали с помощью флеш-хроматографии (гексан-этилацетат, 5:1), в результате чего получали винилстаннат 012А (56 мг, выход 92%) в виде смеси геометрических изомеров (транс:цис, 9:1).
(4) Получение винилиодида 013.
Раствор йода (15 мг, 0,059 мМ) в метиленхлориде (1 мл) по капле добавляли к раствору винилстаннана 012А (56 мг, 0,0517 мМ) в метиленхлориде (4 мл), охлажденном до 0oC. Образовавшийся слегка розоватый раствор размешивали 10 мин при 0oC, а затем разводили метиленхлоридом (20 мл). Полученную смесь промывали раствором тиосульфата натрия (30 мл) и солевым раствором. Органический экстракт осушали твердым сульфатом натрия и концентрировали в вакууме. Маслообразный остаток очищали с помощью флеш-хроматографии (гексан/этилацетат, 5:1), в результате чего получали винилиодид 013 (44 мг, выход 94%) в виде слегка желтого маслообразного продукта.
(5) Окисление спирта 014 по методу Десс-Мартина.
Периодан (реагент Десс-Мартина) (100 мг, 0,235 мМ) порциями добавляли к раствору спирта 014 (44 мг, 0,048 мМ) в метиленхлориде, охлажденном до 0oC. Затем добавляли бикарбонат натрия (8,0 мг) и раствор размешивали 2 ч при 0oC. Смесь разводили простым эфиром (10 мл) и гасили путем добавления насыщенного раствора бикарбоната натрия (8 мл), содержащего 4 растворенных кристалла тиосульфата натрия. Полученный раствор энергично размешивали в течение 1 ч. Затем смесь исчерпывающе экстрагировали простым эфиром и объединенные органические экстракты промывали солевым раствором, осушали твердым сульфатом натрия. Растворитель удаляли в вакууме, а маслообразный остаток очищали с помощью флеш-хроматографии (гексан/этилацетат, 4:1), в результате чего получали альдегид, соединение 12 (40 мг, выход 91%) в виде бесцветного маслообразного продукта.
Синтез соединений, родственных галихондрину.
Имеется много способов присоединения модифицированных левых половин с правой половиной соединения 10 или его С.35-стереоизомера. В схеме 5 проиллюстрированы 5 возможных способов с соответствующими процедурами. Эти реакции присоединения могут быть также осуществлены в отношении правой половины 18 или его С.35-стереоизомера. Кроме того, используя стадии a и b, можно получить С.35- и С.38-сложные и простые диэфиры из соединений 10, 18 и их С.35-изомеров.
2
Пример ацилирования (стадия a).
К раствору соединения 10 (2,0 мг / 0,1 мл безводного метиленхлорида) добавляли TBS O(CH7)2COoH (1,0 мг), DCC (1,0 мг) и DMAP (около 0,01 мг) при 0oC. После размешивания в течение ночи при комнатной температуре реакционную смесь концентрировали при пониженном давлении, а остаток очищали с помощью препаративной ТСХ (гексан/этилацетат, 1: 2), в результате чего получали сложный эфир (2,3 мг, выход 91%) в виде прозрачного масла.
Пример этерификации (стадия b).
К раствору соединения 10 (2,0 мг / 0,1 мл безводного ТГФ) добавляли NaH (5% дисперсия минерального масла, 1,08 мг) и следовое количество имидазола при 0oC. Смесь размешивали при 0oC в течение 30 мин, а затем при комнатной температуре в течение 15 мин. К полученному раствору при 02C по капле добавляли MeO2C(CH7)2OSOoMe (1,14 мг/0,1 мл безводного ТГФ). После 2-часового размешивания при 02C реакцию гасили путем добавления капли метанола. Реакционную смесь разводили метиленхлоридом и промывали солевым раствором. Органический слой отделяли, осушали сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью препаративной ТСХ (гексан/этилацетат, 1,2), и получали простой эфир (1,53 мг, выход 65%) в виде прозрачного маслообразного продукта.
Примеры Ni(II)/Cr(II)-опосредованной реакции сочетания (стадия с).
См. Главы "соединение 1" и "соединение 2", представленные выше.
Пример реакции сочетания для купрата (стадия d).
К размешанному раствору TBSO(CH8)o 1 (370 мг, 1,0 мМ) в пентане (4 мл) по капле и при -78oC добавляли раствор т-бутиллития в пентане (1,17 мл, 2,0 мМ). Через 10 мин при -78oC по капле добавляли 2-тиенилцикнокупрат лития (0,25 М раствор в ТГФ, 4,0 мл, 1,07 мМ). Полученный мутный раствор размешивали 10 мин при -78oC, а затем медленно нагревали (в течение 5 ч) до -10oC. Полученные таким образом 0,1 мл раствора реагента переносили в небольшую колбу, а затем охлаждали до -30oC. К этому раствору добавляли альдегид, полученный из соединения 10 (2,0 мг/0,05 мл ТГФ; см. выше, главу под заголовком "соединение 1"). Реакционную смесь размешивали при 04C в течение 1 ч, а затем нагревали до комнатной температуры. После этого реакцию гасили NH4Cl/NH2OH (10: 1, об./об.) раствором, разводили EtOAc и промывали солевым раствором. Органический слой осушали сульфатом магния и концентрировали при пониженном давлении. Остаток очищали с помощью препаративной ТСХ (гексан/этилацетат, 1:2), в результате чего получали спирты (1,75 мг, выход 69%) в виде светло-желтого маслообразного продукта.
Пример реакции Хорнера-Эммонса (стадия е).
Кетофосфонат TBSO(CH7)2COCH2P(O)(OMe)o (2,58 мг/0,1 мл безводного ТГФ) добавляли к суспензии NaH (5,0% в минеральном масле, 2,17 мг) в ТГФ (0,1 мл). После 35-минутного размешивания при комнатной температуре и в атмосфере аргона смесь охлаждали в ледяной бане и по капле добавляли ТГФ-раствор альдегида, полученного из соединения 10 (2,0 мг/0,1 мл ТГФ, см. также главу "Соединение 1"). После этого смесь продолжали размешивать еще 30 мин при 050C и 30 мин при комнатной температуре. Затем реакционную смесь разводили эфиром, наносили на короткую прокладку из силикагеля и элюировали этилацетатом. Растворитель удаляли, а остаток очищали с помощью препаративной ТСХ (гексан/этилацетат, 1:2), в результате чего получали енон (1,22 мг, непосредственный выход 47%).
Оценка противоопухолевой активности.
(1) Способность к in vitro-ингибированию роста опухоли.
Синтетический галихондрин BI анализировали на ингибирующую активность против различных линий опухолевых клеток, и в результате этого анализа получали величины IC50 (см. табл.1). Для сравнения использовали данные об ингибирующей активности натурального галихондрина В против клеток мышиного лейкоза L1210 при IC50 = 0,3 нМ (полученные Национальным институтом рака; J. Biol. Chem. 266:15882-15889 (1991)), и данные против клеток мышиной меланомы B-16, IC• = 0,08 нМ (Hirata и Uemura; Pure Appl. Chem. 58:701-710 (1986)). Обе эти работы вводятся в настоящее описание во всей полноте посредством ссылки.
Кроме того, были проведены испытания нескольких синтетических соединений, родственных галихондрину. В качестве примера можно отметить, что соединения 13 и 14 обладают значительной рост-ингибирующей активностью против LOX- и HT-1080-клеток, тогда, как соединения 15 и 16 не показали значительной активности (см. табл.2). Структуры соединений 13 - 16 показаны на фиг.3.
(2) Противоопухолевая in vivo-активность синтетического галихондрина В.
Противоопухолевая in vivo-активность синтетического галихондрина BI была продемонстрирована с использованием клеток меланомы LOX, имплантированных "голым" мышам. Ниже показана способность 25 мкг/кг галихондрина В ингибировать рост подкожных опухолей меланомы человека LOX как у самок, так и самцов голых мышей (20 самок, 20 самцов, при этом 10 животных каждого пола использовали в качестве контроля и для НВ-обработки).
Мышам (на день 0) подкожно инъецировали 1 6 10$$$ клеток LOX методом полной рандомизации в отношении пола и обрабатываемых групп. В дни 3-7 и 10-14 экспериментальные мыши (самцы и самки) получали инъекцию 25 мкг/кг синтетического галихондрина BI (i.p.) в физиологическом растворе, при этом контрольные животные получали лишь один физиологический раствор. Обработка галихондрином В почти полностью предупреждала рост опухоли у мышей обоего пола. Эти результаты нельзя объяснить вариабельностью клеточной инокуляции, поскольку начало роста опухоли приходится в основном на 3 день одновременно у всех 4 групп, достигая 90 - 100%-ной пролиферации на 6 день. У мышей, обработанных галихондрином В, опухоли после начального периода развития в видимое, но очень небольшое новообразование, просто прекращали свой рост. У одной самки, обработанной галихондрином В, опухоль полностью исчезла на 14 день, причем это отсутствие опухоли сохранялось вплоть до конца эксперимента (день 17). Важно отметить, что после начального периода развития, наблюдался фактический регресс опухолей у мышей, обработанных галихондрином В.
Другие варианты осуществления изобретения.
Исходя из приведенного описания любой специалист может легко применить изобретение к конкретным условиям и в различных целях, внося при этом различные изменения и модификации, не выходящие за рамки объема и сущности изобретения. Например, полное число атомов углерода в А и В может составлять 50, 50 или выше (без учета углерода в спиртозащитных группах). Кроме того, в той же формуле, число атомов углерода охватывается диапазоном m и n от 0 до 3. Аналогичная ситуация может быть также применена и к другим формулам, представленным в описании.
Таким образом, другие варианты осуществления изобретения также входят в объем формулы изобретения.
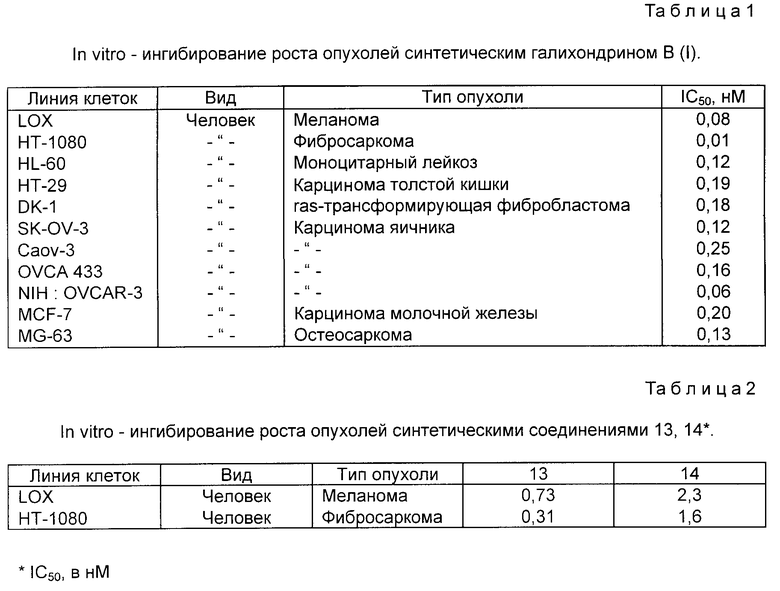

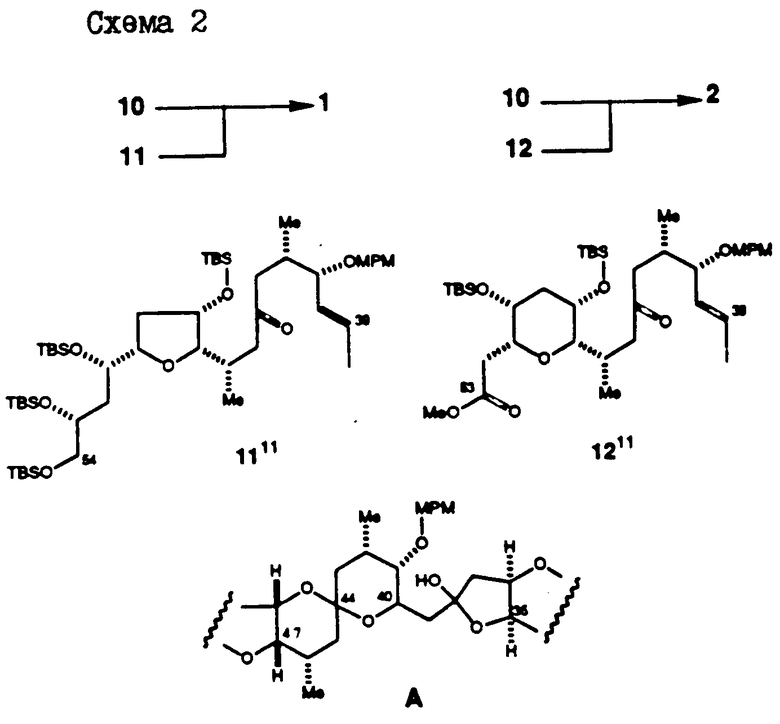
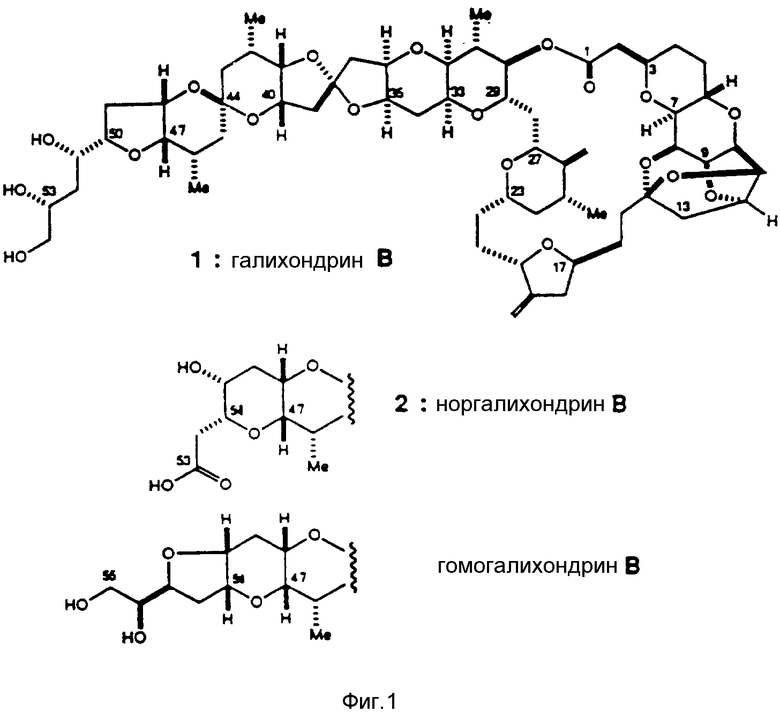


Сущность: производные галихондрина могут быть использованы в медицине, при производстве протвоопухолевых лекарств и имеют структурную формулу I, приведенную в тексте описания, где R1 и R2 - C1-C6-алкил; A - незащищенная или защищенная группа формулы: - CH2CH2OH; B - незащищенная или защищенная OH-группа; m = n = 1. Промежуточные соединения имеют структурную формулу II, приведенную в тексте описания, где R1 - C1-C6-алкил; R2 - защищенная OH-группа, R30 - группа CHO, CO2D, где D - C1-C5-алкил; R4 - OH-группа незащищенная или защищенная; R5 - OH-группа, защищенная или незащищенная или защищенная CH2CH2OH группа, структурную формулу III, приведенную в тексте описания, где X - OH-группа; R1; R2; R3 - защищенная OH-группа; R4 - CO2D, где D -C1-C5-алкил; структурную формулу IV, приведенную в тексте описания, где R1-R5 - защищенная OH-группа; R6 - кислород; R7-R8 - C1-C5-алкил; X - галоген; структурную формулу V, приведенную в тексте описания, где R1-R3 - защищенная OH-группа; R4 -кислород; R5-R6 - C1-C5-алкил; R7 - группа CO2D, где D - C1-C5-алкил; X - галоген, структурную формулу VI, приведенную в тексте описания, где R1-R6 -защищенная OH-группа, R8-R11 - C1-C5-алкил; R12-R13 - метилиден; R14-R15 - кислород, структурную формулу VII, приведенную в тексте описания, где R1-R4 -защищенная OH-группа; R5-R8 - C1-C5-алкил; R9-R10 -метилиден; R11 - группа CO2D, где D - C1-C5-алкил; R12 и R13 - атом кислорода. 7 с. и 9 з.п. ф-лы, 2 табл., 3 ил.
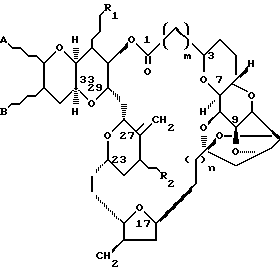
где R1 и R2 - C1-C6-алкил;
A - незащищенная или защищенная группа формулы -CH2CH2OH;
B - незащищенная или защищенная OH-группа;
m и n = 1.
где R6 - R8 - C1-C6-алкил.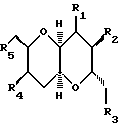
где R1 - C1-C6-алкил;
R2 - защищенная OH-группа;
R3 - группа CHO, CO2D, где D - C1-C5-алкил;
R4 - OH-группа незащищенная или защищенная;
R5 - OH-группа защищенная или незащищенная, или защищенная CH2CH2OH-группа.
где X - OH-группа;
R1, R2, R3 - замещенная OH-гpуппа;
R4 - CO2D-группа, где D - C1-C5-алкил.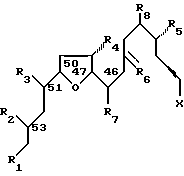
где R1 - R5 - защищенная OH-группа;
R6 - кислород;
R7 и R8 - C1-C5-алкил;
X - галоген.
где R1 - R3 - защищенная OH-группа;
R4 - кислород;
R5 - R6 - C1-C5-алкил;
R7 - группа CO2D, где D - C1-C5-алкил;
X - галоген.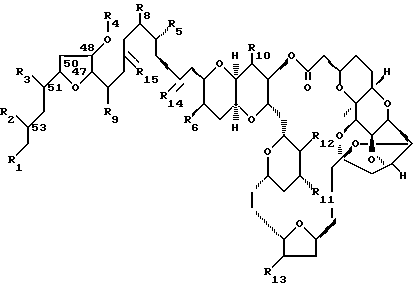
где R1 - R6 - защищенная OH-группа;
R8 - R11 - C1-C5-алкил;
R12 и R13 - метилиден;
R14 и R15 - кислород.
где R1 - R4 - защищенная OH-группа;
R5 - R8 - C1-C5-алкил;
R9 и R10 - метилиден;
R11 - группа CO2D, где D - C1-C5-алкил;
R12 и R13 - кислород.
