Изобретение относится к области аналитических исследований с помощью средств электроники: ионного анализа и детектирования вторично-электронным умножителем макромолекул биополимеров, (например, бактерий, вирусов, клетки и их частей), а именно к дезинтеграционным способам исследования макромолекул (м.м.) с молекулярной массой М > 105 а.е.м.
В аналитической биохимии для определения М биополимерных м.м. в смесях традиционно используются такие методы, как [1]:
- электрофорез, где М определяют по подвижности ионов м.м., дрейфующих в геле под действием электрического поля;
- седиментация, где М определяют по поверхности м.м. в растворе под действием центробежных сил, возникающих при быстром вращении образца в ультрацентрифуге;
- вискозиметрия, где М определяют по вязкости м.м., измеряемой для движущегося раствора м.м.
Известен также способ определения М м.м. по флюоресценции отдельных м.м. [2] . В соответствии с [2] после химического связывания красителем в буфере размеры (длину, а следовательно, и М) м.м. определяют по количеству фотонов, испускаемых при флюоресценции. При этом размеры области детектирования выбирают таким образом, чтобы она содержала в среднем заведомо не более одной м. м. , то есть вероятность нахождения в ней двух и более м.м. пренебрежимо мала.
Низкая точность (несколько %) определения М, большая длительность анализа (несколько часов), а в ряде случаев - и недостаточная чувствительность (например, способ (2) позволяет детектировать только очень большие м.м. с М > 5 - 10 млн. а.е.м.) описанных способов обусловили значительный практический интерес к использованию масс-спектрометров для экспрессного и точного определения М м.м.
В настоящее время наиболее близким техническим решением к заявленному способу, обеспечивающим определение М больших м.м. является метод лазерной десорбции (абляции) при посредничестве матрицы [3].
При этом способе м.м. смешивают с матрицей - хромофором и высушивают до образования равномерного твердого раствора, который затем вводят в вакуум и подвергают лазерной десорбции (абляции).
М м.м. определяют по времени пролета молекулярного иона, образующегося в результате протонирования м.м. в процессе абляции.
Недостатком этого способа является хотя и немного более расширенный по сравнению с другими масс-спектрометрическими способами, но тем не менее ограниченный массовый диапазон из-за чрезвычайно низкой эффективности ионов м. м. на вторично-электронном умножителе (ВЭУ) при М > 200 - 300 тыс. а.е.м. Более того, несмотря на многолетний поиск, пока так и не найдены матрицы, позволяющие определить М для таких важных классов м.м. как, например, олигонуклеотиды и дезоксирибонуклеиновые кислоты (ДНК), при их М > 50 - 100 тыс. а.е.м. (то есть детектируются лишь многочисленные ионы фрагментов).
К недостаткам данного способа относятся также низкая точность определения М при М > 100 тыс. а.е.м. (не выше нескольких %) и ограниченная чувствительность метода вследствие низкой эффективности ионизации м.м., вероятность которой составляет 10-4 - 10-6.
Целью изобретения является значительное повышение чувствительности анализа и верхнего предела измеряемых масс м.м.
Указанная цель достигается за счет отказа от традиционного подхода, заключающегося в необходимости перевода из твердой или жидкой фазы в газовую фазу всей м.м. без ее разрушения с последующим масс-спектрометрическим анализом.
Вместо этого предлагается допускать разрушение м.м., но затем тщательно и с минимальными потерями анализировать при помощи масс-спектрометра все ее фрагменты, и, складывая их массы mi, определять М исходной м.м.
В силу невозможности достижения 100%-ной вероятности детектирования фрагментов, сумма mi всех фрагментов будет неизбежно меньше чем М м.м. Поэтому данный способ требует введения теоретически или экспериментально определяемых поправочных коэффициентов pi > 1. И из-за различной энергетической прочности молекулярных связей pi могут быть различны для различных фрагментов.
С другой стороны, в силу однотипности биополимеров одного класса например, ДНК, состоящих всего из 4 различных типов оснований, pi ожидается весьма воспроизводимым в пределах одного и того же класса при изменении М в широких пределах, вплоть до нескольких порядков.
В итоге М определяют как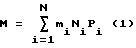
где Ni - количество зарегистрированных фрагментов i-го сорта;
N - общее количество сортов.
Этот способ осуществим, если фрагменты могут быть однозначно соотнесены с индивидуальными исходными м.м. или с известным их числом.
Поскольку при анализе смесей, представляющем наибольший практический интерес, такое соотнесение невозможно, если за один цикл анализа детектируются фрагменты двух и более различных м.м. (так как среди них могут оказаться м.м. разных М), то в каждом цикле должно анализироваться не более одной м.м.
Таким образом, испарение и ионизация м.м. из образца должны быть локализованы до такой степени, что в анализируемом за один цикл объеме содержится не более одной м.м., а вероятность нахождения там двух и более м.м. пренебрежимо мала.
Сопоставительный анализ заявленного способа с прототипом показывает, что заявленный способ отличается от прототипа отсутствием необходимости образования молекулярного иона м.м, что заменено анализом за цикл фрагментов от не более чем одной м.м., а также процедурой определения М по сумме М фрагментов, а не по М протонированной м.м.
Таким образом, заявленный способ соответствует условию изобретения "новизна".
Сравнительный анализ заявленного решения с другими аналогичными по решаемой задаче техническими решениями показывает, что известен способ масс-спектрометрического анализа, по которому возможно определение М м.м. по масс-спектроскопии лишь ее фрагментов, но этот метод так же, как и прототип, обязательно требует образования стабильного молекулярного иона с целью его последующего ускорения и фрагментации [4]. Следовательно, метод неприменим вне того же диапазона М, что и прототип. Более того, даже в этом диапазоне вышеописанный способ неприменим, если образующиеся фрагменты имеют одинаковые массы, а также, если анализируемая смесь молекул дает многочисленные и однотипные фрагменты, поскольку тогда разницы времен пролета фрагментов одинаковы для молекул с различными М, между тем, что именно однотипность фрагментов характерна для ДНК.
Известен также традиционный способ определения М органических молекул, который заключается в определении М сравнением распределений масс-молекулярных фрагментов этих молекул с распределением для известных молекул [5]. Массовый диапазон анализа в нем ограничен вследствие отсутствия молекулярных ионов исследуемых молекул, прогрессирующего уменьшения зависимости распределения фрагментов от М при увеличении М., необходимости сложных и многочисленных калибровок. Как правило, верхний предел масс-анализируемых молекул не превышает 1000 - 2000 а.е.м.
Таким образом, новые свойства, проявляемые указанными в формуле признаками в их неразрывном сочетании, позволяют получить новый технический результат высокоточного анализа м.м. с М > 105 а.е.м., который недостижим во всех вышерассмотренных известных способах, что позволяет сделать вывод о соответствии заявленного способа критерию "изобретательского уровня".
Особенностью заявленного способа является также то, что неизбежные статистические флуктуации числа фрагментов, детектируемых в нем, компенсируются большим числом фрагментов и вносят меньший относительный вклад при увеличении М м. м., что также существенно отличает его от других известных способов.
Требование малой вероятности нахождения двух и более м.м. в анализируемом за один цикл объеме V может быть представлено как необходимость разбавления м.м. в образце до концентрации
C - a/V (м.м./м3), (2)
где a - коэффициент разбавления.
Например, если за счет разбавления или уменьшения площади испаряющего воздействия вероятность нахождения м.м. в объеме V составляет a = 0,001, то вероятность нахождения там двух и более м.м. составляет a2/(1-a) ≈ 1,001 • 10-6, то есть сигнал от двух и более м.м. будет давать фон на уровне a/(1-a) • 100% = 0,1% от полезного сигнала. В силу незначительности требуемого динамического диапазона анализа (не более 10 - 100 практически для всех приложений), этот уровень является удовлетворительным. Однако увеличение a до 0,1 - 0,3 приводит к снижению динамического диапазона анализа до неприемлемо низких величин, (менее 3 - 10), поэтому при a > 0,1 - 0,3 способ теряет практическую ценность.
Высокая вероятность выноса фрагментов из объема V достигается лишь при повышении плотности потока мощности воздействия над некоторым пороговым значением. При распределении м.м. по объему образца необходимо обеспечить разлет материала матрицы, содержащей фрагменты м.м., то есть абляцию матрицы. Порог абляции определяется температурой сублимации матрицы, ее поглощающей способностью для данного воздействия, теплоемкостью, теплопроводностью и т. д.
При распределении м.м. по поверхности достаточно обеспечить высокую вероятность десорбции ее фрагментов, что требует обычно меньших плотностей потока мощности, чем абляция.
Условие границы между поверхностным и объемным распределением м.м. определяется следующим критерием: если вероятность обнаружить м.м. в той же точке поверхности образца, в которой уже был произведен анализ, является малой, то м.м. распределены по поверхности, в противном случае - по объему.
При плотностях потока ниже пороговой анализ в обоих случаях является плохо воспроизводимым и имеет низкую точность. Таким образом требования к плотности потока мощности j могут быть представлены в единой форме
j > j0 • b, (3)
где j0 - пороговая плотность для каждого из описанных случаев.
При дальнейшем увеличении плотности потока фрагментация может быть усилена до любого требуемого уровня, вплоть до отдельных атомов.
Практически важным является вариант квазинепрерывной подачи образца к месту анализа, например, из хроматографа или электрофоретической колонки. При такой подаче скорость поступления образца должна уравновешиваться скоростью его испарения. При невыполнении этого условия образец будет накапливаться в системе ввода, делая анализ нестабильным. Особенно это актуально для водных растворов, обладающих свойством испарительного замораживания при вводе в вакуум. Не будучи предотвращено притоком энергии извне, это способно сделать продолжение анализа невозможным.
Таким образом, тепловая мощность испаряющего воздействия (например, буферного газа, нагревателя, излучателя и т.д.) должна быть достаточна для испарения всего вводимого в источник ионов образца
P>c•ρ•v•q (Вт) (4),
где ρ - плотность образца в потоке, кг/м3;
v - средний поток образца, подлежащий анализу, м3/с;
q - удельная теплота нагрева и испарения образца, Дж/кг;
105 > c > 1 - коэффициент запаса мощности.
Для детектирования образованных при испарении и ионизации м.м. фрагментов предлагается использовать ВЭУ на основе шевронной сборки микроканальных пластин (МКП) [6]. Поскольку количество детектируемых фрагментов ожидается в диапазоне от единиц до сотен и тысяч, то реальный режим работы ВЭУ оказывается промежуточным между счетом ионов и аналоговым. Для перехода к более точному и помехоустойчивому режиму счета и ионов предлагается разделить коллектор ВЭУ на множество независимых секций, каждая из которых работает уже заведомо в режиме счета ионов. То есть количество секций на единицу площади должно превышать количество ионов, одновременно (в течение одного шага дискредитации счетчика ионов) приходящих на эту площадь. Вследствие эффекта "мертвого времени" для каналов МКП [6], количество секций должно быть выбрано с запасом в несколько порядков.
Таким образом, площадь секции должна быть не более f/J
d < s < f/J, (5а)
где J - максимальное количество ионов на МКП за длительность одного шага дискретизации счетчика ионов, (ионов/м2);
f - коэффициент запаса по насыщению ВЭУ. (10-6 - 1);
d - расстояние между центрами каналов МКП (м)
С другой стороны, не может быть меньше площади одного канала МКП.
s > d2 (5b),
где d - расстояние между центрами каналов МКП, м.]
При нарушении хотя бы одного из этих условий ВЭУ выходит из режима счета ионов и точность анализа при вышеуказанном частном использовании способа резко падает.
Дальнейшим усовершенствованием способа является разделение в пространстве и/или времени стадий фрагментации и испарения/ионизации; это может быть достигнуто за счет фрагментации м.м. непосредственно в образце с помощью содержащихся в матрице рестриктаз. Действие этих рестриктаз, в качестве которых могут выступать различные неорганические (кислоты, щелочи, соли) и органические (ферменты и т.д.) молекулы, может активизироваться как с помощью внешних воздействий (нагрев, облучение), так и непосредственно при смешивании.
При этом основным условием их применения остается условие однозначной идентификации м.м. по ее фрагментам.
Таким образом, фрагменты от различных м.м. не должны смешиваться в течение всего времени анализа и не должны интерферировать друг с другом. Это условие выполняется при концентрации м.м. C вида:
C = a/max (V, Vd),(6)
где Vd - максимальный объем, занимаемый фрагментами м.м. непосредственно перед испарением, м3;
Vd - определяется диффузией, перемешиванием, вязкостью и др.
Если Vd > V, то для анализа одной м.м. в среднем требуется более одного цикла анализа, то есть каждой м.м. соответствует последовательность циклов анализа, а сами последовательности разделены сериями "пустых" спектров.
Отдельным частным случаем этого режима является решение, при котором области фрагментации и испарения/ионизации разделены в пространстве настолько, что высота объема Vd (то есть размер в направлении нормали к поверхности образца) много больше, чем высота объема V (тем не менее при этом может оказаться V > Vd).
В этом случае фрагменты от различных м.м. должны однозначно различаться либо за счет выбора достаточно малой площади испаряющего и/или ионизирующего воздействия, либо за счет обеспечения их детектирования как разделенных в пространстве. Таким образом, условие (6) должно быть усилено условием:
R = g•min{1/Sв,1/(πr2)} (м.м./м2) (7),
где R - поверхностная плотность распределения м.м., фрагменты которых могут одновременно анализироваться на поверхности образца (то есть поверхностная плотность скоплений фрагментов, причем каждое из скоплений порождается единственной м.м.),
r - пространственное разрешение всей ионно-оптической системы и детектора в плоскости образца, м;
g < 1 - коэффициент запаса по пространственному разрешению.
При нарушении условия (7) анализ данным способом в вышеописанном частном случае становится невозможным из-за высокой вероятности детектирования фрагментов одновременно от двух и более м.м.
Описанный частный случай может быть реализован, например, путем смешивания объемного раствора м.м. с рестриктазами, активизируемыми за счет нагрева потоком тепла, отводимым внутрь образца и поступающего от испаряющего воздействия.
М. м. могут быть также иммобилизированы с помощью химических или физических связей на поверхности подложки или мембраны (1) и затем подвержены действию рестриктаз (в том числе возможно последовательное разрезание м.м. по длине).
В обоих случаях полученные фрагменты могут быть доставлены к месту испарения/ионизации с помощью, например, электрофореза во внешнем поле или в потоке матрицы.
Все описанные частные случаи анализа могут быть организованы параллельно для многих точек образца, при этом детектирование удобно производить описанным выше многоканальным ВЭУ в режиме ионов.
Такой параллельный анализ возможен только в том случае, если фрагментарные зоны различных м.м. детектируются различными областями ВЭУ, то есть расстояние между любыми двумя точками образца не менее введенного выше пространственного разрешения r всей ионно-оптической системы и детектора.
Необходимость такого пространственного разделения налагает предел на количество точек К, одновременно анализируемых с помощью детектора заданной площади F. Если минимальная площадь изображения от одной точки составляет S
K<h•F/S
10-2 < h < 1 - коэффициент запаса по площади МКП.
Изобретение поясняется чертежом, где на фиг. 1 - 3 приведены основной и частные случаи реализации предложенного способа в виде примеров реализации.
На фиг. 1 изображено устройство реализации способа, когда раствор м.м. в воде с концентрацией 10-15 моль/л в виде капель по 0,5 - 1 мклитр наносят на подложку 1 из инертного металла и высушивают. Затем подложка 1 вводится в вакуумную камеру 2 через порт 3 с помощью манипулятора 4, после чего вакуумная камера 2 откачивается до вакуума 10-6 Topp откачной системой 5. Импульсная десорбция и ионизация фрагментов единичной м.м. производятся азотным лазером 6, который облучает подложку 1 импульсами УФ-излучения с длительностью 3 HC, длиной волны 355 нм и с энергией 1 - 10 мкДж в импульсе. Фокусирующая линза 7 обеспечивает фокусировку излучения в пятно 2 х 5 мкм2 на подложке 1.
Образованные ионы фрагментов 8 ускоряются в ускорительном промежутке 9 и проецируются дефокусирующей ионно-оптической линзой 10 на всю площадь ВЭУ, состоящего из шевронной сборки МКП 11 и многосекционного коллектора 12, содержащего около 10 000 секций. Все секции соединены с каналами многоканального счетчика 13 ионов, обеспечивающего подсчет и запоминание числа детектированных ионов и соответствующие им времена пролета. М м.м. определяют по зависимости (1) с учетом калибровочных данных.
Согласно фиг. 2 раствор м.м. в воде с концентрацией 10 -13 моль/л подается из шприца 14 перистальтическим насосом 15 по капилляру 16 со скоростью 0,1 мклитр/мин (при диаметре капилляра 26 мкм это соответствует скорости потока 4 мм/с и скорости анализа 100 м.м./с). Капилляр 16 подсоединен к вакуумной камере 2 через нагреватель 17, соединенный в свою очередь с камерой 2 через теплоизолятор 18. Нагреватель 17 обеспечивает нагрев малых объектов раствора до температур в несколько сотен oC, что приводит к пиролизу м.м. на малые фрагменты 8 и их выносу в вакуумную камеру 2 в струе водяного пара. Фрагменты м. м. ионизируются в источнике ионов 20 электронным ударом. Полученные ионы анализируются в квадрупольном фильтре масс 21, который настроен на некоторый характерный для исследуемых м.м. фрагментарный ион (например, на ионы P+ или PO+ и т.д. для ДНК). М м.м. определяется с помощью калибровочных данных по количеству ионов, зарегистрированных с помощью динодного ВЭУ 22 и одноканального счетчика ионов 23. При этом в формуле (1) полагают N = 1.
Согласно фиг. 3 м.м. ДНК иммобилизируются на диэлектрической, например, лавсановой, мембране 24 с металлизированными поверхностями 25 внутри ее каналов 26, расположенных с частотой ≈ 106 каналов/см2. Мембрана натягивается на поддерживающий электрод 27, предохраняющий ее от продавливания в вакуумную систему 2, а обращенный от вакуума торец приводится в соприкосновение с буферным раствором 28, подаваемым из шприца 14, перистальтическим насосом 15. Буферный раствор 28 содержит рестриктазы, осуществляющие последовательное отщепление оснований от ДНК, причем образующиеся ионы фрагментов дрейфуют в электрическом поле, создаваемом приложенной к поверхностям 25 разностью потенциалов, в сторону вакуума, а имеющие противоположный знак ионы рестриктаз удерживаются вблизи м.м.
Поверхность буфера 28, обращенная к вакууму, находится в замороженном состоянии из-за эффекта испарительного замораживания при контакте с вакуумом. Импульсное расплавление поверхности буфера одновременно для многих каналов осуществляется импульсами инфракрасного излучения от Nd:YAG лазера 29, сфокусированными линзой 7 в пятно 100 х 100 мкм2 и длительностью 10 нс.
Образующиеся при выдавливании раствора капли 19 порождают ионы фрагментов 8 в процессе электрораспыления в промежутке 9. Эти ионы ускоряются в промежутке 9 после импульсной подачи напряжения и анализируются по времени пролета, при этом ионно-оптическая система 30 и сканирующая система 31 обеспечивают отображение ионов с увеличением 100 на площадь 10 х 10 мм2 входного окна ВЭУ, содержащего шевронную сборку МКП 11 и многосекционный коллектор 12, соединенный с многоканальным счетчиком 13. М м.м. определяют из условия (1) параллельно для 100 каналов мембраны 24 после окончания процесса поступления на счетчик 13 сигналов от ионов фрагментов.
Данный способ обеспечивает высокую точность при анализе м.м. в области аналитической биохимии, в особенности при исследовании ДНК в различных экспериментальных установках.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОПРЕДЕЛЕНИЯ МЕТАЛЛОВ И КОМПЛЕКСНЫХ СОЕДИНЕНИЙ МЕТАЛЛОВ | 2013 |
|
RU2531762C1 |
| СПОСОБ ТРАНСПОРТА ИОНОВ ИЗ ПОЛЯРНОЙ ЖИДКОСТИ В ВАКУУМ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2013 |
|
RU2537961C2 |
| СПОСОБ МАСС-СПЕКТРОМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ ХИМИЧЕСКИХ СОЕДИНЕНИЙ | 2015 |
|
RU2599330C1 |
| МАСС-СПЕКТРОМЕТР ДЛЯ МАКРОМОЛЕКУЛ С КРИОГЕННЫМ ДЕТЕКТОРОМ ЧАСТИЦ | 1995 |
|
RU2189086C2 |
| СПОСОБ ДЕСОРБЦИИ-ИОНИЗАЦИИ ХИМИЧЕСКИХ СОЕДИНЕНИЙ | 2005 |
|
RU2285253C1 |
| УСТРОЙСТВО ДЛЯ АНАЛИЗА ГАЗОВОЙ СМЕСИ | 2004 |
|
RU2272334C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ИЗОТОПНОГО СОСТАВА МЕТАНА | 2010 |
|
RU2461909C2 |
| СПОСОБ ИДЕНТИФИКАЦИИ ИЗОМЕРИЗОВАННОГО АСПАРТАТА В БЕТА-АМИЛОИДЕ | 2009 |
|
RU2431143C2 |
| СПОСОБ АНАЛИЗА ПРИМЕСЕЙ В ЖИДКОСТЯХ ИЛИ ГАЗАХ ПРИ ИХ МИКРОКАНАЛЬНОМ ИСТЕЧЕНИИ В ВАКУУМ ПОД ВОЗДЕЙСТВИЕМ СВЕРХЗВУКОВОГО ГАЗОВОГО ПОТОКА, СОДЕРЖАЩЕГО ИОНЫ И МЕТАСТАБИЛЬНО ВОЗБУЖДЁННЫЕ АТОМЫ, С ФОРМИРОВАНИЕМ И ТРАНСПОРТИРОВКОЙ АНАЛИЗИРУЕМЫХ ИОНОВ В РАДИОЧАСТОТНОЙ ЛИНЕЙНОЙ ЛОВУШКЕ, СОПРЯЖЁННОЙ С МАСС-АНАЛИЗАТОРОМ | 2016 |
|
RU2640393C2 |
| СПОСОБ БЕССТАНДАРТНОЙ ОЦЕНКИ КОЛИЧЕСТВА ФОСФОРОРГАНИЧЕСКОГО ВЕЩЕСТВА В ПРОБЕ | 2015 |
|
RU2610558C1 |


Использование: аналитические исследования с помощью средств электроники: ионного анализа и детектирования вторично-электронным умножителем макромолекул (м. м. ) биополимеров, в особенности ДНК. Сущность изобретения: при определении молекулярной массы м.м. за один цикл анализа производят испарение и ионизацию не более одной м.м. в виде ее фрагментов, при этом плотность потока мощности воздействия устанавливают из условия фрагментации м.м. , а определение молекулярной массы определяют из заданного аналитического выражения. В частных решениях устанавливают и поддерживают следующие параметры процесса: концентрацию м.м., плотность потока мощности воздействия, поверхностную плотность распределения м.м. и параметры оборудования для различных режимов исследования. 7 з.п.ф-лы, 3 ил.
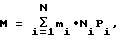 кг
кг
где mi - молекулярная масса фрагмента i-сорта, кг;
Ni - количество зарегистрированных фрагментов i-го сорта;
Pi > 1 - калибровочный коэффициент для фрагмента i-го сорта;
N - общее число сортов регистрируемых фрагментов.
C = a/V, макромолекул/м3,
где a < 0,1 - 0,3 - коэффициент разбавления;
V - испаряемый за один цикл анализа объем образца, м3.
j = j0 • b, Вт/м2,
где j0 - пороговая плотность потока мощности, Вт/м2;
b = 1 - 105 - коэффициент превышения порога.
P = c•ρ•V•q, Вт,
где ρ - плотность раствора, кг/м3;
V - средний расход раствора, м3/c;
q - удельная теплота нагрева и испарения раствора, Дж/кг;
105 > c > 1 - коэффициент запаса по мощности.
d2 < s < f/j, м2,
где d - расстояние между центрами каналов микроканальной пластины, м;
j - максимальное количество ионов, приходящих на микроканальную пластину за промежуток времени, равный временному шагу дискретизации счетчика, ион/м2,
f = 10-6 - 1,0 - коэффициент запаса по насыщению каналов,
причем обеспечивают обслуживание каждой секции отдельным каналом многоканального счетчика ионов.
С = а/mах (V, Vd), макромолекул/м3,
где Vd - максимальный объем, занимаемый фрагментами макромолекулы непосредственно перед испарением, м3.
R = q•min(1/sв, 1/πr2), макромолекул/м2,
где sв - площадь воздействия, м2;
r - пространственное разрешение всей ионно-оптической системы и детектора в плоскости образца, м;
q < 1 - коэффициент запаса по пространственному разрешению.
K = h•F/s
где s
10-2 < h < 1 - коэффициент запаса по площади микроканальной пластины,
а расстояние между любыми двумя указанными точками составляет не менее r.
