Данное изобретение в общем относится к гетероциклическим углеродным соединениям, имеющим лекарственные и био-воздействующие свойства, и к их получению и использованию. В частности, изобретение касается 1,4-дизамещенных пиперазиновых производных, в которых один заместитель представляет 1,2,5-тиадиазол; 1,2,5-тиадиазол-1-оксид, или 1,2,5-тиадиазол-1,1- диоксид-замещенную индол-3-илалкильную группу, а другой фрагмент представляет пиридинильное или пиримидинильное кольцо. Эти соединения обладают уникальным серотонергическим профилем, а также метаболической стабильностью, что делает их полезными при лечении сосудистой головной боли, такой как мигрень или головная боль кластерного типа.
Авторы Dowie и др. описали в опубликованной патентной заявке GB 2124210 ряд производных 3-алкиламино-индола в качестве потенциально полезных при лечении мигрени. Один представитель данного ряда соединений был конкретно заявлен в более поздней патентной заявке Оксфорда GB 2162522, опубликованной 5 февраля 1986 года. Данное конкретное соединение известно в литературе как суматриптан (I).
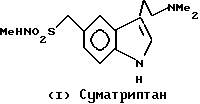
Ряд новых производных индолина был описан авторами Маноури и др., в европейской патентной заявке EPA 354094. Эти соединения описываются как полезные для лечения различных расстройств ЦНС, включающих депрессию, беспокойство или страх и мигрень. В число этих известных соединений входят соединения формулы (II)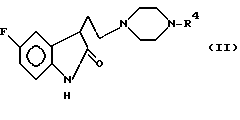
в которой R4 представляет арильный, пиридиновый или хинолиновый фрагмент.
Авторы Смит и др., в патенте США 954502 описали ряд 1,4-дизамещенных пиперазиновых производных формулы (III), которые полезны как антидепрессантные агенты.
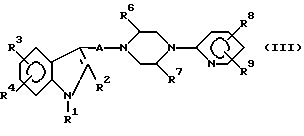
Индолильные заместители R3 и R4 представляли водород, алкил, алкоксиалкилтио, галоген, карбоксамид и трифторметил.
Еще один ряд антидепрессантных 1,4-дизамещенных пиперазинов, в которых вместо пиридина использовались пиримидиновые фрагменты, раскрыты авторами Смит и др. в патенте США 5077293.
Более близким источником известного уровня техники является патент США 5300506, касающийся раннего описания антимигреневых алкоксипиримидиновых производных формулы (IV)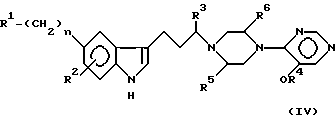
в которой заместители 5-индола (R1) включают амино, алкокси, амидо, алкилсульфониламино и группу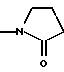
Считается, что наиболее близким источником известного уровня техники является более ранняя заявка на патент настоящих заявителей - заявка США 08/122266, в которой раскрываются антимигреневые соединения формулы (V):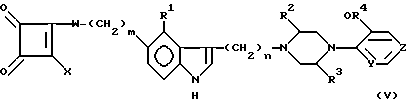
Ни одно из указанных известных соединений не предлагало рассматриваемых новых 5-тиадиазольных (и его оксидных)-замещенных-индол-3-илалкильных производных пиридинил- или пиримидинил-пиперазино для лечения мигрени и головных болей кластерного типа.
Мигрень является одной из широкого класса головных болей, который включает также головные боли кластерного типа, и других головных болей, которые, как считается, имеют причастность сосудов в своей этиологии, т.е. в этиологии которых причастны сосуды. Эти головные боли часто классифицируются как сосудистые головные боли. В качестве обзорной информации о головных болях и их лечении см., например, главу 13; "Drugs Used to Treat Migraine and Other Headaches" в "Drug Evaluations, 6th Edn., 1986, стр. 239 - 253, Американской медицинской ассоциации, W.B.Saunders Co., Филадельфия, PA.
Частые имеющие место нерегулярно случаи головной боли поражают большое количество людей и обычно являются острыми по своему характеру и непродолжительными. Облегчение от головной боли данного типа обычно достигается мягкими анальгетиками; такими как аспирин или ацетаминофен. Такие головные боли являются совершенно обычным явлением, и хотя они являются весьма болезненными и возможно досаждающими, они редко делают человека нетрудоспособным и истощают его здоровье. Хронически повторяющиеся головные боли сосудистой категории, однако, часто заставляют пациента обращаться за консультацией к врачу вследствие остроты боли, которая часто приводит к нетрудоспособности.
Хотя нет никакой универсально принятой системы классификации для головной боли, сосудистая головная боль для целей настоящего изобретения относится главным образом к мигрени и кластерным головным болям. Мигрень является обычным или классическим типом, так же как и варианты мигрени, которые знакомы специалистам в данной области. К категории связанных с сосудами головных болей и подвергаемых лечению с помощью настоящего изобретения могут быть также отнесены другие подтипы, такие как токсические сосудистые и гипертензивные головные боли, хроническая пароксизмальная гемикрания, так же как и некоторые головные боли, связанные с сокращением сердечной мышцы, и объединенные или смешанные сосудисто-мышечные головные боли. Специалистам в данной области понятно, что никакое единое лечение не является эффективным для всех пациентов, которым поставлен диагноз головной боли одного и того же подтипа, и в связи с этим возникают дальнейшие неопределенности с классификацией головных болей.
Лекарства, которые, как сложилось исторически, наиболее обычно использовались для лечения головной боли, относятся к следующей группе:
эргот алкалоиды,
бета-бдокирующие агенты,
агенты, блокирующие кальциевые каналы,
антидепрессанты и
их смеси.
Оказание помощи при повторяющейся сосудистой головной боли осложняется отсутствием единого метода лечения, который эффективен для всех пациентов с одним и тем же типом головной боли. Ставший доступным с недавнего времени антимигреневый препарат (агент) Суматриптан, достиг некоторого успеха в лечении мигрени у пациентов, но все еще обладает недостатками. Дальнейшее осложнение влечет использование против мигрени лекарств, которые могут вызывать зависимость при продолжительном использовании, таких как эрготамин. Еще одним важным фактором для настоящего изобретения является то, что более сильные антимигреневые агенты, используемые в настоящее время, например эрготы и метисергид, дают при длительном использовании сильные ограничивающие их применение побочные эффекты.
Таким образом, испытывается потребность в безопасном и эффективном лекарстве для лечения мигрени и связанных с ней нарушений, которое может использоваться или для устранения угрозы головной боли, или для облегчения уже наступившей боли.
Объекты настоящего изобретения относятся к использованию новых 5-(3,4-диамино-1,2,5-тиадиазол-1-оксид)-замещенных индол-3-илалкильных производных пиридинил- и пиримидинил-пиперазинов для лечения сосудистых головных болей, особенно мигрени и болей кластерного типа; к процессам их получения и к фармацевтическим композициям на их основе и медицинскому использованию.
Способ настоящего изобретения предназначен для облегчения сосудистых или связанных с сосудами головных болей, из которых наиболее хорошо известными конкретными примерами являются мигрень и кластер. Способ включает по существу назначение для приема человеку, нуждающемуся в таком лечении, 5-тиадиазол (или S-оксид)-замещенных индол-3-илалкильных производных пиридинил- или пиримидинил-пиперазина или их фармацевтически приемлемых солей и/или сольватов. Для использования в данном способе предпочитаются оральный и трансназальный методы назначения фармацевтических композиций, содержащих являющиеся предметом обсуждения антимигреневые агенты.
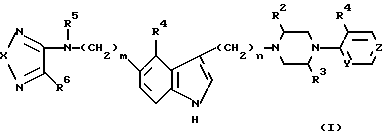
В широком аспекте настоящее изобретение касается индол-3-илалкильных производных пиридинил или пиримидинил-пиперазинов, обладающих полезными противомигреневыми серотонэргическими свойствами и характеризующихся формулой I
В формуле I R1 представляет заместитель, выбранный из водорода, галогена, низшего алкила и низшего алкокси.
R2, R3 и R5 независимо выбраны из водорода и низшего алкила.
В предпочтительных соединениях R2 и R3 не являются одновременно низшим алкилом.
R4 представляет низший алкокси.
R6 выбран из амино, низшего алкиламино, ди-низшего алкиламино и низшего алкокси.
Символ "m" может быть представлен целым числом от 1 до 3 или нулем, в то время как "n" может быть целым числом от 1 до 5. В предпочтительных соединениях m представляет нуль, и n=3.
X выбран из S, SO и SO2. Предпочтительными соединениями являются те, в которых X представляет SO.
Y и Z независимо выбраны из N и CN, при условии, что Y и Z не могут быть одновременно CH.
В дополнение к сказанному соединения формулы I также охватывают все фармацевтически приемлемые кислотно-аддитивные соли и/или сольваты. Настоящее изобретение включает также стереоизомеры, а также оптические изомеры, например, смеси энантиомеров, также как и индивидуальные энантиомеры и диастереомеры, которые возникают как следствие структурной асимметрии в некоторых соединениях данного ряда. Разделение на индивидуальные изомеры достигается с помощью применения различных методов, которые хорошо известны специалистам в данной области.
Термин "низший алкил" относится к углеродным радикалам, как с прямой, так и разветвленной цепью, содержащим от 1 до 4 атомов углерода включительно. Примерами данных радикалов являются углеродные цепи, которыми могут быть метил, этил, пропил, изопропил, 1-бутил, 1-метилпропил, 2-метилпропил.
Низший алкокси относится к (1-4)C алкильным группам, связанным с атомом кислорода. Тиадиазольный фрагмент охватывает также сульфоксидные (SO) и сульфоновые (SO2) производные.
С помощью соответствующего выбора символом Y и Z обозначается или пиридиновое, или пиримидиновое кольцо.
Фармацевтически приемлемыми кислотно-аддитивными солями согласно изобретению являются соли, в которых противо-ион не способствует в значительной степени токсичности или не нарушает фармакологическую активность соли, и как таковые, они являются фармакологическими эквивалентами основной формулы I. Для медицинского применения они обычно предпочитаются. В некоторых случаях они обладают физическими свойствами, которые делают их более желательными для приготовления фармацевтических готовых форм, такими как растворимость, отсутствие гигроскопичности, способность к сжатию, что касается образования таблеток, и совместимость с другими ингредиентами, с которыми вещество может использоваться для фармацевтических целей. Соли обычно получаются путем смешения основной формулы I с выбранной кислотой, предпочтительно, с помощью контактирования в растворе с применением избытка обычно используемых инертных растворителей, таких как вода, эфир, бензол, метанол, этанол, этилацетат и ацетонитрил. Они могут также изготавливаться с помощью метатезиса или реакции обмена или обработки ионообменной смолой в условиях, в которых анион одной соли соединения формулы I заменяется другим анионом, что позволяет отделить желаемые виды в таких условиях, как осаждение из раствора или экстракция в растворитель, или элюирование с ионообменной смолой или удерживание на ионообменной смоле. Фармацевтически приемлемые кислоты для целей образования солей соединений формулы I включают серную, фосфорную, соляную, бромистоводородную, иодистоводородную, лимонную, уксусную, бензойную, коричную, фумаровую, миндальную, азотную, слизевую, малеиновую, изетионовую, пальмитиновую, гептановую и другое.
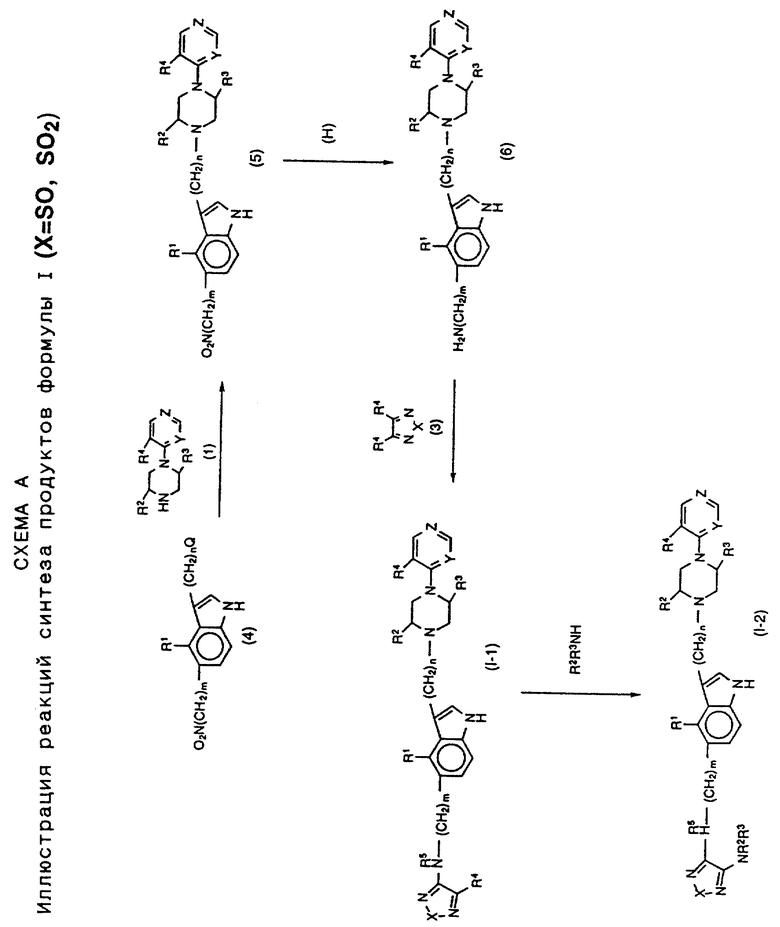
Соединения формулы I могут получаться с применением процессов синтеза и соединений, показанных на Схемах A и B.
Некоторые соединения и способы их синтеза будут представлены также более подробно в разделе конкретных воплощений, представленном ниже. На схемах синтеза символы от R1 до R6, X, Y, Z, m и n имеют значения, определенные выше. Символ Q представляет синтетический органический уходящий или удаляемый фрагмент или группу, такую как тозил, мезил, галогенид, сульфат, фосфат и т. д.
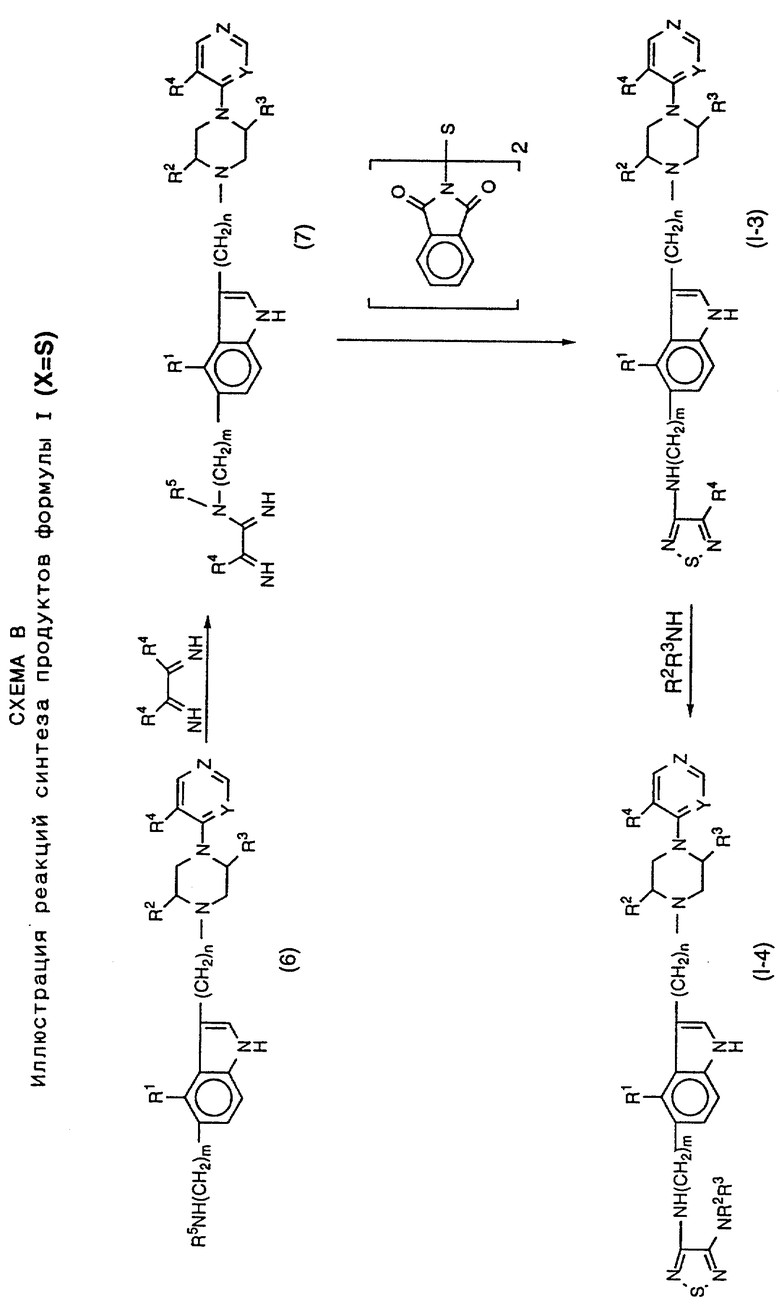
На схеме A представлены процессы синтеза соединений формулы I, в которой X представляет SO или SO2. Схема B иллюстрирует получение соединений, в которых X представляет S. Процессы протекают через 5-амино-замещенные промежуточные соединения формулы (6), в которой гетероциклический-замещенный, пиперазиновый фрагмент уже включен в молекулярную структуру. Модификация пути синтеза может производиться для определения не только желаемого R6-заместителя в тиадиазольном фрагменте (например, R4 или NR2R3, как показано), но также и желаемой идентичности R5 заместителя, или H или алкила. Для синтеза продукта, в котором R5 представляет алкил, промежуточное соединение (6) моно-N-алкилируется перед дальнейшей реакцией, показанной на схемах A и B.
Процессы синтеза, проиллюстрированные на схеме A, по существу включают построение индолилалкилпиперазинилгетероциклического фрагмента с концевой амино группой, присоединенной в 5-положении индольного кольца, например соединения (6). Данное промежуточное соединение подвергается реакции или с 1-оксидной или 1,1-диоксидной формой 3,4-диалкокситиадиазола (3), давая продукт формулы I, в которой R6 представляет алкокси (R4). Данный продукт после вытеснения алкоксидного фрагмента тиадиазола амином дает продукт формулы I, в которой R6 представляет амино группу, например, соединение (1-2). Как указано выше, промежуточное соединение (6) может N-алкилироваться в 5-индолил-амино конце, давая промежуточные соединения и продукты, в которых R5 представляет алкильную группу.
Процессы схемы B включают синтез продуктов, имеющих тиадиазольное кольцо (X представляет только S). Данный процесс, описанный ранее в патентах США 4440933 и 4600778 для ряда антагонистов гистамин-H2-рецепторов, применим в данном случае. Эти патенты США приведены здесь для сведения. По существу схема В иллюстрирует реакцию промежуточного соединения (6) с 1,2-диалкокси-замещенным этандиимидом с получением промежуточного соединения формулы (7). Реакция соединения (7) с N,N'-тиобисфталимидом дает в результате тиадиазольный аналог продукт (1-3). Как и на схеме A, тиадиазольный алкокси заместитель (R6=R4) соединения (1-3) может вытесняться амином, давая продукт формулы (1-4), в котором R6 представляет амино функцию.
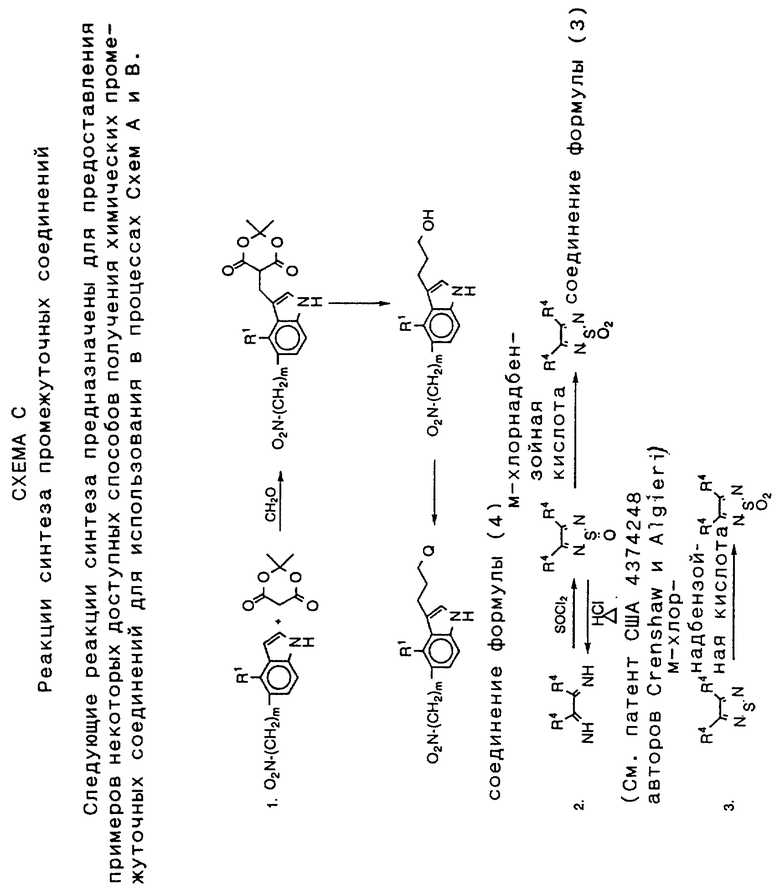
На схеме C даны пути синтеза промежуточных соединений для использования в процессах схем A и B; на ней представлены, например, некоторые типичные методы синтеза, которые дают исходные промежуточные соединения для указанных схем.
Реакции, применяемые на схемах A, B и C, и их применение знакомы специалистам в области органического синтеза, и им легко будут понятны модификации условий и реагентов. Квалифицированный химик синтетик знает, как приспособить данные процессы для получения конкретных соединений формулы I, включая другие соединения, охватываемые данным изобретением, но конкретно не раскрытые в заявке. Для специалиста в данной области также очевидны вариации методов получения тех же самых соединений несколько отличным образом. Для предоставления больших подробностей в описании даются ниже характерные примеры синтеза в разделе конкретных воплощений.
Серотонин связали с патофизиологией мигрени путем накопления свидетельств, включающих повышенную экскрецию метаболитов серотонина после приступа мигрени и снижение содержания серотонина в тромбоцитах крови во время мигрени. Этот последний эффект, по-видимому, является специфичным для мигрени и не является результатом боли или стресса (Anthony и др., "Plasma serotonin in migraine and stress". Arch. Neurol. 1967, 16:544-552). Более важно, внутримышечная инъекция резерпина снижает серотонин в плазме и вызывает головную боль типа обычной мигрени у людей, страдающих мигренью. Данную индуцированную головную боль можно облегчить медленной внутривенной инъекцией серотонинкреатининсульфата (Kimball и др., "Effect of serotonin in migraine patients". Neurology N.Y., 1960, 10:107-111).
Хотя показано, что серотонин является эффективным при лечении приступов мигрени, его использование в случае мигрени устраняется его побочными действиями, такими как беспокойство, тошнота, потеря сознания, гиперстения, прилив крови к лицу и парастезия (Lance и др., "The control of cranial arteries by humoral mechanisms and its relation to the migraine syndrome". Headache. 1967, 7:93-102). По этой причине более специфичные серотониновые агенты, которые лечили бы мигрень, не оказывая всех других действий, являются потенциально полезными антимигреневыми медикаментами. Накапливающиеся находки привели к осознанию того, что соединения с селективностью к 5-HT1D подтипу серотониновых рецепторов были бы клинически эффективными в лечении мигрени. В данном отношении соединения настоящего изобретения демонстрируют сильное сродство к 5-HT1D сайту. Соединения формулы I, представляющие интерес, имеют активность, характеризующуюся тем, что IC50 значения этих соединений составляет менее чем 100-нмолярные. Предпочтительные соединения имеют значения IC50 ниже 10-нмолярной.
Определение свойств связывания с 5-HT1D выполнялось с применением такой методики, как описана авторами Heuring и Peroutka, J. Neurosci. 7(3), 1987, 894-903; лишь с незначительными видоизменениями. Значения IC50 (нМ) в опыте ин витро определялись для соединений данного изобретения с применением тритированного серотонина.
В дополнение к данным испытания на связывание с 5-HT1D, способность соединений данного изобретения вызывать сокращение на модели подкожной вены собак дополнительно свидетельствует о полезности их в лечении сосудистых головных болей. Предпочтительные соединения демонстрируют активность, равную или превышающую таковую самого серотонина. Кроме того, данные соединения показали гораздо большую стабильность ин витро после инкубирования в гомогенате печени крыс, чем известный ряд индолил-скваратных соединений, описанных в заявке США 08/122266. Все эти описанные выше фармакологические испытания указывают на полезное антимигреневое действие соединений данного изобретения. Далее, согласно еще одному аспекту данное изобретение представляет способ лечения страдающих мигренью, который предусматривает системное назначение пациентам терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Предполагается, что соединения формулы I могут назначаться для снятия приступа мигрени на ее ранних стадиях, а также назначаются для лечения уже установившейся сосудистой головной боли.
Можно считать, что назначение и режим дозировки соединений формулы I могут быть в основном такими же, как в случае известного соединения суматриптана, см. Оксфорд, GB 2162522 A. Хотя данные соединения могут назначаться интра-назально или орально, дозы и режим дозировки должны в каждом случае тщательно подбираться с использованием профессионального опыта и с учетом возраста, веса и состояния реципиента, способа назначения и характера и тяжести болезни. Обычно дневная доза составляет примерно от 0,05 до 10 мг/кг, предпочтительно 0,1-2 мг/кг, когда средство назначается парентерально, и примерно от 1 до 50 мг/кг, предпочтительно около 5 - 20 мг/кг, при оральном назначении. В некоторых случаях достаточный терапевтический эффект может достигаться при более низких дозах, хотя в других могут требоваться более высокие дозы. Системное назначение относится к оральному, интра-назальному, ректальному и парентеральному способам (т.е. внутримышечному, внутривенному и подкожному). Найдено, что, когда соединение настоящего изобретения назначается орально, обычно требуется большее количество активного агента для получения того же эффекта, как и в случае меньшего количества, даваемого интра-назально или парентерально. Согласно квалифицированной клинической практике предпочитается назначать данные соединения с уровнем концентрации, который будет давать эффективное противомигреневое действие, не вызывая при этом каких-либо вредных или нежелательных побочных действий.
Соединения настоящего изобретения могут назначаться для антимигреневых целей или в виде индивидуальных терапевтических агентов, или в виде смесей с другими терапевтическими агентами. В терапии они обычно даются в виде фармацевтических композиций, включающих противомигреневое количество соединения формулы I или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель. Фармацевтические композиции, которые обеспечивают примерно от 1 до 500 мг активного ингредиента на дозированную единицу, являются предпочтительными, и обычно они приготавливаются в виде таблеток, лепешек, капсул, порошков, водных или масляных суспензий, сиропов, эликсиров и водных растворов.
Характер применяемых фармацевтических композиций конечно зависит от желаемого способа назначения. Например, оральные композиции могут быть в форме таблеток или капсул и могут содержать обычные эксципиенты, такие как связующие агенты (например, крахмал) и смачивающие агенты (например, лаурилсульфат натрия). Растворы или суспензии соединений формулы I с общепринятыми фармацевтическими носителями применяются для интра-назальных и парентеральных композиций, таких как водные растворы для внутривенной инъекции или масляная суспензия для внутримышечной инъекции.
Соединения, являющиеся предметом данного изобретения, их способы получения и их биологические действия будут более полными с учетом следующих примеров, которые даются лишь для целей иллюстрации, и не должны рассматриваться как ограничивающие изобретение в его сфере или объеме. В следующих примерах, использованных для иллюстрации описанных выше процессов синтеза, температуры выражены в градусах Цельсия, а точки плавления являются нескорректированными. Спектральные характеристики ядерного магнитного резонанса (ЯМР) относятся к химическим сдвигам (δ), выраженным в миллионных долях (м. д. ) против тетраметилсилана (ТМС) как ссылочного стандарта. Относительная область, приведенная для различных сдвигов в 1H ЯМР спектральных данных, соответствует числу атомов водорода конкретного типа группы в молекуле. Характер сдвигов, что касается множественности, приводится как широкий синглет (шир.с.), синглет (с.), мультиплет (м.), гептет (гепт.), квартет (кв.), триплет (т. ) или дублет (д. ). Применяемые сокращения обозначают ДМСО-d6 (дейтеродиметилсульфоксид), CDCl3 (дейтерохлороформ) и являются в остальных отношениях и случаях обычными. Инфракрасные (ИК) спектральные описания включают величины абсорбционной волны (см-1), имеющей идентификационное значение функциональной группы. ИК определения применялись или чистыми, или с использованием бромида калия (KBr) в качестве разбавителя. Элементные анализы приводятся в процентах по весу.
Следующие примеры описывают подробно получение соединений формулы I, а также синтез промежуточных соединений в каждом процессе. Специалистам в данной области совершенно понятно, что видоизменения как материалов, так и методики, позволят получить другое соединения, описанные в заявке. Исходя из предыдущего описания и следующих ниже примеров специалисты в данной области смогут использовать изобретение в наиболее полной степени. Кроме того, описаны примеры синтеза некоторых промежуточных соединений и 1,2,5-тиадиазольных соединений, имеющих амино заместители в 3- и 4-положениях. См. например, Crenshaw и Algieri, США 4374248 и 4600779; Montzka, патент США 4440933. Эти патенты США приведены здесь для сведения, чтобы дать возможность получить некоторые из данных промежуточных продуктов и продуктов.
A. Получение промежуточных соединений
Ниже приводятся некоторые характерные процедуры получения синтетических промежуточных соединений, участвующих в процессах, показанных на схемах. Большинство исходных веществ и некоторые промежуточные продукты являются или промышленно доступными, или процедуры их синтеза свободно доступны из химической литературы, что позволяет специалистам в области химии органического синтеза полностью их использовать.
Пример 1
5-[(5-Нитро-1H-индол-3-ил)метил]-2,2-диметил-1,3-диоксан-4,6-дион
Использовалась процедура, описанная Flaugh1. Так, раствор 5-нитроиндола (50,0 г, 0,32 моля), кислоты Meldrum's (46,0 г, 0,32 моля), 37% водного формальдегида (26,0 мл, 0,32 моля) и пролина (1,8 г, 0,016 моля) в 200 мл ацетонитрила перемешивался при комнатной температуре в течение 18 часов. Получающаяся густая желтая суспензия фильтровалась, и осадок на фильтре промывался ацетонитрилом, затем ацетоном и наконец эфиром. Данное вещество сушилось в вакууме, давая целевое соединение (80.0 г, 81%) в виде ярко-желтого твердого вещества, т. пл. 182oC (разл.). Маточная жидкость концентрировалась, а затем разбавлялась водой, и получающееся твердое вещество собиралось, промывалось и сушилось, как указано выше, давая еще один сбор продукта (7.0 г) в виде более темно-желтого твердого вещества. Общий выход = 87 г (89%); ИК (KBr) 3330, 1767, 1732 см-1;
1H ЯМР (ДМСО-d6, 200 МГц) δ 11.64 (с., 1H), 8.63 (д., J = 2,2 Гц, 1H), 7.96 (дд. , J = 9.0, 2.2 Гц, 1H), 7.49 (д., J = 9.0 Гц, 1H), 7.33 (д., J = 2.2 Гц, 1H), 4.84 (т., J = 4.6 Гц, 1H), 3.45 (д., J = 4.5 Гц, 2H), 1.78 (с., 3H), 1.55 (с., 3H).
Анализ:
Вычислено для C15H14N2O6: C 56.60 H 4.43 N 8.80.
Найдено: C 56.62 H 4.41 N 8.91.
1D. S. Farlow, M.E. Flaugh, S.D. Horvath, E.R. Lavignino, P. Pranc, Org. Prep. Proc. Int. 1981, 13, 39.
Пример 2
Этил 5-нитро-3-(1H-индод)пропионат
К раствору [5-(5-нитроиндол-3-ил)-метил]-2,2-диметил- 1,3-диоксан-4,6-диона (10.0 г, 0.031 моля) в смеси пиридина (80 мл) и абсолютного этанола (20 мл) добавлялось 0.1 г медного порошка, и смесь нагревалась до дефлегмации в атмосфере аргона в течение 2 часов. Охлажденная смесь фильтровалась, и фильтрат выпаривался. Получающийся в результате остаток растирался со смесью эфир-дихлорметан, давая целевое соединение (7.3 г, 89%) в виде твердого вещества, т. пл. 118-121oC; ИК (KBr) 3330, 1730 см-1;
1H ЯМР (ДМСО-d6, 200 МГц) δ 11.59 (шир.с., 1H), 8.53 (д., J = 2.2 Гц, 1H), 7.97 (дд., J = 9.0, 2.3 Гц, 1H), 7.49 (д., J = 9.0 Гц, 1H), 7.40 (д., J = 2.1 Гц, 1H), 4.03 (кв., J = 7.1 Гц, 2H), 3.02 (т., J = 7.4 Гц, 2H), 2.67 (т., J = 7.4 Гц, 2H), 1.13 (т., J = 7.1 Гц, 3H).
Пример 3
3-(3-гидроксипропил)-5-нитро-1H-индол
К суспензии 95% литийалюминийгидрида (2.20 г, 0.058 моля) в 60 мл сухого ТГФ добавлялся раствор этил 5-нитро-3-индолпропионата (7.30 г, 0.028 моля) в 100 мл сухого ТГФ, при 0oC в атмосфере аргона. После перемешивания в течение 20 мин смесь гасилась осторожным добавлением 3 мл воды. Получающаяся в результате суспензия перемешивалась в течение 10 мин, а затем она фильтровалась, и осадок на фильтре промывался дополнительным ТГФ. Фильтрат выпаривался, и остаток брался в эфир, сушился (сульфатом натрия) и выпаривался, и получающееся в результате твердое вещество растиралось с гексаном, давая целевое соединение (4.30 г, 70%) в виде желтого твердого вещества, т. пл. 107-110oC, ИК (KBr) 3480, 3180, 1625 см-1;
1H ЯМР (CDCl3, 200 МГц)  8.60 (д., J = 2.1 Гц, 1H), 8.35 (шир.с., 1H), 8.11 (дд. , J = 9.0, 2.2 Гц, 1H), 7.38 (д., J = 8.9 Гц, 1H), 7.16 (м., 1H), 3.75 (т. , J = 6.2 Гц, 2H), 2.91 (т., J = 7.2 Гц, 2H), 2.07-1.93 (м., 2H), 1.37 (шир.с., 1H).
8.60 (д., J = 2.1 Гц, 1H), 8.35 (шир.с., 1H), 8.11 (дд. , J = 9.0, 2.2 Гц, 1H), 7.38 (д., J = 8.9 Гц, 1H), 7.16 (м., 1H), 3.75 (т. , J = 6.2 Гц, 2H), 2.91 (т., J = 7.2 Гц, 2H), 2.07-1.93 (м., 2H), 1.37 (шир.с., 1H).
Пример 4a
3-(3-Бромпропил)-5-нитро-1H-индол
К раствору трифенилфосфина (6.70 г, 0.025 моля) в 80 мл ацетонитрила добавлялся раствор 3-(3-гидроксипропил)-5-нитро- 1H-индола (пример 3) (4.30 г, 0.020 моля) в 75 мл ацетонитрила с последующим добавлением раствора четырехбромистого углерода (8.00 г, 0.027 моля) в 25 мл ацетонитрила, при 0oC в атмосфере аргона. Смесь перемешивалась при комнатной температуре в течение 3 часов, а затем она выпаривалась, и остаток хроматографировалоя (двуокись кремния/этилацетат-гексан, 1:9 затем 1:4), давая целевое соединение (4.60 г, 84%) в виде твердого вещества, т. пл. 92-95oC; ИК (чистый) 3420, 1330 см-1;
1H ЯМР (CDCl3, 200 МГц) δ 8.59 (д., J = 2.1 Гц, 1H), 8.40 (шир.с., 1H), 8.13 (дд. , J = 9.0, 2.2 Гц, 1H), 7.40 (д., J = 9.1 Гц, 1H), 7.21 (д., J = 2.2 Гц, 1H), 3.45 (т., J = 6.4 Гц, 2H), 2.99 (т., J = 7.2 Гц, 2H), 2.26 (м., 2H).
Пример 4b
3-(3-Иодпропил)-5-нитро-1H-индол
Раствор 3-(3-гидроксипропил)-5-нитро-1H-индола (1.13 г, 5.06 ммоля) в 20 мл ацетонитрила охлаждался до 0oC и обрабатывался последовательно триэтиламином (1.05 г, 7.59 ммоля) и метансульфонилхлоридом (0.43 мл, 5.6 ммоля), и смесь перемешивалась в течение 30 мин. Реакционная смесь гасилась 30 мл воды, и органическое вещество экстрагировалось в этилацетат. Органические экстракты сушились (сульфатом магния) и концентрировались в вакууме. Сырой остаток растворялся в 20 мл ацетонитрила, содержащего KI (1.7 г, 10.1 ммоля) и нагревался до дефлегмации в течение 3 часов. Реакционная смесь охлаждалась и растворитель удалялся в вакууме. Остаток растворялся в 100 мл этилацетата и промывался водой. Этилацетатный слой сушился (сульфатом магния) и концентрировался, а остаток очищался с помощью флэш-хроматографии на колонке (20% этилацетат в гексанах), давая целевое соединение (1.37 г, 4.20 ммоля, 83%) в виде желтого твердого вещества: т. пл. 95-98oC;
1H ЯМР (ДМСО-d6, 300 МГц) δ 8.53 (д., J = 2.3 Гц, 1H), 7.97 (дд., J = 2.3, 9.0 Гц, 1H), 7.51 (д., J = 9.0 Гц, 1H), 7.43 (с., 1H), 3.30 (т., J = 6.7 Гц, 2H), 2.84 (т., J = 7.7 Гц, 2H), 2.11 (м., 2H);
ИК (KBr) 1330, 1510, 810 см-1;
MC (m/e) 330 (M+).
Анализ:
Вычислено для C11H11IN2O2: C 40.02 H 3.36 N 8.48.
Найдено: C 40.26 H 3.27 N 8.51.
Пример 5a
3-[3-[4-(5-Метокси-4-пиримидил)-1-пиперазинил]пропил]-5- нитро-1H-индол
Смесь 5-нитро-3-(3-бромпропил)индол (0.57 г, 2.0 ммоля), 1-(5-метокси-4-пиримидил)пиперазина (0.47 г, 2.4 ммоля), KI (0.40 г, 2.4 ммоля) и диизопропилэтиламина (1.75 мл, 10.0 ммолей) в 20 мл ацетонитрила нагревалась до кипения в атмосфере аргона в течение 6 часов. Охлажденная реакционная смесь разбавлялась этилацетатом и промывалась (водой, солевым раствором). Водная фаза подвергалась обратной экстракции дихлорметаном, и объединенная органическая фаза промывалась (водой, солевым раствором), сушилась (сульфатом натрия) и выпаривалась. Получающийся в результате остаток хроматографировался (двуокись кремния/метиленхлорид-метанол, 95: 5), давая твердое вещество, которое растиралось со смесью дихлорметан-гексан, давая целевое соединение (0.55 г, 70%) в виде желтого твердого вещества, т. пл. 163-166oC; ИК (KBr) 3440, 3175, 1578, 1320 см-1;
1H ЯМР (CDCl3, 200 МГц) δ 8.60 (д., J = 2.1 Гц, 1H), 8.47 (шир.с., 1H), 8.33 (с., 1H), 8.11 (дд., J = 9.0, 2.2 Гц, 1H), 7.89 (с, 1H), 7.38 (д., J = 9.0 Гц, 1H), 7.18 (д., J = 2.0 Гц, 1H), 3.86 (с., 3H), 3.8-3.9 (м., 4H), 2.86 (т., J = 7.4 Гц, 2H), 2.59 (т., J = 4.9 Гц, 4H), 2.50 (т., J = 7.5 Гц, 2H), 2.05-1.90 (м., 2H).
Анализ:
Вычислено для C20H24N6O3 ·H2O·CH2Cl2:
C 57.08 H 6.24 N 19.87.
Найдено: C 57.37 H 5.85 N 19.53.
Пример 5b
3-[3-[4-(3-Метокси-4-пиридинил)-1-пиперазинил] пропил]-5-нитро-1H-индол
Смесь 3-[3-иодпропил]-5-нитро-1H-индола (1.4 г, 4.2 ммоля), 1-(3-метокси-4-пиридинил)-пиперазина (0.98 г, 5.09 ммоля) и карбоната калия (1.4 г, 10.2 ммоля) в 30 мл ацетонитрила нагревалась до кипения в течение 4 часов. Реакционная смесь охлаждалась и перемешивалась в течение 12 часов. Растворитель удалялся, и остаток растворялся в этилацетате и воде, водный слой отделялся и экстрагировался этилацетатом. Органические экстракты сушились (сульфатом магния) и концентрировались, и смолообразный остаток очищался с помощью флэш хроматографии на силикагеле (5% метанол в дихлорметане в качестве элюента), давая 3-[3-[4-(3-метокси-4-пиридинил)-1- пиперазинил]пропил] -5-нитро-1H-индол (0.6 г, 36%) в виде желтого твердого вещества; ИК (KBr) 3600, 2400, 1600, 1520, 1330, 1250, 815 см-1;
1H ЯМР (ДМСО-d6, 300 МГц) δ 8.52 (д., J = 2.2 Гц, 1H), 8.10 (с., 1H), 8.02 (д. , J = 5.3 Гц, 1H), 7.96 (дд., J = 2.3, 9.0 Гц, 1H), 7.48 (д., J = 9.0 Гц, 1H), 7.42 (с., 1H), 6.81 (д., J = 5.4 Гц, 1H), 3.84 (с., 3H), 3.21 (шир. с., 4H), 3.07 (дд., J = 6.4, 14.7 Гц, 2H), 2.79 (т., J = 14.7 Гц, 2H), 2.66 (шир.с., 4H), 1.97 (м., 2H);
MC (m/е) 395 (M+).
Пример 5c
3-[3-[4-(2-Пиридинил)-1-пиперазинил]пропил]-5-нитро-1-индол
Смесь 3-[3-бромпропил]-5-нитро-1H-индола (1.4 г, 4.2 ммоля), 1-(2-пиридинил)-пиперазина, (0.98 г, 5.09 ммоля) и карбоната кадия (1.4 г, 10.2 ммоля) в 30 мл ацетонитрила нагревалась до кипения в течение 4 часов. Реакционная смесь охлаждалась, растворитель удалялся, и остаток растворялся в этилацетате и воде. Водный слой отделялся и экстрагировался этилацетатом. Органические экстракты сушились (над сульфатом магния) и концентрировались, и смолообразный остаток очищался с помощью флэш хроматографии на силикагеле (5% метанол в дихлорметане), давая 3-[3-[4-(2-пиридинил)-1- пиперазинил]пропил] -5-нитро-1H-индол, (0.6 г, 36%) в виде желтого твердого вещества; ИК (KBr) 3182, 1520, 1330 см-1;
1H ЯМР (ДМСО-d6, 300 МГц) δ 8.52 (д., J = 2.24 Гц, 1H), 8.80 (дд., J = 1.8, 4.8 Гц, 1H), 7.95 (дд., J = 2.25, 9.0 Гц, 1H), 7.52-7.49 (м., 2H), 7.41 (с., 1H), 6.79 (д., J= 8.6 Гц, 1H), 6.60 (т., J = 6.6 Гц, 1H), 3.46 (т., J = 4.7 Гц, 4H), 2.78 (т., J = 7.4 Гц, 2H), 2.42 (т., J = 5.0 Гц, 4H), 2.34 (т., J = 6.9 Гц, 4H), 1.82 (дт., J = 7.4, 6.9 Гц, 2H);
MC (m/e) 365 (M+).
Анализ:
Вычислено для C20H23N5O2: C 65.73 H 6.34 N 19.16.
Найдено: C 65.35 H 6.26 N 18.87.
Пример 5d
3-[3-[(3-Метокси-2-пиридинил)-1-пиперазинил]пропил]-5-нитро-1H-индол
Смесь 3-(3-бромпропил)-5-нитро-1H-индола (0.88 г, 3.11 ммоля), карбоната калия (0.43 г, 3.11 ммоля), йодистого калия (0.52 г, 3.11 ммоля) и 1-(3-метокси-2-пиридинил)пиперазина (0.60 г, 3.11 ммоля) в 50 мл ацетонитрила нагревалась до кипения в течение 5 часов. Смесь охлаждалась, фильтровалась и концентрировалась. Остаток очищался с помощью флэш хроматографии на колонке с использованием 5% метанола в дихлорметане в качестве элюента, давая целевое соединение (1.2 г, 99%) в виде желтой пены; ИК (KBr) 3300, 1520, 1330, 1240 см-1;
1H ЯМР (ДМСО-d6, 300 МГц) δ 8.54 (д., J = 2.2 Гц, 1H), 7.97 (дд., J = 2.2, 9.0 Гц, 1H), 7.77 (м., 1H), 7.50 (д., J = 9.0 Гц, 1H), 7.44 (с., 1H), 7.24 (д., J = 7.75 Гц, 1H), 6.90 (м., 1H), 3.78 (с., 3H), 3.33 (шир.с., 2H), 2.80 (т., J = 7.3 Гц, 2H), 1.93 (м., 2H);
MC (m/е) 395 (M+).
Пример 6
3-[3-[4-(5-Метокси-4-пиримидил)-1-пиперазинил]пропил]-5- амино-1H-индол
К раствору 3-[3-[4-(5-метокси-4-пиримидил)-1-пиперазинил] пропил]-5-нитроиндола (0.550 г, 1.39 ммоля) в смеси этанола (120 мл) и ТГФ (40 мл) добавлялся 10% палладий на активированном угле (0.30 г), и смесь гидрировалась в шейкере Парра при 40 фунт./кв. дюйм (2.812 кг/см2) в течение 18 часов. Смесь затем фильтровалась через целит, и катализатор промывался дополнительным количеством смеси этанол-ТГФ. Выпаривание фильтрата дало по существу чистое целевое соединение (0.557 г, 100%) в виде коричневой пены. Образец данного материала (0.143 г) обрабатывался избытком метанольного HCl, и получающийся в результате раствор разбавлялся ацетоном, давая осадок. Осадок отфильтровывался, а затем кристаллизовался из этанола, давая 0.100 г пурпурноватого твердого вещества, т. пл. 192oC (разл); ИК (KBr) 3410, 3200, 1630, 1540 см-1;
1H ЯМР (ДМСО-d6, 200 МГц) δ 11.22 (шир.с., 1H), 10.20 (шир.с., 1H), 8.60 (м. , 1H), 8.20 (с., 1H), 7.55 (д., J = 1.6 Гц, 1H), 7.45 (д., J = 8.6 Гц, 1H), 7.35 (д., J = 2.1 Гц, 1H), 7.07 (дд., J = 8.6, 1.9 Гц, 1H), 4.89-4.82 (м., 2H), 3.91 (с., 3H), 3.8-3.0 (шир.м., 8H), 2.76 (м., 2H).
Анализ:
Вычислено для C20H26N6O·4HCl·H2O:
C 45.29 H 6.08 N 15.85.
Найдено: C 45.32 H 5.97 N 15.59.
Пример 7
4-Метил-5-амино-3-(3-гидроксипропил)индол
A. 4-Метилиндол
Смесь 3-нитро-о-ксилола (13.4 мл, 0.1 моля), диметилформамид-диметилацеталя (40 мл, 0.3 моля) и пирролидина (10 мл, 0.12 моля) в 200 мл сухого ДМФ нагревалась при 120-130oC (температура масляной бани) в атмосфере аргона в течение 21 часа. Охлажденная смесь выливалась в холодную воду (400 мл) и экстрагировалась эфиром (4x200 мл). Эфирный раствор промывался (водой, 4x100 мл), сушился (сульфатом натрия) и выпаривался, давая темно-красное вязкое масло. Данное масло бралось в 150 мл этилацетата, добавлялось 1.5 г 10% палладия на активированном угле, и смесь гидрировалась при 50 фунт/кв.дюйм (3.515 кг/см2) в шейкере Парра в течение 1 часа. Реакционная смесь затем фильтровалась, катализатор промывался дополнительным количеством этилацетата; и фильтрат выпаривался, давая темно-пурпурное масло. Флэш хроматография (двуокись кремния/дихлорметан-петролейный эфир, 1:1) данного масла давала чистый 4-метилиндол (8.85 г, 68%) в виде светлого желто-коричневого масла;
1H ЯМР (ДМСО-d6, 200 МГц) δ 11.04 (шир.с., 1H), 7.29 (т., J = 2.7 Гц, 1H), 7.21 (д. , J = 7.7 Гц, 1H), 6.96 (т., J = 7.2 Гц, 1H), 6.75 (дт., J = 6.9, 0.8 Гц, 1H), 6.43 (м., 1H), 2.46 (с., 3H).
B. 1-Ацетил-4-метилиндолин
К раствору 4-метилиндола (7.433 г, 0.0567 моля) в 100 мл ледяной уксусной кислоты добавлялся NaCNBH3 (7.25 г, 0.12 моля) порциями на протяжении 1.5 часов. Реакционная смесь затем концентрировалась в вакууме, добавлялась вода, и раствор подщелачивался 10 н. NaOH. Получающаяся смесь экстрагировалась этилацетатом (x3), и органический экстракт промывался (солевым раствором), сушился (сульфатом натрия) и выпаривался, давая масло. Флэш хроматография (двуокись кремния/этилацетат-гексан, 1:4) данного масла давала чистый 4-метилиндолин (6.962 г, 92%) в виде масла:
1H ЯМР (ДМСО-d6, 200 МГц) δ 6.78 (т., J = 7.6 Гц, 1H), 6.33 (д., J = 7.4 Гц, 1H), 6.30 (д., J = 7.6 Гц, 1H), 5.36 (шир.с., 1H), 3.38 (т., J = 8.5 Гц, 2H), 2.81 (т., J = 8.5 Гц, 2H), 2.11 (с., 3H).
Получающееся масло (6.945 г, 0.0522 моля) бралось в 10 мл уксусного ангидрида. Происходила экзотермическая реакция, и спустя 15 мин, смесь затвердевала. Летучие вещества впоследствии удалялись в вакууме, давая твердое вещество.
Растирание данного материала с эфиром давало 6.317 г 1-ацетил-4-метилиндолина в виде белого кристаллического твердого вещества, т. пл. 110-111oC. Выпаривание супернатанта и растирание получающегося в результате остатка с гексаном давали дополнительно 2.191 г чистого продукта. Общий выход = 8.508 г (93%); ИК (чистый) 1649 см-1;
1H ЯМР (ДМСО-d6, 200 МГц) δ 7.86 (д., J = 7.9 Гц, 1H), 7.03 (т., J = 7.7 Гц, 1H), 6.80 (l., J = 7.5 Гц, 1H), 4.08 (т., J = 8.5 Гц, 2H), 3.03 (т., J = 8.5 Гц, 2H), 2.18 (с., 3H), 2.13 (с., 3H).
Анализ:
Вычислено для C11H13NO: C 75.39 H 7.48 N 8.00.
Найдено: C 75.41 H 7.53 N 7.95.
C. 4-Метил-5-нитроиндолин
Раствор 1-ацетил-4-метилиндолина (8.260 г, 0.0372 моля) в 50 мл концентрированной серной кислоты охлаждался при 5oC, а затем по каплям добавлялась азотная кислота так, чтобы внутренняя температура поддерживалась при 5-10oC. После того, как добавление завершалось, смесь хранилась при той же температуре в течение 15 мин, а затем выливалась в 500 мл дробленого льда, и получающаяся суспензия перемешивалась до тех пор, пока лед не растаял. Суспензия затем фильтровалась, осадок на фильтре промывался водой, и остаток брался в дихлорметан. Органическая фаза отделялась, а водная фаза повторно экстрагировалась дихлорметаном (x3). Объединенная органическая фаза сушилась (сульфатом натрия) и выпаривалась, давая темно-желтое твердое вещество. Хроматография (9x10 см двуокись кремния/дихлорметан, затем дихлорметан-ацетонитрил, 95: 5) данного твердого вещества давала неразделимую смесь 1-ацетил-4-метил-5-нитроиндолина и 1-ацетил-4-метил-7-нитроиндолина (8.090 г, 78%) в соотношении приблизительно 9:1:
1H ЯМР (ДМСО-d6, 200 МГц) δ 7.98 (д., J = 8.9 Гц, 0.88H), 7.89 (д., J = 8.9 Гц, 0.88H), 7.54 (д., J = 8.3 Гц, 0.12 H), 7.03 (д., J = 8.3 Гц, 0.12H), 4.18 (т., J = 8.7 Гц, 2H), 3.14 (т., J =8.7 Гц, 2H), 2.37 (с., 3H), 2.19 (с. , 3H).
К суспензии 1-ацетил-4-метил-5(7)-нитроиндолина (8.049 г, 0.0366 моля) в 75 мл метанола добавлялось 25 мл щелочи Кляйзена (см. Fieser и Fieser, Reagents for Organiс synthesis, том 1, стр. 153), и получающаяся смесь подогревалась на паровой бане до тех пор, пока она не становилась гомогенной. Охлажденная реакционная смесь концентрировалась, а затем она разбавлялась водой, и получающаяся суспензия фильтровалась, давая оранжево-коричневое твердое вещество. Фильтрат экстрагировался дихлорметаном (x3), и органический экстракт сушился (сульфатом натрия) и выпаривался, давая твердое вещество. Объединенные твердые вещества хроматографировались (двуокись кремния/эфир-гексан, 1:1, затем хлороформ), давая две фракции. Фракция 1 бралась в эфир, и раствор обрабатывался обесцвечивающим древесным углем, фильтровался (целит) и выпаривался, давая 4-метил-7-нитроиндолин (0.575 г, 9%) в виде темно-оранжевого твердого вещества, т. пл. 125-127oC; ИК (KBr) 3395, 1623, 1596 см-1;
1H ЯМР (ДМСО-d6, 200 МГц) δ 7.83 (шир.с., 1H), 7.55 (д., J = 8.8 Гц, 1H), 6.36 (д., J = 8.8 Гц, 1H), 3.75 (т., J = 8.6 Гц, 1H), 2.99 (т., J = 8.6 Гц, 2H), 2.15 (с., 3H).
Анализ:
Вычислено для C9H10N2O2: C 60.66 H 5.66 N 15.72.
Найдено: C 60.99 H 5.71 N 15.48.
Фракция 2 повторно хроматографировалась (хлороформ), давая твердое вещество, которое растиралось с эфиром, давая 4-метил-5-нитроиндоил (4.813 г, 74%) в виде оранжевого кристаллического твердого вещества, т. пл. 160-170oC; ИК (KBr) 3330, 1598 см-1;
1H ЯМР (ДМСО-d6, 200 МГц) δ 7.85 (д., J = 8.8 Гц, 1H), 7.04 (шир.с., 1H), 6.33 (д., J = 8.8 Гц, 1H), 3.63 (т., J = 8.8 Гц, 1H), 2.98 (т., J = 8.8 Гц, 2H), 2.38 (с., 3H).
Анализ:
Вычислено для C9H10N2O2: C 60.66 H 5.66 N 15.72.
Найдено: C 60.66 H 5.47 N 15.74.
D. 4-Метил-5-нитроиндол
К суспензии 4-метил-5-нитроиндолина (4.767 г, 0.0268 моля) в 100 мл метанола добавлялся 2,3-дихлор-5,6-дициано- 1,4-бензохинон (6.697 г, 0.0295 моля) весь сразу, и получающаяся смесь перемешивалась при комнатной температуре в течение 1 часа. Реакционная смесь затем выпаривалась, и остаток брался в дихлорметан. Данный раствор затем промывался насыщенным водным бикарбонатом натрия (x4), сушился (сульфатом натрия) и выпаривался, давая твердое вещество. Кристаллизация данного вещества из смеси этилацетат-гексан (-20oC) давала 4.161 г целевого соединения в виде зеленовато-золотых игл, т. пл. 179-180oC. Хроматография маточной жидкости (двуокись кремния/этилацетат-гексан, 1: 1) давала дополнительно 0.417 г чистого продукта. Общий выход = 4.578 г (97%); ИК (KBr) 3318, 1604, 1585 см-1;
1H ЯМР (ДМСО-d6, 200 МГц) δ 11.02 (шир.с., 1H), 7.81 (д., J =9.0 Гц, 1H), 7.56 (т. , J = 2.8 Гц, 1H), 7.39 (д., J = 8.9 Гц, 1H), 6.79 (м., 1H), 2.75 (c., 3H).
Анализ:
Вычислено для C9H8N2O2: C 61.35 H 4.58 N 15.90.
Найдено: C 61.32 H 4.40 N 15.96.
E. 5-(4-Метил-5-нитроиндол-3-илметил)-2,2-диметил-1,3- диоксан-4,6-дион
Использовалась процедура Flaugh. Так, раствор 4-метил-5-нитроиндола (0.880 г, 5.00 ммолей), кислоты Meldrum (0.864 г, 6.00 ммолей), 37% водного формальдегида (0.5 мл, 6.0 ммолей) и D,L-пролина (0.029 г, 0.25 ммоля) в 25 мл ацетонитрила перемешивался при комнатной температуре в течение 72 часов. Получающаяся в результате желтая суспензия хранилась при -20oC, а затем холодная смесь фильтровалась. Осадок на фильтре промывался холодным ацетонитрилом и эфиром, а затем он сушился в вакууме, давая целевое соединение (1.055 г, 64%) в виде канареечно-желтого твердого вещества, т. пл. 196-198oC (разл.); ИК (KBr) 3338, 1782, 1742 см-1;
1H ЯМР (ДМСО-d6, 200 МГц) δ 11.46 (шир.с., 1H), 7.61 (д., J = 8.9 Гц, 1H), 7.32 (д., J = 8.9 Гц, 1H), 7.25 (д., J = 2.4 Гц, 1H), 4.74 (т., J = 5.0 Гц, 1H), 3.64 (д., J = 4.9 Гц, 1H), 2.80 (с., 3H), 1.84 (с., 3H), 1.69 (с., 3H).
Анализ:
Вычислено для C16H16N2O6: C 57.83 H 4.85 N 8.43.
Найдено: C 57.42 H 4.68 N 8.52.
F. Этил 4-метил-5-нитро-3-индолпропионат
К раствору 5-[(4-метил-5-нитроиндол-3-ил)метил]-2,2-диметил-1,3- диоксан-4,6-диона (1.009 г, 3.04 ммоля) в смеси пиридина (18 мл) и абсолютного этанола (2 мл) добавлялось 0.05 г медного порошка, и смесь нагревалась до кипения в атмосфере аргона в течение 2 часов. Охлажденная смесь фильтровалась, и фильтрат выпаривался в вакууме, давая вязкое коричневое масло. Данное вещество бралось в этилацетат, и раствор промывался (1 н. HCl, насыщенным водным хлоридом аммония, солевым раствором); сушился (сульфатом натрия) и выпаривался, давая желтое твердое вещество. Растирание с эфиром давало 423 мг целевого соединения в виде рыжеватого коричневого твердого вещества. Дополнительные 166 мг продукта могло быть выделено с помощью выпаривания супернатанта и повторного растирания с эфиром. Общий выход = 671 мг (80%). Аналитический образец кристаллизовался из смеси этилацетат-гексан, давая рыжевато-коричневые кристаллы, т. пл. 105-106oC; ИК (KBr) 3340, 1717, 1517, 1335 см-1;
1H ЯМР (ДМСО-d6, 200 МГц) δ 11.47 (шир.с. 1H), 7.63 (д., J = 9.0 Гц, 1H), 7.30 (д., J = 8.6 Гц, 1H), 7.28 (с., 1H), 4.06 (кв., J = 7.1 Гц, 2H), 3.18 (т. , J = 7.1 Гц, 2H), 2.77 (с., 3H), 2.68 (м., 2H), 1.16 (т., J = 7.1 Гц, 3H).
Анализ:
Вычислено для C14H16N2O4: C 60.86 H 5.84 N 10.14.
Найдено: C 60.76 H 5.74 N 10.00.
G. 4-Метил-5-нитро-3-(3-гидроксипропил)индол
К суспензии 95% литийалюминийгидрида (0.378 г, 9.44 ммоля) в 10 мл сухого ТГФ добавлялся раствор этил 4-метил-5-нитро-3-индолпропионата (0.650 г, 2.36 ммоля) в 2 мл сухого ТГФ, при 0oC в атмосфере аргона. Спустя 5 мин, охлаждающая баня удалялась, и перемешивание продолжалось при комнатной температуре в течение 30 мин. Реакционная смесь затем гасилась последовательным добавлением 0.4 мл воды, 0.4 мл 15% водного NaOH и, наконец, 1.2 мл воды. Получающаяся в результате суспензия разбавлялась этилацетатом, а затем фильтровалась, и осадок на фильтре промывался дополнительным количеством этилацетата. Фильтрат выпаривался, а остаток хроматографировался (двуокись кремния/дихлорметан-этилацетат, 2: 1), давая целевое соединение (0.458 г, 83%) в виде твердого вещества. Аналитический образец кристаллизовался из этилацетата, давая ацетат-гексан, давая желто-оранжевые иглы, т. пл. 129- 130oC; ИК (KBr) 3543, 3210, 1616, 1520, 1330 см-1;
1H ЯМР (ДМСО-d6, 200 МГц) δ 11.43 (шир. с., 1H), 7.63 (д., J = 8.9 Гц, 1H), 7.30 (д. , J = 8.7 Гц, 1H), 7.29 (с., 1H), 4.51 (т., J = 5.2 Гц, 1H), 5.20 (дт. , J = 6.2, 5.4 Гц, 2H), 2.91 (т., J = 7.7 Гц, 2H), 2.78 (с., 3H), 1.78 (м., 2H).
Анализ:
Вычислено для C12H14N2O3: C 61.52 H 6.02 N 11.96.
Найдено: C 61.23 H 5.85 N 11.90.
H. 4-Метил-5-амино-3-(3-гидроксипропил)индол
К раствору 4-метил-5-нитро-3-(3-гидроксипропил)индола (0.365 г, 1.56 ммоля) в 20 мл абсолютного этанола добавлялся 10% палладий на угле (0.150 г), и смесь гидрировалась в шейкере Парра при 50 фунт./кв.дюйм (3.515 кг/см2) в течение 0.5 часа. Смесь затем фильтровалась через слой целита, катализатор промывался дополнительно этанолом, и фильтрат выпаривался, давая целевое соединение (0.280 г, 88%) в виде твердого вещества. Аналитический образец кристаллизовался из этилацетата, давая окрашенные в кремовый цвет иглы, т. пл. 141-142oC; ИК (KBr) 3388, 3180, 1618 см-1;
1H ЯМР (ДМСО-d6, 200 МГц) δ 10.20 (шир.с., 1H), 6.87 (д., J = 8.8 Гц, 1H), 6.83 (с. , 1H), 6.50 (д., J = 8.3 Гц, 1H), 4.43 (т., J = 5.2 Гц, 1H), 4.12 (с. , 2H), 3.47 (дт., J = 6.4, 5.3 Гц, 2H), 2.79 (т., J = 7.7 Гц, 2H), 2.31 (с., 3H), 1.73 (м., 2H).
Анализ:
Вычислено для C12H16N2O: С 70.55 H 7.90 N 13.72.
Найдено: C 70.41 H 7.89 N 13.55.
Пример 8
3,4-Диметокси-1,2,5-тиадиазол-1,1-диоксид
Раствор 3,4-диметокси-1,2,5-тиадиазола (1.48 г, 10.1 ммолей) [полученного согласно процедуре, описанной в J. Org. Chеm., 40, 2749 (1975)] в 20 мл хлороформа добавлялся на протяжении периода 1 мин к перемешиваемому раствору м-хлорнадбензойной кислоты (4.11 г, 20.3 ммолей, 85% анализ) в 60 мл хлороформа. После перемешивания при температуре окружающей среды в течение 1 часа, смесь нагревалась до температуры дефлегмации в течение 8 часов, а затем перемешивалась при температуре окружающей среды в течение 1 часа. Реакционная смесь экстрагировалась водным раствором бикарбоната натрия и водой, и органическая фаза сушилась над сульфатом натрия, фильтровалась и выпаривалась при пониженном давлении. Остаток обрабатывался метанолом и фильтровался, давая 1.03 г продукта. Перекристаллизация из метанола давала целевое соединение, т. пл. 200-202oC.
Анализ:
Вычислено для C4H6N2O4S:
С 26.97 H 3.39 N 15.72 S 18.00.
Найдено: С 26.82 H 3.18 N 16.09 S 18.00.
Пример 9
3.4-Диметокси-1,2,5-тиадиазол-1-оксид
Раствор диметилоксальдиимидата(4.0 г, 34.5 ммоля) и пиридина (5.71 мл, 5.58 г, 70.6 ммолей) в 8 мл метиленхлорида добавлялся по каплям к холодному раствору тионилхлорида (2.61 мл, 4.25 г, 34.7 ммолей) в 18 мл метиленхлорида под струей азота с такой скоростью, чтобы температура реакции поддерживалась между 0oC и 15oC. После перемешивания при температуре окружающей среды в течение 20 мин реакционная смесь промывалась двумя 11 мл порциями водного 0.055 н. HCl. Водная фаза экстрагировалась двумя 20 мл порциями метиленхлорида, и объединенная органическая фаза сушилась и выпаривалась досуха при пониженном давлении. Твердый остаток перекристаллизовывался из изопропилового спирта, давая 3.0 г целевого соединения, т. пл. 137-139oC.
B. Продукты формулы I
1. Продукт формулы I-1 (X = SO, SO2)
Пример 10
3-[3-[4-(5-Метокси-4-пиримидил)-1-пиперазинил] пропил]-5-(1-оксо-4-метокси-1,2,5-тиадиазол-3-ил)аминоиндол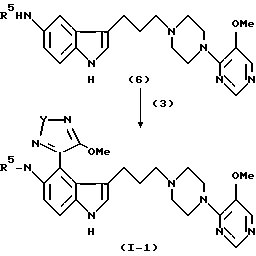
Раствор 3-[3-[4-(5-метокси-4-пиримидил)-1-пиперазинил] пропил]-5-аминоиндола (0.366 г, 1.0 ммоль) и 3,4-диметокси-1,2,5- тиадиазол-1-оксида (3) (0.162 г, 1.0 ммоль) (см. патент США 4374248) в 20 мл метанола перемешивался при комнатной температуре в течение 30 мин, а затем он нагревался до кипения в течение 4 часов. Охлажденная смесь выпаривалась, и остаток растирался с дихлорметаном, давая 0.323 г твердого вещества. Данный материал хроматографировался (двуокись кремния/метиленхлорид-метанол, 95:5, затем метиленхлорид-метанол-гидроокись аммония, 95: 4.5: 0.5), давая целевое соединение (0.150 г, 30%) в виде желтого твердого вещества, т. пл. 164oC (разл.); ИК (KBr) 3330 (шир.), 1605, 1580, 1130 см-1;
1H ЯМР (ДМСО-d6, 400 МГц) δ 10.85 (с., 1H), 10.38 (с., 1H), 8.23 (с., 1H), 8.18 (с., 1H), 8.02 (с., 1H), 7.57 (д., J = 8.7 Гц, 1H), 7.34 (д., J = 8.7 Гц, 1H), 7.17 (с., 1H), 4.17 (с., 3H), 3.83 (с., 3H), 3.68 (шир.с., 4H), 2.69 (т., J = 7.4 Гц, 2H), 2.37 (т., J = 7.4 Гц, 2H), 1.84 (м., 2H).
Анализ:
Вычислено для C23H28N8O3S· H2O:
C 53.68 H 5.88 N 21.78.
Найдено: C 53.87 H 5.88 N 21.63.
2. Продукт формулы I-2
Пример 11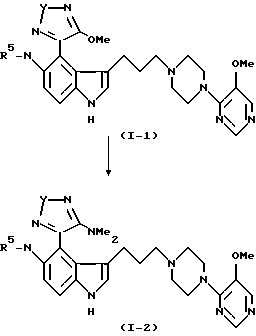
Безводный диметиламин барботировался в 50 мл абсолютного этанола при -10oC в течение приблизительно 30 мин. К данному холодному раствору добавлялся раствор 3-[3-[4-(5-метокси-4-пиримидил)-1-пиперазинил]пропил]-5- (1-оксо-4-метокси-1,2,5-тиадиазол-3-ил)аминоиндола (пример 8) (0.190 г, 0.38 ммоля), и получающийся в результате раствор перемешивался при комнатной температуре в течение 1 часа. Реакционная смесь затем выпаривалась, давая не совсем белое твердое вещество, которое хроматографировалось (двуокись кремния/метиленхлорид-метанол, 95: 5, затем метиленхлорид-метанол-гидроокись аммония 95:4.5:0.5 до 90:9:1), давая целевое соединение (0.130 г, 62%) в виде не совсем белого твердого вещества, т. пл. 150oC (разл.); ИК (KBr) 3310 (шир.), 1628, 1608, 1575 см-1;
1H ЯМР (ДМСО-d6, 200 МГц)  10.88 (шир.с., 0.5H), 9.88 (шир.с., 0.5H), 8.51 (шир. с. , 1H), 8.23 (с.,1H), 8.11 (с., 1H), 8.02 (с., 1H), 7.38 (с., 2H), 7.18 (с. , 1H), 3.83 (с., 3H), 3,70 (шир.с., 4H), 3.00 (с., 3H), 2.71 (м., 2H), 2.50 (м., 9H), 1.86 (м., 2H).
10.88 (шир.с., 0.5H), 9.88 (шир.с., 0.5H), 8.51 (шир. с. , 1H), 8.23 (с.,1H), 8.11 (с., 1H), 8.02 (с., 1H), 7.38 (с., 2H), 7.18 (с. , 1H), 3.83 (с., 3H), 3,70 (шир.с., 4H), 3.00 (с., 3H), 2.71 (м., 2H), 2.50 (м., 9H), 1.86 (м., 2H).
Анализ:
Вычислено для C24H31N9O2S·1,9H2O· 0.04CH2Cl2:
C 52.76 H 6.42 N 23.04.
Найдено: C 52.37 H 6.02 N 23.50.
Пример 12
3-[3-[4-(5-Метокcи-4-пиримидил)-1-пиперазинил] пропил] -5- (1-оксо-4-метиламино-1,2,5-тиадиазол-3-ил)аминоиндол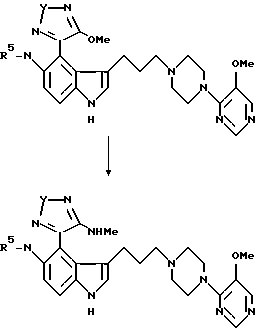
Безводный метиламин барботировался в 50 мл абсолютного этанола при -10oC в течение приблизительно 30 мин. К данному холодному раствору добавлялся 3-[3-[4-(5-метокси-4-пиримидил)-1- пиперазинил] пропил] -5-(1-оксо-4-метокси-1,2,5-тиадиазол-3- ил)аминоиндол (пример 8) (0.200 г, 0.40 ммоля), и получающийся раствор перемешивался при комнатной температуре в течение 3 часов. Реакционная смесь затем выпаривалась, давая твердое вещество, которое растиралось, давая по существу чистый продукт (0.120 г, 60%) в виде бледно-желтого твердого вещества. Данное вещество перекристаллизовывалось из водного ДМСО, давая целевое соединение (0.100 г, 47%) в виде бледно-желтого твердого вещества, т. пл. 150-158oC; ИК (KBr) 3310 (шир.), 1626, 1608, 1575 см-1;
1H ЯМР (ДМСО-d6, 200 МГц) δ 10.86 (шир.с., 1H), 9.84 (шир.с., 1H), 8.47 (шир. с. , 1H), 8.22 (с., 1H), 8.09 (с., 1H), 8.01 (с., 1H), 7.37 (с., 2H), 7.17 (с., 1H), 3.82 (с., 3H), 3.67 (шир.с., 4H), 3.00 (шир.с., 3H), 2.70 (м. , 2H), 2.54-2.33 (м., 6H), 1.83 (м., 2H).
Анализ:
Вычислено для C23H29N9O2S·1.3H2O· 0.15C2H6OS:
C 52.72 H 6.17 N 23.76.
Найдено: C 52.90 H 6.21 N 23.39.
Пример 13
3-[3-[4-(5-Метокси-4-пиримидил)-1-пиперазинил] пропил] -5-(1-оксо-4-амино-1,2,5-тиадиазол-3-ил)аминоиндол
Безводный аммиак барботировался в 40 мл абсолютного этанола при -10oC в течение приблизительно 15 мин. К данному раствору добавлялся раствор 3-[3-[4-(5-метокси-4-пиримидил)-1- пиперазинил] пропил] -5-(1-оксо-4-метокси-1,2,5-тиадиазол-3- ил)аминоиндола (пример 8) (0.175 г, 0.35 ммоля) в 10 мл абсолютного этанола, и получающийся раствор перемешивался при комнатной температуре в течение 1 часа. Реакционная смесь затем выпаривалась, давая желтую смолу. Данное вещество хроматографировалось (двуокись кремния/метиленхлорид-метанол- гидроокись аммония, 90:9:1), давая желтую смолу, которая впоследствии обрабатывалась избытком метанольного HCl. Получающийся раствор выпаривался, а остаток растирался с ацетоном, давая гидрохлорид целевого соединения (0.160 г, 76%) в виде желтого твердого вещества, т. пл. 158oC (разл.); ИК (KBr) 3380 (шир.), 3200 (шир.), 1628, 1574, 1543 см-1;
1H ЯМР (ДМСО-d6, 200 МГц) δ 10.0-11.3 (м., 2H), 8.64 (шир. с., 1H), 8.29 (шир. с., 1H), 8.20 (шир. с., 1H), 7.60 (м.,1H), 7.35 (д., J = 8.7 Гц, 1H), 6.96-7.22 (м. , 1H), 4.89 (м., 2H), 3.90 (с., 3H), 3.62 (м., 4H), 3.16 (м., 4H), 2.77 (м., 2H), 2.14 (м., 2H).
Анализ:
Вычислено для C22H27N9O2S·2HCl·2,2H2O· 0,2CH4O:
C 44.40 H 5.74 N 21.00.
Найдено: C 44.44 H 6.14 N 21.25.
C. Процедуры биологических испытаний
Пример 14
Исследования агонистов на латеральной подкожной вене собаки
Латеральная подкожная вена получалась от анестезированной собаки и очищалась от приставшего материала. Сосуд затем разрезался на кольцевые сегменты размером 2-3 мм и помещался между проволочками из нержавеющей стали в тканевых ваннах, содержащих 20 мл модифицированного буфера Кребса, которые непрерывно аэрировались 5% CO2/95% O2 и поддерживались при 37oC. Натяжение (тонус) в период покоя доводилось вручную до 1 г и поддерживалось до тех пор, пока не будет достигнута стабильная базовая линия в течение периода равновесия 1 час. Раствор тканевой ванны в течение данного равновесия заменяется каждые 15 мин.
В ванны добавляются кетансерин, атропин и пириламин в концентрации 1 мкМ для блокирования 5-HT2, холинеричеcкого и гистаминного эффектов. Спустя 15 мин с антагонистами кумулятивным образом проводится ответная кривая на концентрацию серотонина. В заключение ванны отмываются несколько раз, напряжение повторно доводится до 1 г, и ткани дают возможность возвратиться к состоянию равновесия на протяжении периода 45-60 мин. В ванны снова добавляются антагонисты, и через 15 мин для выбранных испытываемых соединений получаются ответные кривые концентрации. Отдельные сегменты сосуда подвергаются воздействию только одного испытываемого соединения.
Активность испытываемых соединений выражается в виде относительной активности и эффективности по сравнению с 5-HT (значение которого произвольно принято за 1,0) в таком же препарате сосудов.
Описываются новые 1,2,5-тиадиазольные производные индолилалкил-пиримидинил-пиперазинов общей формулы I, в которой R1 выбран из водорода, R2, R3 и R5 независимо выбраны из водорода, R4 представляет низший алкокси; R6 представляет амино, низший алкиламино, ди-низший алкиламино или низший алкокси; Х выбран из S, SO и SO2; Y и Z представляют азот, m выбран из нуля и целых чисел от 1 до 3 и n выбран из целых чисел 1-5. Соединения могут найти применение для использования в облегчении сосудистых головных болей. Описываются также фармацевтические композиции на их основе. 2 с. и 7 з.п.ф-лы.
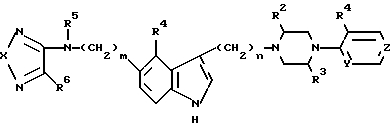
в которой R1 представляет водород;
R2, R3 и R5 представляют водород;
R4 представляет низший алкокси;
R6 представляет амино, низший алкиламино, ди-низший алкиламино или низший алкокси;
X выбран из S, SO и SO2;
Y и Z представляют азот;
m выбран из нуля или целых чисел от 1 до 3;
n выбран из целых чисел от 1 до 5,
или их фармацевтически приемлемые кислотно-аддитивные соли и/или сольваты.
| Огнеупорная бетонная смесь | 1973 |
|
SU464558A1 |
| Способ получения замещенных пирролов или их фармацевтически приемлемых солей | 1989 |
|
SU1799382A3 |
| Грузозахватное устройство для штучных грузов типа каменных блоков | 1974 |
|
SU496222A1 |
| Способ определения содержания водо-рода в металлах | 1974 |
|
SU508723A1 |

