4-Хлор-2-тиофенкарбоновая кислота имеет структуру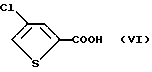
Это соединение имеет большое значение как промежуточный продукт в синтезе соединений, обладающих фармацевтическим действием, таких как, например, 5-фтор-6-хлор-3-(4-хлор-2-теноил)-2-оксииндол-1-карбоксамид, который впервые описан в патенте США 5047554 (Ehrgolt et al.) в примере 72.
Синтез 4-хлор-2-тиофенкарбоновой кислоты (для удобства далее сокращенно называемой "ХТКК") впервые описывается в публикации I. Iriate et al., J. Heterocyclic Chem., 13, 393 1976. В ней сообщается, что ХТКК получена с количественным выходом в виде технического продукта окислением соответствующего 2-альдегида оксидом серебра. Сообщалось, что технический продукт "после повторной перекристаллизации из метанола или дихлорметана" имеет температуру плавления 131 - 132oC. Iriarte с соавторами в этой же статье сообщали о получении СТСА с т.пл. 125 - 126oC омылением этилового эфира 4-хлортиофен-2-карбоновой кислоты в растворе гидроксида калия в метаноле, причем карбоксилат был получен прямым хлорированием этилового эфира тиофен-2-карбоновой кислоты в присутствии хлорида алюминия.
В публикации (Lemaire et al., J. Electroanal. Chem., 281, 293 1990) сообщается о получении 3-хлор-2-триметилсилилтиофена способом с применением реактива Гриньяра, при этом продукт используют в электрополимеризации для получения поли(3-хлортиофена). Никаких других способов синтеза ХТКК описано не было.
Авторами данного изобретения установлено, что ХТКК может быть получена в результате температурно-зависимого региоселективного процесса, в котором возможные сайты реакции блокируются и таким образом предотвращается получение побочных продуктов.
Изобретение предлагает способ получения 4-хлор-2-тиофенкарбоновой кислоты, включающий удаление силильной группы SiR3 из соединения формулы IVa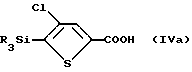
в которой каждую из R-групп независимо выбирают из /C1-C6/-алкила, бензила и фенила.
Разновидностью вышеуказанного способа получения 4-хлор-2-тиофенкарбоновой кислоты является способ, включающий удаление силильной группы SiR2 из соединения формулы IVb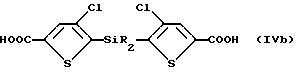
в которой R принимает значения, определенные ранее.
Более точно, способ получения 4-хлор-2-тиофенкарбоновой кислоты включает следующие стадии:
1) обработка 3-хлортиофена при температуре ниже приблизительно -50oC основанием и последующая обработка кремнийорганическим соединением формулы R3-SiX, в котором X является уходящей группой и каждую R-группу независимо выбирают из /C1-C6/алкила, бензила и фенила, с образованием соединения формулы
2) обработка продукта стадии (1) при температуре ниже приблизительно -50oC подходящим основанием для депротонирования хлортиофенового кольца в 5 положении, с образованием соответствующего аниона формулы IIa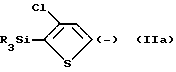
3) Обработка продукта стадии (2) при температуре ниже -50oC диоксидом углерода для образования, соответственно, монокарбоксилата формулы IIIa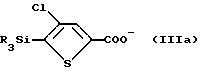
4) превращение продукта стадии (3) в соответствующую кислоту (т.е. формулы IVa) и
5) удаление силильной группы SiR3.
Другой вариант способа прямого получения 4-хлор-2-тиофенкарбоновой кислоты включает следующие стадии:
1) обработка 3-хлортиофена при температуре ниже приблизительно -50oC основанием и последующая обработка кремнийорганическим соединением формулы R2SiX2, в котором X и R принимают значения, определенные ранее, с образованием соединения формулы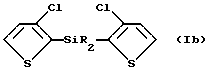
2) обработка продукта стадии (1) при температуре ниже приблизительно -50oC основанием, подходящим для депротонирования в 5 и 5' - положениях двух тиофеновых колец, с образованием дианиона формулы IIb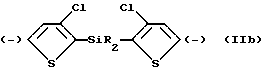
3) обработка продукта стадии (2) при температуре ниже -50oC диоксидом углерода с образованием соответствующего дикарбоксилата формулы IIIb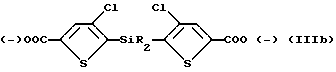
4) превращение продукта стадии (3) в соответствующую дикислоту (т.е. в соединение формулы IVb); и
5) удаление силильной группы SiR2.
В качестве уходящей группы X используют хлор, бром и йод, трифторметансульфонат, трифторацетат, ацетамид, трифторацетамид, 1,2,4-триазол и имидазол, а также некоторые другие группы хорошо известные специалистам данной области химии. Хлор и трифторметансульфонат являются предпочтительными группами, благодаря широкой коммерческой доступности.
Конкретные примеры R-группы, определенной как /C1-C6/алкил, включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил и н-гексил.
ХТКК, полученная вышеуказанным способом, как полагают, является по существу чистым продуктом и имеет температуру плавления 138oC, что указывает на значительно большую чистоту, чем чистота продукта, полученного описанным ранее способом, температура плавления которого составляет 131 - 132oC даже после повторной перекристаллизации.
Данное изобретение предлагает также способы /которые являются разновидностями основного способа/ получения 5-фтор-6-хлор-3-(4-хлор-2-теноил)-2-оксиндол-1-карбоксамида (структуры VIII, схема I ниже), включающие реакцию в присутствии основания 5-фтор-6-хлор-2-оксиндол-1-карбоксамида (структуры VII, схема I) с активированной формой (например, хлорангидридом кислоты, ацилимидазолом или метиловым эфиром) 4-хлор-2-тиофенкарбоновой кислоты, полученной согласно любому из вышеуказанных способов. Предпочтительно использовать либо активированный ацилимидазол, полученный реакцией СТСА с карбонилдиимидазолом, либо хлорид кислоты, полученный взаимодействием СТСА с тионилхлоридом. Оба эти предпочтительные активированные производные могут быть получены удобным способом.
Дальнейший способ получения 5-фтор-6-хлор-3-(4-хлор-2-теноил)-2-оксиндол-1-карбоксамида включает следующие стадии:
1) взаимодействие в присутствии основания 5-фтор-6-хлор-2-оксиндол-1-карбоксамида с активированной монокарбоновой кислотой формулы IVc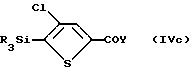
где Y - хлор, имидазол-1-ил или метил, с образованием таким образом 5-фтор-6-хлор-3-(4-хлор-3-тризамещенного силил-2-теноил)-оксиндол-1-карбоксамида формулы Va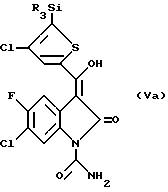
и
2) удаление силильной группы SiR3.
Еще один вариант способа, описанного выше, включает следующие стадии:
1/ взаимодействие в присутствии основания 5-фтор-6-хлор-2-оксиндол-1-карбоксамида с активированной дикарбоновой кислотой формулы IVd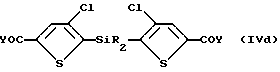
где Y принимает значения, определенные выше с образованием в результате соответствующего соединения формулы Vb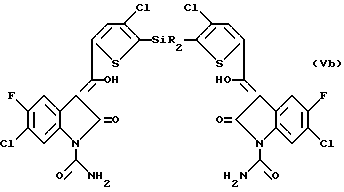
2) удаление силильной группы SiR2.
Соединения формул IVa и IVb, как полагают, также являются новыми и соответственно, представляют собой дополнительный объект изобретения.
Химия данного изобретения с использованием силанов формулы RSiX для простоты может быть кратко представлена в обобщенной схематической форме следующего вида
Схема 1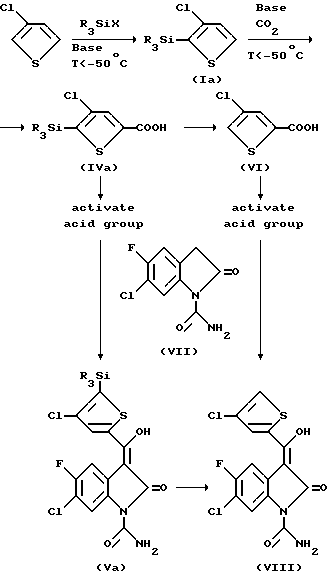
Специалисту данной области будет ясно, что использование соединения формулы R2SiX2 вместо соединения формулы R3SiX приводит, соответственно, к замещению в схеме 1 моно-производного формулы Ia на димеризованный 3-хлортиофен формулы Ib, к замещению моно-кислоты формулы IVa на дикислоту формулы IVb и к замещению моно-производного формулы Va на ди-производное формулы Vb (строение указанных производных показано выше).
Предварительно следует отметить, что все реакции, описанные далее, должны проводиться в инертной атмосфере, предпочтительно в аргоне или азоте. Для удобства силильные блокирующие группы R3Si- и (-R2Si-) в приведенном описании называют "блокирующими группами", и силильные соединения R3SiX (и R2SiX2) называют "блокирующими реагентами". Далее следует отметить, что когда приводится значение температуры менее чем приблизительно -50oC и предпочтительное значение менее -70oC, предпочтительной температурой является температура -78oC вследствие того, что ее легко получить при помощи бани со смесью сухой лед/ацетон. Достижимы и более низкие температуры, например -94oC, которая может быть достигнута охлаждением на бане со смесью гексана с жидким азотом. Однако таким образом получают лишь несущественные преимущества.
3-Хлортиофен, блокированный во втором положении, может быть получен первичным взаимодействием 3-хлортиофена с эквивалентным количеством сильного основания, такого как алкиллитий, при температуре ниже -50oC, предпочтительно ниже -70oC, таким образом, что депротонирование становится региоселективным и направляется во 2 положение. н-Бутиллитий предпочтителен в качестве основания, благодаря его коммерческой доступности. 3-Хлортиофен и основание растворяют в инертных растворителях (термин "инертный" используется в отношении примененных условий реакции) таких как тетрагидрофуран (ТГФ) и гексан соответственно. Реакционную смесь перемешивают в течение периода времени, который может составлять от 30 мин до нескольких часов. Блокирующий реагент может добавляться в эквивалентном количестве или с небольшим (до 10%) избытком. После периода времени от нескольких минут до нескольких часов реакцию гасят после нагревания до приблизительно комнатной температуры или меньшей температуры добавлением воды и/или рассола. Блокированный продукт можно экстрагировать подходящим органическим растворителем, таким как этилацетат, обычным способом, сушить и удобно выделять, например, выпариванием.
2-Блокированный-3-хлортиофен может затем использоваться в реакции в подходящем сухом растворителе, таком как ТГФ, с основанием (в эквивалентном количестве или с небольшим избытком) при температуре менее -50oC, предпочтительно менее -70oC, для селективного депротонирования на остающемся неблокированным атоме углерода, соседнем с атомом серы в тиофеновом цикле. После этого при сохранении температур ниже -50oC газообразный диоксид углерода может использоваться для обработки депротонированного промежуточного продукта, в результате чего получают 4-хлор-5-блокированный-2-тиофенкарбоксилат формулы IIIa или IIIb. Специалисту понятно, что необходимо использовать основание, которое не обладает более сильными нуклеофильными свойствами, чем блокирующая группа, уже присоединенная к тиофеновому циклу. Основания, которые можно применять на данной стадии, включают литийдиалкиламиды, которые можно получить in situ реакцией алкиллитиевого производного с ди-алкиламином. Например, н-бутиллитий может быть обработан при температуре приблизительно 0oC в ТГФ до введения в 2-блокированный-3-хлортиофена, полученного на предыдущей стадии, эквивалентным количеством диалкиламина, такого как диизопропиламин, с образованием диизопропиламида лития. После этого температура может быть понижена до менее -50oC и проведено присоединение диоксида углерода. Карбоксилат превращают в кислоту гашением реакции водным раствором кислоты, например, водной соляной кислотой. Органическая фракция может быть отделена, водный слой можно экстрагировать (например, этилацетатом) с последующим выделением кислоты.
Блокирующая группа в этой точке может быть удалена обработкой продукта фторидом с образованием в результате реакции 4-хлор-2-тиофенкарбоновой кислоты. После этого кислота может быть активирована и введена во взаимодействие с 5-фтор-6-хлор-2-оксиндол-1-карбоксамидом (называемым также "карбоксамидным предшественником"), с получением в результате 5-фтор-6-хлор-3-(4-хлор-2-теноил)-2-оксиндол-1-карбоксамида, обладающего фармацевтической активностью.
При другом способе блокирующую группу не следует удалять, и блокированная кислота может быть активирована и реагировать непосредственно с карбоксамидным предшественником с получением соответствующего блокированного продукта (Va) или (Vb) обладающего фармацевтическими свойствами.
В любом случае блокирующая группа может быть удалена обработкой анионом фтора, при этом конечный фармацевтически активный продукт выделяют при гашении реакции водным раствором (например, соляной) кислоты. Вид источника аниона фтора не играет важной роли и могут использоваться различные реагенты, содержащие анион фтора, в том числе и фториды щелочных металлов (например, фториды натрия, калия, лития и цезия), фториды щелочно-земельных металлов (например, фториды магния и кальция), фторводород как в свободной (HF) так и в связанной форме (например, в виде гидрофторида пиридина) и фториды тетра-низший алкил-аммония. Фториды тетра-низший алкил-аммония являются предпочтительными, причем наиболее предпочтителен фторид тетра-н-бутиламмония вследствие его легкой коммерческой доступности. Для интенсификации реакции может использоваться избыток фторида, например, 2 - 3 эквивалента.
Способы применения 5-фтор-6-хлор-3-(4-хлор-2-теноил)-2-оксиндол- -1-карбоксамида описаны в патенте США, 5047554 /Ehrgott/.
Приведенные ниже примеры иллюстрируют различные аспекты изобретения, без какого бы то ни было ограничения его области. В приведенных примерах данные ЯРМ получены на приборах марки Brooker AM u Brooker АM300. Для H1 ПМР-спектров использовались частоты от 250 MHz или 300 MHz, 13C ЯМР-спектры получены при 62,5 MHz или 75 MHz.
Пример 1а. 2-Триметилсилил-3-хлортиофен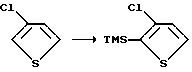
К 3-хлортиофену (5,0 г, 42,16 ммоль), растворенному в 50 мл тетрагидрофурана при перемешивании при температуре -72oC (охлаждение на бане: ацетон/сухой лед) в течение 15 мин добавляют 16,8 мл 2,5 М-ного раствора бутиллития в гексане. В процессе добавления температуру реакционной массы сохраняют ниже -70oC. По окончании добавления н-бутиллития образуется белый осадок. После перемешивания в течение 40 мин при температуре -70oC к полученной реакционной смеси медленно в течение 5 мин добавляют 5,88 мл хлортриметилсилана. После добавления раствор быстро становится прозрачным, а затем опять мутнеет вследствие образования хлорида лития, который является побочным продуктом. Спустя 10 мин реакционной раствор нагревают до 0oC и добавляют 5 мл воды, а затем 25 мл рассола для гашения реакции. Водный раствор экстрагируют этилацетатом (2 х 30 мл). Органический экстракт сушат сульфатом натрия, фильтруют и упаривают, в результате получают 8,0 г соединения, указанного в заглавии, в виде прозрачного масла.
Физические свойства: масс-спектр /EIMS/ m/z = 192 M+ +2,14%/, 190 /M+, 36%/, 177 /M+ +2-CH3, 41%/, 175 /M+-CH3, 100%/; ПМР /CDCl3/ δ 7,49 /1H, д, J = 4,7 Hz/ 7,07 /1H, д, J = 4,7 Hz/ и 0,46 /9H, с/; 13C ЯМР /CDCl3/ δ 132,0; 130,2; 130,1; 129,8 и -0,7.
Пример 1b. 4-Хлор-5-триметилсилил-2-тиофенкарбоновая кислота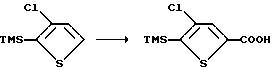
Небольшое количество (4 мг) дифенилуксусной кислоты растворяют в 50 мл тетрагидрофурана и перемешивают при комнатной температуре. К полученному раствору медленно по каплям добавляют н-бутиллитий в виде 2,5 М-ного раствора в гексане до тех пор, пока раствор не приобретет желтый цвет от образовавшегося дианиона ди-фенилуксусной кислоты. Такая последовательность обеспечивает сухой раствор. Затем раствор охлаждают до температуры -72oC (ацетон/сухой лед). При этой температуре добавляют 4,62 мл (11,542 ммоль) 2,5 М-ного раствора бутиллития в гексане с последующим добавлением 1,76 мл (12,592 ммоль) диизопропиламина. Для образования литийдиизопропиламида реакционный раствор нагревают до 0oC (ледяная баня) в течение 20 мин и снова охлаждают до -72oC. К охлажденному реакционному раствору добавляют 2,0 г (10,493 ммоль) 2-триметилсилил-3-хлортиофена в виде 5 мл раствора в тетрагидрофуране в течение 20 мин. Для обеспечения региоселективного депротонирования температуру реакции поддерживают ниже -70oC. После 30 мин реакции через пожелтевший реакционный раствор медленно пропускают газообразный диоксид углерода, сохраняя температуру реакционной массы ниже -55oC. Общее время обработки реакционного раствора диоксидом углерода составляет 10 мин. После этого раствор быстро нагревают до 0oC и реакцию гасят 50 мл IN соляной кислоты, в результате чего происходит выделение некоторого количества газа. В процессе добавления раствора кислоты реакционный раствор нагревают до комнатной температуры, после чего добавляют 50 мл рассола. Органическую фракцию отделяют и водную часть экстрагируют этилацетатом (2 х 50 мл). Органические экстракты соединяют, промывают рассолом (1 х 50 мл), сушат сульфатом натрия, фильтруют и упаривают, в результате получают 3 г белого твердого вещества. Перекристаллизация технического продукта из гептана приводит к получению 1,67 г указанного в заглавии чистого продукта в виде белого кристаллического вещества, т.пл. 206 - 210oC.
Физические свойства: масс-спектр /LSIMS/ m/z = 237 (M + H+ + 2,12%), 235 (M + H+ 10%); ПМР (CDCl3) δ 7,74 (1H, с) и 0,41 (9H, с); 13C ЯМР (CDCl3 + CD3OD) δ 163,1; 141,2; 137,8; 134,7; 131,6 и -1,4.
Пример 1c. 4-Хлор-2-тиофенкарбоновая кислота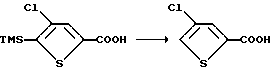
300 мл (1,278 ммоль) 4-хлор-5-триметилсилил-2-тиофенкарбоновой кислоты растворяют в 10 мл тетрагидрофурана с 0,3 мл воды и охлаждают до -50oC (лед/рассол). 2,5 мл 1M-ного раствора фторида тетра-н-бутиламмония медленно добавляют в реакционный раствор. Спустя 4 ч реакционный раствор переносят в 50 мл 5%-ного водного раствора бикарбоната натрия. Полученный реакционный раствор помещают в делительную воронку и промывают этилацетатом (2 х 25 мл). Щелочной водный раствор подкисляют концентрированной соляной кислотой до pH 2 и экстрагируют этилацетатом (3 х 30 мл). Органическую фракцию сушат сульфатом натрия, фильтруют и упаривают с получением 216 мг продукта в виде белого твердого вещества. После перекристаллизации из теплого гептана получают 85 мг чистого кристаллического продукта, т.пл. 138oC.
Физические свойства: масс-спектр (EIMS) m/z = 164 (M+ + 2,30%) и 162 (M+, 100%); ПМР (CDCl3) δ 10,9 (1H, уш. с, заменяемый), 7,74 (1H, д, J = 1,5 Hz) и 7,43 (1H, д, J = 1,5 Hz); 13C ЯМР (CDCl3) 166,1; 134,6; 133,2; 128,3 и 126,5.
Пример 2a. 5-Фтор-6-хлор-2,3-дигидро-3-[гидрокси-2-(4-хлор-5- триметилсилилтиенил)метилен]-2-оксо-1H-индол-1-карбоксамид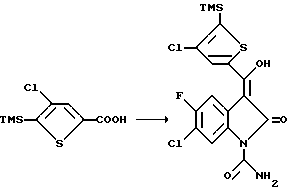
375 мг (1,60 ммоль) 4-хлор-5-триметилсилил-2-тиофенкарбоновой кислоты соединяют с 5 мл тионилхлорида и нагревают до кипения. Спустя 1,5 ч реакция завершается. Колбу медленно охлаждают до комнатной температуры и избыток тионилхлорида упаривают с получением требуемого хлорида в виде коричневого масла. Коричневое масло растворяют в 5 мл N,N'-диметилформамида и медленно добавляют к 15 мл раствора 5-фтор-6-хлор-2,3-дигидро-2-оксо-1H-индол-1-карбоксамида (500 мг, 2,24 ммоль) и 4-(N,N-диметиламино)пиридина (708 мг, 5,8 ммоль) в N,N-диметилформамиде при температуре 0oC и перемешивании. Спустя 1 ч реакционный раствор переносят в 30 мл 1 N-ной соляной кислоты, в результате чего продукт выпадает в виде осадка каштанового цвета. Технический продукт фильтруют и перекристаллизовывают из теплой уксусной кислоты с получением 307 мг (609 ммоль) указанного в заглавии чистого соединения в виде твердого кристаллического вещества желтого цвета, т.пл. 200oC.
Физические свойства: масс-спектр /LSIMS/ m/z = 470 (M - H+ + Na + 4,4%), 468 (M - H+ + Na + 2,16%), 466 (M - H+ + Na, 21%), 448 (M+ + 4,17%), 446 (M+ 2,74%), 444 (M+, 100%), 405 (M+ - CONH + 4,8%), 403 (M+ - CONH + 2,28%) и 401 (M+ - CONH, 39%); ПМР (DMSO-d6) (1H, заменяемый), 8,51 (1H, с), 8,11 (1H, д, J = 7,3 Hz), 7,96 (1H, JM-F = 11,0 Hz), 7,30 (1H, заменяемый), 6,21 (1H, заменяемый) и 0,37 (9H, с).
Пример 2b. 5-Фтор-6-хлор-2,3-дигидро-3-[гидрокси-2-(4-хлортиенил)- метилен]-2-оксо-1Н-индол-1-карбоксамид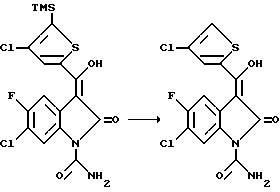
50 мг (0,11 ммоль) 5-фтор-6-хлор-2,3-дигидро-3-[гидрокси-2-(4-хлор-5- триметилсилилтиенил)метилен] -2-оксо-1Н-индол-1-карбоксамида растворяют в 2 мл тетрагидрофурана и охлаждают до 5oC. Небольшую 0,56 мл аликвоту 1M-ного раствора фторида тетра-бутиламмония добавляют шприцем к охлажденному раствору индольного субстрата при перемешивании. Спустя 1 ч к реакционному раствору добавляют 0,25 мл воды. Спустя еще 30 мин реакцию гасят добавлением 5 мл 1N соляной кислоты. Содержимое реакционной колбы выливают в 15 мл воды, что приводит к выпадению продукта из реакционного раствора в виде осадка. После фильтрования получают 25 мл продукта, указанного в заглавии, в виде твердого вещества желтого цвета, после перекристаллизации из уксусной кислоты получают кристаллический продукт, т.пл. 234 - 237 oC.
Физические свойства: ПМР (DMSO - d6) δ 9,10 (1H, заменяемый), 8,69 (1H, д, J = 1,5 Hz), 8,10 (1H, д, JH-F = 7,4 Hz), 8,06 (1H, д, JH-F = 11,4 Hz), 7,65 (1H, d, J = 1,5 Hz) и 7,26 (1H, заменяемый).
Пример 3a. Получение бис(3-хлортиофен-2-ил)-диметилсилана.
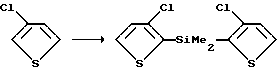
К 3-хлортиофену (5,0 г, 42,16 ммоль), растворенному в тетрагидрофуране при перемешивании на бане (ацетон/сухой лед) при температуре -72oC добавляют 16,8 мл 2,5 М раствора н-бутиллития в гексане в течение 15 мин. Температуру реакции поддерживают - 70oC в течение всего времени добавления. Как только добавление н-бутиллития завершается, образуется осадок. После перемешивания при -70oC в течение 40 мин, медленно добавляют 2,81 мл дихлордиметилсилана в течение 5 мин. После завершения добавления раствор моментально становится прозрачным с последующим появлением помутнения вследствие образования полупродукта литийхлорида. Через 10 мин реакционный раствор нагревают до 0oC. Добавляют 5 мл воды, а затем 25 мл рассола при 0oC для прекращения реакции. Затем экстрагируют водный раствор этилацетатом (2 х 30 мл). Органический экстракт сушат (Na2SO4), фильтруют и выпаривают с получением соединения, указанного в заголовке.
Пример 3b. Получение бис(5-карбокси-3-хлортиофен-2-ил) диметилсилана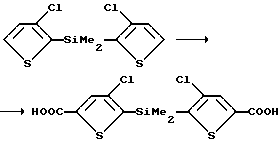
Указанное соединение получают по методике примера 1б, используют вместо 2-триметилсилил-3-хлортиофена, продукт, полученный в примере 3a.
Пример 3c. Получение 4-хлор-2-тиофен карбоновой кислоты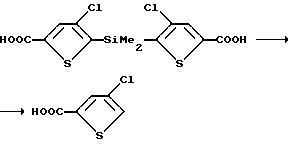
Указанное соединение получают в кристаллической форме по методике примера 1c, используя вместо 4-хлор-3-триметилсилил-2- -тиофен карбоновой кислоты, продукт, полученный в примере 3b.
Пример 4. Получение 5-фтор-6-хлор-2,3-дигидро-3-[гидрокси-2-(4-хлортиенил)метилен]-2-оксо-1H-индол-1-карбоксамида.
Указанный продукт получают по методикам примеров 2a и 2b, используя 1/2 мольного эквивалента (0,8 ммоль) соединения, полученного в Примере 3b вместо 375 мг (1,60 ммоль) 4-хлор-5-триметилсилил-2-тиофен карбоновой кислоты.
4-Хлор-2-тиофенкарбоновую кислоту получают взаимодействием 4-хлор-5-триметилсилил-тиофена с подходящим основанием при температуре ниже -50oC для депротопирования хлортиофенолового кольца в 5 положении с образованием соответствующего аниона. Который обрабатывают диоксидом углерода при температуре ниже -50oC с последующим удалением силильной группы. Продукт используют как промежуточное соединение при получении 5-фтор-6-хлор-3-(4-хлор-2-теноил)-2-оксииндол-1-карбоксамида, обладающего фармацевтической активностью. 5 с. и 5 з.п. ф-лы.
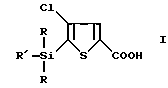
где R - независимо выбирают из C1 - C6-алкила, бензила и фенила;
R' - C1 - C6-алкил, бензил, фенил или группа формулы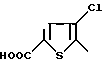
2. Способ получения 4-хлор-2-тиофенкарбоновой кислоты, отличающийся тем, что из соединения формулы IVa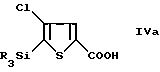
где каждую из групп R независимо выбирают из C1 - C6-алкила, бензила, фенила,
удаляют силильную группу-SiR3.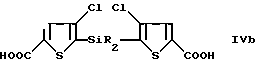
где R независимо выбирают из C1 - C6-алкила, бензила, фенила,
удаляют силильную группу -SiR2-.
R3SiX,
где X - уходящая группа,
каждую R-группу независимо выбирают из C1 - C6-алкила, бензила и фенила,
с образованием соединения формулы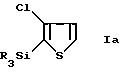
b) обрабатывают продукт стадии (a) при температуре приблизительно ниже -50oC подходящим основанием для депротонирования хлортиофенового цикла в 5-положении с образованием соответствующего аниона формулы IIa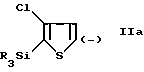
c) обрабатывают продукт стадии (b) при температуре ниже -50oC диоксидом углерода с образованием соответственно монокарбоксилата формулы IIIa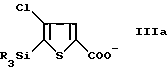
d) превращают продукт стадии (c) в соответствующую кислоту, e) удаляют силильную группу -SiR3.
R2SiX2,
где X - уходящая группа,
каждую R-группу независимо выбирают из C1 - C6-алкила, бензила и фенила,
с образованием соединения формулы Ib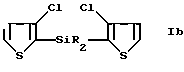
b) обрабатывают продукт стадии (a) при температуре приблизительно ниже -50oC основанием, подходящим для депротонирования в 5- и 5'-положениях в обоих тиофеновых циклах, с образованием дианиона формулы IIb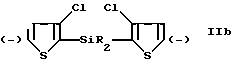
c) обрабатывают продукт стадии (b) при температуре ниже -50oC диоксидом углерода с образованием соответствующего дикарбоксилата формулы IIIb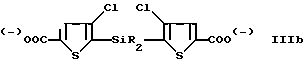
d) превращают продукт формулы IIIb в соответствующую дикислоту и e) удаляют силильную группу -SiR2-.
| J.Heterocyclic chem., 13, 393(1976) | |||
| SU, 1015622, C 07 D 333/38, 1985. |
